

Abstract
Objective
To evaluate the safety and efficacy of a novel formulation of midazolam administered as a single‐dose nasal spray (MDZ–NS) in the outpatient treatment of patients experiencing seizure clusters (SCs).
Methods
This was a phase III, randomized, double‐blind, placebo‐controlled trial (ClinicalTrials.gov NCT01390220) with patients age ≥12 years on a stable regimen of antiepileptic drugs. Following an in‐clinic test dose phase (TDP), patients entered an outpatient comparative phase (CP) and were randomized (2:1) to receive double‐blind MDZ–NS 5 mg or placebo nasal spray, administered by caregivers when they experienced an SC. The primary efficacy end point was treatment success (seizure termination within 10 minutes and no recurrence 10 minutes to 6 hours after trial drug administration). Secondary efficacy end points were proportion of patients with seizure recurrence 10 minutes to 4 hours, and time‐to‐next seizure >10 minutes after double‐blind drug administration. Safety was monitored throughout.
Results
Of 292 patients administered a test dose, 262 patients were randomized, and 201 received double‐blind treatment for an SC (n = 134 MDZ–NS, n = 67 placebo, modified intent‐to‐treat population). A significantly greater proportion of MDZ–NS‐ than placebo‐treated patients achieved treatment success (53.7% vs 34.4%; P = 0.0109). Significantly, fewer MDZ–NS‐ than placebo‐treated patients experienced seizure recurrence (38.1% vs 59.7%; P = 0.0043). Time‐to‐next seizure analysis showed early separation (within 30 minutes) between MDZ–NS and placebo that was maintained throughout the 24‐hour observation period (21% difference at 24 hours; P = 0.0124). Sixteen patients (5.5%) discontinued because of a treatment‐emergent adverse event (TEAE) during the TDP and none during the CP. During the CP, 27.6% and 22.4% of patients in the MDZ–NS and placebo groups, respectively, experienced ≥1 TEAE.
Significance
MDZ–NS was superior to placebo in providing rapid, sustained seizure control when administered to patients experiencing an SC in the outpatient setting and was associated with a favorable safety profile.
Keywords: acute intervention, acute repetitive seizures, benzodiazepine, epilepsy, intranasal, rescue
Key points.
Midazolam nasal spray (MDZ–NS) has been developed for the outpatient treatment of patients with epilepsy who are experiencing seizure clusters
In a phase 3, randomized, double‐blind, placebo‐controlled trial, caregivers administered 5 mg MDZ–NS to 134 patients and placebo to 67
A significantly greater proportion of patients achieved treatment success with MDZ–NS than with placebo (53.7% vs 34.4%; P = 0.0109)
During double‐blind treatment, no patient experienced acute central respiratory depression and none discontinued due to adverse events
Use of MDZ–NS was associated with rapid and sustained seizure control, and a favorable safety profile in patients as young as 12 years
1. INTRODUCTION
Seizure clusters (SCs) or acute repetitive seizures are episodes characterized by the occurrence of seizures that differ from patients’ usual pattern in type, frequency, severity, and duration with an onset easily recognizable by caregivers and physicians.1 SCs can lead to a further decline in patients’ quality of life and increased morbidity, hospitalization, and risk of mortality.2, 3, 4
Most SCs occur outside of hospitals, typically necessitating acute intervention to minimize adverse sequelae, including potentially life‐threatening status epilepticus.5, 6, 7 Intravenous benzodiazepines, the mainstay of treatment in seizure emergencies, cannot be administered by non–health care professionals (HCPs). Diazepam rectal gel (PR‐DZP) is the only drug approved for out‐of‐hospital administration in the United States,8 and buccal midazolam (MDZ) is approved for use in children in some European countries.9 Both, however, have limitations that restrict their use; notably, rectal administration can be limited by physical and social constraints, while buccal MDZ is associated with risk of aspiration, inconsistent absorption due to ictal hypersalivation and buccal secretion, and first‐pass metabolism if swallowed.5, 10
Intranasal administration of the injectable solution of MDZ was first reported in 1996 to be a feasible option for the treatment of children with acute seizures.11 Since then, its effectiveness has been reported in numerous prospective, open‐label studies, whether dripped directly into the nostrils12, 13, 14, 15, 16, 17, 18, 19, 20 or administered via sprays or atomization devices.21, 22 A novel MDZ formulation, optimized for delivery as a single‐dose nasal spray (MDZ–NS), has been developed in conjunction with its delivery device as a combination product. The objective of the current trial was to evaluate the safety and efficacy of MDZ–NS compared with placebo in the outpatient treatment of patients with epilepsy experiencing an SC.
2. METHODS
Acute Rescue Therapy in Epilepsy with Midazolam Intranasal Spray (ARTEMIS‐1) was a phase III, randomized, double‐blind, placebo‐controlled trial, conducted between July 2011 and March 2017 across Australia, New Zealand, Israel, North America, and Europe (ClinicalTrials.gov NCT01390220; EudraCT 2011‐001318‐32). It was conducted in accordance with the ethical principles of the Declaration of Helsinki, the International Council for Harmonization's Good Clinical Practice, and applicable regulatory requirements. The protocol, consent form, and patient materials were approved by the institutional review boards of the participating centers. Participants provided written informed consent before entering the trial.
2.1. Trial design
The trial consisted of two phases (Figure 1). An in‐clinic, open‐label test dose phase (TDP) allowed assessment of drug safety while patients were under observation. The subsequent double‐blind, comparative phase (CP) allowed comparison of MDZ–NS with placebo in the outpatient setting.
Figure 1.
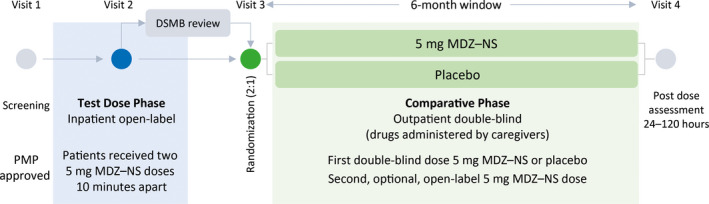
Trial design. At screening, each patient's individualized patient management plan (PMP) was reviewed by a member of the Epilepsy Study Consortium who determined whether the cluster description met the protocol definition and provided final approval for trial participation. A review of safety data from at least the first 25 patients who completed the TDP was required before patients could progress to the CP. The review was conducted by an independent Data and Safety Monitoring Board (DSMB), as were further reviews at periodic intervals during the trial. MDZ–NS, midazolam nasal spray
Patients were prohibited from progressing from TDP to CP if they met certain criteria, such as Observer's Assessment of Alertness/Sedation (OAA/S) responsiveness score 1, or oxygen saturation <90% for >30 seconds. If a patient discontinued because of these, investigators recorded discontinuation due to an adverse event (AE).
