

Summary
Single-cell RNA sequencing (scRNAseq) has been used to assess the intra-tumor heterogeneity and microenvironment of pancreatic ductal adenocarcinoma (PDAC). However, previous knowledge is not fully universalized. Here, we built a single cell atlas of PDAC from six datasets containing over 70 samples and >130,000 cells, and demonstrated its application to the reanalysis of the previous bulk transcriptomic cohorts and inferring cell–cell communications. The cell decomposition of bulk transcriptomics using scRNAseq data showed the cellular heterogeneity of PDAC; moreover, high levels of tumor cells and fibroblasts were indicative of poor-prognosis. Refined tumor subtypes signature indicated the tumor cell dynamics in intra-tumor and their specific regulatory network. We further identified functionally distinct tumor clusters that had close interaction with fibroblast subtypes via different signaling pathways dependent on subtypes. Our analysis provided a reference dataset for PDAC and showed its utility in research on the microenvironment of intra-tumor heterogeneity.
Subject areas: Cancer systems biology, Cancer, Transcriptomics
Graphical abstract
Highlights
-
•
Generation of reference single cell atlas for pancreatic adenocarcinoma
-
•
Decomposition of bulk transcriptomics showed the heterogeneous microenvironment
-
•
Refined tumor subtypes signature indicated the tumor cell dynamics in intra-tumor
-
•
Two subtype of fibroblast support the growth of tumor cell with distinct pathways
Cancer systems biology; Cancer; Transcriptomics
Introduction
Pancreatic ductal adenocarcinoma (PDAC) originates from the ductal epithelial cells of the pancreas and is a lethal disease with limited treatment options and poor survival (the 5-year survival rate was estimated at 9%) (Siegel et al., 2020). This poor-prognosis is attributed to late detection and early metastases; thus, the cancer is often in the unresectable stage at diagnosis (McGuigan et al., 2018).
Major genomic mutations, such as those in KRAS, TP53, SMAD4, and CDKN2A genes, eventually accumulate during the progression from low-grade pancreatic intraepithelial neoplasia to tumorigenesis; however, there are currently no targeted therapies for these driver mutations (Pihlak et al., 2018). In addition to cancer cells, the tumor microenvironment (TME), which is composed of blood vessels, hematopoietic cells, fibroblasts, and extracellular matrices, largely affects tumor heterogeneity and malignancy. Based on the transcriptomic profile of tumor samples, several tumor subtypes were suggested in PDAC (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015; Network, 2017). Furthermore, the estimation of cell composition using empirical cell marker genes has been challenging, in particular for immune cells (Newman et al., 2015; O'Kane et al., 2020). Moreover, non-tumor cells within the TME play a critical role in PDAC progression; however, the elucidation of their role in this context has been challenging because of their complexity, such as stromal hyperplasia with low tumor purity.
The remarkable breakthroughs in single-cell transcriptomics have further advanced the interpretation of the TME at single-cell resolution, which can be used to address the complex cellularity of PDAC via the analysis of the target cells, regardless of cell composition, in a given sample. In addition to cell rate, their state could be proposed based on known markers, such as T cells (e.g., memory, effector, and exhausted cells) (Chen et al., 2021; Lin et al., 2020; Steele et al., 2020; Zhou et al., 2021) and macrophages (e.g., M1 and M2 types) (Chen et al., 2021; Kemp et al., 2021b; Lin et al., 2020). Moreover, beyond conventional cellular analysis methods, such as flow cytometry and/or immunohistochemistry, scRNAseq analysis can indicate the presence of new subpopulations within the TME: Elyada et al. proposed that cancer fibroblasts may be classified into inflammatory, myofibroblast, and antigen-presenting cells (Elyada et al., 2019); conversely, Chen et al. suggested two cancer-associated fibroblasts (CAF) types in classical and complement secreting (Chen et al., 2021). Regarding tumor cells, novel tumor subtypes at cellular levels and their trajectory along differentiation or malignancy have been described (Carstens et al., 2021; Lee et al., 2021; Peng et al., 2019a; Qadir et al., 2020; Raghavan et al., 2021). Furthermore, single-cell transcriptomics allow the interpretation of the communication among different cell types within the TME (Lee et al., 2021).
The tumor subtype is determined in each patient with PDAC (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015; Network, 2017). Moreover, the heterogeneity of tumor subtypes is of great practical importance, and scRNAseq is not a high-throughput assay that deals with a large number of individuals. However, because of tremendous efforts and technological convergence, the data available for various tumors, including PDAC, have been deposited in similar platforms. Peng et al. reported scRNAseq data from 35 patients, including 24 PDAC and 11 control pancreas-adjacent tumors (Peng et al., 2019a), whereas Steele et al. reported data from 16 patients with PDAC and three adjacent tissues (Steele et al., 2020). The secondary use of these data has contributed to the fine-tuning of basic research on PDAC (Carstens et al., 2021; Chen et al., 2021; Dominguez et al., 2020; Kemp et al., 2021a, 2021b). In the present study, we integrated these freely available datasets and built a huge reference dataset containing >130,000 cells, to universalize the scRNAseq data available for PDAC. Furthermore, we reanalyzed the cellular components of a previous bulk RNAseq cohort (i.e., the TCGA) using built data and demonstrated a profound cell–cell communication between the tumor and CAF.
Results
Establishment of a reference single-cell transcriptome from deposited data
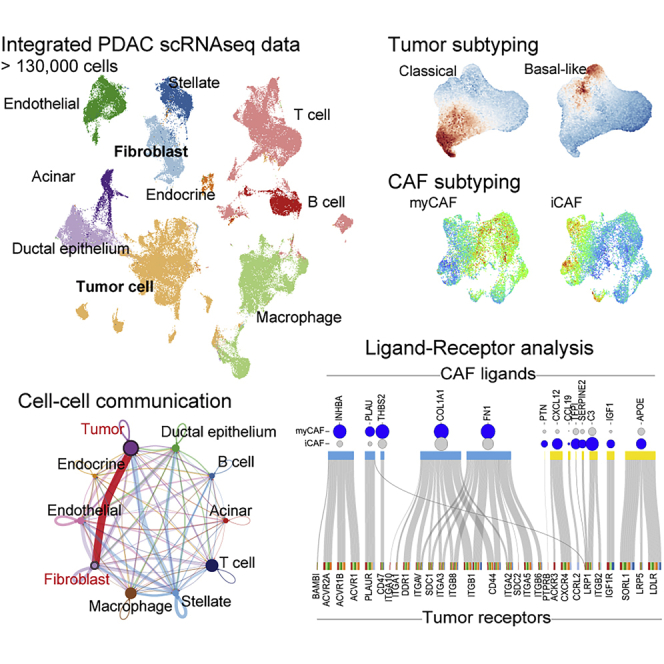
Single-cell transcriptomes for human pancreatic adenocarcinoma (PDAC) have been reported by several groups. However, they hold obvious differences in clinical setting, sampling methods, platform used (10× Genomics or InDrop), and version of the kit/reagent used for scRNAseq. To generalize previous efforts, we tried to build a useful reference tool based on five freely available datasets (PRJCA001063, GSE111672, GSE154778, GSE155698, and GSM4293555) (Lin et al., 2020; Moncada et al., 2020; Peng et al., 2019a; Schlesinger et al., 2020; Steele et al., 2020) and our original data (termed OUGS). These data were largely divergent and biased regarding cell population (Figure S1A). When each dataset was simply merged, cells were divided using a bias of batch effect between datasets, rather than cell-specific features (Figure S1B). Therefore, each dataset was integrated through batch correlation, followed by dimensionality reduction and unsupervised clustering (Figures 1A–1C). Two large datasets, PRJCA001063 and GSE155698, which encompass over 15 patients, including non-cancerous pancreas samples, were used as reference data during the integration of the six datasets. The cell labels were transferred from PRJCA001063 by predicting cell-specific signatures using the Seurat function. According to a previous report (Peng et al., 2019a), clusters for normal ductal epithelial cells and malignant epithelial cells were referred to as ductal cell type 1 and 2, respectively. The integrated data included 10 cell types with reasonable differentially expressed genes (Table S1). Although the proportion of cells varied among patients (Figures S2A and S2B) and each dataset exhibited different ranges of unique molecular index (UMI) counts and types of detected mRNAs, their trends beyond cell type were almost comparable between datasets (Figure S2C), indicating proper correlation.
