

Graphical abstract
Keywords: Multi-omics, Deep learning model, Data integration, Patient stratification, Glioma
Abstract
Effective and precise classification of glioma patients for their disease risks is critical to improving early diagnosis and patient survival. In the recent past, a significant amount of multi-omics data derived from cancer patients has emerged. However, a robust framework for integrating multi-omics data types to efficiently and precisely subgroup glioma patients and predict survival prognosis is still lacking. In addition, effective therapeutic targets for treating glioma patients with poor prognoses are in dire need. To begin to resolve this difficulty, we developed i-Modern, an integrated Multi-omics deep learning network method, and optimized a sophisticated computational model in gliomas that can accurately stratify patients based on their prognosis. We built a survival-associated predictive framework integrating transcription profile, miRNA expression, somatic mutations, copy number variation (CNV), DNA methylation, and protein expression. This framework achieved promising performance in distinguishing high-risk glioma patients from those with good prognoses. Furthermore, we constructed multiple fully connected neural networks that are trained on prioritized multi-omics signatures or even only potential single-omics signatures, based on our customized scoring system. Together, the landmark multi-omics signatures we identified may serve as potential therapeutic targets in gliomas.
1. Introduction
Glioma is currently one of the most common types of primary brain cancer and the incidence has continued to increase worldwide since the 1970s [1], [2]. However, the main treatment strategy for gliomas is surgery with chemotherapy and radiation therapy, which remains particularly challenging because gliomas can be hard to reach and remove completely[3], [4]. Given the high level of heterogeneity in gliomas along with the complex biological molecular markers, the median overall survival for glioma patients with the current standard of medical treatment is<3 years [5], [6]. Therefore, developing computational methods to discover novel therapeutic targets is urgently needed in the community.
To understand the heterogeneity among gliomas, many efforts have been made to identify glioma molecular subtypes [7], [8], [9], [10], [11], [12]. Multiple novel molecular subtypes are identified based on various data sources like histopathology, gene expression profiles, and driver genes [7], [8], [9], [10], [11], [12]. However, these works explore the molecular subtypes without taking survival prognosis into account, making the identified subtypes difficult to translate into clinical practice. Indeed, survival time is a key factor when classifying glioma patients into valuable subtypes with differential prognoses. Moreover, it remains challenging in discovering effective therapeutic targets for gliomas especially high-risk glioma subtypes in the community. Therefore, computational methods to robustly classify glioma patients into survival-associated subtypes and discover potential therapeutic targets are urgently needed.
With the advance of artificial intelligence and high-performance computing, machine learning technologies are playing a more and more active role in computational biology and gaining significant success in biological fields [13], [14], [15], [16], [17]. For example, deep learning has been applied to find genetic variants, DNA methylation, and image analysis, and made substantial breakthroughs in these fields [13], [14], [15], [16], [17]. The application of deep learning to multi-omics data is a promising area. However, existing methods are often limited due to the following reasons: 1. Unsupervised learning with low interpretability in predicted results or classifier labels; 2. Most multi-omics methods focused on 2–3 data types with limited scope, given the increasing amount of data types becoming available for a given sample or patient.
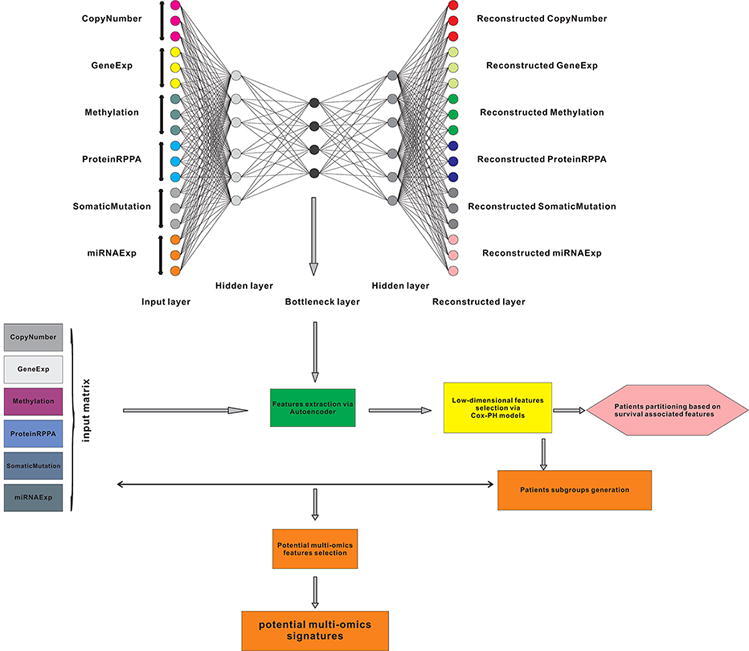
To address the outstanding issues in gliomas, for the first time, we devised a sophisticated deep learning computational framework (i-Modern, an integrated Multi-omics deep learning network method) on a comprehensive multi-omics dataset in gliomas, including mRNA profile, miRNA expression, DNA methylation, somatic mutation, RPPA protein expression, and copy number variation profiles. Considering multi-omics are high-dimensional features, we adopted and optimized one autoencoder framework to remodel multi-omics data using complex functions in the network and extracted low-dimensional informative features from the model [18], [19]. We classified glioma patients into two subgroups with significant differences in survival prognosis based on survival-associated low-dimensional features from the bottleneck in the autoencoder [18], [19]. These two subgroups represent two distinguished survival prognosis states in glioma patient populations, namely the high-risk subgroup and low-risk subgroup. After extracting potential multi-omics features associated with glioma subgroups, we selected the top ranked multi-omics features as landmark signatures. These potential signatures empower us to better understand glioma development and progression from the multi-omics perspective. Furthermore, we trained and optimized multiple fully connected neural networks, and these specified networks are proved to be robust and effective in subgrouping glioma patients compared with other benchmark prediction models based on multi-omics signatures or single-type omics signatures. Lastly, given that fully connected neural networks achieve robust predictions in subgrouping glioma patients, we evaluated and ranked all potential multi-omics signatures based on the intrinsic information in the neural networks, and some of the landmark signatures we prioritized may serve as putative therapeutic targets in gliomas.
2. Methods
2.1. Datasets employed in this study
In this study, we used a comprehensive TCGA LGG cohort as of March 2022. We obtained multi-omics glioma datasets, including RNA sequencing data (TPM normalized gene expression quantification), protein expression data (Reverse Phase Protein Array RPPA), miRNA-seq expression data (reads per million for miRNA mapping to miRbase 20), DNA methylation data (Infinium HumanMethylation450 BeadChip), copy number variation data (Affymetrix SNP Array 6.0) and somatic mutation data (DNA sequencing). For copy number variation data, we calculated a gene-level copy number value as the average copy number of the genomic region of a gene; for the DNA methylation, we mapped CpG islands within 1500 base pairs (bp) ahead of transcriptional start sites (TSS) and averaged their values as the methylation value of a gene; for somatic mutation data, we transformed the data into a matrix, where each row is one specific mutation of a gene, each column is a patient sample pair and each cell includes 0/1 elements, where 1 indicates the patient sample pair has this mutation while 0 indicates not. The data processed above and clinical patient information download were implemented by TCGA-Assembler 2 [20]. When dealing with missing values, two steps were performed. we removed the samples where more than 20% of features are missing and we removed the features that have zero value in more than 20% of patients and then we filled out the missing expression values via impute package according to the comparable criteria in other research [21], [22], [23].
