

Abstract
Gene products can affect the concentrations of small molecules (aka “metabolites”), and conversely, some metabolites can modulate the concentrations of gene transcripts. While many specific instances of this interplay have been revealed, a global approach to systematically uncover human gene-metabolite interactions is still lacking. We performed a metabolome- and transcriptome-wide association study to identify genes influencing the human metabolome using untargeted metabolome features, extracted from 1H nuclear magnetic resonance spectroscopy (NMR) of urine samples, and gene expression levels, quantified from RNA-Seq of lymphoblastoid cell lines (LCL) from 555 healthy individuals. We identified 20 study-wide significant associations corresponding to 15 genes, of which 5 associations (with 2 genes) were confirmed with follow-up NMR data. Using metabomatching, we identified the metabolites corresponding to metabolome features associated with the genes, namely, N-acetylated compounds with ALMS1 and trimethylamine (TMA) with HPS1. Finally, Mendelian randomization analysis supported a potential causal link between the expression of genes in both the ALMS1- and HPS1-loci and their associated metabolite concentrations. In the case of HPS1, we additionally observed that TMA concentration likely exhibits a reverse causal effect on HPS1 expression levels, indicating a negative feedback loop. Our study highlights how the integration of metabolomics, gene expression, and genetic data can pinpoint causal genes modulating metabolite concentrations.
Keywords: transcriptomics, metabolomics, genome-wide association study, ALMS1, NAT8, HPS1, PYROXD2, N-acetylated compounds, trimethylamine
Introduction
Genome-wide association studies (GWAS) have identified thousands of common variants that are associated with complex traits,1 but the regulatory mechanisms behind these associations mostly remain poorly understood. Pinpointing causal variants is difficult since the lead variants associated with a trait are often in high linkage disequilibrium (LD) with other variants in the same region with only a slightly lower association signal. Such associated LD blocks typically contain several genes or functional elements, preventing the accurate identification of causal genes. Furthermore, some trait associated variants fall into intergenic regions of the genome with no obvious functional role at all.
A number of studies reported that trait associated genetic variants are significantly enriched in expression quantitative trait loci (eQTLs), suggesting that many trait associated variants affect the phenotype by altering gene expression.2−5 There is also a growing body of literature highlighting the more pronounced effects of genetic variants on molecular traits compared to phenotypic traits.6−9 This is not surprising since molecular traits representing fundamental biological processes such as gene expression and metabolism are intermediates in the causal chain from genotype to phenotype.
With high-throughput measurements becoming more accessible and widespread, integration of molecular traits into association studies has become a central challenge in the field. Such synthesis allows investigating the interplay between different organizational layers of a biological system. Despite metabolism and gene expression regulation both being fundamental biological processes that are commonly studied as molecular phenotypes, there are very few studies in humans that focus on the interplay between them. Several studies investigated the relationship between serum metabolites and whole blood gene expression in humans,10−12 but to the best of our knowledge, no transcriptome- and metabolome-wide association study has been performed using urine metabolome data of healthy human subjects. Most metabolome- and genome-wide association studies (mGWAS) reporting metabolite quantitative trait loci (mQTL) use targeted approaches where the concentrations of a limited number of metabolites are estimated from the metabolome data generated by mass spectrometry or NMR spectroscopy. This targeted approach is limited to the number of known quantifiable metabolites in the biofluid under study.
In the current study, we adopted an untargeted approach, making use of the entire metabolomic data captured by binned urine 1H NMR spectra as our molecular traits. We use RNAseq data from lymphoblastoid cell lines (LCLs). LCLs have been widely used in genomic studies and proven their usefulness as surrogates of primary tissues for studying both gene expression variation among individuals and the genetic architecture underlying regulatory variation of gene expression.13−16 While LCLs partially reflect the genetic variance of gene expression in primary tissues affecting the urine metabolome, they do have the advantage of not being influenced by immediate environmental factors such as recent changes in the diet or exposure to drugs. Our transcriptome and metabolome data was taken from 555 healthy individuals that were part of the Cohort Lausannoise (CoLaus).17 In addition, using a different NMR platform, metabolomic profiles were generated for a subset of 315 individuals from follow-up urine samples taken after 5 years. We identified several associations between expression levels and urine metabolome features that were partly validated with the follow-up data, allowing us to refine previous links between the corresponding genes and metabolites.
Materials and Methods
Study Samples
CoLaus (Cohorte Lausannoise) is a population-based cross-sectional study of 6188 healthy participants residing in Lausanne, Switzerland.17 Recruitment to the cohort was done on the basis of a simple, non-stratified random selection of the entire Lausanne population aged 35 to 75 in 2003. While all participants were genotyped, a random subset of 1000 participants was selected for NMR spectroscopy of baseline urine samples. For a random subset of 555 participants (limited by available funding and sample preparation success rate), transcriptome analysis by RNA sequencing of lymphoblastoid cells was performed (see below). The mean age of these subjects with transcriptome and metabolome profiles was 55 (min = 35, max = 75) and 53% of them were women. For a further subset of 315 participants NMR spectroscopy was performed on urine samples taken at a follow-up 5 years after the baseline.
Metabolomics Data
Baseline urinary metabolic profiles were generated using one-dimensional proton nuclear magnetic resonance (NMR) spectroscopy. NMR spectra were acquired at 300 K on a Bruker 16.4 T Avance II 700 MHz NMR spectrometer (Bruker Biospin, Rheinstetten, Germany) using a standard 1H detection pulse sequence with water suppression. The spectra were referenced to the TSP signal and phase and baseline corrected. We binned the spectra into chemical shift increments of 0.005 ppm, obtaining metabolome profiles of 2200 metabolome features, of which 1276 remain after filtering for missing values.18 Lastly, the data set was log10-transformed and standardised first across features (thereby normalizing the concentration of each sample) then across samples (thereby making intensities comparable). We used the z-score as a standardization method. We decided to use this statistical normalization approach, as opposed to normalizing to maximum or total creatinine levels, because it resulted in similar associations with transcriptome data, but with higher significance.
The follow-up data were acquired with an Avance III HD 600 NMR spectrometer. Spectra were referenced to the TSP signal and phase and baseline corrected. We binned the chemical shifts into 0.005 ppm bins. After removing water and urea spectral regions (4.55–5.00 ppm and 5.5–6.1 ppm), the data set was log10-transformed and standardised as described above. Our final metabolic data set includes 1289 features.
PCA plots for baseline and follow-up metabolomics data and a comparison between the baseline and follow-up urine NMR data are shown in Figure S1.
Gene Expression Data
Total RNA was extracted from Epstein–Barr-virus-transformed lymphoblastoid cell lines (LCLs) by following the Illumina TruSeq v2 RNA Sample Preparation protocol (Illumina, Inc., San Diego, CA) by the Department of Genetic Medicine and Development at the University of Geneva. Next, mRNA sequencing was performed on the Illumina HiSeq2000 platform producing 49 bp paired-end reads. On average, 17.5 million RNA-Seq reads were sequenced per sample, and its distribution is shown in Figure S2. Paired-end reads were mapped to human genome assembly GRCh37 (hg19) with GEMTools using GENCODE v15 as gene annotation.19 The reads were then filtered for concordant orientation of the two ends and a minimum quality score of 150 while allowing 5 mismatches at both ends. Gene level read counts were quantified with an in-house script. This resulted in expression profiles of 45,470 genes for 555 individuals, which were quantified as RPKM (reads per kilobase per million reads) values. The number of genes with RPKM > 0 was on average 23,000 in each sample (Figure S2). We transformed RPKM values by applying log-transformation [log2(1 + RPKM)] and then standardization (i.e., z-scoring) across samples to make genes comparable. For our analysis, we excluded genes on sex chromosomes as well as on the mitochondrial chromosome, resulting in 43,614 genes to use in the association analysis.