Figure 1.

Integration of the six scRNAseq datasets and reinterpretation of altered gene expression in bulk transcriptomics at the cellular level
(A–C) The integrated data are summarized in the UMAP and color-coded according to cell type (A), dataset (B), and sample type (C).
(D and E) The module scores calculated using the signatures defined in the NMF method on TCGA-PDAC are plotted on UMAP (D) and their violin plot (E).
(F and G) Bubble plot showing the cell origin of the driving genes that were highly upregulated (F) and downregulated (G) in TCGA-PAAD, with cell types indicated in the rows and genes in the columns.
The size of each bubble represents the rate of cells expressing the gene, and the color represents the scaled average expression in their cell type cluster. The bar plot shows the log2 fold change in the TCGA-PAAD sample vs. the control pancreas obtained from GEPIA2.
Differentially expressed genes among cell types were comparable to general cell markers (Table S1). Recent research on TCGA-PAAD using NMF methods identified nine different components (including non-tumor cells), as follows: classical tumor, basal tumor, activated stroma, normal stroma, endocrine, exocrine, immune, histone, and olfactory cells (Peng et al., 2019b). Those authors reported that unique gene sets determined these components. When the scRNAseq data were scored using these gene sets, their component scores agreed with the cell types (Figures 1D and 1E). The scores for endocrine and exocrine types were specifically high in clusters of endocrine and acinar cells, respectively. The immune score reflected the presence of T cells, B cells, and macrophages. Two cancer scores were higher in the malignant cell cluster, and two stromal scores were higher in the clusters of fibroblasts and stellate cells, in accordance with the finding that CAFs originate from stellate cells (Öhlund et al., 2017). These facts supported the validity of this bulk scRNAseq dataset.
Cell origin of the differentially expressed genes identified in TCGA-PAAD
To further understand the previous findings of whole-tissue transcriptome analyses, the cell origin of the differentially expressed genes (DE-Gs) identified in TCGA-PAAD were verified in this scRNAseq dataset. We found that the upregulated genes were mainly derived from malignant cells, although fibroblasts, B cells, and macrophages were also listed as the unique origin of top DE-Gs, which indicated the formation of stromal tissues and infiltration of immune cells (Figure 1F). Conversely, the downregulated genes reflected the loss of acinar cell functions (Figure 1G). Genes having a significant effect on overall survival were also validated that the poor-prognosis genes were found to be upregulated in malignant cells (Figure S3A). However, genes associated with prognosis were less specific among cell types, unlike that observed for DE-Gs. Moreover, genes associated with a good prognosis were not marked clearly, as they were expressed in a low proportion of each cell type (Figure S3B). Genes from T cells, which are important for tumor immunity, were not detected among the top DE-Gs, with the exception of IL2RG; moreover, immune cell features were not ranked among the good-prognosis markers. These findings may reflect the feature of PDAC as a “cold” tumor (Kabacaoglu et al., 2018).
Decomposition of TCGA-PAAD using scRNAseq data
Cell composition in samples can be estimated via decomposition of bulk RNAseq data using cell-specific markers or scRNAseq data (Baron et al., 2016; Jew et al., 2020; Menden et al., 2019). Decomposition of the built scRNAseq dataset revealed the diversity in cell proportions among the patients in TCGA-PAAD (Figure 2A). Based on the patterns of estimated composition, patients were divided into five groups and their survival data were then analyzed (Figure 2B and Table S2). This grouping was not affected by clinical status, genomic mutation, and previously reported cancer subtypes (Figures 2C and 2D). Surprisingly, patients in group 4 had an obviously good prognosis, as they had a high proportion of endocrine, stellate, and T cells and a low proportion of cancer cells (Figures 2E and 2F). However, this good prognosis group was relatively small. Furthermore, similar population could not be identified in other cohorts of PDAC transcriptomics (ICGA-PACA-CA, ICGA-PACA-AU, and E-MTAB-6830 (Dijk et al., 2020)) as patients with a high ratio of endocrine, stellate, and T cells also exhibited cancer cells, which corresponds to group 3 in TCGA-PAAD (Figure S4A and Table S2). T cells and normal pancreatic function may contribute to survival, whereas tumor immunity fails in most patients with PDAC. In fact, the estimation of infiltrated T cells alone does not correlate with prognosis in TCGA-PAAD (Figure S4B).
Figure 2.

Cellular decomposition analysis of the bulk transcriptome using scRNAseq data
(A–D) Heatmap showing the estimated cell ratio of each patient from TCGA-PAAD (A). Hierarchical clustered heatmap for the correlation among patients based on the cell composition (B) with previously defined tumor subtypes, clinical metadata (C), and Oncoprint for major gene mutation (D).
(E) Kaplan–Meier curve for a group of five patients defined by hierarchical clustering of cell composition (p = 0.03 from likelihood ratio test in Cox proportional hazards regression model).
(F) Boxplot (25th percentile, median, and 75th percentile) showing cell composition aggregated for each patient group. Significance was assessed using Pairwise Wilcoxon Rank Sum Tests with Bonferroni correction (∗∗∗∗, p < 0.001; ∗∗∗, p < 0.05; ∗∗, p < 0.01; ∗, p < 0.05. Sample number is indicated in Figure 2E).
(G) Hierarchical clustering for the correlation among cell types based on their pattern. The red frame highlights the positive/negative correlations among stellate cells, endocrine cells, T cells, and fibroblasts.
Interestingly, although group 2 had a lower ratio of malignant cells, their prognosis was poor compared with other clusters comprising a high level of malignant cells. This group contained abundant fibroblasts, which accounted for the hyperplasia of the stroma (Figure 2F). Moreover, the presence of fibroblasts was negatively correlated with T cells, endocrine cells, and stellate cells in all cohorts, suggesting that the presence of CAFs is serious risk factor for PDAC, as well as malignant cells (Figures 2G and S4A).
Estimation of cell–cell communication in PDAC
The signaling interaction within PDAC was explored using CellChat (Jin et al., 2021) based on the expression of the known ligand–receptor pair. The number of signaling patterns indicated that normal pancreatic components, such as acinar and endocrine cells, work independently from other cells as a less outgoing and incoming signaling (Figure 3A). Conversely, malignant cells received signals most frequently, especially from fibroblasts. Moreover, their interaction was the strongest of the cell–cell communications detected in PDAC (Figure 3B). We hypothesized that fibroblast-derived extracellular matrices, such as collagen, laminin, fibronectin, and thrombospondin, provide scaffolds for the TME and interfered with malignant cells via several adhesion molecules, such as integrins, CD44, and SDC4, which in turn transduce inner cellular signals for cell growth, differentiation, and adhesion/migration (Figures 3C and S5) (Harjunpää et al., 2019). These findings supported the notion that malignant cells and CAFs coordinately operate on cancer development.
Figure 3.

Estimation of cell–cell communication within PDAC using CellChat
(A) Heatmap showing the summary of the signaling pathways that contribute to outgoing or incoming communication. The color bar represents the relative signaling strength of a signaling pathway across cell types. The bars indicate the sum of the signaling strength of each cell type or pathway.
(B) Circle plots displaying putative ligand–receptor interactions, with the width of edges representing the strength of the communication.
