

Abstract
Metabolism is dynamically regulated to accompany immune cell function, and altered immunometabolism can result in impaired immune responses. Concomitantly, the pharmacological manipulation of metabolic processes offers an opportunity for therapeutic intervention in inflammatory disorders. The nicotinamide adenine dinucleotide (NAD+) is a critical metabolic intermediate that serves as enzyme cofactor in redox reactions, and is also used as a co‐substrate by many enzymes such as sirtuins, adenosine diphosphate ribose transferases and synthases. Through these activities, NAD+ metabolism regulates a broad spectrum of cellular functions such as energy metabolism, DNA repair, regulation of the epigenetic landscape and inflammation. Thus, the manipulation of NAD+ availability using pharmacological compounds such as NAD+ precursors can have immune‐modulatory properties in inflammation. Here, we discuss how the NAD+ metabolism contributes to the immune response and inflammatory conditions, with a special focus on multiple sclerosis, inflammatory bowel diseases and inflammageing.
LINKED ARTICLES
This article is part of a themed issue on Inflammation, Repair and Ageing. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v179.9/issuetoc
Keywords: ageing, CD38, experimental autoimmune encephalomyelitis, inflammation, lymphocytes, NAD+ , T cells
Abbreviations
- ARTs
adenosine diphosphate (ADP)‐ribose transferases
- cADPR
cyclic ADP‐ribose
- CAR‐T
chimeric antigen receptor T cells
- CRC
colorectal cancer
- DSS
dextran sodium sulphate
- EAE
experimental autoimmune encephalomyelitis
- ecNAMPT
extracellular form of NAMPT
- FAO
fatty acid oxidation
- FoxP3
forkhead box P3
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- IBD
inflammatory bowel diseases
- IDO
indoleamine 2,3‐dioxygenase
- LPS
lipopolysaccharide
- MS
multiple sclerosis
- NA
nicotinic acid
- NAAD
nicotinic acid adenine dinucleotide
- NADS
NAD synthase
- NAM
nicotinamide
- NAMN
nicotinic acid mononucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NAPRT
nicotinic acid phosphoribosyltransferase
- NICD
NAD+‐induced cell death
- NMN
nicotinamide mononucleotide
- NMNATs
nicotinamide mononucleotide adenylyl‐transferases
- NR
nicotinamide riboside
- PBLs
human peripheral blood lymphocytes
- PBMC
peripheral blood mononuclear cell
- PncA
microbe‐encoded nicotinamidase
- RORγt
RAR‐related orphan receptor γ
- TCA
tricarboxylic acid cycle
- TCR
T Cell receptor
- TDO
tryptophan 2,3‐dioxygenase
- Tfam
mitochondrial transcription factor A
- Tregs
regulatory T cells
1. INTRODUCTION
Inflammation is a complex biological response against pathogens and tissue injuries, being involved in the surveillance and clearance of damaged cells and in tissue homeostasis. This protective response is local and tightly regulated to ensure that the inflammation is resolved once the challenge has been eliminated. Nevertheless, failures in the resolution of inflammation trigger a wide variety of chronic inflammatory and autoimmune disorders, and they are linked to many age‐related pathologies, including cardiovascular and neurodegenerative diseases, and the associated low‐grade sterile inflammation known as inflammageing. The differentiation and function of the immune system are closely related to metabolic processes that provide energy and intermediate precursors for the synthesis of macromolecules required for growth and survival. Research developed in the last 15 years have contributed to change our view of metabolic intermediates not just as a source of ATP and building blocks to maintain housekeeping functions, but as essential signalling molecules in the inflammatory process. Immunometabolism controls key features of the immune response such as the activation, proliferation, acquisition of effector function and return to homeostasis, through the regulation of gene expression, epigenetic remodelling and post‐translational modifications (Soto‐Heredero et al., 2020). The metabolic pathways that regulate the maintenance and activation of immune cells are the balanced result of environmental cues (growth factors, nutrient availability) and intracellular processes (internal metabolites, ROS, reducing/oxidising substrates) (Diskin et al., 2021; Mills et al., 2017; Pearce & Pearce, 2013). Accordingly, metabolism is dynamically regulated to accompany immune cell function, and altered immunometabolism leads to impaired immune response and inflammation. Therefore, pharmacological manipulation of metabolic processes offers an opportunity for the therapeutic intervention in inflammatory conditions (Assmann & Finlay, 2016; Patel et al., 2019). Nicotinamide adenine dinucleotide (NAD+) is a critical metabolic intermediate that serves as a universal electron acceptor in hundreds of oxidation–reduction reactions, becoming reduced to NADH in the process. NADH subsequently provides reducing power throughout the cell, including to the complex I of the mitochondrial electron transport chain driving cellular respiration. In addition, NAD+ is used as a co‐substrate by several different types of enzymes such as sirtuins (SIRTs), adenosine diphosphate (ADP)‐ribose transferases (ARTs) and the cyclic ADP‐ribose (cADPR) synthases. Through these activities, NAD+ metabolism and its intermediate metabolites regulate a broad spectrum of cellular functions such as energy metabolism, DNA repair, regulation of the epigenetic landscape and inflammation (Rajman et al., 2018; Verdin, 2015; Xie et al., 2020) (Figure 1). Thus, NAD+ is emerging not only as a major component of cellular metabolism and energy homeostasis but also as a key signalling metabolite, and the manipulation of its availability can have immune‐modulatory properties in inflammatory conditions.
FIGURE 1.
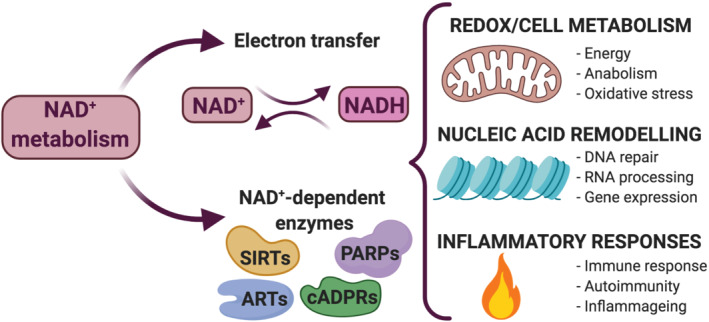
Nicotinamide adenine dinucleotide (NAD+) metabolism controls a wide range of biological processes. NAD+ serves as an enzyme cofactor, becoming reduced to NADH, which subsequently provides reducing power to drive respiration and other metabolic processes. NAD+ is also used as an enzyme co‐substrate by sirtuins (SIRTs), adenosine diphosphate (ADP)‐ribose transferases (ARTs, PARPs) and cyclic ADP‐ribose (cADPRs) synthases. Through these activities, NAD+ metabolism and its intermediate metabolites regulate a broad spectrum of cellular functions such as cell metabolism, nucleic acid remodelling and inflammation
NAD+ is continuously being degraded, synthesised and recycled, and these processes cause changes in its intracellular concentration. These changes in NAD+ availability influence essential cellular processes such as the immune response, inflammatory diseases or inflammageing. Currently, there are three main approaches to modulate NAD+ levels and to study the consequences for the immune response: (i) supplementation with NAD+ precursors (Rajman et al., 2018; Yoshino et al., 2018), (ii) activation of NAD+ biosynthetic pathways (Audrito et al., 2020; Galli et al., 2020) and (iii) altering the expression of NAD+‐consuming enzymes (Audrito et al., 2019; Kar et al., 2020). The most common precursors used to increase NAD+ levels are metabolic intermediates of biosynthetic pathways such as NAD+ itself, nicotinic acid (NA), nicotinamide (NAM), nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN). The uptake and biodistribution of NAD+ precursors have been revised elsewhere (Lautrup et al., 2019; Rajman et al., 2018). It is important to highlight that most cell types are unable to take up NAD+, with the exception of neurons (Rajman et al., 2018). Thus, supplementation with NAD+ itself will increase its extracellular concentration, which can exert different pharmacological effects compared to NAD+ precursors that will modulate the intracellular NAD+ pool.
In this review, we have collected studies that analyse how the manipulation of NAD+ availability using NAD+ precursors and pharmacological compounds modulates the immune response. Furthermore, we discuss how the dysregulation of NAD+ metabolism contributes to inflammatory and autoimmune conditions, with a special focus on multiple sclerosis, inflammatory bowel diseases (IBD), inflammageing and senescence, with data derived primarily from studies addressing the role of NAD+ in immune cells and comments from other fields when relevant.
1.1. NAD+ biosynthesis, consumption and cellular function
NAD+ is a fundamental intermediate in cellular metabolism that serve as enzyme cofactor participating in oxidation‐reduction reactions such as glycolysis, tricarboxylic acid cycle (TCA) and fatty acid oxidation (FAO). In these reactions, the nicotinamide portion of NAD+ receives high‐energy electrons in the form of hydride to generate NADH, and NADH serves as electron donor for ATP synthesis through mitochondrial oxidative phosphorylation. The flux of electrons via the mitochondrial electron transport chain regulates the NAD+/NADH ratio that is essential for maintaining mitochondrial function and energy metabolism (Xie et al., 2020). Recently, some studies have identified a mitochondrial NAD+ transporter (Slc25a51) in mammals (Girardi et al., 2020; Luongo et al., 2020). Following an immune challenge, immune cells up‐regulate mitochondrial metabolism and aerobic glycolysis to meet the increased demand for biosynthetic precursors and energy. As NAD+ plays a critical role in both metabolic processes, the modulation of its intracellular levels has the potential to strongly influence the activation of the immune response (Mills et al., 2017; Pearce & Pearce, 2013). In addition to the role of NAD+ as an intracellular metabolite of energy metabolism, NAD+ is also used as a co‐substrate for different types of enzymes, and there is an emerging body of evidence demonstrating the pivotal role of NAD+ as a signalling molecule in the intracellular environment. Being a major component of both bioenergetic and signalling pathways, the molecule is ideally suited to regulate metabolism and major cellular events. The scope of NAD+‐mediated regulatory processes is wide including enzyme regulation, control of gene expression, DNA repair, cell cycle regulation and calcium signalling (Xie et al., 2020).
As mentioned above, in addition to its role as coenzyme in energy metabolism, NAD+ is used as a co‐substrate for sirtuins (SIRT1‐7) and for different types of nucleotide‐metabolising enzymes: the adenosine diphosphate (ADP)‐ribose transferases (ARTs), the poly (ADP‐ribose) polymerases (PARPs) and the cyclic ADP‐ribose (cADPR) synthases (i.e., CD38 and CD157) (Verdin, 2015). These enzymes cleave NAD+ to generate NAM and ADP‐ribose, and the latter is further used for post‐translational modification of other proteins. NAM, in conjunction with nicotinic acid, also represents an essential vitamin (Vitamin B3 or niacin) required as an exogenous precursor of NAD+ in most living organisms. Globally, the NAD+‐consuming activity of these enzymes influences its intracellular concentration (Figure 2).
FIGURE 2.
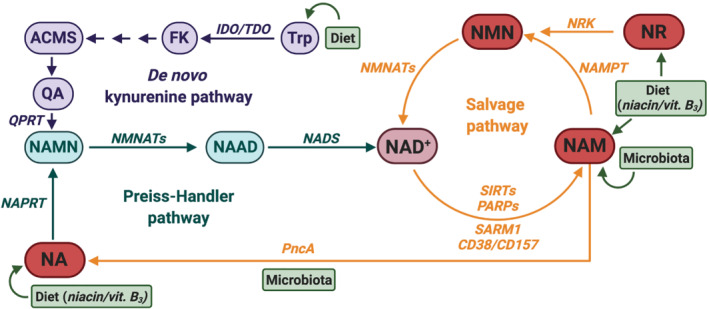
Nicotinamide adenine dinucleotide (NAD+) biosynthetic pathways counteract NAD+ consumption. NAD+ biosynthetic and recycling pathways ensure a balanced intracellular NAD+ concentration to elicit its functions. Most of the NAD+ is recycled from intermediates through the salvage pathway, but it can also be synthesised de novo from different dietary sources. The NAD+ salvage pathway recycles the nicotinamide (NAM) generated by NAD+‐consuming enzymes (i.e., SIRTS, PARPs, SARM1, CD38/CD157). NAM is transformed into nicotinamide mononucleotide (NMN) by the nicotinamide phosphoribo‐syltransferase (NAMPT), and NMN is further converted into NAD+ by the action of nicotinamide mononucleotide adenylyl‐transferases (NMNATs). NMN is also derived from dietary nicotinamide riboside (NR) through the action of the nicotinamide riboside kinase (NRK). Dietary nicotinic acid (NA, niacin/vitamin B3) is transformed into NAD+ in the Preiss‐Handler pathway. NA is first converted into nicotinic acid mononucleotide (NAMN) by the nicotinic acid phosphoribosyltransferase (NAPRT), and further transformed into nicotinic acid adenine dinucleotide (NAAD) by NMNATs. Finally, NAAD is converted into NAD+ by the NAD synthase (NADS). In addition, gut microbiota offers an alternative pathway for synthesising NAD+ through the salvage of NA from NAM by using a microbe‐encoded nicotinamidase (PncA). NAD+ can also be obtained de novo from dietary tryptophan (Trp) through the kynurenine pathway. Trp is converted to N‐formylkynurenine (FK) by the rate‐limiting enzymes indoleamine 2,3‐dioxygenase (IDO) or the tryptophan 2,3‐dioxygenase (TDO), and FK is finally converted to 2‐amino‐3‐carboxymuconate semialdehyde (ACMS). ACMS can spontaneously cyclize into quinolinic acid (QA), which is transformed into NAMN by the quinolinate phosphoribosyl transferase (QPRT) and enters the Preiss–Handler pathway
SIRTs are protein deacylases that remove acyl groups from lysine residues on proteins, using the ADP‐ribose produced upon NAD+ cleavage as acyl acceptor to generate acyl‐ADP‐ribose. SIRTs act as sensors of intracellular concentration of NAD+, and they are capable of transducing these signals via protein deacylation. SIRTs deacylate specific lysine residues on histones and key transcription factors, playing a key role in epigenetic regulation (Bheda et al., 2016; Chalkiadaki & Guarente, 2015). ARTs transfer the ADP‐ribose moiety from NAD+ to protein acceptors (MARylation), which regulate target protein function by altering their enzymic activity or serving as a signalling scaffold for recruitment of binding proteins. PARPs are a relevant subfamily of ARTs that post‐translationally modify proteins, DNA and RNA by transfer of multiple ADP‐ribose moieties, generating poly (ADP‐ribose) chains (PARylation) (Ke et al., 2019). Activation of PARPs upon DNA damage leads to a marked increase of NAD+ consumption and decrease in intracellular NAD+ concentration (Kraus, 2015). Importantly, ADP‐ribose post‐translational modifications by ARTs and PARPs are reversible (Cohen & Chang, 2018). The cADPR synthases CD38 and CD157 produce the calcium‐releasing second messenger cyclic ADP‐ribose (cADPR) (Kar et al., 2020; Lee & Zhao, 2019), and they act as major NAD+ consumers. CD38 and CD157 have been implicated in different aspects of the immune response (Quarona et al., 2013). Interestingly, CD38 deficient mice show increased NAD+ concentrations in different tissues, enhanced energy expenditure and higher metabolic rates, and they are protected against high‐fat diet‐induced obesity (Hogan et al., 2019). SARM1 is a newly discovered NAD+‐consuming enzyme that cleavages NAD+ into ADP‐ribose, cADPR and NAM, whose activity promotes axonal NAD+ depletion and neurodegeneration (Essuman et al., 2017; Jiang et al., 2020). Recent work determined that different enzymes use NAD+ and NADH as a nucleotide analogue in DNA ligation and RNA capping (Bird et al., 2018; Chen & Yu, 2019). To summarise, through their role as coenzyme in energy metabolism and as a co‐substrate for NAD+‐dependent enzymes, NAD+ and its intermediate metabolites regulate a broad spectrum of cellular functions such as energy metabolism, DNA repair and epigenetic modifications (see Xie et al., 2020) (Figure 1).
