

Abstract
Side‐chain‐to‐side‐chain cyclization is frequently used to stabilize the α‐helical conformation of short peptides. In a previous study, we incorporated a lactam bridge between the side chains of Lys‐i and Asp‐i+4 in the nonapeptide 1Y, cyclo‐(2,6)‐(Ac‐VKRLQDLQY‐NH 2 ), an artificial ligand of the inhibitor of DNA binding and cell differentiation (ID) protein with antiproliferative activity on cancer cells. Herein, we show that only the cyclized five‐residue segment adopts a helical turn whereas the C‐terminal residues remain flexible. Moreover, we present nine 1Y analogs arising from different combinations of hydrophobic residues (leucine, isoleucine, norleucine, valine, and tyrosine) at positions 1, 4, 7, and 9. All cyclopeptides except one build a lactam‐bridged helical turn; however, residue‐4 reveals less helix character than the neighboring Arg‐3 and Gln‐5, especially with residue‐4 being isoleucine, valine, and tyrosine. Surprisingly, only two cyclopeptides exhibit helix propagation until the C‐terminus, whereas the others share a remarkable outward tilting of the backbone carbonyl of the lactam‐bridged Asp‐6 (>40° deviation from the orientation parallel to the helix axis), which prevents the formation of the H‐bond between Arg‐3 CO and residue‐7 NH: As a result, the propagation of the helix beyond the lactam‐bridged sequence becomes unfavorable. We conclude that, depending on the amino‐acid sequence, the lactam bridge between Lys‐i and Asp‐i+4 can stabilize a helical turn but deviations from the ideal helix geometry are possible: Indeed, besides the outward tilting of the backbone carbonyls, the residues per turn increased from 3.6 (typical of a regular α‐helix) to 4.2, suggesting a partial helix unwinding.
Keywords: carbonyl tilting, circular dichroism, lactam bridge, NMR spectroscopy, α‐helix
The NMR solution structures of several Lysi ‐Aspi+4 lactam‐bridged short helical peptides showed a significant outward tilt of the backbone carbonyls and 4.2 residues per turn.
1. INTRODUCTION
The α‐helix represents one of the most common secondary structures of bioactive peptides and protein domains, and helical motifs often play a key role in important molecular recognition events. 1 , 2 In order to augment the helicity of short sequences involved in ligand/protein or protein/protein interactions, a variety of covalent constraints have been developed, which include backbone H‐bond mimicry 3 , 4 and side‐chain crosslinking via disulfide, 5 sulfide, 6 , 7 1,4‐triazole, 8 , 9 hydrocarbon, 10 , 11 , 12 , 13 , 14 , 15 diester, 16 and lactam bridges. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 These covalent constraints are also used in combination with unnatural amino acids that prefer the helical structure and/or may increase peptide stability against proteases, like Cα‐tetrasubstituted amino acids and β‐ and γ‐amino acids. 12 , 16 , 30 , 31 , 32
Among the cyclization approaches mentioned above, the conversion of a linear peptide into a macrolactam via two side chains containing an amino and carboxylic group, respectively, is broadly applied, especially because it can be accomplished also by exploiting the natural amino acids lysine and aspartate or glutamate which are readily accessible and affordable on the market with different orthogonal groups. In addition, a comparative study conducted by de Araujo et al. 33 has shown that the lactam bridge between Lys‐i and Asp‐i+4 is the best covalent constraint to induce a nearly ideal α‐helical turn of the model pentapeptide Ac‐KAAAD‐NH 2 in an aqueous environment, followed by the 1,4‐triazole and the unsaturated hydrocarbon bridges. 34 In contrast, the m‐xylene‐ and the perfluorophenyl‐dithioether linkages have not been able to induce an α‐helical turn. In a previous work, the same group had also shown that the lactam bridge between Lys‐i and Glu‐i+4 or between Asp‐i and Lys‐i+4 is not suited to stabilize an α‐helical turn in aqueous solution. 22
Recently, we have used the lactam‐bridge constraint between Lys‐i and Asp‐i+4 to stabilize the helical conformation of octapeptides derived from native helical motifs of the helix‐loop‐helix (HLH) region of the ID2 protein (inhibitor of DNA binding and cell differentiation 2 35 ). Not all cyclized sequences adopted the expected helical conformation, 36 suggesting that the primary structure plays a role in determining the secondary structure despite the presence of covalent constraints. 36 , 37 Among the resulting helical cyclopeptides, cyclo‐(2,6)‐(Ac‐VKRLQDLQ‐NH 2 ) (1) could nicely mimic the side‐chain hydrophobic pattern of the two ID‐protein helices at positions i, i+3, and i+6 (Figure 1A), as shown by the comparison of the NMR solution structure of 1 38 with the crystal structure of the ID2 HLH homodimer. 39 Moreover, 1Y, an analog of 1 containing an additional C‐terminal residue (Tyr‐9, used as internal chromophore) and whose NMR solution structure has been elucidated in this work (Figure 1B), was found to be an ID protein‐binding candidate that negatively modulated the cell‐cycle progression of cancer cells 38 and further achieved synergistic effects in combination with photodynamic therapy. 40
FIGURE 1.
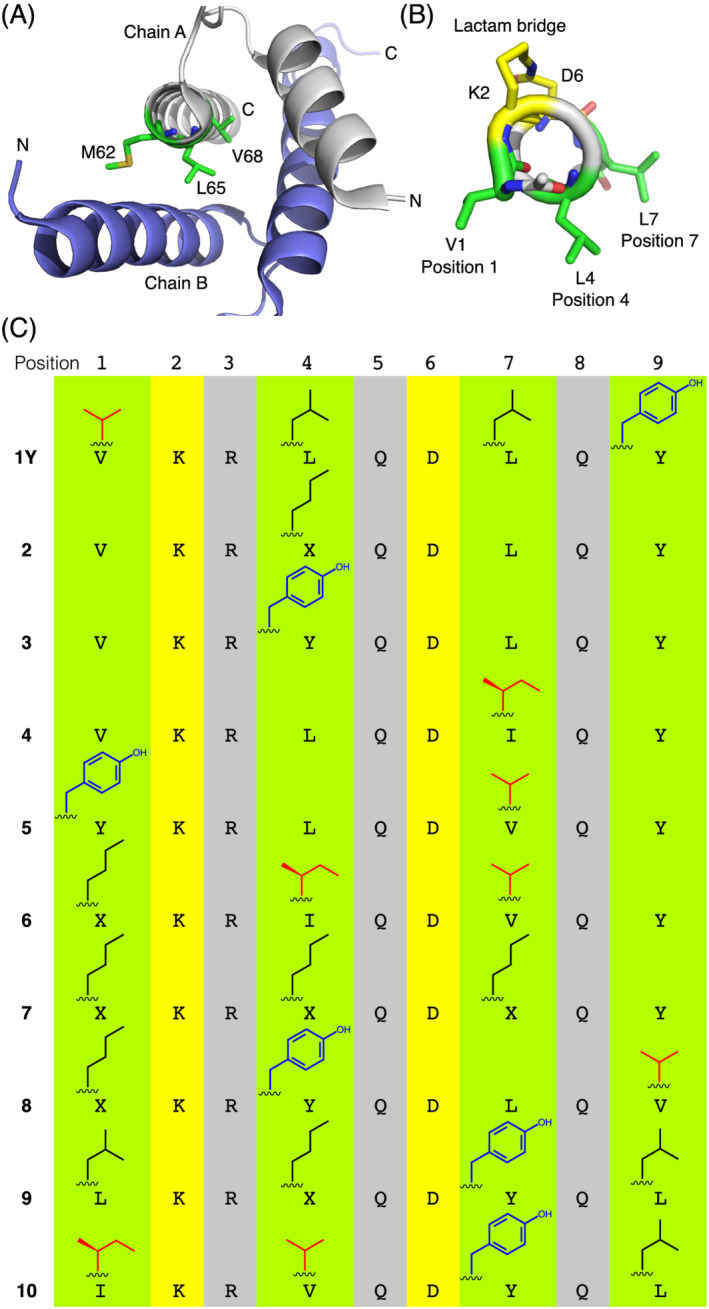
(A) Crystal structure of the ID2 HLH dimer (PDB ID: 4AYA) showing the hydrophobic three‐residue pattern Met‐62, Leu‐65, Val‐68 from the C‐terminal helix of the ID2 HLH domain. (B) NMR solution structure of 1Y showing the hydrophobic three‐residue pattern Val‐1, Leu‐4, Leu‐7 (one of the 20 low‐energy structures is shown. Backbone RMSD: 0.50 ± 0.23 Å). (C) Sequence of 1Y and its analogs derived by modifying the type of hydrophobic residue at positions 1, 4, 7, and 9 (X = norleucine, Nle). The β‐branched residues Val and Ile are shown in red, and the aromatic residue Tyr is shown in blue
Because of its interesting biological properties, 1Y might be used as a lead structure to develop other ID protein‐binding helical peptides. Since the ID proteins undergo protein‐protein interactions via hydrophobic contacts of their native helices, 39 , 41 the optimization of the hydrophobic amino acid pattern of 1Y at positions 1, 4, and 7 (Figure 1B) might result in optimized ligand and inhibitor properties. However, we were wondering if the conformation of the side‐chain‐to‐side‐chain cyclized nonapeptide scaffold would remain unaffected by multiple changes along the sequence. Although it has been proposed that the lactam bridge between Lys‐i and Asp‐i+4 stabilizes the helical turn via a water‐driven n‐π* interaction between the α‐carbonyl oxygen of Lys‐i and the carbonyl carbon of the lactam moiety, 34 it has been also reported that the helicity of such lactam‐bridged scaffolds is sensitive to the amino acid sequence. 24 , 36 Here, we elucidated the NMR solution structure of 1Y and compared it with those of nine analogous sequences that differ in the type of hydrophobic residues at positions 1, 4, 7, and 9 (Figure 1C). Interestingly, most structures reveal a helical conformation in which some backbone carbonyls are tilted outwards compared to the helix axis, which is especially critical for the lactam bridged aspartate and is not compatible with the propagation of the helix until the C‐terminal end.