(C) Chord diagram showing each ligand–receptor pattern and their weights in the interaction between CAFs and malignant cells.
Tumor subtypes in malignant cells and optimization signatures for scRNAseq data
As a classification of PDAC subtypes, classical or basal-like tumors have been proposed via the factorization of bulk transcriptome data (Moffitt et al., 2015), which did not depend on tumor purity compared with the other classification (Network, 2017). Of particular interest in the cell-type scoring described above (Figure 1D), the cancer subtypes appeared to be segmented within malignant cell clusters. To further analyze the heterogeneity of malignant cells, ductal cells (normal and malignant) were extracted and processed for dimensionality reduction using PCA and UMAP. The normal ductal cells were overlapped within the same cluster, whereas malignant cells behaved heterogeneously among patients (Figures 4A and 4B). Furthermore, the score on tumor subtypes showed that these malignant cell types were determined in accordance with individual patients (Figure 4C).
Figure 4.

Re-clustering of ductal cells and their tumor-subtype analysis
(A and B) The subset of ductal cells alone was isolated and further processed using the Seurat pipeline without batch correction. UMAP color-coded according to cell type (A) and individual (B) showing high heterogeneity of malignant cell clusters.
(C) Tumor-subtype scores defined in Figure 1 plotted on UMAP showing the segmented pattern of the score.
(D–G) Pearson’s correlation analysis of the two subtypes of scores indicate weak correlation. The color coding was according to tissue type (D) or new classification (F) (E, G) Modified tumor-subtype scores are shown on the UMAP, which processed new signatures as features for PCA to classify cells according to tumor-subtype (E). Based on their score distribution (F), ductal cells were classified into five groups (G).
(H) Boxplot (25th percentile, median, and 75th percentile) of modified subtype scores aggregated for cells from each tumor sample showing their dependency on the source sample. Significance was assessed using Welch’s t-tests (∗∗∗∗, p < 0.001; ∗∗∗, p < 0.05; ∗∗, p < 0.01; ∗, p < 0.05. N = 26 to 2585 cells per patient.).
(I) Enrichment analysis of differentially expressed genes in cell clusters using Gene Ontology Biological Process. Tree plot showing the hierarchical clustering of enriched terms with word clouds.
(J and K) Tumor-subtype specific regulatory analysis conducted using SCENIC. The heatmap was colored according to the residual sum of squares (RSS) showing the specificity of the activated transcription factor (J). The representative transcript factor is plotted on the UMAP with color attributed according to regulon activity.
Because the current scRNAseq techniques cannot detect expression over an adequate dynamic range, these data are sparse: many genes are undetectable or detected at a lower level. In fact, 41 out of 372 genes in the classical tumor signatures and 34 out of 394 genes in the basal-like tumor signatures were not detected in this scRNAseq data. Moreover, the expression of some genes was not correlated with the subtype scores (Figures S6A and S6B), and two subtype scores in malignant cells were weakly correlated (Figure 4D). Thus, tumor-subtype signatures should be refined for scRNAseq data. To omit unrelated features, clusters containing cells undergoing cell division, tuft cells, contamination of immune cells, and rare cell populations were excluded after correction for their batch effect between each individual (Figures S7A–S7E). To optimize subtype signatures adaptable for scRNAseq data, genes for which the expression was correlated with subtype scores were extracted as new subtype signatures (top-25% genes). Furthermore, genes that were correlated with both scores were omitted as common PDAC features (Table S3). These new signatures could classify the malignant cells into classical or basal-like tumor cells and describe the cellular fate together with their subtypes (Figures 4E–4G, S7F and S7G).
Tumor subtypes were determined in individual patients: 33/58 and 20/58 patients had a significantly higher classical score and basal-like score, respectively (Figure 4H). However, several patients harbored both subtypes of cells and/or intermediate cells, as reported recently (Lee et al., 2021; Raghavan et al., 2021; Zhou et al., 2021) (Figure S8).
Differentially expressed genes and upstream transcription factors among tumor subtypes
DE-Gs were compared among normal ductal cells, classical type cells, basal-like cells, and intermediate cells (lower score of subtypes), which revealed the existence of subtype-specific features (Table S4). An enrichment analysis for a disease database, all three tumor cells that had terms for cancer and basal-like cells exhibited common pan-cancer features (Figure S9A). Regarding cell function and signaling, terms involved in the gastrointestinal epithelium, digestion, glycosylation, mucin, and interferon gamma response were enriched in classical cells. In contrast, terms such as extracellular matrix, cell adhesion, and growth signaling (PI3/Akt, HIF-1, p53, estrogen, VEGF, and YAP/TAZ) were enriched in basal-like cells, which indicated the key features in the epithelial–mesenchymal transition (Figures 4I, S9B–S9D and Table S4).
A regulatory network analysis using SCENIC (Aibar et al., 2017) showed that the subtype-specific transcriptional activities (Figures 4J and 4K, S10A, S10B and Table S5), such as GATA4, GATA6, FOXA1, and HNFs, were activated in classical cells, as reported previously (Brunton et al., 2020; Camolotto et al., 2018; Gong et al., 2018; Kloesch et al., 2021; O'Kane et al., 2020; Roe et al., 2017; Song et al., 2010; Zhou et al., 2021). FOXA3, NR1I2, SREBF2, and ONECUT2 were also reported in a PDAC study, although they have not been well clarified as classical PDAC features. Of note, the transcription factors that are activated in basal-like cells, i.e., RCOR1, RARG, PITX1, and PLAG1, have not been reported in PDAC. RARG-related retinoic acid signaling was reported to be involved in the EMT (Doi et al., 2015; Kim et al., 2017; Kobayashi et al., 2021). RCOR1 interacts with epigenetic factors, such as HDACs (Figure S10C) (Monaghan et al., 2017). These findings may reflect the dynamic epigenetic regulation that occurs during the epithelial–to–mesenchymal transformation (Skrypek et al., 2017).
Unsupervised clustering of malignant cells
As recently pointed out, some patients harbor the two subtypes and/or an intermediate state (Figure S8) (Lee et al., 2021; Raghavan et al., 2021; Zhou et al., 2021). In the present study, a regulatory analysis indicated several activations of transcription factors, such as NF-κB, specifically in intermediate cells (Figure 4H). To identify the functional features of malignant cells, unsupervised clustering based on highly variable genes was performed next. Because re-clustering of ductal subsets showed that cells assigned to clusters 0, 1, and four were representative in patients with PDAC (Figure S7A–S7D), these cells were selected for dimension reduction and classified into eight clusters with removal of the batch effect between patients (Figures 5A and 5B). Without intending to assign along subtypes, the classical score was higher in clusters 0 and one and the basal-like score was higher in cluster 6, indicating that tumor subtype was a prominent feature of malignant cells (Figures 5C–5E). Conversely, cancer cells that were not biased with subtype scores were found in clusters 2, 3, 4, and 5. An enrichment analysis of DE-Gs among clusters showed the activation of RNA splicing in cluster three in GO_BP (Figure S11A) and translation in cluster 4, as evidenced by terms like ribosome and protein folding in the KEGG pathway (Figures 5F, S11B and Table S6). These clusters harbored highly intermediate cells, implying that gene expression was activated in cells with an undefined cellular status (Figure 5E). Terms that were enriched in clusters 0, 1, and six resembled the findings obtained in the subtype classification (Figures 4G and S11A–S11D). Cluster five exhibited the feature involved in inflammation. Although a fraction of cells from non-tumor sites (adjacent normal tissues) was assigned to malignant cells (Figure 5B), that had inflammatory features as marked higher ratio of cluster 5 (Figures 5E and 5G). This implied the involvement of inflammation as a key mediator in precancerous lesions and/or the underlying status in patients with PDAC (Shadhu and Xi, 2019).
Figure 5.

