

Abstract
Objective
Dravet syndrome is a severe developmental and epileptic encephalopathy (DEE) most often caused by de novo pathogenic variants in SCN1A. Individuals with Dravet syndrome rarely achieve seizure control and have significantly elevated risk for sudden unexplained death in epilepsy (SUDEP). Heterozygous deletion of Scn1a in mice (Scn1a +/−) recapitulates several core phenotypes, including temperature‐dependent and spontaneous seizures, SUDEP, and behavioral abnormalities. Furthermore, Scn1a +/− mice exhibit a similar clinical response to standard anticonvulsants. Cholesterol 24‐hydroxlase (CH24H) is a brain‐specific enzyme responsible for cholesterol catabolism. Recent research has indicated the therapeutic potential of CH24H inhibition for diseases associated with neural excitation, including seizures.
Methods
In this study, the novel compound soticlestat, a CH24H inhibitor, was administered to Scn1a +/− mice to investigate its ability to improve Dravet‐like phenotypes in this preclinical model.
Results
Soticlestat treatment reduced seizure burden, protected against hyperthermia‐induced seizures, and completely prevented SUDEP in Scn1a +/− mice. Video–electroencephalography (EEG) analysis confirmed the ability of soticlestat to reduce occurrence of electroclinical seizures.
Significance
This study demonstrates that soticlestat‐mediated inhibition of CH24H provides therapeutic benefit for the treatment of Dravet syndrome in mice and has the potential for treatment of DEEs.
Keywords: anticonvulsants, cholesterol 24‐hydroxylase, CYP46A1, epilepsy, Nav1.1 voltage‐gated sodium channel
Key Points.
Soticlestat is a novel inhibitor of cholesterol 24‐hydroxlase (CH24H), a brain‐specific enzyme responsible for cholesterol catabolism.
Treatment with soticlestat protected against hyperthermia‐induced seizures in two preclinical Dravet mouse models.
Soticlestat treatment reduced seizure burden and completely prevented sudden unexpected death in epilepsy (SUDEP) in the Scn1a +/− mouse model of Dravet syndrome.
The novel investigational compound soticlestat substantially improves the epilepsy phenotypes in a preclinical model of Dravet syndrome.
1. INTRODUCTION
Developmental and epileptic encephalopathies (DEE) are clinically the most severe category of epilepsies and therefore extremely challenging to treat. 1 Dravet syndrome is a severe DEE, considered to be one of the most pharmacoresistant epilepsies. Only 10% of patients achieve complete seizure control, despite most patients being treated with three or more standard antiseizure medications. 2 , 3 In addition to intractable seizures and developmental delays, individuals have elevated risk of sudden unexplained death in epilepsy (SUDEP), with 15%–20% of patients dying by early adulthood. 2
At least 80% of Dravet syndrome cases arise from de novo variants in SCN1A, resulting in heterozygous loss‐of‐function mutations. 2 , 3 Several preclinical models of Dravet syndrome have been generated, including mice with heterozygous deletion of Scn1a (Scn1a +/−) and Scn1aR1407X (Scn1aRX /+) mice with a premature termination codon. 4 , 5 , 6 Scn1a haploinsufficiency models recapitulate many core features of Dravet syndrome, including spontaneous and heat‐induced seizures, SUDEP, and cognitive and behavioral deficits. 4 , 5 , 6 , 7 , 8 , 9 We and others previously demonstrated that treatment of Dravet‐like phenotypes of Scn1a +/− mice correlate with clinical pharmacological responses of individuals with Dravet syndrome. 4 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17
Currently, only three drugs—stiripentol, cannabidiol, and fenfluramine—have US Food and Drug Administration (FDA) labels for the treatment of seizures associated with Dravet syndrome. 18 , 19 , 20 All three are limited in their ability to completely prevent seizures and were approved based on trials where they were used as an add‐on treatment to standard anticonvulsant therapies, exemplifying the crucial need for development of novel therapeutic strategies. One compound, soticlestat (TAK‐935/OV935), is currently in clinical development for DEEs, including Dravet syndrome and Lennox‐Gastaut syndrome. Soticlestat is the first potent, selective and central nervous system (CNS)–penetrant inhibitor of the cholesterol 24‐hydroxylase (CH24H) enzyme, also commonly known as cytochrome P450 46A1 (CYP46A1). CH24H is responsible for the conversion of cholesterol to 24S‐hydroxycholesterol (24HC) in the brain. 21 CH24H is expressed in both cortical and hippocampal neurons and synthesis of 24HC is predominantly neuronal. 21 , 22 CH24H null mice exhibited a 40% reduction in brain cholesterol synthesis, with otherwise normal development. 23 Soticlestat demonstrated disease‐modifying potential in mouse pentylenetetrazol (PTZ) and kainic acid kindling models of epilepsy, as well as in the amyloid precursor protein and presenilin 1 transgenic (APP/PS1‐Tg) Alzheimer model that has co‐occurring seizures. 24 , 25 , 26 , 27 , 28 In the mouse kainic acid kindling model, soticlestat treatment prevented the development of seizures in 31% of mice, whereas 100% of control mice exhibited spontaneous seizures. 25 Furthermore, spontaneous seizure frequency was reduced by 50% compared to controls. 25 The APP/PS‐1‐Tg Alzheimer model has an average 3‐month survival rate of 50% due to seizure‐related sudden death. 27 Soticlestat treatment dramatically improved survival of APP‐1‐Tg mice to 93% at a dose that lowered brain 24HC by ~50%. 27
In the current study, we evaluated the effect of soticlestat on Dravet‐like phenotypes of the Scn1a haploinsufficiency mouse model. We evaluated the effect of soticlestat treatment on temperature thresholds for hyperthermia‐induced seizures in both Scnla +/− and Scn1a RX/+ mouse models. In addition, we assessed spontaneous generalized tonic‐clonic seizure (GTCS) frequency and severity, survival, and open field and zero maze performance in Scn1a +/− mice during subchronic treatment with soticlestat. Treatment of Scn1a +/− mice with soticlestat at a dose that lowered brain 24HC by ~50% resulted in reduced spontaneous seizure burden and completely prevented premature lethality, as well as protected against hyperthermia‐induced seizures. Our data support the novel approach of inhibiting CH24H activity for the treatment of refractory seizures associated with DEEs.
2. METHODS
2.1. Mice
Scn1atm1Kea mice (abbreviated as Scn1a +/− ), with deletion of exon 1, were generated by homologous recombination in TL1 embryonic stem (ES) cells and are maintained and genotyped as described previously. 4 Experimental mice were generated by crossing Scn1a +/− males with C57BL/6J females (#000664, Jackson Laboratory, Bar Harbor, ME), resulting in [129 × B6]F1. Scn1a +/−, abbreviated as Scn1a +/− mice in this article. Mice were maintained in a specific‐pathogen‐free (SPF) barrier facility with a 14‐h light/10‐h dark cycle and access to food and water ad libitum. Both female and male Scn1a +/− mice were used for all experiments.
