

ABSTRACT
Dominance is a basic property of inheritance systems describing the link between a diploid genotype at a single locus and the resulting phenotype. Models for the evolution of dominance have long been framed as an opposition between the irreconcilable views of Fisher in 1928 supporting the role of largely elusive dominance modifiers and Wright in 1929, who viewed dominance as an emerging property of the structure of enzymatic pathways. Recent theoretical and empirical advances however suggest that these opposing views can be reconciled, notably using models investigating the regulation of gene expression and developmental processes. In this more comprehensive framework, phenotypic dominance emerges from departures from linearity between any levels of integration in the genotype‐to‐phenotype map. Here, we review how these different models illuminate the emergence and evolution of dominance. We then detail recent empirical studies shedding new light on the diversity of molecular and physiological mechanisms underlying dominance and its evolution. By reconciling population genetics and functional biology, we hope our review will facilitate cross‐talk among research fields in the integrative study of dominance evolution.
Keywords: gene expression, genotype‐to‐phenotype map, modifier theory, fitness landscape, allele‐specific expression, epistasis, gene network, Haldane's sieve, dominance evolution
I. INTRODUCTION
‘It is obvious that no single theory can explain all dominance mechanisms’
(Wright, 1934)
Genetic dominance describes the relationship between the phenotype and the genotype at a diploid locus in heterozygotes. An allelic variant may behave as dominant when a single copy is sufficient for full phenotypic expression, co‐dominant when the effects of the two alleles are equally apparent, or recessive when a single copy of the allele has no detectable phenotypic effect. These interactions depend on the partner allele in the heterozygous genotype: a dominant allele in one genotype may be recessive when paired with another allelic variant. The dominance relationship among alleles also depends on the focal character: for instance, a pleiotropic allele can cause a dominant effect for one trait but a recessive effect for another trait. Starting from Mendel's observations on peas, it has been repeatedly documented that strict additivity is the exception rather than the rule for many traits. However, we know shockingly little about the actual distribution of dominance coefficients for new mutations (Manna, Martin & Lenormand, 2011), and even less for segregating variants in natural populations (Eyre‐Walker & Keightley, 2007). This lack of empirical data prevents estimating the extent to which mutations are visible to natural selection and therefore severely limits the power of population genomic approaches to predict the adaptive potential of species.
Understanding the biological phenomenon of dominance has been a topic of intense and continued interest, both from the population genetics and the functional biology communities. Nevertheless, these two scientific communities have approached this question in different ways. The evolution of dominance is thus an excellent case study of the sometimes conflictual, yet fruitful interactions between these disciplines. The population genetics community has explored the conditions under which natural selection acts on dominance interactions at the level of organismal fitness. These generic models largely neglected the molecular processes underlying these interactions. By contrast, the functional biology community provided detailed mechanistic models focusing on the genotype‐to‐phenotype map, detailing the molecular processes involved. However, by focusing on the organismal level, they largely ignored the complexity of predicting the evolutionary fate of mutations within populations. In particular, intragenomic conflicts among regulatory elements can lead to counter‐intuitive evolutionary outcomes and were initially ignored. Recent theoretical models have moved towards more integrative approaches, and are now considering the diversity of molecular processes by which gene expression can be linked to fitness through explicit phenotypes. These models are bridging the gap between the two research fields, revealing that just as gene expression and integrated phenotypes are evolvable properties, so are dominance interactions.
To embrace the complexity of dominance relationships and of their evolution, here we provide definitions for the dominance‐related terms used in different fields (Table 1) and specifically review the range of mechanisms that have been proposed to cause variation of dominance at three different levels (Fig. 1). First, at the fitness level, i.e. when dominance relationships result from contrasted evolutionary fates of alleles with a dominance assumed to be fixed. This process was initially formulated by Haldane (1927), and we review recent empirical examples consistent with this phenomenon. Second, at the level of phenotypic integration, whereby differences between homozygotes and heterozygotes translate into different levels of biochemical activity being integrated over successive cellular and developmental processes, ultimately resulting in different organismal phenotypes. This aspect is akin to the ‘physiological’ model proposed by Wright (1934). We review recent studies providing detailed molecular explanations for how dominance could arise as the consequence of such processes, beyond the classical case of enzymes. Third, at the level of allelic expression, through the qualitative modification of gene regulatory networks. This aspect is akin to the controversial model of ‘dominance modifiers’ proposed by (Fisher, 1928), for which recent studies have now provided compelling evidence in specific biological situations.