2.2. Trial population
Patients age ≥12 years with epilepsy and a history of SCs were eligible to participate if they had an adult caregiver able to recognize clusters and administer treatment. Clusters had to be composed of ≥2 seizures (focal or generalized); last ≥10 minutes; and have an observable, stereotyped, and recognizably different pattern from patients’ noncluster seizure activity, with another seizure occurring within 6 hours of cluster onset. The pattern must have been established >3 months before screening, with ≥3 clusters occurring the year before, and ≥1 occurring ≤4 months before screening. Each patient's pattern was described by an investigator in an individualized patient management plan (PMP). All PMPs were reviewed by a member of the Epilepsy Study Consortium, who determined whether the cluster description met the protocol definition, ensuring consistency in enrollment, and provided final approval for trial participation.
Patients had to be on a stable regimen of antiepileptic drugs (AEDs); dose changes were allowed provided the new dose was stable for ≥7 days before trial drug administration. Use of benzodiazepines was allowed as rescue therapy, or for nonepilepsy indications, if used ≤3 days in a 7‐day period, but not for maintenance therapy as an AED. Patients were ineligible if they had status epilepticus due to cluster progression 2 years; psychogenic nonepileptic seizure(s) 5 years; major depressive episode 6 months; or suicidal ideation/attempts, nonpostictal psychosis, or drug or alcohol abuse 1 year before screening. They were also ineligible if they had a diagnosis of acute narrow‐angle glaucoma, or progressive neurologic or severe cardiorespiratory disease. Sexually active women of childbearing potential were required to use contraception. Restrictions on concomitant medications included those with potential relevant pharmacokinetic (eg, significant cytochrome P450 (CYP)3A4 inhibitors or inducers) or pharmacodynamic (eg, opioids) interactions with MDZ.
2.3. Randomization and blinding
Patients were randomized using the Interactive Response Technology System. The randomization code was generated using fixed blocks by a nonblinded statistician not otherwise involved in the trial. Patients, caregivers, investigators, and trial personnel were unaware of treatment assignment, and the sponsor did not have access to the randomization code until after database lock.
2.4. Procedures
During the TDP, patients (in a stable condition and not experiencing seizures) received two 5‐mg MDZ–NS doses 10 minutes apart. Dose selection was based on results of two phase I trials23, 24 and published literature. Initially, patients could not proceed from the TDP to the CP until an independent Data and Safety Monitoring Board (DSMB) reviewed TDP data from ≥25 patients. Enrollment was paused during the review; when resumed, patients progressed directly from TDP to CP. Safety reviews were also conducted after approximately 33, 66, 99, 132, and 165 patients completed the CP.
Caregivers were trained on trial drug administration, safety monitoring, trial‐related measurements, and cardiopulmonary resuscitation. They were provided with information in PMPs on when to administer the trial drug, requirements for administration of the second dose, an emergency rescue protocol, and contact information for trial nurses and emergency services, as well as a diary to record seizure information, trial drug administration, and safety data.
At the start of the CP, patients were randomized 2:1 to receive 5 mg MDZ–NS or placebo administered by their caregiver when they experienced an SC. There was a 6‐month window for patients to receive double‐blind treatment for a qualifying SC. The double‐blind dose could be followed by an optional, open‐label 5‐mg MDZ–NS dose if the cluster did not terminate within 10 minutes, or another seizure occurred up to 6 hours after initial drug administration and the patient did not have <8 breaths/minute, did not require emergency rescue treatment and assisted breathing or intubation, and did not have excessive sedation. Patients and caregivers returned to the clinical site 24–120 hours after drug administration.
2.5. Outcomes
2.5.1. Efficacy outcomes
The primary efficacy outcome was the proportion of patients achieving treatment success, a composite measure defined as seizure termination within 10 minutes of, and no seizure recurrence 10 minutes to 6 hours after, double‐blind administration of trial drug during the CP. There were two secondary efficacy outcomes: proportion of patients with seizure recurrence 10 minutes to 4 hours after trial drug administration, and time‐to‐next seizure occurring >10 minutes after trial drug administration.
Efficacy was further evaluated using three exploratory outcomes: treatment success with the second dose (open‐label 5 mg MDZ–NS), return to baseline functionality within 24 hours of drug administration according to caregiver evaluation, and proportion of patients with seizure recurrence 10 minutes to 24 hours after trial drug administration.
2.5.2. Safety outcomes
Patients were monitored throughout the trial for treatment‐emergent AEs (TEAEs), coded according to Medical Dictionary for Regulatory Activities version 16.1. Given that MDZ has a short half‐life and that there may have been a long interval between the TD and exposure to double‐blind drug during the CP, presentation of TEAEs within 2 days of administration was included to provide a more relevant assessment of TEAEs (independent of investigator causality assessment) that might be attributable to trial drug. Analysis of AEs of special interest (AESI) was also performed to evaluate specific risks and meet regulatory requirements based on the known pharmacology of MDZ, its use as an AED, and route of administration (see supplement for full details). Safety assessments also included vital signs measurements; 12‐lead electrocardiograms; physical, nasal, and neurologic examinations; clinical laboratory tests; and the Columbia‐Suicide Severity Rating Scale (C‐SSRS).
2.6. Statistical analyses
Originally, a sample size of 132 was planned based on a placebo response of 30%, but this was amended following publication of a trial with another benzodiazepine reporting a higher placebo response in the treatment of patients with SCs.25 In the updated plan, treatment success in the placebo arm was assumed to be 40%; therefore a larger sample size of 240 patients (160 MDZ–NS and 80 placebo) completing the CP was deemed necessary to reach 90% power in demonstrating a clinically meaningful difference in treatment success between MDZ–NS and placebo (odds ratio 2.9).
Three interim analyses were planned for when 132, 165, and 204 patients had received ≥1 dose of trial drug during the CP. Patients lost to follow‐up or with missing data were considered treatment failures. At each point, a one‐sided Fisher's exact test for treatment success for possible early stopping was performed and results compared with critical values for the efficacy boundary were calculated according to the Lan‐DeMets Pocock approximation (using R package gsDesign version 2.8‐8). Predictive probability of success at the maximum sample size was also calculated at each interim analysis, and if <10%, the trial was to be stopped for futility. If stopped early, the P value required for success would be the value determined for the final analysis (N = 240) according to the prespecified Lan‐DeMets Pocock boundary (P = 0.0085). Interim analyses were conducted by a monitoring committee, independent of both the sponsor and DSMB.