Scn1a R1407X mice (Scn1a RX/+) with a premature stop code in exon 21 were generated by homologous recombination in ES cells and are maintained and genotyped as described previously. 5 , 8 Experimental Scn1a RX/+ mice for hyperthermia experiments were generated on the inbred 129+TER/SvJcl strain (CLEA Japan, Tokyo, Japan). Scn1a RX/+ mice were maintained on a 12‐h light/dark cycle with ad libitum access to food and water. Male and female mice were used in the hyperthermia assay and differences between sexes were not identified, so groups were collapsed across sex.
Animal care and experimental procedures for Scn1a +/− mice were approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Scn1a RX/+ mice were handled in accordance with the guidelines of the Animal Experiment Committee of RIKEN Brain Science Institute. Principles outlined in the ARRIVE guidelines and the Basel declaration were considered when planning experiments.
2.2. Soticlestat formulation and dosing
For all studies with Scn1a +/− mice, chow pellets were custom formulated by Research Diets (New Brunswick, NJ). Teklad 7912 was used as chow base (metabolizable energy: 3.1 kcal/g of food) (Envigo, Indianapolis, IN) with 0.02% soticlestat (Takeda Pharmaceuticals, Cambridge, MA). No compound was added for vehicle control chow. Based on an average body weight of 15 g and assumed consumption of 3–5 g of chow per day (10–12 kcal/g body weight, Research Diets), the soticlestat dose was estimated at 40–53 mg/kg/day. For blinding purposes, vehicle and soticlestat chows were assigned a random three‐digit number. Unblinding occurred when the study was complete. For hyperthermia studies utilizing Scn1a RX/+ mice, soticlestat chow was custom formulated by Oriental Yeast Co. (Tokyo, Japan). CRF‐1 standard mouse chow was manufactured with 0.02% soticlestat (Takeda Pharmaceutical Co., Tokyo, Japan). No compound was added to vehicle control chow.
2.3. Hyperthermia‐induced seizures
Hyperthermia‐induced seizure thresholds were independently determined in two distinct Dravet mouse models: Scn1aRX /+ and Scn1a +/− mice. Hyperthermia inductions were performed as previously described and detailed in the Supplementary Methods. 10 , 29 For Scn1aRX /+ mice, oral treatment with vehicle or soticlestat chow commenced at 4 weeks of age for 7 days (n = 30/treatment). For Scn1a +/− mice, animals were weaned at P18 and block‐randomized into vehicle (n = 16/treatment) or soticlestat (n = 20/treatment) groups. Scn1a +/− mice had ad libitum access to vehicle or soticlestat chow for 7 days. For both studies, average GTCS threshold temperatures were compared between groups using unpaired t test.
2.4. Spontaneous seizure video monitoring
At P17‐18, male and female Scn1a +/− mice were subjected to a single hyperthermia priming seizure and quickly cooled back to baseline temperature as described previously. 10 If a GTCS did not occur during priming (<1%), the mouse was excluded from the study to ensure that all mice had a similar baseline that included a hyperthermia‐induced seizure. Scn1a +/− mice were then weaned and block randomized into vehicle (target: n = 50) or soticlestat (target: n = 30) groups, and were maintained on vehicle or soticlestat chow for the remainder of the study until ~P40. The vehicle group was scaled to compensate for premature lethality during the course of the study in order to have equivalent group sizes for open field and zero maze assays at P33‐37 (described below). Two to four mice were placed in monitoring cages with ad libitum access to vehicle or soticlestat chow and water. General health was monitored throughout to ensure that mice consumed chow and that deaths were sudden and unexpected death in otherwise healthy appearing mice. Survival of Scn1a +/− mice was monitored until ~40 days of age. Survival was analyzed using time‐to‐event analysis with p‐value determined using Mantel‐Cox LogRank, with sexes considered separately.
Spontaneous GTCS frequency was assessed by continuous video monitoring as previously described 10 and detailed in the Supplementary Methods. Videos were scored offline by reviewers blinded to treatment to determine frequency and severity of spontaneous GTCS. Final group sizes were scaled to reflect the unequal proportion of vehicle‐to‐treated mice enrolled (~1.6) while maintaining equivalent statistical power (β = 0.8, α = 0.05) (vehicle: n = 35 female, n = 36 male; soticlestat: n = 25 female, n = 23 male). Severity of GTCS was assessed using a modified Racine scale adapted for Scn1a +/− mice (Table 1). Seizure frequency was first compared between sexes and no difference was detected (p > 0.63, Mann‐Whitney U test); therefore, groups were collapsed across sex. Treatment groups were compared using Mann‐Whitney U tests for frequency and chi‐square for severity (GraphPad Prism).
TABLE 1.
Modified Racine scale adapted for Scn1a +/− mice
| Stage | Description |
|---|---|
| 1 | Rearing and paddling, Straub tail with no other movement |
| 2 | Rearing and paddling, Straub tail, loss of posture, short bursts of movement, often backwards |
| 3 | Rearing and paddling with wild running and/or jumping (no loss of posture) |
| 4 | Rearing and paddling with wild running and/or jumping with loss of posture |
| 5 | Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic hindlimb extension (HLE) |
| 6 | Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic hindlimb extension (HLE) and death |
2.5. Open field and zero maze
Behavioral testing was performed on surviving Scn1a +/− mice after 14 days of continuous spontaneous seizure monitoring and wild‐type (WT), age‐matched controls on vehicle chow. Assay methods are detailed in the supplementary methods. Total distance traveled was compared separately for each sex between vehicle‐treated Scn1a +/− mice, soticlestat‐treated Scn1a +/− mice, and vehicle‐treated WT controls using one‐way analysis of variance (ANOVA) with Tukey's post hoc tests, with n = 21–31 mice per group. Percentage of time in the open portions of the maze was compared separately for each sex between vehicle‐treated Scn1a +/− mice, soticlestat‐treated Scn1a +/− mice, and vehicle‐treated WT controls using one‐way ANOVA with Tukey's post hoc tests, with n = 22–35 animals per group.