2.2. Generation of low-dimensional transformed features using a deep learning framework
We treated each type of omics data such as RNA-seq as a matrix, where rows represent samples and each column represents the expression of each gene. The same data preprocessing could be applied to the other types of omics, including somatic mutation profiles where each column represents one specific mutation of each gene, protein expression profiles where each column represents the expression of each protein, copy number variation where each column represents CNV of each gene, DNA methylation profiles where each column represents methylation expression of each gene, and miRNA profiles where each column represents the expression of each miRNA. We concatenated and stacked each type of normalized matrices and generated one integrated matrix which could represent high-dimensional multi-omics features for each patient. After that, we took the integrated matrices as input in the autoencoder.
An autoencoder is one type of artificial neural networks and it is proven to learn efficient representation from a mass of features in an unsupervised manner and achieve dimensionality reduction effectively [18], [19]. Here, we took advantage of the autoencoder to transform high-dimensional multi-omics information into low-dimensional features.
2.3. The architecture of the autoencoder
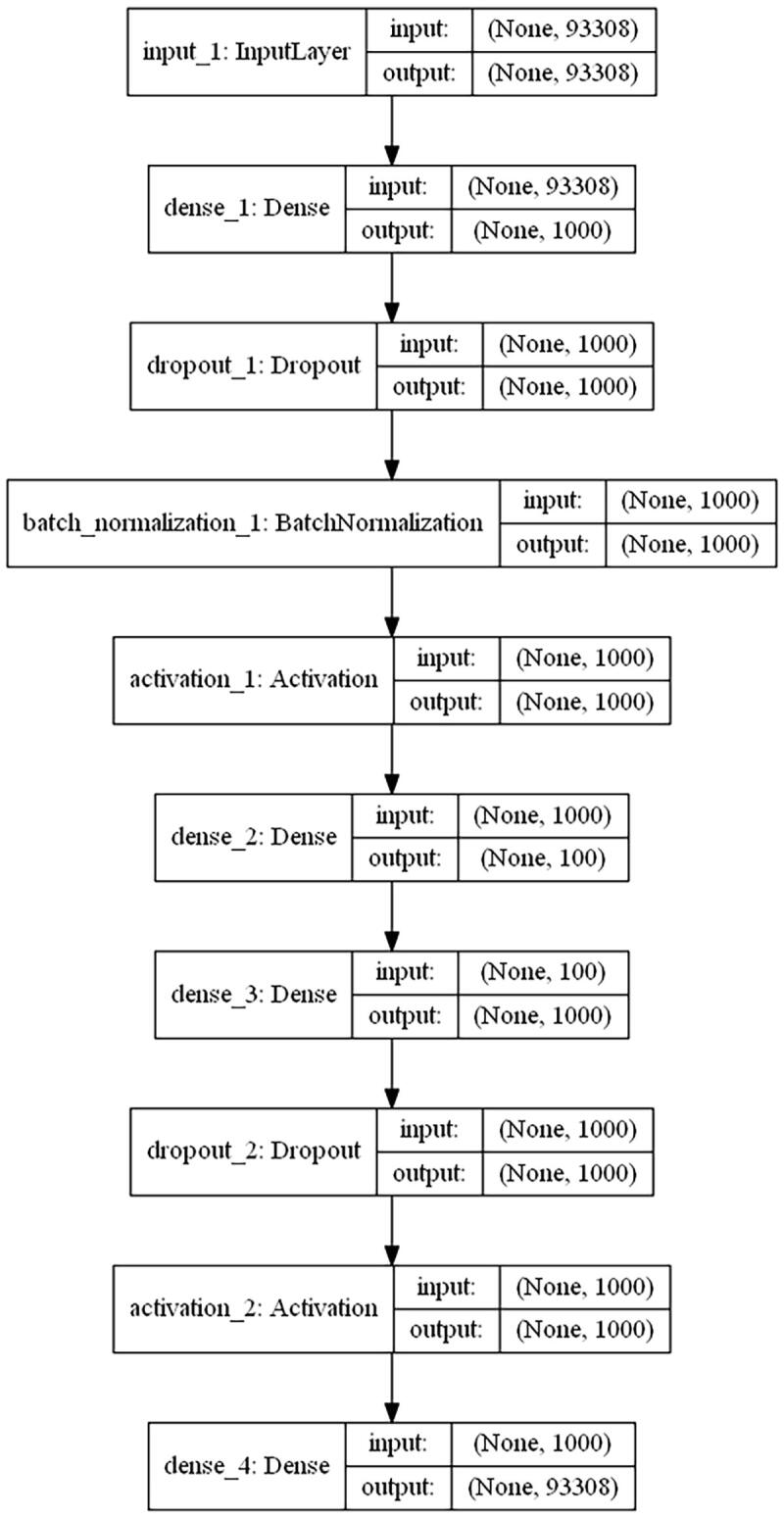
We developed one autoencoder for automatic feature extraction. Briefly speaking, being normalized and scaled, the integrated multi-omics matrix is taken as an input. In the autoencoder, several hyperparameters and parameters needed to be optimized for the best fit during the training process, including the number of neural net layers, dropout rate and the number of neurons of each layer, batch size, etc. The best hyperparameter and parameter combination was validated via 5-fold cross-validation. To get the best combination of hyperparameters in the high-dimensional hyperparameter space effectively, Bayesian optimization was adopted in the training process. Finally, the best autoencoder was obtained based on the integrated matrix data. The optimized autoencoder consists of three dense neural net layers (1000, 100, and 1000 nodes separately) with one batch normalization layer and two dropout layers placed between the dense neural net layers [24], [25]. To note, the hyperparameter space for the unit number of the bottleneck layer was [50, 100, 200, 400] and we finally chose 100 units in the layer according to loss comparison in validation datasets during autoencoder training and optimization. The activation function was tanh from the space [tanh, relu, leaky relu, and sigmoid] in those densely connected layers, and for obtaining better local minima of parameters, adam was adopted as a gradient descent optimizer [26]. The loss function of the autoencoder was mean squared error (MSE) and the autoencoder was trained for a maximum of 300 epochs. Besides, we evaluated the performance of the autoencoder in terms of mean absolute error (MAE) and root mean squared error (RMSE). Finally, as an effective regularization method, early stopping was also adopted to terminate training with 5 epochs of patience once the model performance was no longer improved on the validation dataset to avoid overfitting [27].
After the autoencoder was finalized, we made use of the trained encoder of the autoencoder based on the input omics data and obtained 100 transformed low-dimensional features from the bottleneck layer to represent multi-omics features for each patient.