The primary outcome was analyzed by Fisher's exact test, and a chi‐square test performed as a sensitivity analysis. Patients without sufficient data to confirm treatment success were classified as treatment failures. Fisher's exact test was used to analyze the proportion of patients with seizure recurrence 10 minutes to 4 hours after double‐blind drug administration. The Kaplan‐Meier method was used to analyze time‐to‐next seizure after double‐blind drug administration. Time‐to‐next seizure in treatment groups was compared using log‐rank test.
Statistical comparisons were performed using two‐sided tests at the α = 0.05 significance level (except for interim analyses). For the primary and secondary efficacy outcomes, a statistical gate‐keeping procedure was applied to control for multiplicity. All analyses were conducted using SAS version 9.1.3 or higher (SAS Institute, Inc.).
The safety population included all patients who received ≥1 dose of trial drug, including those who discontinued following the TDP, before randomization. The randomized safety population consisted of randomized patients who received ≥1 trial drug dose during the CP. Analyses of all efficacy outcomes were conducted with data from the modified intent‐to‐treat (mITT) population, which consisted of patients in the randomized safety population who had any posttreatment efficacy assessment. The per protocol population (PPP) included all patients in the mITT population who did not discontinue, or who had excludable protocol deviations. Data from the PPP were used for supportive analyses to assess robustness of the primary analysis.
3. RESULTS
The trial was stopped early, before the third interim analysis, due to acquisition of the original sponsor company. The trial remained blinded until database lock, and the US Food and Drug Administration was informed of the decision. Given early termination, a conservative statistical approach was taken by using the prespecified P value (P = 0.0085, one‐sided Fisher's exact test) required for treatment success for the final target enrollment (N = 240).
Of 402 patients screened, 292 entered the TDP and received ≥1 MDZ–NS dose (safety set; Figure 2). Subsequently, 262 patients were randomized and entered the CP; 201 received ≥1 double‐blind dose of trial drug and had posttreatment efficacy assessments, constituting the mITT set (134 received MDZ–NS, and 67 placebo). Overall, 200 patients completed the trial and 92 discontinued: 30 during the TDP, 61 during the CP, and one after the CP, before completing procedures at visit 4.
Figure 2.
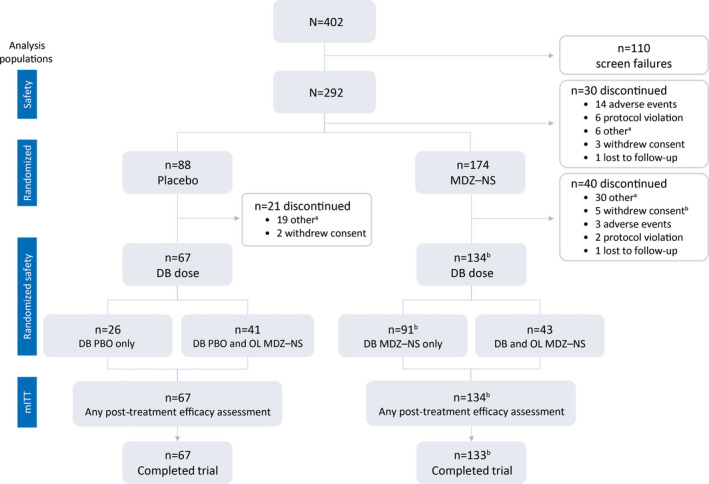
Patient disposition over the course of the trial and analysis populations. DB, double‐blind; MDZ–NS, midazolam nasal spray; mITT, modified intent‐to‐treat; OL, open‐label aOther reasons for discontinuation included: patient did not experience/treat seizure cluster(s) according to trial criteria within protocol‐specified time period; caregiver no longer available; trial drug unavailable at site; patient/caregiver unable to comply with trial procedures/visits; trial terminated; site closure bOne patient received treatment in the CP and had postdose efficacy assessments, but subsequently withdrew consent before completing procedures at visit 4
Demographics and disease characteristics were similar in the treatment arms (Table 1). According to the protocol, all patients were on stable AED regimens (Table S1).
Table 1.
Demographics and baseline disease characteristics
| Safety population | Randomized safety population | ||
|---|---|---|---|
| MDZ–NS (N = 292) | MDZ–NS (n = 134) | Placebo (n = 67) | |
| Age (years) | |||
| Mean (SD) | 33.0 (11.96) | 34.0 (11.23) | 31.5 (12.83) |
| Median (range) | 31.5 (12–65) | 32.0 (14–61) | 27.0 (12–62) |
| Age group, n (%) | |||
| <18 years | 18 (6.2) | 5 (3.7) | 5 (7.5) |
| ≥18 to <65 years | 272 (93.2) | 129 (96.3) | 62 (92.5) |
| ≥65 years | 2 (0.7) | 0 | 0 |
| Sex, n (%) | |||
| Male | female | 146 (50.0) | 146 (50.0) | 68 (50.7) | 66 (49.3) | 33 (44.8) | 37 (55.2) |
| Race, n (%) | |||
| White | 275 (94.2) | 125 (93.3) | 65 (97.0) |
| Black or African American | 7 (2.4) | 3 (2.2) | 1 (1.5) |
| Other | 5 (1.7) | 2 (1.5) | 1 (1.5) |
| Asian | 2 (0.7) | 2 (1.5) | 0 |
| American Indian or Alaska Native | 2 (0.7) | 2 (1.5) | 0 |
| Native Hawaiian or Other Pacific Islander | 1 (0.3) | 0 | 0 |
| Body mass index (kg/m2) | n = 1 (0.3) | n = 0 | n = 0 |
| Mean (SD) | 26.05 (5.924) | 25.89 (5.745) | 25.01 (5.066) |
| Typical duration of cluster (min) | n = 278 | n = 129 | n = 63 |
| Mean (SD) | 259.10 (548.79) | 270.77 (572.75) | 239.34 (528.05) |
| Median (range) | 67.5 (2.5–4320.0) | 65.0 (2.5–4320.0) | 60.0 (8.5–2880.0) |
| Number of years since clusters onset | n = 283 | n = 133 | n = 63 |
| Mean (SD) | 9.24 (9.93) | 9.88 (10.11) | 6.69 (6.30) |
| Median (range) | 6.0 (0.3–62.0) | 5.0 (0.3–48.0) | 5.0 (0.5–32.0) |
| Number of episodes in year before visit 1 | |||
| Mean (SD) | 50.8 (109.99) | 56.3 (128.53) | 49.1 (98.43) |
| Median (range) | 15 (3–999) | 18 (3–999) | 15 (4–600) |
| Number of seizures in cluster episode | n = 291 | ||
| Mean (SD) | 12.23 (21.02) | 12.81 (23.60) | 14.16 (25.57) |
| Median (range) | 6.0 (2–200) | 5.2 (2–200) | 6.0 (2–170) |
| Seizure type(s) in cluster, n (%)a | |||
| Focal impaired awareness (complex partial) | 153 (52.4) | 71 (53.0) | 33 (49.3) |
| Focal to bilateral (secondary generalized) | 97 (33.2) | 46 (34.3) | 20 (29.9) |
| Focal aware (simple partial) | 60 (20.5) | 23 (17.2) | 17 (25.4) |
| Primary generalized tonic‐clonic | 20 (6.8) | 8 (6.0) | 7 (10.4) |
| Tonic | 18 (6.2) | 8 (6.0) | 3 (4.5) |
| Absence | 13 (4.5) | 7 (5.2) | 0 |
| Other | 11 (3.8) | 7 (5.2) | 0 |
| Myoclonic | 10 (3.4) | 3 (2.2) | 3 (4.5) |
| Atonic | 2 (0.7) | 2 (1.5) | 0 |
All values are based on the safety or randomized safety populations unless specified otherwise. Abbreviations: MDZ–NS, midazolam nasal spray; SD, standard deviation
Patients may have reported >1 seizure type.
