

Abstract
Background
Aortic aneurysms (AA) are pathological dilations of the aorta, associated with an overall mortality rate up to 90% in case of rupture. In addition to dilation, the aortic layers can separate by a tear within the layers, defined as aortic dissections (AD). Vascular smooth muscle cells (vSMC) are the predominant cell type within the aortic wall and dysregulation of vSMC functions contributes to AA and AD development and progression. However, since the exact underlying mechanism is poorly understood, finding potential therapeutic targets for AA and AD is challenging and surgery remains the only treatment option.
Methods
In this review, we summarize current knowledge about vSMC functions within the aortic wall and give an overview of how vSMC functions are altered in AA and AD pathogenesis, organized per anatomical location (abdominal or thoracic aorta).
Results
Important functions of vSMC in healthy or diseased conditions are apoptosis, phenotypic switch, extracellular matrix regeneration and degradation, proliferation and contractility. Stressors within the aortic wall, including inflammatory cell infiltration and (epi)genetic changes, modulate vSMC functions and cause disturbance of processes within vSMC, such as changes in TGF‐β signalling and regulatory RNA expression.
Conclusion
This review underscores a central role of vSMC dysfunction in abdominal and thoracic AA and AD development and progression. Further research focused on vSMC dysfunction in the aortic wall is necessary to find potential targets for noninvasive AA and AD treatment options.
Keywords: aortic aneurysm, aortic dissection, pathophysiology, vascular biology, vascular smooth muscle cell
1. INTRODUCTION
Aortic aneurysms (AA) are defined as a progressive weakening of the aortic wall, leading to gradual dilatation. Based on their anatomical location, AA are classified into thoracic aorta aneurysms (TAA) and abdominal aortic aneurysms (AAA), or thoraco‐abdominal AA (Crawford classification) when affecting both locations. Both TAA and AAA share some common pathophysiological characteristics and risk factors, including ageing, male sex and smoking.1, 2 Moreover, TAA is caused by inherited gene mutations in approximately 20% of the patients. 3 In addition to the dilation of the aorta, patients may present with aortic dissections (AD). AD are defined as a tear within the intimal layer of the aorta, causing separation of the intimal and medial layers of the aortic wall. AD are divided into Stanford type A (involving the aortic arch) and type B (aortic disease starts after the left subclavian artery). The aorta of AD patients can likely dilate and develop into AA. 4
The majority of AA patients is diagnosed accidently or after sudden rupture of the aortic wall. Sudden AA rupture is associated with an overall high mortality of 90% due to internal bleeding complications. 5 AD are usually diagnosed acutely; commonly patients report an acute pain between the shoulders. Currently, surgical open or endovascular repair is offered to AA and AD patients with known risk of complications or rupture, mainly based on aneurysm size. 6 Patients are usually also treated with medications for overall cardiovascular risk management. However, the majority of patients with acute type B AD is usually non‐responsive to pharmacological therapy. 7 No further pharmaceutical treatment for prevention, stabilization or reversal of AA and AD is available, since the knowledge about the pathophysiology of aortic wall weakening is limited.
Aortic wall weakening is characterized by loss of vascular smooth muscle cells (vSMCs) due to apoptosis and extracellular matrix (ECM) degradation within the middle layer of the aortic wall. Vascular SMCs are the predominant cell type in the middle aortic layer, called the tunica media. Their embryonic origin is dependent on the location in the aortic wall; in the ascending aorta and the aortic arch, vSMCs are neural crest‐derived, whereas in the descending aortic vSMC originate from somatic mesoderm. 8 vSMCs are paramount for providing structural and functional integrity of the aortic wall and ECM synthesis. They can adapt to environmental stimuli and mechanical stress due to their characteristic vSMC plasticity ‐ being able to switch between a contractile and a synthetic phenotype. 9
Imbalance in vSMC phenotypic switching can contribute to several pathological processes causing cardiovascular diseases, including AA and AD. 10 However, the exact role of vSMC dysregulation in AA and AD development and progression is complex and poorly understood. In this review, we summarize current knowledge on vSMC functions in the aortic wall and describe the potential role of vSMC dysfunction in abdominal and thoracic AA and AD development and progression. Knowledge on vSMC‐mediated pathomechanisms underlying early and advanced stages of aortic wall weakening will aid in the design of nonsurgical treatments for prevention and stabilization of AA and AD. Figure 1 gives an overview of the described vSMC functions, stressors and disturbed processes involved in progressive aortic wall weakening. A systematic database search was performed to select articles for this review and the screening process is depicted in Figure S1.
FIGURE 1.
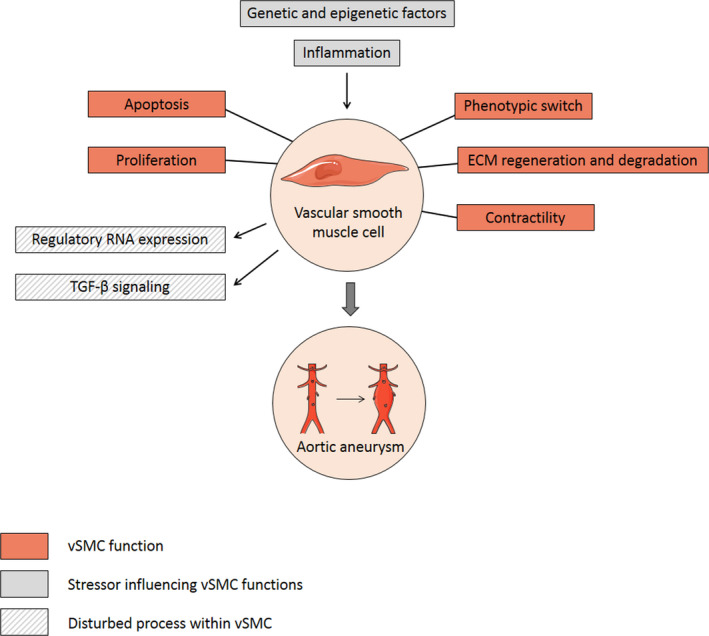
Overview of vSMC functions, stressors influencing vSMC functions and disturbed processes within vSMC during AA and AD development and progression. Apoptosis, phenotypic switch, ECM regeneration and degradation, proliferation and contractility are important functions of vSMC in the aortic wall. Dysregulation of these vSMC functions by infiltrative inflammatory cells and (epi)genetic factors can contribute to AA and AD formation. These pathological conditions activate adaptive responses within vSMC, such as changes in TGF‐β signalling and regulatory RNA expression. Abbreviations: AA, aortic aneurysm; AD, aortic dissection; ECM, extracellular matrix; RNA, ribonucleic acid; TGF‐β, transforming growth factor β; vSMC, vascular smooth muscle cell. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
1.1. vSMC functions
1.1.1. Apoptosis of vSMC
The loss of vSMC in the medial layer of the aortic wall due to apoptosis is an early hallmark of AA development. Decreased vSMC density weakens the aortic wall and limits matrix repair capacity, since vSMCs are essential for ECM regeneration. The decrease in vSMC density and coincident reduction in ECM regeneration make the aortic wall more prone to dilatation and thereby contribute to development and progression of AA and AD. A decrease in vSMC number in AAA compared with healthy human aortic tissue was first demonstrated by López‐Candales et al. 11 High levels of p53 (cell cycle arrest and apoptosis marker) observed in AAA tissue suggested that this decreased vSMC density was caused by vSMC apoptosis. 11 Increased vSMC apoptosis in AAA tissue was described in multiple human12, 13, 14, 15 and rodent studies. 16 Various mechanisms to induce the switch of vSMC to a more senescent phenotype with high susceptibility to apoptosis were reported and are shown in Figure 2. Inflammatory cell infiltration into the human aneurysmal aortic wall contributes to vSMC programmed cell death by production of apoptosis‐promoting molecules, such as cytokines and perforin.13, 17 An important cytokine involved in the inflammatory response in the aneurysmal aortic wall is monocyte chemoattractant protein‐1 (MCP‐1), and elevated MCP‐1 levels increased vSMC apoptosis in different AAA mouse models.17, 18, 19, 20 Oxidative stress, caused by products such as nitric oxide and oxygen‐free radicals produced by atherosclerotic plaques on the inside of the aortic wall and by vSMC, induces vSMC apoptosis in rats. 21 Also, endoplasmic reticulum (ER) stress is demonstrated in human AAA tissue and accompanied by severe vSMC apoptosis. 22
FIGURE 2.
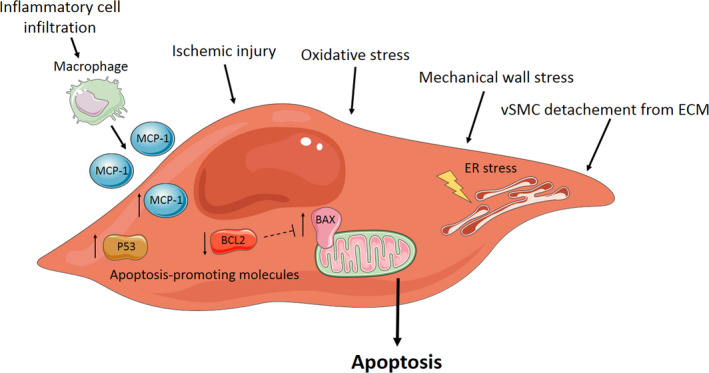
Different mechanisms to induce vSMC apoptosis within the aortic wall. Inflammatory cell infiltration into the aortic wall, ischaemic injury, oxidative stress, mechanical wall stress and detachment of the ECM can induce vSMC apoptosis. ER stress and high expression levels of MCP‐1, P53 and BAX proteins within vSMC promote vSMC apoptosis. BCL2, an inhibitor of BAX, is downregulated. Abbreviations and symbols: BAX, Bcl‐2‐associated X protein; BCL2, B‐cell lymphoma 2; ECM, extracellular matrix; ER, endoplasmic reticulum; MCP‐1, monocyte chemoattractant protein‐1; vSMC, vascular smooth muscle cell; ‐ ‐|, inhibitory effect; ↓, Decreased expression; ↑, Increased expression. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
In the human thoracic aorta, the infiltration of inflammatory cells and ER stress induced by mechanical wall stress are associated with vSMC apoptosis, contributing to TAA and thoracic aortic dissection (TAD) development and progression.23, 24 vSMC isolated from human TAA tissue exposed to oxidative stress demonstrate pro‐apoptotic traits, such as DNA fragmentation, cellular shrinkage, membrane blebbing and chromatin condensation, indicating that oxidative stress contributes to vSMC apoptosis activation in the thoracic aorta.25, 26 vSMC apoptosis in human TAA and TAD tissue is regulated by an imbalance between pro‐apoptotic Bcl‐2‐associated X protein (BAX) and anti‐apoptotic B‐cell lymphoma 2 (BCL2) levels. 27 Mechanical stress induced downregulation of transcriptional regulator yes‐associated protein‐1 (YAP‐1) in human thoracic aortic aneurysm and dissection (TAAD) tissue and was associated with increased vSMC apoptosis. 28 In contrast, upregulation of YAP‐1 in a rat model has a protective effect against TAD formation by decreasing vSMC apoptosis. 29 Heat‐shock protein (HSP)70 30 and HSP27, 31 which are increased in response to cellular oxidative stress in human TAA and TAD tissue respectively, also both have an anti‐apoptotic and protective effect in the thoracic aortic wall.
Apoptosis can be inhibited by transcription factor EB (TFEB), a master regulator of autophagy. However, TFEB is found to be downregulated in both human and mouse AA lesions, thereby increasing vSMC apoptosis and promoting AAA formation in different mouse models. 32 This critical role for autophagy in regulating vSMC death is also regulated by the autophagy protein 5 gene (ATG5). Deletion of this gene causes loss of autophagy in vSMC and increases susceptibility of vSMC death and enhancement of ER stress‐dependent inflammation within the aortic wall. 33
1.1.2. Phenotypic switch of vSMC
Vascular smooth muscle cells have the ability to switch from a contractile to a synthetic phenotype in the healthy aortic wall. Synthetic vSMCs are proliferative, while contractile vSMCs are more differentiated and express a higher number of SMC specific contractile markers. During AA and AD progression, the balance between contractile and synthetic vSMC is shifted towards synthetic vSMC and increased proteolytic enzyme production by those vSMC. These proteolytic enzymes can degrade the ECM, which facilitates the detachment of vSMC from the ECM and promotes vSMC migration and apoptosis. vSMC phenotypic switching is reported in AAA,9, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50 TAA51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61 and TAAD62, 63, 64, 65, 66, 67, 68, 69 human and rodent studies. An overview of the most important changes and inducers of vSMC phenotypic switching is indicated in Figure 3. The switch is characterized by a reduction in vSMC‐specific contractile proteins, such as smooth muscle 22alpha (SM22alpha) and alpha smooth muscle actin (αSMA) in both thoracic and abdominal aneurysmal aortic tissues.9, 39, 51, 53
FIGURE 3.
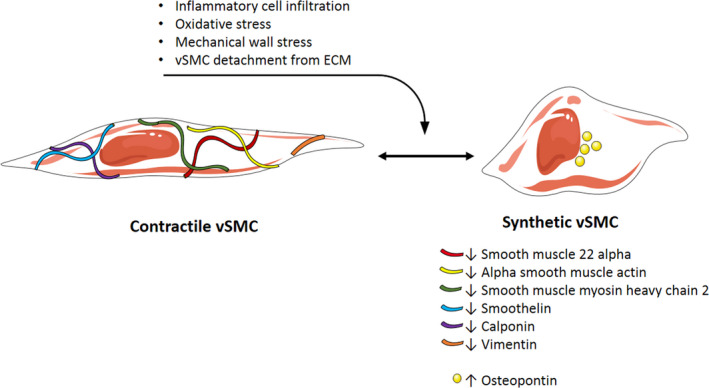
Vascular smooth muscle cell phenotypic switching. vSMC phenotypic switching can be induced by inflammatory cell infiltration, oxidative stress, mechanical wall stress or vSMC detachment from the ECM caused by ECM degradation. The switch from contractile to synthetic vSMC phenotype results in decreased expression of smooth muscle 22 alpha, alpha smooth muscle actin, smooth muscle myosin heavy chain 2, smoothelin, calponin and vimentin are decreased, while osteopontin is upregulated. Abbreviations and symbols: ECM, extracellular matrix; vSMC, vascular smooth muscle cell; ↓, Decreased expression; ↑, Increased expression. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
vSMC phenotypic switching in the abdominal aorta was found to be initiated by inflammatory cell infiltration regulated by MCP‐1 in vitro and in a mouse model. 40 Upregulation of osteoprotegerin (member of the cytokine receptor of tumour necrosis factor (TNF) receptor superfamily) in human AAA biopsies promoted an inflammatory vSMC phenotype with impaired proliferation and increased apoptosis.47, 48 Oxidative stress is another driver for the switch from contractile to synthetic phenotype.39, 41 Furthermore, the Notch1 pathway is known to be involved in AAA formation and vSMC‐specific haploinsufficiency of Notch1 maintains the contractile vSMC phenotype and prevents matrix remodelling in the murine abdominal aorta. 43
In the thoracic aneurysmal aorta, smooth muscle myosin heavy chain 2 (SM‐MHC‐2), smoothelin, calponin and vimentin were also found to be reduced.51, 53, 64 In contrast, the expression of osteopontin, a marker for synthetic vSMC, is increased in human ascending TAA and TAD tissue.61, 64, 65 HSP90 is upregulated in response to cellular stress in human TAD tissue and inhibition of HSP90 in a mouse model has a protective effect on TAD formation by suppressing vSMC phenotypic switching. 69 This is in contrast to the protective effect of upregulated HSP27 and HSP70 against vSMC apoptosis, as described above.30, 31 Hypertension causes vSMC phenotypic switching mediated by reactive oxygen species (ROS) accumulation in the human thoracic aorta, accompanied by increased mechanical wall stress. 54 ECM‐vSMC connection is crucial for maintenance of vSMC phenotype; vSMC‐specific deletion of the ECM fibulin‐4 gene in a mouse model leads to loss of the vSMC contractile phenotype, hyperproliferation and ascending AA formation.58, 59
1.1.3. ECM regeneration and degradation regulated by vSMC
Vascular smooth muscle cells are involved in regulation of the aortic wall elasticity and production of ECM proteins. Together with elastic ECM fibres, such as collagen and elastin, vSMCs form a functional unit vital for maintaining structural and functional integrity. This connection between vSMC and the ECM is essential for regulation of vSMC functions, such as vSMC contractility. However, ECM degradation occurs in aneurysmal tissue, contributing to rupture and dilation of the aortic wall. vSMCs are actively participating in this process through activation and modulation of matrix metalloproteinases (MMP), the A disintegrin and metalloproteinase (ADAM) family, cathepsins and the fibrinolytic pathway. An overview of the role of vSMC in ECM degradation is shown in Figure 4.
