

Abstract
Aims
Whereas intravenous administration of Toll‐like receptor 4 ligand lipopolysaccharide (LPS) to human volunteers is frequently used in clinical pharmacology studies, systemic use of LPS has practical limitations. We aimed to characterize the intradermal LPS response in healthy volunteers, and as such qualify the method as local inflammation model for clinical pharmacology studies.
Methods
Eighteen healthy male volunteers received 2 or 4 intradermal 5 ng LPS injections and 1 saline injection on the forearms. The LPS response was evaluated by noninvasive (perfusion, skin temperature and erythema) and invasive assessments (cellular and cytokine responses) in skin biopsy and blister exudate.
Results
LPS elicited a visible response and returned to baseline at 48 hours. Erythema, perfusion and temperature were statistically significant (P < .0001) over a 24‐hour time course compared to saline. The protein response was dominated by an acute interleukin (IL)‐6, IL‐8 and tumour necrosis factor response followed by IL‐1β, IL‐10 and interferon‐γ. The cellular response consisted of an acute neutrophil influx followed by different monocyte subsets and dendritic cells.
Discussion
Intradermal LPS administration in humans causes an acute, localized and transient inflammatory reaction that is well‐tolerated by healthy volunteers. This may be a valuable inflammation model for evaluating the pharmacological activity of anti‐inflammatory investigational compounds in proof of pharmacology studies.
Keywords: drug development, endotoxin, healthy volunteers, inflammatory model, intradermal LPS
What is already known about this subject
Human intravenous lipopolysaccharide (LPS) challenge is a valuable tool for clinical pharmacology studies.
Intradermally injected UV‐killed Escherichia coli is an important dermal inflammatory model, but it drives inflammation through multiple pathways, including the LPS‐induced Toll‐like receptor 4 pathway.
A local LPS challenge model would greatly contribute to translational comprehension and enable accelerated drug development.
What this study adds
The response to intradermal LPS was objectively quantified by several different methodologies (noninvasive and invasive).
Intradermal LPS administration in healthy volunteers evokes an acute and transient inflammatory response that is safe and well tolerated.
1. INTRODUCTION
Inflammation is a response to damaged tissue or pathogens resulting in the release of inflammatory mediators and cellular activation. Although inflammation is a physiological process, an excessive or poorly regulated inflammatory response can be harmful to the host, which is the case in inflammatory disorders. 1 To study the inflammatory process in humans and the effect of potential anti‐inflammatory drugs, several challenge models have been developed, of which systemic lipopolysaccharide (LPS) administration has been used for decades. 2 , 3 LPS, also known as endotoxin, are large molecules found on the outer membrane of Gram‐negative bacteria such as Escherichia coli and Salmonella. Recognition of LPS by Toll‐like receptor 4 (TLR4) on myeloid cells leads to MyD88‐ and TRIF‐dependent signalling resulting in the secretion of proinflammatory cytokines and type I interferons. The challenge response is characterized by a significant but transient cytokine release, and a clinical response that can include headache, fever, myalgia and tachycardia. 4 Intravenous LPS challenge has several limitations: (i) it only supports the evaluation of systemic anti‐inflammatory investigational compounds; (ii2) it leads to a prolonged state of immunological hypo‐responsiveness to LPS due to systemic innate memory, hampering the possibility of a repeated challenge in the same individual 5 , 6 ; and (iii) it results in a systemic inflammatory response in which it is difficult or impossible to assess the contribution of stromal tissues. 7
A local innate immune challenge in humans could in theory overcome all three limitations mentioned above. Local inflammatory models are mainly limited to inflammation induced in the lungs 7 or skin, such as the UV‐B model driving inflammatory hyperalgesia 8 or the reversible human skin inflammation model based on topical imiquimod application. 9 In 2016 Motwani et al. published a novel dermal inflammatory model with UV‐killed E. coli (UVEk), showing that intradermal administration of UVEk resulted in an acute localized and transient inflammatory reaction, which included inflammatory cell recruitment and cytokine release. 10 This model comprises TLR4 agonist LPS as well as other pathogen associated damage molecules including TLR5 agonist flagellin, 11 TLR9 agonist unmethylated CpG dinucleotides 12 and many more—even noncharacterized—components. While UVEk mimics real‐life bacterial infection including broad activation of the innate immune response and in situ phagocytosis of bacterial particles, it is less suitable for evaluating TLR4‐specific inflammation. LPS is a TLR4‐specific ligand, and, importantly, is commercially available within a quality framework that is required for clinical pharmacology trials.
Basran et al. 13 described the inflammatory response following intradermal LPS injection at doses up to 15 ng per injection. LPS administration induced a local inflammatory response, characterized by a strong neutrophil attraction and the production of interleukin (IL)‐8 and IL‐1β. We used this study as a foundation for a more extensive characterization of the local LPS‐driven inflammatory response in healthy volunteers. We integrated imaging endpoints (local perfusion, temperature, erythema), cellular responses (immune cell attraction), and proteins (cytokines and chemokines), and evaluated the time course of the response in greater detail. Moreover, we collected biopsies and suction blister exudate of the LPS‐inflamed regions, allowing bioanalytical comparison of both matrices, with the overall aim to characterize the intradermal LPS model for future use in clinical pharmacology trials.
2. MATERIALS AND METHODS
This study was conducted from August 2018 to October 2018 at the Centre for Human Drug Research and according to the Dutch Act on Medical Research involving Human Subjects. The study protocol (registered at ToetsingOnline, number: NL65297.056.18) was approved by a Medical Ethics Committee (Stichting Beoordeling Ethiek Biomedisch Onderzoek, Assen, The Netherlands) prior to the start of the clinical phase. Subjects gave written informed consent before any study related procedures were undertaken.
2.1. Study design and subjects
This was an open‐label, saline‐controlled study. In total, 18 nonsmoking healthy males (Fitzpatrick skin type I–III), aged 18–45 years, were included. Our aim was to minimize intersubject variability, therefore only males were included due to known sex differences in response to LPS exposure in vivo and in vitro. 14 , 15 Subjects with any disease associated with immune system impairment, including autoimmune diseases were excluded. To explore the effect of intradermal LPS vs. saline, subjects received either 2 (subjects 1–6) or 4 (subjects 7–18) intradermal LPS injections of 5 ng LPS/50 μL saline per injection and 1 50‐μL saline injection. Subjects 1–6 who received 2 intradermal LPS injections on 1 arm also contributed to baseline biopsy and blister exudate on their contralateral arm. All injections were placed on the volar forearm as depicted in Figure S1.