Table 1.
Definitions
| Additivity (also referred to as semi‐dominance) | Quantitative genetics term naming the trait value of heterozygotes as the mean of trait values of the two corresponding homozygotes. By extension, additivity refers to intermediate phenotypes of heterozygotes as compared to corresponding homozygous phenotypes. |
| Allelic exclusion | Takes place when expression of one allele limits the expression of the other allele(s). It usually results in recessivity of the excluded allele. Allele suppression can be considered as an extreme case of allelic exclusion. |
| Balancing selection | Any selective regime promoting the persistence of several alleles at intermediate frequency within a population. It usually results in an increased proportion of heterozygotes in the population, favouring the evolution of dominance. |
| Cis‐ and trans‐acting factors (see Fig. 3 for examples) |
Regulation in cis: modification of gene or allele expression by a genetic factor located on the same chromosome or from the same genetic background in the case of hybrids. Regulation in trans: modification of gene or allele expression by a genetic factor located on the alternative chromosome or from a different genetic background in the case of hybrids. |
| Domestication syndrome | Refers to the artificial directional selection of traits in bred or cultivated species by humans, triggering hard selective sweeps on the genomic regions controlling the targeted traits and a striking loss of diversity throughout the genomes of these domesticated species. |
| Dominance | Usually describes the link between the diploid genotype at a single locus and the corresponding phenotype. Müller (1932) proposed an early classification of mutations into amorph, hypomorph and hypermorph to refer to various levels of quantitative change to a pre‐existing wild‐type character, antimorph to describe mutual antagonistic interaction of the mutant with the wild type, and neomorph to refer to a mutation encoding an entirely new phenotype not present the wild type. As reviewed in Wilkie (1994), these different genetic properties of mutations can be linked to a variety of underlying molecular mechanisms by which dominance can arise between the mutant and the wild‐type alleles. With recent advances in genetics and molecular tools, the dominance phenotype can be defined at different observation scales, e.g. relative levels of gene expression, amount of protein in a cell or a tissue, organismal trait or even fitness (see Fig. 1), introducing substantial challenges in comparisons of dominance across the literature. |
| Dominant‐negative mutations | Describes mutations with a negative impact on protein function, in both homozygotes and heterozygotes (Herskowitz, 1987). |
| Nucleolar dominance | A common epigenetic phenomenon first described in interspecific hybrids by which the number and identity of expressed 45S ribosomal RNA (rRNA) genes is regulated. |
| Overdominance | A term mainly used by population geneticists to characterize a locus where the fitness of heterozygotes is higher than the fitness of either homozygote. By extension, overdominance may also refer to a locus where the phenotype of heterozygotes departs from either homozygote phenotype (typically at a quantitative trait, when the trait value is higher in heterozygotes as compared to homozygotes). Underdominance then describes the opposite pattern, where fitness or trait values are lower in heterozygotes as compared to both homozygotes. |
| Robustness | Indicates a developmental invariability of phenotype when facing environmental and/or genetic variations. It has a similar meaning to canalization, which usually also implies an adaptive value to this insensitivity to variation (Debat & David, 2001) |
| Standing genetic variation | Genetic variation maintained within a population, resulting from a balance between mutation, drift and selection. |
| Structured populations | Network of populations linked by migration, within which the allelic composition and distribution may vary substantially. |
| Supergene | Unique locus controlling variations of multiple adaptive traits, which might form strikingly different phenotypes. A supergene can be composed of several tightly linked genes, whose dominance is likely to be coordinated through selection on the encoded syndromes. |
| Trans‐dominant effect | Refers to a dominant allele suppressing the function of the alternative allele (Zhou, Yang & Shi, 2017) |
| Transvection | Trans‐acting modification of allele expression triggered by physical interactions or at least proximity of two homologous chromosomes during meiosis. |
Fig 1.