Genotypic Data
Genotyping was performed by using the Affymetrix GeneChip Human Mapping 500 K array set, and the imputation was carried out for HapMap II SNPs. Further details of genotype calling and the imputation can be found in Rueedi et al.18
Association Analysis
All statistical analyses were performed using Matlab.20 Urine metabolome features were transformed to be normally distributed, conserving their rank in order to remove strong outlier effects.
We used a linear regression model for each pair of (transformed) metabolome feature as the response variable and gene expression level as the explanatory variable. The model also included the following common confounding factors: age, sex, the first four principal components of the genotypic data (correcting for population stratification), and the first 50 principal components of the gene expression data (correcting for potential batch effects). We tested 1276 metabolome features for association with the expression of 19,123 protein coding and 24,491 non-coding genes. We decided to not apply any a priori exclusion criteria to remove genes from the analysis, as any such criterion would be arbitrary. Instead, for genes with significant associations, we evaluated the distribution of RPKM values to ensure close to normal distribution for accurate regression estimations. In particular, we kept genes if the maximum RPKM was larger than 1 or if RPKM values were larger than 0 for ≥5% of samples.
We applied a nominal Bonferroni threshold for multiple testing pmax = 0.05/(125 × 1109) = 3.6 × 10–7 by taking into account the effective number of independent tests which we estimated to be 125 for metabolome features and 1109 for genes (i.e., the number of principal components explaining more than 95% of the data, as proposed by Gao et al.21). Associations with p values below pmax were considered significant.
Metabomatching
Metabomatching is a method to identify metabolites underlying associations of SNPs with metabolome features.18,22 It compares the association profile of a given variable with all metabolome features across the full ppm range, so-called pseudospectrum, with NMR spectra of pure metabolites available in public databases such as HMDB.23
While metabomatching was originally developed to use SNP-metabolome associations, we recently showed that it can also identify metabolites based on co-varying features in NMR data.24 In the present study, we use metabomatching to identify metabolites that are associated with gene expression.
Mendelian Randomization
We performed Mendelian randomization (MR) analysis25,26 to assess the causal relationship between gene expression and metabolite concentration, using SNPs as instrumental variables (IVs), gene expression as exposure, and metabolome features as the outcome (or vice versa in the case of testing for reverse causality). For the MR analysis, we used mQTL and eQTL summary statistics from studies with greater statistical power, i.e., from untargeted mQTL study by Raffler et al.27 (n = 3861) and from the largest blood eQTL database eQTLGen Consortium28 (n = 31,684).
Causal effects were estimated by using the Wald method where the effect of a given genetic variant on the outcome is divided by the effect of the same genetic variant on the exposure.29 Next, ratio estimates from different instruments (SNPs) were combined using the inverse variance weighted method (IVW) to calculate the causal estimate.30
We selected significant SNPs from relevant eQTL studies as our IVs. To detect independent SNPs, we used a stepwise pruning approach where we first selected the strongest lead eQTL and then pruned the rest of the SNPs in a stepwise manner if they were correlated with the lead SNP (r2 > 0.2). We repeated the pruning process with the next available SNP until there were no SNPs left to prune. We used Cochran’s Q test to determine heterogeneity among the candidate IVs.31 Heterogeneous SNPs were removed in a stepwise manner from the model until the model did not show any more signs of heterogeneity (Cochran’s Q statistic p value >0.05/# of original instruments). We applied four different meta-analysis (MR inference) methods to evaluate the significance of the causal estimates: Inverse variance weighted, maximum-likelihood, weighted median, and MR-Egger. The latter two methods are robust methods that have more relaxed MR assumptions, can tolerate the violation of the exclusion-restriction assumption for some instruments, and are thus less sensitive to heterogeneity among IVs. When using these robust MR methods, we did not remove any heterogeneous instruments. For all MR analysis, we used the Mendelian randomization package implemented in R.32
Results
Association Analysis Identifies 20 Significant Metabolome- and Transcriptome-Wide Associations
We performed an untargeted metabolome- and transcriptome- wide association study by pairwise linear regression of each of 1276 metabolome features (as response variable) onto each of log-transformed expression levels of each of 43,614 genes (as explanatory variable) quantified in 555 healthy subjects (see Materials and Methods). Metabolome features resulted from binning raw urinary NMR spectra with a bin-size of 0.005 ppm, and rank-normalizing each bin passing quality control (see Materials and Methods). Gene expression levels were quantified as RPKM from RNAseq on lymphoblastoid cell lines derived for each subject (see Materials and Methods).
The QQ-plot of all pairwise associations (Figure 1) is well calibrated, with four highly significant associations (FDR < 0.05) (involving the ALMS1 and HPS1 genes). Applying an adjusted Bonferroni threshold of 3.6 × 10–7 to account for the effective number of independent variables (see Materials and Methods), we identified 20 additional less significant (“suggestive”) feature-gene associations. These 24 association pairs involved 19 unique genes and 21 unique features. As we did not apply any a priori exclusion criteria to remove genes from the analysis, we inspected the expression value distributions of these 19 significant genes in order to identify cases in which the small p value may be due to a problematic distribution of the expression values (see Materials and Methods). This resulted in four genes to be removed, corresponding to four significant association pairs (Figure S3). The remaining 20 gene-feature associations are listed in Table 1 and their distributions are presented in Figure S4.
Figure 1.
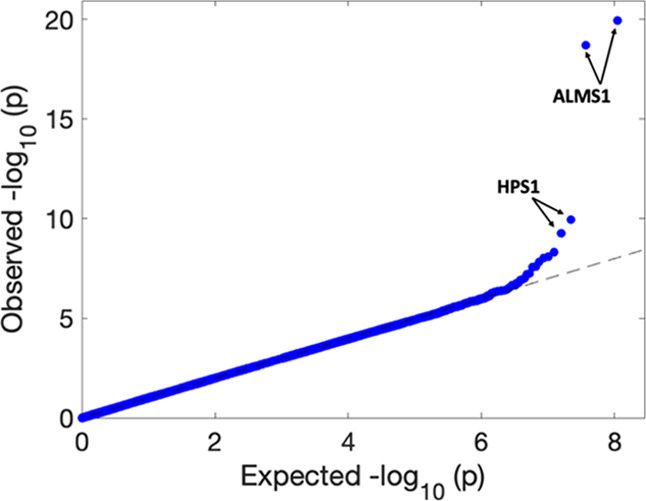
QQ-plot of −log10 (p values) of metabolome- and transcriptome-wide association analysis. The highly significant associations (FDR < 0.05) with ALMS1 expression are ranked 1st and 2nd and with HPS1 expression 3rd and 4th.
Table 1. 20 Study-Wide Significant Associations from Metabolome- and Transcriptome-Wide Association Analysisa.
