

Abstract
The conversion of the native monomeric cellular prion protein (PrPC) into an aggregated pathological β‐oligomeric form (PrPβ) and an infectious form (PrPSc) is the central element in the development of prion diseases. The structure of the aggregates and the molecular mechanisms of the conformational changes involved in the conversion are still unknown. We applied mass spectrometry combined with chemical crosslinking, hydrogen/deuterium exchange, limited proteolysis, and surface modification for the differential characterization of the native and the urea+acid‐converted prion β‐oligomer structures to obtain insights into the mechanisms of conversion and aggregation. For the determination of the structure of the monomer and the dimer unit of the β‐oligomer, we applied a recently‐developed approach for de novo protein structure determination which is based on the incorporation of zero‐length and short‐distance crosslinking data as intra‐ and inter‐protein constraints in discrete molecular dynamics simulations (CL‐DMD). Based on all of the structural‐proteomics experimental data and the computationally predicted structures of the monomer units, we propose the potential mode of assembly of the β‐oligomer. The proposed β‐oligomer assembly provides a clue on the β‐sheet nucleation site, and how template‐based conversion of the native prion molecule occurs, growth of the prion aggregates, and maturation into fibrils may occur.
Keywords: molecular modeling, protein aggregation, protein folding, structural proteomics
Abbreviations
- ABC
ammonium bicarbonate
- AcOH
acetic acid
- BS3
BisSulfoSuccinimidylSuberate
- CBDPS
CyanurBiotinDimercaptoPropionylSuccinimide
- CD
circular dichroism
- CL‐DMD
crosslink‐guided discrete molecular dynamics
- DMTMM
4‐(4,6‐dimethoxy‐1,3,5‐triazin‐2‐yl)‐4‐methylmorpholinium chloride
- DSA
DiSuccinimidylAdipate
- DSG
DiSuccinimidylGlutarate
- DSS
DiSuccinimidylSuberate
- ECD
electron‐capture dissociation
- EDC
1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride
- FTICR
Fourier‐transform ion cyclotron resonance
- MD
molecular dynamics
- NaOAc
sodium acetate
- NMR
nuclear magnetic resonance
- PCASS
pyridine carboxylic acid N‐hydroxysulfosuccinimide ester
- PICUP
photo‐induced crosslinking of unmodified proteins
- PrPC
native monomeric α‐helix rich cellular prion protein
- PrPSc
a pathogenic, fibril‐forming, multimeric, and predominately β‐sheet, prion protein
- REX
replica xchange
- SDA
succinimidyl 4,4'‐azipentanoate
- SDS
sodium dodecyl sulphate
- SDS‐PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- ShPrP
Syrian hamster prion protein
- TATA
2,4,6‐triazido‐1,3,5‐triazine
1. INTRODUCTION
The conversion of the native monomericα‐helix rich cellular prion protein (PrPC) into a pathogenic, fibril‐forming, multimeric, and predominately β‐sheet, prion protein (PrPSc) is the central element in the transmission and development of prion diseases [1, 2, 3]. The major hallmark of the conversion process is the creation of insoluble PrPSc from soluble PrPC [4, 5, 6, 7]. In the central nervous system, these insoluble proteins eventually accumulate as amyloid fibrils. Amyloid fibrils and plaques are the late products of the aggregation pathway and are often treated as the explicit effectors of prion disorders [8]. The intermediates or by‐products of the transition from PrPC to PrPSc, however, may actually be the pathogenic forms. β‐oligomers are one of the intermediate species that may be toxic and which may also be involved in the later stages of the process of PrPSc assembly [8, 9]. The study of the molecular mechanisms involved in the conformational change of PrPC that lead to the assembly and final structure of β‐oligomers are critical for understanding the aggregation process in prion disease [10]. Conversion of PrPC to PrPß oligomers can be studied in vitro using non‐glycosylated recombinantly‐expressed proteins. These oligomers carry structural features which are believed to resemble those existing in vivo during prion disease pathogenesis. Conversion to a β‐rich intermediate form, PrPβ, can be induced by a number of different methods including: low pH with chemical denaturants [11], low pH only [12], shaking [13], salt [11, 14‐17], lipopolysaccharide [18], copper [19, 20], manganese [21], dopamine [22], SDS [23, 24, 25, 26, 27] and other detergents [24], lipid [25, 28‐31], RNA [32], and lipid‐RNA [33, 34]. Conversion of PrPC to PrPβ can be confirmed using multiple methods such as: dynamic light scattering, proteinase‐K resistance assay, 1‐anilinonapthalene‐8‐sulfonate fluorescence and circular dichroism. Previously, we have studied strong acid (pH 1)‐converted [12] β‐oligomers by structural proteomics [35, 36]. Here, we study urea/mild acid (pH 4)‐converted prion protein, because the reaction product may more closely resemble the existing in vivo converted species, which occurs in an acidic environment comparable to that of endocytic vesicles [9, 37]. These β‐rich forms can be inherently challenging to study using conventional structural biology methods, such as, liquid‐state NMR spectroscopy and X‐ray crystallography, due to their large size, poor solubility, and heterogeneity.
Despite these difficulties, significant understanding of prion aggregate structures has been obtained using alternative methods such as protease accessibility [38, 39, 40], X‐ray diffraction [4, 41‐43], spin labeling [44, 45], computer modeling [46, 47, 48], infrared spectroscopy [4, 8, 49, 50], electron microscopy [51, 52], and antibody mapping [38, 53‐55]. Important structural insights have been gained by these methods, but there is still little agreement between the actual prion oligomer structures proposed [42, 47, 56‐59].
To improve the characterization of prion aggregate structures, additional proteomic approaches can be used [60]. Structural proteomics can be defined as a combination of protein chemistry methods, such as, chemical crosslinking, hydrogen/deuterium exchange, limited proteolysis, and surface chemical modification, with contemporary mass spectrometry. By using these methods, specific structural details of proteins and protein complexes can be obtained. These techniques are especially useful in situations such as that of prion aggregates [35, 61].
Numerous publications utilizing a single structural proteomics method for the study of prion structure have been reported [40, 55, 62‐64]. Crosslinking studies, which provide distance information between pairwise crosslinked amino acid residues, were reported for prion proteins using 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide hydrochloride (EDC) [65], BisSulfoSuccinimidylSuberate (BS3) [66], and CyanurBiotinDimercaptoPropionylSuccinimide (CBDPS) [35, 67]. Hydrogen/deuterium exchange (HDX) studies, which provide hydrogen‐bonding status of the amino acid residues in the protein, have been presented for prions using bottom‐up [34, 55, 62, 64, 68‐72] or top‐down [35] strategies. Limited proteolysis, which provides information on the accessibility of the surface of a protein to a large enzymatic probe, was used with chymotrypsin, pepsin, proteinase K, or trypsin for the characterization of both PrPβ and PrPSc [35, 39, 63, 73]. Chemical surface modification provides information on the protein's surface exposure to small molecule chemical modification reagents. This information can be complementary to that obtained by limited proteolysis. Surface modification studies of prions has been previously reported using pyridine carboxylic acid N‐hydroxysulfosuccinimide ester (PCASS) [35], acetic acid N‐hydroxysulfosuccinimide ester [74, 75], nitration and acetylation [76, 77], and methionine oxidation [36, 78].
We and others have shown that, during PrPC to PrPβ conversion, the prion protein undergoes a significant conformational rearrangement and that, for this to occur, there is a disengagement of the H1 α‐helix and a separation of contacts between the β1‐H1‐β2 domain and the H2‐H3 core [35, 41, 42, 68, 76]. This rearrangement is thought to result in changes where previously buried surfaces become exposed to solvent, and from which new inter‐protein contacts can develop. This conversion is also thought to allow the formation of a β‐sheet nucleation site, from which maturation to fibrillar forms can begin.
To further our investigations of prion oligomers, we have now used a comprehensive study using multiple proteomic techniques for the determination of the urea+acid‐induced prion‐oligomer structure. We utilized a panel of crosslinking reagents, including zero‐length and short‐ and long‐range reagents to identify intra‐ and inter‐protein crosslinks by performing the crosslinking reaction on an equimolar mixture of 14N/15N‐metabolically labelled β‐oligomer. We then used the short‐distance crosslinking constraints obtained from these experiments to guide discrete molecular dynamic simulations (CL‐DMD) [79], and obtained a structure of the PrPβ monomer and dimer units. The model was verified using hydrogen‐deuterium exchange, limited proteolysis and surface modification.
HDX was also used to assess changes in secondary structure between PrPC and PrPβ. A panel of proteolytic enzymes was used for limited proteolysis. 12C‐ and 13C‐PCASS was used for differential modification of K, Y, S, and T residues, and isotopically‐labelled hydrogen peroxide was used for the differential oxidation of W and M residues to obtain surface‐exposure changes as a result of the conversion. We used a combination of these structural‐proteomic methods to compare the structure of PrPC before and after conversion to urea+acid‐induced PrPβ oligomers. This approach allowed us to assemble a structure of the β‐oligomer, based on all of the constraints obtained. Our model of the β‐oligomer supports the rearrangement and disassembly of the β1‐H1‐β2 region from the H2‐H3 core which is pivotal to the conversion of PrPC to PrPβ, the formation of new inter‐protein hydrophobic contacts resulting from the change in exposure of hydrophobic residues, and the development of an apparent β‐sheet nucleation site.
2. MATERIALS AND METHODS
2.1. Materials
All chemicals were from Sigma‐Aldrich, unless noted otherwise. Crosslinking reagents azido‐benzoic‐acid‐succinimide (ABAS) [80]. CyanurBiotinDimercaptoPropionylSuccinimide (CBDPS) [81]. DiSuccinimidylAdipate (DSA), DiSuccinimidylGlutarate (DSG), DiSuccinimidylSuberate (DSS), succinimidyl 4,4'‐azipentanoate (SDA) [82], and 2,4,6‐triazido‐1,3,5‐triazine (TATA) [83] were obtained from Creative Molecules Inc.
Statement of significance
The conformational changes leading to the final structure of the misfolded infectious prion protein form is critical for our understanding of the mechanism of prion disease. Here, we determined the structure of urea+acid‐induced misfolded prion oligomers by using experimental short‐distance crosslinking data as constraints in discrete molecular dynamics simulations. The structure obtained was then validated using additional structural proteomic techniques: hydrogen‐deuterium exchange, limited proteolysis, and surface modification experiments. During the conformational change, a structured loop region dissociates from the protein core, resulting in exposure of hydrophobic residues and the creation of a new putative β‐structure nucleation site, which leads to an assembly of stacked subunits in the oligomer. The structure of the prion oligomer explains a likely mechanism of prion aggregation, and may allow the rational design of therapeutics to prevent this aggregation.