2.4. Transformed feature selection and clustering
The autoencoder reduced the initial high-dimensional information to 100 low-dimensional features obtained from the bottleneck layer. Next for each of these transformed features produced by the optimized autoencoder, we combined clinical information and built a univariate Cox-PH model for each feature, and then selected informative features from which a significant Cox-PH model was obtained (both Wald and Log-rank P-value < 0.05). We further used these informative features to cluster the samples using partitioning around medoids, a more robust version of K-means [28]. To determine the optimal number of clusters, we calculated two metrics from multiple testing numbers of clusters, including Silhouette index and Calinski-Harabasz criterion.
2.5. Molecular signatures for glioma subgrouping
After obtaining the labels from partitioning around medoids, we identified the potential multi-omics features that are most correlated with the risk subgroup of patients via the Wilcoxon test for continuous features that include CNV features, gene expression features, methylation features, protein expression features, and miRNA expression features and Chi-square test or Fisher’s exact test for somatic mutation features. We obtained 12,458 potential CNV features, 13,368 potential gene expression features, 16,142 potential methylation features, 108 potential protein expression features, 15 potential somatic mutation features, and 703 potential miRNA expression features. We selected top potential features in P-values ascending order as potential multi-omics signatures, including 1,246 CNV signatures, 1,337 gene expression features, 1,614 methylation features, 108 protein expression signatures, 15 somatic mutation signatures, and 703 miRNA signatures.
2.6. Subgroup prediction via deep neural networks
After getting potential multi-omics signatures, we sought to establish a robust and effective prediction model that could achieve glioma patient stratification well only based on the potential multi-omics signatures or single-omics signatures. More specifically, to predict subgroups of glioma patients based on multi-omics data, we built and optimized subgrouping prediction models from the combination of potential multi-omics signatures. In addition, we devised and optimized subgrouping prediction models from each single-omics data, using the corresponding potential signatures respectively. To guarantee the robust classification of glioma patients, we constructed multiple prediction models, including fully connected neural networks (FCN), support vector machine (SVM), random forest (RF), LogitBoost (LGB), and Naive Bayes (NB). We constructed multiple parameter spaces for each prediction model and carried out grid search to tune model parameters to find the best model with optimal parameters.
2.7. Data partitioning and processing
We randomly split 80% of the glioma samples as training sets for 5-fold cross-validation and 20% of the glioma samples as testing sets to guarantee a reliable performance evaluation. We determined the known labels of the glioma samples by taking advantage of partitioning around medoids based on informative low-dimensional features from the autoencoder. We constructed prediction models using 80% training sets and then predicted the labels of testing sets. To guarantee the robustness of the models, we adopted 5-fold cross-validation for training sets. Besides, we ran grid search cross-validation scheme to tune prediction algorithm parameters and sought the best model training set of parameters. To guarantee a more reliable performance evaluation, we repeated the random split, cross-validation, and grid search for all prediction models 10 times and took account of performance metrics in all evaluation experiments. We applied multiple preprocessing steps on both the training sets and testing sets. For gene expression, DNA methylation, and protein expression data, we adopted a robust scaling on the omics data using the means and the standard deviations and we took advantage of unit-scale normalization for miRNA expression and CNV data according to the similar practice in [23], [29].
2.8. Evaluation metrics
To evaluate the prediction models comprehensively, we adopted multiple metrics, including accuracy, Kappa, balanced accuracy, and ROC AUC. In addition, we adopted the brier score as the mean squared error between the expected probabilities for one specific subgroup and the predicted probabilities. The score ranges from 0 to 1, and a lower score suggests higher classification accuracy.
2.9. Alternative PCA to the autoencoder
To show the robust performances of the autoencoder, we carried out principal components analysis (PCA) as the benchmark. We extracted the same number of principal components (100 PCs) as those low-dimensional features in the bottleneck of the autoencoder. We identified 11 survival-associated PCs using the Cox-PH model with the same wald and log-rank p-value cutoff and then we clustered the samples using the same partitioning around medoids for glioma patients based on these PCs.
2.10. Ranking omics signatures associated with survival prognosis
Although the deep neural network is proved to be effective and robust in subgrouping glioma patients, the network is often perceived as a black box and it’s hard to derive informative biological insights from the model. To tackle the problem, we employed Olden's algorithm in the neural network and extracted and ranked these potential multi-omics signatures according to their relative importance when subgrouping the samples [30]. Finally, we could deem those signatures ranking on the top and bottom as landmark multi-omics signatures contributing to the glioma patient stratification.
3. Results
3.1. Survival-associated glioma subtypes are identified
We developed i-Modern, an integrated Multi-omics deep learning network method, and optimized a sophisticated computational model to stratify patients based on their prognosis in glioma. To do so, from the TCGA project, we obtained 419 patient samples that included clinical information, gene expression, copy number variation (CNV), DNA methylation, protein RPPA expression, miRNA expression, and somatic mutation profiles. These multi-omics data were preprocessed as described in the “Methods” section, and finally, we obtained 18,582 gene expression features, 23,554 CNV features, 20,156 DNA methylation features, 173 protein expression features, 884 miRNA expression features, and 29,951 somatic mutation features in these patients. Our i-Modern workflow is shown in Fig. 1A. After the model training and parameter optimization, we finally adopted the architecture setting (Fig. 1B and Supplemental Fig. S1). To note, early stopping terminated the model training and made the autoencoder avoid overfit according to MSE as shown in Fig. 2B. According to MSE, MAE, and RMSE metrics, the autoencoder managed to compress the original high-dimensional features effectively with lower validation MSE, MAE, and RMSE during training (Fig. 2B and Supplemental Figs. S2A and S2B). Regarding the relationship between the input features and reconstructed features by the autoencoder, the model achieved a promising performance where the Spearman correlation coefficient was 0.789 ± 0.144 (standard deviation, SD), 0.787 ± 0.129 (SD), 0.762 ± 0.188 (SD), 0.678 ± 0.135 (SD), 0.572 ± 0.100 (SD) for CNV features, gene expression features, DNA methylation features, miRNA expression features, protein expression features, respectively, and the accuracy is 0.998 ± 0.002 for somatic mutation features (Supplemental Fig. S2C and Supplemental Table S1). After that, we extracted 100 low-dimensional features from the 100 nodes of the bottleneck as new features (Fig. 2A). To identify survival-associated low-dimensional features, we then built a univariate CoX-PH model on each low-dimensional feature and obtained 76 features significantly associated with survival prognosis (wald and log-rank p-value < 0.05). Based on these 76 features, we performed partitioning around medoids in glioma patients, with cluster testing numbers from 2 to 6. By evaluating silhouette index and calinski-harabasz index, we determined that 2 clusters would be the best setting for clustering the glioma patients (Supplemental Fig. S3A). Interestingly, the 2 clusters classified the patients into 2 subgroups that represent two significantly different survival groups (Log-Rank P-value = 3.59e-07, Cox P-value = 1.18e-07), including low-risk subgroup S1 with a better survival state and high-risk subgroup S2 with worse survival state (Fig. 2C and 2D). Considering S1 and S2 subgroups are informative for clinical practice, we thus determined to adopt these two subgrouping criteria.
Fig. 1.