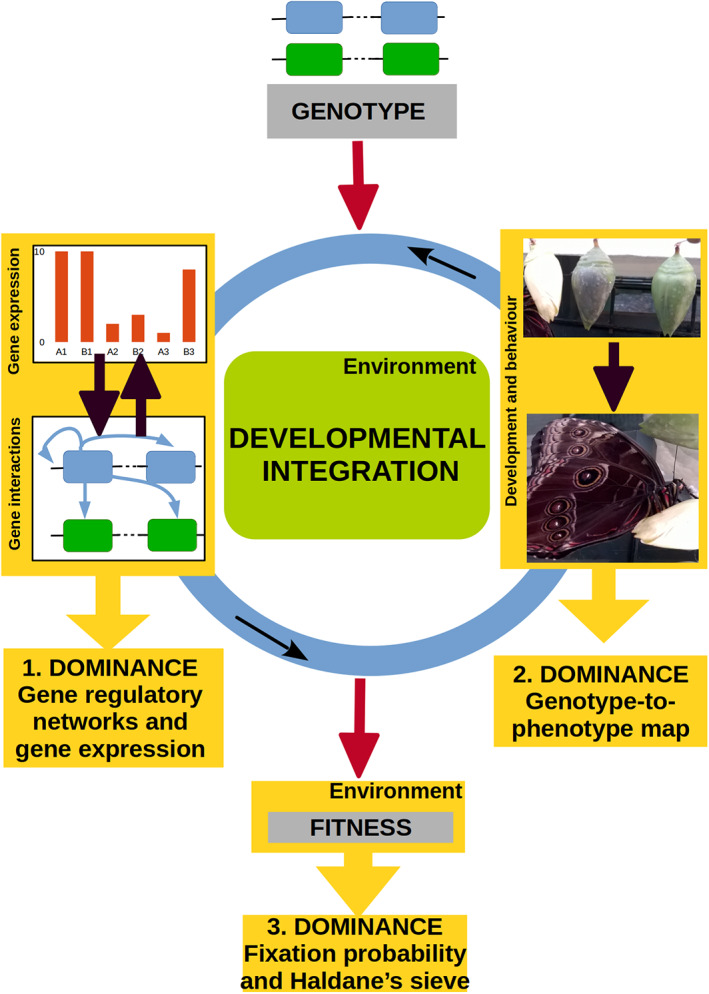
From genes to fitness: three levels of integration giving rise to dominance evolution. Genotypes in an environment produce individual organisms through multiple steps of developmental and ecological integration. (1) Gene regulatory networks and gene expression: dominance evolution arises as a consequence of the evolution of differential expression between alleles. (2) Genotype‐to‐phenotype map: dominance evolution arises as the consequence of non‐linear maps between developmental levels n and n + 1. (3) Haldane's sieve: dominance evolution arises as a consequence of the higher fixation probability of dominant‐favourable alleles.
Altogether, in this review, we argue that recent advances in theoretical modelling of the evolution of gene expression, along with the uncovering of molecular mechanisms involved in dominance, now provide a more comprehensive, less‐polarized view of the evolutionary biology of genetic dominance.
II. NATURAL SELECTION AND THE DISTRIBUTION OF DOMINANCE COEFFICIENTS
(1). The distribution of dominance coefficients in natural populations
Population genetics models generally describe dominance using a coefficient of dominance (h) modulating the effect of the selection coefficient s on the fitness of heterozygous individuals (1–hs). Classically, they considered this parameter as a fixed quantity. Recently, models have provided predictions on the distribution of dominance coefficients expected within populations. For instance, by considering explicit adaptive landscapes assuming concavity near the fitness optimum and studying resident genotypes close to the optimum, deleterious mutations are expected to be recessive, while favourable mutations are generally expected to be dominant (Manna et al., 2011). Such studies based on adaptive landscapes have received much attention for their role in adaptive walks and the distribution of fitness effects, specifically regarding how their dimensionality, level of epistasis and pleiotropy might affect evolution (Fragata et al., 2019), but predictions on how these properties affect the evolution of dominance are still scarce. By explicitly considering the functions linking genotypes to phenotypes, and the functions linking phenotypes to fitness, Martin (2014) also pinpointed that the distribution of fitness effects of mutations vary depending on the levels of pleiotropy considered and on the essentiality of the genes (i.e. the level of lethality provoked by their deletion). Such variation in the fitness landscape can then strongly affect the distribution of dominance coefficients. The current scarcity of data on actual fitness landscapes (e.g. in their ruggedness) is currently limiting our ability to derive general predictions on the distribution of dominance coefficients.
