

Abstract
Background and Aims
RO7062931 is an N‐acetylgalactosamine (GalNAc)‐conjugated single‐stranded locked nucleic acid oligonucleotide complementary to HBV RNA. GalNAc conjugation targets the liver through the asialoglycoprotein receptor (ASGPR). This two‐part phase 1 study evaluated the safety, pharmacokinetics, and pharmacodynamics of RO7062931 in healthy volunteers and patients with chronic hepatitis B (CHB) who were virologically suppressed.
Approach and Results
Part 1 was a single ascending dose study in healthy volunteers randomized to receive a single RO7062931 dose (0.1‐4.0 mg/kg), or placebo. Part 2 was a multiple ascending dose study in patients with CHB randomized to receive RO7062931 at 0.5, 1.5, or 3.0 mg/kg or placebo every month for a total of 2 doses (Part 2a) or RO7062931 at 3.0 mg/kg every 2 weeks, 3.0 mg/kg every week (QW), or 4.0 mg/kg QW or placebo for a total of 3‐5 doses (Part 2b). Sixty healthy volunteers and 59 patients received RO7062931 or placebo. The majority of adverse events (AEs) reported were mild in intensity. Common AEs included self‐limiting injection site reactions and influenza‐like illness. Supradose‐proportional increases in RO7062931 plasma exposure and urinary excretion occurred at doses ≥3.0 mg/kg. In patients with CHB, RO7062931 resulted in dose‐dependent and time‐dependent reduction in HBsAg versus placebo. The greatest HBsAg declines from baseline were achieved with the 3.0 mg/kg QW dose regimen (mean nadir ~0.5 log10 IU/mL) independent of HBeAg status.
Conclusions
RO7062931 is safe and well tolerated at doses up to 4.0 mg/kg QW. Supradose‐proportional exposure at doses of 3.0‐4.0 mg/kg was indicative of partial saturation of the ASGPR‐mediated liver uptake system. Dose‐dependent declines in HBsAg demonstrated target engagement with RO7062931.
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- ASGPR
asialoglycoprotein receptor
- ASO
antisense oligonucleotide
- BMI
body mass index
- CHB
chronic hepatitis B
- Fe
dose fraction excreted
- GalNAc
N‐acetylgalactosamine
- ISR
injection site reaction
- LNA
locked nucleic acid
- NUC
nucleos(t)ide analogue
- PD
pharmacodynamics
- PK
pharmacokinetics
- Q2W
every 2 weeks
- QM
every month
- QW
every week
- RISC
RNA‐induced silencing complex
- RNase‐H
ribonuclease H
- siRNA
small interfering RNA
- ULN
upper limit of normal
Chronic hepatitis B (CHB) infection is a major global health problem, with an estimated prevalence of 257 million people.( 1 ) Nearly 25% of all patients with CHB develop serious liver diseases, such as cirrhosis or HCC, and in 2015, nearly 0.8 million deaths were attributed to the disease.( 1 , 2 ) Current clinical guidelines consider clearance of HBsAg as the optimal endpoint for CHB disease, as it has been shown to be associated with improved survival and protective against disease progression and development of HBV complications including cirrhosis, liver failure, and HCC.( 3 , 4 ) However, currently available treatments including nucleos(t)ide analogues (NUCs) and pegylated interferons (PEG‐IFNs) reduce the risk of CHB sequelae but are associated with very low rates of HBsAg loss in addition to challenges of potentially lifelong treatment with NUCs and the safety and tolerability profile of PEG‐IFNs. Because of the therapeutic limitations of the available treatments, therapeutic modalities are being developed with the goal of a functional cure.( 2 , 5 , 6 ) Functional cure is defined as sustained, undetectable HBsAg with or without seroconversion to hepatitis B surface antibody after completion of a finite course of treatment.( 7 )
An important characteristic of CHB infection that is responsible for preventing viral clearance and recovery from HBV is T‐cell exhaustion. Increased circulating antigens or viral load in CHB stimulates an immunologic environment rich in coinhibitory receptors, such as programmed death receptor 1/programmed death ligand 1 and T‐regulatory cells that contribute to persistent T‐cell exhaustion and loss of an appropriate immune response to HBV infection.( 8 ) High levels of HBsAg expression persist throughout the HBeAg‐positive and HBeAg‐negative phases of the disease,( 2 , 9 ) which is thought to play a prominent role in promoting virus‐specific T‐cell anergy and deletion in chronic HBV infection.( 10 , 11 , 12 ) Other viral antigens, including HBeAg and the HBV protein X, have also been shown to play a role in maintaining CHB by suppressing cellular immune responses to HBV.( 2 ) Therefore, a combination of antiviral agents (that inhibit antigen production) together with immune‐modulating agents are likely to be more efficacious than single agents alone in achieving higher rates of functional cure.
Investigational therapies targeting the shared 3′ region of HBV RNA transcripts prevent the translation of viral antigens, including HBsAg, thereby potentially facilitating restoration of the virus‐specific immune response and functional cure.( 13 , 14 , 15 , 16 , 17 ) HBV gene expression inhibitors in development include double‐stranded small interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs). siRNAs, which work through the RNA‐induced silencing complex (RISC), are being investigated in clinical trials for the treatment of CHB.( 6 , 13 , 17 , 18 , 19 , 20 , 21 ) ASOs, which work through a ribonuclease H (RNase‐H)–mediated mechanism of mRNA degradation, have also shown promising antiviral activity in preclinical studies( 14 , 22 ) and early‐phase clinical trials involving patients with CHB.( 17 , 23 )
RO7062931 is a liver‐targeted locked nucleic acid (LNA)‐containing ASO that is complementary to a highly conserved sequence in the shared 3′ region common to all HBV RNA transcripts. To achieve hepatocyte‐specific targeting, RO7062931 comprises an RNase‐H–directing LNA‐ASO (gapmer) conjugated to three molecules of N‐acetylgalactosamine (GalNAc), which is a high‐affinity ligand of the asialoglycoprotein receptor (ASGPR) predominantly expressed at the hepatocyte cell surface.( 24 , 25 ) Studies in the adeno‐associated virus‐HBV mouse model have shown that GalNAc conjugation results in a distribution shift of LNA‐ASO to liver hepatocytes away from the kidney, leading to potent and durable reduction of viral replication and HBsAg levels relative to unconjugated LNA‐ASO.( 14 ) However, ASGPR uptake processes are saturable, and doses of RO7062931 beyond which ASGPR saturation occurs would result in a proportionally lower liver uptake and increased off‐target exposures (e.g., kidney), with increased potential for off‐target toxicity.( 14 , 26 ) Dose level and dosing frequency should be tailored to minimize ASGPR receptor saturation, thereby maintaining efficient hepatocyte‐targeted delivery of RO7062931.( 26 ) Preclinical data in rodents and monkeys have shown that plasma and urine RO7062931 levels are indicators of ASGPR saturation, with a shift to supradose‐proportional plasma kinetics and appearance of RO7062931 in the urine as the ASGPR liver uptake system begins to saturate with increasing dose (unpublished data).