NAD+ is continuously being consumed and degraded, and these processes are balanced by biosynthetic and recycling pathways that maintain NAD+ cellular concentration. Most of the NAD+ is recycled from intermediates through the nicotinamide (NAM) salvage pathway, but it can also be synthesised de novo from different dietary sources (Rajman et al., 2018) (Figure 2). The NAD+ salvage pathway recycles the NAM generated as an intermediate product of the enzymic activity of NAD+‐consuming enzymes (i.e., SIRTS, PARPs, CD38). NAM is transformed into NAD+ in subsequent steps catalysed by the nicotinamide phosphoribosyltransferase (NAMPT). NAMPT recycles NAM into nicotinamide mononucleotide (NMN), which is further converted into NAD+ by the action of nicotinamide mononucleotide adenylyl‐transferases (NMNATs). The main dietary source for de novo synthesis of NAD+ is nicotinic acid (NA) in the form of niacin (vitamin B3) that is transformed into NAD+ in the Preiss‐Handler pathway. NA is first transformed into nicotinic acid mononucleotide (NAMN) by the nicotinic acid phosphoribosyltransferase (NAPRT), and further transformed into nicotinic acid adenine dinucleotide (NAAD) by NMNATs. Finally, NAAD is converted into NAD+ by the NAD synthase (NADS). NAD+ can also be obtained de novo from dietary tryptophan through the kynurenine pathway. Tryptophan is converted to N‐formylkynurenine by the rate‐limiting enzymes indoleamine 2,3‐dioxygenase (IDO) or the tryptophan 2,3‐dioxygenase (TDO), and N‐formylkynurenine is finally converted to 2‐amino‐3‐carboxymuconate semialdehyde (ACMS). ACMS can spontaneously cyclise into quinolinic acid and transform into NAMN that enters the Preiss‐Handler pathway. Additionally, ACMS can be decarboxylated into 2‐amino‐3‐muconate semialdehyde (AMS) by ACMS decarboxylase (ACMSD), leading to its oxidation into acetyl‐CoA via the TCA cycle. In addition, recent research has uncovered gut microbiota as another player in NAD+ metabolism, since host NAM levels positively correlates to some intestinal‐resident bacteria (Blacher et al., 2019). More strikingly, gut microbiota offers an alternative pathway for synthesising NAD+ through the salvage of NA from NAM by using a microbe‐encoded nicotinamidase (PncA) (Shats et al., 2020).
To summarise, redox reactions and the action of NAD+‐consuming enzymes cause changes in the intracellular concentration of NAD+ that, subsequently, regulate multiple cellular functions through the modulation of the activity of NAD+‐dependent enzymes. Thus, boosting the intracellular concentration of NAD+ using NAD+ precursors or pharmacological compounds has the potential to modulate multiple aspects of the cellular function. Due to the wide range of NAD+‐dependent cellular processes, strategies aimed at increasing NAD+ concentration may have pleiotropic effects that are highly influenced by the metabolic state of specific cells or tissues.
1.2. NAD+ metabolism in the immune response
There is an increasing body of evidence that associates the availability of NAD+ with perturbed immune responses during infections, suggesting the potential of targeting the intracellular levels of NAD+ to manipulate the host defence against pathogens (Singhal & Cheng, 2019; Wei et al., 2017).
Early work showed that exogenously added NAD+ induced apoptosis (NAD+‐induced cell death, NICD) in splenic T cells via the ADP‐ribosyl‐transferase ART2 (Adriouch et al., 2001; Liu et al., 2001). ART2 is a GPI‐anchored ectoenzyme that transfers the ADP‐ribose moiety from NAD+ to specific aminoacids in target proteins, modulating their activity in a post‐translational manner. ART2 senses the local concentration of NAD+, and translates this concentration to corresponding levels of ADP‐ribosylated cell surface proteins. Further work identified the purinergic P2X7 receptor as the ART2 substrate that, upon ART2‐mediated ADP‐ribosylation, initiated the apoptotic programme in peripheral T cells (Seman et al., 2003) (Figure 3a). NICD in T cells has also been observed at physiological concentrations of extracellular NAD+ released during acute inflammation (Adriouch et al., 2007) or by injured cells (Scheuplein et al., 2009). NAD+ levels were increased in an acute inflammation model induced by polyacrylamide beads, and caused T cell depletion in the draining lymph nodes via ART2 and P2X7 receptor activation (Adriouch et al., 2007). In addition to ART2, CD38 is another NAD+‐consuming enzyme implicated in the regulation of immune cell function and NICD. CD38 is a type II transmembrane protein with ecto‐NAD‐glycohydrolase activity (ecto‐NADase) that controls the level of cell surface protein ADP‐ribosylation by hydrolysing extracellular NAD+ and thus limiting substrate availability for ART2 (Figure 3a). CD38 deficient mice show a dramatic loss of tissue‐associated NAD+ glycohydrolase activity in different tissues including lymphoid tissues such as lymph nodes, spleen and bone marrow, indicating that CD38 is a major NADase in lymphoid cells (Cockayne et al., 1998). Lymphoid cell development did not show gross abnormalities in CD38 deficient mice, but they had impaired humoral response, neutrophil chemotaxis to the infection site and dendritic cell trafficking from inflammation site to lymph nodes (Cockayne et al., 1998; Partida‐Sánchez et al., 2001, 2004). CD38‐deficient lymphocytes showed high levels of cell surface ADP‐ribosylation. Remarkably, ART2 deficient mice did not show any ADP‐ribosylation, indicating that ART2 is the main ART activity in T cells (Krebs et al., 2005). Moreover, CD38 null mice showed an exacerbated NICD in T cells upon i.v. injection of NAD+, suggesting that CD38 is involved in the clearance of NAD+ at the inflammation site, reducing NAD+ availability for ART2‐induced apoptosis and thus it is partly protective against NICD (Adriouch et al., 2007).
FIGURE 3.
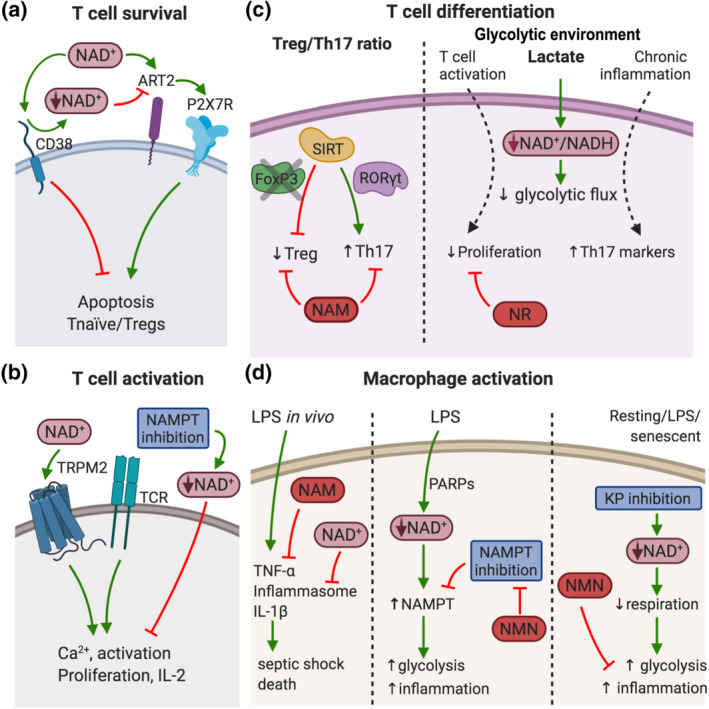
Nicotinamide adenine dinucleotide (NAD+) regulates the immune response. (a) NAD+ controls T cell survival. Increased levels of NAD+ induce apoptosis in naïve T cells and Tregs through the activation of ART2 and P2X7 receptors (P2X7R). CD38 action reduces NAD+ availability for ART2 and protects against NAD+‐induced apoptosis. (b) NAD+ levels regulate T cell activation. NAD+ precursors increase TCR‐mediated Ca2+ mobilisation via the TRPM2 channel, boosting cell proliferation and IL‐2 production. Accordingly, acute depletion of NAD+ by NAMPT inhibition reduced Ca2+ mobilisation. (c) NAD+ concentration affects T cell differentiation. SIRT1 inhibits Treg differentiation via FoxP3 inhibition, and promotes Th17 function via RORγt activation. Treatment with SIRT inhibitor NAM boosts Treg function while supresses Th17 differentiation. Lactate levels increase in glycolytic environments, promote NAD+ reduction to NADH and slows down the glycolytic flux. Lactate supress effector T cell proliferation, and NR administration restores T cell proliferation. In chronic inflammation, lactate uptake increases Th17 pro‐inflammatory markers. (d) NAD+ levels control macrophage activation. High dose of LPS induce TNF‐α production, inflammasome assembly and IL‐1β release by dendritic cells and macrophages. NAM supplementation reduced TNF‐α, and NAD+ precursors inhibited inflammasome and IL‐1β, preventing septic shock and death in mice. LPS challenge also induces a rapid decrease in NAD+ levels in macrophages through PARPs activation. The reduction in NAD+ levels was accompanied by increased NAMPT expression to maintain glycolysis and inflammatory function. NAMPT inhibition impaired the production of pro‐inflammatory mediators, and supplementation with NMN restored glycolysis in these conditions. Moreover, in resting, LPS‐activated and senescent macrophages, inhibition of the kynurenine pathway (KP) leads to reduced cellular NAD+ concentration, impaired respiration, increased glycolysis and inflammation. These defects were restored by exogenous addition of the NAD+ precursor NMN
Interestingly, the developmental stage of T cells influences the sensitivity to NICD. NICD primarily affected naïve T cells, while thymocytes and T cells with activated/memory phenotype were resistant to NICD (Adriouch et al., 2007; Haag et al., 2002; Liu et al., 2001). These results are consistent with previous work showing that ART2 is shed upon T cell activation (Kahl et al., 2000). Moreover, T cell subsets also display a differential sensitivity to NICD, as in vivo administration of NAD+ preferentially induced apoptosis of regulatory T cells (Tregs) via ART2 and P2X7 receptors (Aswad et al., 2005; Hubert et al., 2010). Accordingly, while P2X7 receptor‐deficient mice have increased numbers of Tregs, which were resistant to apoptosis induced by NAD+ injection (Aswad et al., 2005), CD38 deficient mice had lower numbers of Tregs and they were sensitive to NICD at lower doses of NAD+ than wild‐type counterparts, and the administration of an ART2‐blocking antibody protected Tregs from NICD in vivo (Hubert et al., 2010). Interestingly, the higher sensitivity of Tregs to NICD can be used to selectively manipulate Treg function in pathological conditions. NAD+ has been showed to promote Treg conversion into Th17 cells (Elkhal et al., 2016). Tregs are known to suppress anti‐tumour immune response in different tumour models and thus, Treg depletion can serve as a strategy to boost anti‐tumour immunity. Accordingly, NAD+ injection controlled tumour growth in different tumour models (EL4 cell line, EG7 lymphoma and B16 melanoma) and increased the frequency of granzyme B positive CD4+ and CD8+ T cells among tumour‐infiltrating lymphocytes (Hubert et al., 2010).
Exogenously added NAD+ can exert different pharmacological effects from the modulation of NAD+ intracellular levels via supplementation with NAD+ precursors or altering NAD+‐biosynthetic or ‐consuming pathways. In fact, while NAD+ itself induces NICD in naïve and regulatory T cells, NAD+ precursors modulate essential functions in T cell activation and differentiation. In activated T cells, acute depletion of NAD+ using FK866, a pharmacological inhibitor of the rate‐limiting enzyme for NAD+ biosynthesis NAMPT, reduced T cell proliferation and viability, and these defects were rescued by NAM supplementation that restored NAD+ levels (Bruzzone et al., 2009) (Figure 3b). FK866 treatment caused the reduction of anti‐CD3‐induced Ca2+ mobilisation in the Jurkat cell line and in activated human peripheral blood lymphocytes (PBLs). In contrast, exogenously added NAM, NA and NMN increased the intracellular concentration of NAD+, and increased anti‐CD3‐mediated Ca2+ mobilisation in human PBLs via the cyclic ADP‐ribose‐dependent TRPM2 channel gating (Magnone et al., 2012) (Figure 3b). Of note, NAD+ also regulates intracellular calcium levels in human granulocytes and monocytes (Bruzzone et al., 2006; Gerth et al., 2004). In accordance to increased Ca2+ mobilisation in PBLs, pre‐incubation with NAD+ precursors resulted in increased cell proliferation and IL‐2 production (Magnone et al., 2012). Overall, data show that T cells require NAD+ for proper activation and indicate that the intracellular concentration of NAD+ can be manipulated to control T cell activation.