3.1. Efficacy outcomes
Among patients who received MDZ–NS, 53.7% (95% confidence interval [CI] 45.3‐62.2) experienced treatment success compared with 34.3% (23.0‐45.7) who received placebo. The difference was statistically significant (P = 0.0069, one‐sided Fisher's exact test; P = 0.0109, two‐sided). Results of the sensitivity analysis based on PPP data were consistent with those of the mITT analysis, with a significantly greater proportion of MDZ–NS‐treated patients achieving treatment success than placebo‐treated patients (54.4% [45.7, 63.1] vs 32.8% [21.3, 44.3]; P = 0.0056, Fisher's exact test; P = 0.0049, chi‐square test). For the components, seizure(s) termination occurred within 10 minutes in 80.6% of patients treated with MDZ–NS and 70.1% of those treated with placebo; 58.2% of MDZ–NS‐treated and 37.3% of placebo‐treated patients did not experience seizures starting 10 minutes and up to 6 hours after trial drug administration (patients who were administered the second dose of trial drug were assumed to have had a seizure).
Results of the secondary outcomes supported those of the primary outcome. A significantly smaller proportion of patients in the MDZ–NS group experienced seizure recurrence 10 minutes to 4 hours after drug administration than those in the placebo group (38.1% vs 59.7%; P = 0.0043, Fisher's exact test). Time‐to‐next seizure after double‐blind drug administration was longer (P = 0.0124) in the MDZ–NS group than in the placebo group (25th percentile, 2.6 vs 0.9 hours); median time‐to‐next seizure could not be estimated since <50% of cluster episodes for which patients received a first MDZ–NS dose were followed by another seizure within 24 hours. Based on the Kaplan‐Meier analysis, the probability of experiencing no seizures over the 24‐hour observation period after 10 minutes of drug administration was significantly greater in the MDZ–NS group than in the placebo group (58.3% vs 37.1%; P = 0.0124, log‐rank test) (Figure 3). Separation between MDZ–NS and placebo curves was apparent from 30 minutes after double‐blind administration. In this analysis, 50 patients (37.3%) in the MDZ–NS group and 31 (46.3%) in the placebo group experienced a next seizure within 24 hours of drug administration.
Figure 3.
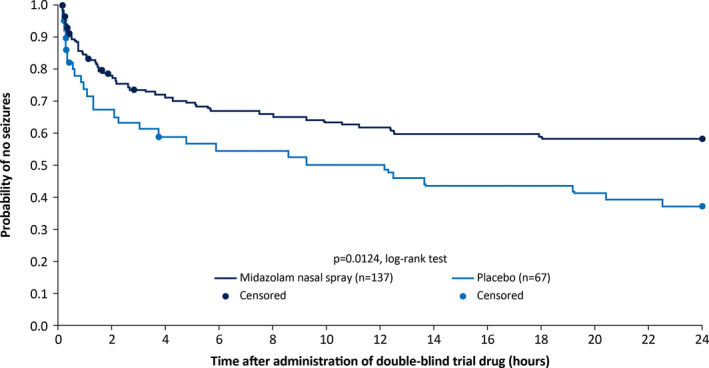
Secondary efficacy outcome: probability of experiencing no seizures over the 24‐hour observation period after 10 minutes of double‐blind trial drug administration based on a Kaplan‐Meier analysis of time‐to‐next seizure in the modified intent‐to‐treat population. Patients who did not have another seizure before the end of the 24‐hour observation period, and who had not been administered the second dose of trial drug, were censored at the end of the observation period. Those administered the second dose of trial drug who did not have a seizure before the administration of the second dose were censored at the time of the second dose
With regard to exploratory outcomes, fewer MDZ–NS‐ than placebo‐treated patients required administration of the open‐label dose (31.3% vs 61.2%). Of patients receiving the open‐label dose, 54.8% (39.7, 69.8) in the MDZ–NS group (5 mg + 5 mg) and 65.9% (51.3, 80.4) in the placebo group (placebo + 5 mg MDZ–NS) achieved treatment success. Based on the assumption that a seizure had occurred if the open‐label dose was administered, 49.3% (40.8, 57.7) of patients in the MDZ–NS group compared with 74.6% (64.2, 85.0) in the placebo group experienced seizure recurrence within 10 minutes to 24 hours from double‐blind administration (P = 0.0008, Fisher's exact test). Among MDZ–NS‐treated patients, 72.4% (64.0, 79.8) had documented return to baseline functionality within 24 hours after double‐blind drug administration compared with 43.3% (31.2, 56.0) of placebo‐treated patients (P < 0.0001, Fisher's exact test).
3.2. Safety outcomes
Of 292 patients in the TDP, 286 received two MDZ–NS doses and six received one dose. During the CP, of 134 patients randomized to MDZ–NS, 91 (67.9%) received a single double‐blind dose of trial drug, and 43 (32.0%) received the double‐blind and open‐label doses. Corresponding values among the 67 patients in the placebo group were 26 and 41 (38.8% and 61.1%).
3.2.1. Test dose phase
During the TDP, 51.4% of patients experienced ≥1 TEAE (Table 2). The most frequently reported TEAEs within 2 days of MDZ–NS administration are summarized in Table 3, and overall in Table S2; most were mild or moderate in intensity. TEAEs considered related to trial drug occurred in 108 patients (37%).
Table 2.