FIGURE 4.
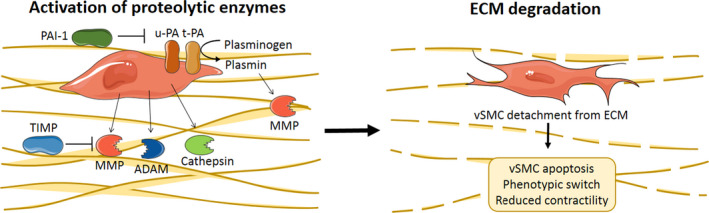
Extracellular matrix degradation by proteolytic enzymes regulated by vSMC. MMP, ADAM and cathepsins, activated and secreted by vSMC, can break down the ECM. Plasminogen can be activated into plasmin by u‐PA or t‐PA, both located on the vSMC membrane. PAI‐1 can inhibit u‐PA, and t‐PA. MMP can be inhibited by TIMP and activated by plasmin. ECM degradation causes vSMC detachment from the ECM and results in vSMC apoptosis, phenotypic switching and reduced contractility. Abbreviations: ADAM, the A disintegrin and metalloproteinase; ECM, extracellular matrix; MMP, matrix metalloprotease; PAI‐1, plasminogen activator inhibitor; TIMP, tissue inhibitors of matrix metalloproteinases; t‐PA, tissue‐type plasminogen activator; u‐PA, urokinase plasminogen activator; vSMC, vascular smooth muscle cell. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
Matrix metalloproteinases (MMP) are endopeptidases capable of degrading ECM proteins, resulting in aortic tissue remodelling. Synthesis levels of MMP by vSMC in AAA tissue are found to be increased compared with control aortic tissues in human and rodent studies, especially of MMP‐270, 71, 72, 73, 74, 75, 76, 77, 78 and MMP‐9.70, 72, 76, 78, 79, 80, 81 Also, other MMP, namely MMP‐1,80, 82, 83 MMP‐3, 83 MMP‐7, 84 MMP‐12 80 and MMP‐13, 85 are expressed by vSMC in AAA tissue. An important MMP regulator found in human AAA tissue is the activator of MMP‐2, called membrane type 1 MMP (MT‐1 MMP).75, 80, 86 In TAA and TAD tissue, increased levels of MMP‐1, MMP‐2 and MMP‐9 expression by vSMC compared with control are reported.62, 87, 88, 89, 90, 91 MMP‐2 has a dual role in ECM turnover in the mouse thoracic aorta: it mediates ECM degradation, as well as ECM synthesis. 92 MMP‐17 loss of function in a genetic mouse model resulted in dysfunctional vSMC, altered ECM and increased TAA susceptibility, which suggested that proteolytic MMP‐17 activity is an important regulator of vSMC function. 93 The secretion and activation of MMP is regulated by tissue inhibitors of matrix metalloproteinases (TIMP) expressed in AAA tissue73, 77, 80, 86, 94, 95, 96 and TAA and TAD tissue.62, 97
The proteolytic function of the ADAM family of metalloproteases regulated by vSMC also plays a role in ECM degradation. Similar expression levels of ADAMs 8, 9, 10, 12, 15 and 17 were found in vSMC of human AAA tissue and control aorta. 94 In contrast, ADAM17 was upregulated in mouse AAA 98 and human TAA tissue, 99 and ADAM17 deficiency in vSMC had a protective function in two different mouse models.98, 99 In human TAAD tissue, increased expression of A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)‐1 and ADAMTS‐4 localized in vSMC was reported compared with control aortic tissue. Increased levels of ADAMTS‐1 and ADAMTS‐4 were associated with more degradation of the ECM proteoglycan versican. 100
The activation of another type of proteases, called cathepsins, can modulate the ECM and vSMC function in the aortic aneurysmal wall as well. Cathepsins B, D, G, L and S were found, localized in vSMC, in higher concentrations in human AAA tissue, compared with control.101, 102, 103, 104 Cathepsins can promote vSMC apoptosis,105, 106 elastin degradation, 105 protease activity106, 107 and inflammatory cell accumulation. 107 Reduced levels of the cysteine protease inhibitor cystatin C were demonstrated in human AAA lesions 108 and complete cystatin C knockout in an AAA mouse model resulted in inflammatory cell accumulation, severe elastin fragmentation and increased vSMC apoptosis. 109
Extracellular matrix breakdown by proteolytic enzymes and vSMC apoptosis increases arterial wall permeability and thereby the advection of plasma proteins interacting with vSMC and ECM in the aortic wall. As a result, high concentrations of plasminogen are present and activate the fibrinolytic pathway in the aortic wall. In the human abdominal aorta, the expression of tissue‐type plasminogen activator (t‐PA) on vSMC membranes can activate plasminogen into plasmin. 110 Once active, plasmin is involved in MMP activation and fibronectin degradation, both leading to vSMC detachment from the ECM. Overexpression of plasminogen activator inhibitor (PAI‐1), an inhibitor of t‐PA and urokinase plasminogen activator (u‐PA), in an AAA rat model prevented aneurysm formation by the inhibition of plasminogen activators and MMP. 111 In human TAA tissue, accumulation of plasminogen, t‐PA, u‐PA and plasmin was also seen, associated with vSMC. 112 Inhibition and clearance of plasmin in human TAA tissue is regulated by plasmin‐protease nexin 1 (PN‐1) complexes internalized via low‐density lipoprotein receptor‐related protein‐1 (LRP‐1) in vSMC. 113 Overexpression of PN‐1 and PAI‐1 mRNA and proteins in human vSMC cultures isolated from TAA tissue compared with TAD and control tissue was associated with plasmin inhibition and protection of vSMC from detachment and death. 114
1.1.4. vSMC proliferation
Vascular smooth muscle cells proliferation increases the number of vSMC within the aortic wall and can therefore have a protective effect and delay AA and AD development. However, diminished proliferative capacity is seen in human AAA‐derived vSMC compared with nonaneurysmal vSMC. 115 Inhibition of vSMC proliferation in human AAA biopsies is caused by an G1 cell cycle arrest, regulated by urocortin‐2. 116 High aortic flow causes aortic mechanical stretch, and this stimulates endothelial cell and vSMC proliferation in an experimental rat model.117, 118 In an AAA rabbit model, interleukin‐10 (IL‐10) treatment inhibited the inflammatory response in the aortic wall and promoted vSMC proliferation. 119 In the tunica media of human thoracic AD patients, SM22 is downregulated, which was associated with promotion of vSMC proliferation in an in vitro assay. 120 Furthermore, upregulated HSP27 has a protective effect on vSMC by promoting vSMC proliferation, suppressing apoptosis and protecting against elevated oxidative stress in human TAD samples. 31
1.1.5. Contractility of vSMC
The ability of vSMC within the aortic wall to contract is important to maintain vSMC/ECM tensegrity and plays a key role in mechanotransduction. Impaired contractility of vSMC of human AAA patients upon ionomycin stimulation was shown compared with control vSMC. 121 Fibulin‐5‐deficient mice showed diminished vSMC contractility of the thoracic aorta in response to potassium loading, suggesting that sufficient vSMC‐ECM connection is essential for vSMC contraction. 122 Contraction assays in LRP‐1‐deficient mice demonstrated attenuated vasoreactivity, which identified LRP‐1 as a critical modulator of vSMC contraction by regulating calcium signalling in the thoracic aorta. 123
1.2. Stressors and disturbed processes influencing vSMC functions
1.2.1. Inflammation
Vascular smooth muscle cells have the ability to attract inflammatory cells and induce a pro‐inflammatory state in the aortic wall. The infiltration of these inflammatory cells into the aortic wall and their release of cytokines and other mediators affect vSMC functions. An overview of the inflammatory response in the aortic wall regulated by vSMC and affecting vSMC functions during AA and AD development is depicted in Figure 5.
FIGURE 5.
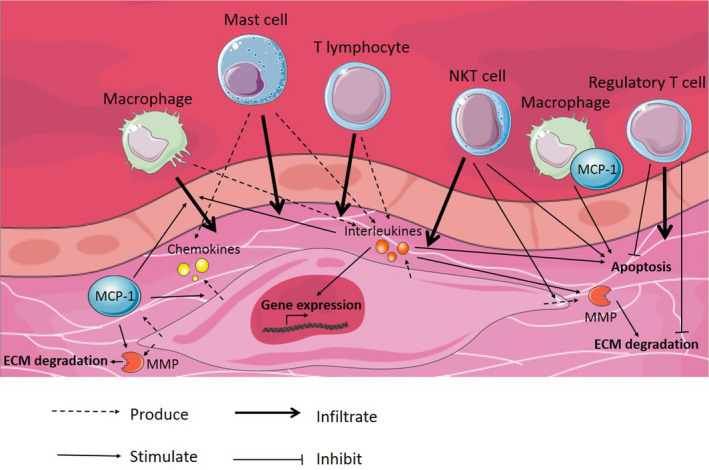
Inflammatory response in the aortic wall regulated by vSMC and affecting vSMC functions. During AA and AD progression, inflammatory cells infiltrate into the aortic wall. These inflammatory cells, together with vSMC, produce chemokines and interleukins, which subsequently activate various processes in the tunica media. vSMC start producing MMP, in turn causing ECM degradation. vSMC apoptosis is induced and gene expression in vSMC is modulated. In contrast, infiltration of regulatory T cells has a protective effect by inhibiting vSMC apoptosis and ECM degradation. Abbreviations: AA, aortic aneurysm; AD, aortic dissection; ECM, extracellular matrix; MCP‐1, monocyte chemoattractant protein‐1; MMP, matrix metalloprotease; NKT cell, natural killer T cell; vSMC, vascular smooth muscle cell. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
MCP‐1 is an important regulator of the inflammatory response during AAA development and is found in increased levels in human 124 and mouse AAA tissue125, 126 compared with control tissue. MCP‐1 in the tunica media can increase MMP‐9 secretion by vSMC 124 and induce more chemokine production by vSMC 125 or macrophage accumulation within the aortic wall. 126 An in vitro vSMC/macrophage co‐culture was used to demonstrate that macrophages primed with MCP‐1 cause higher levels of vSMC apoptosis compared with control macrophages, suggesting that MCP‐1‐primed macrophages are more cytotoxic. 127 Lysyl oxidase (LOX) can suppress secretion of MCP‐1 in cultured vSMC, and enhanced LOX activity in an AAA mouse model prevented macrophage infiltration and AAA progression. 128
The expression of interleukins (IL), such as IL‐5, 129 IL‐6126, 130 and IL‐1beta (IL‐1b), 131 in the aortic wall modulates vSMC functions and is involved in the inflammatory response contributing to AAA development. IL‐5 stimulation of mouse aortic vSMC increased MMP‐2 and MMP‐9, while IL‐5 stimulation of macrophages did not alter MMP expression. 129 Elevated wall tension caused by hypertension resulted in IL‐6 production by vSMC, subsequently causing macrophage infiltration in an AAA mouse model. 126 IL‐1b expression mediates an increased matrix turnover by affecting collagenase and collagen gene expression in human vSMC. 131 Similar findings were reported for TAA tissue, where IL‐1b protein was increased approximately 20‐fold compared with control tissue. Genetic deletion of IL‐1b and IL‐1 receptor in an experimental TAA mouse model demonstrated preserved elastin and vSMC associated with fewer inflammatory cells. 132 In TAD patients, increased IL‐6 downregulated the expression of vSMC contractile proteins alpha‐SMA and SM22alpha and induced autophagy. 133 In human TAD samples, IL‐18 expression was increased, mainly derived from macrophages and partly from T lymphocytes and vSMC. This upregulated IL‐18 expression participated in macrophage‐induced vSMC apoptosis. 134
The infiltration of different inflammatory cell types into the abdominal aortic wall affects vSMC functions. Accumulated mast cells in AAA mouse lesions release pro‐inflammatory cytokines IL‐6 and IFN‐gamma, which may induce vSMC apoptosis, proteolytic enzyme expression and aortic wall remodelling. 135 In vitro studies with human vSMC showed that both T and natural killer T (NKT) cells adhere to vSMC. While CD4+ T cells enhance vSMC proliferation, CD4+ CD161+ NKT cells inhibit vSMC proliferation by inducing apoptosis, 136 which suggest that these cells contribute to AAA development. In contrast, regulatory T cells were shown to have a protective effect in an AAA mouse model. Regulatory T cells decreased pro‐inflammatory cytokines levels, MMP‐2 and MMP‐9 expression, vSMC apoptosis and oxidative stress, while the expression of the anti‐inflammatory IL‐10 and TGF‐β was increased. 137 Furthermore, regulatory T‐cell treatment in an AAA mouse model decreased expression of the pro‐inflammatory protein cyclooxygenase‐2 in vSMC and macrophages and increased vSMC viability. 138 Patients suffering from bicuspid aortic valve (BAV) and tricuspid aortic valve (TAV) disease have an increased risk for TAAD development and rupture of the aortic wall. When comparing gene expression profiles of human BAV and TAV tissue biopsy samples, an immune response activation is only seen in the aortic media of TAV patients, suggesting that inflammation is involved in TAA formation in TAV patients, but not in BAV patients. 139
1.2.2. Genetic and epigenetic factors
Thoracic aortic aneurysm and dissection pathogenesis is often driven by a genetic component, affecting proteins encoding for the vSMC contractile apparatus and the ECM in the tunica media. In contrast to TAAD, little is known about genetic causes of AAA. 140 A highly efficient method to investigate the pathogenicity of variants of vSMC contractile apparatus proteins is the use of SMC‐like cells generated by transdifferentiation of human dermal fibroblasts. 141 Figure 6 and Table 1 show an overview of mutations affecting vSMC functions and causing TAAD formation.