Another example of how the intracellular level of NAD+ affects immune cell function can be found in T cells deficient for the mitochondrial transcription factor A (Tfam) that controls mitochondrial DNA expression, including the mitochondrial respiration complexes I, III, IV and V. Tfam‐deficient CD4+ T cells displayed impaired mitochondrial performance and dysregulated electron transport from NADH due to imbalanced NAD+/NADH ratio. To compensate mitochondrial dysfunction, Tfam null CD4+ T cells rewire their metabolism towards glycolysis, favouring the acquisition of a pro‐inflammatory Th1 phenotype with increased transcription and secretion of IFN‐γ and TNFα, as well as reduced amounts of the immunosuppressive cytokine IL‐10. Interestingly, the restoration of NAD+/NADH balance by treatment with the NAD+ precursor NAM, in vitro, corrected inflammatory defects, with reduced IFN‐γ production and Th1 differentiation (Baixauli et al., 2015). Mitochondrial dysfunction also contributes to several aspects of heart failure. Recently, it has been shown that peripheral blood mononuclear cell (PBMC) from heart failure subjects display a reduced respiratory capacity and elevated expression of pro‐inflammatory markers such as NLRP3, a key inflammasome component, and IL‐1β, IL‐18, TNF‐α (Zhou et al., 2020). Interestingly, this study showed that the oral administration of the NAD+ precursor NR enhanced mitochondrial respiration and reduced expression of pro‐inflammatory genes in PBMC from heart failure subjects, when compared with healthy donors. Thus, boosting NAD+ intracellular levels have promising anti‐inflammatory effects in the context of diseases associated with mitochondrial dysfunction. The NAD+/NADH ratio also influences T cell differentiation through the regulation of SIRTs deacetylase activity. In this context, while initial studies on whole body SIRT1‐deficient mice suggested that SIRT1 suppressed inflammation (Gao et al., 2012; Zhang et al., 2009), further studies analysing the impact of the specific deletion of SIRT1 in Tregs and Th17 suggested that SIRT1 exerts pro‐inflammatory actions in these subpopulations (Beier et al., 2011; Kwon et al., 2012; Lim et al., 2015; van Loosdregt et al., 2010). In whole body SIRT1‐deficient mice, SIRT1 was shown to inhibit the transcriptional activity of AP‐1, a key transcription factor for T cell activation formed by c‐Fos and c‐Jun. Mechanistically, SIRT1 inhibited c‐Jun acetylation, which is required for its activity and accordingly, SIRT1‐deficient T cells were hyperresponsive to TCR stimulation. In addition, SIRT1‐deficient mice developed a severe form of experimental autoimmune encephalomyelitis (EAE) and spontaneous autoimmunity (Zhang et al., 2009), pointing to an anti‐inflammatory action of SIRT1. In contrast, SIRT1 seems to have pro‐inflammatory function in Tregs and Th17 cells. SIRT1 was found to deacetylate FoxP3, leading to its degradation, and treatment with SIRT inhibitor NAM increased FoxP3 expression and boosted Treg suppressive function (van Loosdregt et al., 2010). Supporting the pro‐inflammatory action of SIRT1 expression in Tregs, the specific ablation of SIRT1 in FoxP3‐expressing cells promoted the expression of FoxP3 and increased Treg immunosuppressive features in vitro and in vivo, in a cardiac allograft survival model (Beier et al., 2011) (Figure 3c). SIRT1 also binds and deacetylates the transcription factor RORγt, a master regulator of Th17 differentiation (Lim et al., 2015). In contrast to the action on FoxP3, RORγt deacetylation by SIRT1 was required for optimal transcriptional activity and accordingly, specific deletion of SIRT1 in RORγt‐expressing cells and pharmacological inhibition of SIRT1 using NAM and Ex‐527 showed reduced Th17 cell differentiation in vitro (Figure 3c). More importantly, SIRT1 inhibition by genetic ablation in Th17 and by treatment with Ex‐527 inhibitor were protective in the EAE model (discussed below) (Lim et al., 2015). Thus, through the inhibition of Treg function and the boosting of Th17 differentiation, these results suggest that SIRT1 exert pro‐inflammatory roles in T cells, making it an attractive target for therapeutic intervention of the Treg/Th17 axis in autoimmune and inflammatory diseases (Chadha et al., 2019). In contrast, recent studies in human CD8+ T cells have found that the terminally differentiated memory population (CD8+CD28−) that accumulate with age showed decreased expression of SIRT1 compared to resting CD8+CD28+ cells. The decreased SIRT1 expression promoted a metabolic reprogramming of CD8+CD28− cells that displayed an enhanced capacity to use glycolysis and a higher cytotoxic activity, suggesting an anti‐inflammatory function for SIRT1 (Jeng et al., 2018). Thus, further research is required to dissect the role of SIRTs in different effector immune cells.
NAD+ precursors can have a different effect on effector T cells depending on the environmental conditions. Upon activation, T cells rewire their metabolic profile towards a glycolytic state to cope with the increased demand of energy and intermediate metabolites required to fulfil the effector function. As consequence of the increased glycolysis, there is an enrichment of lactate in inflamed tissues, something that has been also observed in highly glycolytic tumours (Buck et al., 2017; Certo et al., 2021). Lactate was shown to supress effector T cell proliferation (Angelin et al., 2017). Follow up work has recently determined that lactate promotes the reduction of NAD+ to NADH, and the increased NADH slowed down the glycolytic flux via GAPDH inhibition and limited glucose‐derived serine production, as addition of serine fully rescued lactate‐induced T cell suppression (Quinn et al., 2020). This study also showed that the addition of NR to restore NAD+ intracellular levels was sufficient to increase T cell proliferation in low glucose media. Thus, this data suggests that increasing the pool of NAD+ may be used to boost T cell proliferation (Figure 3c). Other work examining the role of lactate in CD4+ T cell function in the context of chronic inflammatory disorders, such as rheumatoid arthritis (Pucino et al., 2019) found that lactate uptake by activated CD4+ T cells via the lactate transporter Slc5a12 reduced glycolytic rate by decreasing NAD+/NADH ratio. In this context, lactate uptake increased pro‐inflammatory markers such as up‐regulated expression of Rorc (encoding RORγt), Il17a and Ifng mRNAs (Figure 3c). Mechanistically, lactate was found to promote nuclear translocation of PKM2 and phosphorylation of STAT3, a key driver of IL‐17 production in Th17 cells, and also to promote the retention of CD4+ T cells in inflamed tissue. Thus, in the context of chronic inflammatory diseases, restoration of NAD+ intracellular levels may result in attenuated pro‐inflammatory features of T cells. Taken together, these studies highlight the relevance of a balanced NAD+/NADH ratio in T cell function (Pucino et al., 2019; Quinn et al., 2020). However, further research is required to dissect the functional consequences of using NAD+ precursors in different immune cells and in specific inflammatory conditions.
Collectively, these studies show that the modulation of NAD+ levels has different effects in distinct T cell subpopulations, inducing apoptosis in naïve and Tregs, increasing activation and proliferation in effector cells while restraining pro‐inflammatory features. These differences may reflect that the intrinsic metabolic state and bioenergetic demands of specific T cell subpopulations influence the overall effect of raising NAD+ levels. Additionally, genetic models may lead to metabolic remodelling to cope with long‐term imbalance of NAD+ levels, while acute changes of NAD+ concentration (i.e., treatment with pharmacological compounds) may result in a different metabolic state and thus, increasing NAD+ may have different effects on T cells in these conditions. Finally, changes in the extracellular concentration of NAD+ (i.e., through the supplementation with NAD+ itself) can exert pharmacological effects, different from those of modulating intracellular levels via supplementation with NAD+ precursors or manipulation of NAD+‐biosynthetic or ‐consuming pathways.
Besides T cells, the intracellular concentration of NAD+ plays a relevant role in the function of other immune cells such as macrophages. For example, the administration of NAM protected mice against a lethal high dose of LPS (Van Gool et al., 2009). Mechanistically, this work determined that NAM supplementation reduced TNF‐α production by dendritic cells and macrophages in a SIRT6‐dependent manner (Figure 3d). Moreover, other studies have shown that nicotine agonists and β‐NAD efficiently inhibited ATP‐induced inflammasome assembly and IL‐1β release in human monocytic cells (Hecker et al., 2015; Hiller et al., 2018). Taking in to account that TNF‐α and IL‐1β production are markers of several inflammatory and autoimmune diseases, these data supports the therapeutic potential of NAD+ precursors in inflammatory diseases.
Interestingly, some recent studies highlighted the functional relevance of NAD+ metabolism in the regulation of pro‐inflammatory response in macrophages (Cameron et al., 2019; Minhas et al., 2019). The study by Minhas et al. showed that the kynurenine pathway is a key source for de novo synthesis of NAD+ in macrophages, and the genetic ablation (in Ido −/− and Qprt −/− mice) or pharmacological disruption (using 1‐methyl‐L‐tryptophan and phthalic acid) of this pathway reduced intracellular NAD+ concentration, impaired mitochondrial respiration and increased glycolysis in human and mouse macrophages in vitro. These metabolic changes were accompanied by an increased expression of the pro‐inflammatory markers CD86 and CD64, decreased expression of the anti‐inflammatory markers CD206 and CD23, and impaired phagocytosis. Interestingly, decreased mitochondrial respiration parameters and increased pro‐inflammatory markers were reverted by exogenous addition of NAD+ precursor NMN (Figure 3d). Moreover, these results were observed in resting macrophages, and recapitulated in vivo in a model of acute inflammation upon LPS challenge, as pharmacological inhibition of de novo NAD + biosynthesis potentiated the effect of LPS on proinflammatory and anti‐inflammatory markers. This work also showed that LPS challenge suppressed de novo NAD+ synthesis, decreasing intracellular NAD+ concentration. Thus, de novo NAD+ synthesis via the kynurenine pathway is essential to maintain anti‐inflammatory functions in macrophages (Minhas et al., 2019). Cameron et al. reported a rapid decrease in NAD+ levels in macrophages upon LPS challenge due to activation‐induced mitochondrial ROS production, which leads to ROS‐mediated DNA damage, and consequently PARP activation with associated NAD+ consumption (Cameron et al., 2019). Other factors such as CD38 expression or sirtuin activity contribute to NAD+ consumption during macrophage activation (Matalonga et al., 2017; Van Gool et al., 2009). The LPS‐induced decrease in NAD+ levels was accompanied by increased NAMPT expression, a key enzyme of the NAD+ salvage pathway. The pharmacological inhibition of NAMPT using FK866 (APO866) led to a reduction in cellular ATP levels and impaired the production of pro‐inflammatory mediators such as IL‐1β, IL‐6 and TNF‐α in vitro. Moreover, supplementation with NMN, the metabolite directly downstream of NAMPT in the NAD+ salvage pathway, restored glycolysis and ATP levels in FK866‐treated macrophages (Cameron et al., 2019) (Figure 3d). Hence, this work indicates that the NAD+ salvage pathway is required to maintain the NAD+ content to drive glycolysis and support the activation of inflammatory macrophages. These two studies highlight the relevance of NAD+ metabolism in macrophages (Cameron et al., 2019; Minhas et al., 2019) but, the data are somewhat contradictory. While inhibition of NAD+ de novo synthesis exacerbates inflammation, and NMN reduces inflammatory markers in macrophages (Minhas et al., 2019), block of the salvage pathway reduces inflammatory features, and NMN restored macrophage activation (Cameron et al., 2019). A potential explanation is that the two different strategies used to decrease NAD+ levels may be accompanied by the generation of different intermediate metabolites, leading to different functional effects on macrophage activation. Additionally, other work suggested that de novo and salvage pathways maybe required at different time points. While low dose endotoxin enhanced NAMPT‐dependent NAD+ salvage and promoted inflammation, high dose endotoxin shifted NAD+ biosynthesis from early salvage pathway to late (IDO1)‐dependent NAD+ de novo pathway and promote immunosuppression in a SIRT1‐dependent manner (Zhang et al., 2019). Thus, stimuli timing, dosage and the resulting cellular metabolic state may determine the global response to increased levels of NAD+.
1.3. NAD+ metabolism in multiple sclerosis
Experimental autoimmune encephalomyelitis (EAE) is the most common animal model of multiple sclerosis (MS), a neuroinflammatory and demyelinating disease characterised by immune cell infiltration in the CNS, which causes severe physical disability in young adults. The infiltration of activated immune cells induce local inflammation, demyelination and axonal degeneration (Compston & Coles, 2008; Dendrou et al., 2015). Thus, targeting immune cell function is a very active field for current and future therapeutic strategies in MS management. The therapeutic potential of NAD+ precursors have been examined in the EAE model (Penberthy & Tsunoda, 2009). The systemic administration of NAD+ precursors may have effects on immune cells as well as direct effects on neurons and glia cells, and different studies have identified protective actions of NAD+ precursors on immune cells and particularly in T cells.
Several reports have shed some light on the relevance of NAD+ levels in EAE and MS pathogenesis (Figure 4a). Low serum NAD+ levels in MS patients correlates with severity and progression of the disease (Braidy et al., 2013), and different mechanisms can contribute to NAD+ decrease. For example, the expression of NAD+‐consuming enzymes such as CD38 was up‐regulated in astrocytes and microglia in a demyelination model induced by cuprizone. CD38 knockouts had increased NAD+ levels, and they showed attenuated demyelination and axonal damage, reduced glia activation and decreased inflammation (Roboon et al., 2019). Other studies have shown protective effects of the administration of NAD+ and NAD+ precursors in EAE progression by different mechanisms. For example, both preventive and therapeutic administration of the NAD+ biosynthesis precursor NAM delayed EAE onset with reduced CD4+ T cell infiltration and axon demyelination (Kaneko et al., 2006). NAD+ also reduced EAE symptoms when administered after the disease onset, highlighting its therapeutic potential. This study found that NAD+ protective effects were mediated by promoting the generation of immunosuppressive IL‐10‐secreting CD4+ T cells, as a results of the induced expression of the mRNA for tryptophan hydroxylase‐1 (Tph1), a rate‐limiting enzyme in the biosynthesis of serotonine (Tullius et al., 2014). Supporting the role of the Tph1 pathway in the protection against EAE development, previous work found that Tph1‐deficient mice displayed exacerbated EAE (Nowak et al., 2012). Preventive administration of NAD+ also ameliorated EAE by decreasing the frequency of Th1 and Th17 cells (Wang et al., 2016), and the combined treatment with NAD+ precursors and atorvastatin boosted NAD+ protection by increasing Tregs frequency and function while reducing IL‐17 secretion (Sun et al., 2020). In line with these findings, NAD+ treatment attenuated infiltration of inflammatory cells, demyelination and microglial activation, and suppressed the activation of the NLRP3 inflammasome in the spinal cord. The protective effects of NAD+ were partially reverted by autophagy blockade using 3‐methyladenine, indicating that NAD+ protection is also mediated by activation of autophagy (Wang et al., 2020). Additionally, beneficial effects of NAD+ precursors in the EAE model have been linked to sirtuins activity. SIRT1 was shown to deacetylate and activate the Th17 master transcription factor RORγt. Accordingly, both treatment with nicotinamide, a sirtuin inhibitor, and T cell–specific deletion of Sirt1 suppressed Th17 differentiation and showed decreased EAE severity (Lim et al., 2015). Likewise, pharmacological inhibition of Sirt6 ameliorated EAE by reducing the migration of dendritic cells to the lymph nodes (Ferrara et al., 2020). Besides the immune system, NAD+ and related metabolites also exert key functions in synaptic plasticity and neuronal stress resistance (Lautrup et al., 2019), and NAD+ precursors have protective effects in axonal degeneration (Nimmagadda et al., 2017). Of note, changes in the extracellular NAD+ may have different pharmacological effects from increasing the intracellular levels of NAD+ using NAD+ precursors, but overall, the systemic administration of NAD+ and NAD+ precursors has pleiotropic beneficial effects by acting both on neurons and immune cells.