2.6. Video‐electroencephalography (EEG) monitoring
A separate cohort of P17‐18 Scn1a +/− mice was surgically implanted with EEG headmounts as previously described, 30 and detailed in the Supplementary Methods. After a 48‐h recovery period, mice (P19‐20) were subjected to a single priming hyperthermia‐induced seizure as described above. Following the hyperthermia‐induced seizure, mice were randomly assigned to vehicle or soticlestat and had ad libitum access to chow and water for the remainder of the study period. Immediately following the hyperthermia‐induced seizure, video‐EEG data were recorded continuously from freely moving mice for >168 h or until death occurred. Digitized data were acquired and reviewed with Sirenia software (Pinnacle Technology). All video‐EEG records were randomly assigned a five‐digit number to blind data analysis. EEG records were scored for GTCS by visual inspection by an observer who was blinded to treatment. GTCS were defined by greater than 15 s of sustained high‐frequency and high amplitude (>2× background) activity and included both generalized paroxysmal fast activity and generalized spike and slow wave discharges. GTCS events were followed by generalized slowing or attenuation of EEG activity. Behavioral correlates during electroclinical seizures were assessed from synchronized video records and scored based on the modified Racine scale adapted for Scn1a +/− mice (Table 1). EEG analysis included mice (vehicle: n = 11/sex; soticlestat: n = 10/sex) with at least 12 h (range 12–243 h/mouse) of EEG data post‐priming. To calculate seizure frequency, the total number of seizures exhibited by each mouse were divided by the total hours monitored and then converted to a daily seizure frequency (seizures/day). The percentage of seizures with HLE was determined for each mouse based on presence or absence of HLE (Racine ≥5) for each GTCS event. Seizure frequency was first compared between sexes and no difference was detected (p > 0.21, Mann‐Whitney U test); therefore, groups were collapsed across sex. Seizure frequency was compared between treatments using Mann‐Whitney U tests (GraphPad Prism). Percentage of mice with seizures and seizure severity were compared between treatments using Fisher exact and chi‐square, respectively.
2.7. Determination of soticlestat and 24HC concentrations in plasma
At the end of the study, Scn1a +/− mice were deeply anesthetized with isoflurane (2%–5%) and blood and brain samples were collected. Blood was obtained by cardiac puncture and immediately transferred to a lithium heparin microtainer (BD #365965, Franklin Lakes, NJ). Plasma was isolated by centrifugation (2000 × g, 10 min at 4°C), snap frozen on dry ice, and stored at −80°C. Whole brains were removed following exsanguination, snap frozen on dry ice, and stored at −80°C. Samples were assayed by liquid chromatography with tandem mass spectrometry (LC‐MS/MS) as described previously. 27
2.8. Statistics
Supplementary Table 1 summarizes statistical tests described above for each assay and includes the computed values. No differences were detected between sexes for seizure frequency, severity, or hyperthermia threshold endpoints; therefore, groups were collapsed across sex for these endpoints. There was a modest difference in survival between male and female Scn1a +/− mice (p = 0.0476; Log‐Rank Mantel Cox); therefore, sexes were considered separately for this endpoint. Sexes were considered separately for open field and zero maze as is conventional for behavioral assays. D’Agostino & Pearson tests were used to assess normality and nonparametric tests were used in cases where there were violations of the Gaussian assumption. Post hoc comparisons are reported in the text and figure legends.
3. RESULTS
3.1. Soticlestat protects against hyperthermia‐induced seizures in Dravet mouse models
Dravet syndrome often presents with a seizure provoked by a febrile illness, and body temperature elevation is a common trigger. 31 Scn1a haploinsufficient mice exhibit enhanced susceptibility to seizures induced by elevated body temperature, similar to those observed in Dravet syndrome patients. 11 , 15 Therefore, we wanted to determine the effect of soticlestat on hyperthermia‐induced seizure thresholds by independently assessing thresholds in two distinct haploinsufficient Dravet mouse models, Scn1a R1407X and Scn1a +/− mice. 4 , 5 Separate cohorts of Scn1a RX/+ and Scn1a +/− mice were randomly assigned to 0.02% soticlestat or vehicle control chow and provided ad libitum access for 7 days. After 1 week of treatment, each mouse was tested to determine the temperature threshold for induction of a GTCS. Soticlestat treatment resulted in an elevated threshold for hyperthermia‐induced GTCS in both Scn1a RX/+ and Scn1a +/− mice (Figure 1). In the Scn1aRX /+ cohort, average seizure temperature was 43.1 ± 0.1°C in soticlestat‐treated mice vs 42.0 ± 0.1°C for vehicle‐treated control mice (Figure 1A) (p < 0.0001). In the Scn1a +/− cohort, average temperature for soticlestat‐treated mice was 42.9 ± 0.1°C compared to 42.0 ± 0.4°C for vehicle controls (Figure 1B) (p < 0.04). Observing a protective effect of soticlestat on threshold temperature in two different Dravet models at separate institutions suggests this is a robust effect.
FIGURE 1.
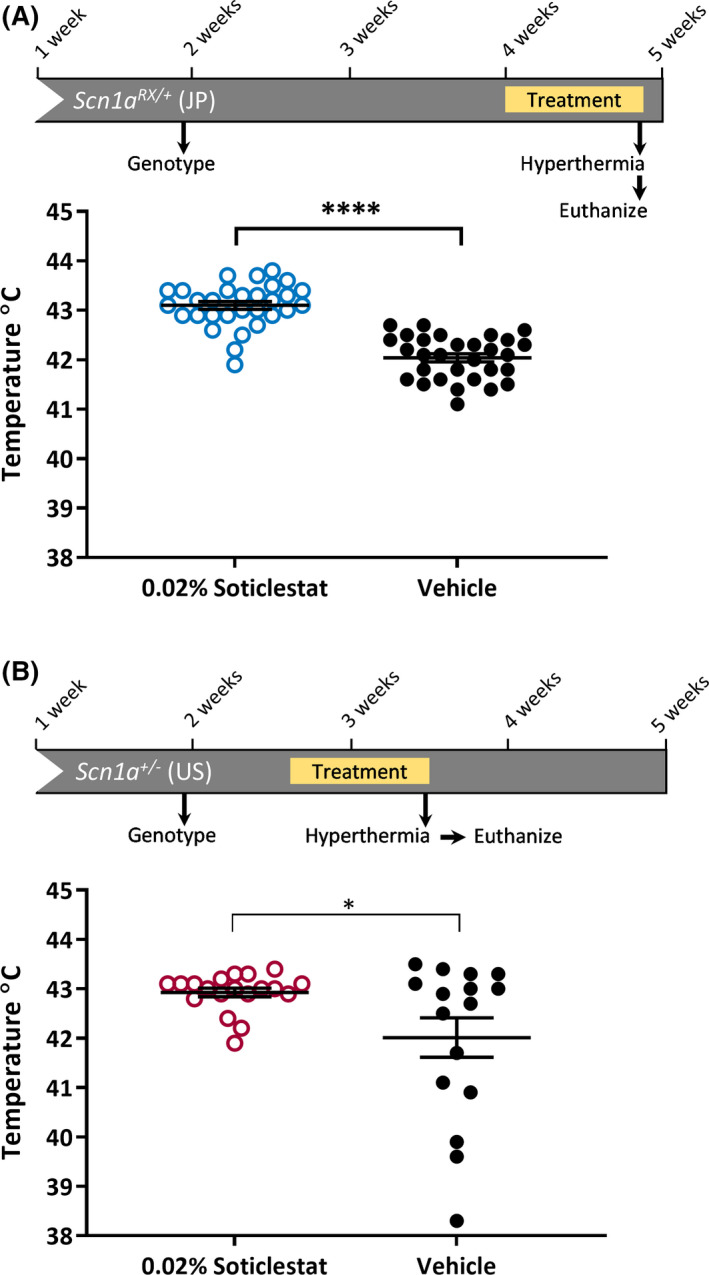