A synthetic gene corresponding to Syrian hamster prion protein residues 90–232 (ShPrP(90‐232)) including a 22‐residue N‐terminal fusion tag with thrombin cleavage site and 6x‐His was obtained from the laboratory of Dr. David Wishart (University of Alberta). ShPrP(90‐232) was expressed with or without minimal 15N‐labeled media in E. coli as previously described [12, 84]. Cells were sonicated, and—in order to purify expressed PrP from inclusion bodies—cell lysates were resuspended in guanidine solubilizing buffer and the protein was loaded onto immobilized nickel‐nitrilotriacetic acid (Ni‐NTA) beads via their fused histidine tail [85]. To ensure the correct folding to native PrPC, bound protein was re‐folded on‐column with a decreasing urea concentration gradient, over a 12‐h time period [12]. Immobilizing PrP on‐column favors formation of intramolecular disulfide bond and prevents protein aggregation [85]. Protein refolded using this method is predominantly in the native PrPC form with little or no β‐isoforms present [11]. Refolded soluble protein was eluted with imidazole in sodium phosphate buffer without urea, and then buffer exchanged and concentrated with 10 kDa cut‐off centrifugal filters (Amicon) and 20 mM NaOAc pH 5.2. SDS‐PAGE was used to determine PrPC purity, and protein can be further characterized by NMR, CD, and fluorescence spectroscopy [85]. PrPC can be dialysed in a low‐salt buffer such as 20 mM sodium acetate (NaOAc) and remains stable for at least 5 weeks at 4°C [11].
The β‐oligomeric form was created as previously described [11] (from 0 to 5 M urea at pH 4.0). After conversion, oligomers were dialyzed in 20 mM NaOAc pH 5.2 (or 10 mM ammonium acetate for HDX experiments) using dialysis buttons (Hampton Research) at 4°C for 180 min three times in 50 ml of 20 mM NaOAc pH 5.2 and the concentration was determined by measuring the absorbance at 280 nm. Oligomer formation was confirmed by comparative crosslinking assay between PrPC and PrPβ using SDA.
2.2. Crosslinking
Crosslinking was performed on an equimolar 14N15N mixture of PrPβ (Figure S1) [86] with a panel of crosslinking reagents [ABAS [80], CBDPS, 4‐(4, 6‐dimethoxy‐1,3,5‐triazin‐2‐yl)‐4‐methylmorpholinium chloride (DMTMM) [87, 88], DSA, DSG, DSS, 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide hydrochloride (EDC) [89], photo‐induced crosslinking of unmodified proteins (PICUP) [90], SDA [80, 82], and TATA [83] differing in reactivity and spacer length (see Table S1 for crosslinking conditions for each reagent). After crosslinking reaction, 1% (final concentration) deoxycholate was added to each sample for 30 min at 25°C. Samples were then digested with proteinase K [79] in 1:25 enzyme: protein substrate ratio for 2.25 h at 37°C 400 rpm then acidified with 0.9% final trifluoroacetic acid for 5 min. Samples were then spun down at 15,000 rpm on a desktop centrifuge for 10 min and supernatant removed to separate microcentrifuge tube prior to C18 stage tip clean up then analyzed on LC/ESI‐MS/MS on an LTQ Orbitrap Velos or Orbitrap Fusion mass spectrometer followed by crosslinking site assignment using 14N15N DXMSMS Match [91], with 2 ppm precursor and 10 ppm fragment mass tolerance, 10 ppm 14N‐15N mass difference tolerance, 50% 15N/14N intensity ratio tolerance, tryptic digestions sites, all possible missed cleavages.
2.3. Modeling
2.3.1. PrPβ dimer simulation
The protocol for the crosslink‐guided discrete molecular dynamics (CL‐DMD) simulations has been described in detail in our previous work [60, 79, 92, 93]. DMD is an enhanced‐sampling molecular‐dynamics method that allows efficient ab initio protein structure prediction [94, 95]. Discretization of inter‐atom interaction potentials and changing from fixed to event‐based time steps significantly optimizes the computational performance of DMD compared to conventional molecular dynamic (MD) approaches [96, 97]. A series of well‐like potentials have been incorporated into the DMD force field to account for the experimentally identified inter‐residue distances. The widths of the potentials correspond to the crosslinker spacer lengths and the side‐chain flexibility [79]. These constraint potentials energetically penalize atom pairs if the distance between them does not satisfy experimentally observed distances.
All‐atom Replica Exchange (REX) [98, 99] simulations of the PrPβ dimer were performed starting from two completely unfolded chains of PrPC. Twenty‐four replicates of the system, with temperatures equally distributed over the range from 0.375 to 0.605 kcal/(mol kB), were simulated for 6 × 106 DMD time steps. REX simulations allow the system to overcome local energy barriers and increase conformational sampling by periodically exchanging the simulation temperatures of the replicates. By monitoring the system energy distribution and the specific heat curve [100], we found that approximately 1.5 × 106 time steps were necessary for the simulations to reach system equilibrium. The time steps before equilibrium was reached were not included in the analysis. As described in [79], we performed distance‐based clustering analysis [101, 102] on the lowest‐energy 10% of the structures obtained during CL‐DMD simulations. The centroids of the clusters were chosen as the computationally predicted models of the PrPβ dimer and were subject to further verification against additional structural‐proteomics data.
2.3.2. β‐oligomer assembly
The monomer subunits, which form the predicted PrPβ dimer, were used as the initial point in predicting the structure of the prion trimer. Starting from three chains, which were folded into the conformations observed in the PrPβ dimer structure, we performed REX simulations according to the procedure described above. The chains were placed far enough from each other to allow them to freely rotate without crashing into another chain. Similar to the structure prediction of the dimer, experimentally determined inter‐ and intra‐chain inter‐atom proximity constraints were used to guide DMD simulations. After 3×106 time steps of REX CL‐DMD simulations, centroids of the most highly populated lowest energy structures were selected as possible computational models for the prion trimer.
2.4. Hydrogen/deuterium exchange
HDX using FTMS‐ECD top‐down analysis was performed on a Bruker 12‐T FTICR mass spectrometer. Experiments were carried out under “exchange‐in” conditions, using a two‐stage continuous‐flow mixer as described previously [35, 103]. Protein solution and D2O from separate syringes were mixed continuously in a 1:4 ratio (80% D2O final) through a three‐way tee connected to a 100 μm × 21 cm capillary, resulting in a 10 s labeling time. The outflow from this capillary was mixed with a quenching solution (0.4% formic acid in 80% D2O) from a third syringe via a second three‐way tee. The outflow from this last capillary was injected into a Bruker 12T Apex‐Qe hybrid Fourier Transform mass spectrometer, equipped with an Apollo II electrospray source. In‐cell ECD fragmentation experiments were performed with an m/z 900–1200 precursor selection range using a grid potential of 12 V and a cathode filament current of 1.2 A. Approximately 1200 scans were accumulated over the m/z range 250–2600, resulting in an approximate acquisition time of 30 min for each ECD spectrum. FT‐MS calibration used the ECD fragments of PrPβ. HDX Match software was used to determine deuteration levels of the amino acid residue's amide groups from centroid masses of the c‐ and z‐ion series [104].
2.5. Circular dichroism
Circular dichroism spectra were recorded on a Jasco J‐720 spectropolarimeter using a temperature‐regulated with 1 mm optical path‐length quartz cell. Spectra were recorded in the UV region (190–250 nm) in 20 mM NaOAc (pH 5.2) at a protein concentration of 0.5 mg/ml. Secondary structure estimation performed using BeStSel [105].
2.6. Limited proteolysis
Limited proteolysis was performed using Arg‐C (in 20 mM Na2HPO4), Asp‐N (in 100 mM ammonium bicarbonate (ABC)), chymotrypsin (in PBS and 10 mM CaCl2), Glu‐C (in either 50 mM ABC or PBS pH 7.4), pepsin (in 20 mM HEPES 6.9), proteinase‐K (in PBS) and trypsin (in PBS pH 7.4). For each experiment, a 15‐μg aliquot of PrPC and PrPβ was incubated with enzyme: substrate ratios at 1:25 for pepsin, 1:500 for proteinase‐K, and 1:50 for all other enzymes at 37°C. Aliquots of 3 μg each were removed at time 0, 1, 5, 10, and 30 min and were immediately mixed with 4x NuPage lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) and heated at 100°C for 10 min and all samples were then separated by SDS‐PAGE. Individual bands, corresponding to proteolytic fragments of the prion protein were individually excised and prepared by in‐gel tryptic digestion [106], and digestion sites were determined by LC/ESI‐MS/MS analysis on an LTQ Orbitrap Velos mass spectrometer using PEAKS Client 7.0 software with 2 ppm precursor and 10 ppm fragment mass tolerance settings.
2.7. Surface modification
PrPC and PrPβ were modified with either the heavy or light form of the surface modification reagent, pyridine carboxylic acid N‐hydroxysulfosuccinimide ester (PCASS‐12C6 and 13C6) (Creative Molecules Inc.) to modify the N‐terminus, K, Y, S, and T residues (Figure S2A) [35], and heavy or light hydrogen peroxide (H2 16O2 or H2 18O2) for modification of W and M residues (Figure S2B) [36].
For PCASS differential surface modification, 6.5 μg aliquots of PrPC or PrPβ were prepared at 4.0 μM. Reaction mixtures containing either 10 mM PCASS light (PCASS‐12C6) or heavy (PCASS‐13C6) and either PrPC or PrPβ were incubated for 30 min at 25°C, and were quenched with 25 mM ABC. Solutions were then acidified to pH 2 with 10% acetic acid. Prior to the addition of pepsin, samples were combined (PrPC + PCASS‐12C6 and PrPβ + PCASS‐13C6, or PrPC + PCASS‐13C6 and PrPβ + PCASS‐12C6) then digested with pepsin at a 1:10 enzyme: substrate ratio for 12 h at 37°C. Digests were analyzed by LC/ESI‐MS/MS on a LTQ Orbitrap Velos mass spectrometer, and peptides containing a modification and their chromatographic peak areas were identified using Peaks Client 7.0 software.
For H2O2 differential oxidation, solutions containing 6.5‐μg aliquots of PrPC or PrPβ at 4.0 μM were bubbled with nitrogen gas prior to the addition of H2O2. PrPC or PrPβ were incubated with either 10 mM H2O2 light (H2 16O2) or heavy (H2 18O2) for 30 min at 25°C, and were quenched with 10% acetic acid to pH 2. Prior to the addition of pepsin, samples were combined (PrPC + H2 16O2 and PrPβ + H2 18O2, or PrPC + H2 18O2 and PrPβ + H2 16O2 light), bubbled with nitrogen gas, and digested with pepsin at a 1:10 enzyme: substrate ratio for 12 h at 37°C. Digests were analyzed by LC/ESI‐MS/MS on an LTQ Orbitrap Velos mass spectrometer, and identification of peptides containing a modification and their chromatographic peak areas were identified using Peaks Client 7.0 software.
For PCASS modification experiments, the mean of the log peak area ratio (PrPC with PCASS‐12C6/ PrPβ with PCASS‐13C6 or PrPC with PCASS‐13C6/ PrPβ with PCASS‐12C6) was calculated for each modified residue. The antilog of the mean of the ratio provided a value indicative of the relative surface exposure of a residue in PrPC and PrPβ.
For H2O2 modification experiments, the log of the peak area ratio (PrPC with H2 16O2/ PrPβ with H2 18O2 or PrPC with H2 18O2/ PrPβ with H2 16O2) was calculated for each modified residue. Differences in the oxidation levels of a residue between PrPC and PrPβ were calculated using a comparative ratio formula [36] which accounts for endogenous oxidation of the residues. The resulting ratio provides a value indicative of the relative exposure of a residue in PrPC and PrPβ.
