

Abstract
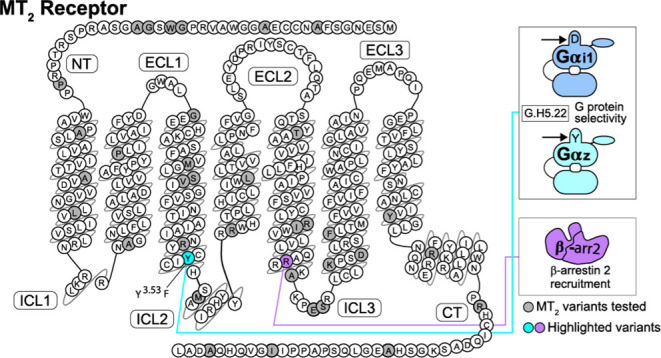
G protein-coupled receptors (GPCRs) can engage distinct subsets of signaling pathways, but the structural determinants of this functional selectivity remain elusive. The naturally occurring genetic variants of GPCRs, selectively affecting different pathways, offer an opportunity to explore this phenomenon. We previously identified 40 coding variants of the MTNR1B gene encoding the melatonin MT2 receptor (MT2). These mutations differently impact the β-arrestin 2 recruitment, ERK activation, cAMP production, and Gαi1 and Gαz activation. In this study, we combined functional clustering and structural modeling to delineate the molecular features controlling the MT2 functional selectivity. Using non-negative matrix factorization, we analyzed the signaling signatures of the 40 MT2 variants yielding eight clusters defined by unique signaling features and localized in distinct domains of MT2. Using computational homology modeling, we describe how specific mutations can selectively affect the subsets of signaling pathways and offer a proof of principle that natural variants can be used to explore and understand the GPCR functional selectivity.
Keywords: melatonin type 2 receptor, functional selectivity, β-arrestin recruitment, G protein activation, structure/activity analysis, clustering
G protein-coupled receptors (GPCRs) represent the largest family of proteins involved in signal transduction across biological membranes.1 GPCRs bind to many ligands acting as agonists, such as hormones, chemokines, neurotransmitters, ions, tastants, and odorants. Upon agonist binding in their ligand-binding pocket, GPCRs undergo conformational changes leading to, and stabilizing, active receptor conformations. In their active conformations, receptors can then engage diverse transducers, including hetero-trimeric G proteins that lead to the activation of downstream effectors such as enzymes and channels generating second messengers and changes in ion fluxes resulting in cellular responses. Once activated, GPCRs also become the substrates for various kinases including GPCR kinases leading to the recruitment of β-arrestins to the receptors. β-Arrestins uncouple the receptors from their G proteins and recruit adaptor proteins involved in receptor endocytosis.2 Ultimately, this leads to signaling arrest at the plasma membrane while promoting signaling from endosomal compartments.3 In recent years, different agonists, presumably stabilizing distinct receptor conformations, were found to differentially activate G protein subtypes and β-arrestins, a phenomenon known as biased signaling.4
Signal propagation from the ligand-binding pocket to the intracellular face of GPCRs is mediated by structural rearrangements of residue contacts in the transmembrane (TM)5 domains involving a redistribution of the water-mediated polar network interactions.6 This allows a large opening of the cytoplasmic side of TM6, increasing the accessibility of G proteins and β-arrestins to the receptor core. As this rearrangement is required for a proper signal transmission, any mutation impairing this process may result in a global change of receptor responsiveness.7−9
Whereas the GPCR conformational rearrangements involved in their general activation are well understood, the structural determinants underlying the selective engagement of specific downstream effectors are still not clear. The ability of ligands to promote the activation of some of the cognate downstream pathways engaged by a given GPCR but not others (i.e., ligand-biased signaling or functional selectivity) has led to the hypothesis that among the various possible receptor conformations that can be stabilized by ligands, some preferentially activate subsets of the signaling repertoire of a receptor.10,11 In a similar fashion to ligands, receptor mutations are also susceptible to stabilize certain conformational states over others and induce biased signaling. Naturally occurring mutations leading to biased signaling have been reported for several GPCRs: calcium-sensing receptor, glucagon-like peptide-1 receptor, angiotensin II type 1 receptor (AT1R), human thyroid stimulating hormone receptor, and melanocortin 3 and 4 receptors.12−18 It is reasonable to expect that the conformations stabilized by mutations that bias signaling are similar to those underlying ligand-biased signaling. However, the structural consequences of such mutations have not been fully explored.
Our group previously identified 40 non-synonymous MT2 variants in Europeans19 and assessed their signaling profiles by monitoring their ability to activate Gαi1, Gαz, and ERK1/2; to inhibit cAMP production; and to promote the recruitment of β-arrestin 2 (βarr2).20 The study revealed a significant association between melatonin-promoted activation of Gαi1 and Gαz, constitutive βarr2 recruitment, and the occurrence of type 2 diabetes.20 However, no analysis of how the mutations affecting distinct signaling pathways group functionally and structurally has been attempted. Here, using the data sets of the full signaling profiles of the 40 naturally occurring genetic variants, we utilized non-negative matrix factorization (NMF) of 13 parameters resulting in the clustering of the variants in 8 distinct groups. When overlaid on the MT2 structure, the mutations belonging to the same groups are not distributed randomly but rather point to mechanistic explanations of the functional selectivity of the variants.
Results
Functional Clustering of MT2 Variant Signaling Signatures Reveals Eight Different Profiles
For wild-type (WT) and MT2 variants, agonist-independent activation of Gαi1 and Gαz, and recruitment of βarr2, as well as concentration–response curves of melatonin-promoted Gαi1 and Gαz activation, inhibition of forskolin-simulated cAMP production, βarr2 recruitment, and ERK phosphorylation have been previously published.20 We used constitutive (or basal) activity (BA), maximal agonist-induced response (M), and the difference of transduction coefficient Δlog(τ/KA) (ΔTC) of βarr2 recruitment, Gαi1 and Gαz activation, as well as M and ΔTC of inhibition of cAMP production, and ERK activation (13 total signaling parameters) for the 40 variants (Table 1). To identify the commonalities among the signaling signatures, we used NMF21 and K-means clustering, providing similarity scores for each pair of variants (see Methods section for details). Two hundred matrices of sampled data were created (40 variants with 13 signaling parameters per variant) by randomly sampling for each signaling parameter a single value from the normal distribution. Using sparse NMF and unsupervised clustering using K-means, feature reduction was independently performed for K = 2 through K = 10. The matrix was deconstructed into two vectors (W and H). W has the dimensions of 40 mutations by k, and H has the dimension k by 13 signaling parameters. The clusters were assigned, and a clustering frequency matrix (f) was created for every K by calculating the similarity scores (how frequently any pair of mutations clustered together). This resulted in a clustering frequency matrix with values ranging from 0 (always clustered together; maximal similarity) to 1 (never clustered together; no similarity). The similarity scores were converted into a heat map in which low similarity scores (high similarity) are represented in blue/cyan and high similarity scores are represented in yellow/green (Figure 1). The variants were grouped together by a cluster, and the level of similarity between each pair of variants is also illustrated in a dendrogram (Figure 1).