2.2. Skin assessments
The skin was assessed predose and at 3, 6, 10, 24 and 48 hours after LPS administration and up to 24 hours after saline administration. Erythema was assessed by multispectral imaging (Antera 3D, Miravex, Dublin, Ireland), perfusion by laser speckle contrast imaging (LSCI; PeriCam PSI System, Perimed Jäfälla, Sweden) and temperature by thermography camera FLIR X6540sc (FLIR Systems Inc., Wilsonville, OR, USA). The performance and analysis of the noninvasive measurements were standardized for all subjects. At the indicated time‐points (Figure S1) a suction blister was made over the marked injection site or untreated (baseline) area, or a 3‐mm skin punch biopsy was taken. The induction of 10‐mm suctions blisters was performed according to the method published by Motwani et al. 10 The performance of a suction blister or skin punch biopsy disqualified that area for further follow‐up with noninvasive measurements.
2.3. Suction blisters
Blister exudate was collected in a V‐bottom plate containing 50 μL 3% sodium citrate (Sigma‐Aldrich, St. Louis, MO, USA) in phosphate‐buffered saline (PBS; Gibco, Thermo Fischer Scientific, Waltham, MA, USA) and kept on ice. The plate was centrifuged, and supernatant was weighed to estimate the volume and then frozen at −80°C for cytokine analysis (Meso Scale Discovery, Rockville, MD, USA). The following cytokines were analysed: IL‐1β, IL‐6, IL‐8, IL‐10, interferon (IFN)‐γ and tumour necrosis factor (TNF). The pellet was resuspended in RoboSep buffer (Stemcell Technologies, Vancouver, Canada). A cocktail of fluorescent antibodies for cell surface markers were added to the cells and incubated for 30 minutes on ice. Stained samples were washed with PBS and measured with a MACSQuant 10 (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Flow cytometry data were analysed with Flowlogic 7.1 (Inivai Technologies, Mentone, VIC, Australia). Parallel to the blister exudate, peripheral blood was collected by venipuncture in a sodium heparin vacutainer (BD, Franklin Lakes, NJ, USA). 100 μL whole blood was treated with red blood cell lysis buffer (eBioscience, Thermo Fischer Scientific) and washed with PBS and resuspended in RoboSep buffer. Staining was similar to previously mentioned blister cells. The following antibodies were used: CD4 PerCP (clone OKT4, cat# 317432; BioLegend, San Diego, CA, USA); CD8 BV510 (clone SK1, cat# 344732; BioLegend); CD56 PE‐Cy7 (clone MEM‐188, cat# 304628; BioLegend); CD14 BV421 (clone M5E2, cat# 301830; BioLegend); CD16 APC‐Cy7 (clone 3G8, cat# 302018; BioLegend); CD66b AF700 (clone G10F5, cat# 305114; BioLegend); CD19 FITC (clone HIB19, cat# 302206; BioLegend); CD20 FITC (clone 2H7, cat# 302304; BioLegend); and HLA‐DR PE (clone REA805, cat# 130–111‐789; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). For an overview of the gating strategy used see Figure S2. Cell populations were classified based on the following profile: HLA‐DR‐ CD66b + CD16 + neutrophils; HLA‐DR + CD14 + CD16‐ classical monocytes; HLA‐DR + CD14 + CD16 + intermediate monocytes; HLA‐DR + CD14‐ CD16 + nonclassical monocytes; HLA‐DR + CD14‐ CD16‐ dendritic cells; HLA‐DR‐ CD56 + NK cells; HLA‐DR‐ CD4 + T helper cells; HLA‐DR‐ CD8 + cytotoxic T cells; and HLA‐DR + CD19 + CD20 + B cells. An overview of the different subsets is in Table S1.
2.4. Skin punch biopsies
Skin punch biopsies (3 mm) were collected after local anaesthesia and immediately snap frozen using liquid nitrogen as previously described. 16 The biopsies were stored at −80°C until analysis. Immunohistochemistry was performed for the following targets: CD1a dendritic cells (Clone EP3622; Cell Marque Sigma‐Aldrich); CD4 T cells (Clone SP35; Ventana, Roche Diagnostics, Rotkreuz, Switzerland); CD8 T cells (Clone SP57; Ventana, Roche Diagnostics); CD14 monocytes (Clone EPR3653; Cell Marque Sigma‐Aldrich); CD19 B cells (clone LE‐CD19; Thermo Fischer Scientific) and myeloperoxidase (MPO) neutrophils (polyclonal 760–2659; Cell Marque Sigma‐Aldrich). The slides were scored qualitatively on a 6‐point scale (negative, minimal, few, moderate, many or excessive) by a blinded dermato‐pathologist. The remaining tissue was used for the determination of IFN‐γ, IL‐1β, IL‐6, IL‐8, IL‐10, MxA and TNF mRNA expression relative to the housekeeping gene ABL by quantitative polymerase chain reaction. An overview of the primers used for quantitative polymerase chain reaction can be found in Table S2.
2.5. Safety
Safety and tolerability were monitored by tracking adverse events, measuring vital signs, and standard laboratory tests (i.e. haematology) at 3, 6, 10, 24 and 48 hours after LPS administration. Circulating cytokines (IL‐1β, IL‐6, IL‐8, IL‐10, IFN‐γ and TNF; Meso Scale Discovery, Rockville, Maryland, USA) were measured in blood samples to detect a possible systemic effect of intradermal LPS administration. The blood samples for cytokine analysis were analysed in 2 batches with each having a different dilution, therefore the lower limit of quantitation (LLOQ) of the samples was the following: IL‐1β 0.298 or 0.745 pg/mL; IL‐6 1.52 or 3.81 pg/mL; IL‐8 1.25 or 3.12 pg/mL; IL‐10 0.702 or 1.76 pg/mL; IFN‐γ 10 or 25 pg/mL; and TNF 0.760 or 1.90 pg/mL.