The few published distributions of dominance coefficients have been mostly based on the characterization of laboratory mutants in a few model organisms such as Drosophila, Caenorhabditis and yeast, and point at partial recessivity as the mean of the distribution (as reviewed in Manna et al., 2011). In a recent study, Huber et al. (2018) took an original approach and compared the site frequency spectra in natural plant populations between the outcrosser Arabidopsis lyrata and the predominant selfer Arabidopsis thaliana. They explored the link between the strength of selection (as estimated by the selection coefficient s) and dominance (as estimated by the dominance coefficient h). In outcrossers, most single nucleotide polymorphisms (SNPs) are in a heterozygous state, and their fitness will therefore depend on the product hs. In selfers however, heterozygotes are expected to be rare such that the frequency at which SNPs segregate should depend on the selection coefficient s only and be largely independent from dominance. Assuming that the distribution of fitness effects of new mutations is identical between the two species, the contrast between the two site frequency spectra can thus be used to reveal general features of the distribution of dominance coefficients h. They showed that, on average, segregating variants significantly depart from additivity: the mean of the distribution was h = 0.46, i.e. most mutations were only slightly recessive. They also showed that assuming a negative correlation between h and s improved the fit to the data, confirming predictions from the fitness landscape models and observations from laboratory mutants that more deleterious alleles generally tend to be more recessive (Orr, 1991). Interestingly, they found that the rate of decay of the h–s relationship was significantly higher for catalytic genes (which only need to be expressed at low levels) than for any other category of genes (such as structural genes that need to be expressed at high levels). This suggests that the cost of gene expression may be an important determinant of dominance.
(2). Natural selection on the dominance of emerging alleles
A large body of theoretical literature has studied the extent to which dominance determines the fate of new alleles, and in return how selective processes have shaped the distribution of dominance coefficients in natural populations over the long term. Haldane (1927) noted that in contrast to the widespread recessivity of deleterious mutations, adaptive alleles are more likely to become fixed within populations when dominant. Indeed, when adaptive variations arise by mutation, they are initially at the heterozygous state with wild‐type alleles, so that the effect of positive selection depends on their expression in the heterozygous state. Hence, fully recessive adaptive variations need to occur in the homozygous state before they induce any beneficial phenotypic change, and therefore have a high risk of being lost by drift before ever being expressed. In the long run, this is expected to lead to a filtering on the dominance level of new adaptive variants (the so‐called Haldane's ‘sieve’) leading to a turnover of increasingly dominant alleles over the course of evolution. However, direct empirical evidence of Haldane's sieve has remained scarce because adaptive alleles usually replace their ancestral versions, preventing direct comparison of dominance between them. Three indirect lines of evidence nevertheless suggest that Haldane's sieve is indeed a potent force in natural populations.
The study of highly polymorphic loci, where several alleles jointly persist within populations over long evolutionary times (as expected, for example when balancing selection is acting) allow such a comparison and offer a first line of evidence. In these systems, the ancestral versus derived status of the different segregating alleles can be determined either by comparing allelic variations in sister species or, in cases of alleles that emerged from chromosomal inversions, by reconstructing the mutational scenario given synteny information from sister species. In many polymorphic loci, the derived alleles have been shown to be dominant over the ancestral ones (Llaurens, Whibley & Joron, 2017), consistent with the action of Haldane's sieve.
A second line of evidence for Haldane's sieve has been obtained by studying the migration of adaptive alleles through different populations. In the case of negative frequency‐dependent selection for instance, migration of dominant alleles across populations might indeed be more effective than that of recessive ones (Pannell, Dorken & Eppley, 2005), as theoretically predicted in the case of increased migration of dominant female‐sterility alleles in gynodioecious plant species (Pannell, 1997). The action of Haldane's sieve on the migration success of dominant alleles has been observed in transition zones between contrasting environments, where clines in the frequency of variants adapted to either environment are typically observed, as for instance between alleles controlling flower colours in connected populations of Anthirrhinum majus (Whibley et al., 2006) or between alleles controlling mimetic coloration in Heliconius erato butterflies (Mallet & Barton, 1989). These clines are maintained by positive frequency‐dependent selection acting at the local scale, whereby locally abundant phenotypes benefit from increased attraction of pollinators or protection against predators in these respective examples. However, the position of the cline is predicted to vary among alleles depending on their dominance (Mallet, 1986). Dominant alleles, by being expressed more frequently, can gradually invade transition zones between populations, leading to a forward movement of the cline towards the population displaying phenotypes controlled by recessive alleles.