3. RESULTS AND DISCUSSION
3.1. Formation of oligomers
Multiple structural proteomic techniques were used in order to characterize the conformational change from PrPC to PrPβ and the arrangement of the subunits in the aggregate. We compared both forms of the N‐terminal histidine tagged Syrian hamster prion protein (90–232), recombinantly expressed in E. coli. The E. coli. construct contains aa90‐aa128 of the aa29‐aa128 flexible and unstructured N‐terminal region [107]. Even with the truncation, the construct contains PrP 27–30, the hallmark proteinase K resistant core region of pathogenic PrPSc identified in vivo [108, 109], and carries all of the necessary features for the conversion and propagation of aggregates [110, 111, 112, 113]. The expressed protein was purified from the inclusion bodies and refolded on‐column with a decreasing urea‐concentration gradient.
Of the multiple conversion methods reported in the literature, we chose the urea+acid conversion, as it uses conditions thought to be comparable to those occurring in vivo [9, 37, 114, 115] and may, therefore, adequately represent the mechanism of aggregation involved in the pathogenesis. To convert the purified soluble PrPC to PrPβ, we incubated the protein in the conversion buffer (5 M urea, 20 mM NaOAc, 200 mM NaCl, 60 mM AcOH) for 12 h at 25°C. The solution was then dialyzed against 20 mM NaOAc pH 5.2. β‐oligomers obtained using this method exhibited an increased resistance to proteinase‐K digestion (Figure S3A) and an increase in β‐sheet structure as observed by CD (11.8% β‐sheet content in PrPC and 20.1% in PrPβ) (Figure S3B). A crosslinking titration assay of PrPC and PrPβ shows marked differential formation of the inter‐protein crosslinked species on SDS‐PAGE gel for PrPβ but not for PrPC (Figure S3C). We performed structural characterization by HDX, crosslinking, limited proteolysis, and surface modification on these aggregated forms.
3.2. Crosslinking
Crosslinking combined with mass spectrometry provides the distance information between pairwise crosslinked amino acid residues. Crosslinked proteins were enzymatically digested and analyzed by LC/MS/MS. Crosslinking reagents can vary in their specificities and can include amine‐reactive (e.g., CBDPS with lysine, serine, tyrosine, threonine, and N‐terminal reactivity), amine to carboxyl specific (e.g., EDC or DMTMM with lysine to aspartic or glutamic acid reactivity), specific photo‐reactive (PICUP with tyrosine to tyrosine), and non‐specific photo‐reactive (e.g., TATA). The length of the spacer‐arm between covalently bound functional groups in crosslinks dictates the distance constraints obtained, which can vary from 14 Å in the case of CBDPS to shorter range constraints (e.g., 8 Å DSG and 5 Å TATA) or zero‐length crosslinks (<1 Å) (e.g., EDC and PICUP).
By crosslinking an equimolar mixture of 14N‐ and 15N‐PrPβ, we were able to determine if the crosslinks occur within the same protein (intra‐protein crosslinks) or between two protein molecules (inter‐protein crosslinks) [86] (Figure S1). Determining whether a crosslink is of intra‐ or inter‐protein origin [91] is critical for guiding the PrPβ subunit structure modeling (intra‐protein crosslinks) and PrPβ assembly (inter‐protein crosslinks).
3.3. Structure of the β‐oligomer dimer unit by CL‐DMD
Using a panel of crosslinking reagents (DMTMM, PICUP, TATA, ABAS, SDA, DSA, DSG, DSS, and CBDPS; Figure S4 and Table S1), we detected 14 intra‐protein and 48 inter‐protein inter‐peptide crosslinks (Table S2 and Table S3). The oligomer model was then built by satisfying all of the zero‐length and short‐distance intra‐protein and inter‐protein constraints using CL‐DMD methodology [79] (Figure 1). Zero‐length crosslinks obtained are especially important in the modeling process as they provide the strictest distance constraints. The intra‐protein DMTMM crosslink D167‐ K204 required rearrangement of the β2‐H2 loop to the H3 to satisfy the constraint, while the PICUP inter‐protein crosslinks of Y225 to Y157, Y162, and Y163 anchor the region of increased HDX protection to the H3C‐terminal end of another subunit (Figure 2). Furthermore, the PICUP inter‐protein crosslinks, Y128 to Y128 and Y128 to Y162 or Y163 form a triangle with Y157, Y162, or Y163 to Y225 and support the rearrangement of the β1‐H1‐β2 and formation of a compact hydrophobic region of inter‐protein contacts (Figure 2A).
FIGURE 1.
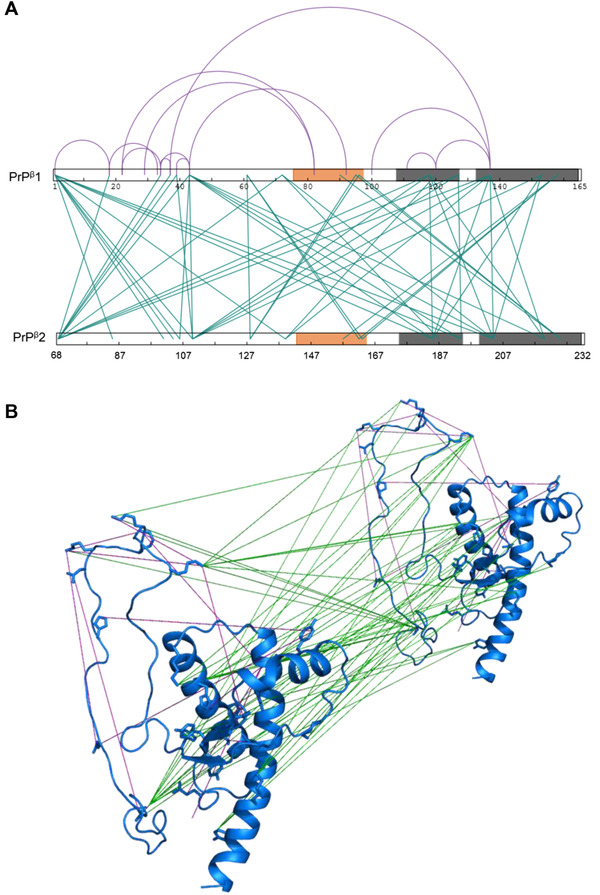
Crosslinking analysis of PrPβ. (A) Representation of all intra‐ and inter‐protein constraints obtained by crosslinking a 1:1 equimolar mixture of 14N‐ and 15N‐PrPβ (Tables S2 and S3). The sequence of two PrPβ monomers is represented. Orange shading in sequence represents newly formed β‐nucleation site and grey shading represents H2‐H3 core α‐helices. Intra‐protein inter‐peptide crosslinks are illustrated as purple (arcs) and inter‐protein inter‐peptide crosslinks are shown as green (lines). The figure was created using xiNet [139]. B) Short‐distance crosslinking constraints guide PrPβ model. All intra‐ and inter‐ protein crosslinks are represented on two PrPβ monomers. Crosslinks are indicated as intra‐protein crosslinks (purple) and inter‐protein crosslinks (green). The intra‐protein crosslinks show the conformational change that takes place under conversion and the inter‐protein crosslinks allow for arrangement of monomers
FIGURE 2.
PrPβ dimer structure obtained by short‐distance crosslinking constraint‐guided discrete molecular dynamics. (A) Intra‐protein crosslinks (magenta) (Table S1) and inter‐protein crosslinks (green) (Table S2). (B) PrPβ monomer unit obtained by CL‐DMD and verified using structural proteomic methods. Residue deuteration values are superimposed on the representative predicted structure of the PrPβ monomer. Intra‐protein crosslinks (magenta) (Table S1). Residues preferentially modified by PCASS or H2O2 in PrPβ and preferentially exposed to proteases in PrPβ (red backbone and spheres and red backbone respectively). Residues equally modified by PCASS or H2O2 between PrPC and PrPβ and equally exposed to proteases in PrPC and PrPβ (marine backbone and spheres and marine backbone respectively). Residues preferentially modified by PCASS or H2O2 in PrPC and preferentially exposed to proteases in PrPC (green backbone and spheres and green backbone, respectively)
An array of inter‐protein crosslinks (especially those found with amine‐reactive reagents, such as DSG, DSA, DSS, and CBDPS) guided the further assembly and placement of the H2‐H3 cores of each subunit which stack together (e.g., CBDPS K194 – K194, K185 – K185, and DSG K204 – K204, K185 – K204, and K204 – K220) and lead to a “twisting” subunit arrangement (Figure 2).
The N‐terminal portion of the protein can be localized in slightly different regions of the oligomer depending on whether intra‐protein or inter‐protein crosslinks are used (Figures 1A and 2). The intra‐protein DSG crosslink K104‐K204 anchors the N‐terminal region to the start of H3, and the SDA crosslinks K110‐N159, E89‐Y149 and H96‐Y149 anchor the N‐terminal region to the rearranged β1‐H1‐β2 region (Figure 2A). Based on the inter‐protein crosslinks, the N‐terminal region of one subunit associates with the newly rearranged β1‐H1‐β2 of an adjacent subunit and the C‐terminal end of H2 (Figure 2A), as indicated by the ABAS crosslinks K110‐K194 (N‐terminal region to C‐terminal end of H2) and M139‐K194 (rearranged β1‐H1 loop to C‐terminal end of H2) (Figure 2A). This observation suggests that the N‐terminal portion of the protein can occupy similar sites on both the subunit it originates from and the adjacent neighboring subunit.
Crosslinking data has allowed us to assemble a structure of the β‐oligomer dimer based on all of the constraints obtained. In the model, region aa144‐aa165—which showed an increased protection in HDX—and the hydrophobic residues from this region (e.g., Y157, Y161, Y162 and Y169) may be involved in the newly formed β‐strands of the putative β‐nucleation site. The hydrophobic residue Y128 is outside the aa151–173 region, but it may also be interacting with this β‐sheet structure, since it is in close proximity (according to PICUP crosslinking) to Y128, Y162 and/or Y163 of the adjacent subunit (Figure 2A). The obtained model was further validated by additional results from both limited proteolysis and surface modification.
3.4. Hydrogen/deuterium exchange
The principle of the hydrogen/deuterium exchange method is that backbone amide hydrogens exchange with deuterium from the solution at different rates based on their hydrogen bonding status. Consequently, the deuteration status of backbone amides will reflect their involvement in secondary structure [116, 117, 118]. In this study, we utilized top‐down ECD‐FTICR mass spectrometry to determine the boundaries of secondary‐structure elements based on the deuteration values of the amino acid residues [103, 119].
PrPβ exhibited an increase in protection of approximately 11 protons compared to PrPC, based on the total deuteration values of the intact proteins (Figures S5 and S6). This increase in the protection of PrPβ, in combination with CD data, suggests an increase in β‐structure, a hallmark of the subsequent maturation of the β‐oligomers to fibrils.