The workflow for this work. (A) Multi-omics data integration and patient stratification. (B) Prediction model training, performance evaluation, and multi-omics signature ranking.
Fig. 2.

The deep learning model for glioma cancer patient stratification. (A) The architecture of the encoder we established. (B) Training and validation MSE loss iteration during model optimization in cross validation. (C)Glioma patient subgrouping visualization based on survival-associated low-dimensional features from the autoencoder. (D) Survival analysis for high-risk subgroup and low-risk subgroup based on survival-associated low-dimensional features from the autoencoder.
To corroborate the superiority of our optimized autoencoder, we also carried out principal component analysis (PCA) on glioma patients. After picking up the top 100 PCs, we filtered and got 11 survival-associated PCs using Cox-PH model (wald and log-rank p-value < 0.05). Using partitioning around medoids, we found that these PCs failed to distinguish two subgroups from each other well (Supplemental Fig. S3B).
3.2. Differential multi-omics profiles characterize high-risk and low-risk subgroups
Based on the two survival-associated subgroups generated by partitioning around medoids, we identified 12,458 potential CNV features, 13,368 potential gene expression features, 16,142 potential methylation features, 108 potential protein expression features, 15 potential somatic mutation features, and 703 potential miRNA expression features that might contribute to the difference between the high-risk subgroup and low-risk subgroup (Wilcoxon test or Independence test, P-value = 0.05). To enable the discovery of potential therapeutic targets in the high-risk glioma group, we selected the top 10% of CNV signatures (1,246), gene expression signatures (1,337), methylation signatures (1,614), and all protein expression signatures (1 0 8), somatic mutation signatures (15), and miRNA signatures (7 0 3) in P-values ascending order as putative multi-omics signatures.
As shown in Fig. 3A, high-risk subgroup S2 harbored less POLR3A copy number variation than low-risk subgroup S1 (Wilcoxon test, P-value < 2.2e-16). Our analysis indicated that decreased CNV of POLR3A was likely to be associated with glioma progression and development, rather than aberrant gene expression, methylation, protein expression, or somatic mutations of POLR3A. In addition, we found that increased MSN gene expression (Fig. 3B; Wilcoxon test, P-value < 2.2e-16), decreased GPX4 methylation (Fig. 3C; Wilcoxon test, P-value < 2.2e-16), increased ANXA1 protein expression (Fig. 3D; Wilcoxon test, P-value = 9.3e-07), increased MIR155 miRNA expression (Fig. 3E; Wilcoxon test, P-value < 2.2e-16), rare occurrence of IDH1,chr2:209113112–209113112,+,Missense_Mutation,SNP,CC > CT (Fig. 3F; Chi-Square test, P-value < 2.2e-16) might contribute to glioma development and progression as well. Remarkably, many of these differential omics genes were found to be possible risk factors in gliomas based on previous reports[31], [32], [33], [34], [35]. This suggests that our findings not only get cross-verified by previous research but also add novel insights or possible mechanisms on how these risk genes may function in glioma progression. For example, it was reported that elevated GPX4 expression is associated with the proliferation and migration of glioma cells[32]. Combined with our finding above, one could infer reasonably that the interaction between increased GPX4 expression and decreased GPX4 methylation was likely to contribute to high-risk gliomas. In addition, there were plenty of potential multi-omics signatures contributing to the difference between high-risk subgroup S2 and low-risk subgroup S1, including decreased CDHR1 CNV (Wilcoxon test, P-value < 2.2e-16), increased CASP8 gene expression (Wilcoxon test, P-value = 2.2e-13), decreased MMP9 methylation (Wilcoxon test, P-value < 2.2e-16), increased TFRC protein expression (Wilcoxon test, P-value < 2.2e-16), increased MIR222 expression (Wilcoxon test, P-value < 2.2e-16), and rare occurrence of TP53,chr17:7577121–7577121,+,Missense_Mutation,SNP,GG > AA (Fisher’s test, P-value = 0.032) in high-risk S2 subgroup (Supplemental Figs. S4A-F). Again, most of them had supporting evidence to be associated with gliomas in previous studies[36], [37], [38], [39], [40], [41], and these research works further corroborate our multi-omics findings and provide inspiration for glioma functional studies further. The detailed differential multi-omics profiles characterizing high-risk and low-risk subgroups is shown in Supplemental Table S2.
Fig. 3.

Profiles for multi-omics landmark signatures between high-risk subgroup and low-risk subgroup. (A) POLR3A CNV (Wilcoxon test, P-value < 2.2e-16). (B) MSN gene expression (Wilcoxon test, P-value < 2.2e-16). (C) GPX4 methylation (Wilcoxon test, P-value < 2.2e-16). (D) ANXA1 protein expression (Wilcoxon test, P-value = 9.3e-07). (E) MIR155 miRNA expression (Wilcoxon test, P-value < 2.2e-16).(F) IDH1,chr2:209113112–209113112,+,Missense_Mutation,SNP,CC > CT (Chi-Square test, P-value < 2.2e-16).
3.3. Fully connected neural networks achieve robust performance based on multi-omics or single-omics data
To explore the value of the multi-omics signatures in subgrouping and predicting survival prognosis for glioma patients, here we developed and optimized multiple specified fully connected neural (FCN) networks customized for classifying glioma patients. We split the samples into 10 bins randomly using a 70/30 ratio where 70% of training sets are for cross-validation and 30% of testing sets are for testing. To exploit the potential performance, we carried out grid search for optimizing parameters for each neural network. Besides, we built multiple traditional machine learning as the benchmark and optimized and evaluated the predictive performance among these models, including support vector machine (SVM), random forest (RF), LogitBoost (LGB), and Naive Bayes (NB). To evaluate the models on testing sets comprehensively, we adopted multiple measures metrics, including accuracy, Kappa, balanced accuracy, specificity, sensitivity, ROC AUC, and Brier score.
The optimized FCN achieved excellent predictive performance when classifying glioma patients into two subgroups based on potential multi-omics signatures. Compared with other prediction models, FCN obtained better performance in these two valuable and practical evaluation metrics: for balanced accuracy, FCN achieved 97.80%, outperforming 82.52% of LGB, 86.31% of NB, 87.93% of RF, 82.01% of SVM; for Kappa, FCN achieved 95.00%, outperforming 74.07% of LGB, 77.71% of NB, 81.58% of RF, 76.64% of SVM (Fig. 4A and Supplemental Table S3). In addition, FCN gained promising predictive performance in terms of accuracy, specificity, and sensitivity as well, suggesting that 2-subgrouping for glioma patients based on multi-omics signatures is effective (Fig. 4A and Supplemental Table S3).
Fig. 4.