Finally, a third line of evidence stems from the emergence of adaptive variants through introgression from closely related species, allowing ancestral variants to be distinguished from their derived forms. For instance, melanism in the grey wolf Canis lupus has been shown to arise from a past hybridization event with domestic dogs (Anderson et al., 2009). The introgressed melanic form is dominant over the ancestral coat colour and reaches high frequency in forest habitats, where it is under positive selection. After hybridization, genomic regions under positive selection might be more easily retained through generations of introgression in the accepting genome when they are dominant, in line with Haldane's hypothesis.
It is unclear how often adaptation proceeds from de novo mutations, migration or introgression, i.e. from alleles that initially arose at low frequency. Instead, it is possible that many adaptive variations emerge from standing genetic variation, i.e. were initially segregating neutrally and became advantageous only after a change in the ecological or genomic environment occurred. Orr & Betancourt (2001) showed that Haldane's sieve predictions no longer hold when adaptive variations emerge from standing genetic variation, because adaptive variants can then be promoted by natural selection even when recessive. Similarly, mechanisms favouring the rapid formation of homozygotes, for instance selfing reproductive mode, should favour the recruitment of recessive or partially recessive adaptive alleles, making Haldane's sieve a less‐prominent process. By studying quantitative trait loci (QTLs) associated with the domestication syndrome in crops, Ronfort & Glemin (2013) showed that adaptive alleles in selfing species are predominantly recessive or partially recessive, while in outcrossing species, adaptive variants are more frequently dominant. This highlights that the filtering of recessive adaptive variants might be counterbalanced when the reproduction regime favours the rapid formation of homozygotes (Hartfield, Bataillon & Glémin, 2017).
Overall, it is clear that dominance strongly affects the fate of genetic variants, from their initial emergence to their spread across spatially structured populations, modulating the effect of natural selection in a variety of ways.
(3). Evolution of dominance through modifiers
While the distribution of dominance can be shaped by direct selection on the mutants themselves, indirect selection through the evolution of dominance modifiers can also play a role. This question of the proximate and ultimate mechanisms underlying the emergence of variation in genetic dominance has been a subject of sustained interest in genetics, leading in particular to the development of a large theoretical literature since the first debate between R.A. Fisher and S. Wright. In brief, Fisher (1928) hypothesized that deleterious mutations are generally recessive because of modifiers of dominance. These genetic elements would be favoured by natural selection because they decrease the disadvantage of deleterious mutations in heterozygous individuals. This hypothesis has been extensively investigated since Fisher's seminal paper, which led to the development of a series of population genetics models collectively referred to as the ‘modifier theory’. The modifier theory relies on multilocus models to determine the conditions for the invasion of alleles at a given locus not selected by themselves (they are generally considered neutral) but because of their effects on other loci (Karlin & McGregor, 1974). Several modifier models have been analysed to study the evolution of dominance after the Fisher–Wright debate (Mayo & Burger, 1997). In particular, Feldman & Karlin (1971) and Bürger (1983) confirmed Wright's (1929) initial claim that the conditions for the invasion of modifiers of dominance are very restrictive: positive selection on such modifiers would be proportional to the frequency of heterozygous individuals in the population, i.e. of the same order as the frequency of deleterious mutations. Precisely because deleterious mutations are expected to be kept at low frequency by purifying selection, dominance modifiers would have little opportunity to play significant roles in natural populations. However, while the evolution of dominance modifiers was unlikely for deleterious mutations because they segregate at low frequencies, Wright (1929) nevertheless acknowledged that dominance modifiers could still evolve when heterozygotes are frequent.