Analysis of the c‐ and z‐ ion fragment series [104] revealed that the aa144‐aa165 region of PrPβ (the region encompassing the H1, the H1‐β2 loop and β2) (Figure S5) was responsible for the increased protection and, therefore, the increased hydrogen bonding of this segment in the aggregated form. These HDX results indicated a rearrangement or restructuring to a β‐sheet structure in the aa144‐aa165 region and may highlight a possible β‐sheet initiation site, which is crucial for the progression of the aggregation process. Indeed, the total numbers of the protected backbone amide protons in PrPC and PrPβ, as measured by HDX of intact proteins, were 55 and 66, respectively. H2 and H3 contain 52 residues and account for 39 (PrPC) and 38 (PrPβ) protected protons (Figure S6). The aa68‐aa142 N‐terminal portion of the protein accounts for 7 protected protons in both forms. H1, β1, and β2 contain 21 residues and account for 13 (PrPC) and 24 (PrPβ) protected protons (Figure S6). Thus, an increase in β‐structure content in PrPβ (up to 20.1% from 11.8% in PrPC), as determined by CD (Figure 3B), points out the disassembly of H1 helix and the formation of new β‐structure in the aa143‐aa164 region (Figure S6D). This rearrangement was consistent with the crosslinking analysis of the PrPβ oligomer.
3.5. Limited proteolysis
Limited proteolysis provides information on the accessibility of a protein's surface to a large (enzyme) probe. The first cleavage sites obtained after short incubation times are restricted to the outermost regions of a protein subunit's surfaces, which are accessible to the active site of the proteolytic enzyme. Under limited proteolysis with pepsin, a different pattern of digestion for PrPC versus PrPβ was observed (Figure 2B, Figure S7). The native form exhibited a rapid accumulation of a C‐terminal ∼15 kDa product. However, for the oligomeric form, virtually no proteolysis was evident. This indicates a lack of enzyme access to the W99 cleavage site in this region of the oligomer (Figures 2B and S7). Limited proteolysis with chymotrypsin also highlighted W99 as being more protected from cleavage in PrPβ than PrPC. Limited proteolysis with chymotrypsin also showed greater protection of residues Y149, Y150, Y162, Y163, F175, and M206 in PrPβ relative to PrPC (Figures 2B and S7). Arg‐C cleavage of the N‐terminal region (R83) was diminished in PrPβ over PrPC (Figures 2B and S7), which is indicative of an N‐terminal region location in the oligomeric form to either the start of H3 or the C‐terminal region of H2, as was evidenced by the crosslinking results (see above). Trypsin also indicated somewhat higher protection of R83 and K110 cleavage sites in PrPβ, which is consistent with the Arg‐C results (Figures 2B and S7). AspN digestion occurred at D227 in the PrP C‐terminal region and was equally available to digestion in both prion forms (Figure S7). These results contrast with the GluC digestion results, which also illustrated PrPC‐terminal region digestion at E221, except that proteolysis occurred more rapidly in PrPβ versus PrPC (Figures 2B and S7). This could be the result of the β2‐H2 loop moving away from H3 in PrPβ. In summary, limited proteolysis data were consistent with the HDX and crosslinking findings for the PrPβ and, therefore, can serve as a positive confirmation of the proposed structure (Figure 2).
3.6. Surface modification
Surface modification provides information on the accessibility of a protein's residues to small (modification reagent) probes. We used isotopically‐coded amine‐reactive PCASS (Figure S2A) [35] and hydrogen‐peroxide reagents for differential surface modification of PrPC versus PrPβ (Figure S2B) [36]. In these experiments, equal amounts of PrPC and PrPβ were modified with either the light or heavy isotopic forms of the reagents. The reactions were quenched, both protein form samples were combined, digested and analyzed by LC–MS/MS. In this experimental design, residues that are equally exposed in both protein forms result in doublets of peaks of equal intensity. Residues which differ in surface accessibility between the two protein states should show a higher degree of modification in one or other of the states, resulting in a doublet of peaks with unequal intensity ratios.
Using the PCASS surface modification, we detected several differentially modified residues between the PrPC and PrPβ forms (Table S4, Figure 2B). Differentially modified tyrosine residues Y149, Y150, Y157, Y162, and Y169 are located within the β1‐H1‐β2 region and showed increased exposure in PrPβ. Tyrosines Y149, Y150, and Y157 contact the internal regions of H2 and H3 and are buried in the native PrPC structure (Figure 2B and Table S4). Interestingly, Y149, Y150, Y162, and Y163 were preferentially modified, but were protected from chymotryptic proteolysis (Figure 2B, Figure S7, Table S4), possibly indicating rather tight packing of this reformed region in the PrPβ form. Y128, which was shown by crosslinking to be in close proximity to this region, exhibited an equal degree of modification between PrPC and PrPβ and must therefore be equally exposed in both prion forms (Figure 2B and Table S4) and may represent adjacent residues to the apparent β‐nucleation site. Both K185 and K220 were less accessible in PrPβ, possibly indicating involvement in the stacking of the subunits and interaction with the relocated N‐terminal part in the oligomer (Figure 2 and Table S4).
Using the isotopically coded hydrogen peroxide surface modification method, we also detected several differentially modified residues between the PrPC and PrPβ forms (Figure 2B and Table S5). The preferentially modified in PrPβ residues—M129, M134, M138, M139, W145, and M154—are located within the rearranged β1‐H1‐β2 region and showed increased exposure in PrPβ (Figure 2B and Table S5). W99 was shown to be less exposed in PrPβ (Table S5). This was in agreement with both the chymotrypsin and pepsin limited‐proteolysis results, indicating that this residue is buried in PrPβ (Figure 2B and Figure S7).
3.7. PrPβ assembly
Our model confirms that the conformational change occurring during urea+acid conversion results from the rearrangement and disassembly of the β1‐H1‐β2 region from the H2‐H3 core. It shows that the H2‐H3 core area becomes involved in contacts with the rearranged aa126‐aa170 region of an adjacent monomer (Figure 2). The aa144 – aa165 region shows increased protection in HDX and is involved in numerous crosslinks (3 intra‐protein, 5 inter‐protein). This region appears to be involved in the evolution of a β‐sheet nucleation site and the formation of new inter‐protein contacts.
The overall shape of the monomer in the dimer unit of the β‐oligomer is reminiscent of an open “pocket knife,” with H2‐H3 forming the “handle” and the mostly open and rearranged β1‐H1‐β2 region forming the “blade” (Figure 2B and Figure S9). Created as a result of conversion internal surfaces of the “handle” and the “blade” constitute newly formed inter‐protein interfaces in the dimer. From the analysis of the dimer unit structure, it became apparent that additional monomer subunits in a similar “blade” conformation could be appended to the structure. This can be accomplished by aligning additional monomer with a twist‐and‐up translocation of the dimer unit with most of the inter‐protein crosslinks still being satisfied (Figure 3).
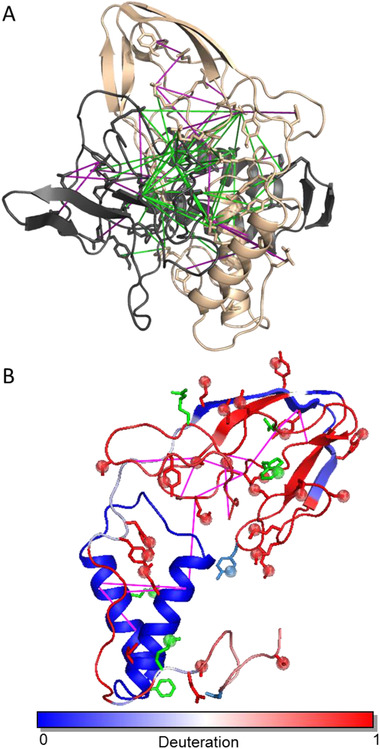
FIGURE 3.
Arrangement of subunits in the PrPβ oligomer. Additional monomer subunits in a conformation similar to the dimer can be appended to the structure with a twist‐and‐up translocation, with most of the inter‐protein crosslinks still being satisfied. Three PrPβ monomers (magenta, green, and blue) are shown in a possible trimer arrangement. β‐strands at the top of the assembly may explain the creation of the β‐nucleation site
This mode of monomer subunit arrangement is also in agreement with the observed electrostatic surface properties of the subunits within the oligomer (Figure S10). Surprisingly, if this assembly is allowed to continue, an assembled from “blades” of “pocket knife” structure resembling a drill bit is observed (Figure 3). The tip of the “drill bit” consists of newly formed β‐strands and contains multiple residues that are critically important for the aggregation. This implies possible involvement of this “drill bit tip” structure in the template‐based conversion and growth of the aggregates. Moreover, an arrangement of the β‐strands in this region is consistent with further formation of the cross‐beta structure during oligomer maturation into fibrils.
Our PrPβ model is consistent with experimental data obtained from numerous other methods. Wille et al. [120] used negative stain electron microscopy to determine recombinant ShPrP 90–231 fibrils as having an average diameter of 79 ± 38 Å, which is comparable to the diameter of our assembled oligomer model, which is approximately 48 Å (Figure 3). The model is also consistent with numerous antibody‐mapping studies of PrPSc, the positions of inhibitory peptides, and contains the residues in critical regions of the assembled structure that are essential for aggregation. For example, Peretz et al. [121] used a panel of recombinant antibodies to ShPrPC, with linear epitopes, to determine that residues within the N‐terminal region (aa90 –aa120) were largely inaccessible in hamster brain derived PrP 27–30 but remained accessible in PrPC (Figure S11A).
Numerous monoclonal antibody studies have also described the N‐terminal domain as important in the conversion of PrPC to PrPSc and implicate this domain as critical to the binding of PrPC to PrPSc (Figure S11A) [122, 123, 124, 125, 126]. These studies indicate that the major change in conformation to PrPSc occurs within this region, which is in agreement with our PrPβ model (Figure 2, Figure 3, Figure S8, and Figure S11). The PrPC‐selective antibody D18 [123] binds to the β1‐H1 loop and the H1 helix (aa132 – aa156) and has no reactivity with PrPSc. This is also consistent with our model, which shows that this region has undergone significant structural rearrangement (Figure 11B). Epitopes for a PrPSc‐specific YYR antibody [127] (aa148‐ aa150 and 161 ‐ aa163) are both exposed in our model (Figure S11C). The PrPSc‐specific monoclonal antibody 6H10 has a discontinuous epitope located at the C‐terminal region of helix H3(aa215 – aa216, aa221‐ aa223, aa228) [128] where our model also shows structural rearrangement of the helix (Figure S11D).
Chabry et al. [129] demonstrated that synthetic peptides corresponding to aa113 ‐ aa120 and aa129 – aa141 can inhibit the formation of protease‐resistant prion protein in vitro. These peptides represent regions of our model which may be implicated in the conversion (Figure S11E). Recombinant antibody antigen‐binding fragments (Fabs) of D18 antibody [123] (described above) has also been shown to inhibit the conversion of PrPC to PrPSc and to inhibit the development of prion disease in vivo [130, 131].
A common polymorphism of human prion protein has either methionine or valine at residue 129. M129V heterozygotes have relative protection from sporadic, acquired, and some inherited forms of prion disease [132, 133]. This protection is thought to occur by inhibiting homotropic protein‐protein interactions [132, 134]. Residue 129 may also directly affect the propagation of some prion strains by conformational selection [135, 136]. Polymorphism at codon 127 (G127V) has been shown to be protective against kuru disease and, interestingly, has only been identified among natives of Papua, New Guinea [137]. Y128‐Y128 PICUP inter‐protein crosslinks between different subunits (Table S3) demonstrate the close inter‐protein proximity of these residue 127 and 129 critical polymorphic variants. M129 was found buried in the core of amyloid fibrils [138]. In our PrPβ assembly model, these residues are buried at the center of the assembly where three subunits converge, which may explain why G127V is important for protective effects against conversion (Figure S11F).
