

Abstract
Insulin is the hormone responsible for maintaining glucose homeostasis in the body, in addition to participating in lipid metabolism, protein synthesis, and the inhibition of gluconeogenesis. These functions are well characterized in the classic organ target cells that are responsible for general energy regulation: the liver, skeletal muscle, and adipose tissue. However, these actions are not restricted to these tissues because insulin has been shown to affect most cells in the body. This review describes the role of insulin in leukocyte signaling pathways, metabolism and functions, and how insulin resistance could affect this signaling and deteriorate leukocyte metabolism and function, in addition to showing evidence that suggests leukocytes may substantially contribute to the development of systemic insulin resistance.
Keywords: insulin resistance, insulin signaling, insulin, leukocytes
Graphical Abstract
Review on the role of insulin in metabolism and leukocyte function
Abbreviations
- AGE
advanced glycation end products
- AKT
protein kinase B
- AP1
activator protein 1
- AS160
AKT substrate of 160 kDa
- ATP
adenosine triphosphate
- CCL3 C‐C
motif chemokine ligand 3
- CD16
cluster of differentiation 16
- CD36
cluster of differentiation 36
- cel/μL
cells/microliter
- c‐Myc
c‐Myc transcription factor
- CRP
C‐reactive protein
- DAG
diacylglycerol
- ERK
1/2 extracelular signal‐regulated kinases 1/2
- ETC
electron transport chain
- FADH2
flavin adenine dinucleotide
- FAO
fatty acids oxidation
- FAT
fatty acids translocase
- FcRI
high affinity IgE receptor
- FFA
free fatty acids
- FMLP
N‐Formyl‐metionyl‐leucyl‐phenylalanine
- FOXO1
forkhead box protein O1
- GLUT
glucosa transporter
- GLUT1
glucose transporter 1
- GLUT3
glucose transporter 3
- GLUT4
glucose transporter 4
- Grb2/SOS
growth factor receptor‐bound protein 2/ son of sevenless complex
- GSK3
glycogen synthase kinase 3
- H2O2
hydrogen peroxide
- HIF‐1α
hypoxia‐inducible factor 1‐alpha
- HK
hexokinase
- IFN‐γ
interferón gamma
- IGF‐1
insulin‐like growth factor 1
- IKK
IkB kinase
- IL‐10
interleukin‐10
- IL1A
interleukin‐1 alpha
- IL‐2
interleukin‐2
- IL‐4
interleukin‐4
- IL‐5
interleukin‐5
- IL‐5
interleukin‐5
- IL‐6
interleukin‐6
- IL‐8
interleukin‐8
- IRS‐1/2
insulin receptor substrate 1/2
- JNK
c‐Jun N‐terminal kinase
- kDa
kilodaltons
- LDH
lactate dehydrogenase
- LDL
low‐density lipoprotein
- LPS
lipopolysaccharide
- Mac‐1
macrophage‐1 antigen
- MAPK
mitogen‐activated protein kinase
- MCP‐1
monocyte chemoattractant
- MEK
mitogen‐activated protein kinase kinase
- MHC II
major histocompatibility complex
- MMP‐9
matrix metalloproteinase 9
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- NAD+
oxidized nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NE
neutrophil elastase
- NET
neutrophil extracelular trap
- NFKBIA
NFκB inhibitor alpha
- NFκB
nuclear factor kappa B
- O2‐
superoxide radical
- OP
oxidative phosphorylation
- PDK1
3‐phosphoinositide‐dependent protein kinase 1
- PFK
posphofructokinase
- PI3K
phosphatidylinositol 3‐kinase
- PIP3
phosphatidylinositol (3, 4, 5)‐trisphosphate
- PKC
protein kinase C
- PPP
pentose phosphate pathway
- PTB
phosphotyrosine‐binding domains
- Rac‐1
Rac family small GTPase 1
- Raf‐1
RAF proto‐oncogene serine/treonine‐protein kinase
- RAGE
advanced glycation end products receptor
- Ras
small GTP‐binding protein Ras
- RAW
264.7 RAW 264.7 cell line
- ROS
rective oxygen species
- RTKs
receptor tirosine kinases
- S6K1
ribosomal protein S6 kinase beta‐1
- SERCA2b
sarco/endoplasmatic reticulum Ca2+‐ATPase
- Shc
adapter protein Shc
- SRA
scavenger receptor class A
- TCA
tricarboxylic acid cycle
- THP‐1
THP‐1 cell line
- TLR4
toll‐like receptor 4
- TNFα
tumor necrosis factor alpha
- TNF‐β
tumor necrosis factor beta
1. INTRODUCTION
Homeostatic regulation of glucose is primarily governed by the action of insulin in adipose, muscle, and liver tissue, which responds by activating signaling pathways involved in the metabolism of glucose and lipids, energy storage, and cell growth. Insulin is a highly effective anabolic hormone whose molecular weight of 5.8 kDa is composed of 2 peptide chains: the A chain, which contains 21 amino acids, and the B chain, which contains 30 amino acids. Both peptide chains are linked by disulfide bridges. 1 Insulin is produced and stored in granules within the beta cells of the islets of Langerhans in the pancreas, from which it is secreted primarily in response to increased plasma glucose concentrations. 2 Its main function is to promote the intake of circulating glucose into skeletal muscle and adipose tissue cells, where it also promotes the formation of glycogen and lipogenesis, respectively, whereas in liver tissue, it inhibits the production of glucose. 3 Other actions of insulin include regulation of growth, cell survival, and protein synthesis. 4 The actions of insulin are governed by the activation of 2 canonical signal transduction pathways that are ubiquitous in practically all cells and that begin once it binds to the insulin receptor present in the plasma membrane. 5 , 6 , 7 Although studies on the actions of insulin have focused on the tissues responsible for energy homeostasis in the body, there is evidence that the hormone participates in the metabolic and functional regulation of other cells, called nonclassical insulin targets, among which are cells of the central nervous system, 8 , 9 epithelial cells, 10 , 11 and even bone cells. 12 , 13 , 14 However, recent evidence describes the relationship between the effects of insulin and the regulation of metabolism and/or the functions of immune cells.
Leukocytes, also known as white blood cells, are the effector cells of the immune system, and their main function is to defend the host against pathogenic microorganisms or against any type of tissue damage. These cells are produced in the bone marrow from hematopoietic stem cells that give rise to 2 cell lineages, the myeloid line and the lymphoid line, from which the mature leukocytes that enter into circulation are derived. 15 , 16
In the bloodstream, leukocytes are present at a concentration of 4,000–10,000 cells/μl under normal conditions. 17 In a routine hematologic analysis, 5 types of leukocytes are morphologically distinguished using conventional histologic stains: neutrophils, eosinophils, basophils, monocytes (belonging to the myeloid line), and lymphocytes (from the lymphoid line). Neutrophils represent 75% of total leukocytes, 18 eosinophils and basophils represent 8% and 2%, respectively (these are known as granulocytes), monocytes represent 5%, whereas lymphocytes make up 20–45% of total leukocytes in circulation. 17
This review describes the role of insulin in the metabolism and functions of leukocytes and how the state of insulin resistance affects its molecular signaling as well as its metabolic and immune function. Similarly, evidence has suggested that leukocytes substantially contribute to the development of systemic insulin resistance. Therefore, the primary objective of this review was to address the molecular and signaling mechanisms of insulin in leukocytes and their relationship with metabolic and functional control, proposing leukocytes as cells with the potential for consideration in the analysis of the molecular processes involved in the development of insulin resistance.
2. INSULIN SIGNALING PATHWAYS IN LEUKOCYTES
Initiation of insulin signal transduction occurs from binding to its receptor. The insulin receptor is a heterotetrameric protein of the receptor tyrosine kinase family that is autophosphorylated in the intracellular region at tyrosine residues, which are recognized by cytoplasmic proteins that serve as scaffolds for the recruitment of other molecules that participate in signaling. 19
One of the pathways activated by insulin in leukocytes is MAPKs, in which insulin receptor substrate proteins 1/2 (IRS‐1/2) or the Shc protein recognize tyrosine residues phosphorylated on the receptor β chain through its phosphotyrosine binding domains, which are also phosphorylated. Subsequently, the protein complex growth factor receptor‐bound protein 2/Son of sevenless (Grb2/SOS) recognizes the phosphorylated residues of IRS‐1/2 or Shc and in turn activates a cascade of Ras, Raf‐1, and MEK kinases. The final result of the pathway is the translocation of ERK 1/2 to the nucleus, regulating migration, proliferation, and cellular growth functions. 20
The other signaling pathway that is activated is PI3K, in which IRS‐1/2 recognizes phosphorylated tyrosine residues in the receptor, which in turn are recognized by the regulatory subunit of PI3K that activates the catalytic subunit, which is capable of phosphorylating membrane phospholipids, such as phosphatidylinositol‐4,5 bisphosphate, to generate phosphoinositol‐3,4,5, triphosphate, allowing the recruitment of protein kinase B (AKT), which is phosphorylated by the 3‐phosphoinositide‐dependent kinase 1 at threonine 308 and by the mTORC2 complex at serine 473, achieving its activation. AKT phosphorylates different substrates that regulate metabolic processes in cells, including the protein glycogen synthase kinase 3, favoring an increase in glycogen synthesis, 21 the mammalian target of rapamycin (mTOR), which regulates protein synthesis through ribosomal protein S6 kinase beta 1 22 and AKT substrate of 160 kDa protein that favor the translocation of GLUT4 transporters from cytosolic vesicles to the plasma membrane to allow glucose entry into cells, 23 , 24 which is the primary effect of insulin in adipose and muscle tissues that contributes to the maintenance of plasma normoglycemia.