Overview of treatment‐emergent adverse events during the TDP (safety population) and CP (randomized safety population)
| Test dose phase | Comparative phase MDZ–NS | Comparative phase placebo | |||
|---|---|---|---|---|---|
| N = 292 | DB only (n = 91) | DB + OL (n = 43) | DB PBO only (n = 26) | DB PBO + OL MDZ–NS (n = 41) | |
| ≥1 TEAE | 150 (51.4) | 24 (26.4) | 13 (30.2) | 6 (23.1) | 9 (22.0) |
| Mild | 81 (27.7) | 17 (18.7) | 9 (20.9) | 5 (19.2) | 4 (9.8) |
| Moderate | 49 (16.8) | 4 (4.4) | 3 (7.0) | 1 (3.8) | 5 (12.2) |
| Severe | 20 (6.8) | 3 (3.3) | 1 (2.3) | 0 | 0 |
| ≥1 Treatment‐related TEAE | 108 (37.0) | 20 (22.0) | 13 (30.2) | 5 (19.2) | 7 (17.1) |
| Mild | 63 (21.6) | 13 (14.3) | 9 (20.9) | 4 (15.4) | 3 (7.3) |
| Moderate | 34 (11.6) | 4 (4.4) | 3 (7.0) | 1 (3.8) | 4 (9.8) |
| Severe | 11 (3.8) | 3 (3.3) | 1 (2.3) | 0 | 0 |
| ≥1 Serious TEAE | 14 (4.8) | 1 (1.1) | 0 | 0 | 1 (2.4) |
| ≥1 Treatment‐related serious TEAE | 3 (1.0) | 0 | 0 | 0 | 0 |
| Discontinuation due to TEAE | 16 (5.5) | 0 | 0 | 0 | 0 |
Adverse events were assigned to TDP if they occurred after administration of test dose and before CP dose, and to CP only at/after the CP dose, not based on randomization
Abbreviations: DB, double‐blind; MDZ–NS, midazolam nasal spray; OL, open‐label; PBO, placebo; TEAE, treatment‐emergent adverse event
Table 3.
Treatment‐emergent adverse events reported for ≥ 5% of patients in any treatment arm within 2 days after trial drug administration during the TD (safety population) and CP (randomized safety population)
| Preferred term, n (%) | Test dose phase | Comparative phase | |||
|---|---|---|---|---|---|
| MDZ–NS | MDZ–NS | Placebo | |||
| N = 292 | DB only (n = 91) | DB + OL (n = 43) | DB PBO only (n = 26) | DB PBO + OL MDZ–NS (n = 41) | |
| Any TEAE | 109 (37.3) | 21 (23.1) | 13 (30.2) | 6 (23.1) | 9 (22.0) |
| Nasal discomfort | 47 (16.1) | 5 (5.5) | 7 (16.3) | 2 (7.7) | 3 (7.3) |
| Somnolence | 29 (9.9) | 9 (9.9) | 4 (9.3) | 1 (3.8) | 4 (9.8) |
| Lacrimation increased | 20 (6.8) | 1 (1.1) | 1 (2.3) | 0 | 1 (2.4) |
| Product taste abnormal | 17 (5.8) | 4 (4.4) | 0 | 0 | 0 |
| Throat irritation | 15 (5.1) | 2 (2.2) | 3 (7.0) | 0 | 1 (2.4) |
| Headache | 1 (0.3) | 6 (6.6) | 1 (2.3) | 0 | 0 |
Adverse events were assigned to TDP if they occurred after administration of test dose and before CP dose, and to CP only at/after the CP dose, not based on randomization.
Abbreviations: DB, double‐blind; MDZ–NS, midazolam nasal spray; OL, open‐label; PBO, placebo; TEAE, treatment‐emergent adverse event.
Fourteen patients (4.8%) experienced 18 SAEs; however, only three patients had events considered treatment‐related by the investigator. One patient each experienced sedation and somnolence. The third patient experienced an SC 9 days after administration of the test dose, likely reflecting underlying disease.
Seventeen patients discontinued due to AEs; one patient had a brain tumor recurrence diagnosis before in‐clinic administration that was not disclosed until postdosing. Among the remaining 16 patients, 13 discontinued for TEAEs considered treatment‐related. Sedation‐type TEAEs (sedation, somnolence, hypersomnia) were the most common, contributing to discontinuation of eight patients. Four of these patients, however, had protocol‐defined exclusions (OAA/S score) prohibiting progression to CP, which were recorded as discontinuations due to TEAEs. Four patients had TEAEs relating to route of administration that contributed to discontinuation. TEAEs in the AESI category of acute central respiratory depression (ACRD) led to discontinuation in one patient but were also contributory factors in the discontinuation of a further 4 of the 13 patients (Table S3). Only two patients who experienced an AESI in the category of ACRD were considered to have experienced events likely indicative of clinically meaningful respiratory depression. One patient, with bradypnea and decreased oxygen saturation, had an intercurrent seizure considered to be due to underlying disease, while the other, with prolonged decreased oxygen saturation, had a history of sleep apnea that was likely contributory.
3.2.2. Comparative phase
After administration of the double‐blind trial drug in the CP, 27.6% and 22.4% of patients in the MDZ‐NS and placebo groups, respectively, experienced ≥1 TEAE (Table 2). The most frequently reported TEAEs within 2 days of MDZ–NS administration during the CP are summarized in Table 3, and overall in Table S4; most were considered mild or moderate in intensity. Four patients in the MDZ–NS group reported five severe TEAEs; these were somnolence, headache, nasal discomfort, nausea, and vomiting. No severe TEAEs were reported by placebo‐treated patients. No notable differences (>10% absolute difference) in the incidence of TEAEs between the MDZ–NS and placebo groups were observed. There was a difference in the reported rate of nasal discomfort between patients in the MDZ‐NS group who received one vs two doses (5.5% vs 16.3%).
Forty‐five patients (22.4%) experienced ≥1 treatment‐related TEAE, including 20 (22.0%) in the MDZ–NS group who received the double‐blind dose only, and 13 (30.2%) who received both doses; corresponding values in the placebo group were 5 (19.2%) and 7 (17.1%). Treatment‐related TEAEs were similar qualitatively to overall TEAEs. Two patients, one in the MDZ–NS group and one in the placebo group who received open‐label MDZ‐NS, had an serious adverse event (SAE); neither SAE was considered treatment‐related. There were no AESIs in the category of ACRD (Tables S5 and S6), and no discontinuations due to TEAEs.
Throughout the trial, no notable safety trends in laboratory parameters; vital signs; C‐SSRS scores; or physical, nasal, and neurologic examinations were observed.
4. DISCUSSION
Results of ARTEMIS‐1, a double‐blind, randomized, placebo‐controlled trial, provide evidence for the safety and efficacy of MDZ–NS as an outpatient therapeutic option for patients requiring acute intervention during an SC. Based on the primary composite end point—seizure termination within 10 minutes of, and no seizure recurrence 10 minutes to 6 hours after trial drug administration—treatment with a 5‐mg MDZ–NS dose during the double‐blind CP provided rapid, sustained seizure control for patients. The observed treatment effect was clinically meaningful and statistically significant (19.4% difference between MDZ–NS and placebo; P = 0.0109). The conservative statistical approach employed for the final analysis ensured integrity of the results, given that the trial was stopped early.