| genes |
metabolite | association |
published as mGWAS | |||
|---|---|---|---|---|---|---|
| ensembl gene ID | Chr | gene symbol | feature(s) | effect size | p value | body fluid |
| ENSG00000116127 | 2 | ALMS1 | 2.0375, 2.0325, 2.0275, 2.0425 | 0.72, 0.69, 0.45, 0.41 | 1.1 × 10–20, 2.0 × 10–19, 7.8 × 10–09, 1.2 × 10–07 | serum,9,33 urine27,34 |
| ENSG00000107521 | 10 | HPS1 | 2.8575, 2.8725 | –0.38, −0.37 | 1.1 × 10–10, 5.6 × 10–10 | serum,9,33 urine27,34 |
| ENSG00000149089 | 11 | APIP | 2.7925 | –0.33 | 4.7 × 10–09 | serum,9,33 urine27 |
| ENSG00000256029 | 1 | RP11-190A12.7 | 3.0925 | 0.26 | 9.7 × 10–09 | serum9 |
| ENSG00000100603 | 14 | SNW1 | 8.1275 | –0.38 | 1.5 × 10–08 | serum33 |
| ENSG00000163016 | 2 | ALMS1P | 2.0325, 2.0375 | 0.27, 0.27 | 2.5 × 10–08, 2.7 × 10–08 | serum,9,33 urine27 |
| ENSG00000219257 | 6 | RP11-14I4.2 | 2.3275 | –0.25 | 5.7 × 10–08 | |
| ENSG00000259357 | 1 | RP11-316M1.12 | 7.7875 | 0.33 | 6.1 × 10–08 | |
| ENSG00000163520 | 3 | FBLN2 | 5.4375 | 0.29 | 9.5 × 10–08 | serum9,33 |
| ENSG00000226430 | 8 | USP17L7 | 2.7075 | –0.24 | 1.8 × 10–07 | |
| ENSG00000219355 | 12 | RPL31P52 | 2.8675 | –0.23 | 2.1 × 10–07 | |
| ENSG00000266795 | 17 | RP11-744K17.9 | 7.2725 | 0.26 | 2.2 × 10–07 | serum9 |
| ENSG00000150593 | 10 | PDCD4 | 5.4075 | 0.42 | 2.2 × 10–07 | serum,9,33 urine27 |
| ENSG00000266805 | 18 | RP11-61L19.1 | 5.3525 | 0.24 | 2.7 × 10–07 | |
| ENSG00000254396 | 9 | RP11-56F10.3 | 3.0925 | 0.23 | 3.6 × 10–07 | |
20 study-wide significant associations involving 15 unique genes and 17 unique features. Associations are grouped by genes and sorted by the lowest association p value for each gene.
Identification of Metabolites Corresponding to Metabolome Features Associated with Gene Expression
To identify metabolites underlying these significant associations between gene expression levels and metabolome features, we used metabomatching,18,22 which has been previously established as an effective tool for prioritizing candidate metabolites underlying profiles of SNP-metabolome feature associations, so-called pseudospectra.18,27 In this study, we used profiles of metabolome features that were associated significantly with one of the 15 identified genes as input to metabomatching.
We found that the pseudospectrum of the strongest associating gene, ALMS1, consists of four neighboring features at 2.0375 ppm (p value = 1 × 10–20), 2.0325 ppm (p value = 2 × 10–19), 2.0275 ppm (p value = 8 × 10–9), and 2.0425 ppm (p value = 1 × 10–7). A peak in this region is a typical feature of N-acetylated compounds (NACs) in their 1H NMR spectra35 but is also found in NMR spectra of other compounds (see Figure 2A). Although NAC was not ranked first by metabomatching, it is the only compound with the strongest peak at 2.0375 ppm, and the association with ALMS1 expression at the strongest peaks of other compounds is not very significant. We therefore believe that ALMS1 is likely associated with one or several NACs. To investigate if we could pinpoint a specific NAC, we built a library consisting of all NAC proton NMR spectra from HMDB and the Biological Magnetic Resonance Data Bank (BMRB). Metabomatching gave similar scores to many of these compounds (Figure S5). For the first ranked compound, acetylglycine, we would have expected a strong association of ALMS1 expression with its other peak at ∼3.7 ppm, which is not the case. In fact, the secondary strong association signal in the pseudospectrum of ALMS1 is at ∼1.7 ppm and this matches a resonance in the NMR spectra of N(alpha)-acetyl-dl-ornithine and N(alpha)-acetyllysine, making these two compounds likely candidates. N-acetyl-l-aspartate (NAA) is a further possible candidate due to matches of its NMR features at around 7.92 and 2.71 ppm (Figure S5) with an association signal at these positions in the ALMS1 pseudospectrum (p value = 4 × 10–3 and 4 × 10–2, respectively).
Figure 2.

Metabomatching22 results for pseudospectra derived from gene expression - metabolome feature associations for ALMS1 (A) and HPS1 (B). Upper panels show the features in each pseudospectrum, color-coded according to the direction of the effect (positive in blue and negative in orange). Lower panels show the highest ranking candidate metabolites with their reference NMR spectra (color coded to indicate their relative peak intensities). Leading features allowing metabolite identification are in (A) at 2.04 ppm, which matches well with the highest intensity peak of the NAA spectrum and in (B) at 2.87 ppm, which matches well with the TMA singlet.
The pseudospectrum of ALMS1P (ALMS1 pseudogene) similarly points to NAC (including N-acetylneuraminate and NAA) as likely matching compounds (Figure S6), although with less significant association p values (see Table 1).
The reference spectrum of NAA in the Urinary Metabolome Database (UMDB) that we used for metabomatching was recorded in water. In order to verify that the peaks of this spectrum are comparable to those of NAA in urine, we spiked NAA into pooled urine samples from our collection at a concentration of 10 mM and recorded its 1H NMR spectrum. Inspecting the 5 multiplet regions of NAA, we concluded that the NAA peak positions are very similar in both solvents (Figure S7).
Two associations, which are the third and fourth strongest in this study, are between HPS1 expression (second strongest associating gene) and two neighboring metabolome features at 2.8575 ppm (p value = 1 × 10–10) and 2.8725 ppm (p value = 6 × 10–10), respectively. This pseudospectrum matched well with the trimethylamine (TMA) NMR spectrum (Figure 2B). Among the top three metabolites suggested by metabomatching, trimethylamine (TMA) is the most plausible metabolite driving the association pattern because all the other metabolites have secondary peaks for which we have no significant association signal, while TMA has just a single peak in the 2.86 ppm region.
For the third strongest associating gene, APIP, metabomatching suggested asparagine as a metabolite (Figure S8), and the pseudospectrum of SNW1 (5th strongest associating gene) matched well with the single peak compound adenine (Figure S9). For the remaining 10 significantly associating genes, which were associated with only one metabolome feature, metabomatching was not able to identify a corresponding metabolite.
Validation of Significant Metabolome–Transcriptome Associations Using Follow-up Urine NMR Data
To the best of our knowledge, there is no independent data set with urine NMR spectra and gene expression data of LCLs of sufficient sample size for proper out-of-sample replication of our results. However, we generated additional NMR spectra from urine samples collected from a subset of 315 CoLaus subjects in a follow-up study conducted 5 years after the baseline data collection. We note that the follow-up NMR data are not independent from the baseline data, yet they were obtained from physically different samples collected at a significantly later time and processed with a different NMR spectrometer and facility. As for the expression data, we only have those from LCLs derived from blood taken at the baseline, so we could only test whether the associations we observed between baseline metabolomics and baseline transcriptomics measurements would persist as associations between follow-up metabolomics and baseline transcriptomics data.
As baseline and follow-up urine NMR data were each processed and binned individually, the features did not correspond one-to-one between the studies (see Materials and Methods). Thus, to validate the association of genes with relevant features, we tested the two nearest features to every top feature associated with a gene in the baseline data set. We used the Benjamini–Hochberg (BH) procedure36 to detect significant replications; more specifically, we considered replications validated if the p value was smaller than 0.05/(2 features x rank of the association in the discovery) for any one of the two tested features. In the follow-up, both ALMS1 and HPS1 gene expression levels were associated significantly with features corresponding to those of the discovery study (Table S1). The third most significantly associating gene with the baseline metabolome data, APIP, yielded an association in the follow-up data with p value = 9.8 × 10–3, which is just above its BH threshold of 0.005. All other genes did not show any significant association with representative features in the follow-up study.
mGWAS for NAC and TMA Indicates Numerous Significant SNPs in the ALMS1 and HPS1 Gene Loci
To get a broader overview of the genetic influences on metabolome features in the NAC and TMA NMR peak regions, we performed an untargeted metabolome- and genome-wide association study using data from 826 individuals of the CoLaus cohort, for whom baseline urinary NMR spectra were available (similar to Rueedi et al.18). SNPs that are significantly associated with the NAC metabolome feature at 2.0375 ppm are correlated with each other (r2 > 0.8) and lie within an LD block containing nine genes, including ALMS1, ALMS1-IT1, NAT8, and ALMS1P (Figure 3A). Out of these nine genes, ALMS1 and ALMS1P have the most significant association results with the 2.0375 ppm feature and with the NAA feature at 7.9225 ppm (Figure 3B). However, we note that the expression levels in LCLs differ between genes; in particular, NAT8 has RPKM = 0 in most samples, which could impact its association results. Similarly, SNPs that associate with the TMA metabolome feature at 2.8725 ppm are also highly correlated and lie in a locus surrounding the HPS1/PYROXD2 genes (Figure 4A). Even though the SNPs with the most significant association with feature 2.8725 are physically located closer to PYROXD2 rather than HPS1, the expression level of PYROXD2 does not show a significant association with this feature (Figure 4B). Notably here, HPSE2, MIR1287, and MIR4685 are very lowly expressed (RPKM = 0 in most samples).