Table 1. Signaling Data Used for Cluster Assignmenta,b.

Activation of Gαi1 and Gαz, cAMP inhibition, ERK phosphorylation, and βarr2 recruitment mediated by MT2 variants were monitored upon increasing concentrations of melatonin stimulation.
Data highlighted in red and green represent values significantly lower and higher than WT MT2, respectively. The following parameters were used: basal activity (BA), maximal agonist-induced response (M), and difference of transduction coefficient Δlog(τ/KA) (ΔTC). The values of BA are expressed as means, while M values were determined from the sigmoidal curves (nonlinear regression with a variable Hill slope shared with WT MT2). The ΔTC values correspond to the difference between the TC of MT2 variants and WT MT2. N/A denotes that the experimental parameter could not be determined due to the lack of a concentration–response curve. For M, data were analyzed by comparing the independent fits with a global fit that shares the selected parameter and for BA and ΔTC by one-sample t-test compared to WT MT2. Gαi1 and Gαz activation was monitored in quadruplicate, and ERK activation and cAMP inhibition were measured in triplicate, while βarr2 recruitment was monitored in duplicate. Data were considered different from WT MT2 (highlighted in red or green) if p < 0.05.
Figure 1.

NMF of MT2 variant signaling signatures. Heatmap of similarity scores between MT2 variants obtained by NMF for the eight clusters. Low similarity scores (high similarity) are represented in blue/cyan, and high similarity scores (low similarity) are represented in yellow/green. Dendrograms illustrate the level of similarity between each pair of variants.
By superimposing the signaling signatures of each variant, we can also appreciate the differences between the eight clusters (CL) (Figure 2a). The members of CL1, CL2, and CL3 (red, orange, and yellow, respectively, in Figure 2a) are characterized by a severe, moderate, and modest impairment of all signaling pathways, respectively. This suggests that the residues from these CLs are part of the amino acids involved in global signaling transmission and in controlling the transition between inactive and active receptor conformations. All these variants are located within the TM domains, except p.G109A3.21 (superscripts indicate the GPCRdb numbering scheme22) which is located at the interface between TM3 and the extracellular loop 1 and p.R316H8.51 in H8 (Figure 2b). In contrast, variants belonging to CL4 to CL7 are characterized by impairments of selective subsets of the receptor signaling repertoire. The CL4 (purple in Figure 2a,b) variants are mostly positioned in the intracellular parts of MT2, but the members of CL7 (green) are found in TM3 and TM5. The variants within CL4 selectively impair agonist-induced βarr2 recruitment with little or no impact on other pathways (Figure 2a,b). Interestingly, these variants are all localized within the third intracellular loop (ICL) and the cytosolic extensions of TM5 and TM6 (except p.L166I4.51 which is found in TM4), delineating this region as important for βarr2 recruitment. Impairment of basal and agonist-induced Gαz activation is the common feature of the three variants belonging to CL7 (Figure 2a,b). The members of CL5 and CL6 (light and dark blue, Figure 2a,b) are localized toward the intracellular parts of TM3 and TM4 and in the receptor carboxy-terminus and are impaired for agonist-induced Gαi1 activation (ΔTC) with heavy (CL5) or slight (CL6) defect of the basal recruitment of βarr2, respectively (Table 1 and Figure 2a,b). Finally, the members of CL8 (black, Figure 2a,b) do not impair the signaling and are all positioned outside the TM bundle, except p.A52T1.44 (Figures 1 and 2a,b).
Figure 2.

Eight phenotypes of MT2 variants and their localization in the receptor sequence. (a) Superimposition of signaling signatures sharing the same cluster for the eight clusters. Basal activities (BA), agonist-mediated maximal efficacies (M), and Δlog(τ/KA) ratios (ΔTC)51 were expressed as normalized differences from WT MT2. (b) Localization of the 40 MT2 variants color-coded according to their respective cluster (CL): red (CL1; severe global signaling impairment), orange (CL2; moderate global signaling impairment), yellow (CL3; slight global signaling impairment), purple (CL4; selective impairment of agonist-mediated βarr2 recruitment), light blue (CL5; selective impairment of Gαi1 activation and basal βarr2 recruitment), dark blue (CL6; Gαi1 activation selective impairment), green (CL7; Gαz activation selective impairment), and black (CL8; no signaling impairment). * indicates the four variants that lose the ability of binding melatonin. The snake plot was created using the tools at www.gpcrdb.org.
Genetic Variants in CL1 and CL2 Disrupt the Key MT2 Activation Switches (DRY, NPxxY, and ICL 4)
In CL1, the lack of responsiveness of p.A42P1.34, p.L60R1.52, p.P95L2.58, and p.Y308S7.53 is explained by a loss of agonist binding to MT2 as previously reported.19 However, four other variants in CL1 bind melatonin with a similar affinity to WT MT2 (p.R138C3.50, p.R138H3.50, p.R138L3.50, and p.S123R3.35) and, therefore, may disturb the overall signal transduction mechanism of the receptor. p.R1383.50 is part of the evolutionary-conserved and functionally crucial DRY motif and its mutation usually alters the function.23 Remarkably, melatonin receptors have a neutral asparagine instead of the conserved negatively charged residue at position 1373.49; still, the variants with mutations at p.R1383.50 are expected to exhibit a strongly distorted network of interactions around this functionally key residue switch. Regarding the p.S123R3.35 variant, the crystal structure of the agonist-bound human MT2 in the inactive state24 reveals that S1233.35 is in the vicinity of D862.50. These residues likely form part of a water-mediated polar network highly conserved in class A GPCRs6 (Figure 3a). Rearrangement of this network is essential for GPCR activation as it allows propagation of conformational changes from the orthosteric agonist-binding site to the G protein coupling interface.6 The p.S123R3.35 variant probably introduces a salt bridge with D862.50, thus preventing local structural rearrangements in this key region and therefore locking MT2 in an inactive state. To test this hypothesis, we introduced a negatively charged residue at position 1233.35 to disrupt the inactive-state interactions at this site. The S123E3.35 mutation partially rescues melatonin-mediated Gαz activation by allowing around 25% of WT MT2 maximal melatonin-induced Gαz activation (Figure 3b), supporting the proposed mechanism for the lack of signaling of the p.S123R3.35 variant. The variants p.I223T5.61 and p.F250V6.34 in CL2 lead to a moderate impairment of all signaling pathways. In the crystal structure of human MT2, these residues are packed against each other immediately below Y2205.58, a highly conserved residue in class A GPCRs involved in a water-mediated polar network involving the key functional residues R1383.50 of the DRY motif and Y3087.49 of the NPxxY motif25 (Figure 3a). Thus, the global loss of signaling in these variants could result from an alteration of the hydrophobic packing around Y2205.58 that disturbs this important activation switch. Finally, p.R222H5.60 and p.R316H8.51 are also part of the globally but moderately impaired variants in CL2 (Figure 2b). The crystal structure of human MT2 shows that R2225.60 and R3168.51 are located on the intracellular side of the receptor facing with membrane phospholipids and nearby key activation switches at the DRY motif and ICL4 (Figure 3c). To test the functional relevance of these sites, we introduced a glutamate to further impair interactions with the nearby residues. We observed that R222E5.60 and, particularly, R316E8.51 cause a significant reduction of the melatonin-mediated Gαz activation compared to p.R222H5.60 and p.R316H8.51, respectively (Figure 3d).