Based on the Kaplan‐Meier analysis of time‐to‐next seizure after drug administration, there was early and clear separation (within approximately 30 minutes) between MDZ–NS and placebo that was maintained throughout the observation period, with a statistically significant and clinically relevant effect size of 21% at 24 hours. The duration of treatment response observed in the double‐blind CP is of note, given that MDZ is a short‐acting benzodiazepine based on its relatively short half‐life in plasma.26 In MDZ–NS clinical trials, the median elimination half‐lives of MDZ and its hydroxy metabolite ranged from 2.1 to 6.2 hours and 2.7 to 7.2 hours, respectively, independent of dose (data on file). Results suggest that the antiseizure effect of MDZ–NS does not correlate with its short half‐life, unlike the sedative effect. This observation was corroborated by results of the time‐to‐return to baseline functionality analysis, which showed that a substantially larger proportion of patients treated with MDZ–NS had documented return to full baseline functionality within 24 hours after drug administration compared with those who received placebo (72.4% vs 43.3%). Rapid return to full functionality and absence of continued AEs such as sedation are especially important considerations for patients (and caregivers) in full‐time employment or education.
The safety profile of MDZ, and other benzodiazepines, is well known; through potentiation of γ‐aminobutyric acid receptor A (GABAA) activity, their global central nervous system (CNS) inhibitory effects lead to diverse AEs, including motor and sensory impairment and cardiorespiratory depression.5, 27, 28 Throughout the trial, TEAEs experienced by most patients were, not unexpectedly, sedation‐type events, and given the route of administration, nasal discomfort. The incidence of events considered likely indicative of clinically meaningful respiratory depression related to the trial drug was low (0.7%) and occur red only during the TDP. It is important to note that no treatment‐related SAEs, discontinuations due to TEAEs, or AESIs in the category of ACRD were reported after double‐blind administration of MDZ–NS during the CP. Finally, there were no TEAEs in the category of depression and suicidality/self‐injury indicative of suicide or attempted suicide, or abuse. These results confirm the acceptable safety profile of MDZ–NS and support the feasibility of administration by non‐HCPs in outpatient settings.
Antiseizure agents that can be administered rapidly and safely by non‐HCPs in the outpatient setting can help patients experiencing SCs achieve seizure cessation, reduce seizure‐related complications, and prevent progression to more serious sequelae.5, 6, 7 Early intervention may help avoid emergency department visits and also provide a greater sense of control for patients and their caregivers, which may have a positive impact on their quality of life.6 In one study, patients who used acute therapeutic intervention experienced significantly fewer injuries and visits to the emergency department over a 1‐year prospective follow‐up.29 After oral lorazepam, intranasal MDZ (IN‐MDZ) was the most frequently used agent.29 In effect, off‐label IN‐MDZ has been used in the treatment of patients with seizure disorders for over two decades; however, randomized controlled trials, as required for regulatory approval, have not been conducted.
Given the difference in routes of administration, an adequately powered, blinded, comparative trial of MDZ–NS and PR‐DZP would be complex. Results of several prospective, randomized, open‐label studies, however, indicate that IN‐MDZ appears to be as effective as PR‐DZP. In two studies, children with acute seizures presenting to emergency care were randomized to treatment with IN‐MDZ or PR‐DZP.16, 18 In both, seizure cessation within 10 minutes of drug administration was observed in a significantly greater proportion of children treated with IN‐MDZ than PR‐DZP.16, 18 Use of IN‐MDZ and PR‐DZP has also been compared in the outpatient setting. In one study, adult patients in a residential epilepsy center experiencing seizure exacerbation received both drugs alternately for six separate episodes, and in another, caregivers were randomized to administer IN‐MDZ or PR‐DZP to children experiencing prolonged seizures.21, 22 Although no detectable differences in the effectiveness or safety profiles of the drugs were noted in either study, most patients and caregivers in both stated a preference for IN administration.21, 22 In noncomparative studies conducted in outpatient settings, caregivers also expressed a preference for IN‐MDZ over PR‐DZP in terms of ease of use and ability to administer in public if necessary.19, 30
In the aforementioned studies, MDZ solution was delivered into the nostrils directly or via an atomization device. However, use of MDZ solution has drawbacks; notably, solution acidity may cause local irritation, and preparing a syringe or dripper from a vial may be challenging for a caregiver while the patient is experiencing a seizure. Furthermore, prefilled syringes of the solution may have limitations in terms of shelf‐life and/or storage requirements. Consequently, a standardized, commercially available system for delivery of MDZ optimized for intranasal administration should facilitate use of this agent.
Use of MDZ–NS in the outpatient setting was associated with rapid and sustained seizure control in patients experiencing SCs, and its safety profile, in patients as young as 12 years, was acceptable. These results demonstrate the potential of MDZ–NS in addressing an important unmet clinical need.
CONFLICT OF INTEREST
T Meng, WE Pullman, DJ Sequeira, and PJ Van Ess are employees of Proximagen LLC. K Detyniecki has received research support to Yale University for investigator‐initiated studies from Eisai, Sunovion, Acorda, and Upsher‐Smith, and consultation fees from UCB Pharma. JW Wheless has received research grants from Aquestive, Eisai, Greenwich, INSYS Inc, LivaNova, Mallinckrodt, Neuralis, NeuroPace, Shainberg Foundation, and Zogenix; served as a consultant for Aquestive, BioMarin, Eisai, Greenwich, Mallinckrodt, Neuralis, NeuroPace, West, Shire, and Supernus; and participated in speaker's bureau for BioMarin, Eisai, Greenwich, LivaNova, Mallinckrodt, and Supernus. The authors confirm having read the Journal's position on ethical publication and affirm that this report is consistent with their guidelines.