Soticlestat treatment lessens the susceptibility to hyperthermia‐induced seizures in Scn1a haploinsufficiency mouse models. (A) Experimental design for hyperthermia seizure threshold assay in Scn1aRX /+ mice treated with 0.02% soticlestat formulated in chow or vehicle control chow. Temperature thresholds for Scn1aRX /+ mice were determined in Japan (JP). Temperature threshold for seizure induction was elevated in Scn1aRX /+ mice treated with 0.02% soticlestat (n = 30) compared to vehicle controls (n = 30) (****p < 0.0001, Student's t‐ test). Average temperature threshold was 43.1 ± 0.1 in 0.02% soticlestat‐treated mice and 42.0 ± 0.1 in vehicle controls. (B) Experimental design for hyperthermia seizure threshold assay in Scn1a +/− mice treated with 0.02% soticlestat formulated in chow or vehicle control chow. Temperature thresholds for Scn1a +/− mice were determined in the United States. Temperature threshold for seizure induction was elevated in Scn1a +/− mice treated with 0.02% soticlestat (n = 19) compared to vehicle controls (n = 16) (*p < 0.04, Welch's t test). Average temperature threshold was 42.9 ± 0.1 in 0.02% soticlestat‐treated mice and 42.0 ± 0.4 in vehicle controls. Symbols represent individual mice, horizontal lines represent the group means, and error bars are standard error of the mean (SEM)
3.2. Soticlestat decreases seizure burden and improves survival in Scn1a+/− mice
As Dravet syndrome progresses, individuals develop a variety of afebrile seizure types, with generalized convulsive seizures being prominent and conferring a high risk for SUDEP. 31 Therefore, we next wanted to determine the effect of soticlestat on spontaneous seizure burden and premature lethality in Dravet mice. We previously showed that induction of a single, brief hyperthermic seizure at weaning enhances subsequent spontaneous seizure incidence and frequency in Scn1a +/− mice, providing better discrimination power for detecting therapeutic benefit. 10 Using a similar paradigm, Scn1a +/− mice were subjected to a hyperthermia‐induced priming seizure before being weaned onto 0.02% soticlestat or vehicle control chow. Mice were then continuously video monitored for 2 weeks or until spontaneous death occurred. At the end of the 2‐week observation period, surviving mice were tested on open field and zero maze assays, and then euthanized (Figure 2A). Bioanalysis performed on the surviving cohort of Scn1a +/− mice showed that subchronic treatment with 0.02% soticlestat resulted in ~50% reduction in brain 24HC levels (Table 2), a similar level of CH24H enzyme inhibition shown to provide seizure protection in kindling models and a survival benefit in the APP/PS‐1 transgenic model. 25 , 26 , 27 In our pilot experiments, the plasma and brain exposure level of soticlestat reached ~8 ng/mL and 30 ng/g, respectively.
FIGURE 2.
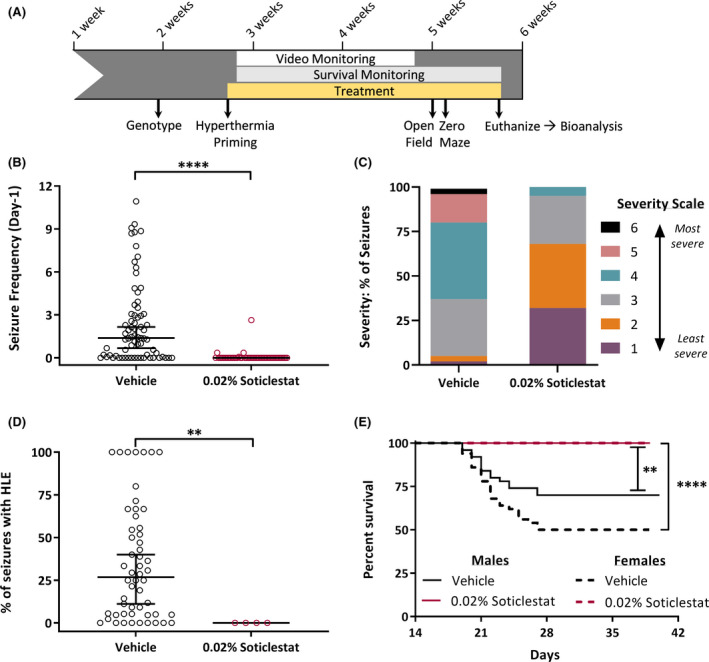
Soticlestat treatment reduces seizure burden and improves survival in Scn1a +/− Dravet mice. (A) Experimental design for spontaneous seizure and survival monitoring in Scn1a +/− mice treated with 0.02% soticlestat formulated in chow or vehicle control chow. Scn1a +/− mice were primed at P17‐18 and immediately weaned onto 0.02% soticlestat or vehicle control chow. Video monitoring commenced on midnight the day of priming and continued for 14 consecutive days. Post video monitoring, open field, and zero maze analysis occurred between P33 and P37. Survival monitoring continued until ~P40. (B) The proportion of Scn1a +/− mice exhibiting spontaneous generalized tonic‐clonic seizure (GTCS) and average GTCS frequency differed between treatment groups. 0.02% soticlestat: 92% seizure free; 0.07 ± 0.06 GTCS/day. Vehicle control: 24% seizure free; 2.4 ± 0.34 GTCS/day. Symbols represent individual mice, horizontal lines represent the median, and error bars represent 95% confidence intervals. 0.02% soticlestat n = 48; Vehicle control n = 71. ****p < 0.0001, Mann‐Whitney. (C) Racine scores of spontaneous seizures differed between treatment groups. Soticlestat treatment (0.02%) reduced seizure severity, with 68% of seizures scoring 1 or 2. Vehicle control treatment resulted in 5% of seizures scoring a 1 or 2. Purple, Severity‐ 1: Rearing and paddling, Straub tail with no other movement. Orange, Severity‐ 2: Rearing and paddling, Straub tail, loss of posture, short bursts of movement, often backwards. Gray, Severity‐ 3: Rearing and paddling with wild running and/or jumping (no loss of posture). Teal, Severity‐ 4: Rearing and paddling with wild running and/or jumping with loss of posture. Pink, Severity‐ 5: Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic hindlimb extension (HLE). Black, Severity‐ 6: Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic HLE and death. 0.02% soticlestat n = 48 seizures; Vehicle control n = 1074 seizures. p < 0.0001, chi‐square. (D) The average percentage of GTCS events that progressed to HLE differed between treatment groups. For vehicle control mice, more seizures progressing to HLE was (median: 26.8%), while none of the seizures progressed to HLE in 0.02% soticlestat‐treated mice. Symbols represent percentage of GTCS events progressing to HLE for each individual mouse (mice with no GTCS are not included). The horizontal line represents the median and error bars represent 95% confidence interval with n = 4 for 0.02% soticlestat and n = 54 for vehicle control. **p < 0.003, Mann‐Whitney. (E) Kaplan Meier plot comparing 40‐day survival between sex and treatment groups. Survival was reduced in vehicle control male (70%) and female (50%) mice compared to 0.02% soticlestat (100%, both sexes). 0.02% soticlestat n = 30/sex; Vehicle control n = 50/sex. **p < 0.0011; ****p < 0.0001, LogRank Mantel‐Cox
TABLE 2.