2. MATERIALS AND METHODS
2.1. Peptide synthesis
The linear peptides were assembled on an automatic peptide synthesizer (Syro I, Biotage, Sweden) by using a Rink‐amide resin (100–200 mesh, loading 0.57 mmol/g; Iris Biotech, Germany) and Fmoc chemistry. The protected amino acids, N,N‐diisopropylethylamine (DIPEA), piperidine, N,N‐dimethylformamide (DMF), N‐methyl‐2‐pyrrolidone (NMP), dichloromethane (DCM), diethylether, and trifluoroacetic acid (TFA) were purchased from Iris Biotech. The amino acid side‐chain protecting groups were tBu for Tyr, alloc for the Lys to be cyclized, allyl for the Asp to be cyclized, Pbf for Arg, Trt for Gln. The Fmoc deprotection was carried out with 25% piperidine in DMF/NMP (70:30, v/v) for 3 min and 12.5% piperidine in DMF/NMP (70:30, v/v) for 12 min. The couplings were accomplished with the mixture Fmoc‐AA‐OH/HOBt/HBTU/DIPEA (5:5:4.8:10 equiv.) for 2 × 40 min. N‐terminal acetylation was performed manually with acetic anhydride/DIPEA (10:10 equiv.) in DMF for 30 min. The allyl/alloc protecting groups were orthogonally removed by repeated treatments (6 × 10 min) with Pd (PPh3)4 (0.5 equiv.) in the presence of PhSiH3 (25 equiv.) in DCM. Side‐chain cyclization was carried out with DIPEA/DEPBT (3:2 equiv.) in DCM/DMF (3:1, v/v) for 72 h. The cyclized peptides were cleaved from the resin with TFA/H2O/TIA/EDT/TIS (90:1:3:3:3; V tot = 1 ml) for about 3 h, precipitated by ice‐cold diethyl ether and recovered by centrifugation at 4°C for 5 min. The homogeneity and identification of the desired cyclopeptides were assessed by analytical HPLC (Ultimate 3000, Thermo Fisher Scientific, Germany) and MALDI‐TOF‐MS (Autoflex, Bruker Daltonics, Germany) (Table S1 and Figures S1 and S2).
2.2. CD spectroscopy
The peptides were dissolved in water, 50 mM phosphate buffer (pH 7.3) or 10 mM acetate buffer (pH 4.5) at the following concentrations (determined by the UV absorbance of tyrosine using the molar extinction coefficient of 1,480 M−1 cm−1 at 276 nm 42 ): 154 μM (1Y), 105 μM (2), 128 μM (3), 152 μM (4), 165 μM (5), 130 μM (6 and 8), 148 μM (7), 140 μM (9), and 135 μM (10). The CD spectra were recorded at 23°C on a Chirascan Plus CD spectrometer (Applied Photophysics, UK) using a 1 mm quartz cell from Hellma Analytics. For each CD spectrum, three scans were accumulated using a step resolution of 1 nm, a bandwidth of 1 nm, and a time‐per‐point of 1 s. The CD spectrum of the solvent was subtracted, and the difference spectrum was normalized to express the ellipticity in mean‐residue molar ellipticity, divided by 103 and represented in the graphs as [θ]R × 10−3 (deg cm2 dmol−1) (Figures 2 and S3–S5).
FIGURE 2.
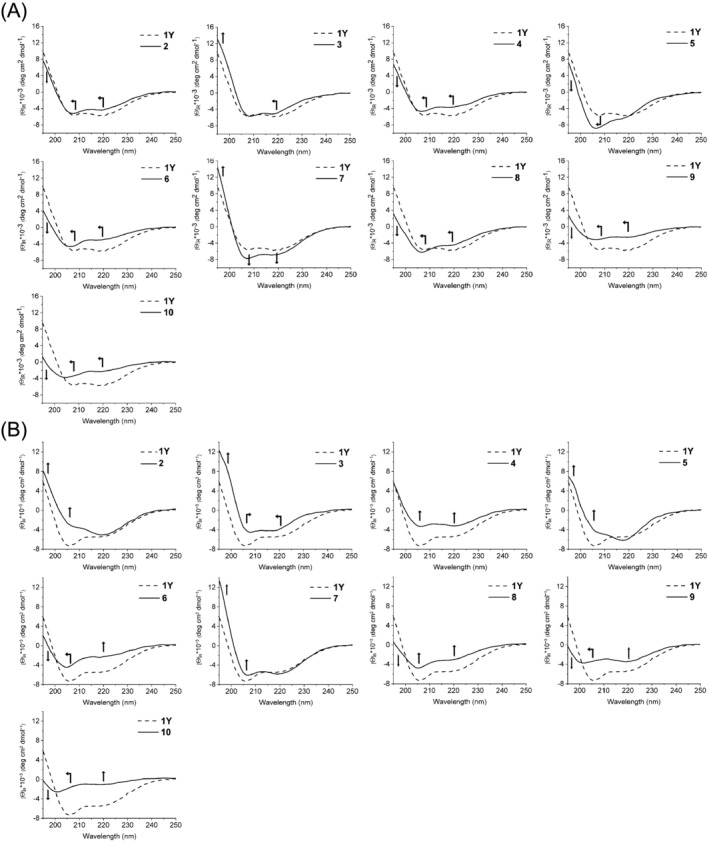
CD spectra of the peptides in (A) phosphate buffer (50 mM, pH 7.3) and (B) water (pH range 3–4). The arrows indicate the change in intensity/position of the CD bands of each cyclopeptide when compared to those of 1Y
2.3. NMR spectroscopy
The NMR spectra were recorded on an AVANCE III HD 600 MHz spectrometer (Bruker Biospin, Germany) equipped with a QXI (1H/13C/15N/31P) probe at 298 K. Samples were dissolved in either D2O (100 atom%D, Armar Europe, Germany) or 7% D2O/93% H2O and measured in 5 mm NMR tubes (Armar Europe, Germany). The solutions were measured at acidic pH, to slow down amide exchange, and without salt or buffer for a high signal‐to‐noise ratio. Standard 2D spectra were recorded: 1H‐1H TOCSY spectra with a mixing time of 120 ms, 1,024 × 256 complex points, spectral widths of 12.9 and 8.33 ppm, four scans, and a recycle delay of 1 s; an identical experiment with a mixing time of 12 ms served as a 1H‐1H COSY; a 1H‐1H ROESY with a mixing time of 200 ms, 1,024 × 350 complex points, spectral widths of 12.9 and 10.0 ppm, 80 scans, and a recycle delay of 1.2 s; a 1H‐13C HSQC optimized for aliphatic side chains with 512 × 125 complex points, spectral widths of 13.9 and 77.0 ppm, 80 scans, a recycle delay of 1.5 s, and a 13C offset of 45 ppm; a 1H‐13C HSQC optimized for aromatic side chains with 512 × 50 complex points, spectral widths of 13.9 and 44.2 ppm, 80 scans, a recycle delay of 1.5 s, and a 13C offset of 128 ppm.
Spectra were referenced to 2,2‐dimethyl‐silapentane sulfonic acid (DSS) using an external sample of 0.5 mM DSS and 2 mM sucrose in H2O/D2O. Data were processed with Topsin 3.6 (Bruker Biospin, Germany) and analyzed using Sparky 3.115. 43 All chemical shift assignments have been deposited at the BioMagResBank 44 under the accession numbers 28119, 28120, 50498–50502, and 50504–50506.