Comparative results about the effects of insulin on glucose metabolism in leukocytes were contradictory, 25 , 26 , 27 , 28 , 29 as glucose uptake by leukocytes may be influenced by different factors, such as interindividual variability, analysis strategy due to leukocyte half‐life, but importantly, because the duration of insulin interaction with the receptor is not known, so there is no clear evidence that such factors directly impact glucose uptake or metabolism. In recent years, the role of insulin in the regulation of metabolism in immune cells has begun to be analyzed with respect to the signaling pathways and metabolic processes involved in energy production, highlighting the different requirements in the quiescent and activated state of leukocytes, as well as the direct impacts on their functions.
3. ENERGY METABOLISM OF LEUKOCYTES
Glucose is the primary energy‐producing molecule. Once it enters cells, 3 main interconnected metabolic pathways are responsible for the generation of ATP from glucose: glycolysis, the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OP). Glycolysis occurs in the cytoplasm and begins with the generation of glucose‐6‐phosphate, a reaction mediated by hexokinase (HK), which prevents glucose from leaving the cell. Subsequent enzymatic reactions generate 2 molecules of NADH, 2 ATP, and 2 pyruvates as final products. 30 The availability of oxygen determines the next step in the metabolic pathways, under hypoxic conditions, hypoxia‐inducible factor 1α (HIF‐1α) is the main driver of glycolysis where pyruvate is converted into lactate by the enzyme lactate dehydrogenase (LDH), which produces oxidized NAD+ molecules from NADH 31 that favor the continuity of glycolysis. Under normoxic conditions, pyruvate is converted to acetyl coA, which enters the TCA in the mitochondria, which, through NADH and reduced flavin adenine dinucleotide, maintains the electron transport chain and, finally, OP, which ultimately produces ATP. 32 A pathway derived from glycolysis and glucose‐6‐phosphate is the pentose phosphate pathway, which is necessary for the production of ribose or NADPH, 33 whereas fatty acid oxidation (FAO) and glutaminolysis replace the acetyl coA and α‐ketoglutarate metabolites, respectively, which can be used during the anabolism of proliferating cells. 34 , 35
Once activated, leukocytes migrate through diapedesis to different sites inflammation, where the availability of oxygen is variable, but they have the ability to rewire their metabolism (aerobic or anaerobic) to carry out their function once they are activated. This phenomenon is known as metabolic plasticity, determined by the measurement of oxygen consumption rate, which is a reflection of mitochondrial energy production via OP, whereas the extracellular acidification rate determines energy production through glycolysis and the generation of lactate. 36
The basal energy metabolism of neutrophils is derived from glycolysis. 37 Once activated, they increase their glucose consumption anaerobically and produce lactate, even when oxygen availability is not limiting, an effect known as Warburg metabolism. This process is mediated by HIF‐1α, a transcription factor that is activated under oxygen‐limited conditions. 38 However, HIF‐1α can also be activated under normoxic conditions by signaling through TLR4 present on leukocytes through the activation of NFκB. 39 As neutrophils depend preferentially on anaerobic glycolysis, the mitochondria present in them do not participate in the generation of ATP but do participate in the production of mitochondrial reactive oxygen species (mtROS) through the NADPH oxidase system, 40 that helps in maintaining mitochondrial membrane potential by importing NAD+ produced by glycerol 3‐phosphate shuttle, which is active in neutrophils. 41
In the case of eosinophils, there are few studies regarding their metabolic requirements, likely due to their low number in circulation and the technical limitations of their isolation; however, it has been discovered that they contain a glycolytic metabolism “similar” to that of neutrophils but with a higher oxygen consumption and greater flexibility toward the use of OP. 42 Finally, with respect to basophils, their metabolic requirements are not yet known.
Circulating monocytes satisfy their basal energetic requirements by producing ATP via OP. 36 Once they migrate to tissues, they become macrophages, where the microenvironment generates local signals that allow them to acquire 2 possible phenotypes: classically activated macrophages (M1 or proinflammatory) or alternatively activated macrophages (M2 or anti‐inflammatory). 43 M1 macrophages have a preferentially anaerobic glycolytic metabolism with the formation of lactate, very similar to that shown by neutrophils 43 ; they also have a low number of mitochondria that do not participate in the generation of energy but are necessary for the production of mtROS. 44 M2 macrophages, on the other hand, have an energy metabolism that depends on both OP 45 and FAO. 46
Lymphocytes are normally under resting conditions, satisfying their energy requirements through OP. 47 When activated, they increase glucose consumption using aerobic glycolysis, but they also maintain active TCA and OP that are necessary to supply the molecules required for proliferation during clonal expansion, a process supported by glutaminolysis that replaces the intermediaries for OP 47 (Fig. 1).
FIGURE 1.
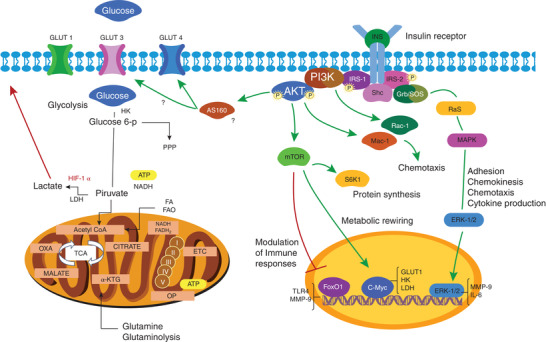
Regulation of metabolism and leukocytes functions by insulin. Leukocytes express the insulin receptor. Once insulin binds to its receptor, it activates two signal transduction pathways. The MAPK pathway results in ERK‐1/2 activation, in leukocytes it participates in adhesion, chemokinesis, chemotaxis and cytokine production. PI3K pathway, activating AKT and phosphoryling different substrates downstream of the signaling cascade in functions such as protein synthesis, inflammatory response, metabolic rewiring, and chemotaxis
4. THE INVOLVEMENT OF INSULIN IN GLUCOSE METABOLISM
The insulin receptor is constitutively expressed on the plasma membrane of both monocytes and neutrophils, and its expression does not change as a function of insulin concentration. 48 There is no evidence to date that had directly evaluated expression of the receptor in eosinophils and basophils, although as they are granulocytes, it is very likely that their expression is similar to that of neutrophils. However, in lymphocytes, expression of the insulin receptor is not constitutive but is positively regulated in response to insulin, 49 and the effect is potentiated during its activation. 50 , 51 , 52 These findings suggest the probable participation of insulin in the regulation of metabolism in these immune cells.
In monocytes, insulin increases the transport and utilization of glucose, 53 , 54 , 55 whereas in neutrophils, insulin does not seem to regulate glucose uptake; however, it shows an influence on glucose metabolism once transported to the cytoplasm, regulating molecules that participate in glycolysis. 56 , 57 Glucose enters the cell by facilitated diffusion through the glucose transporter protein GLUT. Expression of GLUT in the membrane is crucial for increased glucose uptake in activated leukocytes. Fourteen isoforms expressed in humans have been described, of which GLUT1‐4 are the most studied, and leukocytes express the GLUT1 and GLUT3 isoforms independent of insulin stimulation, as well as GLUT4, which has been shown to be dependent of insulin‐activated signaling. 58 , 59
In inactive monocytes, the presence of the insulin receptor appears to be relevant in glucose transport, as it has been shown that insulin stimulation is directly proportional to glucose uptake. 55 , 59 It has been described that in cells isolated from humans, the GLUT3 isoform is the most highly expressed compared with the GLUT1 and GLUT4 isoforms, 55 , 60 and this finding coincides with studies in the monocytic cell lines THP‐1 and RAW 264.7. 60 , 61
Once activated, monocytes differentiate into macrophages; their activation results in an increase in the 3 isoforms, but in the presence of insulin, the translocation of the GLUT3 and GLUT4 isoforms to the plasma membrane is increased. 59 In vitro differentiation of monocytic cell lines toward macrophages has also been shown to increase the expression of GLUT1 and, primarily, GLUT3. 60 , 61
Because monocytes have the ability to increase membrane GLUT4 expression and glucose uptake in the presence of insulin, similar to what occurs in tissues such as adipose and skeletal muscle, they can likely be considered cells for evaluation of systemic insulin sensitivity; however, their probable clinical utility in metabolic pathophysiology, such as insulin resistance, must be further examined. 62
In the inactive state, neutrophils express GLUT1 and GLUT3 transporters, and glucose internalization occurs primarily through GLUT1. 63 Activation of neutrophils has been shown to increase glucose consumption, 64 coinciding with the increase in GLUT1, GLUT3 and GLUT4 transporters in the membrane, and expression of GLUT3 and GLUT4 is observed as a function of insulin concentration via PI3K. 59
Eosinophils differ in their ability to uptake glucose from the medium, being less effective than neutrophils; however, it was also shown that GLUT1, GLUT3, and GLUT4 transporters are involved in transporting glucose into eosinophils. 65 The presence of GLUT4 is interesting as there are no studies related to the effect of insulin on this type of leukocyte. IL‐5 is a factor that stimulates the activation of eosinophils. 66 It was observed that the incubation of eosinophils with IL‐5 substantially increased glucose internalization, and this effect was attenuated when the GLUT 4 transporter, tyrosine kinase activity in general and the specific activity of PI3K were inhibited. 65 Although there is no further evidence in this regard, the results support the idea that glucose uptake in activated eosinophils is regulated by insulin signaling.