Figure 3.

SNP - metabolome feature and SNP - gene expression associations in ALMS1/NAT8 locus. (A) LocusZoom plot for ALMS1/NAT8 locus, where the SNPs are associated with metabolome feature at 2.0375 ppm, LD colored with respect to lead mQTL. (B) Bar plot shows −log10 transformed p values from associating expression values of nine genes in the locus with the five NAA features.
Figure 4.
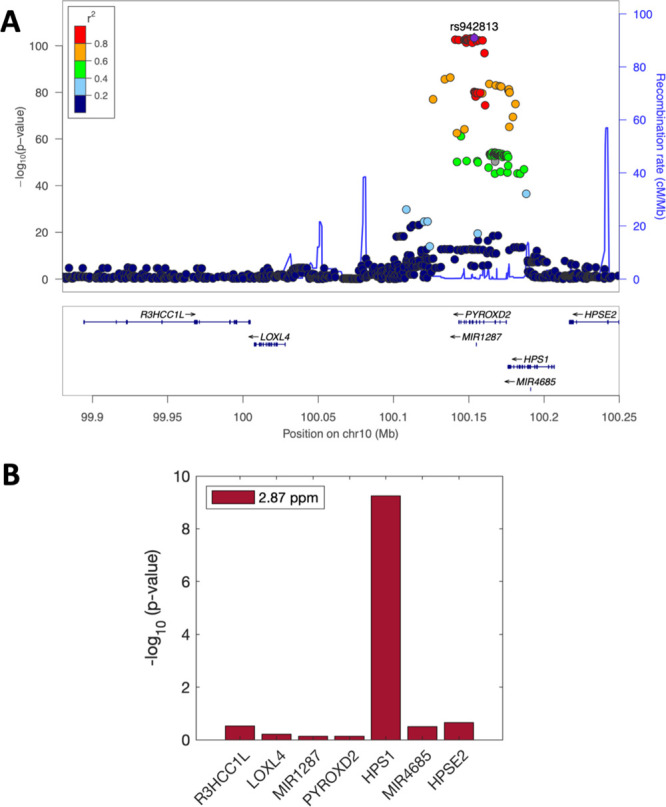
SNP - metabolome feature and SNP - gene expression associations in HPS1/PYROXD2 locus. (A) LocusZoom plot for HPS1/PYROXD2 locus, showing the association significance of SNP with the metabolome feature at 2.8725 ppm. Colors indicate the correlation (LD) to the lead QTL. (B) Bar plot shows −log10 transformed p values from associating expression values of seven genes in the locus with the same feature.
To further evaluate a possible regulation of NAC and TMA by other genes suggested by published mGWAS studies, we investigated the pseudospectra of these using metabomatching. In particular, we investigated genes that either were the target of an eQTL SNP that is a mQTL of NAC/TMA or were within 500 kb of ALMS1/HPS1. However, none of these candidate genes (14 for the ALMS1-NAC and 6 for the HPS1-TMA association pair) produced a pseudospectrum containing even a single nominally significant signal pointing to these metabolites (data not shown).
Inspecting published mGWAS in humans,37 we found that the SNPs in both ALMS1 and HPS1 loci have been previously reported to associate with a number of metabolic traits (Table 2). The ALMS1 locus has been associated with N-acetylated compounds, while the HPS1 locus has been associated with various metabolites including trimethylamine and dimethylamine.18,27,34 In mGWAS studies, the most likely candidate genes are usually inferred based on their physical proximity to the lead mQTL and, if available, their functional annotation. Indeed, published mGWAS studies were not able to distinguish between NAT8 or ALMS1 and HPS1 or PYROXD2 as genes affecting NAC and TMA, respectively. In contrast, our association study using gene expression data from LCLs clearly favors ALMS1 and HPS1 as the relevant genes. However, for NAT8, which is mostly not expressed in LCLs, conclusions on its association with NAC in functionally more relevant tissues are not possible.
Table 2. Previously Reported mGWAS Results for the ALMS1/NAT8 and HPS1/PYROXD2 Locia.
| reference | platform | biofluid | locus | metabolite |
|---|---|---|---|---|
| Nicholson et al. 201134 | MS + NMR | urine + plasma | ALMS1, NAT8 | N-acetylated compounds |
| Montoliu et al. 201344 | NMR | urine | ALMS1 | N-acetylated compounds |
| Rueedi et al. 201418 | NMR | urine | ALMS1 | 2.0375 (suggested as N-acetylated compounds) |
| Raffler et al. 201527 | NMR | urine | NAT8 | 2.031 (suggested as N-acetyl-l-aspartate) |
| Suhre et al. 201138 | MS | serum | NAT8 | N-acetylornithine |
| Yu et al. 201439 | MS | serum | NAT8 | N-acetylornithine |
| Shin et al. 201433 | MS | serum | NAT8 | N-acetyllysine, unknown compounds |
| Nicholson et al. 201134 | MS + NMR | urine + plasma | HPS1, PYROXD2 | trimethylamine (urine), dimethylamine (plasma), |
| Rueedi et al. 201418 | NMR | urine | PYROXD2 | trimethylamine, unknown compound, 2.8575, 1.8025 |
| Raffler et al. 201527 | NMR | urine | PYROXD2 | 2.854 (suggested as trimethylamine) |
| Raffler et al. 201340 | NMR | plasma | PYROXD2 | 2.757 |
| Rhee et al. 201341 | MS | plasma | HPS1 | asymmetric dimethylarginine |
| Krumsiek et al. 201242 | MS | serum | HPS1, PYROXD2 | multiple compounds, unknown compounds |
| Hong et al. 201343 | MS | serum | HPS1 | caprolactam |
| Shin et al. 201433 | MS | serum | PYROXD2 | unknown compounds |
MS: mass spectrometry; numbers in metabolite section refer to NMR spectral shift positions in ppm. Reported genes are mostly based on proximity to the mQTL or based on gene function.
A likely genetically driven relationship between ALMS1 and NAC concentrations measured in the baseline data set, through a shared eQTL and mQTL (SNP rs7566315), is illustrated in Figure 5.
Figure 5.
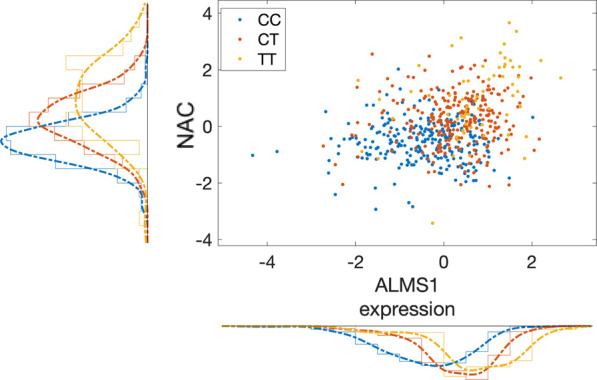
Scatter plot of the mQTL effect of SNP (rs7566315) on NAC and its eQTL effect on ALMS1 gene expression. Each point represents a study sample. NAC concentration is approximated by the feature at 2.0375 ppm that is log10 transformed after feature- and sample-wise z-scoring (y axis). ALMS1 expression is z-scored after log2 transforming RPKM+1 values (x axis). Color code represents the genotype of rs7566315 (legend) that is an eQTL of ALMS1 and mQTL of NAA.