2.4. NOE‐restrained structure calculation
NOE cross‐peaks extracted from 2D ROESY spectra were categorized in small, medium, and strong and converted into distance restraints. Structure calculations based on torsion angle dynamics were conducted using CYANA 3.0. 45 The CYANA library file was extended to include norleucine (NLE), an N‐terminal acetyl (ACE), a C‐terminal amide (NH2), a lysine, and an aspartate forming a lactam bridge (LYL and ASL). Coordinates for these additional residues were extracted from the PDB structures: 5MAS for ACE, 1HJE for NH2, and 2F4K for NLE. Coordinates and topologies of ASL and LYL were based on asparagine and lysine entries of the CYANA library file. A covalent link between ND2 of LYL and CE of ASP was introduced as a restraint of 1.45 Å with a weight of 5.00E+1 (applied as upper and lower limit). To achieve a correct geometry of the peptide bond within the lactam bridge, two restraints, one between LYL CD and ASL ND2, another between LYL CE and ASL CG were introduced (for both a lower limit of 2.40 Å with weight 1.00E+1 and an upper limit of 2.50 Å with weight 1.00E+1). NOE upper limit restraints were applied with a weight of 1. For each peptide, 200 structures were calculated, and the 20 structures with the best target function were selected for the structural ensemble (Table S2). Structures were analyzed, fitted, and illustrated using MolMol. 46
2.5. Calculation of the tilt angle θ and number of residues per turn ρ
The angle θ of a given backbone carbonyl C‐O vector relative to the helix direction vector was obtained from atomic coordinates following the procedure described by Haimov and Srebnik, 47 as was the number of residues per helix turn (ρ). Calculations were carried out in Python (http://python.org) using UCSF ChimeraX 48 and numpy modules (http://numpy.org). Secondary structure assignments by MolMol 46 were used for the definition of the axis of the helices.
3. RESULTS
3.1. Modification of the hydrophobic pattern of 1Y
The hydrophobic pattern of 1Y consists of Val‐1, Leu‐4, and Leu‐7 and reproduces the (i, i+3, i+6)‐hydrophobic pattern of the native helices of the ID proteins (Figure 1A,B). The C‐terminal Tyr‐9 was introduced to serve as internal chromophore. By substitution of one (2–4), two (5), or all three (6 and 7) positions, we generated peptides with increasingly marked differences in the (1, 4, 7)‐hydrophobic pattern with respect to 1Y: For example, the analog with highest similarity to 1Y is represented by 2 that contains norleucine‐4 (Nle‐4) in place of Leu‐4, followed by analogs 3 and 4, in which Leu‐4 or Leu‐7 was replaced by the sterically more demanding Tyr‐4 or Ile‐7, respectively (Figure 1C). In addition, we prepared three analogs, in which also position 9 was changed (8–10). The peptides containing two up to four substitutions are more different from 1Y, except for analog 7 that contains three norleucine residues in place of Val‐1, Leu‐4, and Leu‐7: Indeed, only the substitution Val1Nle (isopropyl versus n‐butyl) can be considered less conservative.
3.2. Replacements within the hydrophobic pattern of 1Y affect its circular dichroism spectroscopic signature
We investigated the conformational properties of the lactam‐bridged peptides by CD spectroscopy in phosphate buffer at pH 7.3. 1Y is characterized by an α‐helix‐like CD spectrum with moderate intensity: the two minima at 220 nm (n‐π*) and 208 nm (||π‐π*) have similar intensities (about −6,000 deg cm2 dmol−1), whereas a positive CD contribution for the ⊥ π‐π* transition is present below 201 nm and reaches about 10,000 deg cm2 dmol−1 at 195 nm (Figures 2A and S3). The subtle substitution Leu4Nle or Leu7Ile in peptides 2 and 4, respectively, leads to a 1‐nm blue shift of the two minima as well as to a moderate loss of intensity of the n‐π* and ⊥ π‐π* bands, especially in the case of the substitution Leu7Ile in peptide 4. For peptide 3 (Leu4Tyr), the main dichroic differences from 1Y are the 2‐nm blue‐shifted n‐π* band and the higher contribution of the ⊥ π‐π* band. The CD spectrum of peptide 7, which contains Nle at the three positions 1, 4, and 7, is more intense than that of 1Y, suggesting a higher helical content. A remarkably different CD shape is shown by peptide 5 (Val1Tyr, Leu7Val), which is characterized by a pronounced, blue‐shifted (to about 206 nm) ||π‐π* band at the expense of the n‐π* band that is detectable as a shoulder near 217 nm. For peptides 6, 8–10, the CD spectra are much less intense than that of 1Y, particularly below 200 nm, which is the region of the ⊥ π‐π* band for an α‐helical motif (Figures 2A and S3).
These CD data suggest that even subtle side‐chain modifications of 1Y (e.g., Leu4Nle in 2 or Leu7Ile in 4) lead to subtle conformational changes that affect the coupling of the dipole moments of the (iso)peptide bonds and, thus, the shape of the CD curve. The importance of the side chains in determining the geometry of the lactam‐bridged peptide backbone is also supported by the observation that the CD spectra of peptides with the same amino acid composition, but scrambled sequences are not superimposable (i.e., 2 with 8 and 4 with 10).
Since the conformational properties of peptides and proteins may change with the environment, we additionally measured the CD spectra of 1Y and its analogs dissolved in pure water (pH range 3–4) (Figures 2B and S3). Unlike peptides 3, 4, 6, and 7 that show comparable CD spectra in buffered and unbuffered solutions (Figure S4), the other peptides display changes iin the intensity of one or both minima. Interestingly, in the case of peptides 1Y, 2 and 5, the n‐π* CD band is not affected by the presence or absence of the buffer, whereas the ||π‐π* band increases in the absence of buffer for 1Y or in the presence of buffer for 2 and 5 (Figures 2B and S4). This observation is quite surprising, especially because 1Y and 2 differ only at position 4, where a leucine (1Y) or a norleucine (2) is present. Since all peptides contain only one basic residue (Arg‐3) and no acidic residues, the different CD curves obtained in phosphate buffer at pH 7.3 or in the absence of buffer at pH 3–4 cannot be attributed to a change in the ionization state of the peptides. To investigate the effect of the buffer ions, we recorded the CD spectra of 2 and 5 in acetate buffer at pH 4.5 (Figure S5); interestingly, the CD curve of the peptides in acetate buffer is more similar to that in phosphate buffer than in water, which excludes a pH effect but supports a salt effect. However, the presence of a different conformation in sodium phosphate and acetate cannot be fully ruled out, since the CD spectra in acetate buffer could not be recorded below 205 nm because of the strong buffer absorbance: In particular, the ||π‐π* CD band is not visible, which might be blue‐shifted below 205 nm or disappear under increased negative contribution, especially in the case of peptide 2, reflecting a modification of the conformation when changing the phosphate ion with the acetate ion.
3.3. The secondary chemical shifts and 3 J HNα values of the lactam‐bridged pentapeptide fragment 2–6 reveal an anomalous behavior of the residue at position 4
We investigated all peptides in water by NMR spectroscopy. A first analysis was performed on the basis of the α‐ and β‐carbon chemical shift deviations from the corresponding random coil values, so called secondary chemical shifts (SCSs), according to Metzler et al.. 49 For norleucine, the random coil Cα and Cβ shifts were obtained by measuring the reference peptide Ac‐GGXGG‐NH 2 in 7 M urea at pH 2.3 and 7.4 (Table S3). For the lactam‐bridged Asp‐6, we used the random coil values of asparagine. Positive SCS values are typical for residues in helical conformation (SCS close or above 2 ppm) or with helix propensity (SCS smaller than 2 ppm). 50 In analogy, negative SCS values are typical for residues in β‐strand conformation or with β‐sheet propensity. As shown in Figure 3A, a common SCS pattern can be recognized, which identifies Lys‐2, Arg‐3, and Gln‐5 as the most helical or helix‐prone residues. Surprisingly, the residue at position 4 and the lactam‐bridged Asp‐6 are characterized by smaller SCSs (or even with opposite sign, as for Val‐4 in peptide 10). The smoothed SCS profiles, which better highlight the presence of secondary structure along the sequence, 49 reveal superior helical character of the N‐terminal over the C‐terminal part of the peptides, which fits with the presence of the lactam bridge between Lys‐2 and Asp‐6 (Figure 3B). However, the effectiveness of this constraint in forming a helix turn is clearly different among the constrained sequences: In fact, except for peptide 7, which is very likely to form a stable N‐terminal helical turn, the other peptides will exist in an ensemble of conformations in equilibrium, in which the helical one should be predominant over residues 1–4. It is interesting to note that peptides 6 and 10, which show the least intense chemical shift deviations for residues 3–5 (Figure 3B), contain the β‐branched residues isoleucine (6) and valine (10) at position 4: This agrees with the knowledge that such residues are less helix‐prone than leucine and methionine, the latter being isosteric to norleucine. 52 This fits also with the CD data, as peptides 6 and 10 in water are characterized by the lowest CD intensity among all peptides (Figure S3d).
FIGURE 3.