Lymphocytes show differential expression of the insulin receptor as inactive lymphocytes do not express the insulin receptor on their membrane, and the GLUT1 isoform is the prevalent isoform in this cell lineage. 60 In B lymphocytes, there are no direct studies on the presence or absence of the insulin receptor when cells are inactive; however, insulin stimulates both glucose uptake and increases the expression of GLUT3 and GLUT4 in the plasma membrane. This effect was suppressed with inhibition of the PI3K pathway, suggesting expression of the insulin receptor in the quiescent state. 59 Inactive T lymphocytes do not express the insulin receptor on their membrane, so in this state, they are insensitive to insulin, 50 but once activated, both subtypes of lymphocytes (B and T) express the receptor and are able to increase glucose uptake. 59 , 67 The influence of insulin on the metabolic regulation of lymphocytes was demonstrated when analyzing diabetic patients, in whom immune cells are mostly activated, demonstrating that treatment with insulin increased glucose uptake in lymphocytes, implicating hormone‐sensitive transporter translocation to the membrane. 68
Pathogenic signals alone can influence the metabolic regulation of lymphocytes; however, they also require costimulatory signals that direct changes in glucose uptake and glucose metabolism necessary to perform their effector function. 69 In this sense, there is evidence that insulin is the extracellular molecule that regulates this process, as elimination of the insulin receptor in these cells leads to impaired glucose transport and aerobic glycolysis. 67 , 70 In activated lymphocytes, glucose and glutamine metabolism are regulated by the transcription factor c‐Myc, which promotes the transcription of glucose transporters and the enzymes that participate in glycolysis, 71 through the PI3K and AKT‐mTOR signaling axes. 72 Lymphocytes lacking the insulin receptor exhibit low levels of c‐Myc expression and decreased glucose absorption and glycolytic capacity, which is related to decreased expression of GLUT1 and the glycolytic enzymes HK, phosphofructokinase and LDH. 73 Therefore, accumulating evidence indicates that insulin plays an important role in the metabolic regulation of leukocytes (Fig. 1).
5. THE ROLE OF INSULIN IN LEUKOCYTE IMMUNE FUNCTION
In leukocytes, insulin not only influences glucose uptake and metabolism but also participates in the regulation of some of its immune functions. Neutrophils are the first leukocytes to reach the site of an infection, and their dynamic capacity is extensive as they migrate from the bloodstream to the sites of injury or inflammation, where their primary function is to phagocytose microorganisms and destroy them intracellularly with the generation of ROS and the fusion of cytoplasmic azurophilic granules; however, neutrophils also have the ability to destroy pathogens extracellularly through secretion of proteolytic enzymes released during degranulation and generation of neutrophil extracellular traps (NETs). 74 , 75
Although insulin does not regulate the transport of glucose in neutrophils, it does mediate effects on the regulation of some of its functions. Insulin per se, in the presence of normal physiologic glucose concentrations, has been shown to stimulate chemokinesis of neutrophils in healthy individuals 76 and to increase migration toward a positive gradient of chemotactic substances. 77 Although the signaling pathways that regulate chemotaxis can be diverse, it has been shown that the PI3K pathway associated with the insulin receptor participates in the regulation of this function. 78 Activation of the insulin‐dependent MAPK pathway produces an increase in the adhesion capacity of neutrophils after stimulation with chemoattractants, such as FMLP. 79 On the other hand, it was shown that insulin is capable of modulating the inflammatory response of neutrophils through the phosphorylation of FoxO1, which remains inactive, preventing the expression of TLR4 and extracellular matrix metalloproteinase 9 (MMP‐9) in response to LPS. 80 Insulin also participates in the regulation of phagocytic and bactericidal capacities of neutrophils. 81 These data show that insulin directly influences some functions of neutrophils, although the role of this hormone in the uptake of glucose by these cells remains to be fully elucidated.
Monocytes are phagocytes and sentinel cells found in circulation, and they have a powerful capacity to migrate toward sites of infection or inflamed tissues in response to chemotactic stimuli. In these cells, insulin‐dependent activation of ERK‐1/2 promotes expression of MMP‐9, which facilitates monocyte chemotaxis in response to MCP‐1. 82 Insulin also stimulates the adhesion and migration capacity of monocytes through the PI3K‐AKT pathway, favoring expression of macrophage antigen 1 in the membrane, 83 in addition to the activation of Rac‐1, an important molecule in the regulation of cell migration. 84 Once monocytes reach the site of infection or injury, they are able to modulate the inflammatory response through cytokine production, and indeed, insulin‐dependent ERK1/2 activation synergistically enhances IL‐6 production induced by saturated fatty acids 85 ; therefore, insulin influences the functions of monocytes, promoting and improving their migration through the secretion of proteolytic enzymes and the production of adhesion molecules and cytokines.
Lymphocytes are the effector cells of the adaptive immune system. There are 2 primary types of lymphocytes, B lymphocytes (or B cells), which produce antibodies, and T lymphocytes (also called T cells), which mediate the destruction of infected or tumoral cells and modulate immune responses. Both types of lymphocytes are capable of proliferating and can give rise to 2 subpopulations, effector and memory lymphocytes. 86
There is limited evidence about the effect of insulin on B cells; however, its effect on the regulation of T lymphocytes is known. Inactive T cells require less energy consumption for their survival and functions, but once activated, become effector T cells, and their activation leads to an increase in the consumption of ATP, which is generated by rewiring their oxidative metabolism to glycolysis. 69 It has been shown that activation and metabolic turnover are accompanied by positive regulation of the insulin receptor, which allows lymphocytes to have a greater capacity for proliferation, cytokine production, and survival, translating into the correct activation of these cells. 70 Similarly, insulin signaling mediated by its receptor exerts a stimulatory effect on T cells, activated by antigenic recognition, enhancing not only metabolism but also proliferation and cytokine production. 73
Eosinophils, a type of immune cell typically associated with allergies and defense against parasitic infections, also regulate the activation state of macrophages in mammalian adipose tissue and may play an important role in metabolic homeostasis. 87 To date, no studies have been reported that directly associate insulin regulation with eosinophil functions.
Basophils are the least abundant leukocytes in the circulation and are known to play an important role in the body's defense against parasitic infections, as well as in allergic reactions. Activation of basophils leads to an increase in the release of histamine and IL‐4. 88 Histamine release has been shown to be regulated by growth factors, such as IGF‐1, as well as by insulin. 89 Basophils share high structural and functional similarity with mast cells, although the latter are not found in circulation but are present in connective tissue and near blood vessels. Insulin in mast cells has been shown not only to promote survival in a PI3K‐dependent manner but also to enhance their degranulation more than activation mediated by the high affinity receptor for IgE (FcRI) alone. 90 , 91
An adequate immune response requires the production and secretion of a large number of proteins, such as cytokines. Insulin also regulates the production of proteins on cells of different tissues, and in this sense, it was observed that in mononuclear cells and neutrophils of adult and elderly individuals, it significantly stimulated the rate of protein synthesis. 92 Therefore, insulin is not only responsible for the homeostatic maintenance of energy in leukocytes but also influences their functional capacities.