FIGURE 6.
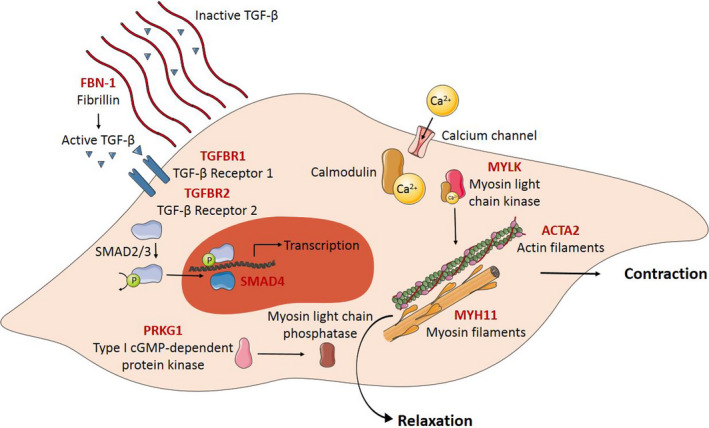
Overview of the contractile apparatus in vSMC and mutations affecting vSMC functions causing TAAD formation. The contraction of vSMC is initiated by Ca2+ influx, binding to calmodulin. This Ca2+/Calmodulin complex binds to myosin light chain kinase, which phosphorylates the myosin filaments and results in contraction. Decrease in the Ca2+ concentration within in the cell inactivates myosin light chain kinase and de‐phosphorylation of the myosin filaments, controlled by type I cGMP‐dependent protein kinase, results in vSMC relaxation. Mutations in genes encoding for vSMC contractile apparatus proteins (indicated in red) result in reduced force generation. Mutations in genes encoding for the large glycoprotein fibrillin‐1 (indicated in red) affect the structure of microfibrils and result in increased active TGF‐β levels. Mutations in the TGF‐β signalling pathway and downstream proteins (indicated in red) can also contribute to TAAD formation by affecting transcription. Abbreviations: Ca2+, calcium ion; P, phosphate; TAAD, thoracic aortic aneurysm and dissection; TGFBR, transforming growth factor β receptor; TGF‐β, transforming growth factor β; vSMC, vascular smooth muscle cell. Elements were modified from Servier Medical Art, licensed under a Creative Common Attribution 3.0 Generic License. https://smart.servier.com/; https://creativecommons.org/licenses/by/3.0/
TABLE 1.
Overview of known genetic causes of familiar TAAD and the affected processes within the aortic wall
| Gene | Affected process within the aortic wall | Reference |
|---|---|---|
| ACTA2 | vSMC contractility | 142, 143, 144, 145, 146, 147, 148, 149, 150, 151 |
| MYH11 | vSMC contractility | 148, 152, 153, 154, 155 |
| PRKG1 | vSMC contractility | 156, 157 |
| MYLK | vSMC contractility | 158 |
| FBN1 (Marfan syndrome) |
ECM organization vSMC phenotypic switching |
|
| TGFB1/2 (Loeys‐Dietz syndrome) |
vSMC contractility vSMC phenotypic switching |
|
| SMAD4 |
vSMC apoptosis vSMC contractility ECM regeneration and degradation |
177, 178 |
| COL3A1 (Ehlers‐Danlos syndrome type IV) | ECM organization | 170 |
| LOX | ECM organization | 171 |
| MFAP5 | ECM organization | 172 |
| FOXE3 | vSMC apoptosis and survival | 183 |
Abbreviations: ECM, extracellular matrix; TAAD, thoracic aortic aneurysm and dissection; vSMC, vascular smooth muscle cell.
Mutations affecting the vSMC contractile apparatus
The vSMC contractile apparatus consists of thin SMC‐specific filaments α‐actin (SM α‐actin, encoded by ACTA2) and thick SMC‐specific filament myosin heavy chain dimer (MHC, encoded by MYH11), assembled with two regulatory light chains (LC) and two essential LC. vSMC contraction is initiated by calcium (Ca2+) influx, binding to calmodulin. This Ca2+/calmodulin complex binds to myosin LC kinase (MLCK, encoded by MYLK), leading to phosphorylation of the regulatory LC of the myosin filaments. The force‐generating cycle is activated by myosin ATPase, and the myosin motor heads move along the actin filaments. After vSMC contraction, a decrease in Ca2+ concentration within the cell inactivates MLCK. The myosin regulatory LC is de‐phosphorylated by myosin light LC phosphatase (MLCP), which is controlled by type I cGMP‐dependent protein kinase (PKG‐1, encoded by PRKG1). A schematic overview of the vSMC contraction is shown in Figure 6.
Mutations in genes encoding the vSMC contractile proteins result in reduced force generation and destabilizing the contractile apparatus, which contributes to the development of TAADs. The most common mutations causing familial TAAD are found in ACTA2, causing 14% of all cases.142, 143, 144, 145, 146, 147, 148, 149, 150, 151 Loss of SM α‐actin filaments in vSMC causes decreased integrin recruitment for cell‐matrix adhesion. This reduced interaction between vSMC and the ECM combined with reduced contractility makes these vSMCs unable to generate force. 144 Mutations in MYH11, encoding the other main protein of the vSMC contractile apparatus myosin, alter contractile vSMC function and may contribute to TAAD development.148, 152, 153, 154, 155 Heterozygous gain‐of‐function mutations in PRKG1 are also associated with TAAD.156, 157 Increased PKG‐1 activity resulted in decreased phosphorylation of myosin regulatory LC and thereby decreased vSMC contraction. 156 Lastly, heterozygous loss‐of‐function mutations in MYLK are associated with familial TAAD. 158
Mutations affecting the ECM
The contractile apparatus in vSMC is anchored to the ECM by integrin‐containing focal adhesions, called dense plaques. Integrin receptors in these dense plaques bind to elastin through microfibrils, with the large glycoprotein fibrillin‐1 (encoded by FBN1) as the main protein in microfibrils. Heterozygous FBN1 mutations cause the connective tissue disorder Marfan syndrome. Marfan patients tend to be tall, slender and have with hyper‐flexible joints, accompanied by serious complications in organs containing connective tissue. Marfan disease also affects the connective tissue in the aortic wall, which makes them susceptible for TAA, but not AAA formation, at younger age compared with nongenetic TAA patients.
The aortic wall in Marfan patients contains low levels of fibrillin‐1 and undifferentiated vSMC. 159 vSMC detachment from the ECM causes vSMC phenotypic switching and increases expression of MMP‐9 and elastin in a Marfan mouse model. 160 vSMC cultured from Marfan mice showed a more mesenchymal‐like phenotype with impaired force‐generating capacity due to impaired cytoskeleton and focal adhesion organization. 161 These phenotypic changes are also seen in human Marfan vSMC, affecting SM α‐actin, smoothelin, SM22 alpha, calponin‐1 and myocardin (transcription factor essential for vSMC‐specific differentiation), and resulting in greater cellular and ECM stiffness. 162 These studies suggested that loss of vSMC attachment caused by the FBN1 mutations activates a nonproductive programme to synthesize and remodel the ECM.
Fibrillin‐1 does not only play a role in the structural integrity of the aortic wall, but also regulates the bioavailability of TGF‐β. Fibrillin‐1 and microfibrils can together create a reservoir for TGF‐β, maintaining TGF‐β there in an inactive from. Mutations in fibrillin‐1 disrupt the biological scaffolding role of fibrillin‐1, causing an increase in active TGF‐β, thereby activating TGF‐β receptor‐mediated Smad signalling. 163 Increased TGF‐β expression is shown in vSMC of Marfan patients compared with healthy subjects,164, 165 leading to the activation of canonical SMAD3 signalling 164 and the noncanonical ERK signalling. 166 The activation of these pathways can model vSMC proliferation, apoptosis, inflammation and phenotypic switching. Furthermore, increased TGF‐β in Marfan patients induced vSMC senescence through excessive ROS generation 165 and increased vSMC apoptosis. 167 The potential role of TGF‐β signalling in AA and AD formation is further described below.
Reactive oxygen species production in vSMC of Marfan mice was found to be anatomically specific, with increased NADPH activity in vSMC derived from the ascending aorta compared with vSMC derived from the descending aorta. 168 ROS produced by vSMC in the tunica media of Marfan patients targets actin‐based cytoskeleton components and regulators of ECM homeostasis. 169
There are more known mutations affecting ECM proteins, subsequently disturbing vSMC functions and initiating AA or AD development. Ehlers‐Danlos syndrome type IV is characterized by an COL3A1 mutation. 170 A mutation in the LOX gene, encoding for an enzyme that normally cross‐links collagen and elastin molecules in the aortic wall, causes a loss of stability and elasticity of the aortic wall. 171 Loss‐of‐function mutations in MFAP5, encoding for microfibril‐associated glycoprotein 2, which is a component of the ECM interacting with fibrillin‐1, also appear as an underlying cause of familiar TAAD development. 172
Mutations affecting TGF‐β signalling
Loeys‐Dietz syndrome is characterized by mutations in genes encoding for components of the TFG‐β pathway, including TGF‐β receptor (TGFBR)‐1 and 2. 173 The role of the altered TGF‐β signalling pathway on vSMC function during AA and AD development and progression is complex, and controversial findings have been reported. vSMC explanted from patients with heterozygous mutations in TGFBR2 (encoded by TGFBR2) showed decreased expression of vSMC contractile proteins, which may contribute to TAAD by affecting the contractile function of vSMC. 174 vSMC‐specific deletion of TGFBR2 in adult mice resulted in mild thoracic aneurysmal degeneration, while vSMC‐specific deletion of TGFBR1 in the same mouse model caused severe thoracic aneurysmal degeneration. 175 In contrast to these findings, vSMC‐specific TGFBR2 disruption in an AAA mouse model prevented AAA formation by reduced elastin degradation, vSMC loss, macrophage infiltration and MMP expression. 176 An explanation for these contrasting effects of TGF‐β on the aortic wall can be the different embryological origins of abdominal and thoracic vSMC. Also, genetic variants of SMAD4, the intracellular secondary messenger of the TGF‐β pathway, are reported as promotors of vSMC apoptosis, proteoglycan degradation and reduced contractile protein gene expression in human TAAD pathogenesis.177, 178 Likewise, SMAD3‐deficient mice developed TAA rapidly by the activation of immune responses, but did not show vSMC loss or MMP activation in vSMC. 179
In human TAD tissue, TGF‐β stimulation was found to contribute to aortic wall weakening by iinducing vSMC phenotypic switching from a contractile to a synthetic phenotype. 180 In human AAA tissue, TGF‐β mRNA and protein expression are found to be increased compared with control tissue.181, 182 This overexpression of TGF‐β was associated with reduced vSMC density, caused by more vSMC apoptosis, while vSMC proliferation was reduced. 182 In contrast, another study reports no changes in proliferation of aneurysmal rat vSMC treated with TGF‐β. 78
Mutations affecting other vSMC functions
FOXE3 encodes for a transcription factor regulating anti‐apoptotic and pro‐survival pathways and FOXE3 mutations were found to contribute to TAAD development. 183 FOXE3‐deficient mice showed reduced vSMC density and impaired vSMC differentiation in the ascending aorta. 183
Epigenetic changes in vSMC
Not only genetic changes but also epigenetic changes in vSMC are related to AAA,184, 185, 186 TAA187, 188 and TAAD 189 pathogenesis. In contrast to genetics, epigenetic changes can modify gene expression without altering the genetic code itself, for example by DNA methylation or histone modification. Sirtuin‐1 is a class III histone deacetylase which is decreased in human AAA samples. 186 Knockout of Sirtuin‐1 in vSMC accelerated formation and rupture of AAA by inducing inflammation and vascular senescence, while Sirtuin‐1 overexpression in vSMC had a protective effect in a mouse model. 186 vSMC collected from human TAA tissue showed increased SMAD2 expression compared with vSMC collected from healthy thoracic tissue, which was dependent on epigenetic regulation of the SMAD2 promotors involving histone modifications.187, 188 Enhancer of zeste homolog 2 (EZH2) is a methyl transferase of histone H3 that functions as a transcriptional repressor and inhibits autophagic cell death of vSMC during TAAD progression. 189 Epigenetic changes in AA pathogenesis are of interest since these changes can be reversible and altered by environmental factors, such as smoking. Epigenetic changes may therefore explain the link between AA risk factors and AA development.
1.2.3. Regulatory RNAs regulating vSMC functions
MicroRNA (miRNA) are RNA strands of 19–24 nucleotides regulating gene expression, which are involved in AA and AD pathogenesis by modulating different vSMC functions. Table 2 gives an overview of miRNA involved in regulating vSMC functions, and how expression level of these miRNA affects vSMC function. The anatomical location and the species in which these miRNAs were studied are also shown. In AAA pathogenesis, miRNA‐21, 190 miRNA‐26a, 191 miRNA‐28‐5p, 192 miRNA‐129‐5p, 193 miRNA‐155‐5p, 194 miRNA‐195 195 and miRNA‐504 196 are reported to be involved in regulation of vSMC apoptosis. miRNA‐155, 197 miRNA‐195, 198 miRNA‐205 199 and miRNA‐516a‐5p 200 are found to be involved in ECM regeneration and degradation. miRNA‐21, 190 miRNA‐129‐5p, 193 miRNA‐155, 197 miRNA‐195 195 and miRNA‐504 196 can regulate vSMC proliferation, while miRNA‐155 197 can regulate vSMC migration. miRNA‐24, 201 miRNA‐33, 202 miRNA‐155 197 and miRNA‐195 198 are involved in regulation of the inflammatory response within the aortic wall.
TABLE 2.