Here, we report the results of a study (BP39405, NCT03038113) designed to evaluate the safety, tolerability, and pharmacokinetics (PK) of a wide range of RO7062931 doses in healthy volunteers and the safety, tolerability, PK, and pharmacodynamics (PD) of multiple RO7062931 dosing regimens in patients with CHB who were NUC‐suppressed.
Patients and Methods
Study Design
This was a randomized, identity‐hidden, placebo‐controlled, adaptive phase 1 study conducted in healthy volunteers (Part 1) and patients with CHB (Part 2) (Fig. 1). The primary objective of the study was to evaluate the safety and tolerability of RO7062931 compared with placebo, with plasma and urine PK assessed as secondary objectives. Part 2 included an additional secondary objective to study HBsAg dynamics after RO7062931 administration. Exploratory objectives of the study were to determine doses of RO7062931 resulting in ASGPR receptor saturation (Part 1) and the effects of RO7062931 on viral parameters other than HBsAg (Part 2). The study protocol is available online at www.clinicaltrials.gov.
FIG. 1.
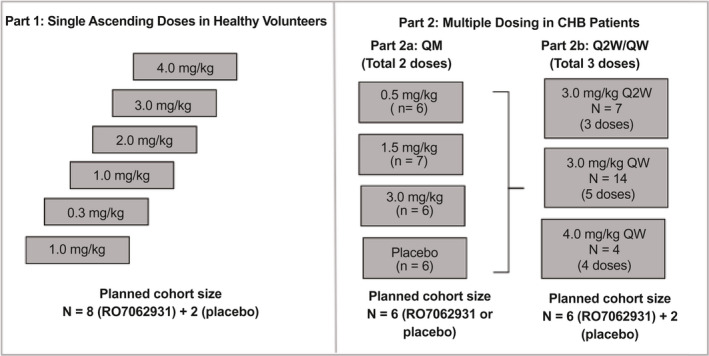
Study design. Part 1: single ascending dose cohorts in healthy volunteers. Parts 2a and 2b: multiple‐dose cohorts in patients with CHB.
The study was performed in accordance with the International Conference on Harmonisation E6 good clinical practice guidelines, the Declaration of Helsinki, and the laws and regulations of the regions in which the research was conducted. The study was approved by the institutional review board/independent ethics committee of the participating sites. All participants gave written informed consent before participation in the study.
Part 1—Single‐Dose Escalation Study in Healthy Volunteers
Part 1 was a single ascending dose study conducted in New Zealand comprising six dose‐escalation cohorts. In each cohort, 10 healthy volunteers were randomized to subcutaneous injection of RO7062931 or placebo in a 4:1 ratio. The dose‐escalation schedule comprised doses of 0.1, 0.3, 1.0, 2.0, 3.0, and 4.0 mg/kg. After screening, volunteers were admitted to the clinic on Day ‑1, received RO7062931 or placebo on Day 1, and were discharged on Day 3. Participants returned for outpatient visits on Days 4, 5, 6, 8, 15, 29, and 85.
Sentinel dosing was performed, whereby 2 participants from each cohort, at least one of whom received RO7062931, were followed for 24 hours after dosing to monitor for acute reactions. The remaining 8 participants were dosed thereafter, contingent on satisfactory safety assessments. Safety was monitored for at least 28 days in the entire cohort before starting the next dose escalation cohort.
Part 2—Multiple‐Dose Study in Patients With CHB
Part 2 was a parallel multiple‐dose study conducted in Australia, Hong Kong, Korea, New Zealand, Taiwan, and Thailand. In Part 2a, patients were randomized 1:1:1:1 to subcutaneous doses of RO7062931 at 0.5, 1.5, or 3.0 mg/kg or placebo administered every month (QM) for a total of 2 doses. In Part 2b, patients were randomized 3:1 to sc RO7062931 or placebo across 3 multiple‐dose cohorts: 3.0 mg/kg every 2 weeks (Q2W) for 3 doses; 3.0 mg/kg every week (QW) for 5 doses; or 4.0 mg/kg QW for 4 doses. Patients in the 4.0 mg/kg QW cohort received only 4 doses because the study protocol specified that the total dose could not exceed 16 mg/kg/month. Part 2c, which was planned to investigate extended QW treatment duration for up to 48 weeks, was not conducted.
Patients were followed up weekly for the first 50/57 days and then monthly up to at least 106/113 days after the last dose of RO7062931, depending on the cohort. Study visits included two optional in‐clinic stays for drug administration (Day 1 and Day 29, or Day 22 for the 4‐dose QW cohort). On other drug administration days, patients received the drug at an outpatient clinic visit.
Participants
Male and female healthy volunteers and patients with CHB aged 18‐65 years were eligible to participate in the study. In Part 1, healthy participants who were nonsmokers with body mass index (BMI) 18‐30 kg/m2 and body weight ≥50 kg were eligible for participation. In Part 2, a diagnosis of CHB (HBsAg‐positive for >6 months before randomization) was required, and patients should have been receiving entecavir, tenofovir, adefovir, or telbivudine treatment for ≥6 months with treatment ongoing throughout the study. Patients were also required to have HBsAg titer ≥1,000 IU/mL at screening and HBV DNA ≤90 IU/mL for ≥6 months. Inclusion criteria also specified alanine aminotransferase (ALT), aspartate aminotransferase, γ‐glutamyltransferase, and alkaline phosphatase to be ≤1.5 × upper limit of normal (ULN) at screening. The required BMI was 18‐32 kg/m2. Additional inclusion and exclusion criteria for Parts 1 and 2 are described in the Supporting Information.
Drug Administration
Study drugs were administered by subcutaneous injection at appropriate sites (i.e., the abdomen or upper thigh). For multiple doses in Part 2, the administration site was rotated. All doses were administered at the study clinic in the morning in the fasted state either 2 hours before or 2 hours after a morning meal.
Safety Assessments
Safety assessments included adverse events (AEs), clinical laboratory tests (hematology, blood chemistry, coagulation, urinalysis), electrocardiogram (ECG), vital signs (blood pressure, heart rate, temperature), and physical examination. AEs were recorded until the last follow‐up visit and classified according to severity as mild, moderate, or severe. The causality of AEs was determined by the investigator. All AEs were followed until resolution, until the condition stabilized, or until the participant was lost to follow‐up.
Abnormalities in laboratory findings or vital signs were reported as AEs if they were accompanied by clinical symptoms, resulted in a change in study treatment, resulted in a medical intervention, or were deemed clinically significant by the investigator.
PK Assessments
Plasma and urine PK were used as surrogate markers to provide exploratory information about single doses that may lead to liver ASGPR saturation (doses at which RO7062931 dose‐dependent PK becomes nonlinear in plasma and urine). Details of PK sample collection are described in the Supporting Information.
PD Assessments
In Part 2, blood samples for quantitative HBsAg assessment were collected from patients with CHB at study visits before and after dosing up to Day 113. HBsAg assessments included change in quantitative HBsAg (log10 IU/mL) from baseline over time and maximum change from baseline in quantitative HBsAg across all time points. HBsAg was assessed in patients who were HBeAg‐positive and patients who were HBeAg‐negative. Exploratory PD assessments included the quantification of HBV RNA and HBcAg. Details of PD methods are described in the Supporting Information.