Supporting information
ACKNOWLEDGMENTS
The authors express their deep gratitude to the patients and their caregivers who took part in the trial. They also express their gratitude to J French, D Friedman, and S Mintzer of the Epilepsy Consortium (https://www.epilepsyconsortium.org ) for providing oversight in patient screening, and the following investigators for their contribution to the trial: S Berkovic, Epilepsy Research Centre, Heidelberg West; T O'Brien, Royal Melbourne Hospital, Parkville, VIC; E Somerville, Prince of Wales Hospital, Randwick, NSW; D Reutens, Royal Brisbane and Women's Hospital, Herston, QLD, Australia; P Tai, Toronto Western Hospital, Toronto, Ontario; D Nguyen, Hospital Notre‐Dame du CHUM Montreal, QC, Canada; C Brandt, Bethel Epilepsy Center, Bielefeld; C Elger, University of Bonn Epileptology Clinic, Bonn; S Arnold, Neurozentrum Nymphenburg, Munich; F Rosenow, Gieβen and Marburg University Clinic, Marburg, Germany; A Altmann, St. John's Hospital and United North Buda Hospitals; A Kelemen, National Institute of Clinical Neurosciences; M Neuwirth, Bethesda Children's Hospital, Budapest; A Bartos, Korház Nonprofit Kft. Kazincbarcika, Hungary; M Herskovitz, Rambam Medical Center, Haifa; I Blatt, Sheba Tel Hashommer, Ramat Gan; H Goldberg‐Stern, Schneider Children's Medical, Beilinson Campus, Petah Tikva; M Neufeld, Tel Aviv Sourasky Medical Center, Tel Aviv; D Eckstein, Hadassah Medical Organization, Jerusalem, Israel; S Striano, Universita degli Studi di Napoli Policlinico, Napoli; CA Galimberti, Istituto Neuroligico Casimiro Mondino Pavia; A Romeo, Ospedale Fatebenefratelli Milano; R Guerrini, Azienda Ospedaliero‐Universitaria, Firenze, Italy; T Anderson, New Zealand Brain Research Institute, Christchurch, New Zealand; J Gawlowicz, WojewOdzki Szpital Specjalistyczny, Lublin; M Mazurkiewicz, Uniwersyteckie Centrum Kliniczne, Gdansk; A Tutaj, Oddzial Udarowy Wojewodzki Szpital Specjalistyczny, Olsztyn; E Motta, Centrum Medyczne Dendryt, Katowice, Poland; A Gil‐Nagel, Hospital Ruber Internacional; I Garcia Morales, Hospital Clinico San Carlos; A Gomez Caicoya, Hospital Universitario Quiron, Madrid; JJ Rodriguez Uranga, Clinica Sagrado Corazôn, Sevilla; A Molins Albanell, Hospital Universitario de Gerona, Gerona; M Martinez Ferri, Hospital Mutua de Tarrassa, Barcelona, Spain; OP Venger, Ternopil Psychoneurological Hospital, Ternopil; M Mulyk, Regional Psychoneurological Hospital, Ivano‐Frankivsk; A Skrypnikov, Regional Psychiatric Hospital, Poltava; T Litovchenko, Central Clinical Hospital, Kharkiv; OA Serebrennikova, Regional Psychiatric Hospital, Vinnitsa; AE Voloshchuk, Regional Psychoneurology Dispensary; E Melnyk, Odessa Regional Medical Centre of Mental Health, Odessa, Ukraine; V Biton, Clinical Trials, Inc.; J Greenfield, University of Arkansas for Medical Sciences, Little Rock, AR; D Labiner, University of Arizona Health Sciences Center, Tucson; J Drazkowski, Mayo Clinic Hospital; S Chung, University Medical Center Phoenix, Phoenix, AZ; P Bhatia, Neuro‐Pain Medical Center, Fresno; M Sazgar, UC Irvine Health, Orange; E Cruz, Mercy Medical Group, Sacramento; R Hutchman, Neurosearch II, Inc., Ventura, CA; L Strom, University of Colorado, Denver, CO; JB Renfroe, NW FL Clinical Research Group, LLC Gulf Breeze; G Li, MEDSOL Clinical Research Center, Port Charlotte; E Liu, Palm Beach Clinical Research Network, LLC, Wellington, FL; R Wechsler, Consultants in Epilepsy and Neurology, PLLC, Boise, ID; M Kohrman, C Marcuccilli, University of Chicago Hospital, Chicago, IL; N Kumar, Precise Clinical Research Solutions Manhattan, KS; M Bensalem‐Owen, Comprehensive Epilepsy Center; T Fakhoury, Bluegrass Epilepsy Research Associates, Lexington, KY; G Krauss, Johns Hopkins University, Baltimore, MD; A Shah, Wayne State University/Detroit Medical Center, Detroit, MI; P Penovich, Minnesota Epilepsy Group, St. Paul, MN; R Hogan, Washington University School of Medicine; W Rosenfeld, Comprehensive Epilepsy Care Center for Children and Adults, St. Louis, MO; A Husain, Duke University Medical Center, Durham; C O'Donovan, Wake Forest Health Sciences, Winston/Salem, NC; E Fertig, Northeast Regional Epilepsy Group, Hackensack, NJ; R Morse, Children's Hospital at Dartmouth, Lebanon, NH; A Boro, Montefiore Medical Center; B Vazquez, NYU Comprehensive Epilepsy Center, P Mullin, Weill Cornell Medical College of Cornell University New York; R Spiegel, Stony Brook University Medical Center, Stony Brook, NY; M Gelfand, J Pollard, University of Pennsylvania Hospital; M Nei, Comprehensive Epilepsy Center, Thomas Jefferson University; D Dlugos, Colket Translational Research Building, Philadelphia, PA; D Spencer, Oregon Health & Science University, Portland, OR; B Abou‐Khalil, Vanderbilt University Medical Center, Nashville, TN; J Harvey, Neurological Clinic of Texas; P Van Ness, University of Texas Southwestern Medical Center, Dallas; AS Ata, AS Clinical Research Consultants of North Texas, PLLC, Greenville; L Morgan, UTHSCSA Neurology San Antonio, TX; MM Frucht, Dean & St. Mary's Outpatient Center, Madison, WI, USA. The authors acknowledge the contribution of T Braun (formerly of Proximagen), in overseeing trial conduct, B Pelgrims, PhD (UCB Pharma, Brussels, Belgium) in publication coordination and A Tofighy, PhD (London, UK) in providing writing support, which was funded by UCB Pharma.
Detyniecki K, Van Ess PJ, Sequeira DJ, Wheless JW, Meng T‐C, Pullman WE. Safety and efficacy of midazolam nasal spray in the outpatient treatment of patients with seizure clusters—a randomized, double‐blind, placebo‐controlled trial. Epilepsia. 2019;60:1797–1808. 10.1111/epi.15159
Funding information
This trial was funded by Proximagen LLC.
Correction added on 10 July 2019, after first online publication: Table 1 and supplementary Table S1 updated.