Overall, our experimentally based model of the β‐oligomer assembly is consistent with the conformational change during urea+acid conversion resulting from the rearrangement of the PrPC β1‐H1‐β2 region and disruption of its original contacts with the H2‐H3 core and formation of new contacts with the rearranged aa126‐aa170 region of an adjacent monomer (Figures 2 and 3). This mode of interaction is preserved in the incorporation of additional subunits to the β‐oligomer. As a result, our final structure of the β‐oligomer is a rod‐like structure composed from monomers assembled in ascending spiral fashion. The tip of the assembly is built from newly formed β‐strands which can be involved into template‐based conversion and further maturation into fibrils.
4. CONCLUSION
Data from multiple structural proteomics approaches combined with constraints‐guided DMD simulations have enabled us to determine the structure of urea+acid converted prion oligomers. The obtained structure explains the mechanism of the conformational change involved in the conversion, the early formation of the β‐structure nucleation site, and describes the possible mode of assembly of the subunits within the oligomer.
CONFLICT OF INTEREST
EVP and CHB are co‐founders of Creative Molecules, Inc., which provides crosslinkers to researchers at cost.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors are grateful to Genome Canada and Genome British Columbia for financial support (project code 264PRO). CHB is also grateful for support from PrioNet Canada, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the Leading Edge Endowment Fund (University of Victoria) and for support from the Segal McGill Chair in Molecular Oncology at McGill University (Montreal, Quebec, Canada). CHB is also appreciative of support from the Terry Fox Research Institute, the Warren Y. Soper Charitable Trust, and the Alvin Segal Family Foundation to the Jewish General Hospital (Montreal, Quebec, Canada). The study was also supported by the MegaGrant of the Ministry of Science and Higher Education of the Russian Federation (Agreement with Skolkovo Institute of Science and Technology, No. 075‐10‐2019‐083). NVD acknowledges support from the National Institutes of Health (1R35 GM134864) and the Passan Foundation. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript
Serpa, J. J. , Popov, K. I. , Petrotchenko, E. V. , Dokholyan, N. V. , & Borchers, C. H. (2021). Structure of prion β‐oligomers as determined by short‐distance crosslinking constraint‐guided discrete molecular dynamics simulations. Proteomics, 21, e2000298. 10.1002/pmic.202000298
Contributor Information
Nikolay V. Dokholyan, Email: dokh@psu.edu.
Christoph H. Borchers, Email: christoph.borchers@mcgill.ca.
DATA AVAILABILITY STATEMENT
Data will be made available upon request.
REFERENCES
- 1. Kupfer, L. , Hinrichs, W. , & Groschup, M. (2009). Prion protein misfolding. Current Molecular Medicine, 9(7), 826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguzzi, A. , & Calella, A. M. (2009). Prions: Protein aggregation and infectious diseases. Physiological Reviews, 89(4), 1105–1152. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen, P. H. , Ramamoorthy, A. , Sahoo, B. R. , Zheng, J. , Faller, P. , Straub, J. E. , Dominguez, L. , Shea, J. ‐ E. , Dokholyan, N. V. , De Simone, A. , Ma, B. , Nussinov, R. , Najafi, S. , Ngo, S. T. , Loquet, A. , Chiricotto, M. , Ganguly, P. , Mccarty, J. , Li, M. S. … Derreumaux, P. (2021). Amyloid oligomers: A joint experimental/computational perspective on Alzheimer's disease, Parkinson's disease, type II diabetes, and amyotrophic lateral sclerosis. Chemical Reviews, 121(4), 2545–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan, K. M. , Baldwin, M. , Nguyen, J. , Gasset, M. , Serban, A. , Groth, D. , Mehlhorn, I. , Huang, Z. , Fletterick, R. J. , & Cohen, F. E. (1993). Conversion of alpha‐helices into beta‐sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences of the United States of America, 90(23), 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prusiner, S. B. (1998). Prions. Proceedings of the National Academy of Sciences of the United States of America, 95(23), 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen, F. E. , & Prusiner, S. B. (1998). Pathologic conformations of prion proteins. Annual Review of Biochemistry, 67, 793–819. [DOI] [PubMed] [Google Scholar]
- 7. Horiuchi, M. (1999). Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease‐resistant state. Embo Journal, 18(12), 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sokolowski, F. , Modler, A. J. , Masuch, R. , Zirwer, D. , Baier, M. , Lutsch, G. , Moss, D. A. , Gast, K. , & Naumann, D. (2003). Formation of critical oligomers is a key event during conformational transition of recombinant Syrian hamster prion protein, Journal of Biological Chemistry, 278(42), 40481–40492. [DOI] [PubMed] [Google Scholar]
- 9. Lu, B. ‐ Y. , & Chang, J. ‐ Y. (2002). Isolation and characterization of a polymerized prion protein, Biochemical Journal, 364(Pt 1), 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding, F. , Larocque, J. J. , & Dokholyan, N. V. (2005). Direct observation of protein folding, aggregation, and a prion‐like conformational conversion, Journal of Biological Chemistry, 280(48), 40235–40240. [DOI] [PubMed] [Google Scholar]
- 11. Baskakov, I. V. , Legname, G. , Prusiner, S. B. , & Cohen, F. E. (2001). Folding of prion protein to its native alpha‐helical conformation is under kinetic control, Journal of Biological Chemistry, 276(23), 19687–19690. [DOI] [PubMed] [Google Scholar]
- 12. Bjorndahl, T. C. , Zhou, G. ‐ P. , Liu, X. , Perez‐Pineiro, R. , Semenchenko, V. , Saleem, F. , Acharya, S. , Bujold, A. , Sobsey, C. A. , & Wishart, D. S. (2011). Detailed biophysical characterization of the acid‐induced PrP(c) to PrP(β) conversion process. Biochemistry, 50(7), 1162–1173. [DOI] [PubMed] [Google Scholar]
- 13. Ladner‐Keay, C. L. , Griffith, B. J. , & Wishart, D. S. (2014). Shaking alone induces de novo conversion of recombinant prion proteins to β‐sheet rich oligomers and fibrils. Plos One, 9(6), e98753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson, G. S. (1999). Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations, Science, 283(5409), 1935–1937. [DOI] [PubMed] [Google Scholar]
- 15. Swietnicki, W. , Morillas, M. , Chen, S. G. , Gambetti, P. , & Surewicz, W. K. (2000). Aggregation and fibrillization of the recombinant human prion protein huPrP90‐231. Biochemistry, 39(2), 424–431. [DOI] [PubMed] [Google Scholar]
- 16. Morillas, M. , Vanik, D. L. , & Surewicz, W. K. (2001). On the mechanism of alpha‐helix to beta‐sheet transition in the recombinant prion protein. Biochemistry, 40(23), 6982–6987. [DOI] [PubMed] [Google Scholar]
- 17. Apetri, A. C. , & Surewicz, W. K. (2003). Atypical effect of salts on the thermodynamic stability of human prion protein. Journal of Biological Chemistry, 278(25), 22187–22192. [DOI] [PubMed] [Google Scholar]
- 18. Saleem, F. , Bjorndahl, T. C. , Ladner, C. L. , Perez‐Pineiro, R. , Ametaj, B. N. , & Wishart, D. S. (2014). Lipopolysaccharide induced conversion of recombinant prion protein. Prion, 8(2), 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stöckel, J. , Safar, J. , Wallace, A. C. , Cohen, F. E. , & Prusiner, S. B. (1998). Prion protein selectively binds copper(II) ions. Biochemistry 37(20), 7185–7193. [DOI] [PubMed] [Google Scholar]
- 20. Wong, E. , Thackray, A. M. , & Bujdoso, R. (2004). Copper induces increased beta‐sheet content in the scrapie‐susceptible ovine prion protein PrPVRQ compared with the resistant allelic variant PrPARR. Biochemical Journal 380(Pt 1), 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown, D. R. (2000). Consequences of manganese replacement of copper for prion protein function and proteinase resistance. Embo Journal, 19(6), 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Da Luz, M. H. M. , Peres, I. T. , Santos, T. G. , Martins, V. R. , Icimoto, M. Y. , & Lee, K. S. (2015). Dopamine induces the accumulation of insoluble prion protein and affects autophagic flux. Frontiers in Cellular Neuroscience, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jansen, K. , Schäfer, O. , Birkmann, E. , Post, K. , Serban, H. , Prusiner, S. B. , & Riesner, D. (2001). Structural intermediates in the putative pathway from the cellular prion protein to the pathogenic form, Biological Chemistry, 382(4), 683–691. [DOI] [PubMed] [Google Scholar]
- 24. Xiong, L. ‐ W. , Raymond, L. D. , Hayes, S. F. , Raymond, G. J. , & Caughey, B. (2001). Conformational change, aggregation and fibril formation induced by detergent treatments of cellular prion protein. Journal of Neurochemistry, 79(3), 669–678. [DOI] [PubMed] [Google Scholar]
- 25. Leffers, K. ‐ W. , Wille, H. , Stöhr, J. , Junger, E. , Prusiner, S. B. , & Riesner, D. (2005). Assembly of natural and recombinant prion protein into fibrils. Biological Chemistry, 386(6), 569–580. [DOI] [PubMed] [Google Scholar]
- 26. Leffers, K. ‐ W. , Schell, J. , Jansen, K. , Lucassen, R. , Kaimann, T. , Nagel‐Steger, L. , Tatzelt, J. , & Riesner, D. (2004). The structural transition of the prion protein into its pathogenic conformation is induced by unmasking hydrophobic sites. Journal of Molecular Biology, 344(3), 839–853. [DOI] [PubMed] [Google Scholar]
- 27. Post, K. , Pitschke, M. , Schäfer, O. , Wille, H. , Appel, T. R. , Kirsch, D. , Mehlhorn, I. , Serban, H. , Prusiner, S. B. , Riesner, D. (1998). Rapid acquisition of beta‐sheet structure in the prion protein prior to multimer formation. Biological Chemistry, 379(11), 1307–1317. [DOI] [PubMed] [Google Scholar]
- 28. Lührs, T. , Zahn, R. , & Wüthrich, K. (2006). Amyloid formation by recombinant full‐length prion proteins in phospholipid bicelle solutions. Journal of Molecular Biology, 357(3), 833–841. [DOI] [PubMed] [Google Scholar]
- 29. Sanghera, N. , & Pinheiro, T. J. T. (2002). Binding of prion protein to lipid membranes and implications for prion conversion. Journal of Molecular Biology, 315(5), 1241–1256. [DOI] [PubMed] [Google Scholar]
- 30. Wang, F. , Yang, F. , Hu, Y. , Wang, Xu , Wang, X. , Jin, C. , & Ma, J. (2007). Lipid interaction converts prion protein to a PrPSc‐like proteinase k‐resistant conformation under physiological conditions. Biochemistry, 46(23), 7045–7053. [DOI] [PubMed] [Google Scholar]