Performance evaluation for deep learning predictive models. (A) Comparison of performance in subgrouping across model methods based on multi-omics signatures in terms of accuracy, balanced accuracy, kappa, sensitivity, and specificity. (B) Comparison of performance in subgrouping across model methods based on gene expression signatures in terms of accuracy, balanced accuracy, kappa, sensitivity, and specificity. (C) Comparison of performance in subgrouping across model methods based on methylation signatures in terms of accuracy, balanced accuracy, kappa, sensitivity, and specificity. (D) Comparison of performance in subgrouping across model methods based on multi-omics signatures in terms of ROC AUC. (E) Comparison of performance in subgrouping across model methods based on gene expression signatures in terms of ROC AUC. (F) Comparison of performance in subgrouping across model methods based on methylation signatures in terms of ROC AUC. (G) Prediction performance in subgrouping for FCNs across multi-omics signatures and single-omics signatures in terms of brier score.
Besides, we explored the scenarios where there were limited available omics signatures. Interestingly, FCN still performed well in these situations and performed better than other models. When it comes to single-gene expression signatures, FCN gained an excellent predictive performance: for balanced accuracy, FCN achieved 97.04%, outperforming 79.11% of LGB, 81.67% of NB, 86.20% of RF, and 82.00% of SVM; for Kappa, FCN achieved 95.02%, outperforming 71.35% of LGB, 71.15% of NB, 81.71% of RF, and 57.73% of SVM (Fig. 4B and Supplemental Table S3). Another robust example is single-methylation signatures where FCN performed better than other models: for balanced accuracy, FCN achieved 95.70%, outperforming 79.88% of LGB, 78.95% of NB, 81.66% of RF, and 84.45% of SVM; for Kappa, FCN achieved 91.28%, outperforming 75.11% of LGB, 74.92% of NB, 78.45% of RF, and 75.02% of SVM (Fig. 4C and Supplemental Table S3). We could get similar trends when we evaluated these models in terms of accuracy, specificity, and sensitivity and the scenario of other single-omics signatures, including single-CNV, single-protein expression, single-somatic mutation, single-miRNA expression, suggesting 2-subgrouping for glioma patients based on single-omics signatures (or incomplete multi-omics) is feasible (Supplemental Figs. S5A-D). For a comprehensive performance comparison based on multi-omics and these six single-omics signatures in these five metrics, please refer to Supplemental Table S3.
Furthermore, we evaluated the optimized FCNs in the aspect of ROC AUC and Brier scores, and these FCNs were proved to be robust. For multi-omics signatures, FCN achieved 0.990 ± 0.005 AUC and 0.037 ± 0.0.007 brier score, suggesting the complex functions in the fully connected neural network could capture the intrinsic relationship between multi-omics signatures and subgroup labels successfully (Fig. 4D and 4G). Besides, FCNs based on single-omics signatures showed good performance. For example, FCN based on single-gene expression signatures achieved 0.971 ± 0.012 AUC and 0.043 ± 0.012 brier score and FCN based on single-methylation achieved 0.971 ± 0.005 AUC and 0.047 ± 0.014 Brier score (Fig. 4E, 4F and 4G). We observed good predictive performance when we evaluated FCNs in the situation of other single-omics signatures (Fig. 4G, Supplemental Figs. S6A-D). All these results suggest that these potential multi-omics signatures can provide promising values and the optimized FCNs manage to capture the intrinsic relationship between these omics signatures and the survival prognosis of glioma patients. For a comprehensive performance comparison based on multi-omics and these six single-omics signatures in these two metrics, please refer to Supplemental Table S4 and Table S5.
3.4. Ranking of landmark omics signatures unveils potential therapeutic targets
Although the optimized FCNs show excellent performance in classifying glioma patients into high-risk and low-risk subgroups, it’s hard to derive informative clues from the black-box models [42]. Here we employed Olden's algorithm to the optimized FCNs we trained from the multi-omics data and extracted importance scores for each potential omics signature [30].
The individual signature of protein expression contributes most to classifying glioma patients into risk-associated subgroups, suggesting aberrant protein expression is more likely to be associated with the survival prognosis of gliomas (Supplemental Figs. 7A and 7B).
For identifying potential therapeutic omics signatures, we ranked multi-omics signatures in each omics layer. In the CNV signature layer, the top-ranked CNV signatures included ZNF28, FAM25C, PSG9, FAM25BP, ANXA8, NLRP2, FAM25G, ZNF285, ST8SIA6, SHLD2, DHTKD1, PTPN20, ZNF419, PSG3, BMS1P1, NUTM2D, GLUD1P2, LENG9, LILRB1, and LOC728218 (Fig. 5A). In the gene expression signature layer, the top-ranked gene expression signatures included BCAN, DLL3, SOX8, SPRY4, MARCKS, EDC3, CIAO1, MCUB, OLIG1, UBQLN4, DKK3, ITM2C, ATP1A1, TUBA1B, NCEH1, TSTD1, PPFIBP1, PPM1J, TPM3, and MAPT (Fig. 5B). In the DNA methylation signature layer, the top-ranked methylation signatures included GNG8, FGF20, MAP3K1, TMEM26, RAB11FIP4, CCNI2, BIRC3, MT3, GSAP, SPAG9, LRMP, KIAA1549, VSIG2, CALHM2, AIFM3, METTL24, PDZK1IP1, MYT1, SLC41A3, and SYNGR2 (Fig. 5C). In the miRNA expression signature layer, the top-ranked miRNA expression signatures included MIR194-2, MIR6804, MIR203A, MIR215, MIR3127, MIR3923, MIR4701, MIR3117, MIR7158, MIR5088, MIR22, MIR5708, LET7A-3, MIR101-1, MIR9-2, MIR6750, MIR127, MIR494, MIR585, and MIR1307 (Fig. 5D). In the protein expression signature layer, the top-ranked protein expression signatures included MAPK3, PRKCA, CTNNB1, SMAD3, YWHAZ, CDK1, ADAR, COPS5, PXN, YWHAB, HSPA1A, PIK3CA, IGFBP2, BCL2A1, BID, ANXA1, RICTOR, PDK1, FN1, and CCNB1 (Fig. 5E). In the somatic mutation signature layer, the top-ranked somatic mutation signatures included IDH1,chr2:209113112–209113112,+,Missense_Mutation,SNP,CC > CT, KTI12,chr1:52499071–52499097,+,In_Frame_Del,DEL,GCCCGCCACCTGAGGTCCCGCGATCGGGCCCGCCACCTGAGGTCCCGCGATCGG>--, FRMPD2,chr10:49380999–49380999,+,Silent,SNP,TT > TC, IDH1,chr2:209113113–209113113,+,Missense_Mutation,SNP,GG > GA, TP53,chr17:7577121–7577121,+,Missense_Mutation,SNP,GG > AA, ARHGAP5,chr14:32561313–32561313,+,Nonsense_Mutation,SNP,CC > CT, ARHGAP5,chr14:32561316–32561316,+,Nonsense_Mutation,SNP,GG > GT, ARHGAP5,chr14:32561340–32561340,+,Missense_Mutation,SNP,GG > GA, RP11-460 N20.5,chr7:64559189–64559189,+,RNA,SNP,GG > GA, DUX4L19,chrY:13488193–13488193,+,RNA,SNP,GG > GT, EGFR,chr7:55233043–55233043,+,Missense_Mutation,SNP,GG > TT, ARHGAP5,chr14:32561296–32561296,+,Missense_Mutation,SNP,TT > TC, ARHGAP5,chr14:32561340–32561340,+,Missense_Mutation,SNP,GG > AA, RNA5-8SP2,chr16:33965459–33965459,+,RNA,SNP,CC > CT, ATG2A,chr11:64666182–64666182,+,Missense_Mutation,SNP,CC > CG, NFKBIZ,chr3:101572635–101572635,+,Missense_Mutation,SNP,TT > TG.