Figure 3.

Disruption of key MT2 activation switches by genetic variants characterized by a global signaling impairment. (a) Side view of the crystal structure of the agonist-bound human MT2 in the inactive state showing the location of conserved residues of the water-mediated polar network—depicted by red dots. S1233.35 (green) is in the vicinity of D862.50, while I2235.61 and F2506.34 (orange) are packed against each other below Y2205.58 and nearby the key functional residues R1383.50 of the DRY motif and Y3087.49 of the NPxxY motif. (b) Melatonin dose–response curves of Gαz activation by WT MT2 (black curve), MT2-S123R3.35 (dotted red curve), MT2-S123E3.35 (dotted blue curve), or in the absence of transfected receptor (Mock; dotted gray curve). (c) Side view of the crystal structure of MT2 showing R2225.60 (orange) and R3168.51 (green) facing the nearby DRY and NPxxY motifs and ICL4. (d) Melatonin dose–response curves of Gαz activation by WT MT2 (black curve), MT2-R222H5.60 or MT2-R316H8.51 (red dotted curve), and MT2-R222E5.60 or MT2-R316E8.51 (blue dotted curve). Each point represents the mean ± SEM of five independent experiments performed in quadruplicate (distinct samples). The statistical differences between melatonin-induced maximal responses (Emax) were assessed by comparing the best-fit values of top (Emax) (*p = 0.0109, **p = 0.0038, and ****p < 0.0001).
Role of ICL3 in βarr2 Recruitment
The CL4 cluster is exclusively composed of genetic variants selectively impaired in their ability to recruit βarr2 upon melatonin stimulation (Figure 2a, purple). Interestingly, most of these residues are located within the third ICL of MT2 or at the cytoplasmic extensions of TM5 and TM6 (Figure 2b, purple). Among them, R2315.69 is predicted to interact with serine 75 (S75) in the β-arrestin finger loop (Figure 4a), according to our model the human MT2/β-arr2 complex (see Methods section). The finger loop is a key domain interacting with the open cytoplasmic region of activated GPCRs26 and essential for β-arrestin recruitment, particularly in receptors having a relatively weak affinity for β-arrestin such as MT2.27 In the p.R231H5.69 variant, a shorter histidine may lead to an impaired interaction with S75 in βarr2. To verify this putative interaction, we mutated S75 in human βarr2 to alanine (βarr2-S75A) (Figure 4b). This mutation significantly reduces βarr2 recruitment to plasma membrane upon stimulation of MT2 with melatonin to a similar extent to the reduction previously reported in the MT2 p.R231H5.69 variant.
Figure 4.

Potential interaction between R2315.69 of MT2 and serine 75 in the finger loop of βarr2. (a) Side view of a homology model of the human MT2/βarr2 complex showing R2315.69 in MT2 cytoplasmic extension of TM5 interacting with serine 75 (S75) located in the finger loop of βarr2. (b) Melatonin dose–response curve of recruitment of human βarr2 or βarr2-S75A to the plasma membrane in the presence of MT2 or MT2-R231H5.69. Each point represents the mean ± SEM of five independent experiments performed in quadruplicate (distinct samples). Statistical differences between melatonin-induced maximal responses (Emax) were assessed by comparing the best-fit values of top (Emax) (*p < 0.05, p = 0.0138).
Role of Y1413.53 of CL5 in Selective Recognition between Gαi1 and Gαz
Variants within CL5 and CL6 are selectively impaired in their melatonin-induced Gαi1 activation (ΔTC) and, for CL5, also basal βarr2 recruitment (Figure 2a). The effects of these variants are remarkable because (except for A342V) they affect Gαi1—but not the related Gαz—signaling. The clearest example of this G protein bias is the p.Y141F3.53 variant in CL5, with the largest difference between the TCs for Gαi1 and Gαz (Figure 5a). In this variant, agonist-induced activation for Gαi1 is right-shifted and blunted compared to WT MT2 (Figure 5a, left panel), but the corresponding response for Gαz is not (Figure 5a, right panel), yielding a significantly different TC (ΔTC = −0.66 for Gαi1 and 0.19 for Gαz). These differences are also in line with the differences in the constitutive activation of Gαi1 and Gαz by p.Y141F3.53, which is reduced for Gαi1 but increased for Gαz (Figure 5b). According to our models of the human MT2/Gαi1 and MT2/Gαz complexes (see Methods section), Y1413.53 may establish different interactions with Gαi1 and Gαz. In these two G protein subtypes, the α-helical 5 (H5) region (the Gα protein region known to interact with the receptor core) differs by only four residues (Figure 6a, blue). Among them, the residue at position H5.22 (D in Gαi1 and Y in Gαz) can interact with Y1413.53 of MT2 (Figure 6b). In the Gαi1 complex, Y1413.53 could form a hydrogen bound with DH5.22, while in the Gαz complex, it could interact with the corresponding YH5.22 through aromatic interactions. Thus, we hypothesized that the hydrophobic mutation in the p.Y141F3.53 variant would impair the interaction with DH5.22 in Gαi1 but not the interaction with YH5.22 (Gαz). To test this conjecture, we replaced DH5.22 of Gαi1 by its Gαz counterpart (tyrosine; Gαi1-DH5.22Y) and YH5.22 of Gαz by its Gαi1 counterpart (aspartate; Gαz-Y5.22D). Remarkably, swapping these residues also swaps the ability of the p.Y141F3.53 variant to activate Gαi1 and Gαz. p.Y141F3.53 activates Gαi1-DH5.22Y in a similar fashion to MT2 in the presence of melatonin (Figure 6c, top panel), as observed for Gαz. Also, the decreased melatonin-induced Gαi1 activation by p.Y141F3.53 is recapitulated with the Gαz-Y5.22D mutation (Figure 6c, central panel). Similarly, the higher constitutive activation of Gαz in the p.Y141F3.53 variant was also recapitulated by testing its activity on Gαi1-DH5.22Y. Reciprocally, the constitutive activity of Y141F3.53 toward Gαi1-DH5.22Y is significantly higher than Gαi1 WT and comparable to that of Gαz (Figure 6c, bottom panel). Similarly, the low basal activation of Gαi1 by p.Y141F3.53 is recapitulated with Gαz-Y5.22D (Figure 6c, bottom panel). These results suggest a role of Y1413.53 in MT2 in the selective recognition of different G protein subtypes. As opposed to variants from CL5, variants from CL7 are characterized by a decreased potency to activate Gαz but not Gαi1. As these mutations are located in TM domains far from the G protein binding region, they most probably affect allosterically the structural elements linking the melatonin binding site to Gαz activation. Additional studies will be required to identify the precise underlying mechanism.