Brain and plasma exposure levels following administration of 0.02% soticlestat
| 0.02% soticlestat (n = 48) | Mean | SD |
|---|---|---|
| Brain (ng/g) | 68.7 | 8.6 |
| Plasma (ng/mL) | 10.9 | 10 |
| Brain 24HC (% of vehicle) | 49.6 | 7.8 |
Treatment of Scn1a +/− mice with soticlestat resulted in lower daily spontaneous seizure frequency with a median of 0 seizures/day (95% confidence interval [CI], 0–0) in the soticlestat‐treated group compared to a median of 1.4 seizures/day (95% CI, 0.7–2.2) in vehicle controls (p < 0.0001) (Figure 2B). Furthermore 92% of soticlestat treated Scn1a +/− mice were seizure‐free, compared to only 24% of vehicle‐treated mice (Figure 2B). To further assess alterations in seizure burden among mice exhibiting seizures, we scored severity of each seizure event using a modified Racine scale adapted for Scn1a +/− mice (Table 1). Among the 8% of soticlestat‐treated mice that did not achieve seizure freedom, seizure severity was ameliorated relative to vehicle controls (p < 0.0001). In soticlestat‐treated mice, the majority of seizures (~68%) scored as 1–2, the lowest end of severity, whereas only ~5% of seizures scored similarly in vehicle‐treated controls (Figure 2C). It is important to note that none of GTCS events in soticlestat‐treated mice advanced to the most severe stages that include tonic hindlimb extension (HLE) (stages 5–6) (Figure 2C, D), which is indicative of brainstem invasion and correlated with increased SUDEP risk. 10 In contrast, for vehicle‐treated mice, the median percentage of events that advanced to the most severe stages (5–6) was 26.8% (95% CI, 11.1–40.0) (Figure 2D). Treatment with soticlestat completely prevented premature lethality in both male and female Scn1a +/− mice, whereas only 50% of female and 70% of male vehicle‐treated controls survived to at least 5 weeks (p < 0.0011, p < 0.0001, respectively) (Figure 2E). Seizure diary plots (Supplementary Figure 1) show detailed record of seizure events, severity, and deaths at the level of individual mice.
At the conclusion of video monitoring, we assessed the effect of soticlestat treatment on open field and zero maze performance in Scn1a +/− mice because previous studies showed elevated levels of activity and anxiety in similar paradigms. 7 , 8 , 13 Consistent with prior studies, Scn1a +/− vehicle mice showed elevated activity in the open field relative to WT controls (Figure 3A). However, soticlestat treatment had no effect relative to Scn1a +/− vehicle mice on distance traveled (Figure 3A). We did not observe any effect of soticlestat treatment on zero maze performance, and we did not observe a difference between Scn1a +/− vehicle and WT mice (Figure 3B). The lack of difference between Scn1a +/− vehicle and WT mice may be due to background strain differences in baseline performance. 32 , 33 Prior reports of elevated anxiety‐like behavior in Scn1a Dravet models used mice congenic on C57BL/6J, which has elevated baseline anxiety‐like behavior relative to 129 strains. 34 , 35 Consistent with our results, Bahceci and colleagues reported no alteration in elevated plus maze performance in Scn1a +/− mice on the same [C57BL/6J × 129S6/SvEvTac]F1 background. 13 It is important to note that we did not observe any evidence of sedation in these assays for the soticlestat‐treated group in agreement with the previous study. 27
FIGURE 3.
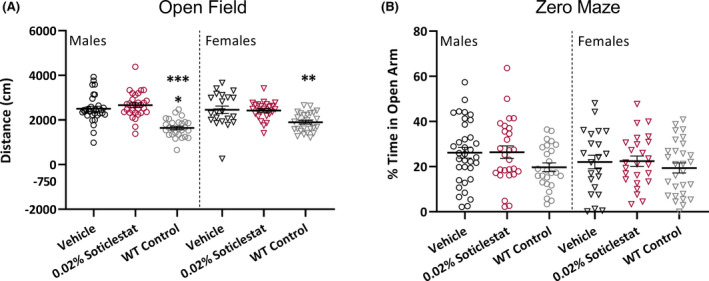
Soticlestat treatment has no effect on open field or zero maze performance of Scn1a +/− mice. (A) Distance traveled in a novel open field environment was not normalized by 0.02% soticlestat treatment. Male (open circles) Scn1a +/− vehicle control and 0.02% soticlestat cohorts traveled 2503 ± 114 cm and 2661 ± 103 cm, respectively, compared to WT vehicle control (1644 ± 78 cm). Female (open triangles) Scn1a +/− vehicle control and 0.02% soticlestat cohorts traveled 2457 ± 168 cm and 2422 ± 81 cm, respectively, compared to WT vehicle control (1898 ± 79 cm). Symbols represent individual mice, horizontal lines represent the group mean, and error bars represent standard error of the mean (SEM). 0.02% soticlestat n = 25–30; Vehicle control n = 21–31; WT vehicle control n = 27–28. ****p < 0.0001, **p < 0.002, Tukey's. (B) Percent time spend in the open arm of a zero maze was unaffected by genotype or 0.02% soticlestat treatment. Symbols represent individual mice, horizontal lines represent the group mean, and error bars represent SEM. 0.02% soticlestat n = 25–27; Vehicle control n = 22–35; WT vehicle control n = 25–29
3.3. Soticlestat decreases electroclinical seizures in Scn1a+/− mice
Following our observation that soticlestat provided strong protection against overt behavioral seizures in Scn1a +/− mice, we sought to evaluate the effect on electroclinical seizures. Video‐EEG is the gold standard for validating efficacy of anti‐seizure compounds to reduce or abolish spontaneous electroclinical seizures. Therefore, we used video‐EEG to confirm that soticlestat reduced electroclinical GTCS in addition to preventing the behavioral component. A separate cohort of Scn1a +/− mice were surgically implanted with EEG headmounts at P17‐18. Following a 48‐hour recovery period, the mice were subjected to a single hyperthermia‐induced priming seizure, and then randomly assigned to 0.02% soticlestat or vehicle control chow. Seizures were monitored by continuous video‐EEG recording for 7 days or until death occurred (Figure 4A).
FIGURE 4.