FIGURE 4.
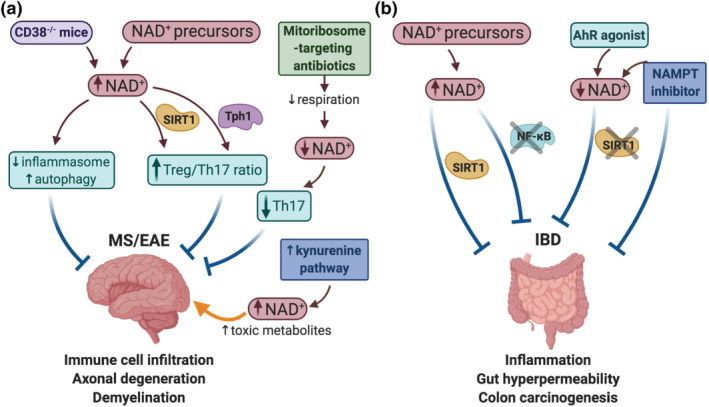
Nicotinamide adenine dinucleotide (NAD+) metabolism alters the development of autoimmune diseases. (a) Therapeutic strategies to modulate NAD+ levels in experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis (MS). MS is characterised by immune cell infiltration, axonal degeneration and demyelination. Strategies aiming to increase NAD+ levels (i.e., CD38 knock‐outs or supplementation with NAD+ precursors) ameliorate EAE severity by different mechanisms: Inhibiting inflammasome assembly and increasing autophagy, and increasing the Treg/Th17 ratio via SIRT1 and Tph1 activation. The administration of mitoribosome‐targeting antibiotics reduced respiration and decreased NAD concentration, leading to reduced Th17 effector function and EAE protection. The kynurenine pathway for de novo synthesis of NAD+ is activated in EAE mice, and long‐term activation of the kynurenine pathway generates an environment with increased concentration of NAD+ together with other toxic metabolites, worsening EAE. (b) Inflammatory bowel diseases (IBD) are characterised by enhanced gut inflammation and permeability, and increased risk to develop colorectal cancer. The therapeutic properties of NAD+ precursors in IBD are exemplified by their role in activating anti‐colitogenic SIRTs and supressing the pro‐inflammatory NF‐κB signalling. Furthermore, pharmacological inhibition of NAMPT as well as modulation of the NAD+/SIRT1 axis using an aryl hydrocarbon receptor (AhR) agonist offers a striking opportunity to fight against IBD
The different studies discussed above shown that the administration of NAD+ precursors ameliorated the development of autoimmune diseases through a variety of mechanisms (Figure 4a). At the same time, other work revealed that a drop in the NAD+/NADH ratio has immunosuppressive effects and thus, potential therapeutic effects in autoimmune and inflammatory diseases. Recently, a study has shown that the administration of linezolid and other ribosome‐targeting antibiotics ameliorated EAE symptoms. These antibiotics impaired mitoribosome function, affecting the expression of key components of the electron transport chain in Th17 cells. The altered mitochondrial respiration hindered the regeneration of NAD+, causing a decrease in the NAD+/NADH ratio and reducing Th17 effector function (Almeida et al., 2020). Thus, this study indicates that limiting cellular NAD+ availability reduces Th17 cell differentiation and function and has protective effects in a model of autoimmune disease, while other data discussed above show that increasing NAD+ concentration through the administration of NAD+ precursors ameliorated EAE. These data suggest that the moment of interference with NAD+ metabolism (i.e., activation vs. effector phase) may have different functional consequences. Furthermore, other research highlights the complexity of NAD+ metabolism in the immune response, indicating that the form of NAD+ supplementation (i.e., NAD+ precursors vs. biosynthesis activation) may have different outcomes. For example, Sundaram et al. determined that the kynurenine pathway for de novo synthesis of NAD+ is activated during EAE and correlates with severity score. Mechanistically, this work found that long‐term activation of the kynurenine pathway generates an environment with increased concentration of toxic metabolites, worsening EAE, and IDO‐1 inhibition using 1‐methyl tryptophan ameliorates EAE progression (Sundaram et al., 2020). Besides MS, the manipulation of NAD+ metabolism has shown beneficial effects in other inflammatory conditions. For example, NAD+ up‐regulation using NAD+ precursors have shown protective effects in inflammatory skin conditions (Radenkovic & Verdin, 2020; Wozniacka et al., 2007). In addition, the CD38/NAD+ axis also plays a relevant role in lupus and other inflammatory conditions (García‐Rodríguez et al., 2018; Kar et al., 2020).
Altogether, most of the available data indicate that increasing the intracellular concentration of NAD+ has protective effects in EAE development, with remarkable exceptions (Almeida et al., 2020; Sundaram et al., 2020). These studies suggest that the moment of interference with NAD+ metabolism (i.e., activation vs. effector phase of the immune response), and the form of NAD+ supplementation (i.e., NAD+ precursors vs. biosynthesis activation) may have different outcomes. In the EAE model, T cells undergo marked steps: TCR stimulation and T cell activation, followed by differentiation towards Th17, CNS infiltration and release of effector functions, and energy and metabolite demands vary in the different stages. Therefore, it is likely that the manipulation of NAD+ metabolism will have different outcomes depending on the moment of interference. For example, while altering early T cell activation will affect the whole immune response, intervention in the effector and contraction phase can have a different result. Further research is required to dissect NAD+ requirements during immune cell activation and function.
1.4. NAD+ metabolism in gut homeostasis and IBD
IBD, which includes Crohn's disease and ulcerative colitis, results from an altered intestinal immune response that, in addition to genetic susceptibility, is highly influenced by host microbiota (Caruso et al., 2020; Ni et al., 2017). Furthermore, gut dysbiosis and the self‐perpetuating chronic inflammation during colitis‐associated diseases favour the development of colorectal cancer (CRC) (Janney et al., 2020), highlighting the importance of addressing this group of pathologies that are becoming a major challenge worldwide. Mitochondria metabolism and bioenergetics, together with the autophagic machinery, are emerging as key regulators of host‐microbiota and gut homeostasis (Larabi et al., 2020; Michaudel & Sokol, 2020). Thereby, the use of NAD+ precursors to modulate the crosstalk between mitochondria and autophagy, as well as the inflammatory response, represents a potential therapeutic strategy for IBD management (Gabandé‐Rodríguez et al., 2019).
Different studies have revealed the relevance of NAD+ metabolism in the maintenance of gut homeostasis and in IBD models (Figure 4b). For example, the expression of the NAD+‐depleting enzyme CD38 is up‐regulated in IBD patients (Ning et al., 2019), and is implicated in intestinal inflammation in mice and humans (Schneider et al., 2015). Moreover, in vitro and in vivo experiments showed that the administration of NAD+ ameliorated inflammation‐related gut hyperpermeability by inhibiting NF‐κB (Han et al., 2003). Mechanistically, further research has highlighted the role of sirtuins as key suppressors of colitis. Decreased levels of SIRT1 were observed in IBD patient biopsies (Caruso et al., 2014), and IL‐10‐deficient mice, which develop spontaneously colitis that progresses to colon carcinogenesis, displayed increased autophagy and down‐regulation of SIRT1 (Talero et al., 2016). Dextran sodium sulphate (DSS) treatment in SIRT2‐deficient mice exacerbated colitis, and macrophages showed a pro‐inflammatory phenotype (Lo Sasso, Menzies, et al., 2014). Accordingly, activating SIRT1 or overexpressing SIRT6 ameliorated DSS‐induced colitis in mice (Liu et al., 2017; Ren et al., 2019; Xu et al., 2020). Other studies have identified a key role for SIRT1 in the regulation of gut microbiota. In this context, while some research indicated that intestinal‐specific loss of SIRT1 protects mice against colitis and CRC by remodelling gut microbiota (Lo Sasso, Ryu, et al., 2014), other works revealed a protective role of intestinal SIRT1 on gut inflammation by mediating host‐microbiota symbiosis (Leber et al., 2018; Wellman et al., 2017). In line with this, some gut‐colonising bacteria have been linked to systemic NAM levels in mice (Blacher et al., 2019) and, interestingly, the gut microbiome offers itself an alternative pathway for synthetizing host NAD+, enhancing the effects of NAM or NR supplementation (Shats et al., 2020). Thereby, biomedicine could take advantage of gut microbiota to treat IBD, given its role on NAD+ metabolism and intestinal inflammation‐related diseases.
In addition to sirtuins, other NAD+‐dependent enzymes such as PARP‐1, which uses NAD+ to facilitate DNA repair, were overactivated in some bowel pathologies leading to NAD+ depletion, SIRT1 down‐regulation and causing mucosal atrophy. Moreover, PARP‐1 inhibition by NAM administration improved intestinal injury in a rat model of necrotizing enterocolitis (Giannone et al., 2011). Additionally, increasing intracellular levels of NAD+ by using dunnione, a NADH:quinone oxidoreductase 1 substrate, up‐regulated SIRT1 and ameliorated cisplatin‐induced intestinal inflammation (Pandit et al., 2015). Other studies have determined that NMN and NAD+ supplementation enhanced the function of intestinal stem cells in aged mice by acting on mTOR and SIRT1 (Igarashi et al., 2019; Igarashi & Guarente, 2016), and NMN suppressed senescence marks in intestinal organoids derived from aged mice (Uchida et al., 2019). Thereby, these findings suggest that strategies aimed to increase NAD+ levels or activating sirtuins can contribute to the maintenance of the intestinal barrier, an essential element in the development of IBD.
Nevertheless, heightening NAD+ levels may not always result in gut homeostasis. For example, a study using norisoboldine, an aryl hydrocarbon receptor agonist that expands epigenetically colonic Treg cells, indicates that DSS‐induced colitis in mice is attenuated by means of diminishing NAD+ and SIRT1 levels in colon (Lv et al., 2018) (Figure 4b). Accordingly, proteomics revealed different expression patterns of proteins involved in NAD+ metabolism in IBD patients compared to controls, finding that NAMPT was overexpressed in IBD (Ning et al., 2019). This enzyme involved in the NAD+ salvage pathway has gained more interest in the context of IBD as this pleiotropic protein can also display pro‐inflammatory and tumorigenic features (Galli et al., 2020). As such, NAMPT is found up‐regulated in T cells during human and mouse acute graft‐versus‐host disease, a major gastrointestinal complication after allogenic haematopoietic stem cell transplantation (Gerner et al., 2020), and nourishes human CRC (Ye et al., 2020). Furthermore, recent studies have unveiled that the extracellular form of NAMPT (ecNAMPT) is elevated in IBD patients and, more intriguingly, in those who fail to respond to anti‐TNFα therapy (Colombo et al., 2020; Neubauer et al., 2019) (Figure 4b). Hence, NAMPT inhibition, using FK866, elicited protection against experimental colitis in human and mice (Colombo et al., 2020; Gerner et al., 2018) and suppressed colon tumourigenesis in mice (Gerner et al., 2018; Ye et al., 2020).
Altogether, evidence shows that a balanced NAD+ biosynthesis and consumption may preserve gut homeostasis. On the one hand, overactivation of the NAD+‐synthetizing enzyme NAMPT feeds tumourigenesis and acts as a pro‐inflammatory cytokine in the gut, unleashing pathology. On the other hand, the NAD+/Sirtuin axis appears to be suppressed in IBD, as supplementation of NAD+ precursors and/or sirtuins activation limit intestinal inflammation. Therefore, fine‐tuned therapies targeting the NAD+ pathway might serve as promising pharmacological tools in IBD.
1.5. NAD+ metabolism in senescence and inflammageing
NAD+ levels are decreased during senescence and tissue ageing. Concomitantly, NAD+ decay leads to mitochondrial dysfunction, oxidative stress and DNA damage, which in turn contribute to the ageing process (Lautrup et al., 2019). Recent research studies have identified different mechanisms that contribute to NAD+ decay during ageing (Covarrubias, Perrone, et al., 2021). Pioneer experiments from Sinclair's lab demonstrated that during ageing there is a specific loss of mitochondrial, but not nuclear, encoded oxidative phosphorylation subunits in skeletal muscle. This event induces a pseudohypoxic state characterised by HIF‐1α stabilisation and reduction in NAD+ levels. Remarkably, this mitochondrial decline and pseudohypoxic state can be reversed with NAD+ precursors (Gomes et al., 2013). In addition, the reduction in NAD+ levels during ageing can be the result of altered activity or expression of enzymes involved in NAD+ biosynthesis and degradation (Covarrubias, Perrone, et al., 2021). For instance, the activation of PARP‐1 in response to DNA damage during ageing may contribute to this drop (Scheibye‐Knudsen et al., 2014). Moreover, NAMPT expression is down‐regulated as we age, contributing to ageing‐related NAD+ decline (Stein & Imai, 2014). Furthermore, the abundance of the protein QPRT, essential in de novo synthesis pathway, decreases during ageing, contributing to a diminished NAD+ pool (Minhas et al., 2019).
One of the shared features in ageing is cellular senescence and, interestingly, cells that undergo senescence, both replicative or associated to mitochondrial dysfunction display lower NAD+/NADH ratios (Wiley et al., 2016). Accordingly, cells resistant to senescence (i.e., Vδ2+ T cells) manifest a transcriptomic signature enriched in NAD+‐related pathways, suggesting an anti‐ageing role of NAD+ metabolism (Xu et al., 2019). Senescence also affects stem cells with age, leading to impaired regenerative capacity and loss of tissue homeostasis (Zhang et al., 2018). Strikingly, research has unveiled that NAD+‐enhancing approaches reprogrammed the age‐associated stem cell dysfunction. For instance, NAD+ and NMN supplementation rejuvenate intestinal stem cells in aged mice (Igarashi et al., 2019; Igarashi & Guarente, 2016) and, more importantly, treatment with NR improves muscle, neural and melanocyte stem cell function in aged mice, expanding their lifespan (Zhang et al., 2016). In line with this, growing evidence support that raising NAD+ levels delay the development of age‐related diseases. As discussed above, different experimental strategies that increase intracellular NAD+ concentration have protective effects in EAE development and prevent neurodegeneration (Lautrup et al., 2019; Verdin, 2015). Furthermore, increasing the activity of the NAD+ salvage pathway enzyme NAMPT by administration of an allosteric activator (P7C3) or by addition of extracellular vesicles containing ecNAMPT showed neuroprotection and improved cognitive function in mice (Yin et al., 2014; Yoshida et al., 2019). NAD+ decline has been observed in mouse models of heart failure, and dietary NR supplementation attenuated disease development (Diguet et al., 2018). Moreover, increasing NAD+ by dietary supplementation with nicotinamide improved diastolic dysfunction in aged mice. More importantly, this recent study associated a cardiac deficit in NAD+ with diastolic dysfunction in humans, and high dietary intake of naturally occurring NAD+ precursors was associated with lower BP and reduced risk of cardiac mortality in a long‐term human cohort study (Abdellatif et al., 2021). Metabolic disorders such as obesity and Type 2 diabetes are age‐related diseases, and in mice on a high fat diet that become prediabetic, NR administration improved glucose tolerance, reduced weight gain, liver damage and the development of hepatic steatosis (Trammell et al., 2016). In addition, long‐term NMN administration suppressed age‐associated body weight gain, enhanced energy metabolism, promoted physical activity and improved insulin sensitivity in mice on normal diet (Mills et al., 2016), and NAM improved glucose homeostasis in mice on a high‐fat diet (Mitchell et al., 2018). The therapeutic benefits of NAD+ raising strategies have showed promising results in muscle wasting and ageing. For example, in a mouse model of Duchenne's muscular dystrophy, NAD+ repletion using dietary NR improved muscle function and heart pathology (Ryu et al., 2016). Moreover, NR and olaparib (a PARP inhibitor) restored muscle homeostasis in different aged animal models and in human aged myotubes (Romani et al., 2021). Therefore, NAD+ repletion through dietary and pharmacological precursors has emerged as a potential therapeutic opportunity to ameliorate age‐related decline and disease.