2.6. Statistics
The sample size of 6 data points per time point for the invasive assessments (suction blister/skin punch biopsy) was based on the paper published by Motwani et al., showing a robust clinical, cellular and molecular inflammatory response to intradermally injected UV‐killed E. coli. 10 Due to practical limitations, the invasive assessments for the saline injection were not performed at all time points and consisted of 3 data points per time point. Data repeatedly measured at the injection site were analysed with a mixed model analysis of variance with fixed factors injection (LPS or saline), time and injection by time, random factors subject, subject by injection and subject by time, with the average prevalue as covariate. The following contrast was calculated within the model: LPS vs. saline up to 24 hours. All calculations were performed using SAS for windows V9.4 (SAS Institute, Inc., Cary, NC, USA).
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 17
3. RESULTS
3.1. Clinical response
To study the classical hallmarks of inflammation (i.e. heat and erythema) the injection site was evaluated by thermography (skin temperature), laser speckle contrast imaging (perfusion) and a multispectral camera (erythema). Three hours after LPS/saline injection a clear inflammatory response was observed at the site of LPS injection marked by erythema (Figure 1A and E), increase in skin perfusion (Figure 1D) and increase in skin temperature (Figure 1C). At 24 hours all 3 parameters peaked with means ± standard deviation of 0.497 AU ± 0.19 (LPS) vs. 0.084 AU ± 0.11 (saline) for erythema, 46.83 AU ± 14.54 (LPS) vs. 0.23 AU ± 3.95 (saline) for perfusion, and 0.71°C ± 0.37 (LPS) vs. 0.09°C ± 0.41 (saline) for control corrected increase in skin temperature. For all 3 parameters the contrast between LPS and saline was calculated up to 24 hours postinjection and was highly significant (P < .0001). The temperature plateaued from 3 to 10 hours, but peaked at 24 hours together with perfusion and erythema (Figure 1C–E). All 3 parameters declined after 24 hours and almost reached baseline levels again after 48 hours (Figure 1C–E). In contrast, saline injected skin showed no or minimal change over a 24‐hour time course (Figure 1C–E).
FIGURE 1.
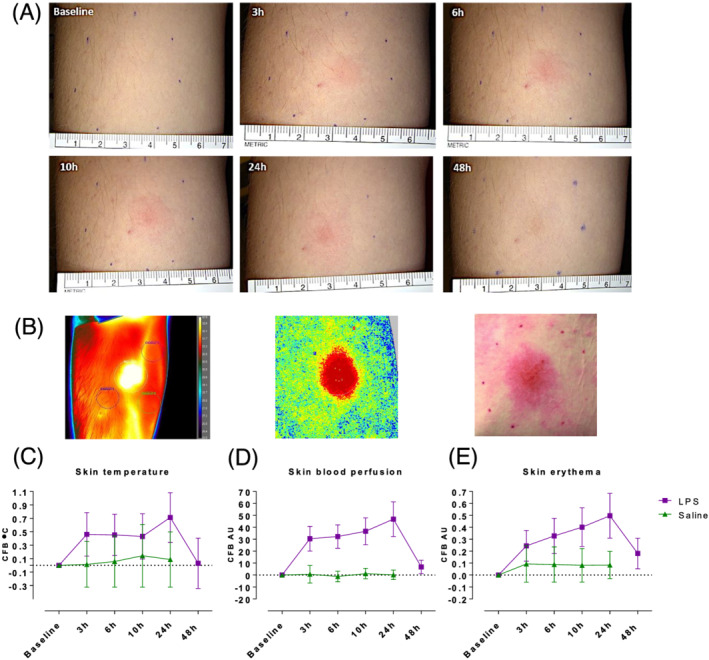
The clinical response after 5 ng lipopolysaccharide (LPS) vs. saline injection. At baseline and at the indicated time‐points post‐LPS or saline injection, skin assessments were performed quantifying temperature, perfusion and erythema. (A) Representative images from standardized photography at the indicated time‐points. (B) Representative images from thermography, laser speckle contrast imaging and erythema photo analysis from LPS injected skin at 24 hours post‐LPS injection. (C–E) LPS increased skin temperature (thermography), blood perfusion (laser speckle contrast imaging) and erythema (photo analysis) in the skin compared to saline injected skin. All data are expressed as change from baseline, means ± standard deviation. For all 3 parameters the contrast between LPS and saline was calculated using mixed model analysis of variance up to 24 hours postinjection and was highly significant (P < .0001)
3.2. Cellular response
The cellular response to intradermal LPS was studied by raising a suction blister or by taking 3 mm skin punch biopsies. The average time for blister induction was 115 ± 34 minutes (standard deviation; data not shown). Blister exudate and skin punch biopsies of untreated skin function as baseline (Figures 2, 3 and 4). In general, blister exudate of untreated skin was characterized by a low level of cell influx with a median of 19 430 cells/mL (Figure 2B). The blister exudate volume ranged from 14 μL (median of saline 6 h) to 98 μL (median at baseline, Figure 2A). Although the blister exudate volume decreased after LPS injection compared to baseline (Figure 2A), cell influx increased after LPS injection with a peak at 24 hours of 177 175 cells per mL (median, Figure 2B), compared to 61 616 cells per mL (median, Figure 2B) at 6 hours after saline injection. LPS induced an influx of neutrophils that was already visible after 3 hours and then gradually declined (Figure 3A). Saline injection caused a delayed influx of neutrophils, which peaked at 24 hours postinjection (Figure 3A). Other innate immune cells such as monocytes and dendritic cells responded later and peaked between 10 and 48 hours after LPS injection (Figure 3B–E). LPS also seemed to initiate an adaptive immune response given the increase in T cells and B cells (Figure 3G–I); however, the saline injection also showed some degree of influx of T cells and B cells (Figure 3G–I). The cellular response was also studied by immunohistochemistry on skin punch biopsies, showing that the inflammatory infiltrate was located in the dermis. There was a high degree of concordance between the results from the cellular response measured in blister exudate and the immunohistochemistry performed on the skin punch biopsies (Figure 4), except for neutrophils in blister exudate (Figure 3A) vs. MPO‐positive cells in the biopsies (Figure 4F). The peak in MPO‐positive skin sections was seen at 24 hours (Figure 4F), whereas the neutrophils in blister exudate peaked at 3 hours (Figure 3A). Figure 5 shows representative images of the MPO‐stained skin sections which shows that intact MPO‐positive cells are present at 3 hours after LPS injection (Figure 5B, D) and at 24 hours no clear intact MPO‐positive cells can be recognized (Figure 5C), but the section shows a strong MPO positivity indicating that neutrophils have degranulated or possibly neutrophil extracellular traps have been formed. There was no statistical analysis performed to analyse the contrast between LPS and saline on the cellular parameters because the saline condition consisted of only 3 subjects per time point and only for 3, 6 and 24 hours post‐saline injection.