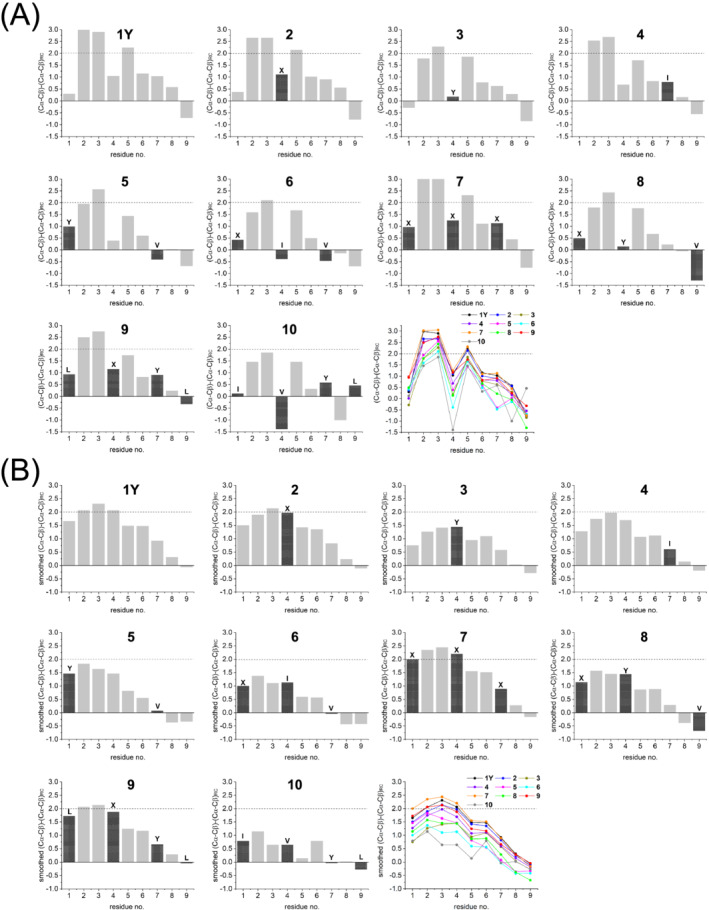
(A) Chemical shift differences (ppm) between the measured and random coil 51 Cα and Cβ shifts for each residue (X = norleucine) of the peptides in water (pH range 3–4). (B) The values were smoothed by using the equation (Δδi‐1 + Δδi + Δδi+1)/3 (except for the end positions, for which only one neighboring residue was considered)
Further, we examined the chemical shifts of the amide protons and their 3 J HNα coupling constants: Especially, the latter depend on the backbone conformation and, thus, are indicative of the presence of ordered or disordered structure. About the amide protons, we noticed that the signal for residue‐4 was significantly upfield (7.3–7.6 ppm) with respect to the others (7.9–8.4 ppm). Although the helical conformation generally induces a moderate upfield shift of the resonances of the backbone amide protons with respect to random coil values, 53 the deviations observed for position 4 are unusually large (−0.8 to −0.6 ppm; Figure S6), so that the amide proton of residue‐4 falls in the region of the C‐terminal amide protons (7.1–7.6 ppm). Position 4 was found to be peculiar also for the 3 J HNα value: Indeed, while the two sequential residues Lys‐2 and Arg‐3 display 3 J HNα values around 4 Hz, which is characteristic of α‐ or 310‐helix conformation, 54 the residue at position 4 adopts significantly higher 3 J HNα values (6–8 Hz), similarly to the C‐terminal residues 6–9. These data agree with the chemical shift deviations reported in Figure 3 and suggest that the formation of an ideal α‐ or 310‐helix spanning over the nine residues of these peptides in water is unlikely.
3.4. The NMR solution structures reveal the presence of tilted backbone carbonyls
We calculated the three‐dimensional structure of the cyclopeptides by using the NOE constraints reported in Figure S7. Figures 5 and 6 summarize the ensembles of 20 low‐energy structures of each peptide and the frequency of helix/bend/turn/coil per residue, as analyzed by the MolMol program 46 on the base of the secondary‐structure element definitions given by Kabsch and Sander: In detail, a helix is defined as the repeat of turns stabilized by backbone H‐bonds between the carbonyl of residue i and the amide hydrogen of residue i+n (n = 3–5), whereas a bend describes a backbone curvature of at least 70° (this angle is built by the Cα atoms of the three residues i‐–2, i, i+2). 55 Notably, peptides 4 and 7 display a high number of helical residues: Indeed, the helix segment 2–8 is present in at least 85% of the 20 low‐energy structures (see Figure 6). In contrast, a shorter helical motif (residues 2–5) is present in all 20 structures of peptides 1Y, 2, 3, and 5, whereas the lactam‐bridged Asp‐6 (and residue 7 in peptides 1Y, 2 and 5) is mainly found in a bend conformation. Regarding peptides 6 and 8–10, the helix structure is moderately or poorly populated, which agrees well with the CD data: Indeed, these four peptides gave much less intense CD spectra than the other more helical analogs (Figure S3a–d).
FIGURE 5.
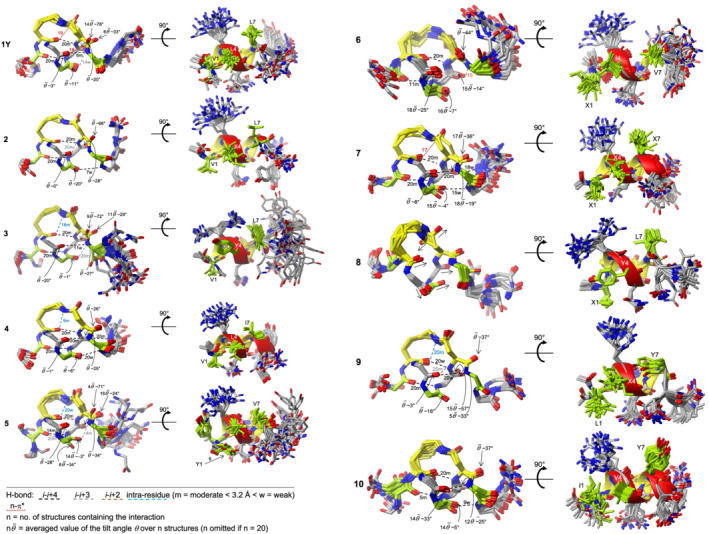
Overlap of the 20 low‐energy NMR solution structures of the peptides in water. For each peptide: On the left, overlayed structural ensembles showing the peptide backbone and the lactam‐bridge (yellow); on the right, the same ensembles with all side chains, rotated by 90° to have the hydrophobic residues in front (green). H‐bond and n‐π* interactions are shown with dashed and dotted lines, respectively. The H‐bonds (O•••N) are shorter (m) or larger (w) than 3.2 Å. The interactions are color‐coded as explained in the legend. The number given at each interaction corresponds to the number of NMR structures containing that interaction. The reported tilt angles of the carbonyl carbons of Arg‐3, residue‐4, Gln‐5, and Asp‐6 are the average of n low‐energy structures (Table S5). The frequency of helix per residue over the 20 structures, which was extrapolated by MolMol and is indicated with the red ribbon, can be found in Figure 6
FIGURE 6.
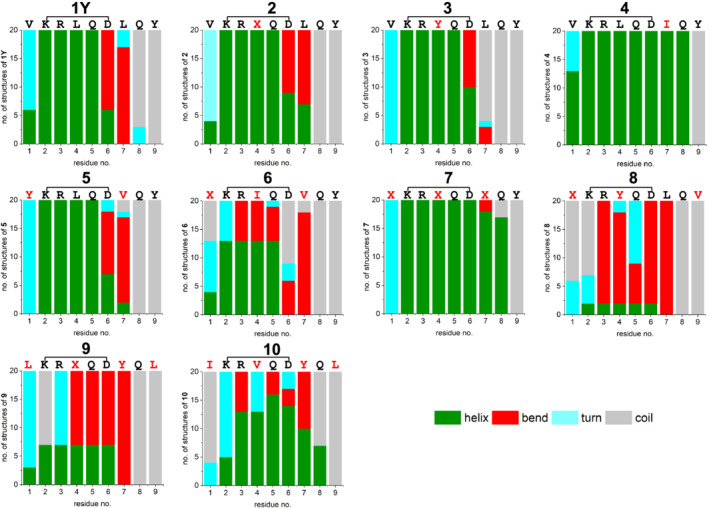
Frequency of helix/bend/turn/coil per residue, as extrapolated from the MolMol program 46 on the base of the secondary‐structure element definitions given by Kabsch and Sander 55
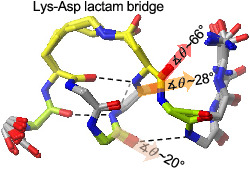
The NMR solution structures of the lactam‐bridged peptides are shown in Figure 5. Besides intramolecular stabilizations, particularly backbone H‐bonds forming 13‐membered (C13) or 10‐membered (C10) rings, distortions of the helical backbone are also evident, as already suggested by the chemical shift deviations and the 3 J HNα coupling constants (Figures 3 and 4). Peptide 1Y shows a helical turn from Lys‐2 to Gln‐5, which is stabilized by two backbone H‐bonds (Val‐1 CO with Gln‐5 NH, and Lys‐2 CO with Asp‐6 NH) (Figure 5). The carbonyl oxygen of the lactam bridge points outwards, and the distance between the Asp‐6 γ‐carbonyl carbon and the backbone‐carbonyl oxygen of Lys‐2 is in the range of 2.8–3.2 Å, suggesting that a n‐π* interaction might additionally stabilize the lactam‐bridged helical turn, as recently proposed by Hoang et al.. 34 Remarkably, the backbone carbonyl of Asp‐6 is tilted outwards and exposed to the solvent: We calculated the tilt angle θ between the C—O vector of the backbone carbonyl of Asp‐6 and the helix axis in all 20 low‐energy structures by using the approach published by Haimov and Srebnik. 47 We observed two clusters, the most populated one (14 out of 20 structures) with a θ value of about 78° and another one with a θ value of about 33° (Table S5). This implies a pronounced deviation of the peptide‐bond plane between Asp‐6 and Leu‐7 from the orientation parallel to the helix axis. Such backbone distortion disfavors the helix propagation beyond Asp‐6 by preventing the formation of the H‐bond between Arg‐3 CO and Leu‐7 NH. Besides Asp‐6, also Gln‐5 shows an accentuated backbone‐carbonyl tilting with a θ value of about 20° (Figure 5).