Statistical Analysis
The planned sample sizes were 10 healthy volunteers per cohort (n = 8 for RO7062931, n = 2 for placebo) in Part 1, 6 patients per cohort in Part 2a, and 8 patients (n = 6 for RO7092931, n = 2 for placebo) per cohort in Part 2b. Healthy volunteers receiving placebo in Part 1 were pooled as one treatment group, as were all patients receiving placebo in Part 2. The safety analysis included study participants who received ≥1 dose of study medication. Study participants who did not significantly deviate from the protocol and had adequate data for PK analysis were included in the PK analysis population. Patients were excluded from the PD analysis if they prematurely discontinued planned study treatment. Descriptive statistics were used to summarize the plasma and urine PK parameters (calculated using WinNonlin software), the change from baseline in log10 quantitative HBsAg, and other PD parameters. The Power model method was used in the assessment of dose proportionality and is described in the Supporting Information.
A post‐hoc analysis evaluated change from baseline in quantitative HBsAg using a Mixed Model for Repeated Measures with treatment, visit, and treatment by visit interaction fitted as fixed effects (between‐participant effects); baseline HBsAg and baseline body weight as covariates; and visit within participant fitted as a random effect (within‐participant effect). Parameters were estimated using restricted maximum likelihood estimators, and the Kenward‐Roger method was used to calculate the denominator degrees of freedom. Differences in adjusted means between active RO7062931 and placebo, of which the associated 90% CI did not contain 0, were considered to be statistically significant and evidence of target engagement.
To simulate longer‐term treatment, a mechanistic PK/PD model to describe RO7062931 viral dynamics was developed (Supporting Fig. S1). Integrating disease processes and a concentration‐driven drug effect, this model could accurately describe the observed individual and mean time course of HBsAg decline as well as the variability in the data. Details of the modeling and simulation work are described in the Supporting Information.
Results
A total of 60 healthy volunteers and 59 patients were enrolled and randomized in Parts 1 and 2, respectively (Fig. 2). All 60 volunteers received RO7062931 (n = 48) or placebo (n = 12) across the Part 1 dose cohorts, and all 59 patients with CHB received at least one dose of RO7062931 (n = 44) or placebo (n = 15) across the Part 2 dose cohorts. For nonsafety reasons, 2 healthy volunteers did not complete follow‐up in Part 1 of the study, and 4 patients did not complete the planned dosing regimens in Part 2. All 60 volunteers and 59 patients were included in the safety and PK analysis populations. In Part 2 of the study, the 4 patients who did not complete the planned dosing regimen were excluded from the PD analysis.
FIG. 2.

Volunteer and patient flow through the study for (A) Part 1 single ascending dose in healthy volunteers and (B) Part 2 multiple dosing in patients with CHB.
In Part 1, all but one of the volunteers were male (98%), mean age was 29 years, mean body weight was 74 kg, and mean BMI was 24 kg/m2. The majority of participants were White (62%), followed by Asian (25%). Dose groups were well balanced for demographic and baseline characteristics (Supporting Table S1).
Demographic and baseline disease characteristics of patients with CHB in Part 2 are summarized in Table 1. With the exception of sex, the treatment groups were well balanced for demographic and baseline characteristics. Overall, 88% of patients were male, mean age was 45 years, mean body weight was 71 kg, and mean BMI was 24 kg/m2. Most patients were Asian (90%). At baseline, 58% of patients were HBeAg‐negative and 86% of patients had an ALT level ≤ULN. Although all patients had ALT ≤1.5 × ULN at screening, 1 patient in each of the 3.0 mg/kg QW and placebo cohorts had elevated ALT levels between 1.5 and 3 × ULN at baseline (Day 1 pretreatment). Baseline HBsAg values were well matched across cohorts with mean values between 3.4 and 3.7 log10 IU/mL. All but 3 patients exhibited HBV DNA values at or below the lower limit of detection in the assay (20 IU/mL). The number of patients who were HBeAg‐positive and patients who were HBeAg‐negative in some cohorts varied but were well balanced in the 3.0 mg/kg QW and placebo groups.
TABLE 1.
Demographic and Baseline Characteristics of Patients With CHB in Part 2 of the Study
| Characteristics | RO7062931 QM Dosing (Part 2a) | RO7062931 Q2W or QW Dosing (Part 2b) | Placebo (n = 15) | ||||
|---|---|---|---|---|---|---|---|
| 0.5 mg/kg QM (n = 6) | 1.5 mg/kg QM (n = 7) | 3.0 mg/kg QM (n = 6) | 3.0 mg/kg QW (n = 14) | 3.0 mg/kg Q2W (n = 7) | 4.0 mg/kg QW (n = 4) | ||
| Age, mean (SD) years | 42 (5) | 43 (1) | 48 (7) | 45 (9) | 46 (12) | 49 (7) | 45 (9) |
| Weight, mean (SD) kg | 76 (15) | 71 (15) | 70 (14) | 71 (12) | 72 (10) | 73 (19) | 70 (10) |
| BMI, mean (SD) kg/m2 | 26 (15) | 24 (3) | 23 (3) | 25 (3) | 24 (4) | 25 (4) | 24 (3) |
| Male, n (%) | 6 (100) | 6 (86) | 4 (67) | 12 (86) | 7 (100) | 3 (75) | 14 (93) |
| Ethnicity, n (%) | |||||||
| Asian | 5 (83) | 6 (86) | 4 (67) | 14 (100) | 6 (86) | 3 (75) | 15 (100) |
| White | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Other* | 1 (17) | 1 (14) | 1 (17) | 0 | 1 (14) | 1 (25) | 0 |
| HBeAg status, n (%) | |||||||
| Negative | 4 (67) | 2 (29) | 2 (33) | 8 (57) | 6 (86) | 4 (100) | 8 (53) |
| Positive | 2 (33) | 5 (71) | 4 (67) | 6 (43) | 1 (14) | 0 | 7 (47) |
| HBV DNA, mean (SD) IU/mL † | 19 (0.0) | 19 (0.0) | 19 (0.0) | 19 (0.0) | 23 (9.5) | 19 (0.0) | 20 (4.7) |
| HBsAg, mean (SD) log10 IU/mL | 3.4 (0.36) | 3.4 (0.29) | 3.4 (0.13) | 3.7 (0.36) | 3.7 (0.38) | 3.7 (0.27) | 3.6 (0.42) |
| ALT ≤ ULN, n (%) ‡ | 4 (67) | 6 (86) | 6 (100) | 11 (79) | 7 (100) | 3 (75) | 14 (93) |
Native Hawaiian or other Pacific Islander.
HBV DNA values reported as <20 IU/mL have been imputed as 19 IU/mL to allow inclusion in summary statistics.
ALT ULN was 41 U/L for male participants and 33 U/L for female participants.