6. INSULIN RESISTANCE
When cells are unable to respond to the transduction of signals derived from the interaction of insulin with its receptor, a pathophysiologic state known as insulin resistance occurs. 93 As a consequence, glucose cannot be transported into cells, causing an increase in plasma levels both when fasting and postprandial. 94 The compensatory mechanism of the pancreas is beta cell hyperplasia and overproduction and hypersecretion of insulin as an attempt to maintain normoglycemia. 95 Constant overexpression of insulin leads to dysfunction of pancreatic beta cells with the consequent appearance of type 2 diabetes, which is considered the primary pathology of systemic insulin resistance. 96
In the state of insulin resistance, cells exhibit alterations in signaling pathways at the level of their receptor or of the proteins involved in the signal transduction cascade. The molecular mechanisms described in the generation of insulin resistance involve chronic low‐grade inflammation, 97 hyperglycemia and advanced glycation end products formation, 98 , 99 increased free fatty acids, 100 oxidative stress and mitochondrial dysfunction, 101 , 102 , 103 endoplasmic reticulum stress, 104 , 105 , 106 and the presence of xenobiotic agents 107 , 108 (Fig. 2)
FIGURE 2.
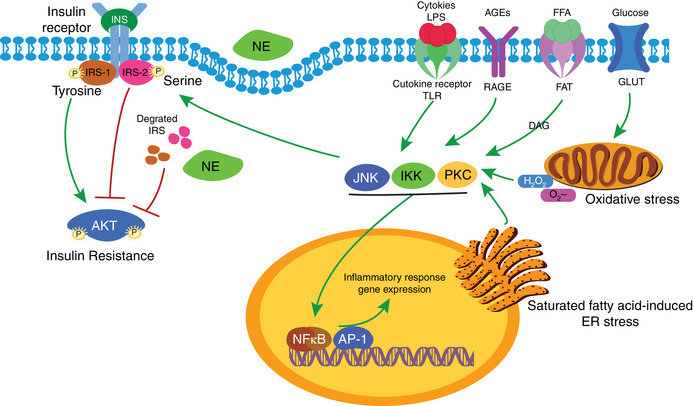
Molecular mechanisms involved in insulin resistance. Insulin signaling requires IRS‐1/2 phosphorylation at tyrosine residues and subsequent AKT phosphorylation at Thr308 and Ser473. Kinases such as JNK, IKK and PKC phosphorylate IRS in serine residues activating transcription factors wich improving pro‐inflammatory genes expression, deteriorating insulin signaling and producing insulin resistance. These kinases are activated by molecular mechanisms involving hyperglycemia, AGEs production, pro‐inflammatory cytokines, LPS, the increasing of free fatty acids and endoplasmic reticulum stress. Another mechanism described involve NE, wich can intake into the cell and degrade IRS, producing insulin resistance
7. INSULIN RESISTANCE IN LEUKOCYTE FUNCTION
The deterioration of intracellular signaling in leukocytes during insulin resistance and its effects on the response mechanisms of the host immune system are poorly understood, and analyzing the leukocyte dysfunction present in the state of insulin resistance as an independent pathophysiology has been complicated. The information obtained is associated with morbidities, such as obesity 109 and diabetes, 110 where the deterioration of immune function has been demonstrated.
Free fatty acids present in circulation during obesity induce insulin resistance; monocytes can capture and internalize these fatty acids through CD36 scavenger receptors. 111 , 112 It has been observed that in obese individuals with insulin resistance, the increase in CD36 receptors on monocytes induces a greater uptake of oxidized LDL, which is associated with the development of atherogenesis linked to insulin resistance. 113 , 114 In addition to these factors, these individuals not only have a higher number of circulating monocytes compared with lean individuals but also exhibit a higher number of CD16+ monocytes, which are considered potent inducers of inflammation. 115
Endoplasmic reticulum stress, in addition to being involved in the development of insulin resistance, also activates signaling pathways that lead to apoptosis. 116 In insulin‐resistant macrophages, reduced AKT phosphorylation interferes with retention of the FOXO1 factor in the cytoplasm, causing it to translocate to the nucleus, where it promotes apoptosis due to cholesterol‐induced stress. 117 It has also been shown that macrophages with deletion of the insulin receptor present decreased AKT phosphorylation and an increase in endoplasmic reticulum stress markers, favoring expression of the class A scavenger receptor and apoptosis when they are exposed to oxidized LDL. 118 Deterioration in MEK–ERK pathway signaling in insulin resistance leads to a decrease in the activity of the SERCA2b pump. This dysfunction causes calcium to be released from the lumen of the endoplasmic reticulum, favoring the increase in apoptosis associated with stress. 119 The direct effects on macrophage apoptosis may also be associated with the defective efferocytosis, a decrease in the ability to phagocytose apoptotic cells, seen in obesity, suggesting that insulin resistance plays an important role in atherogenic pathophysiology. 120
Insulin resistance in macrophages not only leads to reduced phosphorylation of AKT but also favors sustained activation of the mTORC1 protein complex, which causes insulin‐resistant macrophages to elevate glycolysis levels, inducing a phenotype similar to M2 with a decreased bactericidal capacity. 121 In fact, insulin resistance is a condition that favors what is known as trained immunity in macrophages, a form of memory of the cells of the innate immune system that involves changes at the metabolic and epigenetic level and leads to an altered response to damage or pathogens in diseases, such as obesity and diabetes. 122
The molecular and functional effects of insulin on neutrophils are poorly understood; however, it is evident that its function is deregulated as, in diabetic individuals, a decrease in its bactericidal 123 and chemotactic 124 capacity has been observed, whereas in obese individuals, neutrophils show a primed state determined by increased production of superoxide, both in basal and activated states, as well as its chemotactic activity. 125 , 126 These controversial results can be explained because during obesity, the state of low‐grade chronic inflammation determined by the increase in proinflammatory cytokines leads to a priming state in leukocytes, whereas in diabetes, hyperglycemia, or impairment in insulin signaling leads to neutrophil immune dysfunction.
In activated T lymphocytes, elimination of the insulin receptor decreases expression of GLUT 1 and several genes involved in glycolysis, causing a decrease in their ability to internalize glucose and in the inability to rewire their metabolism toward aerobic glycolysis. 73 This attenuates T lymphocytes’ proinflammatory function and in the same way, decreases the proliferative capacity and increases apoptosis, suggesting that the lack of signaling by the insulin receptor affects both the metabolism and immune function of lymphocytes. 70
During insulin resistance, chronic hyperinsulinemia occurs, which may or may not be accompanied by hyperglycemia. 127 It has been difficult to evaluate the effect of chronic exposure of leukocytes to high concentrations of insulin, glucose or both in vitro, as the half‐life of these cells is short, and the results have been contradictory. Acute exposure to hyperinsulinemia has been observed to improve leukocyte function; however, under hyperglycemic conditions, monocytes and neutrophils show a deterioration in functions, such as chemotaxis, phagocytosis and bactericidal capacity. 128
It has been observed in vivo that under conditions of hyperglycemia, neutrophils decrease expression of inflammatory genes, such as NFKBA, IL1A, and CCL3, whereas under conditions of hyperglycemia and hyperinsulinemia, the opposite effect occurs but does not affect the chemotactic, phagocytic, or respiratory burst functions of neutrophils. 129 However, chronic exposure of neutrophils from individuals with diabetes results in reduced chemotaxis. 124 The controversial effects derived from acute or chronic exposure to hyperglycemia, hyperinsulinemia, or both is clear; it seems that acute or short‐term exposure to leukocytes leads to an “improvement” in some of their functions, whereas when exposure is chronic, their functions decrease, probably due to the influence of other metabolic disorders, such as inflammation, dyslipidemia, or arterial hypertension, which can induce expression of various genes that affect the function of leukocytes. 129
8. INSULIN THERAPY IMPROVES LEUKOCYTE FUNCTION
Diabetes is the primary disease caused by altered insulin signaling in insulin‐dependent tissues, and it is clear that there is a dysfunction of the immune system reflected by the increase in infections in diabetic individuals. 130 , 131 The evidence that leukocytes are involved in both the initiation and progression of various diseases, such as diabetes and cardiac complications, has been analyzed 132 , 133 ; however, it has also been shown that insulin treatment in diabetic patients has positive effects, such as longer survival of critically ill patients 134 and improved wound healing. 135 This suggests that insulin may play an immunoregulatory role in these conditions.
Insulin treatment of patients with type 2 diabetes is used to improve glycemic control and reduce complications derived from this pathology. 136 There is evidence that insulin, in addition to improving glycemic control in patients, may have direct effects on the attenuated immune functions of leukocytes, as insulin treatment restores defective chemotaxis, 137 bactericidal function, 123 and the phagocytic activity of neutrophils both in vivo and ex vivo, suggesting that insulin exerts a direct effect on these cells, regardless of its action on glycemic control. 138
Type 1 and type 2 diabetes are associated with the presence of low‐grade inflammation as shown by the high concentration of biomarkers, such as TNFα, IL‐6, and CRP, 139 , 140 , 141 and the immune system plays a crucial role in the incidence and progression of these diseases, especially given its participation in inflammation of pancreatic and adipose tissue, where immune cells with proinflammatory characteristics, such as M1 macrophages and Th1 and Th17 cells, prevail. 142 , 143 Insulin has been shown to exert anti‐inflammatory effects, favoring the polarization of macrophages toward the M2 phenotype 144 and of T cells toward the Th2 phenotype, 67 suggesting that insulin restores the dysregulated inflammatory response in diabetic individuals.