Mendelian Randomization Analysis Suggests ALMS1 Expression in Blood and Confirms NAT8 Expression in Other Tissues to Causally Effect NAC Concentration
To assess a causal relationship between gene expression and metabolite concentration, we performed MR analysis using SNPs as instrumental variables (IVs) selected based on summary statistics from the eQTLGen Consortium28 and Raffler et al.27 for eQTL and untargeted mQTL results, respectively. We investigated both the causal effect of the gene expression on the metabolite concentration and vice versa for the ALMS1-NAC and HPS1-TMA gene-metabolite pairs.
We first investigated the causal effect of the ALMS1 gene on NAC concentration reflected by the NMR peak intensity at 2.0308 ppm.34,44 We considered 86 SNPs that were significant eQTLs (FDR < 0.05) in eQTLGen and that were also identified as mQTLs by Raffler et al. After stepwise pruning (see Materials and Methods), 14 independent SNPs remained as candidate IVs. Next, we performed Cochran’s Q test to detect heterogeneity among these SNPs and removed a further three, resulting in 11 SNPs as potentially valid IVs to use in the MR analysis (see Materials and Methods). Our causal effect estimates given by four meta-analysis methods (inverse variance weighted, weighted median, MR-Egger, maximum-likelihood; see Materials and Methods) are reported in Table 3A. All methods agree on the ALMS1 expression level being causal for NAC concentrations, indicated by p values < 2 × 10–16.
Table 3. MR Results for Testing a Causal Link between ALMS1 Expression and Concentration of N-Acetylated Compoundsa.
| method | causal effect size estimate | std. error | 95% CI | p value | Cochran’s Q-statistic p value | ||
|---|---|---|---|---|---|---|---|
| A | ALMS1 → NAC | inverse variance weighted | 0.967 | 0.061 | 0.847–1.087 | <2 × 10–16 | 0.2323 |
| weighted median | 1.111 | 0.075 | 0.965–1.257 | <2 × 10–16 | NA | ||
| MR - Egger | 0.994 | 0.092 | 0.812–1.175 | <2 × 10–16 | 0.1776 | ||
| maximum-likelihood | 0.999 | 0.065 | 0.872–1.126 | <2 × 10–16 | 0.249 | ||
| B | NAC → ALMS1 | inverse variance weighted | –0.015 | 0.264 | –0.532–0.502 | 0.955 | 0.7443 |
| weighted median | 0.122 | 0.321 | –0.507–0.751 | 0.704 | NA | ||
| MR - Egger | 1.495 | 1.976 | –2.377–5.368 | 0.449 | 0.7256 | ||
| maximum-likelihood | –0.015 | 0.266 | –0.535–0.505 | 0.955 | 0.7443 | ||
| C | ALMS1 → NAC | inverse variance weighted | 0.796 | 0.183 | 0.437–1.155 | <2 × 10–16 | 0.1902 |
| (NAT8 related SNPs removed) | weighted median | 0.668 | 0.242 | 0.193–1.142 | 0.006 | NA | |
| MR - Egger | 1.912 | 0.704 | 0.532–3.291 | 0.007 | 0.4144 | ||
| maximum-likelihood | 0.805 | 0.185 | 0.444–1.167 | < 2 × 10–16 | 0.2199 |
MR results for testing (A) causal effect of ALMS1 gene expression levels on N-acetylated compounds (ALMS1 → NAC), (B) causal effect of N-acetylated compounds on ALMS1 gene expression levels (NAC → ALMS1), (C) causal effect of ALMS1 gene expression levels on N-acetylated compounds (ALMS1 → NAC) when NAT8-related SNPs were removed from the instrument set.
As NAT8 has a known N-acetyltransferase activity and was previously suggested to be involved with NAC concentration (see Table 2), we performed an additional MR analysis with ALMS1 as the exposure where we removed the possible pleiotropic effect of the NAT8 gene. To achieve this, we excluded 10 out of 14 original candidate IVs that were also significant eQTLs of NAT8 or were in LD (R-squared > 0.1) with these NAT8 eQTLs (as there were no NAT8 eQTLs in eQTLGen, these were taken from GTEx consortium (v8), considering all available tissues2 and using a significance cutoff of p value < 10–7). Without these NAT8-related SNPs, the MR results remained significantly causal (Table 3C), indicating that the causal effect of ALMS1 on NAC may not be driven by the pleiotropic effect of NAT8.
Finally, we performed an additional sensitivity analysis by lowering the R-squared threshold in the stepwise LD pruning process to 0.05, resulting in more strictly independent IVs for MR analysis. The significant causal effect of ALMS1 on NAC persisted with more strictly independent instruments, indicated by all four meta-analysis methods (see Table S2, top panel). However, when removing NAT8-related eQTLs, only one meta-analysis method (weighted-median) indicated a significant causal effect of ALMS1 on NAC (see Table S2, bottom panel). We also performed an MR analysis with NAT8 as the exposure, but due to the low number of valid IVs (only two remained after removing two with heterogeneity), we could only use robust MR analysis methods. Both weighted median and MR-Egger indicated a significant causal effect of NAT8 expression on NAC (beta = 0.36 and 0.63, respectively; p values < 10–16). When excluding ALMS1-related SNPs, only two IVs remained and thus we could not test if the causal effect of NAT8 is independent of ALMS1 expression.
For the completeness of the analysis, we also tested a causal effect of NAC on the ALMS1 gene expression level. IVs were selected among the SNPs that were reported as significant mQTLs for the 2.03 ppm feature (p value < 1 × 10–6) in Raffler et al.27 Among the cis-eQTLs of ALMS1 from eQTLGen, we observed strong heterogeneity between their expected and observed effects. To overcome this problem, we sought to use also trans-eQTLs of ALMS1; however, none of the candidate IVs were measured in the trans-eQTL study of eQTLGen. As an alternative, we performed an association study between the candidate IVs and ALMS1 gene expression level as measured for our 555 CoLaus individuals and used these trans-eQTL results. Overall, we identified 26 overlapping significant mQTLs and trans-eQTLs, corresponding to six independent SNPs. Two of the six candidate IVs were removed due to pleiotropic effects resulting in four SNPs to be used as potentially valid IVs in the MR analysis (see Materials and Methods). None of the four meta-analysis methods (Table 3B) gave a significant causal effect estimate, indicating that we have no evidence for NAC concentration causally affecting ALMS1 gene expression.
In conclusion, our MR analysis using SNPs selected from eQTLs in blood tissue suggests a causal effect of ALMS1 expression levels on NAC, which appears independent from NAT8 gene expression (in various GTEx tissues). However, we also detected a significant causal effect of NAT8 expression (in GTex tissues) on NAC concentration.
Mendelian Randomization Analysis Suggests HPS1 as Causal Gene Modulating TMA Concentration and Indicates a Reverse Causal Effect between Both
We performed a similar MR analysis to test a causal relationship between HPS1 gene expression and TMA concentration. As there were no targeted summary statistics for TMA concentration, we used the NMR peak intensity at 2.8541 ppm from Raffler et al.27 as a proxy for TMA concentration. This position is in agreement with the singlet peak position range from 2.79 to 2.99 ppm, according to HMDB. Among the 77 SNPs that were reported as significant eQTLs for HPS1 (FDR < 0.05) in eQTLGen and measured by Raffler et al., only six could be used as valid IVs that were independent and did not exhibit heterogeneity (see Materials and Methods). Causal effects estimated by four different meta-analysis methods were all significant (p value < 0.005; see Table 4A), suggesting that HPS1 gene expression has a causal effect on TMA concentration.