Safety and Tolerability
Overall, AEs were reported in 33 of 48 (68.7%) healthy volunteers in the RO706239 dose groups and 7 of 12 (58.3%) placebo recipients in Part 1 of the study (Supporting Table S2) and in 29 of 44 (65.9%) patients in the RO7062931 treatment groups and 8 of 15 (53.3%) patients receiving placebo in Part 2 (Table 2). Treatment‐related AEs were mainly injection site reactions (ISRs), which were observed in approximately 20.0% of healthy volunteers and patients with CHB. The most common AEs in healthy volunteers were ISRs, followed by upper respiratory tract infection and headache. Influenza‐like illness was most common in patients with CHB, followed by ISR and headache. All cases of upper respiratory tract infection, headache, and influenza‐like illness were considered unrelated to study treatment per the reporting investigators. Although the incidence of influenza was numerically higher in treated subgroups (n = 9/44, 20.5%) than in the placebo group (n = 2/15, 13.3%), this is likely attributed to the presence of nontreatment‐related factors (i.e., AE onset occurred during flu season at one or more sites) as well as imprecise estimates due to small subgroup sample size.
TABLE 2.
AEs Occurring in ≥5% of Patients With CHB Overall in Part 2 of the Study
| Patients With ≥ 1 AE, n (%) | RO7062931 QM Dosing (Part 2a) | RO7062931 Q2W or QW Dosing (Part 2b) | Placebo (n = 15) | ||||
|---|---|---|---|---|---|---|---|
| 0.5 mg/kg QM (n = 6) | 1.5 mg/kg QM (n = 7) | 3.0 mg/kg QM (n = 6) | 3.0 mg/kg QW (n = 14) | 3.0 mg/kg Q2W (n = 7) | 4.0 mg/kg QW (n = 4) | ||
| Total | 2 (33.3) | 6 (85.7) | 4 (66.7) | 10 (71.4) | 4 (57.1) | 3 (75.0) | 8 (53.3) |
| Mild | 2 (33.3) | 6 (85.7) | 3 (50.0) | 10 (71.4) | 4 (57.1) | 3 (75.0) | 8 (53.3) |
| Moderate | 0 | 1 (14.3) | 1 (16.7) | 1 (7.1) | 1 (14.2) | 1 (25.0) | 1 (6.7) |
| Treatment‐related | 0 | 3 (42.8) | 1 (16.7) | 2 (14.3) | 2 (28.6) | 1 (25.0) | 3 (20.0) |
| Mild | 0 | 3 (42.8) | 1 (16.7) | 2 (14.3) | 2 (28.6) | 0 | 2 (13.3) |
| Moderate | 0 | 0 | 0 | 1 (7.1) | 0 | 1 (25.0) | 1 (6.7) |
| Influenza‐like illness | 1 (16.7) | 0 | 1 (16.7) | 3 (21.4) | 2 (28.6) | 2 (50.0) | 2 (13.3) |
| ISR | 0 | 1* (14.3) | 1* (16.7) | 2 † (14.3) | 2* (28.6) | 1 ‡ (25.0) | 0 |
| Headache | 0 | 0 | 0 | 1 (7.1) | 1 (14.2) | 0 | 3 (20.0) |
| URTI | 0 | 0 | 0 | 1 (7.1) | 0 | 0 | 2 (13.3) |
| Dizziness | 0 | 1* (14.3) | 0 | 0 | 0 | 0 | 2 (13.3) |
| Otopharyngeal pain | 0 | 1 (14.3) | 0 | 1 (7.1) | 0 | 0 | 1 (6.7) |
Mild treatment‐related.
Mild‐moderate treatment‐related.
Moderate treatment‐related.
Abbreviation: URTI, upper respiratory tract infection.
In both Parts 1 and 2 of the study, all AEs were of mild or moderate intensity, and there were no dose‐dependent or regimen‐dependent increases in the frequency of AEs. ISRs were generally mild, with no dose‐dependent or regimen‐dependent increase in the frequency or severity, and resolved during the study without sequelae. There were no clinically significant changes in laboratory safety parameters (including hepatic, hematologic, and renal parameters [Fig. 3] or complement activation), ECG parameters, or vital signs. There were no serious AEs, deaths or AEs that resulted in withdrawal from the study, or dose interruption.
FIG. 3.
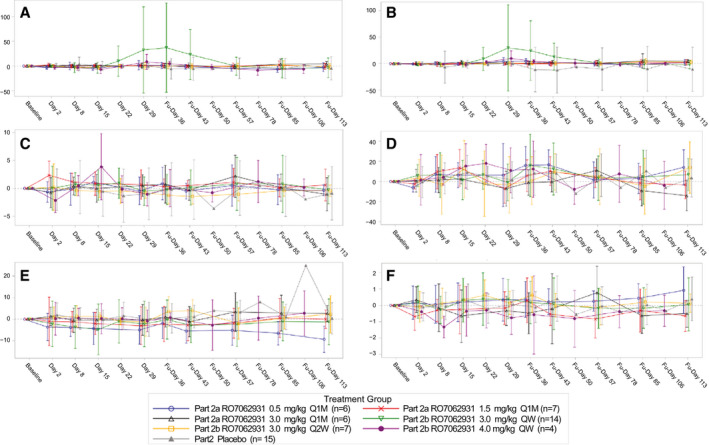
Mean change from baseline in select (hepatic, hematologic, renal) safety laboratory parameters in patients with CHB receiving placebo or RO7062931: (A) ALT (U/L), (B) aspartate aminotransferase (U/L), (C) activated partial thromboplastin time (sec), (D) platelets (109/L), (E) creatinine (μmol/L), and (F) blood urea nitrogen (mmol/L). Abbreviations: Fu, follow‐up; Q1M, every 1 month.
Two patients in the RO7062931 3.0 mg/kg QW dosing cohort had transient treatment‐emergent ALT elevations >3 × ULN (Supporting Fig. S2). In both cases, the ALT elevations peaked at the end of the treatment period (3.5 × ULN at Day 29 and 9.2 × ULN at Day 36) with concurrent declines in HBsAg levels and were not associated with impaired liver excretory/synthetic function or changes in total bilirubin, international normalized ratio, or albumin. In one of these patients, the increased ALT of 9.2 × ULN was reported as an AE.
PK
Single ascending doses of RO7062931 0.1‐4.0 mg/kg were absorbed rapidly with a median t max of 1.75‐3.00 hours in healthy volunteers (Supporting Table S3), and most of the drug was cleared from plasma within 24‐36 hours (Fig. 4A). Biphasic elimination was clearly observed for the 2.0, 3.0, and 4.0 mg/kg doses with a small amount of drug exhibiting an elimination half‐life of 85.70‐115.24 hours (Fig. 4B). This second elimination phase may represent drug redistributed from tissue and was also observed at lower doses, but characterization was limited by drug concentration falling below the limit of detection. RO7062931 exposure increased approximately linearly with increasing dose until the 3.0 mg/kg dose. A trend toward a supradose‐proportional increase in exposure was observed between the 3.0 and 4.0 mg/kg doses (Fig. 4C). In healthy volunteers, the mean fraction of RO7062931 dose excreted in urine (Fe) over 24 hours was approximately 0.9%‐3.2% (Supporting Table S3). To compare with patients with CHB, for whom Fe was only determined in the first 8 hours, Fe was computed for 0‐8 hours in healthy volunteers. In both healthy volunteers and patients with CHB, Fe increased sharply with RO7062931 dose escalation to 3.0‐4.0 mg/kg (Fig. 4D).