Figure 5.

Biased signaling for Gαi1 and Gαz activation by MT2-Y141F3.53. (a) Melatonin dose–response curve of Gαi1 (left panel) and Gαz (right panel) by WT MT2 and MT2-Y141F3.53. (b) Basal Gαi1 and Gαz activation by MT2-Y1413.53 relative to WT. Statistical differences between melatonin-induced maximal responses (Emax) were assessed by comparing the best-fit values of top (Emax) (***p = 0.0006) in (a), while significance compared to WT MT2 was assessed by a one-sample t-test and compared to 100 in (b). Each point represents the mean ± SEM of independent experiments performed in quadruplicate (distinct samples). n denotes the number of experiments performed.
Figure 6.

Potential role of residue H5.22 in the differential activation of Gαi1 and Gαz by MT2-Y141F3.53. (a) Alignment of Gαi1 and Gαz H5 region. Residues are identified according to the common Gα numbering system.52 Residues conserved in all Gα protein subtypes are highlighted in gray, and residues different in Gαi1 and Gαz are highlighted in blue. (b) Side view of human MT2/Gαi1 (green) and MT2/Gαz (purple) complexes showing the residue at position H5.22 (D in Gαi1 and Y in Gαz) interacting with Y1413.53 of MT2 through a hydrogen bond (for D in Gαi1) or through aromatic interactions (for Y in Gαz). (c) Melatonin dose–response curve of Gαi1-DH5.22Y (upper panel) and Gαz-Y5.22D (middle panel) by WT MT2 and the Y1413.53 variant. Basal activation of WT or mutant Gαi1 and Gαz by Y1413.53 (bottom panel). Statistical differences between melatonin-induced maximal responses (Emax) were assessed by comparing the best-fit values of top (Emax) (****p < 0.0001), while a one-way ANOVA, followed by Tukey’s multiple comparisons test, was used to compare the basal WT of mutant Gαi1 and Gαz activation by MT2-Y141F3.53 (*p < 0.05). Each point represents the mean ± SEM of independent experiments performed in quadruplicate (distinct samples). n denotes the number of experiments performed, and N.S. stands for non-significant.
Discussion
MT2 is an example of a GPCR, for which naturally occurring mutations have been found to bias its signaling profile20 as previously reported for other GCPRs.12−18 The biasing effects of the MT2 variants observed in T2D patients have shown that defects in the melatonin-induced activation of Gαi1 and Gαz and in basal β-arrestin recruitment were the most significantly associated with an increased T2D risk already. In the present work, the analysis of 40 naturally occurring variants in MT2 has allowed us to obtain important insights into the structure–function relationship of this receptor. The positions affected by these natural variants include (a) residues crucial for global receptor activation, (b) residues important for βarr2 recruitment and interaction, and (c) residues determining the selectivity between two members of the Gαi/o family: Gαi1 and Gαz.
The total loss of responsiveness of p.S123R3.35 (from CL1) highlights the functional relevance of the water-mediated polar network mediated by D2.50. Likewise, a mutation at position 3.35 (N111W3.35) in AT1R has been reported to abrogate receptor activation.28 In agreement with our hypothesis that the residue 3.35 of MT2 is susceptible to interfere with D862.50, molecular dynamics (MD) simulations have shown that the loss of responsiveness of the N111W3.35 AT1R mutant can be explained by a change of orientation of D2.50 and the interaction between residue 3.35 and this aspartate.29 Almost all human class A GPCRs have a neutral residue at position 3.35, highlighting the requirement for an absence of charge at this position which otherwise could potentially disturb the role of D2.50 in the rearrangement of the water polar network. This observation also supports our results obtained with the S123E3.35 mutation (Figure 3b). The absence of a full rescue of the responsiveness may be explained by the negatively charged glutamate used to replace the positive arginine in p.S123R3.35.
While R2225.60 and R3168.51 (from CL2) seem to be able to interact with the nearby key activation switches at the DRY motif and ICL4 (Figure 3c), these residues are also facing the lipid bilayer. Membrane phospholipids and most importantly phosphatidylinositol-4,5-biphosphate [PtdIns(4,5)P2] have been recently shown to bind to positively charged residues of the intracellular surface of GPCRs.30 These interactions have been reported to contribute to the stabilization of the active state of β1-adrenergic receptor, adenosine A2A receptor, and neurotensin receptor.30 Using coarse-grained MD simulations for nine family A GPCRs, Yen et al. have shown that residues 5.60 and 8.51 interact with PtdIns(4,5)P2 for all tested GPCRs.30 Accordingly, about 40 and 70% of human family A GPCRs have a positively charged residue at these respective positions. Arginine and lysine are predominant, while histidine only accounts for about 4% and less than 1% of positively charged residues at positions 5.60 and 8.51, respectively. This observation suggests that histidine may not be the best positively charged residue to interact with membrane phospholipids as it is shorter and bulkier than arginine and lysine.
One of the most important regions for β-arrestin interaction with activated GPCRs is the finger loop.31 Our data support the interaction between S75 in the finger loop of βarr2 and R2315.69 at the cytoplasmic side of TM5 in MT2. About 75% of GPCRs having an extended TM5 have at least one positively charged amino acid at position comprised between 5.69 and 5.71. Arginine and lysine are more common at this positive “hub”. Only 10% have a histidine at these positions, suggesting that this residue may not be optimal to interact with the finger loop. This is consistent with our results from the p.R231H5.69 variant (from CL4), which showed significantly reduced βarr2 recruitment.