FIGURE 2.
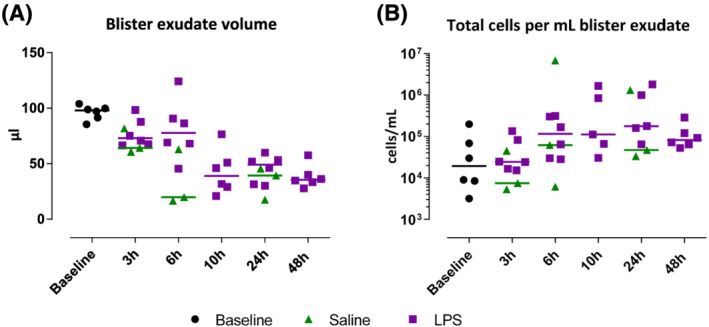
Blister exudate volume in μL and total cell count in blister exudate per mL. Data are presented as individual values with medians. LPS, lipopolysaccharide
FIGURE 3.
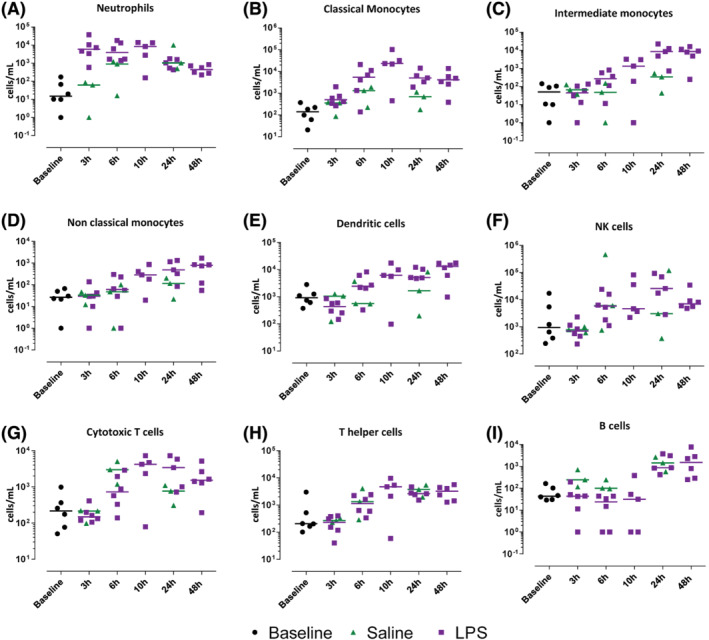
The immune cell influx in the blister exudate after lipopolysaccharide or saline injection quantified by flow cytometry in blister exudate. Data are expressed as individual data points with medians
FIGURE 4.
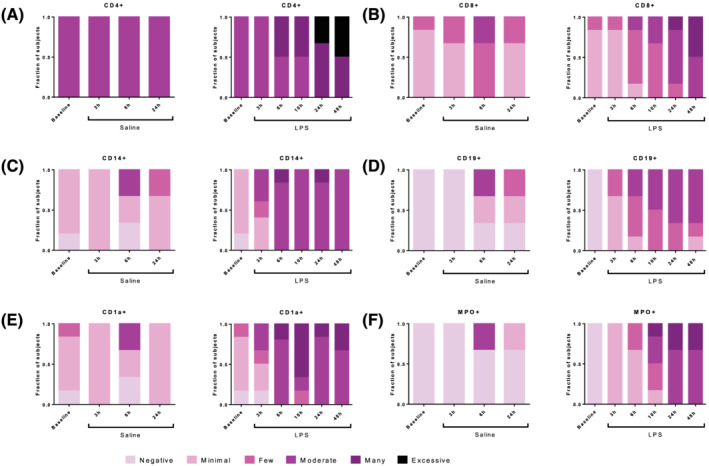
The immune cell influx in the blister exudate after lipopolysaccharide or saline injection quantified by immunohistochemistry in skin punch biopsies
FIGURE 5.
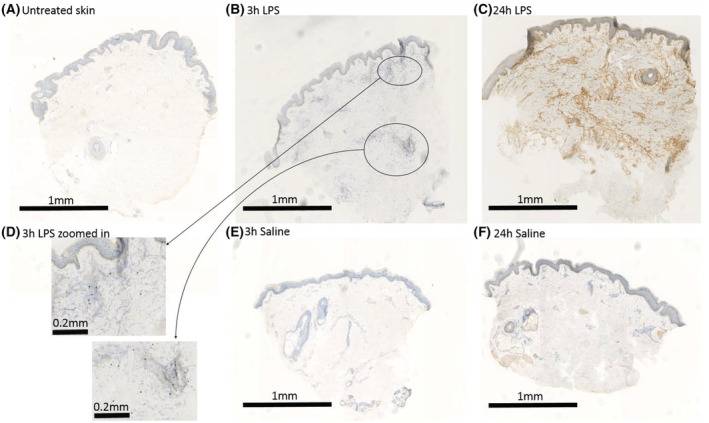
Representative images of Immunohistochemical staining of myeloperoxidase in skin sections of untreated skin (A), skin at 3 hours after lipopolysaccharide (LPS) injection (B and D), 24 hours after LPS injection (C), and after saline injection (E and F)
3.3. Cytokine response
The protein response to intradermal LPS was studied by measuring protein levels of inflammatory cytokines in blister exudate with Meso Scale Discovery and quantifying mRNA levels of inflammatory proteins in skin punch biopsies (Figures 6 and 7). In general, untreated skin (presented as baseline) and saline injected skin showed little to no inflammatory cytokines and low levels of mRNA expression, whereas LPS caused a rapid increase in IL‐6, IL‐8, IL‐1β and TNF (Figure 6C–F) in the blister exudate. A similar pattern of enhanced mRNA expression was observed in skin punch biopsies, showing peak levels already at 3 hours post‐LPS injection (Figure 7C–F), although mRNA levels showed a smaller response window compared to cytokine production. IFN‐γ and IL‐10 showed a slower response and peaked 6 hours post‐LPS injection (Figures 6A, B and 7A, B). Intradermal LPS resulted in a very robust cytokine response as measured in the blister exudate, provided a 10–1000 increase compared to both untreated skin and saline injected skin (Figure 6A–F). There was no statistical analysis performed to analyse the contrast between LPS and saline because the saline condition was evaluated in only 3 subjects, and the cytokine response was minimal (cytokine concentrations either below or slightly above the LLOQ).