Accordingly, the dominance modifier hypothesis was later investigated under various ecological and genetic mechanisms known to promote heterozygosity, at least transiently: in the case of a polymorphic trait such as in Batesian mimicry (Clarke & Sheppard, 1960; O'Donald & Barrett, 1973; Charlesworth & Charlesworth, 1975), during the sweep of a favourable mutant (Wagner & Bürger, 1985), in the case of a trait with heterozygote advantage or in an heterogeneous environment with dispersal (Otto & Bourguet, 1999), in genetic systems under frequency‐dependent selection because of competition for resources (Peischl & Burger, 2008; Peischl & Schneider, 2010), in sporophytic self‐incompatibility systems (Llaurens et al., 2009; Schoen & Busch, 2009) or in the case of sexually antagonistic selection (Spencer & Priest, 2016). This series of models showed that in a wide variety of contexts, heterozygous genotypes can indeed be frequent enough for dominance modifiers to spread in a population. The fundamental mechanism promoting dominance evolution is the mitigation of the deleterious effects in heterozygotes in a specific genetic background, for instance in the emergence of sex‐specific dominance (Spencer & Priest, 2016). The strength of indirect selection on the dominance modifier depends on the fitness of the regulated alleles, as well as on the recombination rate with the locus under direct selection. The recombination rate sometimes dramatically changes the evolutionary outcome. For instance, only strongly linked dominance modifiers invade and go to fixation in sporophytic self‐incompatibility (Schoen & Busch, 2009), while in other cases recombination has no effect (e.g. in the case of sexually antagonistic selection; Spencer & Priest, 2016). The role of recombination can be hard to predict since it depends on the complex interaction between the frequency of the modifier in the population and the level of polymorphism at the locus at which balancing selection acts directly (Wagner & Bürger, 1985; Otto & Bourguet, 1999). Overall, dominance modifiers are expected to evolve in a large variety of genetic and ecological conditions, as long as heterozygotes are frequent, either transiently (in the case of recurrent selective sweeps) or at equilibrium (under various forms of balancing selection).
How often these genetic and ecological conditions are met in nature remains a matter of debate, but empirical case studies have provided evidence of selection acting on dominance, especially at polymorphic loci where several allelic variants are maintained within populations by balancing selection. In the polymorphic butterfly mimetic species Heliconius numata, Arias et al. (2016) showed that the predation risk was higher for intermediate heterozygotes as compared to mimetic homozygotes, highlighting the stringent selection against co‐dominance. Furthermore, when the polymorphic locus is controlling a trait composed of multiple developmental modules, natural selection may promote the coordination of dominance across the different features of the trait. In the butterfly Papilio dardanus for instance, several mimetic wing morphs also segregate, some of which can be shared across natural populations. Wing morphs show strong dominance when crosses are performed between individuals of the same populations: heterozygote phenotypes closely resemble the phenotype of one of the two parents. Strikingly however, crosses between the same wing morphs but between individuals collected from distant populations often result in mosaic phenotypes between parental morphs (Nijhout, 2003). This suggests that natural selection exerted on heterozygote phenotypes promotes the coordination of dominance throughout the whole wing colour pattern: such selection on dominance can occur only when alleles are in geographic contact and thus when heterozygous genotypes can be formed and submitted to natural selection. A similar result was found in the polymorphic mimetic species H. numata, where strict dominance between mimetic alleles is generally observed in sympatry, generating mimetic phenotypes in heterozygotes (Le Poul et al., 2014). One notable exception was observed in some populations of this species, where one pair of mimetic alleles generates an intermediate phenotype in heterozygotes. This intermediate phenotype does not match the mimicry rings corresponding to the parental alleles, but is nevertheless selected positively because of its mimicry towards another mimetic community. This highlights that the fine‐tuning of dominance relationships between alleles of polymorphic loci depends on the associated fitness landscape.