Unsupervised clustering of malignant cells indicates tumor-associated features in addition to subtype-specific functions
(A and B) Unsupervised clustering of the malignant cell fraction plotted on the UMAP and color-coded according to cluster (A), tissue type, and dataset (B).
(C–E) The modified subtype score (C) and cell label defined in Figure 4E are projected on the UMAP. Violin plots showing the aggregation of subtype scores for each cluster (D).
(F) Enrichment analysis of differentially expressed genes in cell clusters using the KEGG database. Gene-Concept Network showing enriched terms of the KEGG database with nodes colored according to malignant cell clusters. The node size indicates the number of leaf nodes, and the characters show the Gene ID.
(G) Heatmap of the proportion of malignant cell clusters in each sample. The normal pancreas sample is highlighted in red.
Two major CAF types and their interaction with malignant cells
As shown above, CAFs possibly orchestrate malignant cells via cell–cell communication (Figure 3). Previous studies have reported several subtypes of CAFs with different terminology, such as inflammatory cells, myofibroblasts, antigen-presenting cells (Elyada et al., 2019), complement-secreting cells, and classical cells (Chen et al., 2021). To validate these subtypes, unsupervised clustering was conducted within the fibroblast cluster of our integrated data. Batch correlation was performed among five datasets (Figures 6A and 6B) because the cell ratio of fibroblasts was largely distinct among patients (Figure S1C), and GSE111672 did not contain fibrotic components (Figure S1A). Among previous reports, the features of myofibroblasts and inflammatory cells were relatively confidential (Han et al., 2021; Raghavan et al., 2021). The expression of representative makers for pan-CAFs (COL1A1 and VIM), myofibroblast CAFs (myCAFs) (TAGLN, ACTA2, and FAP), and inflammatory CAFs (iCAFs) (DCN, PDGFRA, and CXCL12) indicated that cells could be divided along CAF subtypes (Figures 6D and S12A). Moreover, module scores obtained using the CAF-subtype signatures provided in the original report (Elyada et al., 2019) behaved via clear-cut classification as negatively correlated between myCAFs and iCAFs (Figures 6E and 6F). The activated stromal score determined in NMF of bulk RNAseq (Figures 1D and 1E) was mainly reflected as a feature in myCAFs (Figure 6C). Chen et al. suggested two CAF types, classical and complement-secreting cells (Chen et al., 2021), whereas these features were almost comparable to those of myCAFs and iCAFs, respectively (Figure S12B). Regarding antigen-presenting CAFs (apCAFs), cells that were positive for CD74, HLA-DPA1, HLA-HRA, and HLA-DRB1 and negative for PTPRC dispersedly presented in each dataset compared with previous reports (Elyada et al., 2019; Lee et al., 2021) (Figure S12C). However, these markers were remarkable in macrophages and B cells, and the actual roles of apCAFs could not be determined in the present study as apCAFs were not found in other studies (Chen et al., 2021; Lin et al., 2020).
Figure 6.

Re-clustering based on the fraction of cancer-associated fibroblasts and their subtype analysis
(A and B) An unsupervised clustering of the CAF fraction plotted on the UMAP, which was color-coded according to dataset (A) and tissue type (B).
(C–E) UMAP plots showing the stromal scores defined in Figure 1C, the expression of marker genes for CAF subtypes (D), and the subtype score for inflammatory/myofibroblast CAFs (E).
(F) Pearson’s correlation analysis of two CAF scores indicating a negative correlation.
(G) Circle plots displaying putative ligand–receptor interactions, with the width of edges representing the strength of the communication.
(H) Heatmap showing the summary of signaling pathways that contributed to outgoing or incoming communication. The color bar represents the relative signaling strength of a signaling pathway across cell types. The bars show the sum of signaling strength for each cell type or pathway.
(I) Sanky diagram summarizing the interaction between iCAF/myCAF-upregulated ligands and the receptor of malignant clusters. The width of the edges represents the weight value defined in NicheNet, and the color of the output of the edge represents the target malignant cluster. Bubble plot showing the expression levels in myCAFs or iCAFs. The size of each bubble represents the rate of cells expressing a gene, and the color represents the scaled average expression in their cell-type cluster.
The cell–cell interaction estimated among iCAFs, myCAFs, and malignant clusters using CellChat indicated that two CAF types similarly had an effect on each malignant feature (Figure 6G); the extracellular matrices commonly expressed from CAFs supported most of the malignant clusters. Clusters 3 and 4, which were intermediate cells with activated transcription and translation, had less communication, showing that gene expression is a fundamentally independent event. Several communications via growth factors were found, such as TGFb from both CAFs to cluster 6, PDGF from tumor to CAFs, and IGF from iCAFs to tumor cells (Figure 6H). Thus, CAFs and tumors possibly support each other’s growth in the TME. Furthermore, distinct interactions of the two CAFs regulating malignant functions were estimated using NicheNet (Browaeys et al., 2020), which considers not only the expression of receptors, but also downstream expression. Among the upregulated genes of iCAFs, tumor signaling for a lipoprotein receptor, chemokines, and IGF1 were specifically regulated by iCAFs (Figures 6I, S13A and S13B). In contrast, among myCAF highly expressed genes, in addition to strong contribution to the formation of extracellular matrices, INHBA signaling via activin receptors was markedly elevated in the myCAF–tumor interaction.
Discussion
The present study provided a reference scRNAseq dataset for PDAC research and recognized the importance of CAFs in the development of PDAC. As shown here, scRNAseq enables the utilization of previous bulk transcriptomics cohorts. The current cell-estimation tools for bulk transcriptomics often focus on immune cells because their markers, such as surface antigens and transcript factors, have been well studied (Sturm et al., 2019). The estimation of immune cells will provide a diagnostic tool for further immunotherapy. However, despite the promising successes of immune checkpoint inhibitor (ICI) therapies in several cancer types, there have been very limited responses to immunotherapy in patients with PDAC (Morrison et al., 2018; Balachandran et al., 2019). In fact, in patients with infiltration of T cells, the single parameter of their ratio could not predict the prognosis of PDAC because of immune suppression (Figure S4B). In contrast, in addition to T cells, normal pancreatic function and a lower number of fibroblasts were possibly promising markers because of their good prognosis. However, the abundance of CAFs affected other good-prognosis components, as patients with a lower amount of CAFs and a high level of T cells may possibly be sensitive to ICI. Thus, considering the composition of the various cell types will be useful for personalized medicine.
scRNAseq easily provides signatures of undefined subpopulations, which enable the estimation of the whole-cell composition, rather than only of immune cells, from bulk expression. Recent reports demonstrated that not only cell types, but also their detailed state could be indicated by combining scRNAseq and previous bulk transcriptomics (Luca et al., 2021; Steen et al., 2021). Although scRNAseq remains a less high-throughput technique, preparing reference scRNAseq data with a sufficient size will bring added value to bulk expression analyses. However, data quality is a significant problem in practice. In fact, the current scRNAseq data are thought as not being sufficient to comprehend the precise biology of PDAC. Although scRNAseq is expanding, the current available data are often the result of scRNAseq techniques that were custom made or processed with platforms that are currently not in use. We tried to integrate the dataset from GSM4008637, which was processed through Microwell-seq (Han et al., 2020); however, that approach failed to provide extraordinary cluster formation, even after adjusting the Seurat parameter (data not shown). Moreover, two datasets (PRJCA001063 and GSE111672) showed unacceptable expression as insulin was detected in all cells other than common practice (Tosti et al., 2021), which was shown by our ligand–receptor analysis as the insulin signaling was listed. Moreover, several datasets did not contain a sufficient population of CAFs. This is possibly the result of the fact that the specific cell-isolation protocol was not adequate for the dissociation of solid tumors, leading to biases among datasets.