FIGURE 6.
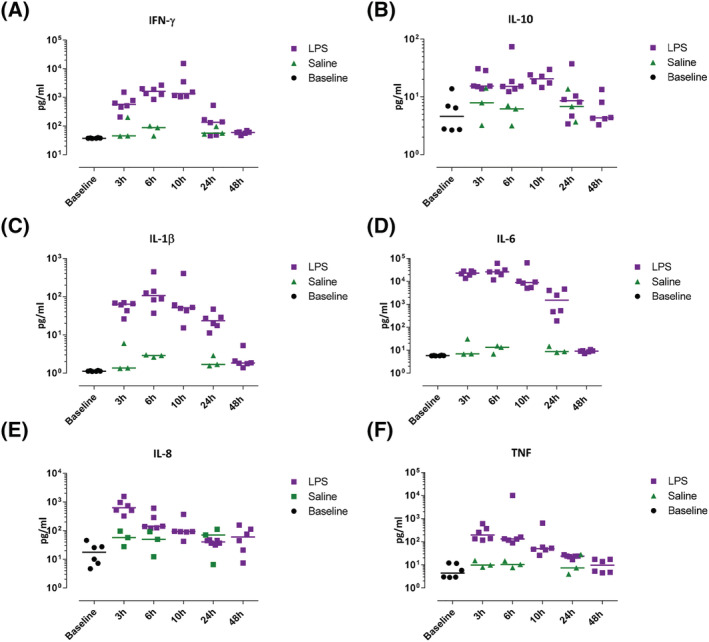
Inflammatory cytokine influx in blister exudate was quantified by Meso Scale Discovery. Data are expressed as individual data points with medians
FIGURE 7.
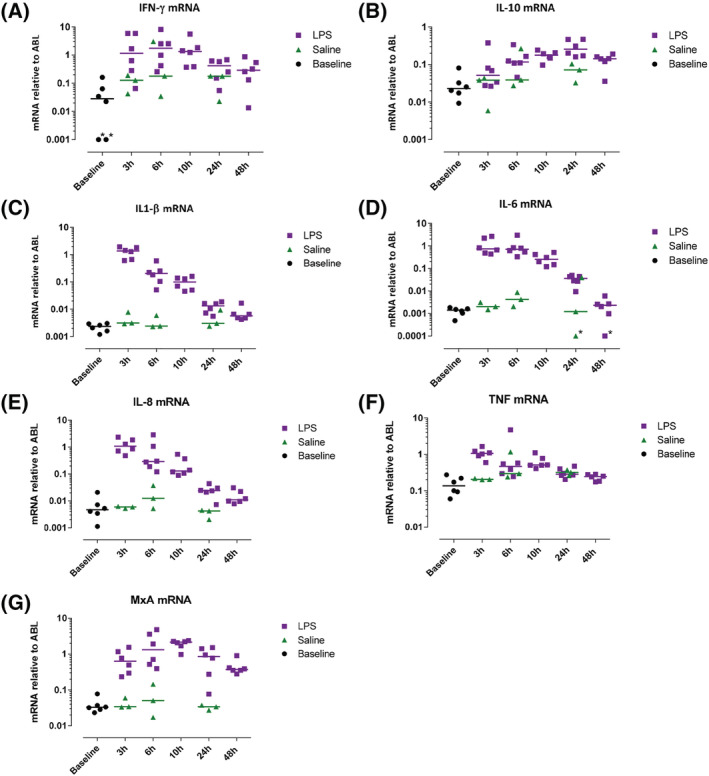
Inflammatory cytokine mRNA in skin punch biopsies were quantified by quantitative polymerase chain reaction. Data are expressed as individual data points with medians, relative to housekeeping gene ABL. *Values were zero but for displaying purposes values were changed to 0.001 (interferon [IFN]‐γ) or 0.0001 (interleukin [IL]‐6). LPS, lipopolysaccharide; TNF, tumour necrosis factor
3.4. Safety
Baseline characteristics including temperature, heart rate, blood pressure and circulating leucocytes can be found in Table S3. A slight decrease in heart rate and systolic blood pressure between 3 to 6 hours post‐LPS injection was observed in both dose groups (Figure S3A, B). Although we cannot definitely conclude the cause of these changes as a control group with subjects who received solely intradermal saline and no LPS was not included in this study, we consider these effects most likely to reflect circadian changes rather than a true effect of LPS. Body temperature increased on average between 0.2 and 0.3°C after LPS administration (Figure S3D). Both the 10 and 20‐ng group showed an increase of circulating leucocytes from 3 to 6 hours after LPS administration (Figure S3E), which plateaued between 6 and 10 hours and declined to baseline levels at 24 hours. The increase in circulating leucocytes was mainly due to an increase in neutrophils (Figure S3F) and to a lesser extent of monocytes (Figure S3G). Changes in circulating lymphocytes were less pronounced (Figure S3H), but increased slightly over the first 10 hours after LPS administration and gradually returned to baseline levels at 24 hours (20‐ng LPS group) or even declined a little below baseline levels (10‐ng LPS group). Circulating IL‐1β, IL‐6, IL‐8, IL‐10, IFN‐γ and TNF were determined at baseline and 3, 6, 10, 24 and 48 hours after LPS administration to further study possible systemic inflammatory effects. All samples showed concentrations below or slightly above LLOQ and no time or dose dependent effects were observed in the samples that were slightly above LLOQ (data not shown). Local tolerance was tested by applying pressure on the injection area with a cotton swab at 3, 6, 10, 24 and 48 hours post‐LPS or saline administration. Subjects were asked to score the tenderness on a 101‐point scale (numerical rating scale, 0 no tenderness, 100 worst pain ever experienced). Subjects reported that the LPS injected site was mildly tender in 108 of 174 occasions (62%) post‐LPS administration, whereas the saline injected site was never reported tender postadministration. The local inflammatory reaction was well tolerated and only resulted in mild tenderness; of the 108 occasions where the LPS injected site was mildly tender, a score below 10 was reported 69/108 times (64%), a score below 20 was reported 32/108 times (30%), a score below 30 was reported 6/108 times (6%), and the highest reported score of 30 was reported only once. Two subjects (11%) experienced an adverse event after LPS administration (oropharyngeal pain, 2 d after LPS administration and fatigue 2.5 h after LPS administration) which were both considered unlikely to be related to LPS administration.