FIGURE 4.
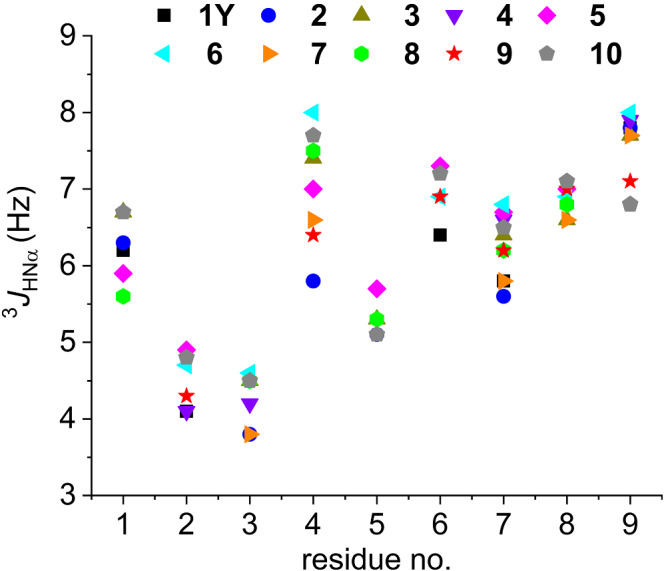
3 J HNα coupling constants of the peptides in water (pH range 3–4). The splitting of resonances suffering from overlap has been omitted. For the 3 J HNα values, see also Table S4
Replacement of Leu‐4 with norleucine (peptide 2) leads to subtle but still detectable changes in the structure: First, the lactam‐bridged helix turn seems to be not stabilized by a n‐π* interaction between the Asp‐6 γ‐carbonyl carbon and the backbone‐carbonyl oxygen of Lys‐2 (distance > 3.2 Å). Second, the peptide‐bond plane between Gln‐5 and Asp‐6 deviates about 28° from the orientation parallel to the helix axis, which, on the one hand, exposes the backbone‐carbonyl oxygen of Gln‐5 to the solvent and, on the other hand, allows the backbone‐amide hydrogen of Asp‐6 making contacts with both Lys‐2 and Arg‐3 backbone‐carbonyl oxygens. Like in 1Y, there is no helix propagation from the C‐terminal side of the lactam bridge, as the peptide bond plane between Asp‐6 and Leu‐7 is rotated of about 66° from the orientation parallel to the helix axis in all 20 low‐energy structures.
Peptide 3, which contains a Leu4Tyr substitution, displays the same H‐bond network of 1Y and the same feature of the peptide bond plane between Asp‐6 and Leu‐7 of being rotated of about 29° or 72° from the orientation parallel to the helix axis in 11 and 9 structures, respectively. This leads to the formation of a weak H‐bond between Arg‐3 CO and Leu‐7 NH in the first case and of a moderate H‐bond between Tyr‐4 CO and Leu‐7 NH in the second case. However, the lactam bridge does not seem to be involved in a n‐π* interaction, like in 1Y, but rather in an intra‐residue H‐bond with the backbone‐carbonyl oxygen of Lys‐2.
Deviations of the peptide‐bond plane between Asp‐6 and Val‐7 from the orientation parallel to the helix axis are also present in peptides 5 (Val1Tyr and Leu7Val) and 6 (Val1Nle, Leu4Ile, and Ile7Val), which lead to the formation of an H‐bond between Leu‐4 CO and Val‐7 NH in four structures of peptide 5, or between Gln‐5 CO and Val‐7 NH in 15 structures of peptide 6. Moreover, the backbone carbonyls of Arg‐3 in both peptides and of Gln‐5 in peptide 5 show accentuated outward tilting (Figure 5).
Peptides 4 (Leu7Ile) and 7 (Val1Nle, Leu4Nle, and Leu7Nle) show not only the lactam‐bridged helical turn but also helix propagation until the C‐terminus, with the five expected backbone H‐bonds (i‐i+4, with i = 1–5). Furthermore, the backbone carbonyls of the fragment 3–5 appear, on average, less tilted than the respective groups in the peptides described above. However, the Asp‐6 backbone carbonyl remains significantly tilted outwards.
Like in peptides 4 and 7, Asp‐6 in peptide 10 adopts a helical conformation in most structures (Figure 6). However, the N‐terminal and C‐terminal ends of the helix are not as well defined as for peptides 4 and 7: Indeed, the H‐bonds Ile‐1 CO with Gln‐5 NH, and Val‐4 CO with Gln‐8 NH are present only in five and nine structures, respectively, whereas the two backbone H‐bonds (i‐i+4, with i = 2, 3) stabilizing the internal helical turn are found more frequently within the 20 structures.
Peptide 9, which contains the four substitutions Val1Leu, Leu4Nle, Leu7Tyr, and Tyr9Leu, displays an unusual H‐bond pattern that stabilizes the N‐terminal part of the structure and consists of the backbone H‐bond between Leu‐1 CO and Gln‐5 NH, followed by the consecutive bifurcated H‐bonds of Lys‐2 CO with both the lactam bridge and Asp‐6 NH, and of Arg‐3 CO with both Asp‐6 NH and Tyr‐7 NH. This generates a C13/C10 turn motif with the coexistence of two i−i+4 H‐bonds (Lys‐2 CO with Asp‐6 NH, and Arg‐3 CO with Tyr‐7 NH) and one i−i+3 H‐bond (Arg‐3 CO and Asp‐6 NH). The latter is made possible by the deviation of the peptide bond plane between Gln‐5 and Asp‐6 from the orientation parallel to the helix axis, which approaches the backbone‐amide hydrogen of Asp‐6 to the backbone‐carbonyl oxygen of Arg‐3, analogous to what was observed in peptide 2.
4. DISCUSSION
The α‐helix is one of the most abundant secondary structures present in peptides and proteins. It may play a structural and/or functional role, contributing to the folding of globular proteins and/or being directly involved in molecular recognition events: Examples are coiled coils 56 and, in general, α‐helix bundles which promote peptide or protein oligomerization, as in the case of HLH and HLH‐leucine zipper transcription factors. 57 The geometry of a regular α‐helix, as described by Pauling et al., 58 is defined by a helical turn, consisting of 3.7 residues and a translation of 5.44 Å along the helix axis, which give rise to the formation of i−i+4 H‐bonds with an oxygen‐nitrogen distance of 2.72 Å and a C—N—H angle of no more than 30°. However, deviations from this ideal geometry are often observed in folded peptides and proteins, which include kinks and curvatures and are induced not only by proline residues, but also by interactions of the backbone with the environment or with the side chains as well as by steric hindrance or packing of neighboring side chains. 59
Helix distortions are often overseen, especially when only the backbone dihedral angles ϕ and ψ are considered. Indeed, the helical basin within the Ramachandran plot spans over a broad range of ϕ and ψ values, which comprises not only ideal α‐helices, whose standard ϕ and ψ values are −64 ± 7° and −42 ± 7°, respectively, but also the 310‐helix and different types of turns. 60 For example, the (ϕ,ψ) pairs adopted by alanine are much closer to those of a regular α‐helix than those adopted by threonine which are more typical for turns. This reflects the different conformational propensities of the natural amino acids, with alanine being the one with the highest α‐helical propensity. 52 , 60 Moreover, the conformational preference of a residue is also affected by neighboring residues. 47 , 60
Recently, Haimov and Srebnik found deviations of helical structures by analyzing residues in the α‐helix basin of the Ramachandran plot. They defined two very useful parameters for describing a helix, namely, the tilt angle θ (with θ > 0 when the backbone‐carbonyl oxygen points outwards) and the number of residues per turn ρ 47 . Since the θ value is particularly sensitive to the H‐bond alignment along the helix axis, it well describes the quality of an α‐helical conformation: θ values in the range of 9–12° are typical of helices with high H‐bond alignment, whereas less ordered helical or even non‐helical conformations display larger θ angles. Interestingly, the β‐branched amino acids valine and isoleucine tend to adopt smaller θ values than other residues. Furthermore, phenylalanine and tyrosine, but not tryptophan, tend to increase the number of residues per turn (ρ value). Considering the dependency of the geometry of a helical conformation from the type of residue itself as well as from its neighboring residues, it is not surprising that the ten cyclopeptides presented here show a significant dispersion of the θ values, despite the presence of a covalent conformational constraint. In detail, the residue at position 4 adopts a tilt angle between −4° and 20° (the average of the most frequent θ values for each peptide is considered, as shown in Table S5), which implies that in some peptides the peptide bond plane between the residue at position 4 and Gln‐5 tends to deviate significantly from the alignment with the helix axis, so that the backbone‐carbonyl oxygen of the residue at position 4 is more exposed to water and forms no or only weak intramolecular H‐bonds. The propensity to outwards tilting of the backbone carbonyl is even higher for Arg‐3 and Gln‐5 in some peptides: For example, Arg‐3 in peptides 3, 5, 6, and 10 adopts a θ value between 20° and 33°. For Gln‐5, all peptides except 6 and 7 show a backbone‐carbonyl tilting of 20° up to 57°. However, the most substantial deviation is that of the peptide bond plane between Asp‐6 and the residue at position 7, which corresponds to a θ value between 44° and 78° in peptides 1Y, 2, 3, and 6. As a result, the helix propagation from the C‐terminus of the lactam‐bridged turn is strongly impaired, except for cyclopeptides 4 and 7 that display a moderate (O•••N distance about 3 Å) backbone H‐bond between Arg‐3 and the residue at position 7 in all 20 low‐energy structures.