As CAFs are considered a therapeutic target, further analysis of CAFs are key for unlocking the door to pancreatic cancer treatment (Geng et al., 2021). Interestingly, the signaling between CAFs and tumor cells was relatively unidirectional, i.e., from CAFs to tumor cells. Our data showed that iCAFs and myCAFs supported malignant cells using distinct growth-factor signaling pathways; moreover, the cancer-suppressive effect of CAFs has also been suggested (Mizutani et al., 2019; Öhlund et al., 2017). Moreover, CAFs affect tumor immunity as CAF-secreted APOE contributes to immune suppression (Kemp et al., 2021a). Macrophages were highly present immune cells in the current scRNAseq data, and these features were highly ranked among the upregulated genes in PDAC (Figures 1F and S2A). Interestingly, the decomposition of the bulk transcriptome showed a positive correlation between macrophages and fibroblasts (Figures 1G and S4A). Furthermore, several CAF ligands that affect tumor cells, such as APOE, C3, FN1, PLAU, and INHBA, were also expressed in the macrophage cluster (Figure S13A). These findings may imply the role of macrophage coupling with fibroblasts and the possibility of their role as a therapeutic target in PDAC (Kemp et al., 2021b).
The classification of the tumor subtypes of PDAC at the individual level remains controversial (Chan-Seng-Yue et al., 2020; Dijk et al., 2020; Martens et al., 2019; Rashid et al., 2020). However, this will yield a future diagnostic tool for personalized medicine (Froeling et al., 2021; Xu et al., 2021). As an advantage of scRNAseq, tumor subtypes could be determined at the cellular resolution and revealed the presence of intra-tumoral heterogeneity. Moreover, our data and recent studies suggested the presence of intermediate cells between the classical and basal-like types and/or their coexistence (Lee et al., 2021; O'Kane et al., 2020; Raghavan et al., 2021; Zhou et al., 2021). A pseudotime analysis indicated their trajectory from the classical to the basal-like type, which was suggestive of the epithelial–mesenchymal transition (Lee et al., 2021). Our results may indicate the features in those undefined cells as the chronic inflammation in tumor-adjacent samples and activated gene expression in intermediate cells. Moreover, the epigenetic dynamism of basal-like cells was also implied in the regulatory network analysis. In contrast, the classical-type-specific regulons shown in this study were known for their roles in retaining their classical features and for the fact that their loss leads to a basal-like (mesenchymal) transition (Brunton et al., 2020; Martinelli et al., 2017; Song et al., 2010).
Limitations of the study
The data presented here could not uncover their full story because of various limitations, such as insufficient quality of the data and missing clinical information. Moreover, the cell proportions and analyzable number of cells were entirely different among datasets and patients. Therefore, the size of the cell clusters and their cell–cell communication in this combined dataset may not represent pancreatic cancer realistically. As the platform of scRNAseq is currently undergoing refinement, and multimodal omics approaches, such as spatial transcriptomes (Moncada et al., 2020) and single-cell mutation analysis (Miles et al., 2020), are rapidly developing, further single-cell chemistry methods and their integration will be continuously required to fully understand PDAC.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Hideshi Ishii (hishii@gesurg.med.osaka-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Study design and patients
The materials sequenced in this study were collected from patients with pancreatic cancer who were admitted to the Osaka University Hospital and underwent surgical treatment from 2019 to 2021. Written informed consent was provided according to the institutional ethical approval of the Osaka University Hospital (approval number 664; chaired by President S. Nishio on December 16, 2018).
Data collection
The processed data of PRJCA001063 (Peng et al., 2019a) were obtained from zenodo [10.5281/zenodo.3969339], which had undergone quality control and annotation with cell labels into 10 cell types. Other public data were obtained from the NIH GEO database. GSE155698 (Steele et al., 2020) provided the output files of the CellRanger (10× Genomics) pipeline. GSE111672 (Moncada et al., 2020), GSE154778 (Lin et al., 2020), and GSM4293555 (Schlesinger et al., 2020) provided expression matrix files. OUGS contained four scRNAseq datasets from patients with PDAC that were newly collected from Osaka University.
Public cohorts for bulk transcriptomics
TCGA-normalized RNAseq gene expression data of PAAD were downloaded from the Broad Institute FIREHOSE portal [https://gdac.broadinstitute.org/] and their clinical data were obtained from cBioPortal [https://www.cbioportal.org/]. Differentially expressed genes between PDAC and normal pancreatic tissues were obtained from GEPIA2 (Tang et al., 2019). Previously assigned tumor subtypes were used (Rashid et al., 2020). Mutation data were obtained using the R package “TCGAbiolinks” (Colaprico et al., 2016). Two additional cohorts (PACA-AU and PACA-CA) were obtained from the ICGA Data Portal [https://dcc.icgc.org/] with a normalized expression table and clinical data. Only patients with RNAseq data were used in the decomposition analysis. A recent cohort in ArrayExpress under project E-MTAB-6830 was downloaded in FASTQ format with clinical data (Dijk et al., 2020). Reads were aligned against the human reference genome (GRCh38.p13) using STAR (ver.2.7.3a), and the gene expression values (transcripts per million) were calculated using RSEM (ver.1.3.3).
Method details
Single-cell preparation
To isolate single cells from surgically resected samples, pancreatic tumor tissues were washed with phosphate-buffered saline (PBS), cut into small pieces, and incubated in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS), 2 mg/mL of collagenase D (Roche, Basel, Switzerland), and 15 μg/mL of DNase I (Roche) for 60 min in a shaking water bath at 37°C. The digested tissues were passed through a 40-μm cell strainer. Next, the isolated cells were washed with RPMI 1640 medium, incubated in 3 mL of ACK buffer for 3 min (to lyse red blood cells), and washed again with RPMI 1640 medium. Pancreatic tumor cells were collected in PBS containing 2% FBS. The isolated cells were stained with surface antibodies for 30 min at 4°C, followed by 7AAD staining (BD Biosciences, Franklin Lakes, NJ). To confirm the proportion of living cells, a flow cytometric analysis and cell sorting were performed using a FACSAria II instrument (BD Biosciences). Data were analyzed using the FlowJo software (Tree Star, San Carlos, CA).
Single-cell RNA sequencing (scRNAseq)
Single-cell suspensions were processed through the 10× Genomics Chromium Controller according to the protocol outlined in the Chromium Single Cell 3' Reagent Kits User Guide. Chromium Next GEM Single Cell 3′ Kit v3.1 (Cat# PN-1000269), Chromium Next GEM Chip G Single Cell Kit (Cat# PN-1000127), and Dual Index Kit TT Set A (Cat# PN-1000215) were applied during the process. Approximately 16,500 live cells per sample, according to the manufacturer’s recommendations, were loaded onto the Chromium controller, to generate 10,000 single-cell gel-bead emulsions for library preparation and sequencing. Oil droplets of encapsulated single cells and barcoded beads (GEMs) were subsequently reverse transcribed in a Veriti Thermal Cycler (Thermo Fisher Scientific), resulting in cDNA tagged with a cell barcode and UMI. Next, the cDNA was amplified to generate single-cell libraries according to the manufacturer’s protocol. Quantification was achieved using an Agilent Bioanalyzer High-Sensitivity DNA assay (Agilent, High-Sensitivity DNA Kit, Cat# 5067-4626). Subsequently, the amplified cDNA was enzymatically fragmented, end repaired, and polyA tagged. Cleanup/size selection was performed on amplified cDNA using SPRIselect magnetic beads (Beckman-Coulter, SPRIselect, Cat# B23317). Next, Illumina sequencing adapters were ligated to the size-selected fragments and cleaned up using SPRIselect magnetic beads. Finally, sample indices were selected and amplified, followed by a double-sided size selection using SPRIselect magnetic beads. The final library quality was assessed using an Agilent Bioanalyzer High-Sensitivity DNA assay. Samples were then sequenced on a NovaSeq 6000 (Illumina) or DNBSEQ-G400RS (MGI) instrument in the paired-end mode. The resulting raw reads were processed by cellranger (10× Genomics).
scRNAseq data processing
The R v4.1 and Seurat v4.0.5 packages were used to process the scRNAseq data. All detailed codes were provided in zenodo [10.5281/zenodo.6024273]. The CellRanger output files were converted into expression matrices using the function Read10x. Expression matrices were transformed to Seurat objects through the “CreateSeuratObject” function. PRJCA001063 was loaded using the “SeuratDisk” package, to convert AnnData into a Seurat object. Counts of transcripts measured as UMIs were normalized to 10,000 counts per cells and log transformed. The detected range varied among the datasets (Figure S2C); therefore, the cutoff value for quality filtering was adjusted according to dataset. Cells with a high percentage of mitochondrial genes (>25%) and outer value of expression genes or UMI counts were digitally filtered out for further analysis.