Beside these metabolic alterations, recent findings suggest that inflammageing could also drive the decay in NAD+ levels during ageing. Inflammatory cytokines, in particular TNF‐α, IL‐6 and IL‐10, secreted by senescent cells as consequence of the senescence‐associated secretory phenotype (SASP) induced macrophages to proliferate and increased expression of the NAD+‐consuming enzyme CD38. CD38 expression in proinflammatory M1‐like macrophages is a key driver of NAD+ loss in the context of inflammageing (Figure 5). Thus, CD38‐knockout mice were protected from the age‐related NAD+ decline and showed enhanced metabolic fitness (Chini et al., 2020; Covarrubias, Kale, et al., 2020). Similarly, we have observed that mice with mitochondrial dysfunction in T cells, by specifically targeting mitochondrial transcription factor A (Tfam), presented premature inflammageing and a drop in the NAD+/NADH ratio in liver, further supporting that inflammageing contributes to NAD+ decline. In addition, these mice displayed accelerated senescence and age‐related multimorbidity, characterised by cardiovascular failure, metabolic dysregulation, and cognitive decline. Importantly, treatment with NR dampened inflammageing and increased resilience to age‐related multimorbidity in these mice (Desdín‐Micó et al., 2020). In contrast, other research has determined that increased intracellular concentration of NAD+ can have potential pro‐tumorigenic effects by enhancing the expression of inflammatory SASP genes. Nacarelli et al. reported that the NAD+ salvage pathway is up‐regulated during senescence, and raising NAD+ levels can also have a pro‐tumorigenic activity (Nacarelli et al., 2019). Therefore, NAD+‐raising strategies through dietary precursors should be considered with caution to balance the beneficial anti‐ageing effects with potential pro‐tumorigenic outcomes.
FIGURE 5.
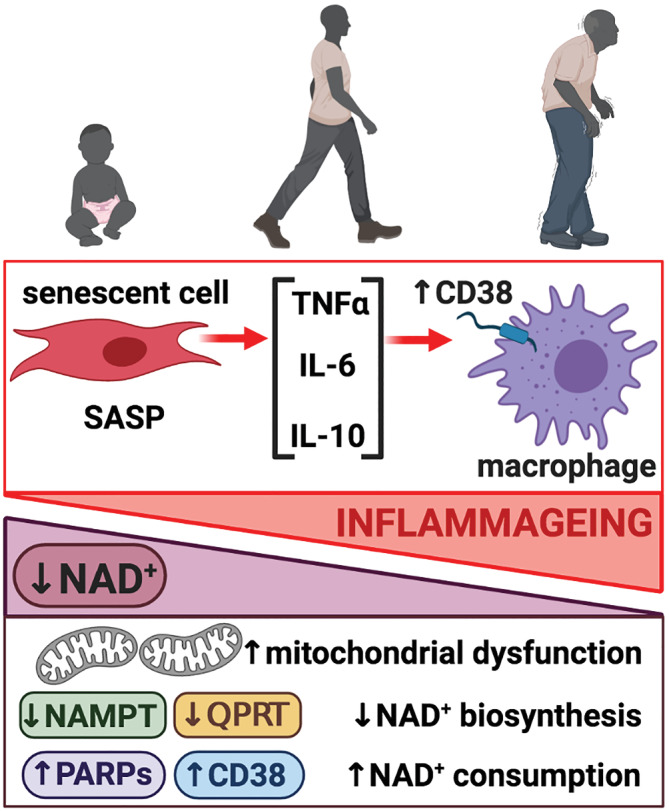
Inflammageing modulates nicotinamide adenine dinucleotide (NAD+) levels. NAD+ levels decrease during ageing as a result of different mechanisms: Mitochondrial dysfunction, decreased expression of enzymes involved in NAD+ biosynthesis (NAMPT, QPRT) and increased expression of NAD+‐consuming enzymes (PARPs, CD38). Inflammageing contributes to the decay in NAD+ levels. Senescent cells secrete TNFα, IL‐6 and IL‐10 as consequence of the senescence‐associated secretory phenotype (SASP). These inflammatory cytokines induce macrophage proliferation and increase expression of the NAD+‐consuming enzyme CD38, which in turn contribute to NAD+ loss during ageing
1.6. Conclusions and perspectives
To summarise, the consumption and biosynthetic pathways induce changes in the NAD+ levels that, subsequently, regulate multiple cellular functions through the modulation of the activity of NAD+‐dependent enzymes and redox reactions. The data discussed here highlight the biological relevance of NAD+ metabolism in normal and altered immune response. While most of the studies show that increasing NAD+ levels have anti‐inflammatory effects, other research also indicates that some strategies to increase NAD+ may increase activation and effector function in immune cells and thus, have deleterious effects in the context of autoimmune diseases. These studies suggest that the moment of interference with NAD+ metabolism (i.e., activation vs. effector phase or chronic inflammation) may result in different outcomes. Therefore, stimuli timing, dosage and the resulting cellular metabolic state, together with the form of NAD+ supplementation (i.e., NAD+ precursors vs. biosynthesis activation) contribute to the global response to increased NAD+. Moreover, NAD+ stoichiometric requirements for the different reactions and signalling pathways are largely unknown and for example, increased NAD+ may not equally affect redox reactions and NAD+‐regulated enzymes. Further research is required to dissect NAD+ requirements during immune cell activation and function.
In addition to its role in the regulation of the immune response, NAD+ metabolism is also emerging as a key modulator of immune cell exhaustion and tumour biology, with a relevant role in immunotherapy. T cell exhaustion facilitates tumour immune evasion, and the rescue of this exhaustion is the aim of inhibitory checkpoint therapies (anti‐PD‐1/PDL1) (Franco et al., 2020). Recently, it has been shown that supplementation with NAD+ precursors improved responsiveness to anti‐PD‐1 treatment in melanoma and colon tumour growth models (Yu et al., 2020) and sensitised immunotherapy‐resistant tumours to anti‐PD‐L1 therapy (Lv et al., 2020). Intratumoral inhibition of NAMPT also potentiated anti‐PD‐1 therapy and increased the survival of glioblastoma‐bearing mice (Li et al., 2020), and these strategies could be of interest in the context of CAR‐T cell therapies, as exhaustion of transferred CAR‐T cells impairs their effector function and persistence (Poorebrahim et al., 2020). Moreover, recent findings have highlighted the crosstalk between NAD+ levels, inflammageing and senescence. For example, NR supplementation reduced neuroinflammation in mouse models of Alzehimer's disease, by the clearance of damaged mitochondria and inhibition of the NLRP3 inflammasome (Fang, 2019; Lautrup et al., 2019). Thus, strategies aimed to increase NAD+ levels provide a promising therapeutic strategy for raising immunotherapy and for the treatment of neuroinflammatory and neurodegenerative disorders.
1.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY (http://www.guidetopharmacology.org) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski et al., 2019; Alexander, Fabbro et al., 2019a, b; Alexander, Kelly et al., 2019a, b; Alexander, Mathie et al., 2019).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
The authors acknowledge all members from María N. Navarro and María Mittelbrunn labs for critical discussion during the preparation of the manuscript. Figures have been created with BioRender.com. The figures were exported under a paid subscription. This work was supported by grants from the Spanish Ministry of Science and Innovation (PID2019‐110511RB‐I00) to M.N.N, and the H2020‐EU.1.1, European Research Council (ERC‐2016‐StG 715322‐EndoMitTalk), Fondo de Investigación Sanitaria del Instituto de Salud Carlos III (PI19/855) to M.M, Fondo Europeo de Desarrollo Regional (FEDER) to M.M and M.N.N. M.M.GH was funded by the Spanish Ministry of Science, Innovation and Universities (FPU19/02576). M.M. is supported by the Miguel Servet program (CPII19/00014, Fundación de Investigación del Hospital 12 de Octubre). Institutional grants from the Fundación Ramón Areces and Banco de Santander to the CBMSO are also acknowledged.
Navarro, M. N. , Gómez de las Heras, M. M. , & Mittelbrunn, M. (2022). Nicotinamide adenine dinucleotide metabolism in the immune response, autoimmunity and inflammageing. British Journal of Pharmacology, 179(9), 1839–1856. 10.1111/bph.15477
Funding information Spanish Ministry of Science and Innovation, Grant/Award Number: PID2019‐110511RB‐I00 to M.N.N; Spanish Ministry of Science, Innovation and Universities, Grant/Award Number: FPU19/02576 to M.M.GH; H2020 European Research Council, Grant/Award Number: ERC‐2016‐StG 715322‐EndoMitTalk to M.M; Fondo de Investigación Sanitaria del Instituto de Salud Carlos III, Grant/Award Number: PI19/855 to M.M; Miguel Servet program, Grant/Award Number: CPII19/00014 to M.M; Fondo Europeo de Desarrollo Regional (FEDER) to M.M and M.N.N; Banco de Santander; Fundación Ramón Areces
Contributor Information
Maria N. Navarro, Email: marian.navarro@cbm.csic.es.
Maria Mittelbrunn, Email: mmittelbrunn@cbm.csic.es.
DATA AVAILABILITY STATEMENT
Data sharing not applicable—no new data generated.
REFERENCES
- Abdellatif, M. , Trummer‐Herbst, V. , Koser, F. , Durand, S. , Adão, R. , Vasques‐Nóvoa, F. , Freundt, J. K. , Voglhuber, J. , Pricolo, M. R. , Kasa, M. , & Türk, C. (2021). Nicotinamide for the treatment of heart failure with preserved ejection fraction. Science Translational Medicine, 13, eabd7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriouch, S. , Hubert, S. , Pechberty, S. , Koch‐Nolte, F. , Haag, F. , & Seman, M. (2007). NAD+ released during inflammation participates in T cell homeostasis by inducing ART2‐mediated death of naive T cells in vivo. Journal of Immunology, 179, 186–194. [DOI] [PubMed] [Google Scholar]
- Adriouch, S. , Ohlrogge, W. , Haag, F. , Koch‐Nolte, F. , & Seman, M. (2001). Rapid induction of naive T cell apoptosis by ecto‐nicotinamide adenine dinucleotide: Requirement for mono (ADP‐Ribosyl)transferase 2 and a downstream effector. Journal of Immunology, 167, 196–203. 10.4049/jimmunol.167.1.196 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Cidlowski, J. A. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear hormone receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019a). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Catalytic receptors. British Journal of Pharmacology, 176, S247–S296. 10.1111/bph.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019b). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019a). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology, 176, S397–S493. 10.1111/bph.14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Buneman, O. P. , Cidlowski, J. A. , Christopoulos, A. , Davenport, A. P. , Fabbro, D. , Spedding, M. , Striessnig, J. , Davies, J. A. , & CGTP Collaborators . (2019b). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Other Protein Targets. British Journal of Pharmacology, 176, S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Sharman, J. L. , Southan, C. , Davies, J. A. , & CGTP Collaborators . (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology, 176, S142–S228. 10.1111/bph.14749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida, L. , Dhillon‐LaBrooy, A. , Castro, C. N. , Adossa, N. , Carriche, G. M. , Guderian, M. , Lippens, S. , Dennerlein, S. , Hesse, C. , Lambrecht, B. N. , & Berod, L. (2020). Ribosome‐targeting antibiotics impair T cell effector function and ameliorate autoimmunity by blocking mitochondrial protein synthesis. Immunity, 54, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelin, A. , Gil‐de‐Gómez, L. , Dahiya, S. , Jiao, J. , Guo, L. , Levine, M. H. , Wang, Z. , Quinn, W. J. III , Kopinski, P. K. , Wang, L. , & Akimova, T. (2017). Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metabolism, 25, 1282–1293.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann, N. , & Finlay, D. K. (2016). Metabolic regulation of immune responses: Therapeutic opportunities. The Journal of Clinical Investigation, 126, 2031–2039. 10.1172/JCI83005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aswad, F. , Kawamura, H. , & Dennert, G. (2005). High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: A role for P2X7 receptors. The Journal of Immunology, 175, 3075–3083. [DOI] [PubMed] [Google Scholar]
- Audrito, V. , Managò, A. , Gaudino, F. , Sorci, L. , Messana, V. G. , Raffaelli, N. , & Deaglio, S. (2019). NAD‐biosynthetic and consuming enzymes as central players of metabolic regulation of innate and adaptive immune responses in cancer. Frontiers in Immunology, 10, 1720. 10.3389/fimmu.2019.01720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audrito, V. , Messana, V. G. , & Deaglio, S. (2020). NAMPT and NAPRT: Two metabolic enzymes with key roles in inflammation. Frontiers in Oncology, 10, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli, F. , Acín‐Pérez, R. , Villarroya‐Beltrí, C. , Mazzeo, C. , Nuñez‐Andrade, N. , Gabandé‐Rodriguez, E. , Ledesma, M. D. , Blázquez, A. , Martin, M. A. , Falcón‐Pérez, J. M. , & Redondo, J. M. (2015). Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metabolism, 22, 485–498. 10.1016/j.cmet.2015.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier, U. H. , Wang, L. , Bhatti, T. R. , Liu, Y. , Han, R. , Ge, G. , & Hancock, W. W. (2011). Sirtuin‐1 targeting promotes Foxp3+ T‐regulatory cell function and prolongs allograft survival. Molecular and Cellular Biology, 31, 1022–1029. 10.1128/MCB.01206-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bheda, P. , Jing, H. , Wolberger, C. , & Lin, H. (2016). The substrate specificity of Sirtuins. Annual Review of Biochemistry, 85, 405–429. 10.1146/annurev-biochem-060815-014537 [DOI] [PubMed] [Google Scholar]
- Bird, J. G. , Basu, U. , Kuster, D. , Ramachandran, A. , Grudzien‐Nogalska, E. , Towheed, A. , Wallace, D. C. , Kiledjian, M. , Temiakov, D. , Patel, S. S. , & Ebright, R. H. (2018). Highly efficient 5′ capping of mitochondrial RNA with NAD+ and NADH by yeast and human mitochondrial RNA polymerase. eLife, 7, e42179. 10.7554/eLife.42179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacher, E. , Bashiardes, S. , Shapiro, H. , Rothschild, D. , Mor, U. , Dori‐Bachash, M. , Kleimeyer, C. , Moresi, C. , Harnik, Y. , Zur, M. , Zabari, M. , Brik, R. B. Z. , Kviatcovsky, D. , Zmora, N. , Cohen, Y. , Bar, N. , Levi, I. , Amar, N. , Mehlman, T. , … Elinav, E. (2019). Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature, 572, 474–480. 10.1038/s41586-019-1443-5 [DOI] [PubMed] [Google Scholar]