Recent work by Hoang et al. 34 suggested that the (1,5)‐lactam‐bridged pentapeptide Ac‐KAAAD‐NH 2 and the (2,6)‐lactam‐bridged heptapeptide Ac‐F (pNO2)KLLLD F (pNO2)‐NH 2 build an α‐helix turn that is stabilized not only by backbone H‐bonds, but also by a medium‐range n‐π* interaction between the Lys α‐carbonyl oxygen and the Asp γ‐carbonyl carbon. This implies a rather rigid conformation of the lactam bridge, as reflected by the splitting of the amide‐proton resonance of the lactam bridge into a doublet of doublets in the solution NMR spectra of the model pentapeptide due to different 3 J HZHE2 and 3 J HZHE3 coupling constants. 1D‐NMR analysis of the ten cyclopeptides presented here revealed a doublet of doublets for the amide‐proton resonance of the lactam group in peptides 1Y, 4, 5, 7, and 9 (Figure 7). Albeit less clearly, peptides 3, 6, and 10 also exhibit a doublet of doublets for the same signal, whereas for peptides 2 and 8, this information could not be obtained due to signal overlap in the 1D‐NMR spectra. The two 3 J coupling constants extrapolated from each doublet of doublets are <5 and >7 Hz for peptides 1Y, 4, 5, 7, and 9, while they both fall in the range of 5–7 Hz for peptides 3, 6, and 10. This suggests that the lactam bridge of the last three peptides adopts a less rigid conformation, which correlates well with the less intense secondary chemical shifts and the higher 3 J HNα coupling constants for the residue at position 4 (>7 Hz).
FIGURE 7.
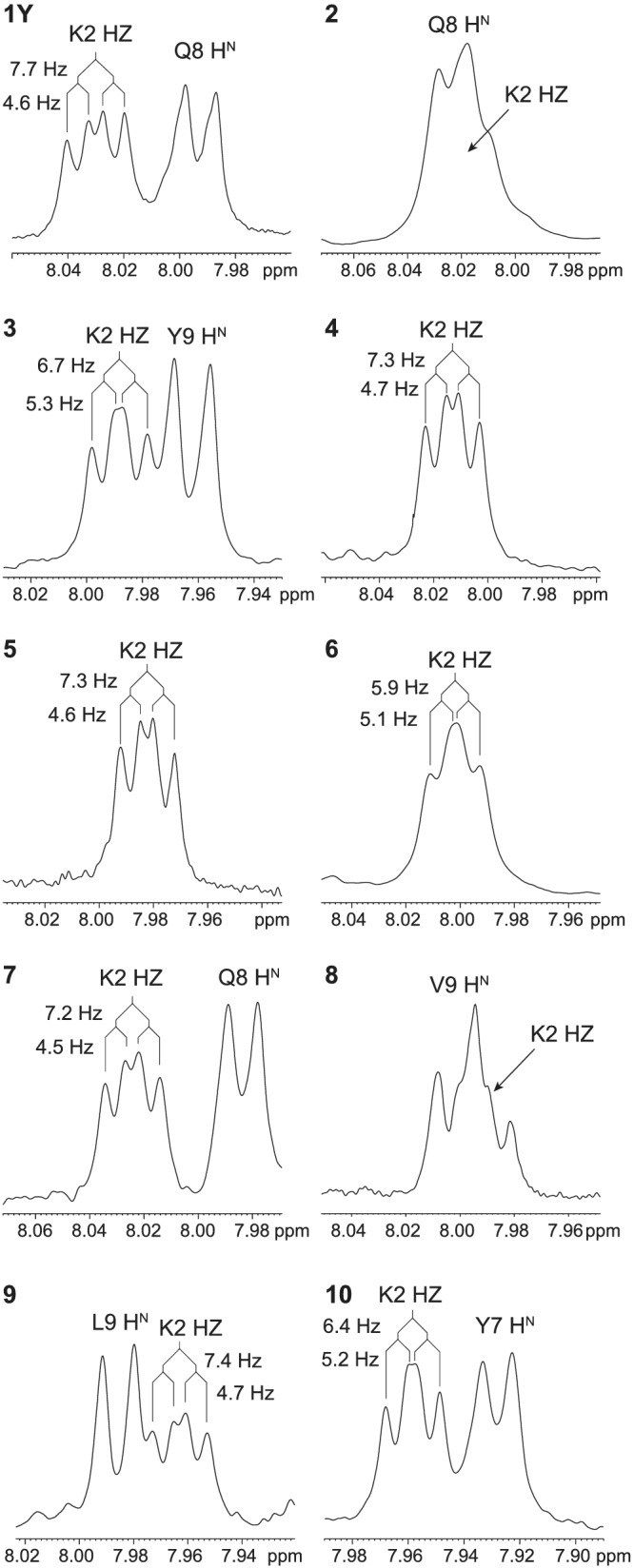
Split of the amide‐proton resonance of the lactam group, as observed in the 1D‐NMR spectra of the cyclopeptides in water (pH 3–4). The complete amide‐proton region of each 1D‐NMR spectrum is reported in Figure S8
5. CONCLUSIONS
In this work, we have investigated a series of analogs of the lactam‐bridged nonapeptide 1Y, which is a ligand of the ID proteins and reduces cancer cell viability. 38 , 40 These analogs, which differ from 1Y in the hydrophobic pattern (residues 1, 4, 7, and 9), were found to be comparable to or less effective than 1Y in the cancer cell viability assay: In particular, the replacement of Leu‐4 with Tyr‐4 was unfavorable (Figure S9). A detailed analysis of NMR secondary chemical shifts and coupling constants of the amide protons revealed an anomalous behavior of the residue at position 4, which is characterized by a weakly positive or even negative secondary chemical shift and by a 3 J HNα value close to 6 Hz or higher. Both NMR parameters are unusual for an ideal α‐helix conformation and indicate that the lactam‐bridged cyclized residues 2–6 do not possess the same degree of helicity. Particularly poor helical character at position 4 has been found for isoleucine, valine, and tyrosine, amino acids that are known to have only moderate helix propensity 36 , 52 or even to disturb the helical geometry. 47 The anomalous NMR parameters of the residue at position 4, especially its 3 J HNα values in the range of 6–8 Hz, might be interpreted as backbone flexibility; however, it is also plausible that they arise from a distortion of the canonical α‐helical turn upon introduction of the lactam‐bridge constraint. In fact, the helical structures of all measured lactam‐bridged peptides display an increment of the number of residues per turn (ρ) in comparison to the ideal α‐helical turn (4.2–4.3 vs. 3.6, as reported in Table S5).
An additional anomaly of the cyclopeptides presented here is the outward tilting of the backbone carbonyls, which is evident especially at Asp‐6 in almost all structures. Outward tilting of the backbone carbonyls is not unusual in native helical structures. 61 Such distortion weakens the α‐helix‐stabilizing H‐bonds and may favor the formation of kinks, as observed at Asp‐6 that corresponds to the stop of the helix in most of the structures. To the best of our knowledge, this type of backbone distortions in lactam‐bridged stabilized helical conformations has not been reported yet. However, by analyzing crystal structures available for this type of constrained helices in the Protein Data Bank (PDB) 62 and in the Cambridge Crystallographic Data Centre (CCDC), 63 we have found significant tilting of the backbone carbonyl either for the lactam‐bridged aspartate or for the residues within the cyclized motif (PDB ID: 5WGD and 5WGQ 64 : CCDC deposition number 1941068 34 ) (Table S6). Moreover, these helices and those of our present work share the same number of residues per turn ranging from 4.0 to 4.2 (Tables S5 and S6), which is significantly higher than that of a regular α‐helix (3.6). Therefore, we tentatively attribute a moderate unwinding effect to the lactam‐bridged constraint between Lys‐i and Asp‐i+4.