Table 4. MR Results for Testing a Causal Link between HPS1 Expression and TMA Concentrationa.
| method | causal effect size estimate | std. error | 95% CI | p value | Cochran’s Q-statistic p value | ||
|---|---|---|---|---|---|---|---|
| A | HPS1 → TMA | inverse variance weighted | 0.266 | 0.094 | 0.082–0.450 | 0.005 | 0.0803 |
| weighted median | 0.311 | 0.072 | 0.170–0.453 | <2 × 10–16 | NA | ||
| MR - Egger | 0.37 | 0.126 | 0.123–0.617 | 0.003 | 0.0852 | ||
| maximum-likelihood | 0.267 | 0.094 | 0.083–0.452 | 0.004 | 0.0829 | ||
| B | TMA → HPS1 | inverse variance weighted | –0.089 | 0.012 | –0.113 to –0.065 | <2 × 10–16 | 0.0958 |
| weighted median | –0.09 | 0.011 | –0.111 to –0.068 | <2 × 10–16 | NA | ||
| MR - Egger | –0.086 | 0.013 | –0.111 to –0.061 | <2 × 10–16 | 0.0758 | ||
| maximum-likelihood | –0.09 | 0.012 | –0.114 to –0.066 | <2 × 10–16 | 0.1258 | ||
| C | HPS1 → TMA | inverse variance weighted | |||||
| (PYROXD2 related SNPs removed) | weighted median | 1.079 | 0.121 | 0.842–1.315 | <2 × 10–16 | NA | |
| MR - Egger | 1.705 | 0.305 | 1.107–2.303 | <2 × 10–16 | 0.5575 | ||
| maximum-likelihood |
MR results for testing (A) causal effect of HPS1 gene expression levels on TMA (HPS1 → TMA), (B) causal effect of TMA on HPS1 gene expression levels (TMA → HPS1), (C) causal effect of HPS1 gene expression levels on TMA (HPS1 → TMA) when PYROXD2-related SNPs were removed from the instrument set.
We performed an additional MR analysis removing the possible pleiotropic effect of the other candidate gene in the locus, PYROXD2, as this gene was previously suggested to be involved in modulating TMA (see Table 2). For this, we removed from the original 77 candidate IVs 54 SNPs that were also significant eQTLs of PYROXD2 (eQTLGen FDR < 0.05) or were in LD with these eQTLs (R-squared >0.1). Stepwise pruning with R-squared thresholds of 0.2 or 0.05 both resulted in 3 SNPs to be used in MR analysis. These 3 SNPs had heterogeneity; however, due to the low number of instruments, we could not further remove any of them. Therefore, we relied on robust MR methods. Both robust MR methods (weighted median and MR-Egger) indicated a significant causal effect of HPS1 on TMA (p value < 2 × 10–16) with estimated causal effect sizes larger than 1.0 (Table 4C). This suggests that the causal effect of HPS1 on TMA is not driven by a pleiotropic effect of PYROXD2. We also attempted an MR analysis testing the causal effect of PYROXD2 on TMA. However, when HPS1-related SNPs were discarded from the candidate IVs, there were no SNPs left for the analysis.
Next, we performed a sensitivity analysis using an R-squared threshold of 0.05 in stepwise LD pruning, which showed a persisting causal effect of HPS1 on TMA, when all HPS1 eQTLs were used as candidate SNPs in the MR analysis (p value < 2 × 10–16 for all four meta-analysis methods; see Table S3, upper panel), and when PYROXD2-related eQTLs were removed from the candidate SNPs (p value < 2 × 10–16 for robust meta-analysis methods; see Table S3, bottom panel).
Finally, we tested the causal effect in the reverse direction, from TMA concentration to HPS1 gene expression. From 87 significant mQTLs in Raffler et al.27 that were also measured in eQTLGen, 18 SNPs remained to be used as IVs in the MR analysis after stepwise pruning and removing the SNPs showing heterogeneity (see Materials and Methods). All four meta-analysis methods agreed on TMA concentration being causal for HPS1 expression (p value < 2 × 10–16; Table 4B). While the estimated causal effect size of HPS1 on TMA ranged between 0.27 and 0.37 for different methods, the causal effect size of TMA on HPS1 was around −0.09, pointing to the existence of a negative feedback loop.
Discussion
In this study, we present a metabolome- and transcriptome-wide association study using RNA-seq from LCLs and NMR urine profiles from 555 subjects of the CoLaus cohort. This is the first study performed on untargeted urine metabolome data from healthy individuals. We identified two genes, ALMS1 and HPS1, whose association with NMR features are highly significant, and 13 additional genes that are associated with metabolome features with lower but still significant p values (see Table 1). Among these 15 genes, 9 are in loci with SNPs that have been previously reported as mQTLs by mGWAS. This shows that our approach can identify likely candidates of metabolically relevant genes, despite a limited sample size.
To search for metabolite candidates underlying gene expression-metabolome feature associations, we used our metabomatching tool that ranks metabolites with known NMR peaks based on their match to a given pseudospectrum.22 We consider NAC as the most likely compounds to be associated with ALMS1 expression, even though NACs were not ranked top by metabomatching. Similarly, for HPS1, we consider TMA, which also did not rank top, as the most likely compound underlying the association with the NMR feature at ∼2.86 ppm. The reason for this was in both cases that the most significant association peaks of ALMS1 and HPS1 matched best with the highest peaks in the reference spectra of NAC and TMA, respectively. Thus, metabomatching was very useful in prioritizing candidate metabolites, but its scoring could be improved by weighting the association signal based on the relative peak height of the reference spectrum, which currently is only visualized.
Our top hit ALMS1 as well as the second strongest association involving HPS1 had previously been implicated by mGWAS linking their loci to compound families. However, in both cases, the reported mQTLs were also in proximity to other genes, leaving the causal gene-metabolite association ambiguous. Specifically, the locus associated through mGWAS with N-acetylated compounds (NAC) includes two genes, ALMS1 and NAT8,18,27,34,44 and the latter seemed to be an appealing candidate due to its known N-acetyltransferase activity. Yet, our association study linked NAC concentrations to ALMS1 and not NAT8 expression in LCLs. This is probably due to the fact that expression of NAT8 in LCLs is very low, so one would need to test its expression in tissues such as liver or kidney, where it is known to be highly expressed, which is however not feasible for a cohort study like CoLaus. A metabolic role of ALMS1 is supported through its known role in Alström syndrome characterized by metabolic deficits54 and kidney health disorder phenotypes.46 Interestingly, in the mGWAS reported by Montoliu et al. using data from a Brazilian cohort, the authors observed the association between NAC and the SNPs located in ALMS1/NAT8 locus with stronger SNP associations in the ALMS1 gene rather than NAT8.(44) They argued that the high ethnic diversity of their study population might have been responsible for breaking down the linkage disequilibrium in the ALMS1/NAT8 region of the genome, resulting in a stronger association for SNPs close to or within the ALMS1 gene compared to other studies.
Intriguingly, NAA, a likely NAC underlying the observed association, is the second most abundant metabolite in the brain and is involved in neural signaling by serving as a source of acetate for lipid and myelin synthesis in oligodendrocytes.47 NAA can be detected in urine in low concentrations,48 and it has a long history of being a surrogate marker of neural health and a broad measure of cognitive performance.49,50 Recently, it has been shown that NAA correlates with neuropsychological performance measures.51 The signals of SNPs in ALMS1 by GWAS with intellectual phenotypes such as a self-reported ability in mathematics52,53 might therefore be due to its role in modulating NAA. This conjecture of course assumes that NAA levels in relevant brain tissues reflect those in urine and that the ALMS1 expression variation and in particular its genetic component, in LCLs or blood, can serve as a proxy for brain tissue.
As for HPS1, our second strongest associating gene, we note that mGWAS previously associated its locus with TMA levels.18,27,34 Yet, most of these studies, including the aforementioned mGWAS using a Brazilian cohort,44 considered the PYROXD2 gene, which is in the same locus, as the most likely modulator of TMA concentrations due to its known function as pyridine nucleotide-disulfide oxidoreductase. While we cannot rule out that PYROXD2 is involved in TMA metabolism, our study finds no evidence for association of PYROXD2 expression levels in LCLs with TMA concentration in urine, indicating that the mQTLs of this locus may act predominantly as eQTLs through HPS1.