FIG. 4.
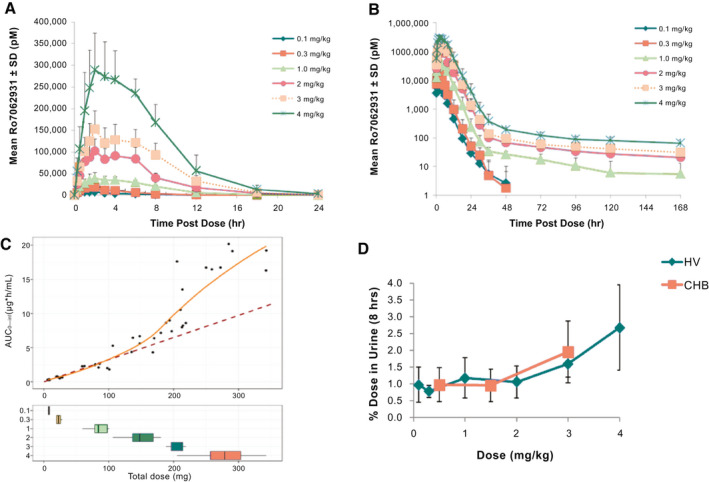
Plasma and urine RO7072931 PK. Plasma concentration‐time profiles for RO7062931 over (A) 24 hours and (B) 168 hours after single doses (0.1‐4.0 mg/kg) in healthy volunteers; (C) mean plasma AUC0‐inf versus total milligram dose in healthy volunteers; the dashed line is a linear regression line of the observed AUC at the doses of 0.1, 0.3, and 1 mg/kg in HVs. The orange line is a loess regression line of the observed AUC in HV. (D) Mean % dose eliminated in urine in the first 8 hours postdose in healthy volunteers and patients with CHB. Error bars indicate standard deviation. Abbreviations: AUC0‐inf, area under concentration‐time curve extrapolated to infinity; HV, healthy volunteers.
PK parameters for multiple dosing with RO7062931 in patients with CHB are shown in Supporting Table S4 and were found to be similar to those in healthy volunteers. No evidence for plasma accumulation with multiple doses was detected. As seen in healthy volunteers, there was a trend for supradose‐proportional increases in RO7062931 plasma exposure and a marked increase in urine Fe (Fig. 4D) with the 3.0 mg/kg dose.
PD
RO7062931 was associated with dose‐dependent and time‐dependent reduction in HBsAg over the 0.5‐3.0 mg/kg (all dosing regimens) dosing range (Fig. 5A,B). The lack of dose dependence in the 4.0 mg/kg cohort is likely due to the smaller sample size. Monthly 1.5 mg/kg and all 3.0 mg/kg regimens showed statistically significant reductions in adjusted mean HBsAg compared with placebo at time points from Day 8 to follow‐up Day 57, inclusive (Supporting Table S5) with associated 90% confidence intervals that did not contain zero. These dose‐dependent and time‐dependent reductions were considered to be evidence of target engagement.
FIG. 5.
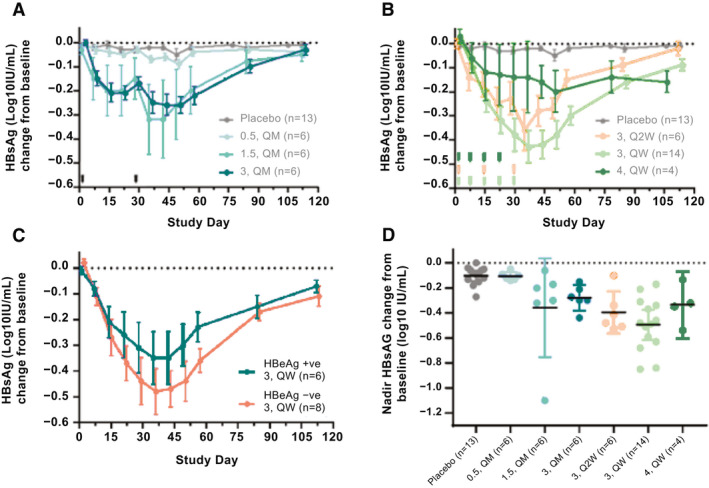
Mean log10 (IU/mL) change in HBsAg from baseline in patients with CHB receiving placebo or (A) RO7062931 0.5‐3.0 mg/kg QM or (B) RO7062931 3.0 mg/kg QW or Q2W or 4.0 mg/kg QW. (C) RO7062931 3.0 mg/kg QW according to HBeAg status. Error bars indicate standard error; (D) maximum log10 change from baseline in HBsAg in patients with CHB receiving RO7062931 (0.5‐3.0 mg/kg QM; 3.0 mg/kg QW or Q2W, or 4.0 mg/kg QW) or placebo. Error bars indicate 95% confidence intervals.
HBsAg levels declined, reaching its minimum (nadir) approximately 2 weeks after starting treatment, and returned to baseline by 12 weeks posttreatment. The 3.0 mg/kg QW regimen resulted in greater HBsAg reductions (mean decline at nadir ‐0.50 log10 IU/mL) than the 3.0 mg/kg Q2W and QM regimens (‐0.39 and ‐0.28 log10 IU/mL, respectively) (Table 3). Mean HBsAg decline at nadir with the 4.0 mg/kg QW regimen was ‐0.34 log10 IU/mL (n = 4). With the exception of one outlier patient receiving RO7062931 1.5 mg/kg QM who had a ≥1 log10 decrease in HBsAg, all patients treated with RO7062931 had an HBsAg decline at nadir ≤1 log10 IU/mL (Fig. 5D). In the 3.0 mg/kg QW dose group (n = 14), 8 patients (57%) had a reduction in HBsAg ≤0.5 log10 IU/mL. The PK/PD model estimated that a mean reduction of 0.7 log10 IU/mL would be achieved after 12 months of treatment with RO7062931 3.0 mg/kg QW, with only 10% of patients reaching a reduction of 1 log10 IU/mL (Supporting Fig. S1C).
TABLE 3.