4. DISCUSSION
The goal of this study was to thoroughly characterize the response to intradermal LPS injection in healthy volunteers. This human inflammatory skin challenge model would not only be useful to increase our understanding of the underlying physiological responses, but could also be a valuable methodological tool in clinical pharmacology studies evaluating the effects of anti‐inflammatory or immuno‐modulating compounds. Our results show very consistently that an intradermal dose of 5 ng LPS elicits an acute, statistically significant, transient and localized inflammatory response as assessed by comprehensive quantification of cellular, molecular and clinical response evaluation. The onset of the clinical response was visible at the first evaluated time point (3 h) after injection, peaked at 24 hours and had normalized at 48 hours postinjection. There was a high concordance between all 3 clinical parameters (local skin temperature, erythema and perfusion), although skin perfusion showed the most sensitive results when compared to the response to saline injection. Intradermal LPS also induced an acute cellular response as measured in suction blisters which consisted of a neutrophil with peak levels at 3 hours followed by classical, intermediate and nonclassical monocytes, and dendritic cells, at the subsequent time points. A potential drawback of our design is that we only included males in order to minimize the potential intersubject variability due to sex differences in LPS response. Wegner et al. investigated sex differences in response to systemic LPS and observed a greater inflammatory response in women compared to men. 14 Future research should elucidate the potential sex differences to intradermal LPS administration. When comparing the cellular response measured in suction blisters to the cellular response measured in skin punch biopsies, a high degree of concordance was observed. However, time courses of neutrophil influx in suction blister exudate and the amount of MPO‐positive cells in skin punch biopsies showed conflicting results. Whereas in blister exudate neutrophils showed a peak influx at 3 hours post‐LPS injection, in skin punch biopsies MPO, a neutrophil granule protein, was most abundant at 24 and 48 hours postinjection. Basran et al. showed that intradermal LPS induced neutrophil influx (indicated by neutrophil elastase positive polymorphonuclear cells) in skin punch biopsies 2–6 hours post‐LPS administration. Other supportive evidence that TLR4‐mediated inflammation induces a rapid dermal neutrophil influx is provided by Motwani et al. who showed that UV‐killed E. coli caused a similar neutrophil response in blister exudate. Therefore, it is more likely that the late rise in MPO‐positive cells in skin punch biopsies, from 10 hours and onwards, represents the delayed degranulation of attracted neutrophils, which is supported by the staining pattern as observed in Figure 5C where no longer intact MPO‐positive cells can be observed. We hypothesize that the relatively low number of neutrophils in blister exudate at 24 and 48 hours, compared to high MPO content in biopsies at these time points, is explained by the formation of neutrophil extracellular traps, fixating neutrophils and neutrophil products (e.g. MPO) in the skin, 18 and inhibiting the extrusion of intact neutrophils in blister exudate. Monocytes were the second largest cell population found in the blister exudate, further distinguished in classical monocytes, intermediate monocytes and nonclassical monocytes. The timing of these different subsets in response to LPS is in line with the current understanding of the different roles that each subset plays: classical monocytes primed for phagocytosis and migration, intermediate monocytes primed for antigen presentation and regulation of apoptosis, and nonclassical monocytes being associated with the wound healing process. 19
We used 2 different methods to study the cellular and cytokine response to LPS: suction blisters and conventional biopsies. We found that suction blisters offer several advantages over conventional biopsies. The procedure to analyse cells via flow cytometry does not require extensive tissue processing (mechanical or enzymatic isolation of single cells) or tissue sectioning, and results in truly quantitative data. Interestingly, the availability of a bed‐side flow cytometry facility allows the collection of real‐time cellular data. Moreover, blister exudate is a great source for the quantitative analysis of inflammatory cytokines and chemokines, which indirectly also provides a functional assessment of skin‐resident immune cells. Next to these advantages, 1 of the theoretical drawbacks of suction blisters is that the blister takes on average 2 hours to form and the induction itself evokes an inflammatory response. 20 This is, however, not an insurmountable problem: our blisters from naïve skin show that when the blister fluid is harvested directly after blister formation, the blister procedure itself only causes minimal inflammatory cell influx and hardly any inflammatory cytokines. These results concur with Davidsson et al., who investigated the inflammatory influx at different time points after blister formation. 21 Harvesting the blisters directly after formation minimizes this potential confounder, and the impact of the blister induction procedure remains limited. However, we did find that the saline injection resulted in a delayed cellular response with increasing neutrophils from 6 to 24 hours postinjection and notably T and B cells showed an increased influx at 24 hours, which is most likely to be the result of a wound and wound healing in response to the saline injection 22 , 23 , 24 This should be taken into account when interpreting the response to LPS. The number of cells per mL blister exudate that we found was less than Motwani et al., observed in their intradermal UVEk challenge. This difference is probably explained by the fact that UVEk challenge drives an inflammatory response via multiple innate immune pathways, while LPS only triggers TLR4. This is supported by Basran et al., who did not observe a significant difference in neutrophil influx nor in the expression of IL‐8, IL‐1α, and IL‐1β between an intradermal dose of 5 and 15 ng LPS. 13 Intradermal LPS resulted in a strong cytokine response that was highly significant compared to low baseline levels and the response to saline injection. IL‐6, IL‐8 and TNF were already present at peak levels at 3 hours post‐LPS injection. Although neutrophils can produce IL‐6, IL‐8 and TNF, 25 , 26 it is more likely that these cytokines were produced by resident dermal cells, driving chemo‐attraction of neutrophils. We hypothesize that the first responders to LPS were resident cells such as dendritic cells, fibroblasts and keratinocytes, which are immunocompetent cells that can produce the observed cytokines in response to LPS. 27 , 28 , 29
Although the main focus of this study was the local response to intradermal LPS, we also assessed whether systemic changes could be observed. We observed mild transient neutrophilia, which did not appear to be dose dependent, and LPS did not lead to any systemic cytokine response, as reported before. 13 Body temperature increased on average between 0.2 and 0.3°C after LPS administration (Figure S3D), which is much less compared to temperature increases of 0.5–1.5°C 3–4 hours after intravenous LPS administration. 4 The observed temperature increase after intradermal LPS administration could be of circadian nature rather than a through LPS effect but we did not investigate this.