Although glycemic control has been shown to reduce the risk of infections, the precise mechanisms through which diseases, such as type 2 diabetes, lead to secondary diseases that deteriorate the prognosis of patients are not known, so additional studies are needed. Exploring how insulin treatment could reverse these complications by improving the prognosis of patients is vital.
9. ROLE OF LEUKOCYTES IN SYSTEMIC INSULIN RESISTANCE
In recent years, foundations have been established due to the discovery of immunometabolism as a field of science that has 2 aspects: the first is to understand how changes in cellular metabolism contribute to regulation of the functions of immune cells, such as their activation, polarization, differentiation, and proliferation, whereas the second focuses on understanding how leukocytes modulate cellular processes in tissues to drive the necessary changes in the body in response to environmental stimuli. 145
Initially, it was considered that in adipose tissue, proinflammatory molecules had a central role in the development of insulin resistance 146 ; however, it was determined that the cells of the immune system infiltrated in these tissues were the ones that significantly modulated the generation. 147 In fact, many of the immune cells, including macrophages, B and T cells, 148 neutrophils and eosinophils, are involved in the production of cytokines during chronic inflammation. 149
Proinflammatory cytokines, such as TNFα and IL‐6, are capable of activating serine/threonine kinases that can impair the insulin signaling pathway through serine phosphorylation of IRS. 150 In this sense, the relationship of leukocytes with insulin resistance has been widely established. The increase in the availability of nutrients generates chemotactic molecules, such as MCP‐1 in adipocytes, 151 that lead to the recruitment of macrophages in adipose tissue, which additionally contributes to potentiating inflammation by releasing additional cytokines and chemoattractants 152 ; therefore, genetic inhibition as the endogenous function of MCP‐1 favors insulin sensitivity, suggesting that the development of insulin resistance related to obesity is promoted through an increase in macrophage infiltration, inducing inflammation in response to chemoattractant stimuli in adipose tissue. 152 In addition, vitamin D deficiency promotes the differentiation of macrophages toward the M2 phenotype through endoplasmic reticulum stress; however, these macrophages exhibit production of proinflammatory cytokines that contributes to the development of insulin resistance. 153 These results suggest that in response to specific signals, leukocytes may exert deleterious effects.
Lymphocytes also participate as mediators of insulin resistance in the presence of obesity, as adipocytes express the major MHC II that activates CD4+ T cells and promotes inflammation. 154 As a consequence, lymphocytes acquire the Th1 phenotype, which produces proinflammatory molecules, such as IFN‐γ, IL‐2, and TNF‐β, which mediate the cellular response and inflammation through the activation of phagocytes. 155 An increase in the number of Th1 cells in adipose tissue and peripheral blood is associated with the presence of insulin resistance. 142 Synergy between T and B lymphocytes can modulate the inflammatory response, as B lymphocytes from individuals with type 2 diabetes secrete reduced IL‐10 than is necessary to modulate Th1 cell differentiation. This imbalance could affect tissue metabolism, increasing inflammation. 156 In fact, during obesity, B cells secrete proinflammatory chemokines, such as IL‐8; they present antigens to T cells that lead to IFN‐γ production, which polarizes M1 macrophages and, in the latter, induces TNFα secretion, 157 suggesting the involvement of lymphocytes in the development of insulin resistance.
Finally, neutrophils play a relevant role in modulating inflammation through the production of various molecules. An enzyme produced by these cells is neutrophil elastase, a protease responsible for the destruction of phagocytosed pathogens, which can be secreted into the environment during degranulation and NET production. Neutrophil elastase is able to enter the cell where it degrades IRS1, which is key in the insulin signaling pathway in adipose and liver tissue cells; therefore, elastase released by neutrophils impairs insulin signaling, directly contributing to insulin resistance 158 , 159 (Fig. 2).
10. CONCLUDING REMARKS
The emergence of immunometabolism as a field of science that relates metabolic regulation and its effect on the cells of the immune system has reoriented the focus of the actions of insulin on leukocytes, proposing insulin as a hormone that regulates the metabolism and function of virtually all cells in the body. Knowledge regarding the molecular mechanisms involved in insulin signaling that guide the control of metabolism and the functions of leukocytes is gradually being uncovered. During this process, insulin resistance, as a condition that affects the mechanisms of signal transduction, is still in the early stages of study, representing a new field to explore. Additionally, the discovery that some leukocytes exhibit insulin sensitivity comparable to that of muscle or adipose cells raises the possibility of considering cells of the immune system as possible targets for the identification for early markers of pathophysiology, such as insulin resistance.
AUTHORSHIP
W.D.C.P. and O.L.G.C. wrote the manuscript and prepared the figures; H.A.R.R. and I.M.G. helped to design the figures and edited the paper; B.I.A. helped to review and design the manuscript; O.L.G.C. and I.P.R. contributed with style correction and review of the manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
ACKNOWLEDGMENT
Cruz‐Pineda WD was a recipient of a CONACyT fellowship (778762).
Cruz‐Pineda WD, Parra‐Rojas I, Rodríguez‐Ruí HA, Illades‐Aguiar B, Matia‐García I, Garibay‐Cerdenares OL. The regulatory role of insulin in energy metabolism and leukocyte functions. J Leukoc Biol. 2022;111:197–208. 10.1002/JLB.2RU1220-847R
Contributor Information
Isela Parra‐Rojas, Email: iprojas@yahoo.com.
Olga Lilia Garibay‐Cerdenares, Email: olgaribayce@conacyt.mx.
REFERENCES
- 1. Fu Z, Gilbert ER, Liu D. Regulation of insulin synthesis and secretion and pancreatic Beta‐cell dysfunction in diabetes. Curr Diabetes Rev. 2013;9(1):25‐53. [PMC free article] [PubMed] [Google Scholar]
- 2. Hou JC, Min L, Pessin JE. Insulin granule biogenesis, trafficking and exocytosis. Vitam Horm. 2009;80:473‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bedinger DH, Adams SH. Metabolic, anabolic, and mitogenic insulin responses: a tissue‐specific perspective for insulin receptor activators. Mol Cell Endocrinol. 2015;415:143‐156. [DOI] [PubMed] [Google Scholar]
- 5. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin‐resistant states. Cold Spring Harb Perspect Biol. 2014;6(1):a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watanabe M, Hayasaki H, Tamayama T, Shimada M. Histologic distribution of insulin and glucagon receptors. Braz J Med Biol Res. 1998;31:243‐256. [DOI] [PubMed] [Google Scholar]
- 7. Watanabe M, Hirose Y, Sugimoto M, Nakanishi M, Watanabe H, Shimada M. The distribution of tissue insulin receptors in the mouse by whole‐body autoradiography. J Recept Res. 1992;12(1):13‐37. [DOI] [PubMed] [Google Scholar]
- 8. Gray SM, Meijer RI, Barrett EJ. Insulin regulates brain function, but how does it get there?. Diabetes. 2014;63(12):3992‐3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heni M, Hennige AM, Peter A, et al. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One. 2011;6(6):e21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Emanuelli T, Burgeiro A, Carvalho E. Effects of insulin on the skin: possible healing benefits for diabetic foot ulcers. Arch Dermatol Res. 2016;308(10):677‐694. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y, Petreaca M, Yao M, Martins‐Green M. Cell and molecular mechanisms of keratinocyte function stimulated by insulin during wound healing. BMC Cell Biol. 2009;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferron M, Wei J, Yoshizawa T, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142(2):296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hou CJ, Liu JL, Li X, Bi LJ. Insulin promotes bone formation in augmented maxillary sinus in diabetic rabbits. Int J Oral Maxillofac Surg. 2012;41(3):400‐407. [DOI] [PubMed] [Google Scholar]
- 14. Klein GL. Insulin and bone: recent developments. World J Diabet. 2014;5(1):14‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ivanovs A, Rybtsov S, Ng ES, Stanley EG, Elefanty AG, Medvinsky A. Human haematopoietic stem cell development: from the embryo to the dish. Development. 2017;144(13):2323. [DOI] [PubMed] [Google Scholar]