Integration of scRNAseq datasets
To perform batch correction among the datasets, data integration was performed using a reciprocal principal component analysis (rPCA) according to the developer’s vignettes [https://satijalab.org/seurat/articles/integration_rpca.html]. Briefly, each dataset was scaled and a PCA was performed using highly variable genes, which were selected using the Seurat functions “FindVariableFeatures” and “SelectIntegrationFeatures”. Subsequently, an anchor was created via the Seurat function “FindIntegrationAnchors” with arguments that 30 principal components, two reference data (PRJCA001063 and GSE155698), and rPCA, followed by six datasets, were integrated through the Seurat function “IntegrateData”. The data were then scaled, analyzed for principal components, and visualized using UMAP.
Re-clustering of scRNAseq data
To subcluster the ductal subpopulation, ductal cells (types 1 and 2) were first isolated from a built scRNAseq data and objects were split into each individual level, followed by integration, as described above. Cell cycling was regressed in the data scaling based on the cell-cycle score determined by the Seurat function “CellCycleScoring”. The remaining clusters mainly consisting of G2M phase or S phase were manually removed. Furthermore, clusters with a suspicion of being contamination of other cell types or taft cells were also eliminated (Figures S7A–S7C). To analyze the features related to tumor subtypes, a PCA was performed using the tumor-subtype signatures refined in this paper (Table S3), followed by a further dimensional reduction UMAP using 1 to 6 principal components. To determine the specific features of malignant cells, clusters included in most tumor samples, but not in normal samples, were isolated, followed by unsupervised clustering with variable features, as described above (Figure S7D).
CAF were collected, with the exception of GSE111672, which did not contain fibroblasts. Batch correction among datasets was performed as described above. Based on the expression of CAF-subtype markers and module scores, cells were classified as inflammatory CAFs or myofibroblast CAFs, and used for further analyses.
Module scoring
Scores for cell cycling and a gene set of interest were calculated based on the average relative expression, as described by Tirosh et al. (Tirosh et al., 2016) and implemented in the Seurat function “AddModuleScore”. Gene sets for tumor subtypes and other PDAC components were obtained from a previous deconvolution study (Peng et al., 2019b). Gene sets for CAF classification were obtained from the original papers (Chen et al., 2021; Elyada et al., 2019).
Decomposition of bulk RNAseq data
Estimation of cell composition in the bulk transcriptome was conducted using the R package “BisqueRNA” and the function “ReferenceBasedDecomposition” (Jew et al., 2020). The required scRNAseq data were randomly extracted into 50,000 cells because of memory limitations. The correlation among individuals based on cell composition was assessed, followed by hierarchical clustering using the R package “pheatmap”.
Differentially expressed genes and their pathway enrichment analysis
Differently expressed genes among cell types or clusters were detected using the Seurat function FindAllMarkers. An enrichment analysis was implemented in the R package clusterProfiler (Wu et al., 2021) using the terms from Gene Ontology Biological Process, KEGG, WikiPathway, ReactomePA, and Disease Ontology (Yu et al., 2016). The visualization functions “dotplot,” “cnetplot,” and “treeplot” provided by clusterProfiler and enrichPlot were used to generate the enriched pathway.
Cell–cell communication
To identify and visualize the cell cross-talk among tumor cells or between malignant clusters and CAFs, the R package “CellChat” was used according to the developer’s vignette [https://github.com/sqjin/CellChat] (Jin et al., 2021). Furthermore, “NicheNet” was used to infer the actual interaction considering the expression of downstream genes (Browaeys et al., 2020).
Regulatory network analysis
SCENIC was used to estimate upstream transcription factors according to the developer’s vignette [https://github.com/aertslab/SCENIC] (Aibar et al., 2017). scRNAseq data were randomly extracted into 10,000 cells because of memory limitations. The specificity of regulons was identified based on the residual sum of squares, which was calculated in the SCENIC pipeline. The protein–protein interactions were further confirmed in the STRING database [https://string-db.org/].
Quantification and statistical analysis
The statistical tests used here are indicated in the relevant figure legends. All analyses were performed using the R software. The Kaplan–Meier survival curves were plotted using the R packages “survival” and “survminer”. The Coxph test was used to determine the hazard ratio (HR) and 95% confidence interval. Fisher’s exact test was used for between-categorical data comparisons. For the comparison of two continuous variables, data were tested by the Wilcoxon rank sum test. Multiple testing correction was performed where necessary using the Bonferroni method.
Acknowledgments
We thank Ms. Otsuka, C. and Ms. Hamano, Y., for fruitful discussion and excellent support of preparation of our research manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (15H05791 [M.M. and H.I.]; 20K18053 [R.C.]; 20H00541 [H.I.]; 21K19526 [H.I.]) and JSPS KAKENHI Grant Number 16H06279 (PAGS) [Y.S. and H.I.]. Partial support was received from Takeda Medical Research Foundation [R.C.], Kowa Life Science Foundation [R.C.], Princess Takamatsu Cancer Research Fund [H.I.], Senshin Medical Research Foundation [H.I.], and Mitsubishi Foundation [H.I.].
Author contributions
Conception, R.C., H.E., and H.I.; Design of the Work, R.C., S.K., D.Y., H.T., T.O., M.T., K.M., M.M., Y.D., H.E., and H.I.; Acquisition and Analysis, C.R., S.K., Y.T., M.K., Y.A., M.N., N.T., K.S., D.Y., Y.T., D.O., D.M., Y.S., and H.I.; Interpretation of Data, R.C., S.K., Y.T., M.K., T.S., D.E., S.T., D.Y., H.T., H.E., and H.I.; Manuscript Writing and Revision, R.C. and H.I.; Final Approval of the Manuscript, All Authors. Agreement to own Contributions, All Authors.
Declaration of interests
Partial institutional endowments were received from Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan), Hirotsu Bio Science Inc. (Tokyo, Japan), Kinshu-kai Medical Corporation (Osaka, Japan), Kyowa-kai Medical Corporation (Osaka, Japan), IDEA Consultants Inc. (Tokyo, Japan), and Unitech Co. Ltd. (Chiba, Japan).
Published: August 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104659.
Contributor Information
Hidetoshi Eguchi, Email: heguchi@gesurg.med.osaka-u.ac.jp.
Hideshi Ishii, Email: hishii@gesurg.med.osaka-u.ac.jp.