In comparison with other inflammatory skin challenges (UVB, topical imiquimod, UVEk, tape stripping, cantharidine) our intradermal LPS model offers several advantages: (i) LPS is the most used antigen in inflammatory and immunological research, making this challenge model ideal for translational research; (ii) intradermal administration results in a standardized administrated dose, which is often questionable with topical application/manipulation (imiquimod, cantharidine, tape stripping); and (iii) the induced inflammatory response is rapid and transient with a negligible burden for the involved subjects. Importantly, intradermally administered LPS evokes a classical tissue inflammation response, characterized by an initial attraction of neutrophils, followed by classical monocytes, intermediate monocytes and nonclassical monocytes, and lymphocyte subsets, combined with a significant nuclear factor κB fingerprint at the level of cytokines. This is not necessarily the case for the earlier mentioned inflammatory skin challenge models. In conclusion, intradermal LPS administration in humans causes an acute, localized and transient inflammatory reaction that is safe and well tolerated by the volunteers, the next step would be to investigate whether the LPS‐induced inflammatory response can be successfully suppressed with known anti‐inflammatory drugs in order to validate the use of intradermal LPS in future proof of pharmacology studies involving potential anti‐inflammatory drugs.
COMPETING INTERESTS
The authors state no conflict of interest.
CONTRIBUTORS
M.M. devised the project and main conceptual ideas together with T.B., G.F., R.R., A.P., S.Y., Y.O. and D.G. T.B., P.H. and M.M. worked out technical details and study design. T.B. coordinated the clinical trial under supervision of M.M., G.F. and J.B. T.B., F.H., I.J. and W.V. performed clinical evaluations. P.H., E.F., H.G., M.S. and M.O. coordinated and performed bioanalysis. J.B. carried the medical responsibility. A.P., D.G., E.L., J.D., M.J., P.H., M.M., S.Y. and T.B. analysed and interpreted data. M.K. carried out statistical analysis. G.F., on behalf of his employer, provided funding for the clinical trial. T.B., M.J. and M.M. wrote the manuscript. All authors provided critical feedback and helped shaping the manuscript.
Supporting information
FIGURE S1 Graphic display of treatment regime and invasive measurements per subject. Subjects 1–6 received 2 LPS injections and 1 saline injection, they also contributed to baseline skin punch biopsies and suction blisters from untreated skin. Subjects 7–18 received 4 LPS injections and 1 saline injection. SB = blister exudate, SPB = skin punch biopsy.
FIGURE S2 Gating strategy used for flow cytometry analysis of the blister exudate.
FIGURE S3 Vital signs and circulating leucocytes were measured pre LPS administration and 3, 6, 10, 24 and 48 hours after administration. Data are presented as means ± SD.
TABLE S1 Overview of different cell subsets characterized by flow cytometry.
TABLE S2 Listed primers used for mRNA analysis by qPCR.
TABLE S3 Baseline characteristics of subjects receiving either 2 intradermal injections of 5 ng LPS (10 ng in total) or 4 5‐ng LPS (20 ng in total)
ACKNOWLEDGMENTS
We would like to thank all volunteers who have participated in this study. Cutanea Life Science, Inc., Wayne, Pennsylvania, USA.
Buters TP, Hameeteman PW, Jansen IME, et al. Intradermal lipopolysaccharide challenge as an acute in vivo inflammatory model in healthy volunteers. Br J Clin Pharmacol. 2022;88(2):680-690. 10.1111/bcp.14999
The authors confirm that the PI for this paper is Matthijs Moerland and the MR Jacobus Burggraaf. Together they had direct clinical responsibility for subjects.
Corrections added October 21, 2021, after first online publication: In the “Methods” section of the abstract, 10 ng LPS injections was changed to 5 ng LPS injections. Similarly, in “Study Design and Subjects”, 10 ng LPS was changed to 5 ng LPS and 100 μL saline was changed to 50 μL saline. In “Safety (3.4)”, 20‐ng and 40‐ng were corrected to 10‐ng and 20‐ng respectively. In “Discussion”, 10 ng LPS was corrected to 5 ng LPS. In the legend of Figure 1, 10 ng LPS was corrected to 5 ng LPS. In the legend of Table S3, 10 ng LPS, 20 ng LPS, and 40 ng LPS were corrected to 5 ng LPS, 10 ng LPS, and 20 ng LPS respectively. These corrections are noted in the text.]