- 16. Kondo M. Lymphoid and myeloid lineage commitment in multipotent hematopoietic progenitors. Immunol Rev. 2010;238(1):37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blumenreich M. The white blood cell and differential count. In: Walker HK, Hall WD, Hurst JW, eds. Clinical Methods: The History, Physical, and Laboratory Examinations. Boston: Butterworths; 1990:724‐727. [PubMed] [Google Scholar]
- 18. Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Adv Neutrophil Biol: Clinical Implications. Chest. 2008;134(3):606‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murakami MS, Rosen OM. The role of insulin receptor autophosphorylation in signal transduction. J Biol Chem. 1991;266(33):22653‐22660. [PubMed] [Google Scholar]
- 20. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213‐1226. [DOI] [PubMed] [Google Scholar]
- 21. McManus EJ, Sakamoto K, Armit LJ, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24(8):1571‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drummond MJ, Bell JA, Fujita S, et al. Amino acids are necessary for the insulin‐induced activation of mTOR/S6K1 signaling and protein synthesis in healthy and insulin resistant human skeletal muscle. Clin Nutr. 2008;27(3):447‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Middelbeek RJW, Chambers MA, Tantiwong P, et al. Insulin stimulation regulates AS160 and TBC1D1 phosphorylation sites in human skeletal muscle. Nutr Diabetes. 2013;3(6):e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sano H, Kane S, Sano E, et al. Insulin‐stimulated phosphorylation of a Rab GTPase‐activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278(17):14599‐14602. [DOI] [PubMed] [Google Scholar]
- 25. Dumm M. Glucose utilization and lactate production by leucocytes of patients with diabetes mellitus. Proc Soc Exp Biol Med. 1957;95(3):571‐574. [DOI] [PubMed] [Google Scholar]
- 26. Esmann V. Effect of insulin on human leucocytes. Diabetes. 1963;12(6):545. [DOI] [PubMed] [Google Scholar]
- 27. Kalant N, Schucher R. Glucose utilization and insulin responsiveness of leukocytes in diabetics. Can J Biochem Physiol. 1962;40(7):899‐903. [PubMed] [Google Scholar]
- 28. Martin SP, McKinney GR, Green R, Becker C. The influence of glucose, fructose, and insulin on the metabolism of leukocytes of healthy and diabetic subjects. J Clin Invest. 1953;32(11):1171‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Englhardt A, Metz T. Investigations on glucose uptake in isolated human leucocytes from normal and diabetic subjects. Diabetologia. 1971;7(3):143‐151. [DOI] [PubMed] [Google Scholar]
- 30. Chaudhry R, Varacallo M. Biochemistry, Glycolysis. In: StatPearls [Internet], ed. Treasure Island (FL). StatPearls Publishing, 2020. [Google Scholar]
- 31. Burgner JW, Ray WJ. On the origin of the lactate dehydrogenase induced rate effect. Biochemistry. 1984;23(16):3636‐3648. [DOI] [PubMed] [Google Scholar]
- 32. Alabduladhem T, Bordoni B. Physiology, Krebs Cycle. In: StatPearls, ed. Treasure Island (FL). StatPearls Publishing, 2020. [PubMed] [Google Scholar]
- 33. Berg J, Tymoczko J, Stryer L. The metabolism of glucose 6‐phosphate by the pentose phosphate pathway is coordinated with glycolysis. In: Freeman WH, ed, Biochemestry. New York. WH Freeman and Company, 2002: Section 20.4. [Google Scholar]
- 34. Le A, Lane AN, Hamaker M, et al. Glucose‐independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang C, Ko B, Hensley CT, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56(3):414‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kramer PA, Ravi S, Chacko B, Johnson MS, Darley‐Usmar VM. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biol. 2014;2:206‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. 1982;70(3):550‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia‐mediated activation of hypoxia‐inducible factor‐1α in cancer. Cancer Sci. 2019;110(5):1510‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jantsch J, Wiese M, Schödel J, et al. Toll‐like receptor activation and hypoxia use distinct signaling pathways to stabilize hypoxia‐inducible factor 1α (HIF1A) and result in differential HIF1A‐dependent gene expression. J Leukoc Biol. 2011;90(3):551‐562. [DOI] [PubMed] [Google Scholar]
- 40. Azevedo EP, Rochael NC, Guimarães‐Costa AB, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril‐ and phorbol 12‐myristate 13‐acetate‐induced neutrophil extracellular trap (NET) formation. J Biol Chem. 2015;290(36):22174‐22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS One. 2008;3(4):e2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Porter L, Toepfner N, Bashant KR, et al. Metabolic profiling of human eosinophils. Front Immunol. 2018;9. 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Domblides C, Lartigue L, Faustin B. Metabolic stress in the immune function of T cells, macrophages and dendritic cells. Cells. 2018;7(7):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vats D, Mukundan L, Odegaard JI, et al. Oxidative metabolism and PGC‐1beta attenuate macrophage‐mediated inflammation. Cell Metab. 2006;4(1):13‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab. 2013;1(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sp Walrand, Guillet C, Boirie Y, Vasson M‐P. Insulin differentially regulates monocyte and polymorphonuclear neutrophil functions in healthy young and elderly humans. J Clin Endocrinol Metab. 2006;91(7):2738‐2748. [DOI] [PubMed] [Google Scholar]
- 49. Helderman JH. Acute regulation of human lymphocyte insulin receptors. Analysis by the glucose clamp. J Clin Invest. 1984;74(4):1428‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brown TJ, Ercolani L, Ginsberg BH. Properties and regulation of the T lymphocyte insulin receptor. J Recept Res. 1983;3(4):481‐494. [DOI] [PubMed] [Google Scholar]
- 51. Helderman JH, Ayuso R, Rosenstock J, Raskin P. Monocyte‐T lymphocyte interaction for regulation of insulin receptors of the activated T lymphocyte. J Clin Invest. 1987;79(2):566‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Helderman JH, Raskin P. The T lymphocyte insulin receptor in diabetes and obesity: an intrinsic binding defect. Diabetes. 1980;29(7):551. [DOI] [PubMed] [Google Scholar]
- 53. Bieger W, Weicker H, Michl J. Transport and utilization of amino acids and glucose in human monocytes: activation of glucose metabolism by insulin. J Clin Endocrinol Metab. 1980;50(6):1121‐1126. [DOI] [PubMed] [Google Scholar]
- 54. Daneman D, Zinman B, Elliott ME, Bilan PJ, Klip A. Insulin‐stimulated glucose transport in circulating mononuclear cells from nondiabetic and IDDM subjects. Diabetes. 1992;41(2):227. [DOI] [PubMed] [Google Scholar]
- 55. Dimitriadis G, Maratou E, Boutati E, Psarra K, Papasteriades C, Raptis SA. Evaluation of glucose transport and its regulation by insulin in human monocytes using flow cytometry. Cytometry A. 2005;64(1):27‐33. [DOI] [PubMed] [Google Scholar]
- 56. Esmann V. The diabetic leukocyte. Enzyme. 1972;13:13‐32. [PubMed] [Google Scholar]
- 57. Esmann V. The polymorphonuclear leukocyte in diabetes mellitus. J Clin Chem Clin Biochem. 1983;21(9):561‐567. [PubMed] [Google Scholar]
- 58. Korgun ET, Demir R, Sedlmayr P, et al. Sustained hypoglycemia affects glucose transporter expression of human blood leukocytes. Blood Cells Mol Dis. 2002;28(2):152‐159. [DOI] [PubMed] [Google Scholar]
- 59. Maratou E, Dimitriadis G, Kollias A, et al. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur J Clin Invest. 2007;37(4):282‐290. [DOI] [PubMed] [Google Scholar]
- 60. Fu Y, Maianu L, Melbert BR, Garvey WT. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: a role for GLUT isoforms . Blood Cells Mol Dis. 2004;32(1):182‐190. [DOI] [PubMed] [Google Scholar]
- 61. Ahmed N, Kansara M, Berridge MV. Acute regulation of glucose transport in a monocyte‐macrophage cell line: glut‐3 affinity for glucose is enhanced during the respiratory burst. Biochem J. 1997;327(Pt 2):369‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mavros Y, Simar D, Singh MAF. Glucose tranporter‐4 expression in monocytes: a systematic review. Diabetes Res Clin Pract. 2009;84(2):123‐131. [DOI] [PubMed] [Google Scholar]
- 63. Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol. 2019;10. 2099‐. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schuster DP, Brody SL, Zhou Z, et al. Regulation of lipopolysaccharide‐induced increases in neutrophil glucose uptake. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L845‐L51. [DOI] [PubMed] [Google Scholar]
- 65. Venge P, Moberg L, Björnsson E, Bergström M, Långström B, Håkansson L. Mechanisms of basal and cytokine‐induced uptake of glucose in normal human eosinophils: relation to apoptosis. Respir Med. 2003;97(10):1109‐1119. [DOI] [PubMed] [Google Scholar]
- 66. Sanderson CJ. Interleukin‐5, eosinophils, and disease. Blood. 1992;79(12):3101‐3109. [PubMed] [Google Scholar]
- 67. Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology. 2007;148(1):346‐353. [DOI] [PubMed] [Google Scholar]