Overview of miRNA and lnc‐RNA involved in regulation of vSMC functions during AAA, TAA, TAAD and TAD development
| vSMC function | Regulated by | Expression level and effect miRNA/lnc‐RNA | Location | Species | Reference |
|---|---|---|---|---|---|
| vSMC apoptosis |
miRNA‐21 miRNA‐26a miRNA‐28‐5p miRNA‐129‐5p miRNA‐155‐5p miRNA‐195 |
Increased during AAA development, miRNA overexpression decreased apoptosis Decreased during AAA development, miRNA upregulation decreased apoptosis Increased during AAA development, miRNA acts as an apoptosis driver Decreased during AAA development, miRNA overexpression enhanced apoptosis Expression during AAA development not mentioned, inhibiting miRNA induced apoptosis Increased during AAA development, miRNA enhanced apoptosis |
AAA AAA AAA AAA AAA AAA |
Humans and mouse Humans Humans Humans and mouse Humans Humans |
|
|
miRNA‐504 miRNA‐26b |
Decreased during AAA development, miRNA overexpression decreased apoptosis Decreased during TAD development, miRNA overexpression decreased apoptosis |
AAA TAD |
Humans Humans |
||
| miRNA‐145 | Decreased during TAD development, miRNA downregulation enhanced apoptosis, 205 while miRNA overexpression decreased apoptosis 206 | TAD |
Humans Humans and rat |
||
| miRNA‐320d | Decreased during TAD development, miRNA overexpression enhanced apoptosis | TAD | Humans | 207 | |
| miRNA‐582 | Decreased during TAD development, miRNA overexpression enhanced apoptosis | TAD | Humans | 207 | |
| lnc‐RNA NEAT1 | Increased during AAA development, lnc‐RNA knockdown decreased apoptosis, while lnc‐RNA overexpression enhanced apoptosis | AAA | Not mentioned | 216 | |
| lnc‐RNA GAS5 | Increased during AAA development, lnc‐RNA overexpression enhanced apoptosis | AAA | Humans and mouse | 217 | |
| lnc‐RNA H19 | Increased during AAA development, lnc‐RNA knockdown decreased apoptosis, while overexpression had the opposite effect | AAA | Mouse | 218 | |
| lnc‐RNA PVT1 | Increased during AAA development, lnc‐RNA knockdown decreased apoptosis | AAA | Humans and mouse | 219 | |
| lnc‐RNA LUCAT1 | Increased during AAA development, lnc‐RNA depletion decreased apoptosis, while lnc‐RNA promotion enhanced apoptosis | AAA | Not mentioned | 220 | |
| lnc‐RNA LINC00473 | Increased during AAA development, lnc‐RNA overexpression enhanced apoptosis | AAA | Humans | 221 | |
| lnc‐RNA LOXL1‐AS | Increased during TAA development, lnc‐RNA overexpression decreased apoptosis | TAA | Humans | 222 | |
| lnc‐RNA MIAT | Increased during TAA development, lnc‐RNA overexpression decreased apoptosis | TAA | Humans | 223 | |
| lnc‐RNA HOTAIR | Decreased during TAA development, lnc‐RNA knockdown enhanced apoptosis | TAA | Humans | 224 | |
| lnc‐RNA HIF 1alpha antisense RNA | Expression during TAA development not mentioned, lnc‐RNA suppression decreased apoptosis | TAA | Humans | 225 | |
| linc‐RNA p‐21 | Increased during TAA development, linc‐RNA overexpression enhanced apoptosis | TAA | Humans | 226 | |
| vSMC phenotypic switch |
miRNA‐124 miRNA‐134‐5p miRNA‐143/145 cluster miRNA‐21 |
Decreased during TAD development, miRNA overexpression enhanced phenotypic switch (decrease in contractile genes) Decreased during TAD development, miRNA overexpression increased contractile phenotype Decreased during TAD development, miRNA knockdown induced phenotypic switch Increased during TAAD development, miRNA knockdown increased switch from contractile to synthetic phenotype, due to dysfunctional TGF‐β signalling |
TAD TAD TAD TAAD |
Humans Humans and mouse Humans Mouse |
|
| ECM regeneration and degradation |
miRNA‐155 miRNA‐195 miRNA‐205 miRNA‐516a‐5p miRNA‐145 miRNA‐30a lnc‐RNA PVT1 lnc‐RNA HOTAIR |
Increased during AAA development, miRNA overexpression enhanced MMP‐2, MPP‐9 protein expression, while inhibition had the opposite effect Increased during AAA development, miRNA overexpression enhanced MMP‐2 and MMP‐9 protein expression Increased during AAA development, miRNA overexpression reduced LRP‐1 protein expression, and subsequent reduced MMP‐9 protein clearance Expression during AAA development not mentioned, miRNA overexpression increased MMP‐2 protein and decreased TIMP‐1 protein expression, while knockdown had the opposite effect Increased during TAA development, miRNA overexpression enhanced OPN and collagen III protein, while inhibition had the opposite effect Increased during TAD development, miRNA overexpression decreased LOX and elastin protein expression Increased during AAA development, lnc‐RNA knockdown suppressed ECM disruption Decreased during TAA development, lnc‐RNA knockdown decreased collagen types I and III mRNA and protein expression |
AAA AAA AAA AAA TAA TAD AAA TAA |
Humans and mouse Humans Humans Humans Humans Humans and rat Humans and mouse Humans |
|
| vSMC proliferation |
miRNA‐21 miRNA‐129‐5p miRNA‐155 miRNA‐195 miRNA‐504 miRNA‐26b miRNA‐124 miRNA‐133 |
Increased during AAA development, miRNA overexpression enhanced proliferation Decreased during AAA development, miRNA overexpression decreased proliferation Increased during AAA development, miRNA overexpression enhanced proliferation, while inhibition had the opposite effect Decreased during AAA development, miRNA inhibited proliferation Decreased during TAD development, miRNA overexpression promoted proliferation Decreased during TAD development, miRNA overexpression enhanced proliferation, while knockdown inhibited proliferation Decreased during TAD development, miRNA overexpression enhanced proliferation Decreased during TAD development, miRNA upregulation decreased proliferation |
AAA AAA AAA AAA AAA TAD TAD TAD |
Humans and mouse Humans and mouse Humans and mouse Humans Humans Humans Humans Humans |
|
|
miRNA‐145 miRNA‐146a‐5p |
Decreased during TAD development, miRNA downregulation enhanced proliferation, 205 and miRNA overexpression promoted proliferation 206 Increased during TAD development, miRNA overexpression enhanced proliferation |
TAD TAD |
Humans Humans and rat Humans |
||
| lnc‐RNA NEAT1 | Increased during AAA development, lnc‐RNA knockdown enhanced proliferation, lnc‐RNA overexpression decreased proliferation | AAA | Not mentioned | 216 | |
|
lnc‐RNA GAS5 lnc‐RNA LUCAT1 |
Increased during AAA development, lnc‐RNA overexpression decreased proliferation Increased during AAA development, lnc‐RNA depletion enhanced proliferation, while lnc‐RNA promotion decreased proliferation |
AAA AAA |
Humans and mouse Not mentioned |
||
| lnc‐RNA LINC00473 | Increased during AAA development, lnc‐RNA overexpression decreased proliferation | AAA | Humans | 221 | |
|
lnc‐RNA LOXL1‐AS lnc‐RNA HOTAIR |
Increased during TAA development, lnc‐RNA overexpression enhanced proliferation Decreased during TAA development, lnc‐RNA knockdown decreased proliferation |
TAA TAA |
Humans Humans |
||
| lnc‐RNA HIF 1alpha antisense RNA | Expression during TAA development not mentioned, lnc‐RNA suppression enhanced proliferation | TAA | Humans | 225 | |
| linc‐RNA p‐21 | Increased during TAA development, linc‐RNA overexpression decreased proliferation | TAA | Humans | 226 | |
| vSMC migration | miRNA‐155 | Increased during AAA development, miRNA overexpression enhanced migration, while inhibition had the opposite effect | AAA | Humans and mouse | 197 |
|
miRNA‐27a miRNA‐133 miRNA‐134‐5p |
Decreased during TAD development, miRNA downregulation decreased migration Decreased during TAD development, miRNA upregulation decreased migration Decreased during TAD development, miRNA overexpression decreased migration |
TAD TAD TAD |
Humans and mouse Humans Humans and mouse |
||
|
miRNA‐145 miRNA‐146a‐5p |
Decreased during TAD development, miRNA downregulation enhanced migration Increased during TAD development, miRNA overexpression enhanced migration |
TAD TAD |
Humans Humans |
||
| Inflammatory response within the aortic wall | miRNA‐24 | Decreased during AAA development, miRNA downregulation enhanced pro‐inflammatory response | AAA | Humans and mouse | 201 |
|
miRNA‐33 miRNA‐155 miRNA‐195 lnc‐RNA PVT1 |
Increased during AAA development, miRNA knockdown reduced MCP‐1 mRNA and protein expression Increased during AAA development, miRNA overexpression enhanced MCP‐1 protein expression, while inhibition had the opposite effect Increased during AAA development, miRNA overexpression enhanced IL‐1β and IL‐6 expression Increased during AAA development, lnc‐RNA knockdown decreased pro‐inflammatory cytokines |
AAA AAA AAA AAA |
Humans and mouse Humans and mouse Humans Humans and mouse |
Abbreviations: AAA, abdominal aortic aneurysm; ECM, extracellular matrix; linc‐RNA, long intergenic noncoding RNA; lnc‐RNA, long noncoding RNA; miRNA, microRNA; RNA, ribonucleic acid; TAA, thoracic aortic aneurysm; TAAD, thoracic aortic aneurysm and dissection; TAD, thoracic aortic dissection; vSMC, vascular smooth muscle cell.
In TAA pathogenesis, only miRNA‐145 203 is reported as regulator of ECM remodelling. In TAD pathogenesis, miRNA‐26b, 204 miRNA‐145,205, 206 miRNA‐320d and miRNA‐582 207 can regulate vSMC apoptosis. miRNA‐124, 208 miRNA‐134‐5p 209 and miRNA‐143/145 gene cluster 210 can have a role in regulation of vSMC phenotypic switching. miRNA‐27a, 211 miRNA‐133, 212 miRNA‐134‐5p, 209 miRNA‐145 205 and miRNA‐146a‐5p 213 are found to be involved in vSMC migration. miRNA‐26b, 204 miRNA‐124, 208 miRNA‐133, 212 miRNA‐145205, 206 and miRNA‐146a‐5p 213 are reported as regulators of vSMC proliferation. miRNA‐30a is found to be involved in ECM remodelling by vSMC. 214 In a TAAD mouse model, knockout of miRNA‐21 induced a switch from a contractile to a synthetic vSMC phenotype, due to dysfunctional TGF‐β signalling. 215
Long noncoding RNA (lnc‐RNA) is defined as transcripts exceeding 200 nucleotides which are not translated into protein. However, they are crucial participants in both AAA and TAA pathogenesis by altering vSMC functions. Table 2 shows an overview of lnc‐RNA involved in regulating vSMC functions during AAA and TAA development and how expression level of these lnc‐RNA affect vSMC function. The anatomical location and the species in which these lnc‐RNA were studied are also shown. In AAA pathogenesis, vSMC apoptosis can be regulated by lnc‐RNA NEAT1, 216 GAS5, 217 H19, 218 plasmacytoma variant translocation 1 (PVT1), 219 LUCAT1 220 and LINC00473. 221 Lnc‐RNA PVT1 is also a regulator of ECM regeneration and degradation, and the inflammatory response within the aortic wall. 219 Regulators of vSMC proliferation are lnc‐RNA NEAT1, 216 GAS5, 217 LUCAT1 220 and LINC00473. 221 In TAA pathogenesis, lnc‐RNA LOXL1‐AS, 222 myocardial infarction associated transcript (MIAT), 223 HOX transcript antisense intergenic RNA (HOTAIR), 224 HIF 1alpha antisense RNA 225 and long intergenic noncoding RNA (linc‐RNA) p‐21 226 are associated with regulation of vSMC apoptosis. Lnc‐RNA LOXL1‐AS, 222 HOTAIR, 224 HIF 1alpha antisense RNA 225 and linc‐RNA p‐21 226 are reported be involved in regulation of vSMC proliferation, and lnc‐RNA HOTAIR is involved in ECM remodelling. 224
2. DISCUSSION AND FUTURE PERSPECTIVES
AA are pathological dilations of the aortic wall, which can rupture and cause internal bleeding. As summarized in this review, previous studies have shown that vSMC dysfunction has a paramount role in aortic wall weakening and subsequent AA and AD formation. Although several attempts have been made to develop pharmacological treatment options for stabilization or prevention of AA and AD, none of them has led to a broadly applicable treatment yet. In case of type B AD, patients are treated with anti‐hypertensive medication but the majority of the patients do not show improvement upon this therapy. 7 Disease progression is unpredictable, which makes it challenging to decide whether patients need surgical repair to prevent rupture. Therefore, more research into the underlying molecular mechanism of vSMC dysfunction in AA and AD progression and development is warranted.
2.1. Treatment options based on rodent studies
Targeting the loss of vSMC within the aortic wall caused by apoptosis is a potential treatment strategy. During endovascular repair, a graft is placed within the aortic wall, and seeding these endovascular grafts with vSMC has been shown to have a protective and reparative effect in a rat model. vSMC graft seeding prevented AAA formation, by decreasing elastin degradation and monocyte infiltration in AAA‐prone rats. 227 Likewise, aneurysm diameter stabilized after endovascular seeding of vSMC with aneurysmal aortic xenografts in an AAA rat model by inhibition of the ECM degradation process. The expression of MMP‐1, MMP‐3, MMP‐7, MMP‐9 and MMP‐12 decreased, while the expression of TIMP‐1, TIMP‐2 and TIMP‐3 increased after vSMC seeding. 228 The importance of vSMC in the aortic wall is highlighted by these studies and demonstrates a protective, paracrine effect of endovascular vSMC seeding on the aortic wall. Endovascular AA stent repair combined with the restoration of the healing capability of vSMC could be a promising therapeutic alternative.
The production and activation of proteolytic enzymes by vSMC contribute to AA progression and could therefore be a potential treatment target. Inhibition of ECM degradation by Doxycycline, a nonspecific inhibitor of MMP, gave promising results in rat and mouse studies.229, 230 However, evidence from clinical studies to confirm this beneficial effect is still lacking and this MMP inhibitor is therefore not used as therapeutic treatment for AA patients yet.231, 232 Upregulation of other inhibitors of proteolytic enzymes, such as PAI‐1 and TIMP, using gene therapy may also decrease ECM degradation within the aortic wall. Local overexpression of TIMP‐1 in a rat aorta resulted in decreased MMP‐9 and activated MMP‐2, which preserved elastin in the tunica media and prevented aneurysmal degeneration and rupture. 233 Another treatment option would be to target external factors which affect vSMC functions, such as inflammation or oxidative stress. Statins, which are lipid‐lowering drugs, have previously been shown to suppress AAA growth in a mouse model, by having an anti‐inflammatory effect on the aortic wall and reducing ER stress and apoptosis. 234 In humans, an association between statin use and a decreased expansion rate of infrarenal AA was also reported, but this still needs to be confirmed by randomized clinical trials. 235 Additionally, the ability of regulatory miRNA and lnc‐RNA expression to alter gene expression and modulate vSMC functions may be explored to improve vSMC function within the aortic wall and prevent AA formation. miRNA activity can be manipulated by anti‐miRs, blockmiRs or miRNA mimics, delivered by viral vectors, nanoparticles or liposomes. This potential of miRNA as a treatment option was previously shown to be effective for other cardiovascular diseases. 236
2.2. Strength of tissue biobanks, patient‐derived vSMC and in vitro aortic models
So far, none of the above‐described proposed therapeutic treatment options are implemented in the clinic. AA is a heterogeneous and multifactorial disorder with differences in origin and disease progression in every patient, which makes the development of therapeutic AA treatments challenging. To address this challenge, it is necessary to perform in vitro experiments and translational studies on a large number of patient samples. Creating a biobank with patient samples, and specifically vSMC, is needed to define the link between defects in vSMC and clinical characteristics in large cohorts of patients. 237 Our group showed in vitro that contraction of vSMC isolated from AAA patients is impaired compared with healthy control vSMC. 121 Impaired vSMC contraction was mainly seen in patients who were current smokers and who underwent open repair surgery after earlier endovascular repair. 121 However, the underlying mechanism of vSMC contractility reduction leading to AA formation still remains unexplored. More research is needed into the exact proteins and pathways contributing to vSMC contractile dysfunction and the link between these molecular defects and other patient characteristics, such as sex, age and diabetes. Functional studies in human vSMC should be combined with multiple –omics studies (RNA‐seq, proteomics) in large numbers of patient compared with control samples to identify proteins and pathways involved in AA progression, which can be used as potential treatment targets.