- Braidy, N. , Lim, C. K. , Grant, R. , Brew, B. J. , & Guillemin, G. J. (2013). Serum nicotinamide adenine dinucleotide levels through disease course in multiple sclerosis. Brain Research, 1537, 267–272. [DOI] [PubMed] [Google Scholar]
- Bruzzone, S. , Fruscione, F. , Morando, S. , Ferrando, T. , Poggi, A. , Garuti, A. , D'Urso, A. , Selmo, M. , Benvenuto, F. , Cea, M. , & Zoppoli, G. (2009). Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS ONE, 4, e7897. 10.1371/journal.pone.0007897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone, S. , Moreschi, I. , Guida, L. , Usai, C. , Zocchi, E. , & De Flora, A. (2006). Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. The Biochemical Journal, 393, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, M. D. , Sowell, R. T. , Kaech, S. M. , & Pearce, E. L. (2017). Metabolic instruction of immunity. Cell, 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, A. M. , Castoldi, A. , Sanin, D. E. , Flachsmann, L. J. , Field, C. S. , Puleston, D. J. , Kyle, R. L. , Patterson, A. E. , Hässler, F. , Buescher, J. M. , & Kelly, B. (2019). Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species‐mediated DNA damage. Nature Immunology, 20, 420–432. 10.1038/s41590-019-0336-y [DOI] [PubMed] [Google Scholar]
- Caruso, R. , Lo, B. C. , & Núñez, G. (2020). Host‐microbiota interactions in inflammatory bowel disease. Nature Reviews. Immunology, 20, 411–426. 10.1038/s41577-019-0268-7 [DOI] [PubMed] [Google Scholar]
- Caruso, R. , Marafini, I. , Franzè, E. , Stolfi, C. , Zorzi, F. , Monteleone, I. , Caprioli, F. , Colantoni, A. , Sarra, M. , Sedda, S. , & Biancone, L. (2014). Defective expression of SIRT1 contributes to sustain inflammatory pathways in the gut. Mucosal Immunology, 7, 1467–1479. 10.1038/mi.2014.35 [DOI] [PubMed] [Google Scholar]
- Certo, M. , Tsai, C.‐H. , Pucino, V. , Ho, P.‐C. , & Mauro, C. (2021). Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nature Reviews Immunology, 21(3), 151–161. [DOI] [PubMed] [Google Scholar]
- Chadha, S. , Wang, L. , Hancock, W. W. , & Beier, U. H. (2019). Sirtuin‐1 in immunotherapy: A Janus‐headed target. Journal of Leukocyte Biology, 106, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalkiadaki, A. , & Guarente, L. (2015). The multifaceted functions of sirtuins in cancer. Nature Reviews. Cancer, 15, 608–624. 10.1038/nrc3985 [DOI] [PubMed] [Google Scholar]
- Chen, S.‐H. , & Yu, X. (2019). Human DNA ligase IV is able to use NAD+ as an alternative adenylation donor for DNA ends ligation. Nucleic Acids Research, 47, 1321–1334. 10.1093/nar/gky1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini, C. C. S. , Peclat, T. R. , Warner, G. M. , Kashyap, S. , Espindola‐Netto, J. M. , de Oliveira, G. C. , Gomez, L. S. , Hogan, K. A. , Tarragó, M. G. , Puranik, A. S. , & Agorrody, G. (2020). CD38 ecto‐enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nature Metabolism, 2, 1284–1304. 10.1038/s42255-020-00298-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockayne, D. A. , Muchamuel, T. , Grimaldi, J. C. , Muller‐Steffner, H. , Randall, T. D. , Lund, F. E. , Murray, R. , Schuber, F. , & Howard, M. C. (1998). Mice deficient for the ecto‐nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood, 92, 1324–1333. 10.1182/blood.V92.4.1324 [DOI] [PubMed] [Google Scholar]
- Cohen, M. S. , & Chang, P. (2018). Insights into the biogenesis, function, and regulation of ADP‐ribosylation. Nature Chemical Biology, 14, 236–243. 10.1038/nchembio.2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, G. , Clemente, N. , Zito, A. , Bracci, C. , Colombo, F. S. , Sangaletti, S. , Jachetti, E. , Ribaldone, D. G. , Caviglia, G. P. , Pastorelli, L. , & De Andrea, M. (2020). Neutralization of extracellular NAMPT (nicotinamide phosphoribosyltransferase) ameliorates experimental murine colitis. Journal of Molecular Medicine (Berlin, Germany), 98, 595–612. 10.1007/s00109-020-01892-0 [DOI] [PubMed] [Google Scholar]
- Compston, A. , & Coles, A. (2008). Multiple sclerosis. Lancet, 372, 1502–1517. 10.1016/S0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- Covarrubias, A. J. , Kale, A. , Perrone, R. , Lopez‐Dominguez, J. A. , Pisco, A. O. , Kasler, H. G. , Schmidt, M. S. , Heckenbach, I. , Kwok, R. , Wiley, C. D. , & Wong, H. S. (2020). Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nature Metabolism, 2, 1265–1283. 10.1038/s42255-020-00305-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias, A. J. , Perrone, R. , Grozio, A. , & Verdin, E. (2021). NAD+ metabolism and its roles in cellular processes during ageing. Nature Reviews. Molecular Cell Biology, 22(2), 119–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendrou, C. A. , Fugger, L. , & Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nature Reviews. Immunology, 15, 545–558. 10.1038/nri3871 [DOI] [PubMed] [Google Scholar]
- Desdín‐Micó, G. , Soto‐Heredero, G. , Aranda, J. F. , Oller, J. , Carrasco, E. , Gabandé‐Rodríguez, E. , Blanco, E. M. , Alfranca, A. , Cussó, L. , Desco, M. , & Ibañez, B. (2020). T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science, 368, 1371–1376. 10.1126/science.aax0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet, N. , Trammell, S. A. J. , Tannous, C. , Deloux, R. , Piquereau, J. , Mougenot, N. , Gouge, A. , Gressette, M. , Manoury, B. , Blanc, J. , & Breton, M. (2018). Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation, 137, 2256–2273. 10.1161/CIRCULATIONAHA.116.026099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin, C. , Ryan, T. A. J. , & O'Neill, L. A. J. (2021). Modification of proteins by metabolites in immunity. Immunity, 54(1), 19–31. [DOI] [PubMed] [Google Scholar]
- Elkhal, A. , Rodriguez Cetina Biefer, H. , Heinbokel, T. , Uehara, H. , Quante, M. , Seyda, M. , Schuitenmaker, J. M. , Krenzien, F. , Camacho, V. , de la Fuente, M. A. , Ghiran, I. , & Tullius, S. G. (2016). NAD+ regulates Treg cell fate and promotes allograft survival via a systemic IL‐10 production that is CD4+ CD25+ Foxp3+ T cells independent. Scientific Reports, 6, 22325. 10.1038/srep22325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman, K. , Summers, D. W. , Sasaki, Y. , Mao, X. , DiAntonio, A. , & Milbrandt, J. (2017). The SARM1 toll/interleukin‐1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron, 93, 1334–1343.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, E. F. (2019). Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy, 15, 1112–1114. 10.1080/15548627.2019.1596497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara, G. , Benzi, A. , Sturla, L. , Marubbi, D. , Frumento, D. , Spinelli, S. , Abbotto, E. , Ivaldi, F. , von Holtey, M. , Murone, M. , Nencioni, A. , Uccelli, A. , & Bruzzone, S. (2020). Sirt6 inhibition delays the onset of experimental autoimmune encephalomyelitis by reducing dendritic cell migration. Journal of Neuroinflammation, 17, 228. 10.1186/s12974-020-01906-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco, F. , Jaccard, A. , Romero, P. , Yu, Y.‐R. , & Ho, P.‐C. (2020). Metabolic and epigenetic regulation of T‐cell exhaustion. Nature Metabolism, 2, 1001–1012. 10.1038/s42255-020-00280-9 [DOI] [PubMed] [Google Scholar]
- Gabandé‐Rodríguez, E. , Gómez de las Heras, M. M. , & Mittelbrunn, M. (2019). Control of inflammation by calorie restriction mimetics: On the crossroad of autophagy and mitochondria. Cell, 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli, U. , Colombo, G. , Travelli, C. , Tron, G. C. , Genazzani, A. A. , & Grolla, A. A. (2020). Recent advances in NAMPT inhibitors: A novel immunotherapic strategy. Frontiers in Pharmacology, 11, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, B. , Kong, Q. , Kemp, K. , Zhao, Y.‐S. , & Fang, D. (2012). Analysis of sirtuin 1 expression reveals a molecular explanation of IL‐2‐mediated reversal of T‐cell tolerance. Proceedings of the National Academy of Sciences of the United States of America, 109, 899–904. 10.1073/pnas.1118462109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Rodríguez, S. , Rosal‐Vela, A. , Botta, D. , Cumba Garcia, L. M. , Zumaquero, E. , Prados‐Maniviesa, V. , Cerezo‐Wallis, D. , Lo Buono, N. , Robles‐Guirado, J. Á. , Guerrero, S. , González‐Paredes, E. , Andrés‐León, E. , Corbí, Á. , Mack, M. , Koch‐Nolte, F. , Merino, R. , Zubiaur, M. , Lund, F. E. , & Sancho, J. (2018). CD38 promotes pristane‐induced chronic inflammation and increases susceptibility to experimental lupus by an apoptosis‐driven and TRPM2‐dependent mechanism. Scientific Reports, 8, 3357. 10.1038/s41598-018-21337-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner, R. R. , Klepsch, V. , Macheiner, S. , Arnhard, K. , Adolph, T. E. , Grander, C. , Wieser, V. , Pfister, A. , Moser, P. , Hermann‐Kleiter, N. , & Baier, G. (2018). NAD metabolism fuels human and mouse intestinal inflammation. Gut, 67, 1813–1823. 10.1136/gutjnl-2017-314241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner, R. R. , Macheiner, S. , Reider, S. , Siegmund, K. , Grabherr, F. , Mayr, L. , Texler, B. , Moser, P. , Effenberger, M. , Schwaighofer, H. , & Moschen, A. R. (2020). Targeting NAD immunometabolism limits severe graft‐versus‐host disease and has potent antileukemic activity. Leukemia, 34, 1885–1897. 10.1038/s41375-020-0709-0 [DOI] [PubMed] [Google Scholar]
- Gerth, A. , Nieber, K. , Oppenheimer, N. J. , & Hauschildt, S. (2004). Extracellular NAD+ regulates intracellular free calcium concentration in human monocytes. The Biochemical Journal, 382, 849–856. 10.1042/BJ20040979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone, P. J. , Alcamo, A. A. , Schanbacher, B. L. , Nankervis, C. A. , Besner, G. E. , & Bauer, J. A. (2011). Poly (ADP‐ribose) polymerase‐1: A novel therapeutic target in necrotizing enterocolitis. Pediatric Research, 70, 67–71. 10.1203/PDR.0b013e31821928ff [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi, E. , Agrimi, G. , Goldmann, U. , Fiume, G. , Lindinger, S. , Sedlyarov, V. , Srndic, I. , Gürtl, B. , Agerer, B. , Kartnig, F. , & Scarcia, P. (2020). Epistasis‐driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nature Communications, 11, 6145. 10.1038/s41467-020-19871-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes, A. P. , Price, N. L. , Ling, A. J. Y. , Moslehi, J. J. , Montgomery, M. K. , Rajman, L. , White, J. P. , Teodoro, J. S. , Wrann, C. D. , Hubbard, B. P. , & Mercken, E. M. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear‐mitochondrial communication during aging. Cell, 155, 1624–1638. 10.1016/j.cell.2013.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag, F. , Freese, D. , Scheublein, F. , Ohlrogge, W. , Adriouch, S. , Seman, M. , & Koch‐Nolte, F. (2002). T cells of different developmental stages differ in sensitivity to apoptosis induced by extracellular NAD. Developmental Immunology, 9, 197–202. 10.1080/10446670310001593514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X. , Uchiyama, T. , Sappington, P. L. , Yaguchi, A. , Yang, R. , Fink, M. P. , & Delude, R. L. (2003). NAD+ ameliorates inflammation‐induced epithelial barrier dysfunction in cultured enterocytes and mouse ileal mucosa. The Journal of Pharmacology and Experimental Therapeutics, 307, 443–449. 10.1124/jpet.103.056556 [DOI] [PubMed] [Google Scholar]
- Hecker, A. , Küllmar, M. , Wilker, S. , Richter, K. , Zakrzewicz, A. , Atanasova, S. , Mathes, V. , Timm, T. , Lerner, S. , Klein, J. , Kaufmann, A. , Bauer, S. , Padberg, W. , Kummer, W. , Janciauskiene, S. , Fronius, M. , Schweda, E. K. H. , Lochnit, G. , & Grau, V. (2015). Phosphocholine‐modified macromolecules and canonical nicotinic agonists inhibit ATP‐induced IL‐1β release. Journal of Immunology, 195, 2325–2334. 10.4049/jimmunol.1400974 [DOI] [PubMed] [Google Scholar]
- Hiller, S. D. , Heldmann, S. , Richter, K. , Jurastow, I. , Küllmar, M. , Hecker, A. , Wilker, S. , Fuchs‐Moll, G. , Manzini, I. , Schmalzing, G. , & Kummer, W. (2018). β‐Nicotinamide adenine dinucleotide (β‐NAD) inhibits ATP‐dependent IL‐1β release from human Monocytic cells. International Journal of Molecular Sciences, 19, 1126. 10.3390/ijms19041126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan, K. A. , Chini, C. C. S. , & Chini, E. N. (2019). The multi‐faceted ecto‐enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Frontiers in Immunology, 10, 1187. 10.3389/fimmu.2019.01187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert, S. , Rissiek, B. , Klages, K. , Huehn, J. , Sparwasser, T. , Haag, F. , Koch‐Nolte, F. , Boyer, O. , Seman, M. , & Adriouch, S. (2010). Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2–P2X7 pathway. The Journal of Experimental Medicine, 207, 2561–2568. 10.1084/jem.20091154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi, M. , & Guarente, L. (2016). mTORC1 and SIRT1 cooperate to foster expansion of gut adult stem cells during calorie restriction. Cell, 166, 436–450. 10.1016/j.cell.2016.05.044 [DOI] [PubMed] [Google Scholar]
- Igarashi, M. , Miura, M. , Williams, E. , Jaksch, F. , Kadowaki, T. , Yamauchi, T. , & Guarente, L. (2019). NAD+ supplementation rejuvenates aged gut adult stem cells. Aging Cell, 18, e12935. 10.1111/acel.12935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janney, A. , Powrie, F. , & Mann, E. H. (2020). Host‐microbiota maladaptation in colorectal cancer. Nature, 585, 509–517. 10.1038/s41586-020-2729-3 [DOI] [PubMed] [Google Scholar]
- Jeng, M. Y. , Hull, P. A. , Fei, M. , Kwon, H.‐S. , Tsou, C.‐L. , Kasler, H. , Ng, C. P. , Gordon, D. E. , Johnson, J. , Krogan, N. , Verdin, E. , & Ott, M. (2018). Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. The Journal of Experimental Medicine, 215, 51–62. 10.1084/jem.20161066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Liu, T. , Lee, C.‐H. , Chang, Q. , Yang, J. , & Zhang, Z. (2020). The NAD+‐mediated self‐inhibition mechanism of pro‐neurodegenerative SARM1. Nature, 588, 658–663. 10.1038/s41586-020-2862-z [DOI] [PubMed] [Google Scholar]
- Kahl, S. , Nissen, M. , Girisch, R. , Duffy, T. , Leiter, E. H. , Haag, F. , & Koch‐Nolte, F. (2000). Metalloprotease‐mediated shedding of enzymatically active mouse ecto‐ADP‐ribosyltransferase ART2.2 upon T cell activation. The Journal of Immunology, 165, 4463–4469. 10.4049/jimmunol.165.8.4463 [DOI] [PubMed] [Google Scholar]
- Kaneko, S. , Wang, J. , Kaneko, M. , Yiu, G. , Hurrell, J. M. , Chitnis, T. , Khoury, S. J. , & He, Z. (2006). Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. The Journal of Neuroscience, 26, 9794–9804. 10.1523/JNEUROSCI.2116-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar, A. , Mehrotra, S. , & Chatterjee, S. (2020). CD38: T cell Immuno‐metabolic modulator. Cell, 9, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, Y. , Zhang, J. , Lv, X. , Zeng, X. , & Ba, X. (2019). Novel insights into PARPs in gene expression: Regulation of RNA metabolism. Cellular and Molecular Life Sciences, 76, 3283–3299. 10.1007/s00018-019-03120-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus, W. L. (2015). PARPs and ADP‐Ribosylation come into focus. Molecular Cell, 58, 901. 10.1016/j.molcel.2015.06.014 [DOI] [PubMed] [Google Scholar]
- Krebs, C. , Adriouch, S. , Braasch, F. , Koestner, W. , Leiter, E. H. , Seman, M. , Lund, F. E. , Oppenheimer, N. , Haag, F. , & Koch‐Nolte, F. (2005). CD38 controls ADP‐ribosyltransferase‐2‐catalyzed ADP‐ribosylation of T cell surface proteins. Journal of Immunology, 174, 3298–3305. 10.4049/jimmunol.174.6.3298 [DOI] [PubMed] [Google Scholar]
- Kwon, H.‐S. , Lim, H. W. , Wu, J. , Schnölzer, M. , Verdin, E. , & Ott, M. (2012). Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. The Journal of Immunology, 188, 2712–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larabi, A. , Barnich, N. , & Nguyen, H. T. T. (2020). New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy, 16, 38–51. 10.1080/15548627.2019.1635384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautrup, S. , Sinclair, D. A. , Mattson, M. P. , & Fang, E. F. (2019). NAD+ in brain aging and neurodegenerative disorders. Cell Metabolism, 30, 630–655. 10.1016/j.cmet.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber, A. , Hontecillas, R. , Tubau‐Juni, N. , Zoccoli‐Rodriguez, V. , Abedi, V. , & Bassaganya‐Riera, J. (2018). NLRX1 modulates Immunometabolic mechanisms controlling the host‐gut microbiota interactions during inflammatory bowel disease. Frontiers in Immunology, 9, 363. 10.3389/fimmu.2018.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. C. , & Zhao, Y. J. (2019). Resolving the topological enigma in Ca2+ signaling by cyclic ADP‐ribose and NAADP. The Journal of Biological Chemistry, 294, 19831–19843. 10.1074/jbc.REV119.009635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Kirtane, A. R. , Kiyokawa, J. , Nagashima, H. , Lopes, A. , Tirmizi, Z. A. , Lee, C. K. , Traverso, G. , Cahill, D. P. , & Wakimoto, H. (2020). Local targeting of NAD+ salvage pathway alters the immune tumor microenvironment and enhances checkpoint immunotherapy in glioblastoma. Cancer Research, 80, 5024–5034. 10.1158/0008-5472.CAN-20-1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, H. W. , Kang, S. G. , Ryu, J. K. , Schilling, B. , Fei, M. , Lee, I. S. , Kehasse, A. , Shirakawa, K. , Yokoyama, M. , Schnölzer, M. , Kasler, H. G. , Kwon, H. S. , Gibson, B. W. , Sato, H. , Akassoglou, K. , Xiao, C. , Littman, D. R. , Ott, M. , & Verdin, E. (2015). SIRT1 deacetylates RORγt and enhances Th17 cell generation. The Journal of Experimental Medicine, 212, 607–617. 10.1084/jem.20132378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. , Bu, H.‐F. , Geng, H. , De Plaen, I. G. , Gao, C. , Wang, P. , Kurowski, J. A. , Yang, H. , Qian, J. , & Tan, X. D. (2017). Sirtuin‐6 preserves R‐spondin‐1 expression and increases resistance of intestinal epithelium to injury in mice. Molecular Medicine, 23, 272–284. 10.2119/molmed.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z.‐X. , Azhipa, O. , Okamoto, S. , Govindarajan, S. , & Dennert, G. (2001). Extracellular nicotinamide adenine dinucleotide induces T cell apoptosis in vivo and in vitro. The Journal of Immunology, 167, 4942–4947. 10.4049/jimmunol.167.9.4942 [DOI] [PubMed] [Google Scholar]
- Lo Sasso, G. , Menzies, K. J. , Mottis, A. , Piersigilli, A. , Perino, A. , Yamamoto, H. , Schoonjans, K. , & Auwerx, J. (2014). SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE, 9, e103573. 10.1371/journal.pone.0103573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Sasso, G. , Ryu, D. , Mouchiroud, L. , Fernando, S. C. , Anderson, C. L. , Katsyuba, E. , Piersigilli, A. , Hottiger, M. O. , Schoonjans, K. , & Auwerx, J. (2014). Loss of Sirt1 function improves intestinal anti‐bacterial defense and protects from colitis‐induced colorectal cancer. PLoS ONE, 9, e102495. 10.1371/journal.pone.0102495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo, T. S. , Eller, J. M. , Lu, M.‐J. , Niere, M. , Raith, F. , Perry, C. , Bornstein, M. R. , Oliphint, P. , Wang, L. , McReynolds, M. R. , & Migaud, M. E. (2020). SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature, 588, 174–179. 10.1038/s41586-020-2741-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv, H. , Lv, G. , Chen, C. , Zong, Q. , Jiang, G. , Ye, D. , Cui, X. , He, Y. , Xiang, W. , Han, Q. , & Tang, L. (2020). NAD+ metabolism maintains inducible PD‐L1 expression to drive tumor immune evasion. Cell Metabolism, 33, 110–127. [DOI] [PubMed] [Google Scholar]
- Lv, Q. , Wang, K. , Qiao, S. , Yang, L. , Xin, Y. , Dai, Y. , & Wei, Z. (2018). Norisoboldine, a natural AhR agonist, promotes Treg differentiation and attenuates colitis via targeting glycolysis and subsequent NAD+/SIRT1/SUV39H1/H3K9me3 signaling pathway. Cell Death & Disease, 9, 258. 10.1038/s41419-018-0297-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnone, M. , Bauer, I. , Poggi, A. , Mannino, E. , Sturla, L. , Brini, M. , Zocchi, E. , de Flora, A. , Nencioni, A. , & Bruzzone, S. (2012). NAD+ levels control Ca2+ store replenishment and mitogen‐induced increase of cytosolic Ca2+ by cyclic ADP‐ribose‐dependent TRPM2 channel gating in human T lymphocytes. The Journal of Biological Chemistry, 287, 21067–21081. 10.1074/jbc.M111.324269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalonga, J. , Glaria, E. , Bresque, M. , Escande, C. , Carbó, J. M. , Kiefer, K. , Vicente, R. , León, T. E. , Beceiro, S. , Pascual‐García, M. , & Serret, J. (2017). The nuclear receptor LXR limits bacterial infection of host macrophages through a mechanism that impacts cellular NAD metabolism. Cell Reports, 18, 1241–1255. 10.1016/j.celrep.2017.01.007 [DOI] [PubMed] [Google Scholar]
- Michaudel, C. , & Sokol, H. (2020). The gut microbiota at the service of immunometabolism. Cell Metabolism, 32, 514–523. 10.1016/j.cmet.2020.09.004 [DOI] [PubMed] [Google Scholar]
- Mills, E. L. , Kelly, B. , & O'Neill, L. A. J. (2017). Mitochondria are the powerhouses of immunity. Nature Immunology, 18, 488–498. 10.1038/ni.3704 [DOI] [PubMed] [Google Scholar]
- Mills, K. F. , Yoshida, S. , Stein, L. R. , Grozio, A. , Kubota, S. , Sasaki, Y. , Redpath, P. , Migaud, M. E. , Apte, R. S. , Uchida, K. , & Yoshino, J. (2016). Long‐term administration of nicotinamide mononucleotide mitigates age‐associated physiological decline in mice. Cell Metabolism, 24, 795–806. 10.1016/j.cmet.2016.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas, P. S. , Liu, L. , Moon, P. K. , Joshi, A. U. , Dove, C. , Mhatre, S. , Contrepois, K. , Wang, Q. , Lee, B. A. , Coronado, M. , Bernstein, D. , Snyder, M. P. , Migaud, M. , Majeti, R. , Mochly‐Rosen, D. , Rabinowitz, J. D. , & Andreasson, K. I. (2019). Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nature Immunology, 20, 50–63. 10.1038/s41590-018-0255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, S. J. , Bernier, M. , Aon, M. A. , Cortassa, S. , Kim, E. Y. , Fang, E. F. , Palacios, H. H. , Ali, A. , Navas‐Enamorado, I. , Di Francesco, A. , & Kaiser, T. A. (2018). Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metabolism, 27, 667–676e4. 10.1016/j.cmet.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacarelli, T. , Lau, L. , Fukumoto, T. , Zundell, J. , Fatkhutdinov, N. , Wu, S. , Aird, K. M. , Iwasaki, O. , Kossenkov, A. V. , Schultz, D. , Noma, K. I. , Baur, J. A. , Schug, Z. , Tang, H. Y. , Speicher, D. W. , David, G. , & Zhang, R. (2019). NAD+ metabolism governs the proinflammatory senescence‐associated secretome. Nature Cell Biology, 21, 397–407. 10.1038/s41556-019-0287-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer, K. , Bednarz‐Misa, I. , Walecka‐Zacharska, E. , Wierzbicki, J. , Agrawal, A. , Gamian, A. , & Krzystek‐Korpacka, M. (2019). Oversecretion and overexpression of nicotinamide Phosphoribosyltransferase/pre‐B Colony‐enhancing factor/visfatin in inflammatory bowel disease reflects the disease activity, severity of inflammatory response and hypoxia. International Journal of Molecular Sciences, 20, 166. 10.3390/ijms20010166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, J. , Wu, G. D. , Albenberg, L. , & Tomov, V. T. (2017). Gut microbiota and IBD: Causation or correlation? Nature Reviews. Gastroenterology & Hepatology, 14, 573–584. 10.1038/nrgastro.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmagadda, V. K. C. , Makar, T. K. , Chandrasekaran, K. , Sagi, A. R. , Ray, J. , Russell, J. W. , & Bever, C. T. Jr. (2017). SIRT1 and NAD+ precursors: Therapeutic targets in multiple sclerosis a review. Journal of Neuroimmunology, 304, 29–34. 10.1016/j.jneuroim.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning, L. , Shan, G. , Sun, Z. , Zhang, F. , Xu, C. , Lou, X. , Li, S. , Du, H. , Chen, H. , & Xu, G. (2019). Quantitative proteomic analysis reveals the deregulation of nicotinamide adenine dinucleotide metabolism and CD38 in inflammatory bowel disease. BioMed Research International, 2019, 3950628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak, E. C. , de Vries, V. C. , Wasiuk, A. , Ahonen, C. , Bennett, K. A. , Le Mercier, I. , Ha, D. G. , & Noelle, R. J. (2012). Tryptophan hydroxylase‐1 regulates immune tolerance and inflammation. The Journal of Experimental Medicine, 209, 2127–2135. 10.1084/jem.20120408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, A. , Kim, H.‐J. , Oh, G.‐S. , Shen, A. , Lee, S.‐B. , Khadka, D. , Lee, S. , Shim, H. , Yang, S. H. , Cho, E. Y. , & Kwon, K. B. (2015). Dunnione ameliorates cisplatin‐induced small intestinal damage by modulating NAD(+) metabolism. Biochemical and Biophysical Research Communications, 467, 697–703. 10.1016/j.bbrc.2015.10.081 [DOI] [PubMed] [Google Scholar]
- Partida‐Sánchez, S. , Cockayne, D. A. , Monard, S. , Jacobson, E. L. , Oppenheimer, N. , Garvy, B. , Kusser, K. , Goodrich, S. , Howard, M. , Harmsen, A. , Randall, T. D. , & Lund, F. E. (2001). Cyclic ADP‐ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nature Medicine, 7, 1209–1216. 10.1038/nm1101-1209 [DOI] [PubMed] [Google Scholar]
- Partida‐Sánchez, S. , Goodrich, S. , Kusser, K. , Oppenheimer, N. , Randall, T. D. , & Lund, F. E. (2004). Regulation of dendritic cell trafficking by the ADP‐Ribosyl cyclase CD38: Impact on the development of humoral immunity. Immunity, 20, 279–291. 10.1016/S1074-7613(04)00048-2 [DOI] [PubMed] [Google Scholar]
- Patel, C. H. , Leone, R. D. , Horton, M. R. , & Powell, J. D. (2019). Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nature Reviews. Drug Discovery, 18, 669–688. 10.1038/s41573-019-0032-5 [DOI] [PubMed] [Google Scholar]
- Pearce, E. L. , & Pearce, E. J. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity, 38, 633–643. 10.1016/j.immuni.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penberthy, W. T. , & Tsunoda, I. (2009). The importance of NAD in multiple sclerosis. Current Pharmaceutical Design, 15, 64–99. 10.2174/138161209787185751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorebrahim, M. , Melief, J. , de Coaña, Y. P. , Wickström, S. L. , Cid‐Arregui, A. , & Kiessling, R. (2020). Counteracting CAR T cell dysfunction. Oncogene, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucino, V. , Certo, M. , Bulusu, V. , Cucchi, D. , Goldmann, K. , Pontarini, E. , Haas, R. , Smith, J. , Headland, S. E. , Blighe, K. , & Ruscica, M. (2019). Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metabolism, 30, 1055–1074e8. 10.1016/j.cmet.2019.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarona, V. , Zaccarello, G. , Chillemi, A. , Brunetti, E. , Singh, V. K. , Ferrero, E. , Funaro, A. , Horenstein, A. L. , & Malavasi, F. (2013). CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytometry. Part B, Clinical Cytometry, 84, 207–217. 10.1002/cyto.b.21092 [DOI] [PubMed] [Google Scholar]