Soticlestat treatment reduces electroclinical seizure burden in Scn1a +/− Dravet mice. (A) Experimental design for video‐electroencephalography (EEG) monitoring in Scn1a +/− mice treated with 0.02% soticlestat formulated in chow or vehicle control chow. P17‐18 Scn1a +/− mice were surgically implanted with EEG headmounts. At P19‐20, mice were primed and immediately weaned onto 0.02% soticlestat or vehicle control chow and video‐EEG recordings commenced and continued for 7 consecutive days. (B) Representative two‐channel cortical traces from Scn1a +/− mice during ictal events. Black EEG traces represent a vehicle control‐treated mouse during a generalized tonic‐clonic seizure (GTCS) with a severity score of 5. Red traces represent a 0.02% soticlestat‐treated mouse during a GTCS with a severity score of 4. All electroclinical seizures corresponded to observable GTCS on video. Y axis displays µV scale. Each trace represents 2 min. (C) The proportion of Scn1a +/− mice exhibiting electroclinical GTCS events and GTCS frequency differed between treatment groups. 0.02% soticlestat: 60% seizure free; 0.7 ± 0.4 GTCS/day. Vehicle control: 33% seizure free; 2.5 ± 0.5 GTCS/day. Symbols represent individual mice, horizontal lines represent the median and error bars represent 95% confidence interval. 0.02% soticlestat n = 20; Vehicle control n = 22. **p < 0.0017, Mann‐Whitney. (D) Racine scores of electroclinical GTCS events differed between treatment groups. Soticlestat treatment (0.02%) reduced seizure severity, with 66% of seizures scoring 1 or 2. Vehicle control treatment resulted in 20% of seizures scoring a 1 or 2. Purple, Severity‐ 1: Rearing and paddling, Straub tail with no other movement. Orange, Severity‐ 2: Rearing and paddling, Straub tail, loss of posture, short bursts of movement, often backwards. Gray, Severity‐ 3: Rearing and paddling with wild running and/or jumping (no loss of posture). Teal, Severity‐ 4: Rearing and paddling with wild running and/or jumping with loss of posture. Pink, Severity‐ 5: Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic hindlimb extension (HLE). Black, Severity‐ 6: Rearing and paddling with wild running and/or jumping with loss of posture, ending in tonic HLE and death. 0.02% soticlestat n = 55 seizures; Vehicle control n = 239 seizures. p < 0.0001, chi‐square. (E) The percentage of GTCS events that progressed to HLE differed between treatment groups. 0.02% soticlestat median: 14%. Vehicle control median: 46% of seizures. Symbols represent percentage of GTCS events progressing to HLE for each individual mouse (mice with no GTCS are not included). The horizontal line represents the median and error bars represent 95% confidence interval with n = 8 for 0.02% soticlestat and n = 17 for vehicle control. **p < 0.0025, Mann‐Whitney
EEG records of representative GTCS events are shown for both treatment groups in Figure 4B. All electroclinical seizures had an observable behavioral component on video, consistent with our previous reports. 10 , 30 , 36 Seizure frequency and severity was again improved with soticlestat treatment compared to vehicle controls, although there was no change in seizure duration (soticlestat: 40.8 ± 2.5 s; vehicle: 40.0 ± 0.9 s). Approximately 60% of soticlestat‐treated Scn1a +/− mice were seizure‐free, compared to 33% of vehicle controls (p < 0.03) (Figure 4C). Spontaneous seizure frequency of Scn1a +/− mice was lower in soticlestat‐treated mice, with a median of 0 seizures/day (95% CI, 0–0.9) compared to 2.2 seizures/day (95% CI, 0.3–3.3) in vehicle controls (p = 0.0017) (Figure 4C). Seizures in Scn1a +/− mice treated with soticlestat were less severe relative to vehicle controls (p < 0.0001). The majority of seizures (~66%) in soticlestat‐treated mice were scored as 1–2, the lowest end of severity, whereas only ~20% of vehicle control seizures scored similarly (Figure 4D). Furthermore, when soticlestat‐treated Scn1a +/− mice experienced a GTCS, it was less likely to progress to HLE relative to vehicle controls (p < 0.0025) (Figure 4E). Finally, 95% of Scn1a +/− mice treated with soticlestat survived for the duration of the experiment, compared to 55% of vehicle control‐treated mice (p < 0.0037). The single death in the soticlestat treatment group occurred within 12 h of commencing treatment (~60 h post‐surgery). Individual‐level event details are shown in seizure diary plots (Supplementary Figure 2).
4. DISCUSSION
Dravet syndrome is a severe infantile‐onset epilepsy that responds poorly to available treatments. Uncontrolled seizures negatively affect brain development and are a leading risk factor for SUDEP. 37 , 38 , 39 Thus novel treatments that provide better seizure control are needed to improve outcomes for individuals with Dravet syndrome.
In this study, we demonstrated that treatment with soticlestat, a novel potent and highly selective brain‐specific inhibitor of the CH24H enzyme, significantly improved Dravet‐like phenotypes of Scn1a Dravet mouse models. Acute soticlestat treatment provided consistent protection against hyperthermia‐induced seizures in two independent Dravet models on different genetic backgrounds at two different institutions, indicating a robust and reproducible effect. Subchronic treatment of Scn1a +/− mice with soticlestat resulted in a 97% reduction in frequency of spontaneous seizures and lessened severity, indicating an overall lower seizure burden. Favorable response was also demonstrated in Scn1a +/− mice that underwent EEG surgery and monitoring, although the effect was slightly less robust, with a 72% reduction in seizure frequency. The difference in seizure reduction is likely due to differences in study design, most prominently, mice underwent a craniotomy 48 h prior to commencing treatment, which coincided with the start of recording. It is possible that consumption of chow was lower than in video‐only mice that did not undergo surgery (Figure 2), particularly during the first days of recording that overlapped post‐surgical recovery. Consistent with this, among the eight soticlestat‐treated mice that exhibited seizures, four had seizures within the first 48 h of monitoring that subsequently resolved except for a single late seizure of modest severity in one mouse (stage 2). Despite small differences in study design, soticlestat lessened seizure severity and reduced frequency by >70% in these two independent subchronic experiments. Together, these studies are indicative of a robust and reproducible anti‐seizure effect, suggesting that soticlestat may provide therapeutic benefit in Dravet syndrome.