REFERENCES
- 1. Cereghino JJ. Identification and treatment of acute repetitive seizures in children and adults. Curr Treat Options Neurol. 2007;9:249–55. [DOI] [PubMed] [Google Scholar]
- 2. Sillanpää M, Schmidt D. Seizure clustering during drug treatment affects seizure outcome and mortality of childhood‐onset epilepsy. Brain. 2008;131:938–44. [DOI] [PubMed] [Google Scholar]
- 3. Haut SR. Seizure clusters: characteristics and treatment. Curr Opin Neurol. 2015;28:143–50. [DOI] [PubMed] [Google Scholar]
- 4. Penovich PE, Buelow J, Steinberg K, Sirven J, Wheless J. Burden of seizure clusters on patients with epilepsy and caregivers: survey of patient, caregiver, and clinician perspectives. Neurologist. 2017;22:207–14. [DOI] [PubMed] [Google Scholar]
- 5. Kälviäinen R. Intranasal therapies for acute seizures. Epilepsy Behav. 2015;49:303–6. [DOI] [PubMed] [Google Scholar]
- 6. McKee HR, Abou‐Khalil B. Outpatient pharmacotherapy and modes of administration for acute repetitive and prolonged seizures. CNS Drugs. 2015;29:55–70. [DOI] [PubMed] [Google Scholar]
- 7. Jafarpour S, Hirsch LJ, Gaínza‐Lein M, Kellinghaus C, Detyniecki K. Seizure cluster: Definition, prevalence, consequences, and management. Seizure 2018. pii: S1059‐1311(18)30112‐2. 10.1016/j.seizure.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 8. Valeant Pharmaceuticals . Diastat prescribing information. [cited 2019 Mar 27] Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/020648s008lbl.pdf
- 9. Shire . Buccolam Summary of Product Characteristics 2018. [cited 2019 Mar 27] Available from https://www.ema.europa.eu/en/documents/product-information/buccolam-epar-product-information_en.pdf
- 10. Mula M. New non‐intravenous routes for benzodiazepines in epilepsy: a clinician perspective. CNS Drugs. 2017;31:11–17. [DOI] [PubMed] [Google Scholar]
- 11. O'Regan ME, Brown JK, Clarke M. Nasal rather than rectal benzodiazepines in the management of acute childhood seizures? Dev Med Child Neurol. 1996;38:1037–45. [DOI] [PubMed] [Google Scholar]
- 12. Jeannet PY, Roulet E, Maeder‐Ingvar M, Gehri M, Jutzi A, Deonna T. Home and hospital treatment of acute seizures in children with nasal midazolam. Eur J Paediatr Neurol. 1999;3:73–7. [DOI] [PubMed] [Google Scholar]
- 13. Kutlu NO, Yakinci C, Dogrul M, Durmaz Y. Intranasal midazolam for prolonged convulsive seizures. Brain Dev. 2000;22:359–61. [DOI] [PubMed] [Google Scholar]
- 14. Lahat E, Goldman M, Barr J, Bistritzer T, Berkovitch M. Comparison of intranasal midazolam with intravenous diazepam for treating febrile seizures in children: prospective randomised study. BMJ. 2000;8(321):83–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scheepers M, Scheepers B, Clarke M, Comish S, Ibitoye M. Is intranasal midazolam an effective rescue medication in adolescents and adults with severe epilepsy? Seizure. 2000;9:417–22. [DOI] [PubMed] [Google Scholar]
- 16. Fişgin T, Gürer Y, Teziç T Senbil N, Zorlu P, Okuyaz C, Akgün D. Effects of intranasal midazolam and rectal diazepam on acute convulsions in children: prospective randomized study. J Child Neurol. 2002;17:123–6. [DOI] [PubMed] [Google Scholar]
- 17. Mahmoudian T, Zadeh MM. Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy Behav. 2004;5:253–5. [DOI] [PubMed] [Google Scholar]
- 18. Bhattacharyya M, Kalra V, Gulati S. Intranasal midazolam vs rectal diazepam in acute childhood seizures. Pediatric Neurol. 2006;34:355–9. [DOI] [PubMed] [Google Scholar]
- 19. Harbord MG, Kyrkou MR, Kay D, Coulthard KP. Use of intranasal midazolam to treat acute seizures in paediatric community settings. J Paediatr Child Health. 2004;40:556–8. [DOI] [PubMed] [Google Scholar]
- 20. Thakker A, Shanbag P. A randomized controlled trial of intranasal midazolam versus intravenous‐diazepam for acute childhood seizures. J Neurol. 2013;260:470–4. [DOI] [PubMed] [Google Scholar]
- 21. de Haan GJ, van der Geest P, Doelman G, Bertram E, Edelbroek P. A comparison of midazolam nasal spray and diazepam rectal solution for the residential treatment of seizure exacerbations. Epilepsia. 2010;51:478–82. [DOI] [PubMed] [Google Scholar]
- 22. Holsti M, Dudley N, Schunk J, Adelgais K, Greenberg R, Olsen C, Healy A, Firth S, Filloux F. Intranasal midazolam vs rectal diazepam for the home treatment of acute seizures in pediatric patients with epilepsy. Arch Pediatr Adoles Med. 2010;164:747–53. [DOI] [PubMed] [Google Scholar]
- 23. Bancke LL, Dworak HA, Rodvold KA, Halvorsen MB, Gidal BE. Pharmacokinetics, pharmacodynamics, and safety of USL261, a midazolam formulation optimized for intranasal delivery, in a randomized study with healthy volunteers. Epilepsia. 2015;56:1723–31. [DOI] [PubMed] [Google Scholar]
- 24. Hayes RJ, Bancke LL, Halvorsen MB. Pharmacokinetics of USL261, a novel formulation of intranasal midazolam optimized for intranasal administration, in subjects with epilepsy. American Epilepsy Society (AES) 2012 Annual Meeting Abstract Database. AESnet.org
- 25. Abou‐Khalil B, Wheless J, Rogin J, Wolter KD, Pixton GC, Shukla RB, Sherman NA, Sommerville K, Goli V, Roland CL. A double‐blind, randomized, placebo controlled trial of a diazepam auto‐injector administered by caregivers to patients with epilepsy who require intermittent intervention for acute repetitive seizures. Epilepsia. 2013;54:1968–76. [DOI] [PubMed] [Google Scholar]
- 26. Fresenius Kabi . Midazolam Prescribing information 2017. [cited 2019 Mar 27] Available from https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208878Orig1s000lbl.pdf
- 27. Guina J, Merrill B. Benzodiazepines I: Upping the care on downers: The evidence of risks, benefits and alternatives. J Clin Med. 2018;7(2):E17. 10.3390/jcm7020017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Humphries LK, Eiland LS. Treatment of acute seizures: is intranasal midazolam a viable option? J Pediatr Pharmacol Ther. 2013;18:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Detyniecki K, O'Bryan J, Choezom T, Rak G, Ma C, Zhang S, Bonito J, Hirsch LJ. Prevalence and predictors of seizure clusters. A prospective observational study of adult patients with epilepsy. Epilepsy Behav. 2018;88:349–56. [DOI] [PubMed] [Google Scholar]
- 30. Kyrkou M, Harbord M, Kyrkou N, Kay D, Coulthard K. Community use of intranasal midazolam for managing prolonged seizures. J Intellect Dev Disabil. 2006;31:131–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials