

Abstract
Introduction
White matter hyperintensities (WMH) are often described in Alzheimer's disease (AD), but their topography and specific relationships with cognition remain unclear.
Methods
Regional WMH were estimated in 54 cognitively impaired amyloid beta–positive AD (Aβpos‐AD), compared to 40 cognitively unimpaired amyloid beta–negative older controls (Aβneg‐controls) matched for vascular risk factors. The cross‐sectional association between regional WMH volume and cognition was assessed within each group, controlling for cerebral amyloid burden, global cortical atrophy, and hippocampal atrophy.
Results
WMH volume was larger in Aβpos‐AD compared to Aβneg‐controls in all regions, with the greatest changes in the splenium of the corpus callosum (S‐CC). In Aβpos‐AD patients, larger total and regional WMH volume, especially in the S‐CC, was strongly associated with decreased cognition.
Discussion
WMH specifically contribute to lower cognition in AD, independently from amyloid deposition and atrophy. This study emphasizes the clinical relevance of WMH in AD, especially posterior WMH, and most notably S‐CC WMH.
Keywords: Alzheimer's disease, amyloid positron emission tomography, cognition, corpus callosum, executive functions, fluid‐attenuated inversion recovery, memory, magnetic resonance imaging, splenium, white matter hyperintensities
1. BACKGROUND
White matter hyperintensities (WMH), defined with magnetic resonance imaging (MRI) as an hyperintense signal on T2‐weighted and fluid‐attenuated inversion recovery (T2‐weighted FLAIR) images, are frequent in cognitively unimpaired older adults. They are associated with vascular risk factors (VRF), for example, hypertension or diabetes, 1 and with worse cognitive performance, particularly executive functions and processing speed. 2 , 3 , 4
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed. Greater white matter hyperintensities (WMH) volume is associated with risk and progression of clinical Alzheimer's disease (AD), as well as cognitive decline in AD. The regional specificities in AD‐related WMH increases and the links with cognition remain unclear.
Interpretation: Our findings in a group of amyloid beta (Aβ)‐positive AD patients compared to Aβ‐negative controls matched for the main vascular risk factors highlight the clinical relevance of posterior WMH, and particularly WMH in the splenium of the corpus callosum (S‐CC). S‐CC WMH are the most strongly associated with cognitive performance in AD, independently from Aβ burden and gray matter loss.
Future directions: The current findings emphasize the clinical relevance of posterior WMH in AD and motivate future research on the nature and etiology of WMH in AD. They argue for S‐CC WMH being a specific pathological manifestation of AD.
In patients with Alzheimer's disease (AD), WMH volume appears to be larger than in cognitively unimpaired older adults, 5 particularly in periventricular 6 , 7 , 8 and posterior regions. These include parieto‐occipital regions 9 , 10 and the splenium of the corpus callosum (S‐CC). 7
AD and cerebrovascular diseases share common VRF and often coexist. Cerebrovascular lesions such as WMH, which are partially due to small vessel disease, 11 are associated with an increased risk of clinical AD. They are likely to add to AD lesions (i.e., amyloid deposition and neurofibrillary tangles), therefore lowering the threshold for cognitive impairment, 12 , 13 and/or directly contribute to AD pathophysiology. 14
Studies assessing the relationship between WMH and cognition in AD are scarce, and findings are inconsistent, showing associations in some 5 , 6 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 but not all 8 , 22 , 23 , 24 , 25 , 26 studies. A recent meta‐analysis showed small to medium‐sized associations between WMH and cognition in patients with AD across all major cognitive domains, with larger effects on attention and executive functions. 27 Little is known, however, about the regional specificity of these associations as most studies in AD analyzed either global WMH volume 5 , 15 , 17 , 18 , 19 , 20 , 22 , 23 , 24 or periventricular versus deep WMH, 6 , 16 , 25 but rarely lobar WMH. 28 , 29 , 30 , 31 This is particularly relevant as the spatial distribution of WMH seems to be associated with partially distinct etiologies. In fact, while anterior WMH have been associated with physiological aging and VRF, posterior WMH would be more specifically associated with AD. 10 , 32 , 33 Moreover, periventricular WMH seem to be more strongly associated with worse cognition than deep WMH. 4 , 27 , 34 , 35 Finally, studies also showed stronger links with cognition for WMH in strategic white matter tracts, such as the forceps minor or anterior thalamic radiation, than for total WMH burden. 36 , 37 , 38 , 39
As another caveat, previous WMH studies mainly used clinical criteria for AD diagnosis, without inclusion of biomarkers, which can lead to clinical misdiagnosis, particularly between AD and vascular dementia. Studies of WMH in the context of AD using a biomarker‐based definition of AD 40 are still needed.
The question remains as to if and how the amount and topography of WMH in AD differ from age‐related WMH, and whether WMH have region‐specific effects on cognition in amyloid beta (Aβ)‐positive AD patients. The present study aims to address these questions by (1) highlighting the regional distribution of WMH in Aβ‐positive patients with AD clinical syndrome (Aβpos‐AD), by comparing them to VRF‐matched Aβ‐negative controls (Aβneg‐controls), considering both lobar and callosal subregions but also periventricular versus deep WMH, and (2) assessing the specific relationships between regional WMH and cognitive performance, and studying whether these associations remain after controlling for cortical amyloid burden, hippocampal, and total gray matter volumes.
2. METHODS
2.1. Study participants
Ninety‐four participants from the IMAP+ (Multimodal Neuroimaging of Early Alzheimer's Disease) cohort were included in the present study. 41 , 42 The participants consisted of 54 cognitively impaired patients with AD clinical syndrome (Mini‐Mental State Examination [MMSE] = 23.9±4.75), 40 including 21 with dementia and 33 with amnestic mild cognitive impairment (MCI; details in Table S1 in supporting information). Clinical criteria were used to define MCI and demented patients. 43 , 44 , 45 , 46 All patients were Aβ‐positive (Aβpos‐AD, see section 2.4.), that is, in the AD continuum, as defined by the National Institute on Aging‐Alzheimer's Association (NIA‐AA) 2018 criteria 40 and corresponding to typical AD, as defined by the International Working Group 2 (IWG‐2) criteria. 47 They were all recruited from memory clinics. For the sake of comparison, we also included data from 40 Aβ‐negative cognitively unimpaired controls (Aβneg‐controls) from IMAP+. These controls were selected automatically from a group of 62 cognitively unimpaired Aβ‐negative participants to match Aβpos‐AD patients in terms of age, sex, years of education, systolic blood pressure (BP), diastolic BP, and glycated hemoglobin (HbA1C; using the R package MatchIt). All controls were recruited from the community, with no history or clinical evidence of major neurological or psychiatric disorders, and had normal performance on neuropsychological tests. The IMAP+ study was approved by local ethics committee (CPP Nord‐Ouest III) and registered at http://clinicaltrials.gov (NCT01638949). All participants gave their written informed consent to the study prior to examinations.
2.2. Cognitive and clinical assessment
The participants underwent a comprehensive neuropsychological assessment, described elsewhere. 41 , 48 Global cognition was evaluated with the Mattis Dementia Rating Scale (Mattis‐DRS). 49 One outlier was identified for the Mattis‐DRS, and analyses were performed with and without this outlier (without substantial differences). To obtain robust proxies of cognitive performance and reduce the issue of multiple comparisons, composite scores were computed. Scores at each individual test were z‐scored, using the Aβneg‐controls as a reference, and averaged by cognitive domains: episodic memory (sum of the Free and Cued Selective Reminding Test 50 free recalls; free recalls from the “encoding, storage, retrieval” paradigm 51 ), working memory (forward and backward digit span 52 ) and executive functions (letter verbal fluency test 53 ; Trail Making Test 53 flexibility score, calculated as the time difference between parts B and A, divided by time at part A; Stroop Test 53 interference score, representing the time difference between interference and naming tasks, divided by time at naming). The Trail Making Test and Stroop Test z‐scores were inverted so that higher values always reflect better performance. Sociodemographic variables consisted of age, sex, and years of education. As they are known to be related to WMH, 1 the following VRF were measured and controlled for in our analyses: systolic and diastolic BP and HbA1C, which corresponds to the 3‐month averaged blood sugar level and is related to diabetes mellitus risk. BP measures were averaged over six assessments (three consecutive measures at two different timepoints).
2.3. Structural MRI
MRI scans were acquired on a Philips Achieva 3T scanner. All exams were performed at the Cyceron Center (Caen, France). A high‐resolution fast‐field echo sequence T1‐weighted scan (3D‐T1‐FFE sagittal; repetition time TR = 20 ms; echo time TE = 4.6 ms; flip angle = 10°; 180 slices with no gap; slice thickness = 1 mm; field of view = 256 × 256 mm2; in plane resolution = 1 × 1 mm2) and a high‐resolution T2‐weighted FLAIR scan (3D‐IR sagittal; TR/TE/TI [inversion time] = 8000/348/2400 ms; flip angle = 90°; 90 slices with no gap; slice thickness = 2 mm; field of view = 250 × 250 mm2; in‐plane resolution = 0.78 × 0.78 mm2; Figure 1) were acquired. Preprocessing was performed using Statistical Parametric Mapping 12 (SPM12, MatLab v. 2018b; MathWorks), unless stated otherwise.
FIGURE 1.
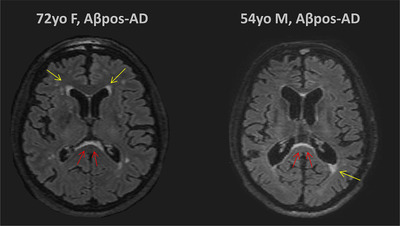
Examples of periventricular white matter hyperintensities (WMH) and subependymal WMH in the region of the splenium of the corpus callosum (S‐CC). The image on the left corresponds to the axial T2‐weighted fluid‐attenuated inversion recovery magnetic resonance imaging (FLAIR MRI) of a 72‐year‐old amyloid beta–positive Alzheimer's disease (Aβpos‐AD) woman with mild cognitive impairment. The image on the right corresponds to the axial T2‐weighted FLAIR MRI of a 54‐year‐old Aβpos‐AD man with dementia. Yellow arrows show mild periventricular WMH and red arrows show subependymal hyperintensities in the region of the S‐CC
For the automatic segmentation of WMH, raw FLAIR images were coregistered onto their corresponding T1‐weighted scan. Then, WMH were segmented in the MRI‐native space, using the lesion prediction algorithm (LPA; Schmidt, Chapter 6.1 54 ) as implemented in the Lesion Segmentation Toolbox version 2.0.15 (www.statistical‐modelling.de/lst.html) for SPM. The algorithm calculates a lesion probability score for each voxel. A minimum extend threshold was set to 0.01 cm³ and lesion probability maps were binarized by applying a threshold of 0.5. Resulting images were corrected for false positives in corticospinal tracts if necessary, using a corticospinal tract mask specific to each participant (corticospinal hyperintensities being common artifacts in WMH segmentations). 55
Anatomic atlases were inversely normalized to native space of each individual binary lesion map to extract the summed WMH volumes in the frontal, temporal, parietal, and occipital lobes (Hammers atlas 56 ), in the corpus callosum and its subregions including the genu, the body and the splenium (JHU white matter adult atlases 57 ), and in the periventricular versus deep regions (using the standard limit of 10 mm from the lateral ventricles to define periventricular vs. deep WMH 34 ). WMH volume in cm³ was defined as the voxel size multiplied by the total number of voxels labeled as lesions. 48
Total gray matter volume (tGMV) and total intracranial volume (TIV), calculated by summing the tGMV, white matter volume, and cerebrospinal fluid volume, were estimated. Hippocampal volumes were obtained using the ASHS‐T1 pipeline (https://sites.google.com/view/ashs‐dox/home) and corresponded to the sum of the anterior and the posterior bilateral hippocampus volumes. All segmentations were visually inspected. Failed segmentations were manually edited when feasible or were discarded. WMH, tGMV, and hippocampal volumes were normalized by the TIV, to account for inter‐individual variability in head size. TIV‐corrected WMH were then log‐transformed (after adding a constant of 0.01 to all values to avoid log transformation of zero values). All references to WMH in what follows correspond to TIV‐corrected and log‐transformed volumes, unless otherwise specified.
2.4. Amyloid PET
Florbetapir‐PET (F18‐AV‐45) scans were acquired on a Discovery RX VCT 64 PET‐CT device (General Electric Healthcare) at the Cyceron Center (Caen, France). Participants underwent a 20‐min positron emission tomography (PET) scan, beginning 50 min after the intravenous injection of Florbetapir adapted to the weight of the participants (≈ 4 MBq/kg). Scans were acquired with a resolution of 3.76 × 3.76 × 4.9 mm3 (field of view = 157 mm). Forty‐seven planes were obtained with a voxel size of 1.95 × 1.95 × 3.27 mm3. A transmission scan was performed for attenuation correction before acquisition. After correction for randoms, decay, deadtime, attenuation, and scatter, raw data were reconstructed using OSEM 2D and a Gaussian post‐filter (fwhm 2.14 mm) using Discovery VCTHD‐64 GE Medical Systems version 1.23.V40. PET images were corrected for partial volume effects (PVE) with PMOD Technologies Ltd, coregistered onto the corresponding T1‐weighted image and spatially normalized using the deformation parameters derived from the MRI procedure, previously computed with SPM. The resulting images were scaled, using cerebellar gray matter as a reference to obtain standardized uptake value ratio (SUVR) images. Finally, Florbetapir SUVR images were masked to exclude non–gray matter voxels. Global cortical SUVR was extracted from normalized, scaled, and masked Florbetapir‐PET images using a neocortical mask (including the entire gray matter, except the cerebellum, occipital and sensory motor cortices, hippocampi, amygdala, and basal nuclei). 58 Aβ‐positivity was defined using a threshold of 0.99, corresponding to the 99.9th percentile of SUVR distribution among cognitively unimpaired young adults (< 40 years old, n = 45). 59
Of note, PVE‐corrected and PVE‐uncorrected SUVR were highly correlated and findings remained largely unchanged when using PVE‐uncorrected SUVR. As a result, only PVE‐corrected data are presented here.
2.5. Statistical analyses
Statistical analyses were performed using the R software version 3.5.2 (R Core Team, www.R‐project.org). We compared sociodemographic, clinical, biological, and neuroradiological variables between the two groups using Student's t tests or nonparametric Wilcoxon Mann‐Whitney tests for continuous variables and Pearson χ² for categorical data.
WMH were then compared between Aβpos‐AD patients and Aβneg‐controls using Student's t tests, and Cohen's d effect size were calculated. The same analyses were replicated using multivariate linear models to adjust for age, sex, systolic BP, diastolic BP, and HbA1C. Analyses of sensitivity and specificity were performed for each regional raw WMH volume to classify participants as Aβneg‐controls or Aβpos‐AD patients (package pROC).
Then, to assess the relationships between regional WMH and cognition in Aβpos‐AD patients, multivariate linear models were performed with cognitive performance (Mattis‐DRS and the three composite scores) as dependent variables, and each regional WMH measure separately as independent variables, controlling for age, sex, and years of education. Analyses were repeated adjusting for cortical Aβ load (Florbetapir SUVR), hippocampal volume, tGMV, and VRF. Finally, analyses were conducted adding the interaction between WMH and disease stage (MCI vs. dementia), to assess if the association between WMH and cognition differed according to disease stage. The same analyses were repeated in the Aβneg‐controls and are presented in the supporting information.
Multiple comparisons errors were controlled for using the false discovery rate (FDR) correction (accounting for multiple testing across the four cognitive domains). In case of missing data, the participant was excluded from the corresponding analysis. The significance level was set to P < .05.
3. RESULTS
3.1. Clinical, demographic, and neuroimaging characteristics of the population
Participants’ characteristics are summarized in Table 1 and detailed in Table S1. By design, Aβpos‐AD patients did not differ from Aβneg‐controls in terms of age, sex, years of education, systolic BP, diastolic BP, or HbA1C. As expected, Aβpos‐AD patients had lower performance than Aβneg‐controls on all cognitive tests (Table 1). Aβpos‐AD patients were mildly impaired with an averaged MMSE of ≈ 24/30 (normal range = 26–30). Most (42/54; 78%) showed mild deficits (MMSE > 20/30), while 11/54 (20%) were moderately impaired (MMSE between 10 and 20), and no patients were severely impaired (MMSE score < 10/30; one missing data). Compared to Aβneg‐controls, Aβpos‐AD patients showed larger total WMH, along with lower hippocampal volume and tGMV. Within Aβpos‐AD, patients with MCI were older than patients with dementia but they did not differ in terms of WMH (Table S1).
TABLE 1.
Summary of demographics, cognitive, and imaging data in the Aβpos‐AD and Aβneg‐controls
| Aβpos‐AD | Aβneg‐controls | P | |
|---|---|---|---|
| Demographics and vascular risk factors | |||
| N | 54 | 40 | |
| Age | 71.02 ± 9.06 | 69.72 ± 6.87 | .43 |
| Sex (% male) | 59.3 | 50.0 | .37 |
| Years of education | 11.57 ± 3.73 | 12.47 ± 3.97 | .30 |
| Systolic BP (mmHg) | 143.19 ± 22.08 | 144.26 ± 21.58a | .82 |
| Diastolic BP (mmHg) | 79.19 ± 12.67 | 82.41 ± 12.46a | .23 |
| HbA1c (%) | 5.81 ± 0.68b | 5.72 ± 0.34d | .48 |
| Cognition | |||
| MMSE (raw score; max = 30) | 23.94 ± 4.75a | 28.77 ± 1.23 | < .001 |
| MMSE (range) | [12–30] | [26–30] | |
| Mattis‐DRS (raw score; max = 144) | 126.10 ± 11.51c | 141.47 ± 2.86 | < .001 |
| Mattis‐DRS (range) | [84–142] | [132–144] | |
| Episodic Memory (z‐score) | –3.23 ± 1.10b | 0 ± 0.82 | < .001 |
| Working Memory (z‐score) | –0.67 ± 0.88a | 0 ± 0.83 | < .001 |
| Executive Functions (z‐score) | –1.17 ± 1.07 | 0 ± 0.70 | < .001 |
| Neuroimaging | |||
| WMH (raw volume, cm³) | 13.08 ± 13.54 | 5.91 ± 7.56 | < .001 |
| WMH (% of TIV) | 0.95 ± 0.98 | 0.43 ± 0.56 | .001 |
| WMH (log) | –0.59 ± 1.17 | –1.37 ± 1.05 | <.001 |
| Amyloid burden (SUVR) | 1.45 ± 0.29 | 0.87 ± 0.05 | < .001 |
| tGMV (dm³) | 0.63 ± 0.07 | 0.65 ± 0.06 | .007* |
| Hippocampal volume (cm³) | 2.90 ± 0.46a | 3.31 ± 0.36 | < .001* |
| TIV (dm³) | 1.37 ± 0.14 | 1.37 ± 0.14 | .89 |
Notes: Values are reported as mean ± standard deviation or percentage.
Statistical analyses were performed using Student's t tests (for age, systolic BP, diastolic BP, Hba1C, MDRS, Episodic memory, Working memory, Executive function, TIV, WMH [log], tGMV, and hippocampal volume), Wilcoxon rank tests (for level of education, MMSE, raw WMH, WMH [TIV‐corrected], and Aβ), and Pearson χ² (for sex). *Raw tGMV and hippocampal volumes were reported in the table, but statistical analyses were performed with TIV‐corrected volumes. a One missing data; b 2 missing data; c 3 missing data ; d 4 missing data. Significant p values are in bold.
Abbreviations: Aβ, amyloid beta; BP, blood pressure; MMSE, Mini‐Mental State Examination; Mattis‐DRS, Mattis Dementia Rating Scale; PET, positron emission tomography SUVR, standardized uptake value ratio (Florbetapir‐PET); tGMV, total gray matter volume; TIV, total intracranial volume; WMH, white matter hyperintensities.
3.2. Comparisons of regional WMH between Aβpos‐AD and Aβneg‐controls
Aβpos‐AD patients showed larger WMH than Aβneg‐controls in all brain regions (Table 2, Figure S1 in supporting information). The greatest effect size was observed for the S‐CC. Overall, a posteroanterior gradient was found with higher effects in posterior than anterior regions. This was true both when considering the CC (S‐CC > CC body > CC genu) or the lobar distribution (largest effect in occipital vs. lowest effect in frontal lobes; Table 2). Larger WMH concerned both periventricular and deep WMH with a slightly higher effect in the latter. The S‐CC WMH showed the best accuracy to differentiate Aβpos‐AD patients from Aβneg‐controls, with a sensitivity of 80% and a specificity of 65% for a threshold of 0.12 cm³ for WMH volume (area under the curve [AUC] = 74.5%, details in Figure S2).
TABLE 2.
Total and regional WMH in Aβpos‐AD compared to Aβneg‐controls
| Aβpos‐AD | Aβneg‐controls | P * | d (effect size) | |
|---|---|---|---|---|
| Total | 13.08 (13.54) | 5.91 (7.56) | < .001 | 0.71 |
| Periventricular | 8.28 (6.71) | 4.25 (4.75) | .002 | 0.67 |
| Deep | 4.63 (7.81) | 1.54 (3.04) | < .001 | 0.78 |
| Frontal | 4.29 (5.73) | 2.18 (3.12) | .022 | 0.48 |
| Parietal | 4.12 (5.04) | 1.56 (2.35) | .001 | 0.68 |
| Temporal | 1.60 (1.57) | 0.69 (0.77) | < .001 | 0.78 |
| Occipital | 1.44 (1.37) | 0.66 (0.93) | < .001 | 0.89 |
| Corpus callosum | 1.66 (1.42) | 0.77 (1.02) | < .001 | 0.75 |
| Genu | 0.39 (0.42) | 0.23 (0.30) | .010 | 0.54 |
| Body | 0.69 (0.73) | 0.32 (0.44) | .003 | 0.64 |
| Splenium | 0.58 (0.56) | 0.22 (0.33) | < .001 | 0.91 |
Notes: Values are reported as mean (standard deviation) of raw WMH volumes (cm3). *P‐values of the Student's t tests and Cohen's d (effect size) are indicated for TIV‐corrected and log‐transformed WMH. All P values remain < .05 after correction for age, sex, systolic BP, diastolic BP, and HbA1C. Significant p values are in bold.
Abbreviations: Aβ, amyloid beta; BP, blood pressure; TIV, total intracranial volume; WMH, white matter hyperintensities.
3.3. Associations between regional WMH and cognition
The associations between cognition and regional WMH in Aβpos‐AD patients, adjusted for age, sex, and years of education, are summarized in Table 3. The strongest association was found between larger S‐CC WMH and lower global cognitive performance (Table 3, Figure 2). The relationships between S‐CC WMH and global cognition remained significant after controlling for cortical Aβ load (P FDR‐corrected < .001), hippocampal volume (P FDR‐corrected < .001), tGMV (P FDR‐corrected = .004) separately or all together (P FDR‐corrected = .008; Table 3).
TABLE 3.
Association between total and regional WMH and cognition in Aβpos‐AD patients
| Global cognition | Episodic memory | |||||
|---|---|---|---|---|---|---|
| β | 95%CI | P | β | 95%CI | P | |
| Total | –0.50 | [–0.80, –0.19] | .01 *†‡§ | –0.09 | [–0.45, 0.27] | .62 |
| Periventricular | –0.53 | [–0.83, –0.23] | .00* †‡§ | –0.12 | [–0.48, 0.24] | .51 |
| Deep | –0.39 | [–0.70, –0.08] | .06¶*† | –0.01 | [–0.35, 0.32] | .93 |
| Frontal | –0.38 | [–0.71, –0.05] | .08¶ | 0.00 | [–0.37, 0.37] | .99 |
| Parietal | –0.48 | [–0.79, –0.18] | .01* †‡§ | –0.02 | [–0.37, 0.33] | .91 |
| Temporal | –0.46 | [–0.76, –0.15] | .02* †‡§ | –0.12 | [–0.47, 0.23] | .50 |
| Occipital | –0.47 | [–0.74, –0.20] | .00* †‡ | –0.15 | [–0.46, 0.16] | .34 |
| Corpus callosum | –0.47 | [–0.75, –0.19] | .01* †‡§ | –0.16 | [–0.50, 0.17] | .33 |
| Genu | –0.24 | [–0.54, 0.06] | .14 | –0.04 | [–0.37, 0.29] | .81 |
| Body | –0.41 | [–0.70, –0.11] | .03* † | –0.15 | [–0.49, 0.20] | .40 |
| Splenium | –0.54 | [–0.79, –0.29] | .00* †‡§ | –0.19 | [–0.50, 0.12] | .22 |
| Working memory | Executive functions | |||||
|---|---|---|---|---|---|---|
| β | 95%CI | P | β | 95%CI | P | |
| Total | –0.30 | [–0.61, 0.01] | .08 | –0.35 | [–0.67, –0.03] | .07¶ |
| Periventricular | –0.26 | [–0.57, 0.05] | .14 | –0.35 | [–0.67, –0.02] | .07¶ |
| Deep | –0.31 | [–0.61, –0.02] | .07¶ | –0.30 | [–0.61, 0.02] | .08 |
| Frontal | –0.33 | [–0.65, –0.02] | .08¶ | –0.27 | [–0.61, 0.08] | .17 |
| Parietal | –0.27 | [–0.58, 0.04] | .11 | –0.35 | [–0.67, –0.03] | .07¶* |
| Temporal | –0.16 | [–0.47, 0.15] | .40 | –0.24 | [–0.57, 0.08] | .27 |
| Occipital | –0.24 | [–0.52, 0.04] | .11 | –0.29 | [–0.57, 0.00] | .10 |
| Corpus callosum | –0.29 | [–0.57, 0.00] | .07 | –0.40 | [–0.69, –0.10] | .02* † |
| Genu | –0.26 | [–0.55, 0.03] | .14 | –0.32 | [–0.63, ‐0.02] | .14¶ |
| Body | –0.26 | [–0.56, 0.03] | .11 | –0.37 | [–0.67, –0.06] | .04* † |
| Splenium | –0.24 | [–0.52, 0.03] | .11 | –0.39 | [–0.66, –0.11] | .01* † |
Notes: Standardized betas (β) with 95% confidence interval (95%CI) and corrected P‐values are reported from regression models where cognitive scores are regressed onto WMH (TIV‐corrected and log‐transformed), adjusted for age, sex, and level of education. Significant p values are in bold.
* Corrected P‐values < .05 when models were also adjusted for cortical Aβ.
† Corrected P‐values < .05 when models were also adjusted for hippocampal volume.
‡ Corrected P‐values < .05 when models were also adjusted for tGMV.
§ Corrected P‐values < .05 when models were also adjusted for cortical Aβ, hippocampal volume, and tGMV.
¶ P‐values < .05 before correction for multiple tests.
Abbreviations: Aβ, amyloid beta; tGMV, total gray matter volume; TIV, total intracranial volume; WMH, white matter hyperintensities.
FIGURE 2.
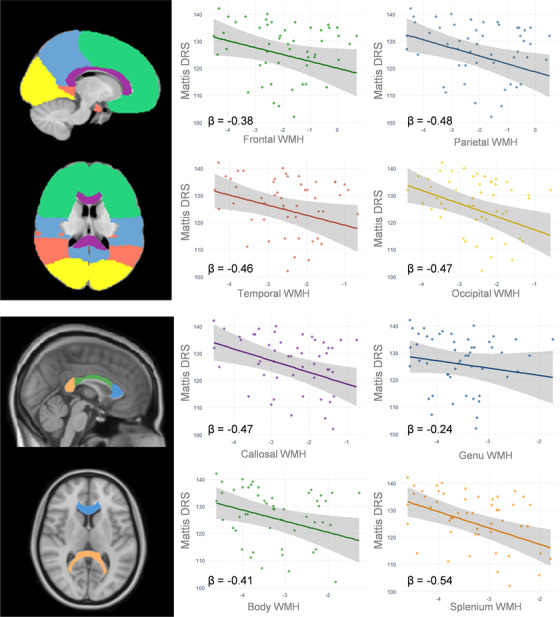
Associations between global cognition and regional white matter hyperintensities (WMH) in amyloid beta–positive Alzheimer's disease (Aβpos‐AD). Association between regional WMH and global cognition (Mattis Dementia Rating Scale [DRS]). Plots represent global cognition regressed onto regional WMH (total intracranial volume–corrected and log‐transformed), controlling for age, sex, level of education (with 95% confidence intervals). Standardized betas (β) were added. For the sake of illustration one outlier (Mattis‐DRS = 84/144) was removed from the figure (results were unchanged with or without the outlier). Anatomic atlases were represented on the left panel for illustration purposes (Hammers atlas for lobes at the top and JHU atlas for CC at the bottom)
The associations between global cognition and total WMH or other regional WMH were also significant, except for frontal, deep, and CC genu regions. Moreover, larger WMH in the CC (total, CC body, and S‐CC) were associated with lower executive functions. These relationships remained after controlling for cortical Aβ load and hippocampal volume, but not after controlling for tGMV (Table 3). After adjusting for VRF (i.e., systolic BP, diastolic BP, and HbA1C), results remained mainly unchanged (Table S2 in supporting information).
Finally, there was an interaction between WMH and disease stage (MCI vs. dementia) in the total, periventricular, temporal, occipital, CC, and S‐CC regions, with stronger associations between WMH and cognition in demented patients (Figure S3 in supporting information).
For the sake of comparison, analyses were repeated in the group of Aβneg‐controls. No significant relationships were found after correction for multiple comparisons (Table S3 in supporting information).
4. DISCUSSION
Our aim was to assess the regional distribution of WMH and the association with cognitive deficits in Aβpos‐AD patients (i.e., Aβ‐positive patients with AD clinical syndrome) compared to Aβneg‐controls, matched for VRF. We found that (1) WMH were larger in Aβpos‐AD patients, particularly in the S‐CC and posterior regions, while differences were more subtle in the frontal lobe and CC genu and (2) larger WMH were associated with worse cognitive performances in Aβpos‐AD patients, especially for the S‐CC, periventricular and posterior regions, and independently from cortical Aβ load, gray matter loss, or VRF.
The overall larger volume of WMH in AD has been reported in several previous studies. 5 , 6 , 7 , 8 , 9 The regional specificity of WMH has rarely been investigated. A few studies showed a posterior predominance of WMH in AD. 7 , 9 , 10 Moreover, WMH in posterior regions were found to predict incident AD 32 , 60 , 61 and to increase several years before the expected symptom onset in autosomal‐dominant AD. 62 In line with these previous reports, we showed that parietal, temporal, and occipital WMH were more strongly associated with AD than frontal WMH. We observed the same posteroanterior gradient in the CC, with a predominance of WMH in the posterior section of the CC (i.e., S‐CC) in AD patients. This specific finding is consistent with a previous study that reported a larger volume of S‐CC WMH in AD patients compared to cognitively unimpaired participants, without significant differences in the CC genu. 7 WMH in the anterior subependymal region of the S‐CC (Figure 1) are sometimes mentioned in the literature 63 , 64 , 65 but rarely further assessed. Our study provides new evidence that posterior WMH, and more particularly S‐CC WMH, are a core feature of AD. They seem to occur relatively early in the disease progression, as most of our patients were only mildly impaired (MMSE of 24 on average) and there were no differences in WMH between MCI and dementia. Interestingly, studies using diffusion tensor imaging evidenced early microstructural alterations of the S‐CC in AD. 66 , 67 , 68 , 69 , 70 , 71 One previous study specifically showed a greater involvement of the S‐CC, compared to the rest of the CC, in AD and proposed that this subregion could be particularly relevant to discriminate AD from vascular dementia, 67 as the CC genu is more vulnerable to cerebrovascular injuries. 67 , 72 , 73 Finally, we observed group differences both in periventricular WMH, as previously described, 6 , 7 , 8 and deep WMH.
Regarding the relationships with cognition, stronger links were found in the periventricular and posterior regions. As for callosal subregions, S‐CC WMH showed the strongest association with cognitive deficits, independently from amyloid burden, total gray matter, and hippocampal atrophy. Our results align with a very recent study highlighting voxel‐wise associations between S‐CC WMH and cognition (particularly attention deficits), in AD. 74 In the present study, global cognition was associated with periventricular WMH, while it was associated with deep WMH only when adjusting for amyloid burden or hippocampal volume. This is in line with previous studies highlighting the relevance of deep versus periventricular WMH distinction, 4 , 27 , 34 which usually found a stronger link with cognitive deficits for periventricular WMH than for deep WMH. 4 , 27 This might reflect the fact that the S‐CC (in which WMH were strongly associated with cognition) is included in periventricular regions. We found WMH to be associated with global cognition and executive functions (to a lesser extent), but not with episodic or working memory. This is consistent with previous studies showing the strongest effect size for the association with executive functions. 27 As regard to episodic memory, a link was found in some previous studies, but the effect size was lower, 27 which might explain why it was not replicated here. Despite early development of posterior WMH in AD (see above), associations between WMH and cognition were stronger in demented patients compared to MCI patients, that is, the downstream effects of WMH on cognition seem to appear at the dementia stage. Future longitudinal studies with larger sample size will be needed to further address this question.
Several hypotheses can be proposed to account for the larger volume of WMH in AD (compared to controls), particularly in the S‐CC, and their associations with cognition. WMH, as other cerebrovascular lesions, could either add up to or interact with AD pathology, leading to AD clinical syndrome. 14 Alternatively, WMH in AD could reflect specific non‐vascular mechanisms, including neurodegeneration caused by AD pathology, as previously suggested by neuropathological 75 , 76 , 77 or neuroimaging studies, 71 particularly in posterior white matter. In line with this hypothesis, a recent study showed that anterior WMH were associated with VRF, while posterior WMH were associated with Aβ‐positivity. 10 Moreover, larger posterior WMH were found in middle‐aged autosomal dominant AD patients 62 and patients with Down syndrome, 78 independently from VRF, reinforcing the idea that posterior WMH lesions could have, at least partially, a nonvascular origin and be involved in AD pathophysiology. Our results align well with this hypothesis for two main reasons. First, the CC is known to be relatively resistant to hypoperfusion, 79 and the two groups of participants (Aβpos‐AD and Aβneg‐controls) were matched in terms of VRF, making vascular mechanisms less likely to be responsible for the observed differences. Second, the S‐CC is an important hub in AD, located at the vicinity of brain areas that are the most sensitive to AD pathology (e.g., posterior cingulate cortex and posterior hippocampus). 42 It is therefore possible that S‐CC WMH are secondary to AD‐pathology, reflecting for instance Wallerian degeneration due to neurofibrillary tangles in the hippocampal region. Studies assessing WMH per white matter tracts would further test this hypothesis, that is, show whether WMH in AD occur more specifically in tracts emanating from medio‐temporal regions. Although not directly assessing this question, Rizvi et al. showed an association between memory function and larger WMH within association and projection tracts rather than commissural tracts, but not with the cingulum‐hippocampus tract. 39
Studies assessing the association between S‐CC WMH and AD pathologies (amyloid or tau) provided mixed results. A link was found between S‐CC WMH and cerebrospinal fluid (CSF) Aβ1‐42 80 and amyloid‐PET, 81 but not CSF phosphorylated tau, 80 while a recent study found a significant association between global WMH and plasma total tau. 82 Cerebral amyloid angiopathy may at least partially explain the association between WMH and AD as well as the posteroanterior gradient of WMH in AD. 83 , 84 , 85 However, previous studies showed that the larger volume of WMH in AD was not entirely mediated by cerebral amyloid angiopathy, 86 and the relationship between CSF Aβ levels and posterior WMH was independent from the presence of lobar cerebral microbleeds, a key marker of cerebral amyloid angiopathy. 80 Finally, there are emerging evidences from animal 82 , 87 and human studies 82 , 88 that associate cerebrovascular disease with tau pathology, and it is possible that WMH, as a cerebrovascular marker, promote tau accumulation. Further studies, assessing the regional relationships between cerebral tau burden, amyloid load, and WMH—especially in the S‐CC and/or in relevant white matter tracts—are needed to address these issues. Altogether, our results highlight regional variability in the etiology of WMH, with differential effects on cognition. Indeed, whatever the specific nature (vascular and/or neurodegenerative) of posterior WMH and notably of S‐CC WMH, the predominance of these lesions in Aβpos‐AD patients and their strong association with cognitive deficits shown in the present study highlight their clinical relevance in AD.
The present study has some strengths and limitations. First, the cross‐sectional and observational design of the study prevents us from assessing the dynamic evolution of WMH in AD and from making causal interpretations. Second, VRF other than age, sex, BP, and HbA1c levels, such as cholesterol level and smoking habits, were not considered; also, other biomarkers of small vessel disease and cerebral amyloid angiopathy, including cerebral microbleeds or lacunes, were not assessed. Our cohort was relatively young (≈70 years), all White, highly educated, with a higher range of VRF and WMH than in previous studies 1 , 33 , 89 and all patients were recruited from memory clinics (and not from the community). For these reasons, findings cannot be generalized to the general population of older adults. Third, our sample size was relatively limited, which could have prevented us from showing subtler effects (e.g., associations between WMH and episodic memory). Finally, the lack of tau measurement prevented us from assessing whether our patients were tau‐positive or not 40 and did not allow us to address the influence of tau pathology on WMH and their links with cognition. On the other hand, strengths of this study include the systematic assessment of WMH topography in terms of lobar and depth distribution, and across the CC longitudinal axis. Moreover, patients were selected to be in the AD continuum as defined by the NIA‐AA 2018 Research Framework, 40 that is, with a significant amount of amyloid deposition in the brain as measured with Florbetapir‐PET, thus increasing the likelihood of AD etiology and decreasing the risk of clinical misdiagnosis, particularly between AD and vascular dementia. Controls were selected to be Aβ‐negative, which reduced the risk of including individuals in the preclinical phase of AD, 47 , even if we cannot exclude the presence of tau pathology in these participants. A major concern about the WMH evidenced in AD patients is that they may be driven by VRF. This issue has been addressed in our study by selecting VRF‐matched controls and by further adjusting our models for VRF. Finally, special attention has been given to the statistical framework, adjusting the models for possible confounding factors (i.e., cortical Aβ, hippocampal volume, and tGMV, in addition to age, sex and education) and correcting for multiple comparisons (accounting for multiple testing across the four cognitive domains, but not for the multiple brain regions analyzed).
In this study, we assessed the topography of WMH in AD and the links with cognition in a well‐characterized group of Aβ‐positive patients with AD clinical syndrome, compared to a group of Aβ‐negative controls, matched for the main VRF. We highlighted the clinical relevance of posterior WMH, and particularly of S‐CC WMH, as the region of most significant increase in AD, and of strongest associations with cognition, independently from cortical Aβ, gray matter loss, and VRF. Those results reinforce the importance of WMH in AD and pave the way for further investigation of S‐CC WMH in AD, and more specifically the nature of these lesions, to better understand their role in the pathophysiological cascade of the disease.
CONFLICTS OF INTEREST
No conflicts of interest were reported by any author.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors are grateful to G. Rauchs, E. Arenaza‐Urquijo, A. Bejanin, C. Tomadesso,, S. Egret, M. Fouquet, R. La Joie, M. Leblond, C. Malle, K. Mevel, J. Mutlu, V. Ourry, A. Pelerin, A. Perrotin, G. Poisnel, S. Segobin, N. Villain, A. Manrique, L. Paly, E. Touron, I. Moulinet, S. Rehel, V. Lefranc, A. Cognet, F. Viader, M. Gaubert, A. Abbas, L. Barré, D. Guilloteau, S. Belliard, A. Lutz, F. Eustache, B. Desgranges, and the Cyceron MRI‐PET staff members and to the participants of the study. This work was supported by the Institut National de la Santé et de la Recherche Medicale (INSERM), the Programme Hospitalier de Recherche Clinique (PHRCN 2011‐A01493‐38 and PHRCN 2012 12‐006‐0347), the Agence Nationale de la Recherche (ANR LONGVIE 2007), Fondation Plan Alzheimer (Alzheimer Plan 2008‐2012), Fondation LECMA‐Vaincre Alzheimer (grant number 13732), Fondation Alzheimer, Fondation d'entreprise MMA des Entrepreneurs du Futur, Association France Alzheimer et maladies apparentées, the Région Basse‐Normandie, the Agence régionale de santé Auvergne‐Rhône‐Alpes, and the European Union's Horizon 2020 Research and Innovation Program (grant number 667696). Funding sources were not involved in the study design, data acquisition and interpretation, analysis, manuscript writing, and in the decision to submit the article for publication.
Garnier‐Crussard A, Bougacha S, Wirth M, et al. White matter hyperintensity topography in Alzheimer's disease and links to cognition. Alzheimer's Dement. 2022;18:422–433. 10.1002/alz.12410
REFERENCES
- 1. Cox SR, Lyall DM, Ritchie SJ, et al. Associations between vascular risk factors and brain MRI indices in UK Biobank. Eur Heart J. 2019;40:2290‐2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11:157‐165. [DOI] [PubMed] [Google Scholar]
- 3. Kloppenborg RP, Nederkoorn PJ, Geerlings MI, van den Berg E. Presence and progression of white matter hyperintensities and cognition: a meta‐analysis. Neurology. 2014;82:2127‐2138. [DOI] [PubMed] [Google Scholar]
- 4. Bolandzadeh N, Davis JC, Tam R, Handy TC, Liu‐Ambrose T. The association between cognitive function and white matter lesion location in older adults: a systematic review. BMC Neurol. 2012;12:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y‐L, Chen W, Cai W‐J, et al. Associations of white matter hyperintensities with cognitive decline: a longitudinal study. J Alzheimers Dis. 2020;73:759‐768. [DOI] [PubMed] [Google Scholar]
- 6. Burns JM, Church JA, Johnson DK, et al. White matter lesions are prevalent but differentially related with cognition in aging and early Alzheimer disease. Arch Neurol. 2005;62:1870‐1876. [DOI] [PubMed] [Google Scholar]
- 7. Yoshita M, Fletcher E, Harvey D, et al. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology. 2006;67:2192‐2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Damulina A, Pirpamer L, Seiler S, et al. White matter hyperintensities in Alzheimer's disease: a lesion probability mapping study. J Alzheimers Dis. 2019;68:789‐796. [DOI] [PubMed] [Google Scholar]
- 9. Gootjes L, Teipel SJ, Zebuhr Y, et al. Regional distribution of white matter hyperintensities in vascular dementia, Alzheimer's disease and healthy aging. Dement Geriatr Cogn Disord. 2004;18:180‐188. [DOI] [PubMed] [Google Scholar]
- 10. Pålhaugen L, Sudre CH, Tecelao S, et al. Brain amyloid and vascular risk are related to distinct white matter hyperintensity patterns. J Cereb Blood Flow Metab. 2020:0271678×20957604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vemuri P, Lesnick TG, Przybelski SA, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain. 2015;138:761‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lorius N, Locascio JJ, Rentz DM, et al. Vascular disease and risk factors are associated with cognitive decline in the Alzheimer Disease Spectrum. Alzheimer Dis Assoc Disord. 2015;29:18‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bracco L, Piccini C, Moretti M, et al. Alzheimer's disease: role of size and location of white matter changes in determining cognitive deficits. Dement Geriatr Cogn Disord. 2005;20:358‐366. [DOI] [PubMed] [Google Scholar]
- 17. Brickman AM, Honig LS, Scarmeas N, et al. Measuring cerebral atrophy and white matter hyperintensity burden to predict the rate of cognitive decline in Alzheimer disease. Arch Neurol. 2008;65:1202‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heo J‐H, Lee S‐T, Chu Kon, Park H‐J, Shim J‐Y, Kim M. White matter hyperintensities and cognitive dysfunction in Alzheimer disease. J Geriatr Psychiatry Neurol. 2009;22:207‐212. [DOI] [PubMed] [Google Scholar]
- 19. Carmichael O, Schwarz C, Drucker D, et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol. 2010;67:1370‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lo RY, Jagust WJ. Vascular burden and Alzheimer disease pathologic progression. Neurology. 2012;79:1349‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Capizzano AA. White matter hyperintensities are significantly associated with cortical atrophy in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:822‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsiskaridze A, Shakarishvili R, Janelidze M, Vashadze T, Chikhladze M. Cognitive correlates of leukoaraiosis in the early stages of Alzheimer's disease. Funct Neurol. 1998;13:17‐25. [PubMed] [Google Scholar]
- 23. Hirono N, Kitagaki H, Kazui H, Hashimoto M, Mori E. Impact of white matter changes on clinical manifestation of Alzheimer's disease: a quantitative study. Stroke. 2000;31:2182‐2188. [DOI] [PubMed] [Google Scholar]
- 24. Claus JJ, Coenen M, Staekenborg SS, et al. Cerebral white matter lesions have low impact on cognitive function in a large elderly memory clinic population. J Alzheimers Dis. 2018;63:1129‐1139. [DOI] [PubMed] [Google Scholar]
- 25. Kono I, Mori S, Nakajima K, et al. Do white matter changes have clinical significance in Alzheimer's disease?. Gerontology. 2004;50:242‐246. [DOI] [PubMed] [Google Scholar]
- 26. Starkstein SE, Sabe L, Vazquez S, et al. Neuropsychological, psychiatric, and cerebral perfusion correlates of leukoaraiosis in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1997;63:66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van den Berg E, Geerlings MI, Biessels GJ, Nederkoorn PJ, Kloppenborg RP. White matter hyperintensities and cognition in mild cognitive impairment and Alzheimer's disease: a domain‐specific meta‐analysis. J Alzheimers Dis. 2018;63:515‐527. [DOI] [PubMed] [Google Scholar]
- 28. Kaskikallio A, Karrasch M, Rinne JO, Tuokkola T, Parkkola R, Grönholm‐Nyman P. Cognitive effects of white matter pathology in normal and pathological aging. J Alzheimers Dis. 2019;67:489‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaskikallio A, Karrasch M, Koikkalainen J, et al. White matter hyperintensities and cognitive impairment in healthy and pathological aging: a quantified brain MRI study. Dement Geriatr Cogn Disord. 2019;48:297‐307. [DOI] [PubMed] [Google Scholar]
- 30. Vipin A, Foo HJL, Lim JKW, et al. Regional white matter hyperintensity influences grey matter atrophy in mild cognitive impairment. J Alzheimers Dis. 2018;66:533‐549. [DOI] [PubMed] [Google Scholar]
- 31. van der Vlies AE, Staekenborg SS, Admiraal‐Behloul F, et al. Associations between magnetic resonance imaging measures and neuropsychological impairment in early and late onset Alzheimer's disease. J Alzheimers Dis. 2013;35:169‐178. [DOI] [PubMed] [Google Scholar]
- 32. Brickman AM. Contemplating Alzheimer's disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep. 2013;13:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Habes M, Sotiras A, Erus G, et al. White matter lesions: spatial heterogeneity, links to risk factors, cognition, genetics, and atrophy. Neurology. 2018;91:e964‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Griffanti L, Jenkinson M, Suri S, et al. Classification and characterization of periventricular and deep white matter hyperintensities on MRI: a study in older adults. Neuroimage. 2018;170:174‐181. [DOI] [PubMed] [Google Scholar]
- 35. Armstrong NJ, Mather KA, Sargurupremraj M, et al. Common genetic variation indicates separate causes for periventricular and deep white matter hyperintensities. Stroke. 2020;51:2111‐2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang J, Paradise M, Liu T, et al. The association of regional white matter lesions with cognition in a community‐based cohort of older individuals. Neuroimage Clin. 2018;19:14‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duering M, Gesierich B, Seiler S, et al. Strategic white matter tracts for processing speed deficits in age‐related small vessel disease. Neurology. 2014;82:1946‐1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Biesbroek JM, Weaver NA, Biessels GJ. Lesion location and cognitive impact of cerebral small vessel disease. Clin Sci. 2017;131:715‐728. [DOI] [PubMed] [Google Scholar]
- 39. Rizvi B, Lao PJ, Colón J, et al. Tract‐defined regional white matter hyperintensities and memory. Neuroimage Clin. 2020;25:102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Besson FL, La Joie R, Doeuvre L, et al. Cognitive and brain profiles associated with current neuroimaging biomarkers of preclinical Alzheimer's disease. J Neurosci. 2015;35:10402‐10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. La Joie R, Perrotin A, Barré L, et al. Region‐Specific hierarchy between atrophy, hypometabolism, and β‐amyloid (Aβ) load in Alzheimer's disease dementia. J Neurosci. 2012;32:16265‐16273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol. 2001;58(12):1985‐92. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 44. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 46. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol. 2014;13:614‐629. [DOI] [PubMed] [Google Scholar]
- 48. Garnier‐Crussard A, Bougacha S, Wirth M, et al. White matter hyperintensities across the adult lifespan: relation to age, Aβ load, and cognition. Alz Res Therapy. 2020;12:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mattis S. In: Bellack L and Karusu TB, Eds Geriatric Psychiatry, eds. Mental status examination for organic mental syndrome in the elderly patients. New York: Grune&Stratton; 1976. [Google Scholar]
- 50. Grober E, Buschke H, Crystal H, Bang S, Dresner R. Screening for dementia by memory testing. Neurology. 1988;38:900‐900. [DOI] [PubMed] [Google Scholar]
- 51. Eustache F, Laisney M, Lalevée C, et al. Une nouvelle épreuve de mémoire épisodique : l’épreuve ESR‐forme réduite (ESR‐r), adaptée du paradigme ESR (encodage, stockage, récupération). Rev Neuropsychol. 2015;7:217‐225. [Google Scholar]
- 52. Petermann F, Wechsler DJ. Wechsler Adult Intelligence Scale ‐ fourth edition. 2012. [Google Scholar]
- 53. Godefroy O. Fonctions exécutives et pathologies neurologiques et psychiatriques. Évaluation En Pratique Clinique Solal. Marseille. 2008. [Google Scholar]
- 54. Schmidt P. Bayesian inference for structured additive regression models for large‐scale problems with applications to medical imaging. Text.PhDThesis. Ludwig‐Maximilians‐Universität München; 2017. [Google Scholar]
- 55. Wardlaw JM, Valdés Hernández MC, Muñoz‐Maniega S. What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc. 2015;4:e001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hammers A, Allom R, Koepp MJ, et al. Three‐dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:224‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wakana S, Caprihan A, Panzenboeck MM, et al. Reproducibility of quantitative tractography methods applied to cerebral white matter. Neuroimage. 2007;36:630‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. André C, Tomadesso C, de Flores R, et al. Brain and cognitive correlates of sleep fragmentation in elderly subjects with and without cognitive deficits. Alzheimers Dement. 2019;11:142‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brickman AM, Provenzano FA, Muraskin J, et al. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol. 2012;69:1621‐1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brickman AM, Zahodne LB, Guzman VA, et al. Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer's disease incidence. Neurobiol Aging. 2015;36:27‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee S, Viqar F, Zimmerman ME, et al. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network: white matter hyperintensities in familial AD. Ann Neurol. 2016;79:929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo G, Wu R, Ter Brugge K, Mikulis DJ. Focal lesion in splenium of corpus callosum on FLAIR MRI: common findings in aged patients. Neuroradiol J. 2006;19:301‐305. [DOI] [PubMed] [Google Scholar]
- 64. Pekala JS, Mamourian AC, Wishart HA, Hickey WF, Raque JD. Focal lesion in the Splenium of the corpus callosum on FLAIR MR images: a common finding with aging and after brain radiation therapy. AJNR Am J Neuroradiol. 2003;24:855‐861. [PMC free article] [PubMed] [Google Scholar]
- 65. Blaauw J, Meiners LC. The splenium of the corpus callosum: embryology, anatomy, function and imaging with pathophysiological hypothesis. Neuroradiology. 2020;62:563‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee DY, Fletcher E, Martinez O, et al. Regional pattern of white matter microstructural changes in normal aging, MCI, and AD. Neurology. 2009;73:1722‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Palesi F, De Rinaldis A, Vitali P, et al. Specific patterns of white matter alterations help distinguishing Alzheimer's and vascular dementia. Front Neurosci. 2018;12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Naggara O, Oppenheim C, Rieu D, et al. Diffusion tensor imaging in early Alzheimer's disease. Psychiatry Res. 2006;146:243‐249. [DOI] [PubMed] [Google Scholar]
- 69. Acosta‐Cabronero J, Alley S, Williams GB, Pengas G, Nestor PJ. Diffusion tensor metrics as biomarkers in Alzheimer's disease. PLoS ONE. 2012;7:e49072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Di Paola M, Spalletta G, Caltagirone C. In vivo structural neuroanatomy of corpus callosum in Alzheimer's disease and mild cognitive impairment using different MRI techniques: a review. J Alzheimers Dis. 2010;20:67‐95. [DOI] [PubMed] [Google Scholar]
- 71. Lee DY, Fletcher E, Martinez O, et al. Vascular and degenerative processes differentially affect regional interhemispheric connections in normal aging, mild cognitive impairment, and Alzheimer disease. Stroke. 2010;41:1791‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vemuri P, Lesnick TG, Przybelski SA, et al. Development of a cerebrovascular magnetic resonance imaging biomarker for cognitive aging. Ann Neurol. 2018;84:705‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Raghavan S, Przybelski SA, Reid RI, et al. Reduced fractional anisotropy of the genu of the corpus callosum as a cerebrovascular disease marker and predictor of longitudinal cognition in MCI. Neurobiology of Aging. 2020;96:176‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huynh K, Piguet O, Kwok J, et al. Clinical and biological correlates of white matter hyperintensities in patients with behavioral‐variant frontotemporal dementia and Alzheimer disease. Neurology. 2021;96:e1743‐54. [DOI] [PubMed] [Google Scholar]
- 75. Leys D, Pruvo JP, Parent M, et al. Could Wallerian degeneration contribute to “leuko‐araiosis” in subjects free of any vascular disorder?. J Neurol Neurosurg Psychiatry. 1991;54:46‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. McAleese KE, Firbank M, Dey M, et al. Cortical tau load is associated with white matter hyperintensities. Acta Neuropathol Commun. 2015;3:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McAleese KE, Walker L, Graham S, et al. Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017;134:459‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lao PJ, Gutierrez J, Keator D, et al. Alzheimer‐related cerebrovascular disease in Down syndrome. Ann Neurol. 2020;88:1165‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Moody DM, Bell MA, Challa VR. The corpus callosum, a unique white‐matter tract: anatomic features that may explain sparing in Binswanger disease and resistance to flow of fluid masses. AJNR Am J Neuroradiol. 1988;9:1051‐1059. [PMC free article] [PubMed] [Google Scholar]
- 80. Weaver NA, Doeven T, Barkhof F, et al. Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol Aging. 2019;84:225‐234. [DOI] [PubMed] [Google Scholar]
- 81. Gaubert M, Lange C, Garnier‐Crussard A, et al. Topographic patterns of white matter hyperintensities are associated with multimodal neuroimaging biomarkers of Alzheimer's disease. Alz Res Therapy. 2021;13:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Laing KK, Simoes S, Baena‐Caldas GP, et al. Cerebrovascular disease promotes tau pathology in Alzheimer's disease. Brain Commun. 2020;fcaa132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Graff‐Radford J, Arenaza‐Urquijo EM, Knopman DS, et al. White matter hyperintensities: relationship to amyloid and tau burden. Brain. 2019;142:2483‐2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Greenberg SM, Charidimou A. Diagnosis of cerebral amyloid angiopathy: evolution of the Boston Criteria. Stroke. 2018;49:491‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thanprasertsuk S, Martinez‐Ramirez S, Pontes‐Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83:794‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee S, Zimmerman ME, Narkhede A, et al. White matter hyperintensities and the mediating role of cerebral amyloid angiopathy in dominantly‐inherited Alzheimer's disease. PLoS One. 2018;13:e0195838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Raz L, Bhaskar K, Weaver J, et al. Hypoxia promotes tau hyperphosphorylation with associated neuropathology in vascular dysfunction. Neurobiol Dis. 2019;126:124‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tosto G, Zimmerman ME, Hamilton JL, Carmichael OT, Brickman AM. The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimers Dement. 2015;11:1510‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dufouil C, de Kersaint‐Gilly A, Besancon V, et al. Longitudinal study of blood pressure and white matter hyperintensities: the EVA MRI Cohort. Neurology. 2001;56:921‐926. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information