- Quinn, W. J. , Jiao, J. , TeSlaa, T. , Stadanlick, J. , Wang, Z. , Wang, L. , Akimova, T. , Angelin, A. , Schäfer, P. M. , Cully, M. D. , & Perry, C. (2020). Lactate limits T cell proliferation via the NAD(H) redox state. Cell Reports, 33, 108500. 10.1016/j.celrep.2020.108500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenkovic, D. , & Verdin, E. (2020). Clinical evidence for targeting NAD therapeutically. Pharmaceuticals (Basel), 13, 247. 10.3390/ph13090247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajman, L. , Chwalek, K. , & Sinclair, D. A. (2018). Therapeutic potential of NAD‐boosting molecules: The in vivo evidence. Cell Metabolism, 27, 529–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, M.‐T. , Gu, M.‐L. , Zhou, X.‐X. , Yu, M.‐S. , Pan, H.‐H. , Ji, F. , & Ding, C. Y. (2019). Sirtuin 1 alleviates endoplasmic reticulum stress‐mediated apoptosis of intestinal epithelial cells in ulcerative colitis. World Journal of Gastroenterology, 25, 5800–5813. 10.3748/wjg.v25.i38.5800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roboon, J. , Hattori, T. , Ishii, H. , Takarada‐Iemata, M. , Le, T. M. , Shiraishi, Y. , Ozaki, N. , Yamamoto, Y. , Sugawara, A. , Okamoto, H. , & Higashida, H. (2019). Deletion of CD38 suppresses glial activation and neuroinflammation in a mouse model of demyelination. Frontiers in Cellular Neuroscience, 13, 258. 10.3389/fncel.2019.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani, M. , Sorrentino, V. , Oh, C.‐M. , Li, H. , de Lima, T. I. , Zhang, H. , Shong, M. , & Auwerx, J. (2021). NAD+ boosting reduces age‐associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Reports, 34, 108660. 10.1016/j.celrep.2020.108660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, D. , Zhang, H. , Ropelle, E. R. , Sorrentino, V. , Mázala, D. A. G. , Mouchiroud, L. , Marshall, P. L. , Campbell, M. D. , Ali, A. S. , Knowels, G. M. , & Bellemin, S. (2016). NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Science Translational Medicine, 8, 361ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye‐Knudsen, M. , Mitchell, S. J. , Fang, E. F. , Iyama, T. , Ward, T. , Wang, J. , Dunn, C. A. , Singh, N. , Veith, S. , Hasan‐Olive, M. M. , & Mangerich, A. (2014). A high‐fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metabolism, 20, 840–855. 10.1016/j.cmet.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuplein, F. , Schwarz, N. , Adriouch, S. , Krebs, C. , Bannas, P. , Rissiek, B. , Seman, M. , Haag, F. , & Koch‐Nolte, F. (2009). NAD+ and ATP released from injured cells induce P2X7‐dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. Journal of Immunology, 182, 2898–2908. 10.4049/jimmunol.0801711 [DOI] [PubMed] [Google Scholar]
- Schneider, M. , Schumacher, V. , Lischke, T. , Lücke, K. , Meyer‐Schwesinger, C. , Velden, J. , Koch‐Nolte, F. , & Mittrücker, H. W. (2015). CD38 is expressed on inflammatory cells of the intestine and promotes intestinal inflammation. PLoS ONE, 10, e0126007. 10.1371/journal.pone.0126007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seman, M. , Adriouch, S. , Scheuplein, F. , Krebs, C. , Freese, D. , Glowacki, G. , Deterre, P. , Haag, F. , & Koch‐Nolte, F. (2003). NAD‐induced T cell death: ADP‐ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity, 19, 571–582. 10.1016/S1074-7613(03)00266-8 [DOI] [PubMed] [Google Scholar]
- Shats, I. , Williams, J. G. , Liu, J. , Makarov, M. V. , Wu, X. , Lih, F. B. , Deterding, L. J. , Lim, C. , Xu, X. , Randall, T. A. , & Lee, E. (2020). Bacteria boost mammalian host NAD metabolism by engaging the deamidated biosynthesis pathway. Cell Metabolism, 31, 564–579.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal, A. , & Cheng, C. Y. (2019). Host NAD+ metabolism and infections: Therapeutic implications. International Immunology, 31, 59–67. 10.1093/intimm/dxy068 [DOI] [PubMed] [Google Scholar]
- Soto‐Heredero, G. , Gómez de Las Heras, M. M. , Gabandé‐Rodríguez, E. , Oller, J. , & Mittelbrunn, M. (2020). Glycolysis—A key player in the inflammatory response. The FEBS Journal, 287, 3350–3369. 10.1111/febs.15327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, L. R. , & Imai, S. (2014). Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. The EMBO Journal, 33, 1321–1340. 10.1002/embj.201386917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H. , Wang, J. , Guo, L. , Wang, Y. , Zhang, J. , Wang, J. , Quan, M. , & Li, B. (2020). The combined treatment of NAD+ and atorvastatin ameliorates the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Journal of Neuroimmunology, 350, 577429. [DOI] [PubMed] [Google Scholar]
- Sundaram, G. , Lim, C. K. , Brew, B. J. , & Guillemin, G. J. (2020). Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression. Journal of Neuroinflammation, 17, 176. 10.1186/s12974-020-01844-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talero, E. , Alcaide, A. , Ávila‐Román, J. , García‐Mauriño, S. , Vendramini‐Costa, D. , & Motilva, V. (2016). Expression patterns of sirtuin 1‐AMPK‐autophagy pathway in chronic colitis and inflammation‐associated colon neoplasia in IL‐10‐deficient mice. International Immunopharmacology, 35, 248–256. 10.1016/j.intimp.2016.03.046 [DOI] [PubMed] [Google Scholar]
- Trammell, S. A. J. , Weidemann, B. J. , Chadda, A. , Yorek, M. S. , Holmes, A. , Coppey, L. J. , Obrosov, A. , Kardon, R. H. , Yorek, M. A. , & Brenner, C. (2016). Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Scientific Reports, 6, 26933. 10.1038/srep26933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius, S. G. , Biefer, H. R. C. , Li, S. , Trachtenberg, A. J. , Edtinger, K. , Quante, M. , Krenzien, F. , Uehara, H. , Yang, X. , Kissick, H. T. , & Kuo, W. P. (2014). NAD+ protects against EAE by regulating CD4+ T‐cell differentiation. Nature Communications, 5, 5101. 10.1038/ncomms6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, R. , Saito, Y. , Nogami, K. , Kajiyama, Y. , Suzuki, Y. , Kawase, Y. , Nakaoka, T. , Muramatsu, T. , Kimura, M. , & Saito, H. (2019). Epigenetic silencing of Lgr5 induces senescence of intestinal epithelial organoids during the process of aging. NPJ Aging and Mechanisms of Disease, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gool, F. , Gallí, M. , Gueydan, C. , Kruys, V. , Prevot, P.‐P. , Bedalov, A. , Mostoslavsky, R. , Alt, F. W. , De Smedt, T. , & Leo, O. (2009). Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin‐dependent manner. Nature Medicine, 15, 206–210. 10.1038/nm.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt, J. , Vercoulen, Y. , Guichelaar, T. , Gent, Y. Y. , Beekman, J. M. , van Beekum, O. , Brenkman, A. B. , Hijnen, D. J. , Mutis, T. , Kalkhoven, E. , & Prakken, B. J. (2010). Regulation of Treg functionality by acetylation‐mediated Foxp3 protein stabilization. Blood, 115, 965–974. 10.1182/blood-2009-02-207118 [DOI] [PubMed] [Google Scholar]
- Verdin, E. (2015). NAD+ in aging, metabolism, and neurodegeneration. Science, 350, 1208–1213. 10.1126/science.aac4854 [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhao, C. , Kong, P. , Sun, H. , Sun, Z. , Bian, G. , Sun, Y. , & Guo, L. (2016). Treatment with NAD(+) inhibited experimental autoimmune encephalomyelitis by activating AMPK/SIRT1 signaling pathway and modulating Th1/Th17 immune responses in mice. International Immunopharmacology, 39, 287–294. 10.1016/j.intimp.2016.07.036 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Li, B. , Liu, L. , Zhang, L. , Ma, T. , & Guo, L. (2020). Nicotinamide adenine dinucleotide treatment alleviates the symptoms of experimental autoimmune encephalomyelitis by activating autophagy and inhibiting the NLRP3 inflammasome. International Immunopharmacology, 90, 107092. [DOI] [PubMed] [Google Scholar]
- Wei, J. , Raynor, J. , Nguyen, T.‐L. M. , & Chi, H. (2017). Nutrient and metabolic sensing in T cell responses. Frontiers in Immunology, 8, 247. 10.3389/fimmu.2017.00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman, A. S. , Metukuri, M. R. , Kazgan, N. , Xu, X. , Xu, Q. , Ren, N. S. X. , Czopik, A. , Shanahan, M. T. , Kang, A. , Chen, W. , Azcarate‐Peril, M. A. , Gulati, A. S. , Fargo, D. C. , Guarente, L. , & Li, X. (2017). Intestinal epithelial Sirtuin 1 regulates intestinal inflammation during aging in mice by altering the intestinal microbiota. Gastroenterology, 153, 772–786. 10.1053/j.gastro.2017.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, C. D. , Velarde, M. C. , Lecot, P. , Liu, S. , Sarnoski, E. A. , Freund, A. , Shirakawa, K. , Lim, H. W. , Davis, S. S. , Ramanathan, A. , Gerencser, A. A. , Verdin, E. , & Campisi, J. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metabolism, 23, 303–314. 10.1016/j.cmet.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniacka, A. , Szajerski, P. , Adamus, J. , Gebicki, J. , & Sysa‐Jedrzejowska, A. (2007). In search for new antipsoriatic agents: NAD topical composition. Skin Pharmacology and Physiology, 20, 37–42. 10.1159/000096170 [DOI] [PubMed] [Google Scholar]
- Xie, N. , Zhang, L. , Gao, W. , Huang, C. , Huber, P. E. , Zhou, X. , Li, C. , Shen, G. , & Zou, B. (2020). NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduction and Targeted Therapy, 5, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K. , Guo, Y. , Ping, L. , Qiu, Y. , Liu, Q. , Li, Z. , & Wang, Z. (2020). Protective effects of SIRT6 overexpression against DSS‐induced colitis in mice. Cell, 9, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Monaco, G. , Wong, E. H. , Tan, W. L. W. , Kared, H. , Simoni, Y. , Tan, S. W. , How, W. Z. , Tan, C. T. , Lee, B. T. , & Carbajo, D. (2019). Mapping of γ/δ T cells reveals Vδ2+ T cells resistance to senescence. EBioMedicine, 39, 44–58. 10.1016/j.ebiom.2018.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, C. , Qi, L. , Li, X. , Wang, J. , Yu, J. , Zhou, B. , Guo, C. , Chen, J. , & Zheng, S. (2020). Targeting the NAD+ salvage pathway suppresses APC mutation‐driven colorectal cancer growth and Wnt/β‐catenin signaling via increasing Axin level. Cell Communication and Signaling: CCS, 18, 16. 10.1186/s12964-020-0513-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, T. C. , Britt, J. K. , De Jesús‐Cortés, H. , Lu, Y. , Genova, R. M. , Khan, M. Z. , Voorhees, J. R. , Shao, J. , Katzman, A. C. , Huntington, P. J. , & Wassink, C. (2014). P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Reports, 8, 1731–1740. 10.1016/j.celrep.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, M. , Satoh, A. , Lin, J. B. , Mills, K. F. , Sasaki, Y. , Rensing, N. , Wong, M. , Apte, R. S. , & Imai, S. I. (2019). Extracellular vesicle‐contained eNAMPT delays aging and extends lifespan in mice. Cell Metabolism, 30, 329–342.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino, J. , Baur, J. A. , & Imai, S.‐I. (2018). NAD+ intermediates: The biology and therapeutic potential of NMN and NR. Cell Metabolism, 27, 513–528. 10.1016/j.cmet.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y.‐R. , Imrichova, H. , Wang, H. , Chao, T. , Xiao, Z. , Gao, M. , Rincon‐Restrepo, M. , Franco, F. , Genolet, R. , Cheng, W. C. , & Jandus, C. (2020). Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nature Immunology, 21, 1540–1551. 10.1038/s41590-020-0793-3 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Menzies, K. J. , & Auwerx, J. (2018). The role of mitochondria in stem cell fate and aging. Development, 145, dev143420. 10.1242/dev.143420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Ryu, D. , Wu, Y. , Gariani, K. , Wang, X. , Luan, P. , D'Amico, D. , Ropelle, E. R. , Lutolf, M. P. , Aebersold, R. , & Schoonjans, K. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science, 352, 1436–1443. 10.1126/science.aaf2693 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Lee, S.‐M. , Shannon, S. , Gao, B. , Chen, W. , Chen, A. , Divekar, R. , McBurney, M. W. , Braley‐Mullen, H. , Zaghouani, H. , & Fang, D. (2009). The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. The Journal of Clinical Investigation, 119, 3048–3058. 10.1172/JCI38902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Tao, J. , Ling, Y. , Li, F. , Zhu, X. , Xu, L. , Wang, M. , Zhang, S. , McCall, C. E. , & Liu, T. F. (2019). Switch of NAD salvage to de novo biosynthesis sustains SIRT1‐RelB‐dependent inflammatory tolerance. Frontiers in Immunology, 10, 2358. 10.3389/fimmu.2019.02358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B. , Wang, D. D.‐H. , Qiu, Y. , Airhart, S. , Liu, Y. , Stempien‐Otero, A. , O'Brien, K. D. , & Tian, R. (2020). Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. The Journal of Clinical Investigation, 130, 6054–6063. 10.1172/JCI138538 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable—no new data generated.