The present work shows that the incorporation of a constraint aiming at the stabilization of a helical conformation is no guarantee for a helical structure and does not exclude the occurrence of backbone distortions. In the case of the lactam‐bridge constraint between Lys‐i and Asp‐i+4, we have identified altered helix character for the residue at position i+2 as well as outward tilting of the backbone carbonyls (particularly of the lactam‐bridged aspartate residue).
AUTHOR CONTRIBUTIONS
V. S. and C. H. carried out the peptide syntheses. V. S. performed the analytical characterization of the peptides and the CD experiments. A. M., A. H., and M. S. performed NMR measurements, assignment, and structure calculation. M. W. performed the calculations of θ and ρ values. C. R. and A. Z. performed the cell‐based experiments. C. C. and M. S. wrote the manuscript with contributions of all authors.
CONFLICT OF INTEREST
There is no conflict to declare.
Supporting information
Supporting Information S1. Backbone distortions in lactam‐bridged helical peptides
ACKNOWLEDGMENTS
The authors acknowledge the Land Salzburg and the University of Salzburg for funding. The authors also thank Sabine Ullrich for technical assistance. A. M. thanks the Ministry of Science and Technology of Iran and University of Tehran for financial support.
Moazzam A, Stanojlovic V, Hinterholzer A, et al. Backbone distortions in lactam‐bridged helical peptides. J Pep Sci. 2022;28(7):e3400. doi: 10.1002/psc.3400
Ali Moazzam and Vesna Stanojlovic contributed equally to this work.
Funding information Land Salzburg; University of Salzburg; Ministry of Science and Technology of Iran and University of Tehran
Contributor Information
Mario Schubert, Email: mario.schubert@plus.ac.at.
Chiara Cabrele, Email: chiara.cabrele@plus.ac.at.
REFERENCES
- 1. Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α‐helix‐mediated protein‐protein interactions using designed molecules. Nat Chem. 2013;5(3):161‐173. doi: 10.1038/nchem.1568 [DOI] [PubMed] [Google Scholar]
- 2. Edwards TA, Wilson AJ. Helix‐mediated protein‐protein interactions as targets for intervention using foldamers. Amino Acids. 2011;41(3):743‐754. doi: 10.1007/s00726-011-0880-8 [DOI] [PubMed] [Google Scholar]
- 3. Cabezas E, Satterthwait AC. The hydrogen bond mimic approach: Solid‐phase synthesis of a peptide stabilized as an α‐helix with a hydrazone link. J Am Chem Soc. 1999;121(16):3862‐3875. doi: 10.1021/ja983212t [DOI] [Google Scholar]
- 4. Henchey LK, Kushal S, Dubey R, Chapman RN, Olenyuk BZ, Arora PS. Inhibition of hypoxia inducible factor 1—transcription coactivator interaction by a hydrogen bond surrogate α‐helix. J Am Chem Soc. 2009;132(3):941‐943. doi: 10.1021/ja9082864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. General approach to the synthesis of short α‐helical peptides. J Am Chem Soc. 1991;113(24):9391‐9392. doi: 10.1021/ja00024a067 [DOI] [Google Scholar]
- 6. Diderich P, Bertoldo D, Dessen P, et al. Phage selection of chemically stabilized α‐helical peptide ligands. ACS Chem Biol. 2016;11(5):1422‐1427. doi: 10.1021/acschembio.5b00963 [DOI] [PubMed] [Google Scholar]
- 7. Cheloha RW, Watanabe W, Dean T, Gellman SH, Gardella TJ. Backbone modification of a parathyroid hormone receptor‐1 antagonist/inverse agonist. ACS Chem Biol. 2016;11(10):2752‐2762. doi: 10.1021/acschembio.6b00404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cantel S, Halperin JA, Chorev M, et al. Side chain‐to‐side chain cyclization by intramolecular click reaction‐building blocks, solid phase synthesis and conformational characterization. In: Valle SD, Escher E, Lubell WD, eds. Peptides for Youth. Advances in Experimental Medicine and Biology, Vol. 611. New York, NY: Springer; 2009. doi: 10.1007/978-0-387-73657-0_80 [DOI] [PubMed] [Google Scholar]
- 9. Cantel S, Isaad Ale C, Scrima M, et al. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+4 intramolecular side‐chain to side‐chain azide‐alkyne 1,3‐dipolar cycloaddition. J Org Chem. 2008;73(15):5663‐5674. doi: 10.1021/jo800142s [DOI] [PubMed] [Google Scholar]
- 10. Blackwell HE, Sadowsky JD, Howard RJ, et al. Ring‐closing metathesis of olefinic peptides: design, synthesis, and structural characterization of macrocyclic helical peptides. J Org Chem. 2001;66(16):5291‐5302. doi: 10.1021/jo015533k [DOI] [PubMed] [Google Scholar]
- 11. Kim YW, Kutchukian PS, Verdine GL. Introduction of all‐hydrocarbon i,i+3 staples into α‐helices via ring‐closing olefin metathesis. Org Lett. 2010;12(13):3046‐3049. doi: 10.1021/ol1010449 [DOI] [PubMed] [Google Scholar]
- 12. Schafmeister CE, Po J, Verdine GL. An all‐hydrocarbon cross‐linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122(24):5891‐5892. doi: 10.1021/ja000563a [DOI] [Google Scholar]
- 13. Shim SY, Kim YW, Verdine GL. A new i, i+3 peptide stapling system for α‐helix stabilization. Chem Biol Drug Des. 2013;82(6):635‐642. doi: 10.1111/cbdd.12231 [DOI] [PubMed] [Google Scholar]
- 14. Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3‐33. doi: 10.1016/B978-0-12-396962-0.00001-X [DOI] [PubMed] [Google Scholar]
- 15. Hilinski GJ, Kim YW, Hong J, et al. Stitched α‐helical peptides via bis ring‐closing metathesis. J Am Chem Soc. 2014;136(35):12314‐12322. doi: 10.1021/ja505141j [DOI] [PubMed] [Google Scholar]
- 16. Caporale A, Sturlese M, Gesiot L, Zanta F, Wittelsberger A, Cabrele C. Side chain cyclization based on serine residues: synthesis, structure, and activity of a novel cyclic analogue of the parathyroid hormone fragment 1‐11. J Med Chem. 2010;53(22):8072‐8079. doi: 10.1021/jm1008264 [DOI] [PubMed] [Google Scholar]
- 17. Bracken C, Gulyas J, Taylor J, Baum J. Synthesis and nuclear magnetic resonance structure determination of an α‐helical, bicyclic, lactam‐bridged hexapeptide. J Am Chem Soc. 1994;116(14):6431‐6432. doi: 10.1021/ja00093a052 [DOI] [Google Scholar]
- 18. Osapay G, Taylor JW. Multicyclic polypeptide model compounds. 1. Synthesis of a tricyclic amphiphilic α‐helical peptide using an oxime resin, segment‐condensation approach. J Am Chem Soc. 1990;112(16):6046‐6051. doi: 10.1021/ja00172a021 [DOI] [Google Scholar]
- 19. Osapay G, Taylor JW. Multicyclic polypeptide model compounds. 2. Synthesis and conformational properties of a highly alpha‐helical uncosapeptide constrained by three side‐chain to side‐chain lactam bridges. J Am Chem Soc. 1992;114(18):6966‐6973. doi: 10.1021/ja00044a003 [DOI] [Google Scholar]
- 20. Phelan JC, Skelton NJ, Braisted AC, McDowell RS. A general method for constraining short peptides to an α‐helical conformation. J Am Chem Soc. 1997;119(3):455‐460. doi: 10.1021/ja9611654 [DOI] [Google Scholar]
- 21. Schievano E, Mammi S, Bisello A, Rosenblatt M, Chorev M, Peggion E. Conformational studies of a bicyclic, lactam‐constrained parathyroid hormone‐related protein‐derived agonist. J Pept Sci. 1999;5(7):330‐337. doi: [DOI] [PubMed] [Google Scholar]
- 22. Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. Single turn peptide alpha helices with exceptional stability in water. J Am Chem Soc. 2005;127(9):2974‐2983. doi: 10.1021/ja0456003 [DOI] [PubMed] [Google Scholar]
- 23. Shepherd NE, Hoang HN, Desai VS, Letouze E, Young PR, Fairlie DP. Modular α‐helical mimetics with antiviral activity against respiratory syncitial virus. J Am Chem Soc. 2006;128(40):13284‐13289. doi: 10.1021/ja064058a [DOI] [PubMed] [Google Scholar]
- 24. Taylor JW. The synthesis and study of side‐chain lactam‐bridged peptides. Biopolymers (Pept Sci). 2002;66(1):49‐75. doi: 10.1002/bip.10203 [DOI] [PubMed] [Google Scholar]
- 25. Yu C, Taylor JW. Synthesis and study of peptides with semirigid i and i+7 side‐chain bridges designed for α‐helix stabilization. Bioorg Med Chem. 1999;7(1):161‐175. doi: 10.1016/S0968-0896(98)00232-6 [DOI] [PubMed] [Google Scholar]