Another explanation for the absence of therapeutic AA treatments is the lack of adequate in vitro models. These models, mimicking human disease, can be used to investigate disease progression and effects of therapeutic interventions. vSMC can be generated from stem cells, but this is a time‐consuming and expensive process. Segments of isolated aortic tissue from large animals, maintained in organ culture, can also be used as an in vitro model. This model has the advantage that laminar shear stress can be applied, but the disadvantages that these isolated aortic segments can only be kept in culture for a limited time period. 238 Preclinical 3D cell culture models seeded with patient‐specific SMC can recreate the complex micro‐environment of the aorta in vitro. This model gives the option to study mechanical properties, fibre orientation, ECM production by vSMC and the interaction with other cell types in the aortic wall, such as endothelial cells. 239 However, in this model, the cells lose their original tubular form. Therefore, the complexity of this model can be increased by generation of tubular grafts using a bioreactor to study additional parameters, such as flow. 240 Another method to study aneurysmal disease in vitro is by transdifferentiation of fibroblasts, isolated from skin tissue, into vSMC‐like cells. After conversion of these dermal fibroblasts, mRNA expression of SMC markers within the vSMC‐like cells is comparable to primary human aortic SMC. 141 This noninvasive method is highly efficient to detect gene mutations and molecular defects in vSMC‐like cells, already at an early disease stage before the patient develops AA. Specific molecular defects within individual patients can be diagnosed, and targeting these patient‐specific defects can improve patient outcome. 241 Furthermore, functional measurements of vSMC‐like cells could have the potential to eventually predict characteristics of AA progression, such as aneurysm size growth and chance of rupture, in patients with early AA who have not yet undergone surgery. The above‐mentioned in vitro models, using either biological materials or biological combined with synthetic materials, have an important role in ‘proof of concept studies’, but they require further validation to optimize in vitro aneurysmal disease research.
A last important point to underscore is that recent studies suggest that also endothelial cells play a key role in development and progression of AA and AD. Endothelial dysfunction can activate aortic wall remodelling by release of proteases, or induction of inflammatory and oxidative stress responses.242, 243, 244 Therefore, we want to highlight the importance to consider the interaction between endothelial cells and vSMC while studying aneurysmal disease.
3. CONCLUSION
In conclusion, our review illustrates the central role of impaired vSMC function in AA and AD development and progression and highlights the stressors and molecular pathways which may be targets for therapeutic interventions. Future research should focus on studies in models, which mimic the individual AA patient and define effects of stressors and interventions in these specific patients. This way, new potential targets for pharmaceutical treatments can be found and better disease progression prediction can be made for both AA and AD in order to improve patient outcome.
CONFLICT OF INTEREST
Not applicable.
AUTHORS’ CONTRIBUTIONS
KR designed and performed the database searches, selected articles found in the database search, wrote the manuscript and designed the figures. TM selected articles found in the database search and revised the manuscript. JCFK designed and performed the database searches. NB, JvV and KKY helped, advised and assisted on the selection of the articles, writing of the manuscript and revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig S1
ACKNOWLEDGEMENTS
Not applicable.
Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest. 2022;52:e13697. doi: 10.1111/eci.13697
Funding information
This research was funded by the Dutch Heart Foundation/Hartstichting, Dekkerbeurs 2019T065 Senior clinical scientist grant.
REFERENCES
- 1. Lederle FA, Johnson GR, Wilson SE, et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Ann Intern Med. 1997;126:441. [DOI] [PubMed] [Google Scholar]
- 2. Blanchard JF, Armenian HK, Friesen PP. Risk factors for abdominal aortic aneurysm: results of a case‐control study. Am J Epidemiol. 1999;151(6):575‐583. [DOI] [PubMed] [Google Scholar]
- 3. Milewicz DM, Guo DC, Tran‐Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283‐302. [DOI] [PubMed] [Google Scholar]
- 4. Sailer AM, Nelemans PJ, Hastie TJ, et al. Prognostic significance of early aortic remodeling in acute uncomplicated type B aortic dissection and intramural hematoma. J Thorac Cardiovasc Surg. 2017;154(4):1192‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Assar AN, Zarins CK. Ruptured abdominal aortic aneurysm: a surgical emergency with many clinical presentations. Postgrad Med J. 2009;85(1003):268‐273. [DOI] [PubMed] [Google Scholar]
- 6. Wanhainen A, Verzini F, Van Herzeele I, et al. European Society for Vascular Surgery (ESVS) 2019 Clinical Practice Guidelines on the management of abdominal aorto‐iliac artery aneurysms. Eur J Vasc Endovasc Surg. 2019;57(1):8‐93. [DOI] [PubMed] [Google Scholar]
- 7. Durham CA, Cambria RP, Wang LJ, et al. The natural history of medically managed acute type B aortic dissection. J Vasc Surg. 2015;61(5):1192‐1198. [DOI] [PubMed] [Google Scholar]
- 8. Sinha S, Iyer D, Granata A. Embryonic origins of human vascular smooth muscle cells: implications for in vitro modeling and clinical application. Cell Mol Life Sci. 2014;71(12):2271‐2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ailawadi G, Moehle CW, Pei H, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138(6):1392‐1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell‐driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. 2018;52:48‐64. [DOI] [PubMed] [Google Scholar]
- 11. Lopez‐Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150(3):993‐1007. [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J, Schmidt J, Ryschich E, Schumacher H, Allenberg JR. Increased apoptosis and decreased density of medial smooth muscle cells in human abdominal aortic aneurysms. Chin Med J (Engl). 2003;116(10):1549‐1552. [PubMed] [Google Scholar]
- 13. Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99(1):96‐104. [DOI] [PubMed] [Google Scholar]
- 14. Satta J, Mennander A, Soini Y. Increased medial TUNEL‐positive staining associated with apoptotic bodies is linked to smooth muscle cell diminution during evolution of abdominal aortic aneurysms. Ann Vasc Surg. 2002;16(4):462‐466. [DOI] [PubMed] [Google Scholar]
- 15. Rowe VL, Stevens SL, Reddick TT, et al. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J Vasc Surg. 2000;31(3):567‐576. [PubMed] [Google Scholar]
- 16. Yamanouchi D, Morgan S, Stair C, et al. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg. 2012;56(2):455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Q, Shu C, Su J, Li X. A crosstalk triggered by hypoxia and maintained by MCP‐1/miR‐98/IL‐6/p38 regulatory loop between human aortic smooth muscle cells and macrophages leads to aortic smooth muscle cells apoptosis via Stat1 activation. Int J Clin Exp Pathol. 2015;8(3):2670‐2679. [PMC free article] [PubMed] [Google Scholar]
- 18. Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin II‐induced abdominal aortic aneurysm in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol. 2010;30(4):702‐707. [DOI] [PubMed] [Google Scholar]
- 19. Xue M, Li G, Li D, et al. Up‐regulated MCPIP1 in abdominal aortic aneurysm is associated with vascular smooth muscle cell apoptosis and MMPs production. Biosci Rep. 2019;39(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morgan S, Yamanouchi D, Harberg C, et al. Elevated protein kinase C‐delta contributes to aneurysm pathogenesis through stimulation of apoptosis and inflammatory signaling. Arterioscler Thromb Vasc Biol. 2012;32(10):2493‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sigala F, Papalambros E, Kotsinas A, et al. Relationship between iNOS expression and aortic cell proliferation and apoptosis in an elastase‐induced model of aorta aneurysm and the effect of 1400 W administration. Surgery. 2005;137(4):447‐456. [DOI] [PubMed] [Google Scholar]
- 22. Ni XQ, Lu WW, Zhang JS, et al. Inhibition of endoplasmic reticulum stress by intermedin 1–53 attenuates angiotensin II‐induced abdominal aortic aneurysm in ApoE KO Mice. Endocrine. 2018;62(1):90‐106. [DOI] [PubMed] [Google Scholar]
- 23. He R, Guo DC, Estrera AL, et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006;131(3):671‐678. [DOI] [PubMed] [Google Scholar]
- 24. Jia LX, Zhang WM, Zhang HJ, et al. Mechanical stretch‐induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J Pathol. 2015;236(3):373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Acilan C, Serhatli M, Kacar O, et al. Smooth muscle cells isolated from thoracic aortic aneurysms exhibit increased genomic damage, but similar tendency for apoptosis. DNA Cell Biol. 2012;31(10):1523‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adiguzel Z, Arda N, Kacar O, et al. Evaluation of apoptotic molecular pathways for smooth muscle cells isolated from thoracic aortic aneurysms in response to oxidized sterols. Mol Biol Rep. 2014;41(12):7875‐7884. [DOI] [PubMed] [Google Scholar]
- 27. Durdu S, Deniz GC, Balci D, et al. Apoptotic vascular smooth muscle cell depletion via BCL2 family of proteins in human ascending aortic aneurysm and dissection. Cardiovasc Ther. 2012;30(6):308‐316. [DOI] [PubMed] [Google Scholar]
- 28. Jiang WJ, Ren WH, Liu XJ, et al. Disruption of mechanical stress in extracellular matrix is related to Stanford type A aortic dissection through down‐regulation of Yes‐associated protein. Aging (Albany NY). 2016;8(9):1923‐1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu T, Xu J, Guo JL, et al. YAP1 up‐regulation inhibits apoptosis of aortic dissection vascular smooth muscle cells. Eur Rev Med Pharmacol Sci. 2017;21(20):4632‐4639. [PubMed] [Google Scholar]
- 30. Rizos IK, Tsoporis JN, Toumpoulis IK, et al. Antiapoptotic effect of beta1 blockers in ascending thoracic aortic smooth muscle cells: the role of HSP70 expression. J Cardiovasc Pharmacol. 2018;72(2):86‐96. [DOI] [PubMed] [Google Scholar]
- 31. Zou S, Liao M, Yang J, et al. Heat shock protein 27 plays a protective role in thoracic aortic dissection by promoting cell proliferation and inhibiting apoptosis. Cell Mol Biol Lett. 2017;22:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu H, Sun J, Liang W, et al. Cyclodextrin prevents abdominal aortic aneurysm via activation of vascular smooth muscle cell transcription factor EB. Circulation. 2020;142(5):483‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clement M, Chappell J, Raffort J, et al. Vascular smooth muscle cell plasticity and autophagy in dissecting aortic aneurysms. Arterioscler Thromb Vasc Biol. 2019;39(6):1149‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farrell K, Simmers P, Mahajan G, et al. Alterations in phenotype and gene expression of adult human aneurysmal smooth muscle cells by exogenous nitric oxide. Exp Cell Res. 2019;384(1):111589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riches K, Clark E, Helliwell RJ, et al. Progressive development of aberrant smooth muscle cell phenotype in abdominal aortic aneurysm disease. J Vasc Res. 2018;55(1):35‐46. [DOI] [PubMed] [Google Scholar]
- 36. Busch A, Hartmann E, Grimm C, et al. Heterogeneous histomorphology, yet homogeneous vascular smooth muscle cell dedifferentiation, characterize human aneurysm disease. J Vasc Surg. 2017;66(5):1553‐1564.e1556. [DOI] [PubMed] [Google Scholar]
- 37. Lai CH, Chang CW, Lee FT, et al. Targeting vascular smooth muscle cell dysfunction with xanthine derivative KMUP‐3 inhibits abdominal aortic aneurysm in mice. Atherosclerosis. 2020;297:16‐24. [DOI] [PubMed] [Google Scholar]
- 38. Liang ES, Cheng W, Yang RX, Bai WW, Liu X, Zhao YX. Peptidyl‐prolyl isomerase Pin1 deficiency attenuates angiotensin II‐induced abdominal aortic aneurysm formation in ApoE(‐/‐) mice. J Mol Cell Cardiol. 2018;114:334‐344. [DOI] [PubMed] [Google Scholar]
- 39. Zhong L, He X, Si X, et al. SM22alpha (Smooth Muscle 22alpha) prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching through suppressing reactive oxygen species/NF‐kappaB (nuclear factor‐kappaB). Arterioscler Thromb Vasc Biol. 2019;39(1):e10‐e25. [DOI] [PubMed] [Google Scholar]
- 40. Moehle CW, Bhamidipati CM, Alexander MR, et al. Bone marrow‐derived MCP1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. J Thorac Cardiovasc Surg. 2011;142(6):1567‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng H, Zhang K, Liu Z, et al. VPO1 modulates vascular smooth muscle cell phenotypic switch by activating extracellular signal‐regulated kinase 1/2 (ERK 1/2) in abdominal aortic aneurysms. J Am Heart Assoc. 2018;7(17):e010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qin X, He L, Tian M, et al. Smooth muscle‐specific Gs alpha deletion exaggerates angiotensin II‐induced abdominal aortic aneurysm formation in mice in vivo. J Mol Cell Cardiol. 2019;132:49‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sachdeva J, Mahajan A, Cheng J, et al. Smooth muscle cell‐specific Notch1 haploinsufficiency restricts the progression of abdominal aortic aneurysm by modulating CTGF expression. PLoS One. 2017;12(5):e0178538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaskovic I, Lackovic V, Radak D, et al. Ultrastructural characteristics of the vascular wall components of ruptured atherosclerotic abdominal aortic aneurysm. Arch Biol Sci. 2013;65(4):1271‐1278. [Google Scholar]
- 45. Wang S, Jia C. TRPV1 inhibits smooth muscle cell phenotype switching in a mouse model of abdominal aortic aneurysm. Channels (Austin). 2020;14(1):59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mourmoura E, Vasilaki A, Giannoukas A, Michalodimitrakis E, Pavlidis P, Tsezou A. Evidence of deregulated cholesterol efflux in abdominal aortic aneurysm. Acta Histochem. 2016;118(2):97‐108. [DOI] [PubMed] [Google Scholar]
- 47. Moran CS, McCann M, Karan M, Norman P, Ketheesan N, Golledge J. Association of osteoprotegerin with human abdominal aortic aneurysm progression. Circulation. 2005;111(23):3119‐3125. [DOI] [PubMed] [Google Scholar]