Fig. 5.

Ranking scheme of multi-omics signatures. (A) Copy number variation signatures ranking. (B) Gene expression signatures ranking. (C) DNA methylation signatures ranking. (D) miRNA expression signatures ranking. (E) Protein expression signatures ranking. (F) Somatic mutation signatures ranking.
(Fig. 5F). To note, a significant portion of these top-ranked top signatures we identified were already found to be involved in glioma signaling in various biological pathways or mechanisms. For example, of top 20 gene expression signatures, more than half have been reported to be involved in gliomas, including BCAN, DLL3, SOX8, SPRY4, MARCKS, EDC3, MCUB, OLIG1, DKK3, ATP1A1, TUBA1B, PPFIBP1, TPM3, and MAPT[43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56]. For a comprehensive ranking of landmark omics signatures in patient classification, please refer to Supplemental Table S6.
3.5. Putative therapeutic targets identified from multi-omics deep learning
Although there were more than 5,000 potential multi-omics signatures in our result, the top-ranked signatures were thought to be more informative and valuable. Here, we selected the top 80 landmark multi-omics signatures, and these included 10 gene expression signatures, 29 CNV signatures, 29 methylation signatures, 9 protein expression signatures, and 3 miRNA signatures. Besides, we also considered those 16 somatic mutation signatures as potential targets.
We performed univariate Cox-PH models using each of the top 80 omics signatures and 16 somatic mutation signatures and identified 74 survival-associated multi-omics signatures out of these signatures (Log-Rank P-value < 0.05), including 5 gene expression signatures, 2 protein expression signatures, 28 methylation signatures, 2 miRNA expression signatures, 29 CNV signatures and 8 somatic mutation signatures (Supplemental Fig. S8). For example, we found that low copy number variation of CEACAM1 (Fig. 6A, Log-Rank P-value = 0.00118), high gene expression of DLL3 (Fig. 6B, Log-Rank P-value = 6.23e-06), somatic mutation of IDH1,chr2:209113112–209113112,+,Missense_Mutation,SNP,CC > CT (Fig. 6C, Log-Rank P-value = 0.0475), high protein expression of IGFBP2 (Fig. 6D, Log-Rank P-value = 0.00181), high methylation of of MAP3K1 (Fig. 6E, Log-Rank P-value = 4.14e-05), and low miRNA expression of MIR22 (Fig. 6F, Log-Rank P-value P = 0.0047) were all associated with significantly better glioma prognosis. Reassuringly, all these potential targets were previously proved to be involved in glioma progression and even associated with prognosis in some way [44], [57], [58], [59], [60], [61]. For detailed information regarding the relationship between the top 80 omics signatures and somatic mutation signatures and survival prognosis, please refer to Supplemental Table S7. Another interesting observation is that all survival-associated CNV signatures and somatic mutation signatures are positive with good prognoses, which may be good guidelines to develop therapeutic targets in glioma.
Fig. 6.

Multi-omics-based identification of potential therapeutic targets for gliomas. (A) CEACAM1 CNV (Log-Rank P-value = 0.00118). (B) DLL3 gene expression (Log-Rank P-value = 6.23e-06). (C) IDH1,chr2:209113112–209113112,+,Missense_Mutation,SNP,CC > CT (Log-Rank P-value = 0.0475). (D) IGFBP2 protein expression (Log-Rank P-value = 0.00181). (E) MAP3K1 methylation (Log-Rank P-value = 4.14e-05). (F) MIR22 miRNA expression (Log-Rank P-value = 0.0047).
Together, all these results further corroborate our multi-omics findings and may gain new insights into glioma development and accelerate precision drug discovery in the future.
4. Discussion
Heterogeneity is one of the main limitations for understanding and diagnosing glioma. Effective subgrouping method to classify glioma patients into high-risk and low-risk subgroups is promising in clinical practice in the field. To our knowledge, we are the first to use the deep learning framework to integrate comprehensive multi-omics, including gene expression, miRNA expression, CNV, protein expression, somatic mutations, and methylation in glioma. Our high-risk and low-risk subgrouping strategy will benefit the diagnosis of glioma and provide a survival prognosis guideline for patients.
The optimized FCN models from multi-omics data achieve robust predictive performance for classifying glioma patients not only on multi-omics but also single-omics data, making our deep learning framework more precise and feasible in the reality. Based on the prediction model, we derived plenty of interesting biomarkers associated glioma risk and progression. For example, CDH11 is already found to be a risk factor in gliomas, however, the mechanism of how CDH11 may play in gliomas is still unknown [62]. We discovered inhibitive CDH11 methylation will contribute to poor prognosis in gliomas rather than proliferative CDH11 methylation, aberrant CDH11 gene expression, protein expression, somatic mutation, and CNV. Other biological results from our subgrouping will benefit the understanding of gliomas in the multi-omics setting as well.
We evaluated and ranked the potential landmark multi-omics signatures and explored the importance of these signatures overall or from an individual perspective. Based on ranking of these multi-omics signatures, we discovered quite a few promising multi-omics signatures that are significant with survival prognosis and may serve as potential therapeutic targets to treating high-risk gliomas in the future.
Key Points.
-
•
A robust deep learning model to integrate multi-omics data in glioma systematically and explore glioma patient subtypes with survival prognosis.
-
•
Accurate classification of gliomas is critical with significant clinical implication, and our prediction models could robustly predict patient cancer risks even with incomplete omics data.
-
•
Our deep learning model could identify landmark omics signatures associated with cancer development, and provide novel and effective therapeutic targets for hard-to-treat cancer subtypes.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
The authors are grateful to contributions from TCGA Research Network Analysis Working Group, and also acknowledge the Biomedical Research Computing Facility at UT Austin, and Texas Advanced Computing Center (TACC) for assistance.
Funding
This work was supported by the National Institutes of Health grants GM133658 (to S.Y.) and GM137836 (to N.S.). N.S. is a CPRIT Scholar in Cancer Research with funding from the Cancer Prevention and Research Institute of Texas (CPRIT) New Investigator Grant RR160021 and Research Grant RP220292. S.Y. was also supported by a Scialog program sponsored jointly by Research Corporation for Science Advancement and the Gordon and Betty Moore Foundation (Award# 28418).
CRediT authorship contribution statement
Xingxin Pan: Investigation, Data curation, Formal analysis, Writing - original draft, Writing - review & editing. Brandon Burgman: Writing - review & editing. Erxi Wu: Writing - review & editing. Jason H. Huang: Writing - review & editing. Nidhi Sahni: Funding acquisition, Project administration, Writing - review & editing. S. Stephen Yi: Funding acquisition, Project administration, Supervision, Writing - original draft, Writing - review & editing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.06.058.