The functional selectivity among members of different G protein families is well documented. However, the functional selectivity among members from the same G protein family has only been reported very recently due to the significant improvement of the resolution of tools to characterize GPCR functionality. This is particularly well exemplified by a recent study exploring the landscape of 100 GPCRs using an effector translocation-based BRET platform.32 In this extensive study, seven GPCRs that robustly couple to Gαi1 but not to Gαz were identified (5-HT1D, A3, FFA2, GPR39, LPA1, LPA2, and GPR68). Conversely, five receptors robustly coupling to Gαz but not Gαi1 were found (5-HT2B, β1AR, D1, FP, and Y1).32 Interestingly, none of these five receptors harbor a hydroxyl-containing residue (serine, threonine, or tyrosine) at position 3.53, supporting the notion that the lack of such residue may disfavor the Gαi1 engagement versus Gαz. However, 85% of the GPCRs found to interact with Gαi1 did not harbor a serine, threonine, or tyrosine at position 3.53, indicating that additional residues can contribute to Gαi1 engagement consistent with the notion that multiple interactions between the receptor and their cognate G proteins determine selectivity. Although selectivity between Gαz and Gαi1 was also found in another large-scale GPCR profiling,33 namely, for LPA1, LPA2, and A3, such selectivity was not detected as widely as in the study of Avet et al.32 The difference between the extent of the selectivity detected may result from the different assays used to profile the signaling repertoires of the receptors, namely, the use of a chimeric G protein in Inoue’s paper that may affect the selectivity. Avet et al.32 highlight the fact that subtle differences between the last residues of the H5 region of Gα proteins from the same family are sufficient to drive functional selectivity. Our data show that only one residue is sufficient and, to our knowledge, our study is the first to show a receptor mutation inducing functional selectivity between G protein isoforms from the Gαi/o family. We found that residue H5.22 (D in Gαi1; Y in Gαz) drives the functional selectivity observed in the p.Y141F3.53 MT2 variant (from CL5). An equivalent interaction can be observed in the cryo-EM structure of the human adenosine A1 receptor (A1R)–Gαi2 complex (PDB ID: 6D9H), in which R3.53 of A1R interacts with DH5.22 of Gαi2.34 In fact, the interaction between residue 3.53 in GPCRs and Gα proteins is not unique to MT2 and A1R, as a recent analysis of 11 structures of GPCR–G protein complexes reported that, while only three positions in GPCRs always make contacts with Gα, residue 3.53 is one of them.35 The differences observed between the Gαi1 and Gαz responsiveness in the Y141F3.53 variant can explain why certain Gα proteins can tolerate more easily sequence variability in the GPCR without any reduction of their coupling to it. Similar to Gαi1, all Gαq/11 and the other Gαi/o isoforms (except Gαo and Gαz) have a negatively charged residue (aspartate or glutamate) at position H5.22. Gαo and Gαz have a glycine and a tyrosine at this position, respectively. Consequently, for these two isoforms, receptor/Gα protein interactions at this position are probably less important than they are for other Gα proteins, and therefore, these two isoforms could tolerate more easily certain variability in receptor sequence without any impact on their ability to couple to GPCRs than the other Gαi/o and Gαq/11 isoforms.
It will be of interest in future studies to match the function of MT2 in different brain regions with the expression profiles of Gαi1 and Gαz to obtain insights into region-specific differences of the MT2 function.
Altogether, our findings demonstrate that natural variants provide new insights into study structure–function relationships. By analyzing the functional data using structural bioinformatics tools, we have furthered our understanding of the structural basis of GPCR activation and functional selectivity.
Methods
Signaling Pathway Measurements and Generation of Signaling Signatures
The melatonin dose–response curves of WT and mutant Gαi1 and Gαz activation, inhibition of forskolin-simulated cAMP production, WT βarr2 recruitment, and ERK phosphorylation were performed using the methods previously described.20 The measurement of mutant βarr2 recruitment upon melatonin stimulation was performed by monitoring the enhanced bystander BRET between Renilla luciferase II (RlucII)-tagged mutant βarr2 and Renilla green fluorescent protein (rGFP) tagged with the CAAX domain of the plasma membrane located protein KRas (rGFP–CAAX) as previously described.36 For these experiments, human MT2-ARMS2-ProLink 2 from DiscoverX was co-transfected with the EbBRET biosensors. Procedures to obtain radial graphs representing the signaling signature of MT2 variants were previously described.20 Briefly, for WT and MT2 variants, 13 signaling parameters were measured as indicated in the results. Dose–response curves were generated using a four-parameter sigmoidal curve equation. Maximal agonist-induced responses (M) of the variants were determined from the sigmoidal curves and expressed as a percent of WT. The basal activity (BA) of variants was also expressed as a percent of WT. The TCs [log(τ/KA)] were also assessed using an operational model of agonism designed by Kenakin and Christopoulos.37 The difference of TC between each variant and WT [Δlog(τ/KA)] was calculated, and the antilog of Δlog(τ/KA) was then extracted (ΔTC). The values obtained for BA, M, and ΔTC were normalized to fit a −1 to +1 scale using the following formula: (variant – WT)/(variant + WT). These normalized values were plotted on a radial web.
Signaling Signature Clustering
We utilized an adaptation of the method outlined in Schönegge et al. to cluster 40 MT2 variants based on phenotypic signaling response using BRET-based biosensors.38 As previously described, our method iteratively samples values from the normal distribution around each mean phenotype parameter [e.g., basal response (BA), agonist-mediated maximal response (M), and Δlog(τ/KA) (ΔTC) calculated from the dose–response curve replicates] to group mutations with similar signaling patterns. To achieve this purpose, we created 200 matrices of sampled data—each 40 variants with 13 phenotype data points per variant—by randomly sampling, for each data point, a single value from the normal distribution with the mean and standard deviation estimated from the variant replicates. To avoid biases caused by large-scale differences between measures (e.g., M vs ΔTC), each column (phenotype measure) was standardized between 0 and 1 based on standard value = (valuei,j – Minj)/(Maxj – Minj), where Minj and Maxj are the column-specific minimum and maximum scores. Feature reduction—using sparse NMF—and unsupervised clustering—using K-means—were independently performed 100 times on each of the 200 sampled matrices for K = 2 through K = 10, where K = K for NMF and K-means clustering. We specifically used sparse NMF to deconstruct the resulting matrix into two basis vectors, [W, H] (where W has the dimensions 40 mutations by k, and H has the dimension k by 13 signaling parameters), using the multiplicative algorithm of NMF with 200 replicates to ignore data points which were impossible to define or that were not obtained via phenotypic testing. The K-means clustering was used to convert the W matrix to cluster assignments using the same K for the K-means as NMF resulting in 100 cluster assignments for every K (2–10) for every 200 sampled matrices. Using these 100 cluster assignments, we created a clustering frequency matrix (f) for every K, of dimensions 40 mutations × 40 mutations, by calculating how frequently any pair of mutations clustered together over the 100 replicates. This resulted in a clustering frequency matrix. The values therefore ranged from 0 (never clustered together) to 1 (mutations always clustered together). To obtain a consensus over all 200 frequency matrices (for a given K), we further used K-means clustering to obtain cluster assignments from individual frequency matrices. The K used in this step matched the K used to obtain the frequency matrix (f). The final result was therefore nine frequency matrices (FK), one for each K (K = 2 through K = 10) quantifying how frequently any two variants (i, j) clustered together across all the 200 sampled matrices. These final frequency matrices are therefore relative quantifications of the similarity between each of the variants. The final heatmap and dendrogram for each K were obtained by calculating the Pearson correlation distance between all pairs of mutations within FK. The final cluster assignment was obtained by subdividing the final tree into K branches by reductively cutting the tree beginning at the most diverged vertex. The number of clusters (K) was determined by selecting a value big enough to allow sufficient resolution between the phenotypical differences but small enough to exclude any situation for which non-statistical features would define a cluster. According to these criteria, the optimal value of K obtained was 8.