- 48. Moran CS, Jose RJ, Biros E, Golledge J. Osteoprotegerin deficiency limits angiotensin II‐induced aortic dilatation and rupture in the apolipoprotein E‐knockout mouse. Arterioscler Thromb Vasc Biol. 2014;34(12):2609‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Salmon M, Johnston WF, Woo A, et al. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. 2013;128(11 Suppl 1):S163‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mukherjee K, Gitlin JM, Loftin CD. Effectiveness of cyclooxygenase‐2 inhibition in limiting abdominal aortic aneurysm progression in mice correlates with a differentiated smooth muscle cell phenotype. J Cardiovasc Pharmacol. 2012;60(6):520‐529. [DOI] [PubMed] [Google Scholar]
- 51. Malashicheva A, Kostina D, Kostina A, et al. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int J Vasc Med. 2016;2016:3107879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mao N, Gu T, Shi E, Zhang G, Yu L, Wang C. Phenotypic switching of vascular smooth muscle cells in animal model of rat thoracic aortic aneurysm. Interact Cardiovasc Thorac Surg. 2015;21(1):62‐70. [DOI] [PubMed] [Google Scholar]
- 53. Forte A, Della Corte A, Grossi M, et al. Differential expression of proteins related to smooth muscle cells and myofibroblasts in human thoracic aortic aneurysm. Histol Histopathol. 2013;28(6):795‐803. [DOI] [PubMed] [Google Scholar]
- 54. Branchetti E, Poggio P, Sainger R, et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res. 2013;100(2):316‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zheng H, Liu J, Shan J, Xue S. Expression of gax in thoracic aortic aneurysm and its effect on phenotypic transformation of thoracic aortic smooth muscle cells. Int J Clin Exp Med. 2019;12(12):13265‐13273. [Google Scholar]
- 56. Liu R, Lo L, Lay AJ, et al. ARHGAP18 protects against thoracic aortic aneurysm formation by mitigating the synthetic and proinflammatory smooth muscle cell phenotype. Circ Res. 2017;121(5):512‐524. [DOI] [PubMed] [Google Scholar]
- 57. Chiarini A, Onorati F, Marconi M, et al. Studies on sporadic non‐syndromic thoracic aortic aneurysms: 1. Deregulation of Jagged/Notch 1 homeostasis and selection of synthetic/secretor phenotype smooth muscle cells. Eur J Prev Cardiol. 2018;25(1_suppl):42‐50. [DOI] [PubMed] [Google Scholar]
- 58. Huang J, Yamashiro Y, Papke CL, et al. Angiotensin‐converting enzyme‐induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin‐4‐deficient mice. Sci Transl Med. 2013;5(183):183ra158, 181‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang J, Davis EC, Chapman SL, et al. Fibulin‐4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res. 2010;106(3):583‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nogi M, Satoh K, Sunamura S, et al. Small GTP‐binding protein GDP dissociation stimulator prevents thoracic aortic aneurysm formation and rupture by phenotypic preservation of aortic smooth muscle cells. Circulation. 2018;138(21):2413‐2433. [DOI] [PubMed] [Google Scholar]
- 61. Meng YH, Tian C, Liu L, Wang L, Chang Q. Elevated expression of connective tissue growth factor, osteopontin and increased collagen content in human ascending thoracic aortic aneurysms. Vascular. 2014;22(1):20‐27. [DOI] [PubMed] [Google Scholar]
- 62. Lesauskaite V, Tanganelli P, Sassi C, et al. Smooth muscle cells of the media in the dilatative pathology of ascending thoracic aorta: morphology, immunoreactivity for osteopontin, matrix metalloproteinases, and their inhibitors. Hum Pathol. 2001;32(9):1003‐1011. [DOI] [PubMed] [Google Scholar]
- 63. Li G, Wang M, Caulk AW, et al. Chronic mTOR activation induces a degradative smooth muscle cell phenotype. J Clin Invest. 2020;130(3):1233‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang J, Wang L, Fu W, et al. Smooth muscle cell phenotypic diversity between dissected and unaffected thoracic aortic media. J Cardiovasc Surg (Torino). 2013;54(4):511‐521. [PubMed] [Google Scholar]
- 65. An Z, Liu Y, Song ZG, Tang H, Yuan Y, Xu ZY. Mechanisms of aortic dissection smooth muscle cell phenotype switch. J Thorac Cardiovasc Surg. 2017;154(5):1511‐1521.e1516. [DOI] [PubMed] [Google Scholar]
- 66. Liu K, Fang C, Shen Y, et al. Hypoxia‐inducible factor 1a induces phenotype switch of human aortic vascular smooth muscle cell through PI3K/AKT/AEG‐1 signaling. Oncotarget. 2017;8(20):33343‐33352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67. Yan Y, Tan MW, Xue X, Ding XY, Wang GK, Xu ZY. Involvement of Oct4 in the pathogenesis of thoracic aortic dissection via inducing the dedifferentiated phenotype of human aortic smooth muscle cells by directly upregulating KLF5. J Thorac Cardiovasc Surg. 2016;152(3):820‐829.e824. [DOI] [PubMed] [Google Scholar]
- 68. Wu HP, Cheng J, Huang W, et al. The roles of phenotypic transformation of vascular smooth muscle cells regulated by AGEs in the thoracic aortic dissection in neonatal rats. Int J Clin Exp Pathol. 2016;9(3):2758‐2764. [Google Scholar]
- 69. Zhao Z, Wang Y, Li S, et al. HSP90 inhibitor 17‐DMAG effectively alleviated the progress of thoracic aortic dissection by suppressing smooth muscle cell phenotypic switch. Am J Transl Res. 2019;11(1):509‐518. [PMC free article] [PubMed] [Google Scholar]
- 70. Patel MI, Melrose J, Ghosh P, Appleberg M. Increased synthesis of matrix metalloproteinases by aortic smooth muscle cells is implicated in the etiopathogenesis of abdominal aortic aneurysms. J Vasc Surg. 1996;24(1):82‐92. [DOI] [PubMed] [Google Scholar]
- 71. Crowther M, Goodall S, Jones JL, Bell PR, Thompson MM. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J Vasc Surg. 2000;32(3):575‐583. [DOI] [PubMed] [Google Scholar]
- 72. Airhart N, Brownstein BH, Cobb JP, et al. Smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin. J Vasc Surg. 2014;60(4):1033‐1041; discussion 1041–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase‐2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104(3):304‐309. [DOI] [PubMed] [Google Scholar]
- 74. Goodall S, Porter KE, Bell PR, Thompson MM. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP‐2 in patients with aortic aneurysms. Eur J Vasc Endovasc Surg. 2002;24(1):72‐80. [DOI] [PubMed] [Google Scholar]
- 75. Sinha I, Hannawa KK, Eliason JL, et al. Early MT‐1 MMP expression following elastase exposure is associated with increased cleaved MMP‐2 activity in experimental rodent aortic aneurysms. Surgery. 2004;136(2):176‐182. [DOI] [PubMed] [Google Scholar]
- 76. Gacchina CE, Deb P, Barth JL, Ramamurthi A. Elastogenic inductability of smooth muscle cells from a rat model of late stage abdominal aortic aneurysms. Tissue Eng Part A. 2011;17(13–14):1699‐1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McMillan WD, Patterson BK, Keen RR, Pearce WH. In situ localization and quantification of seventy‐two‐kilodalton type IV collagenase in aneurysmal, occlusive, and normal aorta. J Vasc Surg. 1995;22(3):295‐305. [DOI] [PubMed] [Google Scholar]
- 78. Kothapalli CR, Gacchina CE, Ramamurthi A. Utility of hyaluronan oligomers and transforming growth factor‐beta1 factors for elastic matrix regeneration by aneurysmal rat aortic smooth muscle cells. Tissue Eng Part A. 2009;15(11):3247‐3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vasic N, Glumac S, Pejic S, et al. Expression of matrix metalloproteinases and endogenous inhibitors in abdominal aortic aneurysm and aortoiliac occlusive disease (syndrome leriche). Folia Biol (Praha). 2017;63(5–6):209‐216. [DOI] [PubMed] [Google Scholar]
- 80. Annabi B, Shedid D, Ghosn P, et al. Differential regulation of matrix metalloproteinase activities in abdominal aortic aneurysms. J Vasc Surg. 2002;35(3):539‐546. [DOI] [PubMed] [Google Scholar]
- 81. McMillan WD, Patterson BK, Keen RR, Shively VP, Cipollone M, Pearce WH. In situ localization and quantification of mRNA for 92‐kD type IV collagenase and its inhibitor in aneurysmal, occlusive, and normal aorta. Arterioscler Thromb Vasc Biol. 1995;15(8):1139‐1144. [DOI] [PubMed] [Google Scholar]
- 82. Verschuren L, Lindeman JH, van Bockel JH, Abdul‐Hussien H, Kooistra T, Kleemann R. Up‐regulation and coexpression of MIF and matrix metalloproteinases in human abdominal aortic aneurysms. Antioxid Redox Signal. 2005;7(9–10):1195‐1202. [DOI] [PubMed] [Google Scholar]
- 83. Reeps C, Pelisek J, Seidl S, et al. Inflammatory infiltrates and neovessels are relevant sources of MMPs in abdominal aortic aneurysm wall. Pathobiology. 2009;76(5):243‐252. [DOI] [PubMed] [Google Scholar]
- 84. Lyon CA, Williams H, Bianco R, et al. Aneurysm severity is increased by combined Mmp‐7 deletion and N‐cadherin mimetic (EC4‐Fc) over‐expression. Sci Rep. 2017;7(1):17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mao D, Lee JK, VanVickle SJ, Thompson RW. Expression of collagenase‐3 (MMP‐13) in human abdominal aortic aneurysms and vascular smooth muscle cells in culture. Biochem Biophys Res Commun. 1999;261(3):904‐910. [DOI] [PubMed] [Google Scholar]
- 86. Crowther M, Goodall S, Jones JL, Bell PR, Thompson MM. Localization of matrix metalloproteinase 2 within the aneurysmal and normal aortic wall. Br J Surg. 2002;87(10):1391‐1400. [DOI] [PubMed] [Google Scholar]
- 87. Wang C, Chang Q, Sun X, et al. Angiotensin II induces an increase in matrix metalloproteinase 2 expression in aortic smooth muscle cells of ascending thoracic aortic aneurysms through JNK, ERK1/2, and p38 MAPK activation. J Cardiovasc Pharmacol. 2015;66(3):285‐293. [DOI] [PubMed] [Google Scholar]
- 88. Wang C, Qian X, Sun X, Chang Q. Angiotensin II increases matrix metalloproteinase 2 expression in human aortic smooth muscle cells via AT1R and ERK1/2. Exp Biol Med (Maywood). 2015;240(12):1564‐1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J Vasc Surg. 2012;56(6):1698‐1709, 1709.e1691. [DOI] [PubMed] [Google Scholar]
- 90. Ren W, Liu Y, Wang X, et al. The complement C3a–C3aR axis promotes development of thoracic aortic dissection via regulation of MMP2 expression. J Immunol. 2018;200(5):1829‐1838. [DOI] [PubMed] [Google Scholar]
- 91. Yuan Y, Wang C, Xu J, Tao J, Xu Z, Huang S. BRG1 overexpression in smooth muscle cells promotes the development of thoracic aortic dissection. BMC Cardiovasc Disord. 2014;14:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Shen M, Lee J, Basu R, et al. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(4):888‐898. [DOI] [PubMed] [Google Scholar]
- 93. Martin‐Alonso M, Garcia‐Redondo AB, Guo D, et al. Deficiency of MMP17/MT4‐MMP proteolytic activity predisposes to aortic aneurysm in mice. Circ Res. 2015;117(2):e13‐26. [DOI] [PubMed] [Google Scholar]
- 94. Lipp C, Lohoefer F, Reeps C, et al. Expression of a disintegrin and metalloprotease in human abdominal aortic aneurysms. J Vasc Res. 2012;49(3):198‐206. [DOI] [PubMed] [Google Scholar]
- 95. Zhai H, Qi X, Li Z, et al. TIMP3 suppresses the proliferation and migration of SMCs from the aortic neck of atherosclerotic AAA in rabbits, via decreased MMP2 and MMP9 activity, and reduced TNFalpha expression. Mol Med Rep. 2018;18(2):2061‐2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bumdelger B, Kokubo H, Kamata R, et al. Induction of Timp1 in smooth muscle cells during development of abdominal aortic aneurysms. Hiroshima J Med Sci. 2013;62(3):63‐67. [PubMed] [Google Scholar]
- 97. Ishii T, Asuwa N. Collagen and elastin degradation by matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase in aortic dissection. Hum Pathol. 2000;31(6):640‐646. [DOI] [PubMed] [Google Scholar]
- 98. Kawai T, Takayanagi T, Forrester SJ, et al. Vascular ADAM17 (a disintegrin and metalloproteinase domain 17) is required for angiotensin II/beta‐aminopropionitrile‐induced abdominal aortic aneurysm. Hypertension. 2017;70(5):959‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shen M, Hu M, Fedak PWM, Oudit GY, Kassiri Z. Cell‐specific functions of ADAM17 regulate the progression of thoracic aortic aneurysm. Circ Res. 2018;123(3):372‐388. [DOI] [PubMed] [Google Scholar]
- 100. Ren P, Zhang L, Xu G, et al. ADAMTS‐1 and ADAMTS‐4 levels are elevated in thoracic aortic aneurysms and dissections. Ann Thorac Surg. 2013;95(2):570‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Klaus V, Schmies F, Reeps C, et al. Cathepsin S is associated with degradation of collagen I in abdominal aortic aneurysm. Vasa. 2018;47(4):285‐293. [DOI] [PubMed] [Google Scholar]
- 102. Liu J, Sukhova GK, Yang JT, et al. Cathepsin L expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis. 2006;184(2):302‐311. [DOI] [PubMed] [Google Scholar]
- 103. Lohoefer F, Reeps C, Lipp C, et al. Quantitative expression and localization of cysteine and aspartic proteases in human abdominal aortic aneurysms. Exp Mol Med. 2014;46:e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lohoefer F, Reeps C, Lipp C, et al. Histopathological analysis of cellular localization of cathepsins in abdominal aortic aneurysm wall. Int J Exp Pathol. 2012;93(4):252‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sun J, Sukhova GK, Zhang J, et al. Cathepsin K deficiency reduces elastase perfusion‐induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2012;32(1):15‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Qin Y, Cao X, Guo J, et al. Deficiency of cathepsin S attenuates angiotensin II‐induced abdominal aortic aneurysm formation in apolipoprotein E‐deficient mice. Cardiovasc Res. 2012;96(3):401‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sun J, Sukhova GK, Zhang J, et al. Cathepsin L activity is essential to elastase perfusion‐induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2500‐2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Shi GP, Sukhova GK, Grubb A, et al. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J Clin Invest. 1999;104(9):1191‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Schulte S, Sun J, Libby P, et al. Cystatin C deficiency promotes inflammation in angiotensin II‐induced abdominal aortic aneurisms in atherosclerotic mice. Am J Pathol. 2010;177(1):456‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Louwrens HD, Kwaan HC, Pearce WH, Yao JS, Verrusio E. Plasminogen activator and plasminogen activator inhibitor expression by normal and aneurysmal human aortic smooth muscle cells in culture. Eur J Vasc Endovasc Surg. 1995;10(3):289‐293. [DOI] [PubMed] [Google Scholar]
- 111. Allaire E, Hasenstab D, Kenagy RD, Starcher B, Clowes MM, Clowes AW. Prevention of aneurysm development and rupture by local overexpression of plasminogen activator inhibitor‐1. Circulation. 1998;98(3):249‐255. [DOI] [PubMed] [Google Scholar]
- 112. Borges LF, Gomez D, Quintana M, et al. Fibrinolytic activity is associated with presence of cystic medial degeneration in aneurysms of the ascending aorta. Histopathology. 2010;57(6):917‐932. [DOI] [PubMed] [Google Scholar]