- 31. Deleault, N. R. , Piro, J. R. , Walsh, D. J. , Wang, F. , Ma, J. , Geoghegan, J. C. , & Supattapone, S. (2012). Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proceedings of the National Academy of Sciences of the United States of America, 109(22), 8546–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deleault, N. R. , Lucassen, R. W. , & Supattapone, S. (2003). RNA molecules stimulate prion protein conversion. Nature, 425(6959), 717–720. [DOI] [PubMed] [Google Scholar]
- 33. Deleault, N. R. , Harris, B. T. , Rees, J. R. , & Supattapone, S. (2007). Formation of native prions from minimal components in vitro. Proceedings of the National Academy of Sciences of the United States of America, 104(23), 9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miller, M. B. , Wang, D. W. , Wang, F. , Noble, G. P. , Ma, J. , Woods, V. L., Jr. , Li, S. , & Supattapone, S. (2013). Cofactor molecules induce structural transformation during infectious prion formation. Structure, 21(11), 2061–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Serpa, J. J. , Patterson, A. P. , Pan, J. , Han, J. , Wishart, D. S. , Petrotchenko, E. V. , & Borchers, C. H. (2013). Using multiple structural proteomics approaches for the characterization of prion proteins. Journal of Proteomics, 81, 31–42. [DOI] [PubMed] [Google Scholar]
- 36. Serpa, J. J. , Makepeace, K. A. , Borchers, T. H. , Wishart, D. S. , Petrotchenko, E. V. , & Borchers, C. H. (2014). Using isotopically‐coded hydrogen peroxide as a surface modification reagent for the structural characterization of prion protein aggregates. Journal of Proteomics, 100, 160–166. [DOI] [PubMed] [Google Scholar]
- 37. Baskakov, I. V. , Legname, G. , Baldwin, M. A. , Prusiner, S. B. , & Cohen, F. E. (2002). Pathway complexity of prion protein assembly into amyloid. Journal of Biological Chemistry, 277(24), 21140–21148. [DOI] [PubMed] [Google Scholar]
- 38. Rezaei, H. , Eghiaian, F. , Perez, J. , Doublet, B. , Choiset, Y. , Haertle, T. , & Grosclaude, J. (2005). Sequential generation of two structurally distinct ovine prion protein soluble oligomers displaying different biochemical reactivities. Journal of Molecular Biology, 347(3), 665–679. [DOI] [PubMed] [Google Scholar]
- 39. Sajnani, G. , Silva, C. J. , Ramos, A. , Pastrana, M. A. , Onisko, B. C. , Erickson, M. L. , Antaki, E. M. , Dynin, I. , Vázquez‐Fernández, E. , Sigurdson, C. J. , Carter, J. M. , & Requena, J. R. (2012). PK‐sensitive PrP is infectious and shares basic structural features with PK‐resistant PrP. Plos Pathogens, 8(3), e1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vázquez‐Fernández, E. , Alonso, J. , Pastrana, M. A. , Ramos, A. , Stitz, L. , Vidal, E. , Dynin, I. , Petsch, B. , Silva, C. J. , & Requena, J. R. (2012). Structural organization of mammalian prions as probed by limited proteolysis. Plos One, 7(11), e50111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wille, H. , Michelitsch, M. D. , Guénebaut, V. , Supattapone, S. , Serban, A. , Cohen, F. E. , Agard, D. A. , & Prusiner, S. B. (2002). Structural studies of the scrapie prion protein by electron crystallography. Proceedings of the National Academy of Sciences of the United States of America, 99(6), 3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Govaerts, C. , Wille, H. , Prusiner, S. B. , & Cohen, F. E. (2004). Evidence for assembly of prions with left‐handed beta‐helices into trimers, Proceedings of the National Academy of Sciences of the United States of America, 101(22), 8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wille, H. , Govaerts, C. , Borovinskiy, A. , Latawiec, D. , Downing, K. H. , Cohen, F. E. , & Prusiner, S. B. (2007). Electron crystallography of the scrapie prion protein complexed with heavy metals. Archives of Biochemistry and Biophysics, 467(2), 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cobb, N. J. , Sonnichsen, F. D. , Mchaourab, H. , & Surewicz, W. K. (2007). Molecular architecture of human prion protein amyloid: A parallel, in‐register beta‐structure. Proceedings of the National Academy of Sciences of the United States of America, 104(48), 18946–18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang, C. , Lo, W. ‐ L. , Kuo, Y. ‐ H. , Sang, J. C. , Lee, C‐Yu , Chiang, Y. ‐ W. , & Chen, R. P. ‐ Y. (2015). Revealing structural changes of prion protein during conversion from α‐helical monomer to β‐oligomers by means of ESR and nanochannel encapsulation. Acs Chemical Biology, 10(2), 493–501. [DOI] [PubMed] [Google Scholar]
- 46. Huang, Z. , Gabriel, J. M. , Baldwin, M. A. , Fletterick, R. J. , Prusiner, S. B. , & Cohen, F. E. (1994). Proposed three‐dimensional structure for the cellular prion protein. Proceedings of the National Academy of Sciences of the United States of America, 91(15), 7139–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Demarco, M. L. , & Daggett, V. (2004). From conversion to aggregation: Protofibril formation of the prion protein, Proceedings of the National Academy of Sciences of the United States of America, 101(8), 2293–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van der Kamp, M. W. , & Daggett, V. (2011). Molecular dynamics as an approach to study prion protein misfolding and the effect of pathogenic mutations. Topics in Current Chemistry, 305, 169–197. [DOI] [PubMed] [Google Scholar]
- 49. Caughey, B. W. , Dong, A. , Bhat, K. S. , Ernst, D. , & Hayes, S. F. , Caughey, W. S. (1991). Secondary structure analysis of the scrapie‐associated protein PrP 27‐30 in water by infrared spectroscopy. Biochemistry, 30(31), 7672–7680. [DOI] [PubMed] [Google Scholar]
- 50. Gasset, M. , Baldwin, M. A. , Fletterick, R. J. , & Prusiner, S. B. (1993). Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proceedings of the National Academy of Sciences of the United States of America, 90(1), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nguyen, J. T. , Inouye, H. , Baldwin, M. A. , Fletterick, R. J. , Cohen, F. E. , Prusiner, S. B. , & Kirschner, D. A. (1995). X‐ray diffraction of scrapie prion rods and PrP peptides. Journal of Molecular Biology, 252(4), 412–422. [DOI] [PubMed] [Google Scholar]
- 52. Safar, J. , Wille, H. , Itri, V. , Groth, D. , Serban, H. , Torchia, M. , Cohen, F. E. , & Prusiner, S. B. (1998). Eight prion strains have PrP(Sc) molecules with different conformations. Nature Medicine, 4(10), 1157–1165. [DOI] [PubMed] [Google Scholar]
- 53. Kosmač, M. , Koren, S. , Giachin, G. , Stoilova, T. , Gennaro, R. , Legname, G. , & Šerbec, V. Č. (2011). Epitope mapping of a PrP(Sc)‐specific monoclonal antibody: Identification of a novel C‐terminally truncated prion fragment. Molecular Immunology, 48(5), 746–750. [DOI] [PubMed] [Google Scholar]
- 54. Kang, H. ‐ E. , Weng, C. C. , Saijo, E. , Saylor, V. , Bian, J. , Kim, S. , Ramos, L. , Angers, R. , Langenfeld, K. , Khaychuk, V. , Calvi, C. , Bartz, J. , Hunter, N. , & Telling, G. C. (2012). Characterization of conformation‐dependent prion protein epitopes. Journal of Biological Chemistry, 287(44), 37219–37232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Noble, G. P. , Wang, D. W. , Walsh, D. J. , Barone, J. R. , Miller, M. B. , Nishina, K. A. , Li, S. , & Supattapone, S. (2015). A structural and functional comparison between infectious and non‐infectious autocatalytic recombinant PrP conformers. PLoS Pathogens, 11(6), e1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schlepckow, K. , & Schwalbe, H. (2013). Molecular mechanism of prion protein oligomerization at atomic resolution. Angewandte Chemie (International Edition In English), 52(38), 10002–10005. [DOI] [PubMed] [Google Scholar]
- 57. Singh, J. , & Udgaonkar, J. B. (2015). Molecular mechanism of the misfolding and oligomerization of the prion protein: Current understanding and its implications. Biochemistry, 54(29), 4431–4442. [DOI] [PubMed] [Google Scholar]
- 58. Kunes, K. C. , Clark, S. C. , Cox, D. L. , & Singh, R. R. P. (2008). Left handed beta helix models for mammalian prion fibrils. Prion, 2(2), 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Langedijk, J. P. M. , Fuentes, G. , Boshuizen, R. , & Bonvin, A. (2006). Two‐rung model of a left‐handed beta‐helix for prions explains species barrier and strain variation in transmissible spongiform encephalopathies. Journal of Molecular Biology, 360(4), 907–920. [DOI] [PubMed] [Google Scholar]
- 60. Dokholyan, N. V. (2020). Experimentally‐driven protein structure modeling. Journal of Proteomics, 220, 103777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Silva, C. J. (2014). Applying the tools of chemistry (mass spectrometry and covalent modification by small molecule reagents) to the detection of prions and the study of their structure. Prion, 8(1), 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nazabal, A. , Hornemann, S. , Aguzzi, A. , & Zenobi, R. (2009). Hydrogen/deuterium exchange mass spectrometry identifies two highly protected regions in recombinant full‐length prion protein amyloid fibrils. Journal of Mass Spectrometry, 44(6), 965–977. [DOI] [PubMed] [Google Scholar]
- 63. Sajnani, G. , Pastrana, M. A. , Dynin, I. , Onisko, B. , & Requena, J. R. (2008). Scrapie prion protein structural constraints obtained by limited proteolysis and mass spectrometry. Journal of Molecular Biology, 382(1), 88–98. [DOI] [PubMed] [Google Scholar]
- 64. Lu, X. , Wintrode, P. L. , & Surewicz, W. K. (2007). Beta‐sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proceedings of the National Academy of Sciences of the United States of America, 104(5), 1510–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaimann, T. , Metzger, S. , Kuhlmann, K. , Brandt, B. , Birkmann, E. , Höltje, H. ‐ D. , & Riesner, D. (2008). Molecular model of an alpha‐helical prion protein dimer and its monomeric subunits as derived from chemical cross‐linking and molecular modeling calculations. Journal of Molecular Biology, 376(2), 582–596. [DOI] [PubMed] [Google Scholar]
- 66. Onisko, B. , Fernandez, E. G. , Freire, M. L. , Schwarz, A. , Baier, M. , Camina, F. , Garcia, J. R. , Rodriguez‐Segade Villamarin, S. , & Requena, J. R. (2005). Probing PrPSc structure using chemical cross‐linking and mass spectrometry: Evidence of the proximity of Gly90 amino termini in the PrP 27‐30 aggregate. Biochemistry, 44(30), 10100–10109. [DOI] [PubMed] [Google Scholar]