- 68. Piątkiewicz P, Czech A, Tatoń J. Glucose transport in human peripheral blood lymphocytes influenced by type 2 diabetes mellitus. Arch Immunol Ther Exp (Warsz). 2007;55(2):119‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maclver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84(4):949‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fischer HJ, Sie C, Schumann E, et al. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol. 2017;198(5):1910. [DOI] [PubMed] [Google Scholar]
- 71. Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Swords RT, Schenk T, Stengel S, et al. Inhibition of the PI3K/AKT/mTOR pathway leads to down‐regulation of c‐Myc and overcomes resistance to ATRA in acute myeloid leukemia. Blood. 2015;126(23):1363. [Google Scholar]
- 73. Tsai S, Clemente‐Casares X, Zhou AC, et al. Insulin receptor‐mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 2018;28(6):922‐934. [DOI] [PubMed] [Google Scholar]
- 74. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532‐1535. [DOI] [PubMed] [Google Scholar]
- 75. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. 2005;23:197‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cavalot F, Anfossi G, Russo I, et al. Insulin stimulates the polymorphonuclear leukocyte chemokinesis. Horm Metab Res. 1993;25(6):321‐322. [DOI] [PubMed] [Google Scholar]
- 77. Cavalot F, Anfossi G, Russo I, et al. Insulin, at physiological concentrations, enhances the polymorphonuclear leukocyte chemotactic properties. Horm Metab Res. 1992;24(05):225‐228. [DOI] [PubMed] [Google Scholar]
- 78. Oldenborg P‐A, Sehlin J. Insulin‐stimulated chemokinesis in normal human neutrophils is dependent on D‐glucose concentration and sensitive to inhibitors of tyrosine kinase and phosphatidylinositol 3‐kinase. J Leukoc Biol. 1998;63(2):203‐208. [DOI] [PubMed] [Google Scholar]
- 79. Pillinger MH, Feoktistov AS, Capodici C, et al. Mitogen‐activated protein kinase in neutrophils and enucleate neutrophil cytoplasts: evidence for regulation of cell‐cell adhesion. J Biol Chem. 1996;271(20):12049‐12056. [DOI] [PubMed] [Google Scholar]
- 80. Zhang Z, Amorosa LF, Coyle SM, et al. Insulin‐dependent regulation of mTORC2‐Akt‐FoxO suppresses TLR4 signaling in human leukocytes: relevance to Type 2 diabetes. Diabetes. 2016;65(8):2224‐2234. [DOI] [PubMed] [Google Scholar]
- 81. Walrand S, Guillet C, Boirie Y, Vasson MP. In vivo evidences that insulin regulates human polymorphonuclear neutrophil functions. J Leukoc Biol. 2004;76(6):1104‐1110. [DOI] [PubMed] [Google Scholar]
- 82. Kappert K, Meyborg H, Clemenz M, et al. Insulin facilitates monocyte migration: a possible link to tissue inflammation in insulin‐resistance. Biochem Biophys Res Commun. 2008;365(3):503‐508. [DOI] [PubMed] [Google Scholar]
- 83. Jin SY, Kim EK, Ha JM, et al. Insulin regulates monocyte trans‐endothelial migration through surface expression of macrophage‐1 antigen. Biochim Biophys Acta. 2014;1842(9):1539‐1548. [DOI] [PubMed] [Google Scholar]
- 84. Liu Y, Dhall S, Castro A, Chan A, Alamat R, Martins‐Green M. Insulin regulates multiple signaling pathways leading to monocyte/macrophage chemotaxis into the wound tissue. Biol Open. 2018;7(1):bio026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bunn RC, Cockrell GE, Ou Y, Thrailkill KM, Lumpkin CK, Fowlkes JL. Palmitate and insulin synergistically induce IL‐6 expression in human monocytes. Cardiovasc Diabetol. 2010;9(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alberts B, Johnson A, Lewis J. Limphocytes and the cellular basis of adaptative immunity. In: Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD, eds. Molecular Biology of Cell. New York. Garland Science, 2002. [Google Scholar]
- 87. Zhu L, Su T, Xu M, et al. Eosinophil inversely associates with type 2 diabetes and insulin resistance in Chinese adults. PLoS One. 2013;8(7). e67613‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hübner MP, Larson D, Torrero MN, et al. Anti‐FcεR1 antibody injections activate basophils and mast cells and delay Type 1 diabetes onset in NOD mice. Clin Immunol. 2011;141(2):205‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hirai K, Miyamasu M, Yamaguchi M, et al. Modulation of human basophil histamine release by insulin‐like growth factors. J Immunol. 1993;150(4):1503. [PubMed] [Google Scholar]
- 90. Kettner A, Di Matteo M, Santoni A. Insulin potentiates FcɛRI‐mediated signaling in mouse bone marrow‐derived mast cells. Mol Immunol. 2010;47(5):1039‐1046. [DOI] [PubMed] [Google Scholar]
- 91. Lessmann E, Grochowy G, Weingarten L, et al. Insulin and insulin‐like growth factor‐1 promote mast cell survival via activation of the phosphatidylinositol‐3‐kinase pathway. Exp Hematol. 2006;34(11):1532‐1541. [DOI] [PubMed] [Google Scholar]
- 92. Walrand Sp, Guillet C, Gachon P, et al. Insulin regulates protein synthesis rate in leukocytes from young and elderly healthy humans. Clin Nutr. 2005;24(6):1089‐1098. [DOI] [PubMed] [Google Scholar]
- 93. Freeman A, Pennings N. Insulin Resistance. In: StatPearls, ed. Treasure Island (FL). StatPearls Publishing, 2020. [Google Scholar]
- 94. Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2004;56(1):45‐62. [DOI] [PubMed] [Google Scholar]
- 95. Wilcox G. Insulin and insulin resistance. Clin Biochem Rev. 2005;26(2):19‐39. [PMC free article] [PubMed] [Google Scholar]
- 96. Cerf ME. Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne). 2013;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dandona P, Aljada A, Bandyopadhyay A. Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol. 2004;25(1):4‐7. [DOI] [PubMed] [Google Scholar]
- 98. Pinto‐Junior DC, Silva KS, Michalani ML, et al. Advanced glycation end products‐induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci Rep. 2018;8(1):8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Unoki H, Yamagishi S. Advanced glycation end products and insulin resistance. Curr Pharm Des. 2008;14(10):987‐989. [DOI] [PubMed] [Google Scholar]
- 100. Vazquez‐Jimenez G, Chavez‐Reyes J, Romero‐Garcia T, et al. Palmitic acid but not palmitoleic acid induces insulin resistance in a human endothelial cell line by decreasing SERCA pump expression. Cell Signal. 2016;28(1):53‐59. [DOI] [PubMed] [Google Scholar]
- 101. Fazakerley DJ, Minard AY, Krycer JR, et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J Biol Chem. 2018;293(19):7315‐7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J. 2017;40(5):257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ohta Y, Kinugawa S, Matsushima S, et al. Oxidative stress impairs insulin signal in skeletal muscle and causes insulin resistance in postinfarct heart failure. Am J Physiol Heart Circ Physiol. 2011;300(5):H1637‐H44. [DOI] [PubMed] [Google Scholar]
- 104. Boden G, Duan X, Homko C, et al. Increase in endoplasmic reticulum stress‐related proteins and genes in adipose tissue of obese, insulin‐resistant individuals. Diabetes. 2008;57(9):2438‐2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chan SMH, Sun R‐Q, Zeng X‐Y, et al. Activation of PPARα ameliorates hepatic insulin resistance and steatosis in high fructose‐fed mice despite increased endoplasmic reticulum stress. Diabetes. 2013;62(6):2095‐2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Villalobos‐Labra R, Subiabre M, Toledo F, Pardo F, Sobrevia L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol Aspects Med. 2019;66:49‐61. [DOI] [PubMed] [Google Scholar]
- 107. Lasram MM, Dhouib IB, Annabi A, El Fazaa S, Gharbi N. A review on the molecular mechanisms involved in insulin resistance induced by organophosphorus pesticides. Toxicology. 2014;322(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 108. Liang Y, Zhan J, Liu D, et al. Organophosphorus pesticide chlorpyrifos intake promotes obesity and insulin resistance through impacting gut and gut microbiota. Microbiome. 2019;7(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Andersen CJ, Murphy KE, Fernandez ML. Impact of obesity and metabolic syndrome on immunity. Adv Nutr. 2016;7(1):66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Geerlings SE, Hoepelman AIM. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol. 1999;26(3‐4):259‐265. [DOI] [PubMed] [Google Scholar]
- 111. Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A‐I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277(51):49982‐49988. [DOI] [PubMed] [Google Scholar]
- 112. Zingg JM, Ricciarelli R, A A. Scavenger receptors and modified lipoproteins: fatal attractions?. IUBMB Life. 2000;49(5):397‐403. [DOI] [PubMed] [Google Scholar]
- 113. Kashyap SR, Ioachimescu AG, Gornik HL, et al. Lipid‐induced insulin resistance is associated with increased monocyte expression of scavenger receptor CD36 and internalization of oxidized LDL. Obesity. 2009;17(12):2142‐2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sarigianni M, Bekiari E, Tsapas A, et al. Effect of leptin and insulin resistance on properties of human monocytes in lean and obese healthy participants. Angiology. 2010;61(8):768‐774. [DOI] [PubMed] [Google Scholar]