Nadir HBsAg (log10 IU/mL) Decline Following RO7062931 3.0 mg/kg (QM, Q2W, and QW) or Placebo in Patients With CHB and According to HBeAg Status
| Maximum Decrease of HBsAg (log10 IU/mL) | RO7062931 3.0 mg/kg | Placebo | ||
|---|---|---|---|---|
| QM | Q2W | QW | ||
| All patients | ||||
| n | 6 | 6 | 14 | 13 |
| Mean (SD) | −0.28 (0.099) | −0.39 (0.159) | −0.50 (0.209) | −0.10 (0.071) |
| Median | −0.29 | −0.45 | −0.48 | −0.09 |
| Range | −0.44 to −0.15 | −0.53 to −0.11 | −0.85 to −0.17 | −0.27 to −0.01 |
| HBeAg‐positive | ||||
| n | 4 | 1 | 6 | 6 |
| Mean (SD) | −0.27 (0.043) | −0.48 | −0.43 (0.171) | −0.11 (0.086) |
| Median | −0.29 | −0.48 | −0.43 | −0.07 |
| Range | −0.30 to −0.21 | −0.48 to −0.48 | −0.63 to −0.17 | −0.27 to −0.04 |
| HBeAg‐negative | ||||
| n | 2 | 5 | 8 | 7 |
| Mean (SD) | −0.30 (0.205) | −0.38 (0.170) | −0.54 (0.233) | −0.10 (0.064) |
| Median | −0.30 | −0.41 | −0.49 | −0.09 |
| Range | −0.44 to −0.15 | −0.53 to −0.11 | −0.85 to −0.21 | −0.18 to −0.01 |
Reductions in HBsAg were similar regardless of baseline HBeAg status. Mean HBsAg declines at nadir for patients who were HBeAg‐negative and patients who were HBeAg‐positive receiving RO7062931 3.0 mg/kg QW were ‐0.54 and ‐0.43 log10 IU/mL, respectively (Fig. 5C and Table 3).
Finally, treatment with RO7062931 was associated with reductions in HBV RNA levels that were less marked and more variable than reductions in HBsAg (Supporting Fig. S3). This less profound effect observed on HBV RNA may be due to the large proportion of patients (54.5%, i.e., 30 of 55 patients who were PD‐evaluable) with a baseline HBV RNA level below the limit of quantification of the assay (10 copies/mL), which is more often observed in patients who were HBeAg‐negative than patients who were HBeAg‐positive. Moreover, RO7062931 did not result in any consistent changes in HBcAg (Supporting Fig. S3) or HBeAg levels (data not shown).
Discussion
In this study, single and multiple doses of the ASO RO7062931 up to 4.0 mg/kg were found to be safe and well tolerated in healthy volunteers and patients with CHB. Treatment‐related AEs observed were primarily ISRs that resolved without sequelae during the course of the study. Two observed cases of transient ALT elevations are suggestive of immune‐mediated hepatocyte damage, as they were concurrent to HBsAg decline.( 27 , 28 ) Apart from ISRs, no other oligonucleotide class effects, such as thrombocytopenia or reduced renal function, were noted with RO7062931. Considering that the vast majority of enrolled study participants were male (93%), it is not possible to adequately generalize the study results to female participants at this time.
This study provides proof of RO7062931 target engagement in patients with CHB as demonstrated by the dose‐dependent and time‐dependent reductions in HBsAg that were clearly differentiated from placebo and independent of baseline HBeAg status. However, the overall effect of RO7062931 on HBsAg was modest, even at a QW dose regimen, and does not compare favorably with other molecules, particularly the GalNAc‐conjugated siRNAs. Less profound reduction in other PD markers (HBV RNA, HBcAg, HBeAg) may be due to differences in baseline levels between HBsAg and these other PD markers. For example, only 45.5% of patients had a quantifiable level of HBV RNA at baseline, which is in line with the low and often undetectable HBV RNA levels in patients who were NUC‐treated as observed in the literature.( 29 , 30 ) Another factor could be the limited performance characteristics of these exploratory assays (e.g., HBcAg has a lower limit of quantification of 3.0 log10 U/mL). Lastly, any differences in the prevalence of intracellular mRNA species and/or efficiency in ASO‐targeted degradation of these species could also be a contributing factor.
In this study, RO7062931 was evaluated for a treatment duration of only 4 weeks with a maximum of 5 doses. Although the short duration of treatment was a limitation of the study, PK/PD modeling analyses conducted had predicted that only 10% of patients would exceed a 1 log10 IU/mL reduction in HBsAg levels with a 48‐week RO7062931 QW dosing regimen (data not shown). Given that RO7062931 is unlikely to be a first‐in‐class or best‐in‐class HBV treatment, the clinical development of RO7062931 was discontinued to focus on development of the in‐licensed GalNAc‐conjugated siRNA RG6346, previously known as DCR‐HBVS.( 19 )
Clinical data characterizing the various RNA targeting approaches have begun to emerge and predominantly consist of double‐stranded siRNAs participating in RISC‐mediated RNA cleavage and single‐stranded ASOs using RNase‐H–mediated RNA breakdown.( 31 ) Two examples of GalNAc‐conjugated siRNAs include JNJ‐3989 (ARO‐HBV) and RG6346 (DCR‐HBVS). Compared with the mean reduction of HBsAg at nadir of 0.5 log10 IU/mL obtained with RO7062931 at 3.0 mg/kg QW, JNJ‐3989 achieved mean HBsAg reductions of 1.52 and 1.62 log10 IU/mL in patients who were NUC‐suppressed and HBeAg‐positive (n = 11) and HBeAg‐negative (n = 37), respectively, 2 months after 3 monthly doses of 100 or 200 mg.( 32 ) Furthermore, sustained reductions in HBsAg (≥1 log10 IU/mL) were observed through to 48 weeks after the last dose of JNJ‐3989,( 33 ) as opposed to the HBsAg rebound to baseline levels observed by 12 weeks posttreatment in the current study. Similarly, RG6346 showed a mean maximal HBsAg reduction of 1.88 log10 IU/mL in patients who were NUC‐suppressed with CHB after 4 monthly doses of 3.0 mg/kg, which was sustained up to Day 336 without rebound.( 19 )
Compared with the GalNAc‐conjugated siRNA molecules, GSK3389404, another GalNAc‐conjugated ASO with a similar underlying mechanism of action to RO7062931, exhibited a modest mean HBsAg decline of 0.75 log10 IU/mL observed during 12 weeks of treatment at 120 mg QW in patients who were NUC‐treated with CHB.( 34 )
At first glance, it appears that as a class, GalNAc‐conjugated ASO drugs may tend to be less effective than GalNAc‐conjugated siRNA drugs in causing robust and sustained reduction of HBsAg in patients with CHB. However, a mechanistic reason for the different efficacy of single‐stranded and double‐stranded oligonucleotides in the HBV field has not yet been elucidated. Possible explanations may include differences in intrahepatic distribution or endosomal trafficking that may have an impact on intrahepatic drug exposures or may include differences in the efficiency of RISC versus RNase‐H–mediated mRNA degradation. Taken together, these factors may result in differences in durability of action. Supporting the concept that different exposures of the active agent at the target site can impact overall activity, a partial rebound of HBsAg can be observed between QM doses of RO7062931 but not between QW dosing. This suggests a suboptimal exposure in hepatocytes for this single‐stranded oligonucleotide with the QM dosing regimen that was not seen with the QW regimen or with equivalent QM dosing for siRNAs such as JNJ‐3989 (ARO‐HBV) described above. Interestingly, in the mouse efficacy model, a GalNAc‐conjugated HBV ASO showed a decrease of HBsAg in serum of up to 3 log after repetitive dosing,( 14 ) which did not translate to the responses seen in this study. This lack of translatability may be attributed to one or more factors, including species‐specific drug potency, HBsAg half‐life, or hepatocyte uptake.