- 67. Petrotchenko, E. V. , Serpa, J. J. , Hardie, D. B. , Berjanskii, M. , Suriyamongkol, B. P. , Wishart, D. S. , Borchers, C. H. (2012). Use of proteinase K nonspecific digestion for selective and comprehensive identification of interpeptide cross‐links: Application to prion proteins. Molecular and Cellular Proteomics, 11(7), M111.013524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eghiaian, F. , Daubenfeld, T. , Quenet, Y. , Van Audenhaege, M. , Bouin, A. ‐ P. , Van Der Rest, G. , Grosclaude, J. , & Rezaei, H. (2007). Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proceedings of the National Academy of Sciences of the United States of America, 104(18), 7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smirnovas, V. , Baron, G. S. , Offerdahl, D. K. , Raymond, G. J. , Caughey, B. , & Surewicz, W. K. (2011). Structural organization of brain‐derived mammalian prions examined by hydrogen‐deuterium exchange. Nature Structural and Molecular Biology, 18(4), 504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Singh, J. , Sabareesan, A. T. , Mathew, M. K. , & Udgaonkar, J. B. (2012). Development of the structural core and of conformational heterogeneity during the conversion of oligomers of the mouse prion protein to worm‐like amyloid fibrils. Journal of Molecular Biology, 423(2), 217–231. [DOI] [PubMed] [Google Scholar]
- 71. Sabareesan, A. T. , Singh, J. , Roy, S. , Udgaonkar, J. B. , & Mathew, M. K. (2016). The pathogenic A116V mutation enhances ion‐selective channel formation by prion protein in membranes. Biophysical Journal, 110(8), 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72. Cobb, N. J. , Apostol, M. I. , Chen, S. , Smirnovas, V. , & Surewicz, W. K. (2014). Conformational stability of mammalian prion protein amyloid fibrils is dictated by a packing polymorphism within the core region. Journal of Biological Chemistry, 289(5), 2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rezaei, H. , Marc, D. , Choiset, Y. , Takahashi, M. , Hui Bon Hoa, G. , Haertlé, T. , Grosclaude, J. , & Debey, P. (2000). High yield purification and physico‐chemical properties of full‐length recombinant allelic variants of sheep prion protein linked to scrapie susceptibility. European Journal of Biochemistry Febs, 267(10), 2833–2839. [DOI] [PubMed] [Google Scholar]
- 74. Silva, C. J. (2012). Using small molecule reagents to selectively modify epitopes based on their conformation. Prion, 6(2), 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Silva, C. J. , Erickson‐Beltran, M. L. , & Dynin, I. C. (2016). Covalent surface modification of prions: A mass spectrometry‐based means of detecting distinctive structural features of prion strains. Biochemistry, 55(6), 894–902. [DOI] [PubMed] [Google Scholar]
- 76. Gong, B. , Ramos, A. , Vázquez‐Fernández, E. , Silva, C. J. , Alonso, J. , Liu, Z. , & Requena, J. R. (2011). Probing structural differences between PrP(C) and PrP(Sc) by surface nitration and acetylation: Evidence of conformational change in the C‐terminus. Biochemistry, 50(22), 4963–4972. [DOI] [PubMed] [Google Scholar]
- 77. Lennon, C. W. , Cox, H. D. , Hennelly, S. P. , Chelmo, S. J. , & Mcguirl, M. A. (2007). Probing structural differences in prion protein isoforms by tyrosine nitration. Biochemistry, 46(16), 4850–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Requena, J. R. , Dimitrova, M. N. , Legname, G. , Teijeira, S. , Prusiner, S. B. , & Levine, R. L. (2004). Oxidation of methionine residues in the prion protein by hydrogen peroxide. Archives of Biochemistry and Biophysics, 432(2), 188–195. [DOI] [PubMed] [Google Scholar]
- 79. Brodie, N. I. , Popov, K. I. , Petrotchenko, E. V. , Dokholyan, N. V. , & Borchers, C. H. (2017). Solving protein structures using short‐distance cross‐linking constraints as a guide for discrete molecular dynamics simulations. Science Advances, 3(7), e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Brodie, N. I. , Makepeace, K. A. T. , Petrotchenko, E. V. , & Borchers, C. H. (2015). Isotopically‐coded short‐range hetero‐bifunctional photo‐reactive crosslinkers for studying protein structure. Journal of Proteomics, 118, 12–20. [DOI] [PubMed] [Google Scholar]
- 81. Petrotchenko, E. V. , Serpa, J. J. , & Borchers, C. H. (2011). An isotopically coded CID‐cleavable biotinylated cross‐linker for structural proteomics. Molecular and Cellular Proteomics S1–S8, 10(2), M110001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gomes, A. F. , & Gozzo, F. C. (2010). Chemical cross‐linking with a diazirine photoactivatable cross‐linker investigated by MALDI‐ and ESI‐MS/MS. Journal of Mass Spectrometry, 45(8), 892–899. [DOI] [PubMed] [Google Scholar]
- 83. Brodie, N. I. , Petrotchenko, E. V. , & Borchers, C. H. (2016). The novel isotopically coded short‐range photo‐reactive crosslinker 2,4,6‐triazido‐1,3,5‐triazine (TATA) for studying protein structures. Journal of Proteomics, 149, 69–76. [DOI] [PubMed] [Google Scholar]
- 84. Ladner, C. L. , & Wishart, D. S. (2012). Resolution‐enhanced native acidic gel electrophoresis: A method for resolving, sizing, and quantifying prion protein oligomers. Analytical Biochemistry, 426(1), 54–62. [DOI] [PubMed] [Google Scholar]
- 85. Zahn, R. , Von Schroetter, C. , & Wüthrich, K. (1997). Human prion proteins expressed in Escherichia coli and purified by high‐affinity column refolding. Febs Letters, 417(3), 400–404. [DOI] [PubMed] [Google Scholar]
- 86. Taverner, T. , Hall, N. E. , O'hair, R. A. J. , & Simpson, R. J. (2002). Characterization of an antagonist interleukin‐6 dimer by stable isotope labeling, cross‐linking, and mass spectrometry. Journal of Biological Chemistry, 277(48), 46487–46492. [DOI] [PubMed] [Google Scholar]
- 87. Leitner, A. , Joachimiak, L. A. , Unverdorben, P. , Walzthoeni, T. , Frydman, J. , Förster, F. , & Aebersold, R. (2014). Chemical cross‐linking/mass spectrometry targeting acidic residues in proteins and protein complexes, Proceedings of the National Academy of Sciences of the United States of America, 111(26), 9455–9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kunishima, M. , Kawachi, C. , Monta, J. , Terao, K. , Iwasaki, F. , & Tani, S. (1999). 4‐(4,6‐dimethoxy‐1,3,5‐triazin‐2‐yl)‐4‐methyl‐morpholinium chloride: An efficient condensing agent leading to the formation of amides and esters, Tetrahedron, 55(46), 13159–13170. [Google Scholar]
- 89. Novák, P. , & Kruppa, G. H. (2008). Intra‐molecular cross‐linking of acidic residues for protein structure studies, European Journal of Mass Spectrometry, 14(6), 355–365. [DOI] [PubMed] [Google Scholar]
- 90. Fancy, D. A. , & Kodadek, T. (1999). Chemistry for the analysis of protein‐protein interactions: Rapid and efficient cross‐linking triggered by long wavelength light. Proceedings of the National Academy of Sciences of the United States of America, 96(11), 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Petrotchenko, E. V. , Serpa, J. J. , Makepeace, K. A. , Brodie, N. I. , & Borchers, C. H. (2014). (14)N(15)N DXMSMS Match program for the automated analysis of LC/ESI‐MS/MS crosslinking data from experiments using (15)N metabolically labeled proteins. Journal of Proteomics, 109, 104–110. [DOI] [PubMed] [Google Scholar]
- 92. Popov, K. I. , Makepeace, K. A. T. , Petrotchenko, E. V. , Dokholyan, N. V. , & Borchers, C. H. (2019). Insight into the structure of the “unstructured” tau protein. Structure (London, England), 27(11), 1710–1715.e4. [DOI] [PubMed] [Google Scholar]
- 93. Brodie, N. I. , Popov, K. I. , Petrotchenko, E. V. , Dokholyan, N. V. , & Borchers, C. H. (2019). Conformational ensemble of native α‐synuclein in solution as determined by short‐distance crosslinking constraint‐guided discrete molecular dynamics simulations. Plos Computational Biology 15(3), e1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ding, F. , Tsao, D. , Nie, H. , & Dokholyan, N. V. (2008). Ab initio folding of proteins with all‐atom discrete molecular dynamics. Structure, 16(7), 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Shirvanyants, D. , Ding, F. , Tsao, D. , & Ramachandran, S. , Dokholyan, N. V. (2012). Discrete molecular dynamics: An efficient and versatile simulation method for fine protein characterization. Journal of Physical Chemistry B, 116(29), 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dokholyan, N. V. , Buldyrev, S. V. , Stanley, H. E. , & Shakhnovich, E. I. (1998). Discrete molecular dynamics studies of the folding of a protein‐like model. Folding and Design, 3(6), 577–587. [DOI] [PubMed] [Google Scholar]
- 97. Proctor, E. A. , Ding, F. , & Dokholyan, N. V. (2011). WIREs Computational Molecular Science, 1(1), 80–92. [Google Scholar]
- 98. Okamoto, Y. (2004). Generalized‐ensemble algorithms: Enhanced sampling techniques for Monte Carlo and molecular dynamics simulations. Journal of Molecular Graphics and Modelling, 22(5), 425–439. [DOI] [PubMed] [Google Scholar]
- 99. Proctor, E. A. , Fee, L. , Tao, Y. , Redler, R. L. , Fay, J. M. , Zhang, Y. , Lv, Z. , Mercer, I. P. , Deshmukh, M. , Lyubchenko, Y. L. , & Dokholyan, N. V. (2016). Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 113(3), 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chodera, J. D. , Swope, W. C. , Pitera, J. W. , Seok, C. , & Dill, K. A. (2007). Use of the weighted histogram analysis method for the analysis of simulated and parallel tempering simulations. Journal of Chemical Theory and Computation, 3(1), 26–41. [DOI] [PubMed] [Google Scholar]
- 101. Pronk, S. , Páll, S. , Schulz, R. , Larsson, P. , Bjelkmar, P. , Apostolov, R. , Shirts, M. R. , Smith, J. C. , Kasson, P. M. , Van Der Spoel, D. , Hess, B. , & Lindahl, E. (2013). GROMACS 4.5: A high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29(7), 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Daura, X. , Gademann, K. , Jaun, B. , Seebach, D. , Van Gunsteren, W. F. , & Mark, A. E. (1999). Peptide folding: When simulation meets experiment, Angewandte Chemie (International Edition), 38(1‐2), 236–240. [Google Scholar]
- 103. Pan, J. , Han, J. , Borchers, C. H. , & Konermann, L. (2009). Hydrogen/deuterium exchange mass spectrometry with top‐down electron capture dissociation for characterizing structural transitions of a 17 kDa protein, Journal of the American Chemical Society, 131(35), 12801–12808. [DOI] [PubMed] [Google Scholar]