- 26. Zhao H, Liu QS, Geng H, et al. Crosslinked aspartic acids as helix‐nucleating templates. Angew Chem Int Ed. 2016;55(39):12088‐12093. doi: 10.1002/anie.201606833 [DOI] [PubMed] [Google Scholar]
- 27. Carpenter KA, Schmidt R, Yue SY, et al. The glycine‐1 residue in cyclic lactam analogues of galanin(1−16)‐NH2 is important for stabilizing an N‐terminal helix. Biochemistry. 1999;38(46):15295–15304. doi: 10.1021/bi991081i [DOI] [PubMed] [Google Scholar]
- 28. Geistlinger TR, Guy RK. An inhibitor of the interaction of thyroid hormone receptor β and glucocorticoid interacting protein 1. J Am Chem Soc. 2001;123(7):1525‐1526. doi: 10.1021/ja005549c [DOI] [PubMed] [Google Scholar]
- 29. Tsomaia N, Pellegrini M, Hyde K, Gardella TJ, Mierke DF. Toward parathyroid hormone minimization: Conformational studies of cyclic PTH(1−14) analogues. Biochemistry. 2004;43(3):690‐699. doi: 10.1021/bi035703i [DOI] [PubMed] [Google Scholar]
- 30. Karle IL, Balaram P. Structural characteristics of α‐helical peptide molecules containing Aib residues. Biochemistry. 1990;29(29):6747‐6756. doi: 10.1021/bi00481a001 [DOI] [PubMed] [Google Scholar]
- 31. Checco JW, Kreitler DF, Thomas NC, et al. Targeting diverse protein‐protein interaction interfaces with α/β‐peptides derived from the Z‐domain scaffold. Proc Natl Acad Sci USA. 2015;112(15):4552‐4557. doi: 10.1073/pnas.1420380112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Checco JW, Lee EF, Evangelista M, et al. α/β‐Peptide foldamers targeting intracellular protein‐protein interactions with activity in living cells. J Am Chem Soc. 2015;137(35):11365‐11375. doi: 10.1021/jacs.5b05896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Araujo AD, Hoang HN, Kok WM, et al. Comparative α‐Helicity of cyclic pentapeptides in water. Angew Chem Int Ed. 2014;53(27):6965‐6969. doi: 10.1002/anie.201310245 [DOI] [PubMed] [Google Scholar]
- 34. Hoang HN, Wu C, Hill TA, et al. A novel long‐range n to pi* interaction secures the smallest known α‐helix in water. Angew Chem Int Ed. 2019;58(52):18873‐18877. doi: 10.1002/anie.201911277 [DOI] [PubMed] [Google Scholar]
- 35. Roschger C, Cabrele C. The Id‐protein family in developmental and cancer‐associated pathways. Cell Commun Signal. 2017;15(1):7. doi: 10.1186/s12964-016-0161-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neukirchen S, Krieger V, Roschger C, Schubert M, Elsasser B, Cabrele C. Impact of the amino acid sequence on the conformation of side chain lactam‐bridged octapeptides. J Pept Sci. 2017;23(7‐8):587‐596. doi: 10.1002/psc.2997 [DOI] [PubMed] [Google Scholar]
- 37. Lathbridge A, Mason JM. Combining constrained heptapeptide cassettes with computational design to create coiled‐coil targeting helical peptides. ACS Chem Biol. 2019;14(6):1293‐1304. doi: 10.1021/acschembio.9b00265 [DOI] [PubMed] [Google Scholar]
- 38. Roschger C, Neukirchen S, Elsasser B, et al. Targeting of a helix‐loop‐helix transcriptional regulator by a short helical peptide. ChemMedChem. 2017;12(18):1497‐1503. doi: 10.1002/cmdc.201700305 [DOI] [PubMed] [Google Scholar]
- 39. Wong MV, Jiang S, Palasingam P, Kolatkar PR. A divalent ion is crucial in the structure and dominant‐negative function of ID proteins, a class of helix‐loop‐helix transcription regulators. PloS One. 2012;7(10):e48591. doi: 10.1371/journal.pone.0048591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roschger C, Verwanger T, Krammer B, Cabrele C. Reduction of cancer cell viability by synergistic combination of photodynamic treatment with the inhibition of the Id protein family. J Photochem Photobiol B. 2018;178:521‐529. doi: 10.1016/j.jphotobiol.2017.11.038 [DOI] [PubMed] [Google Scholar]
- 41. Chavali GB, Vijayalakshmi C, Salunke DM. Analysis of sequence signature defining functional specificity and structural stability in helix‐loop‐helix proteins. Proteins. 2001;42(4):471‐480. doi: [DOI] [PubMed] [Google Scholar]
- 42. Mach H, Middaugh CR, Lewis RV. Statistical determination of the average values of the extinction coefficients of tryptophan and tyrosine in native proteins. Anal Biochem. 1992;200(1):74‐80. doi: 10.1016/0003-2697(92)90279-G [DOI] [PubMed] [Google Scholar]
- 43. Goddard TD, Kneller DG. SPARKY 3, University of California, San Francisco.
- 44. Ulrich EL, Akutsu H, Doreleijers JF, et al. BioMagResBank. Nucleic Acids Res. 2008;36(Database):D402‐D408. doi: 10.1093/nar/gkm957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guntert P. Automated NMR structure calculation with CYANA, Downing AK. In: Methods Mol Biol. 2004;278:353‐378. doi: 10.1385/1-59259-809-9:353 [DOI] [PubMed] [Google Scholar]
- 46. Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14(1):51‐55. doi: 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 47. Haimov B, Srebnik S. A closer look into the α‐helix basin. Sci Rep. 2016;6(1):38341. doi: 10.1038/srep38341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70‐82. doi: 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Metzler WJ, Constantine KL, Friedrichs MS, et al. Characterization of the three‐dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry. 1993;32(50):13818‐13829. doi: 10.1021/bi00213a010 [DOI] [PubMed] [Google Scholar]
- 50. Marsh JA, Singh VK, Jia Z, Forman‐Kay JD. Sensitivity of secondary structure propensities to sequence differences between α‐ and γ‐synuclein: implications for fibrillation. Protein Sci. 2006;15(12):2795‐2804. doi: 10.1110/ps.062465306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schwarzinger S, Kroon GJ, Foss TR, Wright PE, Dyson HJ. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMRView. J Biomol NMR. 2000;18(1):43‐48. doi: 10.1023/A:1008386816521 [DOI] [PubMed] [Google Scholar]
- 52. Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422‐427. doi: 10.1016/S0006-3495(98)77529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222(2):311‐333. doi: 10.1016/0022-2836(91)90214-Q [DOI] [PubMed] [Google Scholar]
- 54. Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; 1986. [Google Scholar]
- 55. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers. 1983;22(12):2577‐2637. doi: 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 56. Truebestein L, Leonard TA. Coiled‐coils: the long and short of it. Bioessays. 2016;38(9):903‐916. doi: 10.1002/bies.201600062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murre C, Bain G, van Dijk MA, et al. Structure and function of helix‐loop‐helix proteins. Biochim Biophys Acta. 1994;1218(2):129‐135. doi: 10.1016/0167-4781(94)90001-9 [DOI] [PubMed] [Google Scholar]
- 58. Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen‐bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA. 1951;37(4):205‐211. doi: 10.1073/pnas.37.4.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barlow DJ, Thornto JM. Helix geometry in proteins. J Mol Biol. 1988;201(3):601‐619. doi: 10.1016/0022-2836(88)90641-9 [DOI] [PubMed] [Google Scholar]
- 60. Jha AK, Colubri A, Zaman MH, Koide S, Sosnick TR, Freed KF. Helix, sheet, and polyproline II frequencies and strong nearest neighbor effects in a restricted coil library. Biochemistry. 2005;44(28):9691‐9702. doi: 10.1021/bi0474822 [DOI] [PubMed] [Google Scholar]
- 61. Blundell T, Barlow D, Borkakoti N, Thornton J. Solvent‐induced distortions and the curvature of α‐helices. Nature. 1983;306(5940):281‐283. doi: 10.1038/306281a0 [DOI] [PubMed] [Google Scholar]
- 62. Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235‐242. doi: 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Groom CR, Bruno IJ, Lightfoot MP, Ward SC. The Cambridge structural database. Acta Crystallogr B. 2016;72(2):171‐179. doi: 10.1107/S2052520616003954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Speltz TE, Mayne CG, Fanning SW, et al. A "cross‐stitched" peptide with improved helicity and proteolytic stability. Org Biomol Chem. 2018;16(20):3702‐3706. doi: 10.1039/C8OB00790J [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1. Backbone distortions in lactam‐bridged helical peptides