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428‐435. [DOI] [PubMed] [Google Scholar]
- 2. Bahador M, Cross AS. From therapy to experimental model: a hundred years of endotoxin administration to human subjects. J Endotoxin Res. 2007;13(5):251‐279. [DOI] [PubMed] [Google Scholar]
- 3. Patel AA, Zhang Y, Fullerton JN, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. 2017;214(7):1913‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dillingh MR, van Poelgeest EP, Malone KE, et al. Characterization of inflammation and immune cell modulation induced by low‐dose LPS administration to healthy volunteers. J Inflamm. 2014;11:28. 10.1186/s12950-014-0028-1 [DOI] [Google Scholar]
- 5. Seeley JJ, Ghosh S. Molecular mechanisms of innate memory and tolerance to LPS. J Leukoc Biol. 2017;101(1):107‐119. [DOI] [PubMed] [Google Scholar]
- 6. Greisman SE, Woodward WE. Mechanisms of Endotoxin tolerance. 3. The refractory state during continuous intravenous infusion of endotoxin. J Exp Med. 1965;121(6):911‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brooks D, Barr LC, Wiscombe S, McAuley DF, Simpson AJ, Rostron AJ. Human lipopolysaccharide models provide mechanistic and therapeutic insights into systemic and pulmonary inflammation. Eur Respir J. 2020;56(1):1901298. [DOI] [PubMed] [Google Scholar]
- 8. Siebenga PS, van Amerongen G, Klaassen ES, de Kam ML, Rissmann R, Groeneveld GJ. The ultraviolet B inflammation model: Postinflammatory hyperpigmentation and validation of a reduced UVB exposure paradigm for inducing hyperalgesia in healthy subjects. Eur J Pain (London, England). 2019;23:874‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Kolk T, Assil S, Rijneveld R, et al. Comprehensive, Multimodal Characterization of an Imiquimod‐Induced Human Skin Inflammation Model for Drug Development. Clin Transl Sci. 2018;11(6):607‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Motwani MP, Flint JD, De Maeyer RP, et al. Novel translational model of resolving inflammation triggered by UV‐killed E coli. J Pathol Clin Res. 2016;2(3):154‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dalpke A, Frank J, Peter M, Heeg K. Activation of toll‐like receptor 9 by DNA from different bacterial species. Infect Immun. 2006;74(2):940‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basran A, Jabeen M, Bingle L, et al. Roles of neutrophils in the regulation of the extent of human inflammation through delivery of IL‐1 and clearance of chemokines. J Leukoc Biol. 2013;93(1):7‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wegner A, Benson S, Rebernik L, et al. Sex differences in the pro‐inflammatory cytokine response to endotoxin unfold in vivo but not ex vivo in healthy humans. Innate Immun. 2017;23(5):432‐439. [DOI] [PubMed] [Google Scholar]
- 15. Aomatsu M, Kato T, Kasahara E, Kitagawa S. Gender difference in tumor necrosis factor‐α production in human neutrophils stimulated by lipopolysaccharide and interferon‐γ. Biochem Biophys Res Commun. 2013;441(1):220‐225. [DOI] [PubMed] [Google Scholar]
- 16. Niemeyer‐van der Kolk T, Assil S, Buters TP, et al. Omiganan Enhances Imiquimod‐Induced Inflammatory Responses in Skin of Healthy Volunteers. Clin Transl Sci. 2020;13(3):573‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander SPH, Fabbro D, Kelly E, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. Br J Pharmacol. 2019;176(Suppl 1):S247‐s96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Odobasic D, Kitching AR, Holdsworth SR. Neutrophil‐Mediated Regulation of Innate and Adaptive Immunity: The Role of Myeloperoxidase. J Immunol Res. 2016;2016:2349817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kapellos TS, Bonaguro L, Gemünd I, et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front Immunol. 2019;10:2035. 10.3389/fimmu.2019.02035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niedzwiecki MM, Samant P, Walker DI, et al. Human Suction Blister Fluid Composition Determined Using High‐Resolution Metabolomics. Anal Chem. 2018;90(6):3786‐3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davidsson L, Björkman L, Christenson K, et al. A simple skin blister technique for the study of in vivo transmigration of human leukocytes. J Immunol Methods. 2013;393(1‐2):8‐17. [DOI] [PubMed] [Google Scholar]
- 22. Li Y, Wu J, Luo G, He W. Functions of Vγ4 T Cells and Dendritic Epidermal T Cells on Skin Wound Healing. Front Immunol. 2018;9. 10.3389/fimmu.2018.01099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sîrbulescu RF, Boehm CK, Soon E, et al. Mature B cells accelerate wound healing after acute and chronic diabetic skin lesions. Wound Repair Regen. 2017;25(5):774‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Debes GF, McGettigan SE. Skin‐Associated B Cells in Health and Inflammation. J Immunol. 2019;202(6):1659‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cassatella MA, Tamassia N, Crepaldi L, et al. Lipopolysaccharide primes neutrophils for a rapid response to IL‐10. Eur J Immunol. 2005;35(6):1877‐1885. [DOI] [PubMed] [Google Scholar]
- 26. Tecchio C, Micheletti A, Cassatella MA. Neutrophil‐derived cytokines: facts beyond expression. Front Immunol. 2014;5:508. 10.3389/fimmu.2014.00508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perrin‐Cocon L, Aublin‐Gex A, Sestito SE, et al. TLR4 antagonist FP7 inhibits LPS‐induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci Rep. 2017;7:40791. 10.1038/srep40791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X‐P, Liu P, Li Y‐F, Zhang G‐L, Zeng D‐S, Liu D‐L. LPS induces activation of the TLR4 pathway in fibroblasts and promotes skin scar formation through collagen I and TGF‐β in skin lesions. Int J Clin Exp Pathol. 2019;2121‐2129. [PMC free article] [PubMed] [Google Scholar]
- 29. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front Immunol. 2018;9:1549. 10.3389/fimmu.2018.01549 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Graphic display of treatment regime and invasive measurements per subject. Subjects 1–6 received 2 LPS injections and 1 saline injection, they also contributed to baseline skin punch biopsies and suction blisters from untreated skin. Subjects 7–18 received 4 LPS injections and 1 saline injection. SB = blister exudate, SPB = skin punch biopsy.
FIGURE S2 Gating strategy used for flow cytometry analysis of the blister exudate.
FIGURE S3 Vital signs and circulating leucocytes were measured pre LPS administration and 3, 6, 10, 24 and 48 hours after administration. Data are presented as means ± SD.
TABLE S1 Overview of different cell subsets characterized by flow cytometry.
TABLE S2 Listed primers used for mRNA analysis by qPCR.
TABLE S3 Baseline characteristics of subjects receiving either 2 intradermal injections of 5 ng LPS (10 ng in total) or 4 5‐ng LPS (20 ng in total)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.