Modeling of Active MT2 Bound to Gαi1/Gαz Proteins and β-Arrestin
The sequences of the human MT2 and β2-adrenergic receptors were aligned using Clustal Omega.39 This initial alignment was manually refined using Chimera40 to adjust some of the gaps in the loop regions. Using this alignment, the active state of MT2 was modeled with Modeller41 using the crystal structure of the active β2-adrenergic (3SN6)42 as a template. This choice was forced by the lack of GPCR/Gi complexes at that moment. When these became available, we observed that our model satisfactorily captured the active conformation of the receptor [RMSD = 1.9 Å (Cα atoms) when compared to the structure of the MT1/Gi complex].43 As a result, the key features relevant to our analysis were well described in our model. In particular, the position of Y1413.53 in our MT2 model is equivalent to that of Y1283.53 in the MT1/Gi complex (Figure S1). Residues missing in the template were refined using the loop optimization method in Modeller, and a disulfide bridge was added between residues Cys1133.25 and Cys19045.50. All models were subjected to 300 iterations of variable target function method optimization and thorough MD and simulated annealing optimization within Modeller and scored using the discrete optimized protein energy potential. The 20 best-scoring models were analyzed visually, and a suitable model (in terms of low score and structure of the loops) was selected. We added ordered water molecules in the TM bundle, as in the high-resolution structure of the adenosine A2A receptor.44 The protonation state of titratable groups was calculated using PROPKA45 at pH 7.0 as implemented in PDB2PQR46 to optimize the hydrogen bond network. The CHARMM-GUI Membrane Builder47 was used to embed the receptor in a hexagonal lipid bilayer composed by 162 POPC molecules, which was hydrated with a layer of approximately 35 Å on each side. Sodium and chloride ions were added to a concentration of 150 mM NaCl, and then additional ions were added to achieve charge neutrality. The final system contains a total of approximately 72,000 atoms. The system was equilibrated with NAMD48 using the protocol from the CHARMM-GUI Membrane Builder and subjected to 200 ns (2 × 100 ns) of unrestrained MD. Simulations were carried out with NAMD 2.10 with the c36 CHARMM force field49 in the NPT ensemble, and using Langevin dynamics to control temperature at 300 K, and at a time step of 2 fs, while constraining all bonds between hydrogen and heavy atoms. The resulting equilibrated model of active MT2 was aligned to the structures of the adenosine A1 receptor bound to Gi2 (PDB ID: 6D9H(34)) or the β1 adrenergic receptor bound to β-arrestin-1 (PDB ID: 6TKO(50)), and models of Gαi1, Gαz, and βarr2 bound to MT2 were built—by homology modeling with Modeller, using the approach described above—with the structures of GPCR-bound Gi2 (PDB ID: 6D9H) and β-arrestin-1 (PDB ID: 6TKO) as templates. MD simulations were carried out at the Paul Scherrer Institute high-performance computing facilities and at the Swiss National Supercomputing Centre (CSCS).
Glossary
Abbreviations
- GPCRs
G protein-coupled receptors
- MT2
melatonin MT2 receptor
- TM
transmembrane
- βarr2
β-arrestin 2
- NMF
non-negative matrix factorization
- WT
wild-type
- ΔTC
Δlog(τ/KA)
- CL
clusters
- ICL
intracellular loop
- H
α-helical
- AT1R
angiotensin II type 1 receptor
- MD
molecular dynamics
- PtdIns(4,5)P2
phosphatidylinositol-4,5-biphosphate
- A1R
adenosine A1 receptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00239.
Superposition of the MT2 and MT1 structures (PDF)
Author Contributions
B.P. and A.K. contributed equally to this work. B.P., A.K., R.J., and M.B. designed the study; B.P., A.K., S.H., C.A.D. and J.L.G. conducted the assays; A.B. and P.F. performed the genomic sequencing and identified the genetic variants, T.F. and X.D. performed the computational modeling experiments; J.M.G. and O.L. performed the clustering analysis; C.L.G. designed some of the BRET-based sensors used and provided technical guidance; B.P. and M.B. wrote the original draft; and B.P., A.K., T.F., J.M.G., O.L., X.D., R.J., and M.B. participated in the writing, reviewing, and editing. All co-authors approved the manuscript.
This work was supported by the Canadian Institutes of Health Research (CIHR) Operating (MOP10501; M.B.) and Foundation (FDN148431; M.B.) grants, Fonds de Recherche du Québec—Santé (26657 “MELA-BETES”, M.B.), the ANR-2011-META (“MELA-BETES”; R.J. and P.F.), the Agence Nationale de la Recherche (ANR-2011-BSV1-012-01 “MLT2D”; R.J., ANR-12-RPIB-0016 “MED-HET-REC-2”; R.J., ANR-10-LABX-46; P.F. and A.B., ANR-10-EQPX-07-01; P.F. and A.B.), the Fondation de la Recherche Médicale (Equipe FRM DEQ20130326503; R.J.), Institut National de la Santé et de la Recherche Médicale (INSERM; R.J.), Centre National de la Recherche Scientifique (CNRS; R.J.) and the “Who am I?” laboratory of excellence (ANR-11-LABX-0071; R.J.) funded by the French Government through its “Investments for the Future” program operated by the French National Research Agency (ANR-11-IDEX-0005-01; R.J.), the European Research Council (ERC Reg-Seq—715575; A.B.), the National Center for Precision Diabetic Medicine (PreciDIAB; P.F. and A.B.) jointly supported by the French National Agency for Research (ANR-18-IBHU-0001), the European Union (FEDER), the Hauts-de-France Regional Council and the European Metropolis of Lille (MEL), the Swiss National Science Foundation (SNF grant 192780; X.D.), the National Institutes of Health (NIH GM066099; O.L.), a Post-Doctoral Fellowship Award from CIHR and Diabetes Canada, as well as an Early-Career Small Grant for Basic Scientists from Diabetes UK to B.P., a Post-Doctoral Fellowship from the Swiss Federal Institute of Technology in Zürich (ETHZ; T.F.), and a PhD scholarship from the Department for Education (DfE) and Queen’s University Belfast (DfE; C.A.D.). M.B. holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
The authors declare the following competing financial interest(s): M.B. is the president of the scientific advisory Board of Domain Therapeutics which licensed-in some of the BRET-based biosensors, used in the present study, for their commercial use.
Notes
⧫ X.D., R.J., and M.B. are co-senior authors.
Notes
Data availability: All data needed to evaluate the conclusions in the paper are present in the paper. All biosensors are available for academic non-commercial studies through regular Material Transfer Agreements and can be requested by email: michel.bouvier@umontreal.ca.