In order to study the causal relationship between gene expression and metabolite concentrations, we conducted Mendelian randomization analyses. Our analyses supported a causal relationship between ALMS1 expression in blood and NAC concentration in urine that appears independent of NAT8 expression. Yet, our MR analysis also confirmed a causal effect of NAT8 expression in other tissues (than LCLs or blood) on NAC concentration in urine. For HPS1, we observed a causal effect of its expression level in blood on TMA concentration in urine that appears independent of PYROXD2 expression in addition to a negative feedback loop between HPS1 expression levels and TMA concentrations. However, the causal links identified through MR analysis using mQTLs from urine and eQTLs mostly from blood may not represent very well the actual effects in the metabolically relevant tissues. Thus, based on the available data, MR analysis cannot unambiguously pinpoint the causal gene in the ALMS1/NAT8- and HPS1/PYROXD2-loci, whose expression drives the respective metabolite concentrations.
We acknowledge that our study has several limitations: First, the transcriptome data from LCLs may only poorly reflect the expression of genes in more functionally relevant cells and tissues. Furthermore, the mRNA expression levels may not correlate well with the protein concentrations of the enzymes involved in the metabolic reactions, as these are additionally shaped by translation regulation, post-translational modifications, or decay. Second, the metabolome data from urine may depend on food intake and may only provide a poor proxy for the metabolite concentrations in other relevant biosamples. Thus, relating LCL expression levels with urine metabolite concentrations might not allow detecting all regulations that occur in more functionally relevant tissues. This is likely the case for NAT8, which is barely expressed in LCLs but has high expression levels in, e.g., kidney and liver. Finally, our study is limited by the sample size of studied CoLaus data and by the available eQTLs and mQTLs, and the LD reference panel, used for MR. More highly powered studies are needed to provide a more definite answer on some casualties.
Nevertheless, our study shows a proof of concept for how global association of sets of two or more distinct molecular traits observed in the same cohort can facilitate new discoveries of how different molecular entities affect each other. Such analyses, when applied to sizable datasets, will be instrumental in unravelling the complex regulatory relationships underlying human metabolism.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant FN 310030_152724/1) and the NIH (grant R03 CA211815).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00585.
Figure S1: Principal component analysis of baseline and follow-up metabolomics data; Figure S2: Overview of RNA-Seq read count and quantifiable genes in 555 individuals; Figure S3: Scatter plots of removed associations; Figure S4: Scatter plots of 20 study-wide significant metabolome feature - gene expression associations; Figure S5: Metabomatching figure showing the pseudospectrum derived from ALMS1 gene expression - metabolome features associations; Figure S6: Metabomatching figure showing the pseudospectrum derived from ALMS1P gene expression - metabolome features associations; Figure S7: NMR profiles of 3 different spike-in experiments; Figure S8: Metabomatching figure showing the pseudospectrum derived from APIP gene expression - metabolome feature associations; Figure S9: Metabomatching figure showing the pseudospectrum derived from SNW1 gene expression - metabolome feature associations; Table S1: Validation of all associations discovered in CoLaus baseline; Table S2: MR results of testing causal effect of ALMS1 gene expression levels on N-acetylated compounds, using R-squared threshold of 0.05; Table S3: MR results of testing causal effect of HPS1 gene expression levels on trimethylamine, using R-squared threshold of 0.05 (PDF)
Author Contributions
⊥ A.B. and S.B. are equal last authors.
Author Contributions
R.S.F., R.R., and S.B. designed the project. R.S.F. carried out the computational analysis and prepared the results, with help from A.B. B.K. and R.R. provided data and feedback on the metabolomics analysis. Z.K. designed and supervised the MR analysis. All authors discussed the results and provided feedback on the manuscript that was written by R.S.F., B.K., A.B., and S.B.
The authors declare no competing financial interest.
Notes
Processed transcriptome and metabolome data are accessible at Zenodo (www.zenodo.org, doi:10.5281/zenodo.5106119). This data set includes RPKM levels in LCLs for 45,484 genes and 555 individuals, binned and normalized NMR peak intensities for 1276 bins in 555 individuals from baseline urine samples, and 1289 bins in 315 individuals from follow-up urine samples.
Supplementary Material
References
- Buniello A.; MacArthur J. A. L.; Cerezo M.; Harris L. W.; Hayhurst J.; Malangone C.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M. T.; Humbert R.; Rynes E.; Thurman R. E.; Haugen E.; Wang H.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae D. L.; Gamazon E.; Zhang W.; Duan S.; Dolan M. E.; Cox N. J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010, 6, e1000888. 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward L. D.; Kellis M. Interpreting non-coding variation in complex disease genetics. Nat. Biotechnol. 2012, 30, 1095. 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones L. R.; Holloway A.; McRae A.; Yang J.; Small K.; Zhao J.; et al. The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 2017, 100, 228–237. 10.1016/j.ajhg.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S. B.; Sammeth M.; Gutierrez-Arcelus M.; Lach R. P.; Ingle C.; Nisbett J.; et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 2010, 464, 773. 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. A.; Sullivan P. F.; Brooks A. I.; Zou F.; Sun W.; Xia K.; et al. Heritability and genomics of gene expression in peripheral blood. Nat. Genet. 2014, 46, 430. 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K.; Wallaschofski H.; Raffler J.; Friedrich N.; Haring R.; Michael K.; et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011, 43, 565–569. 10.1038/ng.837. [DOI] [PubMed] [Google Scholar]
- Bartel J.; Krumsiek J.; Schramm K.; Adamski J.; Gieger C.; Herder C.; et al. The Human Blood Metabolome-Transcriptome Interface. PLoS Genet. 2015, 11, e1005274. 10.1371/journal.pgen.1005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt R.; Kirsten H.; Beutner F.; Holdt L. M.; Gross A.; Teren A.; et al. Integration of Genome-Wide SNP Data and Gene-Expression Profiles Reveals Six Novel Loci and Regulatory Mechanisms for Amino Acids and Acylcarnitines in Whole Blood. PLoS Genet. 2015, 11, e1005510. 10.1371/journal.pgen.1005510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye M.; Kettunen J.; Soininen P.; Silander K.; Ripatti S.; Kumpula L. S.; Hämäläinen E.; Jousilahti P.; Kangas A. J.; Männistö S.; Savolainen M. J.; Jula A.; Leiviskä J.; Palotie A.; Salomaa V.; Perola M.; Ala-Korpela M.; Peltonen L. Metabonomic, transcriptomic, and genomic variation of a population cohort. Mol. Syst. Biol. 2010, 6, 441. 10.1038/msb.2010.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullaughey K.; Chavarria C. I.; Coop G.; Gilad Y. Expression quantitative trait loci detected in cell lines are often present in primary tissues. Hum. Mol. Genet. 2009, 18, 4296–4303. 10.1093/hmg/ddp382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çalışkan M.; Cusanovich D. A.; Ober C.; Gilad Y. The effects of EBV transformation on gene expression levels and methylation profiles. Hum. Mol. Genet. 2011, 20, 1643–1652. 10.1093/hmg/ddr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas A. S.; Deutsch S.; Stranger B. E.; Montgomery S. B.; Borel C.; Attar-Cohen H.; et al. Common regulatory variation impacts gene expression in a cell type–dependent manner. Science 2009, 325, 1246–1250. 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J.; Gudjonsson J. E.; Liang L.; Stuart P. E.; Li Y.; Chen W.; et al. Gene expression in skin and lymphoblastoid cells: Refined statistical method reveals extensive overlap in cis-eQTL signals. Am. J. Hum. Genet. 2010, 87, 779–789. 10.1016/j.ajhg.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmann M.; Mayor V.; Vidal P. M.; Bochud M.; Pécoud A.; Hayoz D.; et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc. Disord. 2008, 8, 6. 10.1186/1471-2261-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueedi R.; Ledda M.; Nicholls A. W.; Salek R. M.; Marques-Vidal P.; Morya E.; et al. Genome-wide association study of metabolic traits reveals novel gene-metabolite-disease links. PLoS Genet. 2014, 10, e1004132. 10.1371/journal.pgen.1004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Sola S.; Sammeth M.; Guigó R.; Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185. 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- MATLAB 8.5.0.197613 (R2015a); The MathWorks Inc.: Natick, Massachusetts, 2015. [Google Scholar]