Supplemental information
Data and code availability
-
•
The authors declare that all data supporting the findings of this study are available within the article, the supplementary data, and the data repository or from the corresponding author upon reasonable request. The processed data for integrated, tumor subtype classified, malignant cell subsets, and CAF subsets are deposited with all custom codes in zenodo (https://zenodo.org/record/6024273#.Yg2eTJZUtaY). The doi listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J.C., Quinn M.C., et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Balachandran V.P., Beatty G.L., Dougan S.K. Broadening the impact of immunotherapy to pancreatic cancer: Challenges and opportunities. Gastroenterology. 2019;156:2056–2072. doi: 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M., et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- Brunton H., Caligiuri G., Cunningham R., Upstill-Goddard R., Bailey P.J., Bailey U.-M., Garner I.M., Nourse C., Dreyer S., Jones M., et al. Using chromatin accessibility to delineate therapeutic subtypes in pancreatic cancer patient-derived cell lines. Cell Rep. 2020;1:100079. doi: 10.1016/j.xpro.2020.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camolotto S.A., Belova V.K., Snyder E.L. The role of lineage specifiers in pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2018;9:1005–1013. doi: 10.21037/jgo.2018.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens J.L., Yang S., Correa de Sampaio P., Zheng X., Barua S., McAndrews K.M., Rao A., Burks J.K., Rhim A.D., Kalluri R. Stabilized epithelial phenotype of cancer cells in primary tumors leads to increased colonization of liver metastasis in pancreatic cancer. Cell Rep. 2021;35:108990. doi: 10.1016/j.celrep.2021.108990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Seng-Yue M., Kim J.C., Wilson G.W., Ng K., Figueroa E.F., O’Kane G.M., Connor A.A., Denroche R.E., Grant R.C., McLeod J., et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet. 2020;52:231–240. doi: 10.1038/s41588-019-0566-9. [DOI] [PubMed] [Google Scholar]
- Chen K., Wang Q., Li M., Guo H., Liu W., Wang F., Tian X., Yang Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine. 2021;66:103315. doi: 10.1016/j.ebiom.2021.103315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T.S., Malta T.M., Pagnotta S.M., Castiglioni I., et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson E.A., Sadanandam A., Olson P., Gibb W.J., Truitt M., Gu S., Cooc J., Weinkle J., Kim G.E., Jakkula L., et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk F., Veenstra V.L., Soer E.C., Dings M.P.G., Zhao L., Halfwerk J.B., Hooijer G.K., Damhofer H., Marzano M., Steins A., et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020;10:337. doi: 10.1038/s41598-019-56826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A., Ishikawa K., Shibata N., Ito E., Fujimoto J., Yamamoto M., Shiga H., Mochizuki H., Kawamura Y., Goshima N., et al. Enhanced expression of retinoic acid receptor alpha (RARA) induces epithelial-to-mesenchymal transition and disruption of mammary acinar structures. Mol. Oncol. 2015;9:355–364. doi: 10.1016/j.molonc.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C.X., Müller S., Keerthivasan S., Koeppen H., Hung J., Gierke S., Breart B., Foreman O., Bainbridge T.W., Castiglioni A., et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020;10:232–253. doi: 10.1158/2159-8290.cd-19-0644. [DOI] [PubMed] [Google Scholar]
- Elyada E., Bolisetty M., Laise P., Flynn W.F., Courtois E.T., Burkhart R.A., Teinor J.A., Belleau P., Biffi G., Lucito M.S., et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.cd-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froeling F.E.M., Casolino R., Pea A., Biankin A.V., Chang D.K. Molecular subtyping and precision medicine for pancreatic cancer. J. Clin. Med. 2021;10:149. doi: 10.3390/jcm10010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Chen H., Zhao L., Hu J., Yang W., Li G., Cheng C., Zhao Z., Zhang T., Li L., Sun B. Cancer-associated fibroblast (CAF) heterogeneity and targeting therapy of CAFs in pancreatic cancer. Front. Cell Dev. Biol. 2021;9:655152. doi: 10.3389/fcell.2021.655152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y., Zhang L., Zhang A., Chen X., Gao P., Zeng Q. GATA4 inhibits cell differentiation and proliferation in pancreatic cancer. PLoS One. 2018;13:e0202449. doi: 10.1371/journal.pone.0202449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., DePinho R.A., Maitra A. Single-cell RNA sequencing in pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2021;18:451–452. doi: 10.1038/s41575-021-00471-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W., et al. Construction of a human cell landscape at single-cell level. Nature. 2020;581:303–309. doi: 10.1038/s41586-020-2157-4. [DOI] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjunpää H., Llort Asens M., Guenther C., Fagerholm S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019;10:1078. doi: 10.3389/fimmu.2019.01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jew B., Alvarez M., Rahmani E., Miao Z., Ko A., Garske K.M., Sul J.H., Pietiläinen K.H., Pajukanta P., Halperin E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020;11:1971. doi: 10.1038/s41467-020-15816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabacaoglu D., Ciecielski K.J., Ruess D.A., Algül H. Immune checkpoint inhibition for pancreatic ductal adenocarcinoma: current limitations and future options. Front. Immunol. 2018;9:1878. doi: 10.3389/fimmu.2018.01878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S.B., Carpenter E.S., Steele N.G., Donahue K.L., Nwosu Z.C., Pacheco A., Velez-Delgado A., Menjivar R.E., Lima F., The S., et al. Apolipoprotein E promotes immune suppression in pancreatic cancer through NF-κB-Mediated production of CXCL1. Cancer Res. 2021;81:4305–4318. doi: 10.1158/0008-5472.can-20-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S.B., Steele N.G., Carpenter E.S., Donahue K.L., Bushnell G.G., Morris A.H., The S., Orbach S.M., Sirihorachai V.R., Nwosu Z.C., et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Science Alliance. 2021;4:e202000935. doi: 10.26508/lsa.202000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.M., Choi J.E., Hur W., Kim J.-H., Hong S.W., Lee E.B., Lee J.H., Li T.Z., Sung P.S., Yoon S.K. RAR-related orphan receptor gamma (ROR-γ) mediates epithelial-mesenchymal transition of hepatocytes during hepatic fibrosis. J. Cell. Biochem. 2017;118:2026–2036. doi: 10.1002/jcb.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesch B., Ionasz V., Paliwal S., Hruschka N., Martinez de Villarreal J., Öllinger R., Mueller S., Dienes H.P., Schindl M., Gruber E.S., et al. A GATA6-centred gene regulatory network involving HNFs and ΔNp63 controls plasticity and immune escape in pancreatic cancer. Gut. 2021:766–777. doi: 10.1136/gutjnl-2020-321397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y., Tokuda K., Yamashiro C., Higashijima F., Yoshimoto T., Ota M., Ogata T., Ashimori A., Hatano M., Kobayashi M., et al. Inhibition of epithelial-mesenchymal transition in retinal pigment epithelial cells by a retinoic acid receptor-α agonist. Sci. Rep. 2021;11:11842. doi: 10.1038/s41598-021-90618-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.J., Bernard V., Semaan A., Monberg M.E., Huang J., Stephens B.M., Lin D., Rajapakshe K.I., Weston B.R., Bhutani M.S., et al. Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single-cell transcriptomics in real-world pancreatic cancer biopsies. Clin. Cancer Res. 2021;27:5912–5921. doi: 10.1158/1078-0432.ccr-20-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Noel P., Borazanci E.H., Lee J., Amini A., Han I.W., Heo J.S., Jameson G.S., Fraser C., Steinbach M., et al. Single-cell transcriptome analysis of tumor and stromal compartments of pancreatic ductal adenocarcinoma primary tumors and metastatic lesions. Genome Med. 2020;12:80. doi: 10.1186/s13073-020-00776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca B.A., Steen C.B., Matusiak M., Azizi A., Varma S., Zhu C., Przybyl J., Espín-Pérez A., Diehn M., Alizadeh A.A., et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell. 2021;184:5482–5496.e28. doi: 10.1016/j.cell.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S., Lefesvre P., Nicolle R., Biankin A.V., Puleo F., Van Laethem J.L., Rooman I. Different shades of pancreatic ductal adenocarcinoma, different paths towards precision therapeutic applications. Ann. Oncol. 2019;30:1428–1436. doi: 10.1093/annonc/mdz181. [DOI] [PubMed] [Google Scholar]