Contributor Information
Erxi Wu, Email: erxi.wu@bswhealth.org.
Jason H. Huang, Email: jason.huang@bswhealth.org.
Nidhi Sahni, Email: nsahni@mdanderson.org.
S. Stephen Yi, Email: stephen.yi@austin.utexas.edu.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary figure 1.
Supplementary figure 2.

Supplementary figure 3.

Supplementary figure 4.

Supplementary figure 5.

Supplementary figure 6.

Supplementary figure 7.

Supplementary figure 8.

References
- 1.Lönn S., Klaeboe L., Hall P., Mathiesen T., Auvinen A., Christensen H.C., et al. Incidence trends of adult primary intracerebral tumors in four Nordic countries. Int J Cancer. 2004;108:450–455. doi: 10.1002/ijc.11578. [DOI] [PubMed] [Google Scholar]
- 2.Hess KR, Broglio KR, Bondy ML: Adult glioma incidence trends in the United States, 1977–2000.Cancer: Interdiscipl Int J Am Cancer Soc 2004, 101:2293-2299. [DOI] [PubMed]
- 3.Nelson D., Diener-West M., Horton J., Chang C., Schoenfeld D., Nelson J. Combined modality approach to treatment of malignant gliomas. Natl Cancer Inst Monogr. 1988;6:279–284. [PubMed] [Google Scholar]
- 4.Choucair A.K., Levin V.A., Gutin P.H., Davis R.L., Silver P., Edwards M.S., et al. Development of multiple lesions during radiation therapy and chemotherapy in patients with gliomas. J Neurosurg. 1986;65:654–658. doi: 10.3171/jns.1986.65.5.0654. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R., Mason W.P., Van Den Bent M.J., Weller M., Fisher B., Taphoorn M.J., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 6.Wen P.Y., Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 7.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips H.S., Kharbanda S., Chen R., Forrest W.F., Soriano R.H., Wu T.D., et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 9.Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R., et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsons D.W., Jones S., Zhang X., Lin J.-C.-H., Leary R.J., Angenendt P., et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiestler B., Capper D., Holland-Letz T., Korshunov A., Von Deimling A., Pfister S.M., et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013;126:443–451. doi: 10.1007/s00401-013-1156-z. [DOI] [PubMed] [Google Scholar]
- 13.Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K., et al. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347 doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronneberger O., Fischer P. In International Conference on Medical image computing and computer-assisted intervention. Springer. 2015. Brox T: U-net: Convolutional networks for biomedical image segmentation; pp. 234–241. [Google Scholar]
- 15.Singh R., Lanchantin J., Robins G., Qi Y. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics. 2016;32:i639–i648. doi: 10.1093/bioinformatics/btw427. [DOI] [PubMed] [Google Scholar]
- 16.Pan X., Liu B., Wen X., Liu Y., Zhang X., Li S., et al. D-GPM: a deep learning method for gene promoter methylation inference. Genes. 2019;10:807. doi: 10.3390/genes10100807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B., Liu Y., Pan X., Li M., Yang S., Li S.C. DNA methylation markers for pan-cancer prediction by deep learning. Genes. 2019;10:778. doi: 10.3390/genes10100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan J., Ung M., Cheng C., Greene C.S. In Pacific symposium on biocomputing co-chairs. World Scientific. 2014. Unsupervised feature construction and knowledge extraction from genome-wide assays of breast cancer with denoising autoencoders; pp. 132–143. [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Q., Song X., Yamada H., Shibasaki R. In Thirtieth AAAI conference on artificial intelligence. 2016. Learning deep representation from big and heterogeneous data for traffic accident inference. [Google Scholar]
- 20.Wei L., Jin Z., Yang S., Xu Y., Zhu Y., Ji Y. TCGA-assembler 2: software pipeline for retrieval and processing of TCGA/CPTAC data. Bioinformatics. 2018;34:1615–1617. doi: 10.1093/bioinformatics/btx812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11:333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 22.Xiang Q., Dai X., Deng Y., He C., Wang J., Feng J., et al. Missing value imputation for microarray gene expression data using histone acetylation information. BMC Bioinf. 2008;9:1–17. doi: 10.1186/1471-2105-9-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhary K., Poirion O.B., Lu L., Garmire L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24:1248–1259. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ioffe S, Szegedy C: Batch normalization: Accelerating deep network training by reducing internal covariate shift. In International conference on machine learning. PMLR; 2015: 448-456.
- 25.Srivastava N., Hinton G., Krizhevsky A., Sutskever I., Salakhutdinov R. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15:1929–1958. [Google Scholar]
- 26.Kingma DP, Ba J: Adam: A method for stochastic optimization.arXiv preprint arXiv:14126980 2014.
- 27.Prechelt L. Automatic early stopping using cross validation: quantifying the criteria. Neural Networks. 1998;11:761–767. doi: 10.1016/s0893-6080(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 28.Van der Laan M., Pollard K., Bryan J. A new partitioning around medoids algorithm. J Stat Comput Simul. 2003;73:575–584. [Google Scholar]
- 29.Angermueller C., Pärnamaa T., Parts L., Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12:878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olden J.D., Jackson D.A. Illuminating the “black box”: a randomization approach for understanding variable contributions in artificial neural networks. Ecol Model. 2002;154:135–150. [Google Scholar]
- 31.Qin Y., Chen W., Liu B., Zhou L., Deng L., Niu W., et al. MiR-200c inhibits the tumor progression of glioma via targeting moesin. Theranostics. 2017;7:1663. doi: 10.7150/thno.17886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H., Ji B., Chen J., Huang Q., Lu X. Gpx 4 is involved in the proliferation, migration and apoptosis of glioma cells. Pathol-Res Pract. 2017;213:626–633. doi: 10.1016/j.prp.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 33.Wei L, Li L, Liu L, Yu R, Li X, Luo Z: Knockdown of annexin-A1 inhibits growth, migration and invasion of glioma cells by suppressing the PI3K/Akt signaling pathway. ASN neuro; 2021, 13:17590914211001218. [DOI] [PMC free article] [PubMed]
- 34.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q., Zou R., Zhou R., Gong C., Wang Z., Cai T., et al. miR-155 regulates glioma cells invasion and chemosensitivity by p38 isforms in vitro. J Cell Biochem. 2015;116:1213–1221. doi: 10.1002/jcb.25073. [DOI] [PubMed] [Google Scholar]