Soticlestat completely prevented premature lethality in Scn1a +/− mice during 14‐day video monitoring, and Scn1a +/− mice that underwent surgery and EEG monitoring exhibited a 95% survival rate. We previously demonstrated that survival extension can be an independent efficacy endpoint that is separable from spontaneous seizure frequency, with lamotrigine providing an example of separability. Treatment of Scn1a +/− mice with lamotrigine abolished the sudden death phenotype; however, there was a 3.5‐fold elevation in seizure frequency, consistent with seizure worsening observed clinically in individuals with Dravet syndrome treated with lamotrigine. 3 , 10 , 40 Sudden death of Scn1a +/− mice is tightly associated with the occurrence of severe seizures with tonic HLE, whereas the absolute number of seizures is not necessarily a key determinant of mortality. 10 , 41 Examination of the vehicle‐treated group in our video monitoring illustrates this relationship. During the 14‐day monitoring period, vehicle‐treated Scn1a +/− mice that died had a range of 1–44 seizures, whereas some surviving vehicle‐treated Scn1a +/− mice had up to 50–100 seizures (Supplementary Figure 1). Thus prevention/reduction in lethality with soticlestat treatment is likely due to preventing progression of seizures to the most severe stage of tonic HLE in the video‐monitoring study and low occurrence in the EEG study. In contrast, Scn1a +/− mice treated with clobazam, a first‐line therapy for Dravet syndrome, had reduced spontaneous seizure frequency, but did not show any improvement in survival and had no difference in the percentage of seizures that progressed to HLE compared to vehicle‐treated mice. 10 Seizures with tonic HLE involve brainstem, a critical component of the autonomic nervous system that modulates cardiovascular and respiratory function. 41 , 42 Our results demonstrate that in addition to providing anti‐seizure properties, soticlestat may prevent spread of seizures from forebrain to brainstem. This suggests that soticlestat may reduce SUDEP risk in addition to providing anti‐seizure benefit.
Of interest, with 14‐day video monitoring we did observe seizures in 8% of soticlestat‐treated Scn1a +/− mice, but onset was delayed by ~1 week compared to vehicle controls. This delay, together with the observed seizure freedom in 92% of mice, suggests the possibility that inhibition of CH24H may interfere with epileptogenesis in the Scn1a +/− mouse model and improve phenotypic outcome through a disease‐modifying effect. Soticlestat also demonstrated disease‐modifying potential in mouse models of PTZ kindling and kainic acid–induced temporal lobe epilepsy. 25 , 26 24HC affects various biological functions, including brain cholesterol homeostasis, inflammation, oxidative stress, and N‐methyl‐d‐aspartate (NMDA) signaling. 43 , 44 , 45 , 46 The mechanism that accounts for the anti‐seizure benefit of soticlestat remains elusive, but it is presumably mediated by lowering 24HC. Considering that 24HC is a cholesterol metabolite, it could be involved in a wide range of brain functions as seen in neuroactive steroids. 47 It is interesting to note that 24HC was reported as a modulator of estrogen receptors. 48 The discovery of therapeutic potential of soticlestat for Dravet syndrome may raise an important argument that epileptic seizures can be controlled by a mechanism that is not necessarily known for a genetic link to epilepsy, thereby opening up a new avenue for safer and more effective seizure medications. Future studies will investigate the biological functions affected by soticlestat to better understand the mechanism(s) underlying the beneficial effect in Scn1a +/− mice.
Overall, our data provide evidence of the ability of soticlestat to drastically improve on core epilepsy phenotypes in the Scn1a +/− mouse model through the novel approach of inhibiting CH24H activity. Based on prior demonstration of safety and target engagement, 27 , 49 clinical translation of soticlestat is ongoing. It should be noted that the soticlestat dosing regimen used in this study yielded clinically relevant levels of pharmacodynamic effects. 50 The current study supports further investigation of therapeutic potential in Dravet syndrome.
CONFLICTS OF INTEREST
TN and SK are employees of Takeda Pharmaceuticals and authors of a patent describing Treatment of CNS conditions (WO‐2019045121). BA and MD were/are employees of Ovid Therapeutics. MD holds equity interest in Ovid Therapeutics. NAH, MJ, TTT, SED, LB, TT, KY, and JAK have no conflicts of interest to disclose.
ETHICAL APPROVAL
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank Nicole Zachwieja, Alexandra Huffman, and Samantha Duarte for technical assistance. This work was supported by Ovid Therapeutics and Takeda Pharmaceuticals (to JAK).
Hawkins NA, Jurado M, Thaxton TT, Duarte SE, Barse L, Tatsukawa T, et al. Soticlestat, a novel cholesterol 24‐hydroxylase inhibitor, reduces seizures and premature death in Dravet syndrome mice. Epilepsia. 2021;62:2845–2857. 10.1111/epi.17062
REFERENCES
- 1. Scheffer IE, Liao J. Deciphering the concepts behind "Epileptic encephalopathy" and "Developmental and epileptic encephalopathy". Eur J Paediatr Neurol. 2020;24:11–4. [DOI] [PubMed] [Google Scholar]
- 2. Samanta D. Changing landscape of dravet syndrome management: an overview. Neuropediatrics. 2020;51:135–45. [DOI] [PubMed] [Google Scholar]
- 3. Wheless JW, Fulton SP, Mudigoudar BD. Dravet syndrome: a review of current management. Pediatr Neurol. 2020;107:28–40. [DOI] [PubMed] [Google Scholar]
- 4. Miller AR, Hawkins NA, McCollom CE, et al. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014;13:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin‐positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–9. [DOI] [PubMed] [Google Scholar]
- 7. Han S, Tai C, Westenbroek RE, et al. Autistic‐like behaviour in Scn1a+/‐ mice and rescue by enhanced GABA‐mediated neurotransmission. Nature. 2012;489:385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito S, Ogiwara I, Yamada K, et al. Mouse with Nav1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis. 2013;49:29–40. [DOI] [PubMed] [Google Scholar]
- 9. Ricobaraza A, Mora‐Jimenez L, Puerta E, et al. Epilepsy and neuropsychiatric comorbidities in mice carrying a recurrent Dravet syndrome SCN1A missense mutation. Sci Rep. 2019;9:14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hawkins NA, Anderson LL, Gertler TS, et al. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin Transl Neurol. 2017;4:326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ogiwara I, Iwasato T, Miyamoto H, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure‐associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bahceci D, Anderson LL, Occelli Hanbury Brown CV, et al. Adolescent behavioral abnormalities in a Scn1a+/− mouse model of Dravet syndrome. Epilepsy Behav. 2020;103:106842. [DOI] [PubMed] [Google Scholar]
- 14. Anderson LL, Absalom NL, Abelev SV, et al. Coadministered cannabidiol and clobazam: Preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia. 2019;60:2224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao D, Ohtani H, Ogiwara I, et al. Efficacy of stiripentol in hyperthermia‐induced seizures in a mouse model of Dravet syndrome. Epilepsia. 2012;53:1140–5. [DOI] [PubMed] [Google Scholar]