- 113. Boukais K, Borges LF, Venisse L, et al. Clearance of plasmin‐PN‐1 complexes by vascular smooth muscle cells in human aneurysm of the ascending aorta. Cardiovasc Pathol. 2018;32:15‐25. [DOI] [PubMed] [Google Scholar]
- 114. Gomez D, Kessler K, Borges LF, et al. Smad2‐dependent protease nexin‐1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler Thromb Vasc Biol. 2013;33(9):2222‐2232. [DOI] [PubMed] [Google Scholar]
- 115. Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res. 2000;92(1):85‐95. [DOI] [PubMed] [Google Scholar]
- 116. Emeto TI, Moxon JV, Biros E, et al. Urocortin 2 is associated with abdominal aortic aneurysm and mediates anti‐proliferative effects on vascular smooth muscle cells via corticotrophin releasing factor receptor 2. Clin Sci (Lond). 2014;126(7):517‐527. [DOI] [PubMed] [Google Scholar]
- 117. Hoshina K, Sho E, Sho M, Nakahashi TK, Dalman RL. Wall shear stress and strain modulate experimental aneurysm cellularity. J Vasc Surg. 2003;37(5):1067‐1074. [DOI] [PubMed] [Google Scholar]
- 118. Sho E, Sho M, Nanjo H, Kawamura K, Masuda H, Dalman RL. Comparison of cell‐type‐specific vs transmural aortic gene expression in experimental aneurysms. J Vasc Surg. 2005;41(5):844‐852. [DOI] [PubMed] [Google Scholar]
- 119. Zhu H, Qu X, Zhang C, Yu Y. Interleukin‐10 promotes proliferation of vascular smooth muscle cells by inhibiting inflammation in rabbit abdominal aortic aneurysm. Int J Clin Exp Pathol. 2019;12(4):1260‐1271. [PMC free article] [PubMed] [Google Scholar]
- 120. Sun Y, Zhao Z, Hou L, et al. The regulatory role of smooth muscle 22 on the proliferation of aortic smooth muscle cells participates in the development of aortic dissection. J Vasc Surg. 2017;66(3):875‐882. [DOI] [PubMed] [Google Scholar]
- 121. Bogunovic N, Meekel JP, Micha D, Blankensteijn JD, Hordijk PL, Yeung KK. Impaired smooth muscle cell contractility as a novel concept of abdominal aortic aneurysm pathophysiology. Sci Rep. 2019;9(1):6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Murtada SI, Ferruzzi J, Yanagisawa H, Humphrey JD. Reduced biaxial contractility in the descending thoracic aorta of fibulin‐5 deficient mice. J Biomech Eng. 2016;138(5):051008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Au DT, Ying Z, Hernandez‐Ochoa EO, et al. LRP1 (low‐density lipoprotein receptor‐related protein 1) regulates smooth muscle contractility by modulating Ca(2+) signaling and expression of cytoskeleton‐related proteins. Arterioscler Thromb Vasc Biol. 2018;38(11):2651‐2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yang CQ, Li W, Li SQ, et al. MCP‐1 stimulates MMP‐9 expression via ERK 1/2 and p38 MAPK signaling pathways in human aortic smooth muscle cells. Cell Physiol Biochem. 2014;34(2):266‐276. [DOI] [PubMed] [Google Scholar]
- 125. Colonnello JS, Hance KA, Shames ML, et al. Transient exposure to elastase induces mouse aortic wall smooth muscle cell production of MCP‐1 and RANTES during development of experimental aortic aneurysm. J Vasc Surg. 2003;38(1):138‐146. [DOI] [PubMed] [Google Scholar]
- 126. Akerman AW, Stroud RE, Barrs RW, et al. Elevated wall tension initiates interleukin‐6 expression and abdominal aortic dilation. Ann Vasc Surg. 2018;46:193‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wang Q, Ren J, Morgan S, Liu Z, Dou C, Liu B. Monocyte chemoattractant protein‐1 (MCP‐1) regulates macrophage cytotoxicity in abdominal aortic aneurysm. PLoS One. 2014;9(3):e92053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Onoda M, Yoshimura K, Aoki H, et al. Lysyl oxidase resolves inflammation by reducing monocyte chemoattractant protein‐1 in abdominal aortic aneurysm. Atherosclerosis. 2010;208(2):366‐369. [DOI] [PubMed] [Google Scholar]
- 129. Xu J, Ehrman B, Graham LM, Eagleton MJ. Interleukin‐5 is a potential mediator of angiotensin II‐induced aneurysm formation in apolipoprotein E knockout mice. J Surg Res. 2012;178(1):512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mamun A, Yokoyama U, Saito J, et al. A selective antagonist of prostaglandin E receptor subtype 4 attenuates abdominal aortic aneurysm. Physiol Rep. 2018;6(18):e13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Keen RR, Nolan KD, Cipollone M, et al. Interleukin‐1 beta induces differential gene expression in aortic smooth muscle cells. J Vasc Surg. 1994;20(5):774‐784; discussion 784–776. [DOI] [PubMed] [Google Scholar]
- 132. Johnston WF, Salmon M, Pope NH, et al. Inhibition of interleukin‐1beta decreases aneurysm formation and progression in a novel model of thoracic aortic aneurysms. Circulation. 2014;130(11 Suppl 1):S51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. An Z, Qiao F, Lu Q, et al. Interleukin‐6 downregulated vascular smooth muscle cell contractile proteins via ATG4B‐mediated autophagy in thoracic aortic dissection. Heart Vessels. 2017;32(12):1523‐1535. [DOI] [PubMed] [Google Scholar]
- 134. Hu H, Zhang G, Hu H, et al. Interleukin‐18 expression increases in the aorta and plasma of patients with acute aortic dissection. Mediators Inflamm. 2019;2019:8691294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sun J, Sukhova GK, Yang M, et al. Mast cells modulate the pathogenesis of elastase‐induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117(11):3359‐3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chan WL, Pejnovic N, Hamilton H, et al. Atherosclerotic abdominal aortic aneurysm and the interaction between autologous human plaque‐derived vascular smooth muscle cells, type 1 NKT, and helper T cells. Circ Res. 2005;96(6):675‐683. [DOI] [PubMed] [Google Scholar]
- 137. Meng X, Yang J, Zhang K, et al. Regulatory T cells prevent angiotensin II‐induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension. 2014;64(4):875‐882. [DOI] [PubMed] [Google Scholar]
- 138. Liu B, Kong J, An G, Zhang K, Qin W, Meng X. Regulatory T cells protected against abdominal aortic aneurysm by suppression of the COX‐2 expression. J Cell Mol Med. 2019;23(10):6766‐6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Folkersen L, Wagsater D, Paloschi V, et al. Unraveling divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation: the ASAP study. Mol Med. 2011;17(11–12):1365‐1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. van de Luijtgaarden KM, Heijsman D, Maugeri A, et al. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum Genet. 2015;134(8):881‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bogunovic N, Musters RJP, Micha D, et al. Transdifferentiation of human dermal fibroblasts to smooth muscle like cells: a novel method to study the effect of MYH11 and ACTA2 variants in the aortic aneurysm wall. Vascular. 2016;24(1):31.25757609 [Google Scholar]
- 142. Chen J, Peters A, Papke CL, et al. Loss of smooth muscle alpha‐actin leads to NF‐kappaB‐dependent increased sensitivity to angiotensin II in smooth muscle cells and aortic enlargement. Circ Res. 2017;120(12):1903‐1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lu H, Fagnant PM, Bookwalter CS, Joel P, Trybus KM. Vascular disease‐causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc Natl Acad Sci USA. 2015;112(31):E4168‐4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Massett MP, Bywaters BC, Gibbs HC, et al. Loss of smooth muscle alpha‐actin effects on mechanosensing and cell‐matrix adhesions. Expe Biol Med. 2020;245(4):374‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cheng J, Zhou X, Jiang X, Sun T. Deletion of ACTA2 in mice promotes angiotensin II induced pathogenesis of thoracic aortic aneurysms and dissections. J Thorac Dis. 2018;10(8):4733‐4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Guo DC, Pannu H, Tran‐Fadulu V, et al. Mutations in smooth muscle alpha‐actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488‐1493. [DOI] [PubMed] [Google Scholar]
- 147. Malloy LE, Wen KK, Pierick AR, et al. Thoracic aortic aneurysm (TAAD)‐causing mutation in actin affects formin regulation of polymerization. J Biol Chem. 2012;287(34):28398‐28408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Renard M, Callewaert B, Baetens M, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2013;165(2):314‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Papke CL, Cao J, Kwartler CS, et al. Smooth muscle hyperplasia due to loss of smooth muscle alpha‐actin is driven by activation of focal adhesion kinase, altered p53 localization and increased levels of platelet‐derived growth factor receptor‐beta. Hum Mol Genet. 2013;22(15):3123‐3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Guo DC, Papke CL, Tran‐Fadulu V, et al. Mutations in smooth muscle alpha‐actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hoffjan S, Waldmuller S, Blankenfeldt W, et al. Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. Eur J Hum Genet. 2011;19(5):520‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38(3):343‐349. [DOI] [PubMed] [Google Scholar]
- 153. Pannu H, Tran‐Fadulu V, Papke CL, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin‐like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16(20):2453‐2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Kuang SQ, Kwartler CS, Byanova KL, et al. Rare, nonsynonymous variant in the smooth muscle‐specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012;110(11):1411‐1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bellini C, Wang S, Milewicz DM, Humphrey JD. Myh11(R247C/R247C) mutations increase thoracic aorta vulnerability to intramural damage despite a general biomechanical adaptivity. J Biomech. 2015;48(1):113‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Guo DC, Regalado E, Casteel DE, et al. Recurrent gain‐of‐function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93(2):398‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gago‐Diaz M, Blanco‐Verea A, Teixido G, et al. PRKG1 and genetic diagnosis of early‐onset thoracic aortic disease. Eur J Clin Invest. 2016;46(9):787‐794. [DOI] [PubMed] [Google Scholar]
- 158. Wang L, Guo DC, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87(5):701‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Grewal N, Gittenberger‐de Groot AC. Pathogenesis of aortic wall complications in Marfan syndrome. Cardiovasc Pathol. 2018;33:62‐69. [DOI] [PubMed] [Google Scholar]
- 160. Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88(1):37‐43. [DOI] [PubMed] [Google Scholar]
- 161. Nolasco P, Fernandes CG, Ribeiro‐Silva JC, et al. Impaired vascular smooth muscle cell force‐generating capacity and phenotypic deregulation in Marfan Syndrome mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(1):165587. [DOI] [PubMed] [Google Scholar]
- 162. Crosas‐Molist E, Meirelles T, Lopez‐Luque J, et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):960‐972. [DOI] [PubMed] [Google Scholar]
- 163. Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin‐1 regulates the bioavailability of TGFbeta1. J Cell Biol. 2007;176(3):355‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Nataatmadja M, West J, Prabowo S, West M. Angiotensin II receptor antagonism reduces transforming growth factor beta and smad signaling in thoracic aortic aneurysm. Ochsner J. 2013;13(1):42‐48. [PMC free article] [PubMed] [Google Scholar]
- 165. You W, Hong Y, He H, et al. TGF‐beta mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany NY). 2019;11(11):3574‐3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pedroza AJ, Koyano T, Trojan J, et al. Divergent effects of canonical and non‐canonical TGF‐beta signalling on mixed contractile‐synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med. 2020;24(3):2369‐2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Nataatmadja M, West J, West M. Overexpression of transforming growth factor‐beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aortic aneurysm. Circulation. 2006;114(1 Suppl):I371‐377. [DOI] [PubMed] [Google Scholar]
- 168. Emrich F, Penov K, Arakawa M, et al. Anatomically specific reactive oxygen species production participates in Marfan syndrome aneurysm formation. J Cell Mol Med. 2019;23(10):7000‐7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jimenez‐Altayo F, Meirelles T, Crosas‐Molist E, et al. Redox stress in Marfan syndrome: dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic Biol Med. 2018;118:44‐58. [DOI] [PubMed] [Google Scholar]
- 170. D'Hondt S, Van Damme T, Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers‐Danlos syndrome: a systematic review. Genet Med. 2018;20(6):562‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lee VS, Halabi CM, Hoffman EP, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci USA. 2016;113(31):8759‐8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Barbier M, Gross MS, Aubart M, et al. MFAP5 loss‐of‐function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95(6):736‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Van Laer L, Dietz H, Loeys B. Loeys‐dietz syndrome. Adv Exp Med Biol. 2014;802:95‐105. [DOI] [PubMed] [Google Scholar]
- 174. Inamoto S, Kwartler CS, Lafont AL, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88(3):520‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yang P, Schmit BM, Fu C, et al. Smooth muscle cell‐specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci Rep. 2016;6:35444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Gao F, Chambon P, Offermanns S, et al. Disruption of TGF‐beta signaling in smooth muscle cell prevents elastase‐induced abdominal aortic aneurysm. Biochem Biophys Res Commun. 2014;454(1):137‐143. [DOI] [PubMed] [Google Scholar]
- 177. Wang Y, Huang HY, Bian GL, et al. A functional variant of SMAD4 enhances thoracic aortic aneurysm and dissection risk through promoting smooth muscle cell apoptosis and proteoglycan degradation. EBioMedicine. 2017;21:197‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Duan XY, Guo DC, Regalado ES, et al. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur J Hum Genet. 2019;27(7):1054‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. van der Pluijm I, van Vliet N, von der Thusen JH, et al. Defective connective tissue remodeling in Smad3 mice leads to accelerated aneurysmal growth through disturbed downstream TGF‐beta signaling. EBioMedicine. 2016;12:280‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zhu SB, Zhu J, Zhou ZZ, Xi EP, Wang RP, Zhang Y. TGF‐beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res. 2015;7(12):2764‐2774. [PMC free article] [PubMed] [Google Scholar]