- Martinelli P., Carrillo-de Santa Pau E., Cox T., Sainz B., Jr., Dusetti N., Greenhalf W., Rinaldi L., Costello E., Ghaneh P., Malats N., et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut. 2017;66:1665–1676. doi: 10.1136/gutjnl-2015-311256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan A., Kelly P., Turkington R.C., Jones C., Coleman H.G., McCain R.S. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018;24:4846–4861. doi: 10.3748/wjg.v24.i43.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menden K., Marouf M., Oller S., Dalmia A., Kloiber K., Heutink P., Bonn S. Deep-learning-based cell composition analysis from tissue expression profiles. bioRxiv. 2019 doi: 10.1101/659227. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles L.A., Bowman R.L., Merlinsky T.R., Csete I.S., Ooi A.T., Durruthy-Durruthy R., Bowman M., Famulare C., Patel M.A., Mendez P., et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587:477–482. doi: 10.1038/s41586-020-2864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani Y., Kobayashi H., Iida T., Asai N., Masamune A., Hara A., Esaki N., Ushida K., Mii S., Shiraki Y., et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79:5367–5381. doi: 10.1158/0008-5472.can-19-0454. [DOI] [PubMed] [Google Scholar]
- Moffitt R.A., Marayati R., Flate E.L., Volmar K.E., Loeza S.G., Hoadley K.A., Rashid N.U., Williams L.A., Eaton S.C., Chung A.H., et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015;47:1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan C.E., Nechiporuk T., Jeng S., McWeeney S.K., Wang J., Rosenfeld M.G., Mandel G. REST corepressors RCOR1 and RCOR2 and the repressor INSM1 regulate the proliferation–differentiation balance in the developing brain. Proc. Natl. Acad. Sci. USA. 2017;114:E406–E415. doi: 10.1073/pnas.1620230114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada R., Barkley D., Wagner F., Chiodin M., Devlin J.C., Baron M., Hajdu C.H., Simeone D.M., Yanai I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020;38:333–342. doi: 10.1038/s41587-019-0392-8. [DOI] [PubMed] [Google Scholar]
- Morrison A.H., Byrne K.T., Vonderheide R.H. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4:418–428. doi: 10.1016/j.trecan.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network T.C.G.A.R. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Kane G.M., Grünwald B.T., Jang G.-H., Masoomian M., Picardo S., Grant R.C., Denroche R.E., Zhang A., Wang Y., Miller J.K., et al. GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res. 2020;26:4901–4910. doi: 10.1158/1078-0432.ccr-19-3724. [DOI] [PubMed] [Google Scholar]
- Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Sun B.F., Chen C.Y., Zhou J.Y., Chen Y.S., Chen H., Liu L., Huang D., Jiang J., Cui G.S., et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29:725–738. doi: 10.1038/s41422-019-0195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.L., Moffitt R.A., Torphy R.J., Volmar K.E., Yeh J.J. De novo compartment deconvolution and weight estimation of tumor samples using DECODER. Nat. Commun. 2019;10:4729. doi: 10.1038/s41467-019-12517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlak R., Weaver J.M.J., Valle J.W., McNamara M.G. Advances in molecular profiling and categorisation of pancreatic adenocarcinoma and the implications for therapy. Cancers. 2018;10:17. doi: 10.3390/cancers10010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir M.M.F., Álvarez-Cubela S., Klein D., van Dijk J., Muñiz-Anquela R., Muñiz-Anquela R., Moreno-Hernández Y.B., Moreno-Hernández Y.B., Lanzoni G., Sadiq S., et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA. 2020;117:10876–10887. doi: 10.1073/pnas.1918314117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S., Winter P.S., Navia A.W., Williams H.L., DenAdel A., Kalekar R.L., Galvez-Reyes J., Lowder K.E., Mulugeta N., Raghavan M.S., et al. The tumor microenvironment drives transcriptional phenotypes and their plasticity in metastatic pancreatic cancer. bioRxiv. 2021 doi: 10.1101/2020.08.25.256214. Preprint at. [DOI] [Google Scholar]
- Rashid N.U., Peng X.L., Jin C., Moffitt R.A., Volmar K.E., Belt B.A., Panni R.Z., Nywening T.M., Herrera S.G., Moore K.J., et al. Purity independent subtyping of tumors (PurIST), A clinically robust, single-sample classifier for tumor subtyping in pancreatic cancer. Clin. Cancer Res. 2020;26:82–92. doi: 10.1158/1078-0432.ccr-19-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J.S., Hwang C.I., Somerville T.D.D., Milazzo J.P., Lee E.J., Da Silva B., Maiorino L., Tiriac H., Young C.M., Miyabayashi K., et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell. 2017;170:875–888.e20. doi: 10.1016/j.cell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger Y., Yosefov-Levi O., Kolodkin-Gal D., Granit R.Z., Peters L., Kalifa R., Xia L., Nasereddin A., Shiff I., Amran O., et al. Single-cell transcriptomes of pancreatic preinvasive lesions and cancer reveal acinar metaplastic cells’ heterogeneity. Nat. Commun. 2020;11:4516. doi: 10.1038/s41467-020-18207-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadhu K., Xi C. Inflammation and pancreatic cancer: an updated review. Saudi J. Gastroenterol. 2019;25:3. doi: 10.4103/sjg.sjg_390_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Skrypek N., Goossens S., De Smedt E., Vandamme N., Berx G. Epithelial-to-Mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet. 2017;33:943–959. doi: 10.1016/j.tig.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Song Y., Washington M.K., Crawford H.C. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010;70:2115–2125. doi: 10.1158/0008-5472.can-09-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele N.G., Carpenter E.S., Kemp S.B., Sirihorachai V.R., The S., Delrosario L., Lazarus J., Amir E.-a.D., Gunchick V., Espinoza C., et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat. Can. (Que.) 2020;1:1097–1112. doi: 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen C.B., Luca B.A., Esfahani M.S., Azizi A., Sworder B.J., Nabet B.Y., Kurtz D.M., Liu C.L., Khameneh F., Advani R.H., et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell. 2021;39:1422–1437.e10. doi: 10.1016/j.ccell.2021.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm G., Finotello F., Petitprez F., Zhang J.D., Baumbach J., Fridman W.H., List M., Aneichyk T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 2019;35:i436–i445. doi: 10.1093/bioinformatics/btz363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosti L., Hang Y., Debnath O., Tiesmeyer S., Trefzer T., Steiger K., Ten F.W., Lukassen S., Ballke S., Kühl A.A., et al. Single-nucleus and in situ RNA–sequencing reveal cell topographies in the human pancreas. Gastroenterology. 2021;160:1330–1344.e11. doi: 10.1053/j.gastro.2020.11.010. [DOI] [PubMed] [Google Scholar]
- Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Hu K., Bailey P., Springfeld C., Roth S., Kurilov R., Brors B., Gress T., Buchholz M., An J., et al. Clinical impact of molecular subtyping of pancreatic cancer. Front. Cell Dev. Biol. 2021;9:743908. doi: 10.3389/fcell.2021.743908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., He Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Yan G.-R., He Q.-Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2014;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- Zhou D.C., Jayasinghe R.G., Herndon J.M., Storrs E., Mo C.-K., Wu Y., Fulton R.S., Wyczalkowski M.A., Fronick C.C., Fulton L.A., et al. Spatial drivers and pre-cancer populations collaborate with the microenvironment in untreated and chemo-resistant pancreatic cancer. bioRxiv. 2021 doi: 10.1101/2021.01.13.426413. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The authors declare that all data supporting the findings of this study are available within the article, the supplementary data, and the data repository or from the corresponding author upon reasonable request. The processed data for integrated, tumor subtype classified, malignant cell subsets, and CAF subsets are deposited with all custom codes in zenodo (https://zenodo.org/record/6024273#.Yg2eTJZUtaY). The doi listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.