As a counterpoint to this view, ISIS 505358/GSK3228836, which is the naked ASO metabolite of GSK3389404, was able to achieve mean ± SD HBsAg declines from baseline of 1.56 ± 1.38 and 2.51 ± 1.57 log10 IU/mL for patients who were treatment naïve and patients who were virologically suppressed, respectively, with 2 patients exhibiting prolonged HBsAg loss.( 23 ) However, the likely explanation for the magnitude of effect for this ASO is the relatively high total dose (300 mg × 6 = 1,500 mg) administered over the 4‐week treatment period, which was almost 10‐fold higher than the total dose administered with JNJ‐3989 (200 mg) or RG6346 (3 mg/kg; ~180‐210 mg) over a 4‐week period.
RO7062931 exhibited similar declines in HBsAg regardless of the HBeAg status of patients receiving RO7062931 at 3.0 mg/kg QW, suggesting that RO7062931 is able to target HBsAg transcripts derived from covalently closed circular DNA and host integrated HBV DNA, thereby avoiding issues with the HBV integration site that was elegantly elucidated with the ARC520 experience.( 17 , 35 ) Unlike RO7062931, which targets a region of HBV RNA upstream from the HBV integration site, the target site of the siRNA ARC520 is located in a region that is commonly deleted on HBV integration.( 17 , 35 )
RO7062931 PK were predictable from preclinical models and similar to observations with other GalNAc‐conjugated ASOs.( 36 ) In monkeys, as the dose increases, plasma clearance can decrease due to a decrease in specific liver uptake by the ASGPR shuttle as it begins to saturate. This effect has been noted but not clearly described in humans because the evaluated range of doses has not been wide enough.( 37 )
Data from this study provide a relatively simple approach to understanding liver uptake of GalNAc‐conjugated ASOs or even siRNAs that use the ASGPR liver shuttle. As the ASGPR liver uptake system begins to saturate with increasing dose, it is hypothesized that relatively more drug will be detected in plasma and urine, resulting in disproportional plasma and urine kinetics. Indeed, PK data from this study suggest that ASGPR saturation had commenced between the 3.0 and 4.0 mg/kg doses. Furthermore, modeling of preclinical (cynomolgus monkeys) and clinical (this study) data indicate that 60%‐70% ASGPR saturation occurs at doses of 3.0‐4.0 mg/kg (manuscript in preparation). It is plausible that with GalNAc‐conjugated agents, a range of doses may be chosen to evaluate the ASGPR interaction, as has been done here. Subsequently, a similar modeling approach could then be used to identify optimum dosing regimens (i.e., determine upper dose limits that maximize PD effects within the liver while preventing ASGPR saturation and potential extrahepatic toxicities).
In summary, RO7062931 was safe and well tolerated at doses up to 4.0 mg/kg QW. Analyses of PK data have identified a modeling approach that may identify optimum dosing regimens that balance tissue exposures with PD activity. RO7062931 exhibited evidence of target engagement and proof of mechanism in patients with CHB although its impact on HBsAg reduction was modest. Given the evolving clinical development landscape of HBV gene expression inhibitors, further preclinical work is warranted to better understand potential differences between the siRNA and ASO platforms.
Author Contributions
All authors contributed to the study in terms of concept, design and implementation, and all authors contributed to the writing and/or editing of this article.
Supporting information
Supplementary Material
Acknowledgment
Editorial and medical writing support was provided by Weber Shandwick Hong Kong, funded by F. Hoffmann‐La Roche. Siijie Lu from Roche Innovation Centre, Shanghai, China provided support in the final stages of PK analysis and review during this study. Avinash Patil from Parexel International India, an external business partner working on behalf of Roche, provided support with undertaking and corroborating the statistical analyses.
Supported by F. Hoffmann‐La Roche.
Clinical trial number: NCT03038113.
Potential conflict of interest: Dr. Gane consults for, advises, and is on the speakers’ bureau for AbbVie. He consults for and advises Roche. He advises Assembly. Dr. Chan advises and is on the speakers’ bureau for Gilead and Roche. He advises AbbVie, Aligos, Arbutus, Hepion, GlaxoSmithKline, Janssen, Merck, Vir, Vaccitech, and VenatoRx. He is on the speakers’ bureau for Mylan. Dr. Surujbally is employed by and owns stock in Roche. Dr. Pavlovic is employed by Roche. Dr. Das is employed by Roche. Dr. Triyatni is employed by and owns stock in Roche. Dr. Kazma is employed by and owns stock in Roche. He owns stock in Novartis and Alcon. Dr. Krippendorff is employed by and owns stock in Roche. He owns stock in Arrowhead and Alnylam. Dr. Mueller is employed by Roche. Dr. Zhang is employed by Roche. Dr. Y. Lim consults for, advises, is on the speakers’ bureau for, and received grants from Gilead. Dr. Tanwandee received grants from Roche and Merck. Dr. Wat is employed by and owns stock in Roche.
References
Author names in bold designate shared co‐first authorship.