- 115. de Matos MA, Duarte TC, Ottone V, et al. The effect of insulin resistance and exercise on the percentage of CD16(+) monocyte subset in obese individuals. Cell Biochem Funct. 2016;34(4):209‐216. [DOI] [PubMed] [Google Scholar]
- 116. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006;7(9):880‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Senokuchi T, Liang C‐P, Seimon TA, et al. Forkhead transcription factors (FoxOs) promote apoptosis of insulin‐resistant macrophages during cholesterol‐induced endoplasmic reticulum stress. Diabetes. 2008;57(11):2967‐2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Han S, Liang C‐P, DeVries‐Seimon T, et al. Macrophage insulin receptor deficiency increases ER stress‐induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3(4):257‐266. [DOI] [PubMed] [Google Scholar]
- 119. Liang C‐P, Han S, Li G, Tabas I, Tall AR. Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin‐resistant macrophages and are reversed by exenatide treatment. Diabetes. 2012;61(10):2609‐2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tabas I, Tall A, Accili D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ Res. 2010;106(1):58‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ieronymaki E, Theodorakis EM, Lyroni K, et al. Insulin resistance in macrophages alters their metabolism and promotes an M2‐Like phenotype. J Immunol. 2019;202(6):1786‐1797. [DOI] [PubMed] [Google Scholar]
- 122. Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol. 2019;10:1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Naghibi M, Smith RP, Baltch AL, et al. The effect of diabetes mellitus on chemotactic and bactericidal activity of human polymorphonuclear leukocytes. Diabetes Res Clin Pract. 1987;4(1):27‐35. [DOI] [PubMed] [Google Scholar]
- 124. Shetty N, Thomas B, Ramesh A. Comparison of neutrophil functions in diabetic and healthy subjects with chronic generalized periodontitis. J Indian Soc Periodontol. 2008;12(2):41‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Brotfain E, Hadad N, Shapira Y, et al. Neutrophil functions in morbidly obese subjects. Clin Exp Immunol. 2015;181(1):156‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Trottier MD, Naaz A, Kacynski K, Yenumula PR, Fraker PJ. Functional capacity of neutrophils from class III obese patients. Obesity. 2011;20(5):1057‐1065. [DOI] [PubMed] [Google Scholar]
- 127. Kim SH, Reaven GM. Insulin resistance and hyperinsulinemia: you can't have one without the other. Diabetes Care. 2008;31(7):1433‐1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Neethi Raj P, Shaji BV, Haritha VH, Anie Y. Neutrophil secretion modulates neutrophil and monocyte functions during hyperglucose and/or hyperinsulin conditions in vitro. J Cell Immunother. 2018;4(2):65‐70. [Google Scholar]
- 129. Stegenga ME, van der Crabben SN, Dessing MC, et al. Effect of acute hyperglycaemia and/or hyperinsulinaemia on proinflammatory gene expression, cytokine production and neutrophil function in humans. Diabet Med. 2008;25(2):157‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 diabetes and its impact on the immune system. Curr Diabetes Rev. 2020;16(5):442‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: a review of pathogenesis. Indian J Endocrinol Metab. 2012;16(Suppl1):S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Dronge AS, Perkal MF, Kancir S, Concato J, Aslan M, Rosenthal RA. Long‐term glycemic control and postoperative infectious complications. Arch Surg. 2006;141(4):375‐380. [DOI] [PubMed] [Google Scholar]
- 133. Tsalamandris S, Antonopoulos AS, Oikonomou E, et al. The role of inflammation in diabetes: current concepts and future perspectives. Eur Cardiol. 2019;14(1):50‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Van den Berghe G, Wilmer A, Milants I, et al. Intensive insulin therapy in mixed medical/surgical intensive care units. Diabetes. 2006;55(11):3151. [DOI] [PubMed] [Google Scholar]
- 135. Vatankhah N, Jahangiri Y, Landry GJ, Moneta GL, Azarbal AF. Effect of systemic insulin treatment on diabetic wound healing. Wound Repair Regen. 2017;25(2):288‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. UK Prospective Diabetes Study (UKPDS) Group . Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837‐853. [PubMed] [Google Scholar]
- 137. Mowat AG, Baum J. Chemotaxis of polymorphonuclear leukocytes from patients with diabetes mellitus. N Engl J Med. 1971;284(12):621‐627. [DOI] [PubMed] [Google Scholar]
- 138. Yano H, Kinoshita M, Fujino K, et al. Insulin treatment directly restores neutrophil phagocytosis and bactericidal activity in diabetic mice and thereby improves surgical site Staphylococcus aureus infection. Infect Immun. 2012;80(12):4409‐4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ebtehaj S, Gruppen EG, Parvizi M, Tietge UJ, Dullaart RP. The anti‐inflammatory function of HDL is impaired in type 2 diabetes: role of hyperglycemia, paraoxonase‐1 and low grade inflammation. Cardiovasc Diabetol. 2017;16(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kengne AP, Batty GD, Hamer M, Stamatakis E, Sb Czernichow. Association of C‐reactive protein with cardiovascular disease mortality according to diabetes status: pooled analyses of 25,979 participants from four U.K. prospective cohort studies. Diabetes Care. 2012;35(2):396‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Mirza S, Hossain M, Mathews C, et al. Type 2‐diabetes is associated with elevated levels of TNF‐alpha, IL‐6 and adiponectin and low levels of leptin in a population of Mexican Americans: a cross‐sectional study. Cytokine. 2012;57(1):136‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. McLaughlin T, Liu L‐F, Lamendola C, et al. T‐cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol. 2014;34(12):2637‐2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Szablewski L. Role of immune system in type 1 diabetes mellitus pathogenesis. Int Immunopharmacol. 2014;22(1):182‐191. [DOI] [PubMed] [Google Scholar]
- 144. Yu T, Gao M, Yang P, et al. Insulin promotes macrophage phenotype transition through PI3K/Akt and PPAR‐γ signaling during diabetic wound healing. J Cell Physiol. 2019;234(4):4217‐4231. [DOI] [PubMed] [Google Scholar]
- 145. Man K, Kutyavin V, Chawla A. Tissue immunometabolism: development, physiology, and pathobiology. Cell Metab. 2017;25(1):11‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor‐alpha: direct role in obesity‐linked insulin resistance. Science. 1993;259(5091):87. [DOI] [PubMed] [Google Scholar]
- 147. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity‐related insulin resistance. J Clin Invest. 2003;112(12):1821‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Burman A, Haworth O, Bradfield P, et al. The role of leukocyte‐stromal interactions in chronic inflammatory joint disease. Joint Bone Spine. 2005;72(1):10‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Khansari N, Shakiba Y, Mahmoudi M. Chronic inflammation and oxidative stress as a major cause of age‐related diseases and cancer. Recent Pat Inflamm Allergy Drug Discov. 2009;3(1):73‐80. [DOI] [PubMed] [Google Scholar]
- 150. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kanda H, Tateya S, Tamori Y, et al. MCP‐1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116(6):1494‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72(1):219‐246. [DOI] [PubMed] [Google Scholar]
- 153. Oh J, Riek AE, Darwech I, et al. Deletion of macrophage Vitamin D receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell Rep. 2015;10(11):1872‐1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Deng T, Lyon CJ, Minze LJ, et al. Class II major histocompatibility complex plays an essential role in obesity‐induced adipose inflammation. Cell Metab. 2013;17(3):411‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2014;74(1):5‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. van Exel E, Gussekloo J, de Craen AJ, Frölich M, Bootsma‐van der Wiel A, Westendorp RG. Low production capacity of interleukin‐10 associates with the metabolic syndrome and type 2 diabetes. Diabetes. 2002;51(4):1088. [DOI] [PubMed] [Google Scholar]
- 157. Winer DA, Winer S, Shen L, Chng MHY, Engleman EG. B lymphocytes as emerging mediators of insulin resistance. Int J Obes Suppl. 2012;2(Suppl 1):S4‐S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mansuy‐Aubert V, Zhou QL, Xie X, et al. Imbalance between neutrophil elastase and its inhibitor α1‐antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013;17(4):534‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Talukdar S, Oh DY, Bandyopadhyay G, et al. Neutrophils mediate insulin resistance in mice fed a high‐fat diet through secreted elastase. Nat Med. 2012;18(9):1407‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]