- 181. Doyle AJ, Redmond EM, Gillespie DL, et al. Differential expression of Hedgehog/Notch and transforming growth factor‐beta in human abdominal aortic aneurysms. J Vasc Surg. 2015;62(2):464‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Fukui D, Miyagawa S, Soeda J, Tanaka K, Urayama H, Kawasaki S. Overexpression of transforming growth factor beta1 in smooth muscle cells of human abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2003;25(6):540‐545. [DOI] [PubMed] [Google Scholar]
- 183. Kuang SQ, Medina‐Martinez O, Guo DC, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. 2016;126(3):948‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Toghill BJ, Saratzis A, Freeman PJ, Sylvius N, Bown MJ. SMYD2 promoter DNA methylation is associated with abdominal aortic aneurysm (AAA) and SMYD2 expression in vascular smooth muscle cells. Clin Epigenetics. 2018;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Vinh A, Gaspari TA, Liu HB, Dousha LF, Widdop RE, Dear AE. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II‐infused apolipoprotein E‐deficient mice. J Vasc Res. 2008;45(2):143‐152. [DOI] [PubMed] [Google Scholar]
- 186. Chen HZ, Wang F, Gao P, et al. Age‐associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. 2016;119(10):1076‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Gomez D, Kessler K, Michel JB, Vranckx R. Modifications of chromatin dynamics control Smad2 pathway activation in aneurysmal smooth muscle cells. Circ Res. 2013;113(7):881‐890. [DOI] [PubMed] [Google Scholar]
- 188. Gomez D, Coyet A, Ollivier V, et al. Epigenetic control of vascular smooth muscle cells in Marfan and non‐Marfan thoracic aortic aneurysms. Cardiovasc Res. 2011;89(2):446‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Li R, Yi X, Wei X, et al. EZH2 inhibits autophagic cell death of aortic vascular smooth muscle cells to affect aortic dissection. Cell Death Dis. 2018;9(2):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Maegdefessel L, Azuma J, Toh R, et al. MicroRNA‐21 blocks abdominal aortic aneurysm development and nicotine‐augmented expansion. Sci Transl Med. 2012;4(122):122ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Peng J, He X, Zhang L, Liu P. MicroRNA26a protects vascular smooth muscle cells against H2O2 induced injury through activation of the PTEN/AKT/mTOR pathway. Int J Mol Med. 2018;42(3):1367‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Yue J, Zhu T, Yang J, et al. CircCBFB‐mediated miR‐28‐5p facilitates abdominal aortic aneurysm via LYPD3 and GRIA4. Life Sci. 2020;253:117533. [DOI] [PubMed] [Google Scholar]
- 193. Zhang Y, Liu Z, Zhou M, Liu C. MicroRNA‐129‐5p inhibits vascular smooth muscle cell proliferation by targeting Wnt5a. Exp Ther Med. 2016;12(4):2651‐2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhao L, Ouyang Y, Bai Y, Gong J, Liao H. miR‐155‐5p inhibits the viability of vascular smooth muscle cell via targeting FOS and ZIC3 to promote aneurysm formation. Eur J Pharmacol. 2019;853:145‐152. [DOI] [PubMed] [Google Scholar]
- 195. Liang B, Che J, Zhao H, Zhang Z, Shi G. MiR‐195 promotes abdominal aortic aneurysm media remodeling by targeting Smad3. Cardiovasc Ther. 2017;35(6):e12286. [DOI] [PubMed] [Google Scholar]
- 196. Cao X, Cai Z, Liu J, et al. miRNA504 inhibits p53 dependent vascular smooth muscle cell apoptosis and may prevent aneurysm formation. Mol Med Rep. 2017;16(3):2570‐2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zhang Z, Liang K, Zou G, et al. Inhibition of miR‐155 attenuates abdominal aortic aneurysm in mice by regulating macrophage‐mediated inflammation. Biosci Rep. 2018;38(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ma X, Yao H, Yang Y, et al. miR‐195 suppresses abdominal aortic aneurysm through the TNF‐alpha/NF‐kappaB and VEGF/PI3K/Akt pathway. Int J Mol Med. 2018;41(4):2350‐2358. [DOI] [PubMed] [Google Scholar]
- 199. Chan CY, Chan YC, Cheuk BL, Cheng SW. Clearance of matrix metalloproteinase‐9 is dependent on low‐density lipoprotein receptor‐related protein‐1 expression downregulated by microRNA‐205 in human abdominal aortic aneurysm. J Vasc Surg. 2017;65(2):509‐520. [DOI] [PubMed] [Google Scholar]
- 200. Chan CYT, Cheuk BLY, Cheng SWK. Abdominal aortic aneurysm‐associated MicroRNA‐516a‐5p regulates expressions of methylenetetrahydrofolate reductase, matrix metalloproteinase‐2, and tissue inhibitor of matrix metalloproteinase‐1 in human abdominal aortic vascular smooth muscle cells. Ann Vasc Surg. 2017;42:263‐273. [DOI] [PubMed] [Google Scholar]
- 201. Maegdefessel L, Spin JM, Raaz U, et al. miR‐24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat Commun. 2014;5:5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Nakao T, Horie T, Baba O, et al. Genetic ablation of MicroRNA‐33 attenuates inflammation and abdominal aortic aneurysm formation via several anti‐inflammatory pathways. Arterioscler Thromb Vasc Biol. 2017;37(11):2161‐2170. [DOI] [PubMed] [Google Scholar]
- 203. Pei H, Tian C, Sun X, et al. Overexpression of MicroRNA‐145 promotes ascending aortic aneurysm media remodeling through TGF‐beta1. Eur J Vasc Endovasc Surg. 2015;49(1):52‐59. [DOI] [PubMed] [Google Scholar]
- 204. Yang P, Wu P, Liu X, et al. MiR‐26b suppresses the development of stanford type A aortic dissection by regulating HMGA2 and TGF‐beta/Smad3 signaling pathway. Ann Thorac Cardiovasc Surg. 2020;26(3):140‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Huang W, Huang C, Ding H, et al. Involvement of miR‐145 in the development of aortic dissection via inducing proliferation, migration, and apoptosis of vascular smooth muscle cells. J Clin Lab Anal. 2020;34(1):e23028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Li T, Liu C, Liu L, et al. Regulatory mechanism of MicroRNA‐145 in the pathogenesis of acute aortic dissection. Yonsei Med J. 2019;60(4):352‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Shen H, Lu S, Dong L, et al. hsa‐miR‐320d and hsa‐miR‐582, miRNA biomarkers of aortic dissection, regulate apoptosis of vascular smooth muscle cells. J Cardiovasc Pharmacol. 2018;71(5):275‐282. [DOI] [PubMed] [Google Scholar]
- 208. Tang Y, Yu S, Liu Y, Zhang J, Han L, Xu Z. MicroRNA‐124 controls human vascular smooth muscle cell phenotypic switch via Sp1. Am J Physiol Heart Circ Physiol. 2017;313(3):H641‐h649. [DOI] [PubMed] [Google Scholar]
- 209. Wang Y, Dong CQ, Peng GY, et al. MicroRNA‐134‐5p regulates media degeneration through inhibiting VSMC phenotypic switch and migration in thoracic aortic dissection. Mol Ther Nucleic Acids. 2019;16:284‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zhang M, Wang Z. Downregulation of miR143/145 gene cluster expression promotes the aortic media degeneration process via the TGF‐beta1 signaling pathway. Am J Transl Res. 2019;11(1):370‐378. [PMC free article] [PubMed] [Google Scholar]
- 211. Sun Y, Xiao Y, Sun H, et al. miR‐27a regulates vascular remodeling by targeting endothelial cells’ apoptosis and interaction with vascular smooth muscle cells in aortic dissection. Theranostics. 2019;9(25):7961‐7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Ma RW, Wang JY, Wu XJ, et al. Diagnostic value of miR‐133 in aortic dissection and its mechanism. Int J Clin Exp Pathol. 2017;10(3):2804‐2813. [Google Scholar]
- 213. Xue L, Luo S, Ding H, et al. Upregulation of miR‐146a‐5p is associated with increased proliferation and migration of vascular smooth muscle cells in aortic dissection. J Clin Lab Anal. 2019;33(4):e22843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Yu Y, Shi E, Gu T, et al. Overexpression of microRNA‐30a contributes to the development of aortic dissection by targeting lysyl oxidase. J Thorac Cardiovasc Surg. 2017;154(6):1862‐1869. [DOI] [PubMed] [Google Scholar]
- 215. Huang X, Yue Z, Wu J, et al. MicroRNA‐21 knockout exacerbates angiotensin II‐induced thoracic aortic aneurysm and dissection in mice with abnormal transforming growth factor‐beta‐SMAD3 signaling. Arterioscler Thromb Vasc Biol. 2018;38(5):1086‐1101. [DOI] [PubMed] [Google Scholar]
- 216. Cai B, Yang B, Huang D, et al. STAT3‐induced up‐regulation of lncRNA NEAT1 as a ceRNA facilitates abdominal aortic aneurysm formation by elevating TULP3. Biosci Rep. 2020;40(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. He X, Wang S, Li M, et al. Long noncoding RNA GAS5 induces abdominal aortic aneurysm formation by promoting smooth muscle apoptosis. Theranostics. 2019;9(19):5558‐5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Li DY, Busch A, Jin H, et al. H19 induces abdominal aortic aneurysm development and progression. Circulation. 2018;138(15):1551‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Zhang Z, Zou G, Chen X, et al. Knockdown of lncRNA PVT1 inhibits vascular smooth muscle cell apoptosis and extracellular matrix disruption in a murine abdominal aortic aneurysm model. Mol Cells. 2019;42(3):218‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Xia Q, Zhang L, Yan H, Yu L, Shan W, Jiang H. LUCAT1 contributes to MYRF‐dependent smooth muscle cell apoptosis and may facilitate aneurysm formation via the sequestration of miR‐199a‐5p. Cell Biol Int. 2020;44(3):755‐763. [DOI] [PubMed] [Google Scholar]
- 221. Tian Z, Sun Y, Sun X, Wang J, Jiang T. LINC00473 inhibits vascular smooth muscle cell viability to promote aneurysm formation via miR‐212‐5p/BASP1 axis. Eur J Pharmacol. 2020;873:172935. [DOI] [PubMed] [Google Scholar]
- 222. Huang B, Lu S, Lai H, Li J, Sun Y, Wang C. LncRNA LOXL1‐AS is up‐regulated in thoracic aortic aneurysm and regulated proliferation and apoptosis of aortic smooth muscle cells. Biosci Rep. 2019;39(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Chen S, Chen H, Yu C, et al. Long noncoding RNA myocardial infarction associated transcript promotes the development of thoracic aortic by targeting microRNA‐145 via the PI3K/Akt signaling pathway. J Cell Biochem. 2019;120(9):14405‐14413. [DOI] [PubMed] [Google Scholar]
- 224. Guo X, Chang Q, Pei H, et al. Long non‐coding RNA‐mRNA correlation analysis reveals the potential role of HOTAIR in pathogenesis of sporadic thoracic aortic aneurysm. Eur J Vasc Endovasc Surg. 2017;54(3):303‐314. [DOI] [PubMed] [Google Scholar]
- 225. Wang S, Zhang X, Yuan Y, et al. BRG1 expression is increased in thoracic aortic aneurysms and regulates proliferation and apoptosis of vascular smooth muscle cells through the long non‐coding RNA HIF1A‐AS1 in vitro. Eur J Cardiothorac Surg. 2015;47(3):439‐446. [DOI] [PubMed] [Google Scholar]
- 226. Hu W, Wang Z, Li Q, Wang J, Li L, Jiang G. Upregulation of lincRNA‐p21 in thoracic aortic aneurysms is involved in the regulation of proliferation and apoptosis of vascular smooth muscle cells by activating TGF‐beta1 signaling pathway. J Cell Biochem. 2019;120(3):4113‐4120. [DOI] [PubMed] [Google Scholar]
- 227. Allaire E, Muscatelli‐Groux B, Mandet C, et al. Paracrine effect of vascular smooth muscle cells in the prevention of aortic aneurysm formation. J Vasc Surg. 2002;36(5):1018‐1026. [DOI] [PubMed] [Google Scholar]
- 228. Allaire E, Muscatelli‐Groux B, Guinault AM, et al. Vascular smooth muscle cell endovascular therapy stabilizes already developed aneurysms in a model of aortic injury elicited by inflammation and proteolysis. Ann Surg. 2004;239(3):417‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase‐induced rat model of abdominal aortic aneurysm: Preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg. 1996;23(2):336‐346. [DOI] [PubMed] [Google Scholar]
- 230. Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter T. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;47(1):166‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Meijer CA, Stijnen T, Wasser MNJM, Hamming JF, van Bockel JH, Lindeman JHN. Doxycycline for stabilization of abdominal aortic aneurysms. A randomized trial. Ann Intern Med. 2013;159:815‐823. [DOI] [PubMed] [Google Scholar]
- 232. Baxter BT, Matsumura J, Curci JA, et al. Effect of doxycycline on aneurysm growth among patients with small infrarenal abdominal aortic aneurysms: a randomized clinical trial. JAMA. 2020;323(20):2029‐2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Allaire E, Forough R, Clowes M, Starcher B, Clowes AW. Local overexpression of TIMP‐1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest. 1998;102(7):1413‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Li Y, Lu G, Sun D, Zuo H, Wang DW, Yan J. Inhibition of endoplasmic reticulum stress signaling pathway: a new mechanism of statins to suppress the development of abdominal aortic aneurysm. PLoS One. 2017;12(4):e0174821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Schouten O, van Laanen JH, Boersma E, et al. Statins are associated with a reduced infrarenal abdominal aortic aneurysm growth. Eur J Vasc Endovasc Surg. 2006;32(1):21‐26. [DOI] [PubMed] [Google Scholar]
- 236. Bernardo BC, Ooi JY, Lin RC, McMullen JR. miRNA therapeutics: a new class of drugs with potential therapeutic applications in the heart. Future Med Chem. 2015;7(13):1771‐1792. [DOI] [PubMed] [Google Scholar]
- 237. Jalalzadeh H, Indrakusuma R, Blankensteijn JD, et al. Design and protocol of a comprehensive multicentre biobank for abdominal aortic aneurysms. BMJ Open. 2019;9(8):e028858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Wills A, Thompson MM, Crowther M, et al. Elastase‐induced matrix degradation in arterial organ cultures: an in vitro model of aneurysmal disease. J Vasc Surg. 1996;24(4):667‐679. [DOI] [PubMed] [Google Scholar]
- 239. Bogunovic N, Meekel JP, Majolee J, et al. Patient‐specific 3‐dimensional model of smooth muscle cell and extracellular matrix dysfunction for the study of aortic aneurysms. J Endovasc Ther. 2021;28(4):604‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Yokoyama U, Tonooka Y, Koretake R, et al. Arterial graft with elastic layer structure grown from cells. Sci Rep. 2017;7(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Burger J, Bogunovic N, de Wagenaar NP, et al. Molecular phenotyping and functional assessment of smooth muscle like‐cells with pathogenic variants in aneurysm genes ACTA2, MYH11, SMAD3 and FBN1. Hum Mol Genet. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. van de Pol V, Kurakula K, DeRuiter MC, Goumans MJ. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: what is the role of endothelial cells? Front Physiol. 2017;8:938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Ramella M, Boccafoschi F, Bellofatto K, et al. Endothelial MMP‐9 drives the inflammatory response in abdominal aortic aneurysm (AAA). Am J Transl Res. 2017;9(12):5485‐5495. [PMC free article] [PubMed] [Google Scholar]
- 244. Sun J, Deng H, Zhou Z, Xiong X, Gao L. Endothelium as a potential target for treatment of abdominal aortic aneurysm. Oxid Med Cell Longev. 2018;2018:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1