- 1. World Health Organization . Global hepatitis report 2017. https://www/who.int/publications/i/item/global‐hepatitis‐report‐2017. Accessed September 16, 2020. [Google Scholar]
- 2. Yuen MF, Chen DS, Dusheiko GM, Janssen HL, Lau DT, Locarnini SA, et al. Hepatitis B virus infection. Nat Rev Dis Primer 2018;4:18035. [DOI] [PubMed] [Google Scholar]
- 3. Anderson RT, Choi HS, Lenz O, Peters MG, Janssen HL, Mishra P, et al. Association between seroclearance of hepatitis B surface antigen and long‐term clinical outcomes of patients with chronic hepatitis B virus infection: systematic review and meta‐analysis. Clin Gastroenterol Hepatol 2021;19:463‐472. [DOI] [PubMed] [Google Scholar]
- 4. European Association for the Study of the Liver . EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67:370‐398. [DOI] [PubMed] [Google Scholar]
- 5. Zoulim F, Durantel D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb Perspect Med 2015;5:a021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee HM, Banini BA. Updates on chronic HBV: current challenges and future goals. Curr Treat Options Gastroenterol 2019;17:271‐291. [DOI] [PubMed] [Google Scholar]
- 7. Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: from discovery to regulatory approval. J Hepatol 2017;67:847‐861. [DOI] [PubMed] [Google Scholar]
- 8. Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T‐cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis 2015;6:e1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seto WK, Lo YR, Pawlotsky JM, Yuen MF. Chronic hepatitis B virus infection. Lancet 2018;392:2313‐2324. [DOI] [PubMed] [Google Scholar]
- 10. Fisicaro P, Barili V, Rossi M, Montali I, Vecchi A, Acerbi G, et al. Pathogenetic mechanisms of T cell dysfunction in chronic HBV infection and related therapeutic approaches. Front Immunol 2020;11:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lebossé F, Testoni B, Fresquet J, Facchetti F, Galmozzi E, Fournier M, et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J Hepatol 2017;66:897‐909. [DOI] [PubMed] [Google Scholar]
- 12. Kondo Y, Ninomiya M, Kakazu E, Kimura O, Shimosegawa T. Hepatitis B surface antigen could contribute to the immunopathogenesis of hepatitis B virus infection. ISRN Gastroenterol 2013;2013:935295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther 2020;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Javanbakht H, Mueller H, Walther J, Zhou X, Lopez A, Pattupara T, et al. Liver‐targeted anti‐HBV single‐stranded oligonucleotides with locked nucleic acid potently reduce HBV gene expression in vivo. Mol Ther Nucleic Acids 2018;11:441‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schluep T, Lickliter J, Hamilton J, Lewis DL, Lai CL, Lau JY, et al. Safety, tolerability, and pharmacokinetics of ARC‐520 injection, an RNA interference‐based therapeutic for the treatment of chronic hepatitis B virus infection, in healthy volunteers. Clin Pharmacol Drug Dev 2017;6:350‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thi EP, Dhillon AP, Ardzinski A, Bidirici‐Ertekin L, Cobarrubias KD, Cuconati A, et al. ARB‐1740, a RNA interference therapeutic for chronic hepatitis B infection. ACS Infect Dis 2019;5:725‐737. [DOI] [PubMed] [Google Scholar]
- 17. Yuen MF, Schiefke I, Yoon JH, Ahn SH, Heo J, Kim JH, et al. RNA interference therapy with ARC‐520 results in prolonged hepatitis B surface antigen response in patients with chronic hepatitis B infection. Hepatology 2020;72:19‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gish RG, Yuen MF, Chan HL, Given BD, Lai CL, Locarnini SA, et al. Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antiviral Res 2015;121:97‐108. [DOI] [PubMed] [Google Scholar]
- 19. Dicerna . Corporate overview: maximizing the impact of RNAi on medicine. https://investors.dicerna.com/static‐files/19a25d4b‐3f5b‐4973‐a99c‐3daf3b1c8437. Accessed November 2, 2020. [Google Scholar]
- 20. Gane E, Lim YS, Tangkijvanich P, O’Beirne J, Lim TH, Bakardjiev A, et al. Preliminary safety and antiviral activity of VIR‐2218, an X‐targeting HBV RNAi therapeutic, in chronic hepatitis B patients. J Hepatol 2020;73:S50‐S51. [Google Scholar]
- 21. Arbutus Biopharma . AB‐729 (GalNAc‐RNAi). http://arbutusbio.com/portfolio/ab‐729‐galnac‐rnai.php. Accessed November 2, 2020. [Google Scholar]
- 22. Billioud G, Kruse RL, Carrillo M, Whitten‐Bauer C, Gao D, Kim A, et al. In vivo reduction of hepatitis B virus antigenemia and viremia by antisense oligonucleotides. J Hepatol 2016;64:781‐789. [DOI] [PubMed] [Google Scholar]
- 23. Yuen MF, Heo J, Jang JW, Yoon JH, Kweon YO, Park SJ, et al. Hepatitis B virus surface antigen inhibition with GSK3228836 (ISIS 505358) in chronic hepatitis B patients on stable nucleos(t)ide analogue regimen and in nucleos(t)ide analogue‐naive patients: a phase 2a, randomized, double‐blind, placebo‐controlled study. J Hepatol 2020;73:S49‐S50. [Google Scholar]
- 24. Tanowitz M, Hettrick L, Revenko A, Kinberger GA, Prakash TP, Seth PP. Asialoglycoprotein receptor 1 mediates productive uptake of N‐acetylgalactosamine‐conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res 2017;45:12388‐12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Y. Preclinical and clinical advances of GalNAc‐decorated nucleic acid therapeutics. Mol Ther Nucleic Acids 2017;6:116‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bon C, Hofer T, Bousquet‐Mélou A, Davies MR, Krippendorff BF. Capacity limits of asialoglycoprotein receptor‐mediated liver targeting. mAbs 2017;9:1360‐1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang ML, Liaw YF. Hepatitis B flares in chronic hepatitis B: pathogenesis, natural course, and management. J Hepatol 2014;61:1407‐1417. [DOI] [PubMed] [Google Scholar]
- 28. Heo NY. Is alanine aminotransferase flare‐up in nucleos(t)ide analogue treatment of chronic hepatitis B a promising, rather than a devastating, sign? Clin Mol Hepatol 2017;23:125‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liao H, Liu Y, Li X, Wang J, Chen X, Zou J, et al. Monitoring of serum HBV RNA, HBcrAg, HBsAg and anti‐HBc levels in patients during long‐term nucleoside/nucleotide analogue therapy. Antivir Ther 2019;24:105‐115. [DOI] [PubMed] [Google Scholar]
- 30. Rong F, Jie P, Qing X, Deming T, Min X, Junqi N, et al.; Chronic Hepatitis B Study Consortium . Combining hepatitis B virus RNA and hepatitis B core‐related antigen: guidance for safely stopping nucleos(t)ide analogues in hepatitis B e antigen‐positive patients with chronic hepatitis B. J Infect Dis 2020;222:611‐618. [DOI] [PubMed] [Google Scholar]
- 31. Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA‐targeted therapeutics. Cell Metab 2018;27:714‐739. [DOI] [PubMed] [Google Scholar]
- 32. Gane EJ, Locarnini S, Lim TH, Strasser S, Sievert W, Cheng W, et al. Dose response with the RNA interference therapy JNJ‐3989 combined with nucleos(t)ide analogue treatment in expanded cohorts of patients with chronic hepatitis B. Hepatology 2019;70:434A (Abstract 696).30724374 [Google Scholar]
- 33. Gane E, Locarnini S, Lim TH, Strasser S, Sievert W, Cheng W, et al. Short‐term treatment with RNA interference therapy, JNJ‐3989, results in sustained hepatitis B surface antigen supression in patients with chronic hepatitis B receiving nucleos(t)ide analogue treatment. J Hepatol 2020;73:S20 (Abstract GS10). [Google Scholar]
- 34. Yuen MF, Heo J, Kumada H, Suzuki F, Suzuki Y, Xie Q, et al. Results after 12 weeks treatment of multiple doses of GSK3389404 in chronic hepatitis B (CHB) subjects on stable nucleos(t)ide therapy in a phase 2a double‐blind, placebo‐controlled study. Hepatology 2019;70:433A (Abstract 695). [Google Scholar]
- 35. Wooddell CI, Yuen MF, Chan HY, Gish RG, Locarnini SA, Chavez D, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med 2017;9:eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol 2009;5:381‐391. [DOI] [PubMed] [Google Scholar]
- 37. Wang Y, Yu RZ, Henry S, Geary RS. Pharmacokinetics and clinical pharmacology considerations of GalNAc3‐conjugated antisense oligonucleotides. Expert Opin Drug Metab Toxicol 2019;15:475‐485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material