- 36.Wang H., Wang X., Xu L., Lin Y., Zhang J., Cao H. Low expression of CDHR1 is an independent unfavorable prognostic factor in glioma. J Cancer. 2021;12:5193. doi: 10.7150/jca.59948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bethke L., Sullivan K., Webb E., Murray A., Schoemaker M., Auvinen A., et al. The common D302H variant of CASP8 is associated with risk of glioma. Cancer Epidemiol Prevent Biomark. 2008;17:987–989. doi: 10.1158/1055-9965.EPI-07-2807. [DOI] [PubMed] [Google Scholar]
- 38.Xue Q., Cao L., Chen X.Y., Zhao J., Gao L., Li S.Z., et al. High expression of MMP9 in glioma affects cell proliferation and is associated with patient survival rates. Oncol Lett. 2017;13:1325–1330. doi: 10.3892/ol.2017.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behr S.C., Villanueva-Meyer J.E., Li Y., Wang Y.-H., Wei J., Moroz A., et al. Targeting iron metabolism in high-grade glioma with 68Ga-citrate PET/MR. JCI insight. 2018;3 doi: 10.1172/jci.insight.93999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gillet E., Alentorn A., Doukouré B., Mundwiller E., van Thuij H., Reijneveld J.C., et al. TP53 and p53 statuses and their clinical impact in diffuse low grade gliomas. J Neurooncol. 2014;118:131–139. doi: 10.1007/s11060-014-1407-4. [DOI] [PubMed] [Google Scholar]
- 41.Zhang C.-Z., Zhang J.-X., Zhang A.-L., Shi Z.-D., Han L., Jia Z.-F., et al. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Molecular cancer. 2010;9:1–9. doi: 10.1186/1476-4598-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benítez J.M., Castro J.L., Requena I. Are artificial neural networks black boxes? IEEE Trans Neural Networks. 1997;8:1156–1164. doi: 10.1109/72.623216. [DOI] [PubMed] [Google Scholar]
- 43.Cook P.J., Thomas R., Kannan R., de Leon E.S., Drilon A., Rosenblum M.K., et al. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun. 2017;8:1–11. doi: 10.1038/ncomms15987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spino M., Kurz S.C., Chiriboga L., Serrano J., Zeck B., Sen N., et al. Cell surface notch ligand dll3 is a therapeutic target in isocitrate dehydrogenase–mutant glioma. Clin Cancer Res. 2019;25:1261–1271. doi: 10.1158/1078-0432.CCR-18-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takouda J., Katada S., Imamura T., Sanosaka T., Nakashima K. SoxE group transcription factor Sox8 promotes astrocytic differentiation of neural stem/precursor cells downstream of Nfia. Pharmacol Res Perspect. 2021;9:e00749. doi: 10.1002/prp2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y., Wang D., Pang Q. Long noncoding RNA SPRY4-IT1 is a prognostic factor for poor overall survival and has an oncogenic role in glioma. Eur Rev Med Pharmacol Sci. 2016;20:3035–3039. [PubMed] [Google Scholar]
- 47.Jarboe J.S., Anderson J.C., Duarte C.W., Mehta T., Nowsheen S., Hicks P.H., et al. MARCKS regulates growth and radiation sensitivity and is a novel prognostic factor for glioma. Clin Cancer Res. 2012;18:3030–3041. doi: 10.1158/1078-0432.CCR-11-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park J.-M., Lee T.-H., Kang T.-H. Roles of tristetraprolin in tumorigenesis. Int J Mol Sci. 2018;19:3384. doi: 10.3390/ijms19113384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu R., Han M., Xu Y., Zhang X., Zhang C., Zhang D., et al. Coiled-coil domain containing 109B is a HIF1α-regulated gene critical for progression of human gliomas. J Transl Med. 2017;15:1–14. doi: 10.1186/s12967-017-1266-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohnishi A., Sawa H., Tsuda M., Sawamura Y., Itoh T., Iwasaki Y., et al. Expression of the oligodendroglial lineage-associated markers Olig1 and Olig2 in different types of human gliomas. J Neuropathol Exp Neurol. 2003;62:1052–1059. doi: 10.1093/jnen/62.10.1052. [DOI] [PubMed] [Google Scholar]
- 51.Mizobuchi Y., Matsuzaki K., Kuwayama K., Kitazato K., Mure H., Kageji T., et al. REIC/Dkk-3 induces cell death in human malignant glioma. Neuro-oncology. 2008;10:244–253. doi: 10.1215/15228517-2008-016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Y., Chen C., Huo G., Deng J., Zhao H., Xu R., et al. ATP1A1 integrates Akt and ERK signaling via potential interaction with Src to promote growth and survival in glioma stem cells. Front Oncol. 2019;9:320. doi: 10.3389/fonc.2019.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu H.-J., Wang L., Kang L., Du J., Li S., Cui H.-X. Sulforaphane-N-acetyl-cysteine induces autophagy through activation of ERK1/2 in U87MG and U373MG cells. Cell Physiol Biochem. 2018;51:528–542. doi: 10.1159/000495274. [DOI] [PubMed] [Google Scholar]
- 54.Dong C., Li X., Yang J., Yuan D., Zhou Y., Zhang Y., et al. PPFIBP1 induces glioma cell migration and invasion through FAK/Src/JNK signaling pathway. Cell Death Dis. 2021;12:1–11. doi: 10.1038/s41419-021-04107-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao T., Shi Y., Han D., Luan W., Qian J., Zhang J., et al. TPM3, a strong prognosis predictor, is involved in malignant progression through MMP family members and EMT-like activators in gliomas. Tumor Biology. 2014;35:9053–9059. doi: 10.1007/s13277-014-1974-1. [DOI] [PubMed] [Google Scholar]
- 56.Zaman S., Chobrutskiy B.I., Sikaria D., Blanck G. MAPT (Tau) expression is a biomarker for an increased rate of survival for low-grade glioma. Oncol Rep. 2019;41:1359–1366. doi: 10.3892/or.2018.6896. [DOI] [PubMed] [Google Scholar]
- 57.Han S., Liu Y., Cai S.J., Qian M., Ding J., Larion M., et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020;122:1580–1589. doi: 10.1038/s41416-020-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J., Liu X., Duan Y., Wang H., Su W., Wang Y., et al. Abnormal expression of circulating and tumor-infiltrating carcinoembryonic antigen-related cell adhesion molecule 1 in patients with glioma. Oncol Lett. 2018;15:3496–3503. doi: 10.3892/ol.2018.7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips L.M., Zhou X., Cogdell D.E., Chua C.Y., Huisinga A. R Hess K, Fuller GN, Zhang W: Glioma progression is mediated by an addiction to aberrant IGFBP2 expression and can be blocked using anti-IGFBP2 strategies. J Pathol. 2016;239:355–364. doi: 10.1002/path.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J., Zuo J., Wahafu A. Wang Md, Li Rc, Xie Wf: Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci Ther. 2020;26:297–308. doi: 10.1111/cns.13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tu J., Fang Y., Han D., Tan X., Xu Z., Jiang H., et al. MicroRNA-22 represses glioma development via activation of macrophage-mediated innate and adaptive immune responses. Oncogene. 2022;41:2444–2457. doi: 10.1038/s41388-022-02236-7. [DOI] [PubMed] [Google Scholar]
- 62.Chen J.-H., Huang W.-C., Bamodu O.A. Chang PM-H, Chao T-Y, Huang T-H: Monospecific antibody targeting of CDH11 inhibits epithelial-to-mesenchymal transition and represses cancer stem cell-like phenotype by up-regulating miR-335 in metastatic breast cancer, in vitro and in vivo. BMC cancer. 2019;19:1–13. doi: 10.1186/s12885-019-5811-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.