- 16. Kaplan JS, Stella N, Catterall WA, et al. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2017;114:11229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patra PH, Serafeimidou‐Pouliou E, Bazelot M, et al. Cannabidiol improves survival and behavioural co‐morbidities of Dravet syndrome in mice. Br J Pharmacol. 2020;177:2779–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug‐resistant seizures in the dravet syndrome. N Engl J Med. 2017;376:2011–20. [DOI] [PubMed] [Google Scholar]
- 19. Eschbach K, Knupp KG. Stiripentol for the treatment of seizures in Dravet syndrome. Expert Rev Clin Pharmacol. 2019;12:379–88. [DOI] [PubMed] [Google Scholar]
- 20. Lagae L, Sullivan J, Knupp K, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2019;394:2243–54. [DOI] [PubMed] [Google Scholar]
- 21. Russell DW, Halford RW, Ramirez DM, et al. Cholesterol 24‐hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem. 2009;78:1017–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun MY, Izumi Y, Benz A, et al. Endogenous 24S‐hydroxycholesterol modulates NMDAR‐mediated function in hippocampal slices. J Neurophysiol. 2016;115:1263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lund EG, Xie C, Kotti T, et al. Knockout of the cholesterol 24‐hydroxylase gene in mice reveals a brain‐specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–8. [DOI] [PubMed] [Google Scholar]
- 24. Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nishi T, Salamone A, Terrone G, Di Sapia R, Balosso S, Ra T. TAK‐935 (OV935) exerts neuroprotective and disease‐modifying effects in a murine model of epileptogenesis. TAK‐935 (OV935) exerts neuroprotective and disease‐modifying effects in a murine model of epileptogenesis. AES 2019 Annual Meeting Abstract Database. AESnet.org. Available at: https://www.aesnet.org/abstractslisting/copyright‐notice/
- 26. Nishi T, Fujimoto S, Hasegawa S, Watanabe S, Kondo S. Inhibition of cholesterol 24‐hydroxylase is a novel pharmacological strategy for epilepsy treatment. Inhibition of cholesterol 24‐hydroxylase is a novel pharmacological strategy for epilepsy treatment. AES 2017 Annual Meeting Abstract Database. AESnet.org. Available at: https://www.aesnet.org/abstractslisting/copyright‐notice/
- 27. Nishi T, Kondo S, Miyamoto M, et al. Soticlestat, a novel cholesterol 24‐hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci Rep. 2020;10:17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ziyatdinova S, Gurevicius K, Kutchiashvili N, et al. Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 2011;94:75–85. [DOI] [PubMed] [Google Scholar]
- 29. Tatsukawa T, Ogiwara I, Mazaki E, et al. Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin‐expressing cells. Neurobiol Dis. 2018;112:24–34. [DOI] [PubMed] [Google Scholar]
- 30. Hawkins NA, Lewis M, Hammond RS, et al. The synthetic neuroactive steroid SGE‐516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci Rep. 2017;7:15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3–9. [DOI] [PubMed] [Google Scholar]
- 32. Cook MN, Crounse M, Flaherty L. Anxiety in the elevated zero‐maze is augmented in mice after repeated daily exposure. Behav Genet. 2002;32:113–8. [DOI] [PubMed] [Google Scholar]
- 33. Kulkarni SK, Singh K, Bishnoi M. Elevated zero maze: a paradigm to evaluate antianxiety effects of drugs. Methods Find Exp Clin Pharmacol. 2007;29:343–8. [DOI] [PubMed] [Google Scholar]
- 34. Mulligan MK, Abreo T, Neuner SM, et al. Identification of a functional non‐coding variant in the GABA (A) receptor α2 subunit of the C57BL/6J mouse reference genome: major implications for neuroscience research. Front Genet. 2019;10:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holmes A, Lit Q, Murphy DL, et al. Abnormal anxiety‐related behavior in serotonin transporter null mutant mice: the influence of genetic background. Genes Brain Behav. 2003;2:365–80. [DOI] [PubMed] [Google Scholar]
- 36. Hawkins NA, Zachwieja NJ, Miller AR, et al. Fine mapping of a dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA‐Seq. PLOS Genet. 2016;12:e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Whitney R, Donner EJ. Risk factors for sudden unexpected death in epilepsy (SUDEP) and their mitigation. Curr Treat Options Neurol. 2019;21:7. [DOI] [PubMed] [Google Scholar]
- 38. Berg AT, Smith SN, Frobish D, et al. Longitudinal assessment of adaptive behavior in infants and young children with newly diagnosed epilepsy: influences of etiology, syndrome, and seizure control. Pediatrics. 2004;114:645–50. [DOI] [PubMed] [Google Scholar]
- 39. O'Reilly H, Eltze C, Bennett K, et al. Cognitive outcomes following epilepsy in infancy: A longitudinal community‐based study. Epilepsia. 2018;59:2240–8. [DOI] [PubMed] [Google Scholar]
- 40. Wirrell EC. Treatment of dravet syndrome. Can J Neurol Sci. 2016;43(Suppl 3):S13–8. [DOI] [PubMed] [Google Scholar]
- 41. Applegate CD, Samoriski GM, Burchfiel JL. Evidence for the interaction of brainstem systems mediating seizure expression in kindling and electroconvulsive shock seizure models. Epilepsy Res. 1991;10:142–7. [DOI] [PubMed] [Google Scholar]
- 42. Chever O, Zerimech S, Scalmani P, et al. GABAergic neurons and NaV1.1 channel hyperactivity: a novel neocortex‐specific mechanism of Cortical Spreading Depression. bioRxiv 2020:2020.2003.2014.991158.
- 43. Paul SM, Doherty JJ, Robichaud AJ, et al. The major brain cholesterol metabolite 24(S)‐hydroxycholesterol is a potent allosteric modulator of N‐methyl‐D‐aspartate receptors. J Neurosci. 2013;33:17290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Noguchi N, Saito Y, Urano Y. Diverse functions of 24(S)‐hydroxycholesterol in the brain. Biochem Biophys Res Commun. 2014;446:692–6. [DOI] [PubMed] [Google Scholar]
- 45. Alexandrov P, Cui JG, Zhao Y, et al. 24S‐hydroxycholesterol induces inflammatory gene expression in primary human neural cells. NeuroReport. 2005;16:909–13. [DOI] [PubMed] [Google Scholar]
- 46. Sun M‐Y, Linsenbardt AJ, Emnett CM, et al. 24(S)‐hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist. 2016;22:132–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mellon SH, Griffin LD. Neurosteroids: biochemistry and clinical significance. Trends Endocrinol Metab. 2002;13:35–43. [DOI] [PubMed] [Google Scholar]
- 48. Umetani M, Domoto H, Gormley AK, et al. 27‐Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat Med. 2007;13:1185–92. [DOI] [PubMed] [Google Scholar]
- 49. Bialer M, Johannessen SI, Koepp MJ, et al. Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. Drugs in preclinical and early clinical development. Epilepsia 2018;59:1811–41. [DOI] [PubMed] [Google Scholar]
- 50. Wang S, Chen G, Merlo Pich E, Affinito J, Cwik M, Faessel H. Safety, tolerability, pharmacokinetics, pharmacodynamics, bioavailability and food effect of single doses of soticlestat in healthy subjects. British Journal of Clinical Pharmacology 2021. Online ahead of print. 10.1111/bcp.14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material