- 104. Petrotchenko, E. V. , & Borchers, C. H. (2015). HDX match software for the data analysis of top‐down ECD‐FTMS hydrogen/deuterium exchange experiments, Journal of the American Society for Mass Spectrometry, 26(11), 1895–1898. [DOI] [PubMed] [Google Scholar]
- 105. Micsonai, A. , Wien, F. , Kernya, L. , Lee, Y‐Ho , Goto, Y. , Réfrégiers, M. , & Kardos, J. (2015). Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proceedings of the National Academy of Sciences of the United States of America, 112(24), E3095–E3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shevchenko, A. , Tomas, H. , Havli, J. , Olsen, J. V. , & Mann, M. (2006). In‐gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols, 1(6), 2856–2860. [DOI] [PubMed] [Google Scholar]
- 107. Donne, D. G. , Viles, J. H. , Groth, D. , Mehlhorn, I. , James, T. L. , Cohen, F. E. , Prusiner, S. B. , Wright, P. E. , & Dyson, H. J. (1997). Structure of the recombinant full‐length hamster prion protein PrP(29‐231): The N terminus is highly flexible. Proceedings of the National Academy of Sciences of the United States of America, 94(25), 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Prusiner, S. B. , Groth, D. F. , Bolton, D. C. , Kent, S. B. , & Hood, L. E. (1984). Purification and structural studies of a major scrapie prion protein. Cell, 38(1), 127–134. [DOI] [PubMed] [Google Scholar]
- 109. Basler, K. , Oesch, B. , Scott, M. , Westaway, D. , Wälchli, M. , Groth, D. F. , Mckinley, M. P. , Prusiner, S. B. , & Weissmann, C. (1986). Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell, 46(3), 417–428. [DOI] [PubMed] [Google Scholar]
- 110. Legname, G. (2004). Synthetic mammalian prions. Science, 305(5684), 673–676. [DOI] [PubMed] [Google Scholar]
- 111. Colby, D. W. , Giles, K. , Legname, G. , Wille, H. , Baskakov, I. V. , Dearmond, S. J. , & Prusiner, S. B. (2009). Design and construction of diverse mammalian prion strains. Proceedings of the National Academy of Sciences of the United States of America, 106(48), 20417–20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ghaemmaghami, S. , Colby, D. W. , Nguyen, H. ‐ O. B. , Hayashi, S. , Oehler, A. , Dearmond, S. J. , & Prusiner, S. B. (2013). Convergent replication of mouse synthetic prion strains. The American Journal of Pathology, 182(3), 866–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Le, N. T. , Narkiewicz, J. , Aulic, S. , Salzano, G. , Tran, H. T. , Scaini, D. , Moda, F. , Giachin, G. , & Legname, G. (2015). Synthetic prions and other human neurodegenerative proteinopathies. Virus Research, 207, 25–37. [DOI] [PubMed] [Google Scholar]
- 114. McKinley, M. P. , Taraboulos, A. , Kenaga, L. , Serban, D. , Stieber, A. , DeArmond, S. J. , Prusiner, S. B. , & Gonatas, N. (1991). Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells, Laboratory Investigation, 65(6), 622–630. [PubMed] [Google Scholar]
- 115. Arnold, J. E. , Tipler, C. , Laszlo, L. , Hope, J. , Landon, M. , & Mayer, R. J. (1995). The abnormal isoform of the prion protein accumulates in late‐endosome‐like organelles in scrapie‐infected mouse brain, Journal of Pathology, 176(4), 403–411. [DOI] [PubMed] [Google Scholar]
- 116. Tsutsui, Y. , & Wintrode, P. (2007). Hydrogen/deuterium exchange‐mass spectrometry: A powerful tool for probing protein structure, dynamics and interactions, Current Medicinal Chemistry, 14(22), 2344–2358. [DOI] [PubMed] [Google Scholar]
- 117. Kaltashov, I. A. , & Eyles, S. J. (2002). Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrometry Reviews, 21(1), 37–71. [DOI] [PubMed] [Google Scholar]
- 118. Konermann, L. , Pan, J. , & Liu, Y. H. (2011). Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev., 40(3), 1224–1234. [DOI] [PubMed] [Google Scholar]
- 119. Pan, J. , Han, J. , Borchers, C. H. , & Konermann, L. (2010). Characterizing short‐lived protein folding intermediates by top‐down hydrogen exchange mass spectrometry. Analytical Chemistry, 82(20), 8591–8597. [DOI] [PubMed] [Google Scholar]
- 120. Wille, H. , Bian, W. , Mcdonald, M. , Kendall, A. , Colby, D. W. , Bloch, L. , Ollesch, J. , Borovinskiy, A. L. , Cohen, F. E. , Prusiner, S. B. , & Stubbs, G. (2009). Natural and synthetic prion structure from X‐ray fiber diffraction. Proceedings of the National Academy of Sciences of the United States of America, 106(40), 16990–16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Peretz, D. , Williamson, R. A. , Matsunaga, Y. , Serban, H. , Pinilla, C. , Bastidas, R. B. , Rozenshteyn, R. , James, T. L. , Houghten, R. A. , Cohen, F. E. , Prusiner, S. B. , & Burton, D. R. (1997). A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. Journal of Molecular Biology, 273(3), 614–622. [DOI] [PubMed] [Google Scholar]
- 122. Yuan, F. F. , Biffin, S. , Brazier, M. W. , Suarez, M. , Cappai, R. , Hill, A. F. , Collins, S. J. , Sullivan, J. S. , Middleton, D. , Multhaup, G. , Geczy, A. F. , & Masters, C. L. (2005). Detection of prion epitopes on PrPc and PrPsc of transmissible spongiform encephalopathies using specific monoclonal antibodies to PrP. Immunology and Cell Biology, 83(6), 632–637. [DOI] [PubMed] [Google Scholar]
- 123. Williamson, R. A. , Peretz, D. , Pinilla, C. , Ball, H. , Bastidas, R. B. , Rozenshteyn, R. , Houghten, R. A. , Prusiner, S. B. , & Burton, D. R. (1998). Mapping the prion protein using recombinant antibodies. Journal of Virology, 72(11), 9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kim, C. ‐ L. , Umetani, A. , Matsui, T. , Ishiguro, N. , Shinagawa, M. , & Horiuchi, M. (2004). Antigenic characterization of an abnormal isoform of prion protein using a new diverse panel of monoclonal antibodies. Virology, 320(1), 40–51. [DOI] [PubMed] [Google Scholar]
- 125. Eghiaian, F. , Grosclaude, J. , Lesceu, S. , Debey, P. , Doublet, B. , Treguer, E. , Rezaei, H. , & Knossow, M. (2004). Insight into the PrPC ‐‐> PrPSc conversion from the structures of antibody‐bound bovine prion scrapie‐susceptibility variants. Proceedings of the National Academy of Sciences of the United States of America, 101(28), 10254–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Trevitt, C. R. , Hosszu, L. L. P. , Batchelor, M. , Panico, S. , Terry, C. , Nicoll, A. J. , Risse, E. , Taylor, W. A. , Sandberg, M. K. , Al‐Doujaily, H. , Linehan, J. M. , Saibil, H. R. , Scott, D. J. , Collinge, J. , Waltho, J. P. , & Clarke, A. R. (2014). N‐terminal domain of prion protein directs its oligomeric association. Journal of Biological Chemistry, 289(37), 25497–25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Paramithiotis, E. , Pinard, M. , Lawton, T. , Laboissiere, S. , Leathers, V. L. , Zou, W. ‐ Q. , Estey, L. A. , Lamontagne, J. , Lehto, M. T. , Kondejewski, L. H. , Francoeur, G. P. , Papadopoulos, M. , Haghighat, A. , Spatz, S. J. , Head, M. , Will, R. , Ironside, J. , O'rourke, K. , Tonelli, Q. … Cashman, N. R. (2003). A prion protein epitope selective for the pathologically misfolded conformation. Nature Medicine, 9(7), 893–899. [DOI] [PubMed] [Google Scholar]
- 128. Saijo, E. , Hughson, A. G. , Raymond, G. J. , Suzuki, A. , Horiuchi, M. , & Caughey, B. (2016). PrPSc‐specific antibody reveals C‐terminal conformational differences between prion strains. Journal of Virology, 90(10), 4905–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chabry, J. , Caughey, B. , & Chesebro, B. (1998). Specific inhibition of in vitro formation of protease‐resistant prion protein by synthetic peptides. Journal of Biological Chemistry, 273(21), 13203–13207. [DOI] [PubMed] [Google Scholar]
- 130. White, A. R. , Enever, P. , Tayebi, M. , Mushens, R. , Linehan, J. , Brandner, S. , Anstee, D. , Collinge, J. , & Hawke, S. (2003). Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature, 422(6927), 80–83. [DOI] [PubMed] [Google Scholar]
- 131. Peretz, D. , Williamson, R. A. , Kaneko, K. , Vergara, J. , Leclerc, E. , Schmitt‐Ulms, G. , Mehlhorn, I. R. , Legname, G. , Wormald, M. R. , Rudd, P. M. , Dwek, R. A. , Burton, D. R. , & Prusiner, S. B. (2001). Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature, 412(6848), 739–743. [DOI] [PubMed] [Google Scholar]
- 132. Wu, X. , Chavez, J. D. , Schweppe, D. K. , Zheng, C. , Weisbrod, C. R. , Eng, J. K. , Murali, A. , Lee, S. A. , Ramage, E. , Gallagher, L. A. , Kulasekara, H. D. , Edrozo, M. E. , Kamischke, C. N. , Brittnacher, M. J. , Miller, S. I. , Singh, P. K. , Manoil, C. , & Bruce, J. E. (2016). In vivo protein interaction network analysis reveals porin‐localized antibiotic inactivation in Acinetobacter baumannii strain AB5075. Nature Communications, 7, 13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Collinge, J. (2005). Molecular neurology of prion disease. Journal of Neurology, Neurosurgery and Psychiatry, 76(7), 906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lanni, C. , Racchi, M. , Mazzini, G. , Ranzenigo, A. , Polotti, R. , Sinforiani, E. , Olivari, L. , Barcikowska, M. , Styczynska, M. , Kuznicki, J. , Szybinska, A. , Govoni, S. , & Memo, M. , Uberti, D. (2008). Conformationally altered p53: A novel Alzheimer's disease marker? Molecular Psychiatry, 13(6), 641–647. [DOI] [PubMed] [Google Scholar]
- 135. Collinge, J. (1999). Variant Creutzfeldt‐Jakob disease. Lancet, 354(9175), 317–323. [DOI] [PubMed] [Google Scholar]
- 136. Collinge, J. , & Clarke, A. R. (2007). A general model of prion strains and their pathogenicity. Science, 318(5852), 930–936. [DOI] [PubMed] [Google Scholar]
- 137. Mead, S. , Whitfield, J. , Poulter, M. , Shah, P. , Uphill, J. , Campbell, T. , Al‐Dujaily, H. , Hummerich, H. , Beck, J. , Mein, C. A. , Verzilli, C. , Whittaker, J. , Alpers, M. P. , & Collinge, J. (2009). A novel protective prion protein variant that colocalizes with kuru exposure. New England Journal of Medicine, 361(21), 2056–2065. [DOI] [PubMed] [Google Scholar]
- 138. Skora, L. , Fonseca‐Ornelas, L. , Hofele, R. V. , Riedel, D. , Giller, K. , Watzlawik, J. , Schulz‐Schaeffer, W. J. , Urlaub, H. , Becker, S. , & Zweckstetter, M. (2013). Burial of the polymorphic residue 129 in amyloid fibrils of prion stop mutants. Journal of Biological Chemistry, 288(5), 2994–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Combe, C. W. , Fischer, L. , & Rappsilber, J. (2015). xiNET: Cross‐link network maps with residue resolution. Molecular and Cellular Proteomics, 14(4), 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Data will be made available upon request.