Supplementary Material
References
- Hilger D. The role of structural dynamics in GPCR-mediated signaling. FEBS J. 2021, 288, 2461–2489. 10.1111/febs.15841. [DOI] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. 10.3389/fphar.2019.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R.; von Zastrow M. GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 2014, 27, 109–116. 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Neto C. M.; Parreiras-e-Silva L. T.; Bouvier M. A Pluridimensional View of Biased Agonism. Mol. Pharmacol. 2016, 90, 587–595. 10.1124/mol.116.105940. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Deupi X.; Lebon G.; Heydenreich F. M.; Flock T.; Miljus T.; Balaji S.; Bouvier M.; Veprintsev D. B.; Tate C. G.; Schertler G. F. X.; Babu M. M. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 2016, 536, 484–487. 10.1038/nature19107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Ma A. K.; Fonseca R.; Latorraca N. R.; Kelly B.; Betz R. M.; Asawa C.; Kobilka B. K.; Dror R. O. Diverse GPCRs exhibit conserved water networks for stabilization and activation. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 3288–3293. 10.1073/pnas.1809251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalatskaya I.; Schüssler S.; Blaukat A.; Müller-Esterl W.; Jochum M.; Proud D.; Faussner A. Mutation of tyrosine in the conserved NPXXY sequence leads to constitutive phosphorylation and internalization, but not signaling, of the human B2 bradykinin receptor. J. Biol. Chem. 2004, 279, 31268–31276. 10.1074/jbc.M401796200. [DOI] [PubMed] [Google Scholar]
- Liu R.; Nahon D.; le Roy B.; Lenselink E. B.; IJzerman A. P. Scanning mutagenesis in a yeast system delineates the role of the NPxxY(x)(5,6)F motif and helix 8 of the adenosine A(2B) receptor in G protein coupling. Biochem. Pharmacol. 2015, 95, 290–300. 10.1016/j.bcp.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Scott C. E.; Abrol R.; Ahn K. H.; Kendall D. A.; Goddard W. A. 3rd. Molecular basis for dramatic changes in cannabinoid CB1 G protein-coupled receptor activation upon single and double point mutations. Protein Sci. 2013, 22, 101–113. 10.1002/pro.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallaert W.; Christopoulos A.; Bouvier M. Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin. Drug Discovery 2011, 6, 811–825. 10.1517/17460441.2011.586691. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Signaling bias in drug discovery. Expert Opin. Drug Discovery 2017, 12, 321–333. 10.1080/17460441.2017.1297417. [DOI] [PubMed] [Google Scholar]
- Gorvin C. M.; Babinsky V. N.; Malinauskas T.; Nissen P. H.; Schou A. J.; Hanyaloglu A. C.; Siebold C.; Jones E. Y.; Hannan F. M.; Thakker R. V. A calcium-sensing receptor mutation causing hypocalcemia disrupts a transmembrane salt bridge to activate beta-arrestin-biased signaling. Sci. Signaling 2018, 11, eaan3714 10.1126/scisignal.aan3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole C.; Wootten D.; Simms J.; Valant C.; Miller L. J.; Christopoulos A.; Sexton P. M. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: allosteric rescue of loss of function mutation. Mol. Pharmacol. 2011, 80, 486–497. 10.1124/mol.111.072884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y.; LeGouill C.; Kumar S.; Cao Y.; Teixeira L. B.; Lukasheva V.; Giubilaro J.; Simões S. C.; Longpré J.-M.; Devost D.; Hébert T. E.; Piñeyro G.; Leduc R.; Costa-Neto C. M.; Bouvier M.; Laporte S. A. Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signaling 2018, 11, eaat1631 10.1126/scisignal.aat1631. [DOI] [PubMed] [Google Scholar]
- Leach K.; Wen A.; Davey A. E.; Sexton P. M.; Conigrave A. D.; Christopoulos A. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology 2012, 153, 4304–4316. 10.1210/en.2012-1449. [DOI] [PubMed] [Google Scholar]
- Yang F.; Huang H.; Tao Y.-X. Biased signaling in naturally occurring mutations in human melanocortin-3 receptor gene. Int. J. Biol. Sci. 2015, 11, 423–433. 10.7150/ijbs.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasberger H.; Van Sande J.; Hag-Dahood Mahameed A.; Tenenbaum-Rakover Y.; Refetoff S. A familial thyrotropin (TSH) receptor mutation provides in vivo evidence that the inositol phosphates/Ca2+ cascade mediates TSH action on thyroid hormone synthesis. J. Clin. Endocrinol. Metab. 2007, 92, 2816–2820. 10.1210/jc.2007-0366. [DOI] [PubMed] [Google Scholar]
- Lotta L. A.; Mokrosiński J.; Mendes de Oliveira E.; Li C.; Sharp S. J.; Luan J. a.; Brouwers B.; Ayinampudi V.; Bowker N.; Kerrison N.; Kaimakis V.; Hoult D.; Stewart I. D.; Wheeler E.; Day F. R.; Perry J. R. B.; Langenberg C.; Wareham N. J.; Farooqi I. S. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e9. 10.1016/j.cell.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A.; Clement N.; Fawcett K.; Yengo L.; Vaillant E.; Guillaume J. L.; Dechaume A.; Payne F.; Roussel R.; Czernichow S.; Hercberg S.; Hadjadj S.; Balkau B.; Marre M.; Lantieri O.; Langenberg C.; Bouatia-Naji N.; Charpentier G.; Vaxillaire M.; Rocheleau G.; Wareham N. J.; Sladek R.; McCarthy M. I.; Dina C.; Barroso I.; Jockers R.; Froguel P. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012, 44, 297–301. 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamitri A.; Plouffe B.; Bonnefond A.; Chen M.; Gallion J.; Guillaume J.-L.; Hegron A.; Boissel M.; Canouil M.; Langenberg C.; Wareham N. J.; Le Gouill C.; Lukasheva V.; Lichtarge O.; Froguel P.; Bouvier M.; Jockers R. Type 2 diabetes-associated variants of the MT2 melatonin receptor affect distinct modes of signaling. Sci. Signaling 2018, 11, eaan6622 10.1126/scisignal.aan6622. [DOI] [PubMed] [Google Scholar]
- Kim H.; Park H. Sparse non-negative matrix factorizations via alternating non-negativity-constrained least squares for microarray data analysis. Bioinformatics 2007, 23, 1495–1502. 10.1093/bioinformatics/btm134. [DOI] [PubMed] [Google Scholar]