- Gao X.; Starmer J.; Martin E. R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. 10.1002/gepi.20310. [DOI] [PubMed] [Google Scholar]
- Rueedi R.; Mallol R.; Raffler J.; Lamparter D.; Friedrich N.; Vollenweider P.; et al. Metabomatching: Using genetic association to identify metabolites in proton NMR spectroscopy. PLoS Comput. Biol. 2017, 13, e1005839. 10.1371/journal.pcbi.1005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Marcu A.; Guo A. C.; Liang K.; Vázquez-Fresno R.; et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili B.; Tomasoni M.; Mattei M.; Parera R. M.; Sonmez R.; Krefl D.; et al. Automated analysis of large-scale NMR data generates metabolomic signatures and links them to candidate metabolites. J. Proteome Res. 2019, 613935. 10.1021/acs.jproteome.9b00295. [DOI] [PubMed] [Google Scholar]
- Burgess S.; Small D. S.; Thompson S. G. A review of instrumental variable estimators for Mendelian randomization. Stat. Methods Med. Res. 2017, 26, 2333–2355. 10.1177/0962280215597579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey Smith G.; Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?. Int. J. Epidemiol. 2003, 32, 1–22. 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- Raffler J.; Friedrich N.; Arnold M.; Kacprowski T.; Rueedi R.; Altmaier E.; et al. Genome-wide association study with targeted and non-targeted NMR metabolomics identifies 15 novel loci of urinary human metabolic individuality. PLoS Genet. 2015, 11, e1005487. 10.1371/journal.pgen.1005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Võsa U.; Claringbould A.; Westra H.-J.; Bonder M. J.; Deelen P.; Zeng B.; Kirsten H.; Saha A.; Kreuzhuber R.; Brugge H.; et al. Large-scale cis-and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald A. The fitting of straight lines if both variables are subject to error. Ann. Math. Stat. 1940, 11, 284–300. 10.1214/aoms/1177731868. [DOI] [Google Scholar]
- Hartung J.; Knapp G.; Sinha B. K.; Sinha B. K.. Statistical meta-analysis with applications; Wiley: New York, 2008. [Google Scholar]
- Greco M. F. D.; Minelli C.; Sheehan N. A.; Thompson J. R. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat. Med. 2015, 34, 2926–2940. 10.1002/sim.6522. [DOI] [PubMed] [Google Scholar]
- Yavorska O. O.; Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 2017, 46, 1734–1739. 10.1093/ije/dyx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.-Y.; Fauman E. B.; Petersen A.-K.; Krumsiek J.; Santos R.; Huang J.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543. 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G.; Rantalainen M.; Li J. V.; Maher A. D.; Malmodin D.; Ahmadi K. R.; et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 2011, 7, e1002270. 10.1371/journal.pgen.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke U. F.; Liebrand-van Sambeek M. L.; De Jong J. G.; Leroy J. G.; Morava E.; Smeitink J. A.; et al. N-acetylated metabolites in urine: proton nuclear magnetic resonance spectroscopic study on patients with inborn errors of metabolism. Clin. Chem. 2004, 50, 58–66. 10.1373/clinchem.2003.020214. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Royal Stat. Soc.: B 1995, 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Kastenmüller G.; Raffler J.; Gieger C.; Suhre K. Genetics of human metabolism: an update. Hum. Mol. Genet. 2015, 24, R93–R101. 10.1093/hmg/ddv263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K.; Shin S. Y.; Petersen A. K.; Mohney R. P.; Meredith D.; Wägele B.; et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011, 477, 54–60. 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Zheng Y.; Alexander D.; Morrison A. C.; Coresh J.; Boerwinkle E. Genetic determinants influencing human serum metabolome among African Americans. PLoS Genet. 2014, 10, e1004212 10.1371/journal.pgen.1004212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffler J.; Römisch-Margl W.; Petersen A. K.; Pagel P.; Blöchl F.; Hengstenberg C.; et al. Identification and MS-assisted interpretation of genetically influenced NMR signals in human plasma. Genome Med. 2013, 5, 13. 10.1186/gm417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee E. P.; Ho J. E.; Chen M. H.; Shen D.; Cheng S.; Larson M. G.; et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013, 18, 130–143. 10.1016/j.cmet.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumsiek J.; Suhre K.; Evans A. M.; Mitchell M. W.; Mohney R. P.; Milburn M. V.; et al. Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 2012, 8, e1003005. 10.1371/journal.pgen.1003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M. G.; Karlsson R.; Magnusson P. K.; Lewis M. R.; Isaacs W.; Zheng L. S.; et al. A Genome-Wide Assessment of Variability in Human Serum Metabolism. Hum. Mutat. 2013, 34, 515–524. 10.1002/humu.22267. [DOI] [PubMed] [Google Scholar]
- Montoliu I.; Genick U.; Ledda M.; Collino S.; Martin F.-P.; le Coutre J.; et al. Current status on genome–metabolome-wide associations: an opportunity in nutrition research. Gene Nutr. 2013, 8, 19. 10.1007/s12263-012-0313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J. C.; Zhang W.; Lord G. M.; Van Der Harst P.; Lawlor D. A.; Sehmi J. S.; et al. Genetic loci influencing kidney function and chronic kidney disease. Nat. Genet. 2010, 42, 373–375. 10.1038/ng.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons M. L.; Frondoza C. G.; Coyle J. T. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neuroscience 1991, 45, 37–45. 10.1016/0306-4522(91)90101-S. [DOI] [PubMed] [Google Scholar]
- Masaharu M.; Hideo M.; Mutsuhiko M.; Yasuo K. N-acetyl-l-aspartic acid, N-acetyl-α-l-aspartyl-l-glutamic acid and β-citryl-l-glutamic acid in human urine. Clin. Chim. Acta 1982, 120, 119–126. 10.1016/0009-8981(82)90082-1. [DOI] [PubMed] [Google Scholar]
- Barker P. B. N-acetyl aspartate—a neuronal marker?. Ann. Neurol. 2001, 49, 423–424. 10.1002/ana.90. [DOI] [PubMed] [Google Scholar]
- Jung R. E.; Brooks W. M.; Yeo R. A.; Chiulli S. J.; Weers D. C.; Sibbitt W. L. Jr. Biochemical markers of intelligence: a proton MR spectroscopy study of normal human brain. Proc. R. Soc. London, Ser. B 1999, 266, 1375–1379. 10.1098/rspb.1999.0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T.; Blyth J. C.; Griffiths G.; Kelly D.; Talcott J. B. Moderate relationships between NAA and cognitive ability in healthy adults: implications for cognitive spectroscopy. Front. Hum. Neurosci. 2014, 8, 39. 10.3389/fnhum.2014.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies G.; Lam M.; Harris S. E.; Trampush J. W.; Luciano M.; Hill W. D.; et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun. 2018, 9, 2098. 10.1038/s41467-018-04362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. J.; Wedow R.; Okbay A.; Kong E.; Maghzian O.; Zacher M.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn T. ALMS1 and Alström syndrome: a recessive form of metabolic, neurosensory and cardiac deficits. J. Mol Med. 2019, 97, 1–7. 10.1007/s00109-018-1714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.