- Isberg V.; de Graaf C.; Bortolato A.; Cherezov V.; Katritch V.; Marshall F. H.; Mordalski S.; Pin J.-P.; Stevens R. C.; Vriend G.; Gloriam D. E. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. 10.1016/j.tips.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovati G. E.; Capra V.; Neubig R. R. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol. Pharmacol. 2007, 71, 959–964. 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- Johansson L. C.; Stauch B.; McCorvy J. D.; Han G. W.; Patel N.; Huang X.-P.; Batyuk A.; Gati C.; Slocum S. T.; Li C.; Grandner J. M.; Hao S.; Olsen R. H. J.; Tribo A. R.; Zaare S.; Zhu L.; Zatsepin N. A.; Weierstall U.; Yous S.; Stevens R. C.; Liu W.; Roth B. L.; Katritch V.; Cherezov V. XFEL structures of the human MT2 melatonin receptor reveal the basis of subtype selectivity. Nature 2019, 569, 289–292. 10.1038/s41586-019-1144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis W. I.; Kobilka B. K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P.; Sommer M. E. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 2017, 45, 160–169. 10.1016/j.sbi.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Cahill T. J. 3rd; Thomsen A. R. B.; Tarrasch J. T.; Plouffe B.; Nguyen A. H.; Yang F.; Huang L.-Y.; Kahsai A. W.; Bassoni D. L.; Gavino B. J.; Lamerdin J. E.; Triest S.; Shukla A. K.; Berger B.; Little J.; Antar A.; Blanc A.; Qu C.-X.; Chen X.; Kawakami K.; Inoue A.; Aoki J.; Steyaert J.; Sun J.-P.; Bouvier M.; Skiniotis G.; Lefkowitz R. J. Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 2562–2567. 10.1073/pnas.1701529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger-Messier M.; Clement M.; Lanctot P. M.; Leclerc P. C.; Leduc R.; Escher E.; Guillemette G. The constitutively active N111G-AT1 receptor for angiotensin II maintains a high affinity conformation despite being uncoupled from its cognate G protein Gq/11alpha. Endocrinology 2003, 144, 5277–5284. 10.1210/en.2003-0677. [DOI] [PubMed] [Google Scholar]
- Cabana J.; Holleran B.; Beaulieu M.-È.; Leduc R.; Escher E.; Guillemette G.; Lavigne P. Critical hydrogen bond formation for activation of the angiotensin II type 1 receptor. J. Biol. Chem. 2013, 288, 2593–2604. 10.1074/jbc.M112.395939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H.-Y.; Hoi K. K.; Liko I.; Hedger G.; Horrell M. R.; Song W.; Wu D.; Heine P.; Warne T.; Lee Y.; Carpenter B.; Plückthun A.; Tate C. G.; Sansom M. S. P.; Robinson C. V. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559, 423–427. 10.1038/s41586-018-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A. K.; Westfield G. H.; Xiao K.; Reis R. I.; Huang L.-Y.; Tripathi-Shukla P.; Qian J.; Li S.; Blanc A.; Oleskie A. N.; Dosey A. M.; Su M.; Liang C.-R.; Gu L.-L.; Shan J.-M.; Chen X.; Hanna R.; Choi M.; Yao X. J.; Klink B. U.; Kahsai A. W.; Sidhu S. S.; Koide S.; Penczek P. A.; Kossiakoff A. A.; Woods V. L. Jr.; Kobilka B. K.; Skiniotis G.; Lefkowitz R. J. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222. 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avet C.; Mancini A.; Breton B.; Le Gouill C.; Hauser A. S.; Normand C.; Kobayashi H.; Gross F.; Hogue M.; Lukasheva V.; St-Onge S.; Carrier M.; Héroux M.; Morissette S.; Fauman E.; Fortin J.-P.; Schann S.; Leroy X.; Gloriam D. E.; Bouvier M. Effector membrane translocation biosensors reveal G protein and βarrestin coupling profiles of 100 therapeutically relevant GPCRs. bioRxiv 2020, 2020.04.20.052027. 10.1101/2020.04.20.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A.; Raimondi F.; Kadji F. M. N.; Singh G.; Kishi T.; Uwamizu A.; Ono Y.; Shinjo Y.; Ishida S.; Arang N.; Kawakami K.; Gutkind J. S.; Aoki J.; Russell R. B. Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell 2019, 177, 1933–1947.e1925. 10.1016/j.cell.2019.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Khoshouei M.; Thal D. M.; Liang Y.-L.; Nguyen A. T. N.; Furness S. G. B.; Venugopal H.; Baltos J.-A.; Plitzko J. M.; Danev R.; Baumeister W.; May L. T.; Wootten D.; Sexton P. M.; Glukhova A.; Christopoulos A. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 2018, 558, 559–563. 10.1038/s41586-018-0236-6. [DOI] [PubMed] [Google Scholar]
- García-Nafría J.; Tate C. G. Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol. Cell. Endocrinol. 2019, 488, 1–13. 10.1016/j.mce.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Namkung Y.; Laporte S. A. Methods to Monitor the Trafficking of beta-Arrestin/G Protein-Coupled Receptor Complexes Using Enhanced Bystander BRET. Methods Mol. Biol. 2019, 1957, 59–68. 10.1007/978-1-4939-9158-7_3. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery 2013, 12, 205–216. 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Schönegge A.-M.; Gallion J.; Picard L.-P.; Wilkins A. D.; Le Gouill C.; Audet M.; Stallaert W.; Lohse M. J.; Kimmel M.; Lichtarge O.; Bouvier M. Evolutionary action and structural basis of the allosteric switch controlling beta2AR functional selectivity. Nat. Commun. 2017, 8, 2169. 10.1038/s41467-017-02257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Webb B.; Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinf. 2014, 47, 5.6.1–5.6.32. 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- Rasmussen S. G. F.; DeVree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T. A.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H. H.; Miyauchi H.; Inoue A.; Raimondi F.; Tsujimoto H.; Kusakizako T.; Shihoya W.; Yamashita K.; Suno R.; Nomura N.; Kobayashi T.; Iwata S.; Nishizawa T.; Nureki O. Cryo-EM structure of the human MT1-Gi signaling complex. Nat. Struct. Mol. Biol. 2021, 28, 694–701. 10.1038/s41594-021-00634-1. [DOI] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Cheng X.; Jo S.; Rui H.; Song K. C.; Dávila-Contreras E. M.; Qi Y.; Lee J.; Monje-Galvan V.; Venable R. M.; Klauda J. B.; Im W. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E. M.; Mittal J.; Feig M.; Mackerell A. D. Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Warne T.; Nehmé R.; Pandey S.; Dwivedi-Agnihotri H.; Chaturvedi M.; Edwards P. C.; García-Nafría J.; Leslie A. G. W.; Shukla A. K.; Tate C. G. Molecular basis of beta-arrestin coupling to formoterol-bound beta1-adrenoceptor. Nature 2020, 583, 862–866. 10.1038/s41586-020-2419-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen E. T.; Breton B.; Christopoulos A.; Bouvier M. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 2014, 85, 492–509. 10.1124/mol.113.088880. [DOI] [PubMed] [Google Scholar]
- Flock T.; Ravarani C. N. J.; Sun D.; Venkatakrishnan A. J.; Kayikci M.; Tate C. G.; Veprintsev D. B.; Babu M. M. Universal allosteric mechanism for Galpha activation by GPCRs. Nature 2015, 524, 173–179. 10.1038/nature14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.