Other cases of coordinated dominance are observed in loci controlling the male and female components of a trait. In Primula plants for instance, two genetically controlled morphs of flowers co‐exist: pin flowers with a short style and long anthers, and thrum flowers with conversely a long style but short anthers. The two morphs also differ in the size of the pollen grains they produce. For each of these three components of the complex trait (length of the style, position of anther and size of pollen grains), strong dominance is observed, each time in the same direction: the short‐style morph is dominant over the long‐style morph (Nowak et al., 2015). A similar pattern is observed in the self‐incompatibility locus (S‐locus) of Brassicacea where a large number of genetically encoded recognition specificities stably segregate in natural populations. The S‐locus contains the tightly linked genes SCR (encoding for a cysteine‐rich protein) and SRK (encoding for a receptor kinase). Male specificities are determined by SCR ligand proteins displayed in the pollen coat and female specificities are determined by SRK receptors in the pistil. The genes encoding the male and female recognition specificities segregate as a single genetic unit, but they are clearly distinct from one another. Yet, dominance between alleles of the SCR and SRK genes is coordinated, with an overall highly consistent pattern of dominance between pollen and pistil phenotypes and no case of opposite dominance between the two genes for any given pair of specificities (Llaurens et al., 2008). The dominance relationships are however not fully identical between the two genes. While a strictly linear hierarchy is observed for the pollen phenotype, co‐dominance is often observed in the pistil phenotype (i.e. heterozygous genotypes jointly express their two S‐alleles), allowing rejection of a greater number of pollen phenotypes by the pistil. This difference may stem from the difference in reproductive investment between the male and female functions: seed production comes at a high metabolic cost, so avoiding the formation of inbred seeds is advantageous even in the face of rejection of a higher number of potential mates. By contrast, the male fitness through pollen increases with access to a greater number of mates, promoting the phenotypic expression of a single self‐incompatibility allele, i.e. strict dominance. Strikingly, pollen dominance seems to be the rule in such systems, with instances of co‐dominant expression being rare and restricted to pairs of alleles that are already high in the dominance hierarchy [S13S20 in Arabidopsis halleri (Llaurens et al., 2008) and class I S‐alleles in Brassica (Hatakeyama et al., 1998)]. More generally, sexually antagonistic selection acting within a locus has been shown to promote the evolution of dominance. For instance, in the Atlantic salmon Salmo salar, selection favours an earlier age of maturity in males than in females. The gene VGLL3, an adiposity regulator, controls a large part of variations in size and age at maturity and is highly polymorphic within salmon populations. Sex‐dependent dominance is observed at this gene, whereby heterozygotes males have the precocious age at maturity, while heterozygous females display late maturity phenotypes (Barson et al., 2015). This sex‐specific dominance may have evolved in response to the sexually antagonistic selection exerted on this trait.
Overall, recent theoretical frameworks have been developed that provide more detailed predictions for how the shape of fitness landscapes should translate into general patterns of dominance, but the distribution of dominance coefficients has been empirically documented in a systematic manner in only a very limited number of cases. Recently, theoretical and empirical studies have shown that natural selection can modify patterns of dominance, either through direct selection of allelic variants, or through indirect selection on dominance modifiers in a number of genetic and ecological situations. A central tenet of the predictions and observations above is that the mechanistic details of how dominance arises is of central importance to predict whether and how natural selection can act on it, either directly or indirectly. We thus review below the main features of the mechanistic models for dominance, and detail several empirical examples that demonstrate the variety of molecular mechanisms by which genetic dominance can arise.
III. DOMINANCE EMERGING FROM THE SHAPE OF GENOTYPE‐TO‐PHENOTYPE MAPS
(1). Regulatory network models describing dominance as a by‐product of the genotype‐to‐phenotype map
Wright (1934) was the first to argue that dominance is an inherent property of biological systems. He focused on enzymatic reactions, where biochemical fluxes typically show a saturating relationship with enzyme concentration. This saturating function is justified by biochemical reactions themselves, as fluxes are limited by the quantity of free enzymes. The biochemical theory of dominance then developed in many directions, sometimes with contradicting conclusions and interpretations. Kacser & Burns (1981) showed that biochemical reactions due to a sequence of enzymatic reactions should necessarily generate dominance because variations in the efficiency of one focal enzyme in the pathway only have a small effect on the general flux. Mutations decreasing enzymatic efficiency would then necessarily be recessive (assuming that the enzymatic flux generates a concave genotype‐to‐phenotype curve; Fig. 2). Kacser & Burns (1981) and followers (e.g. Orr, 1991; Keightley, 1996; Porteous, 1996) therefore concluded that dominance was due to metabolic properties, neglecting the effect of selective processes that could act directly on its evolution.
Fig 2.
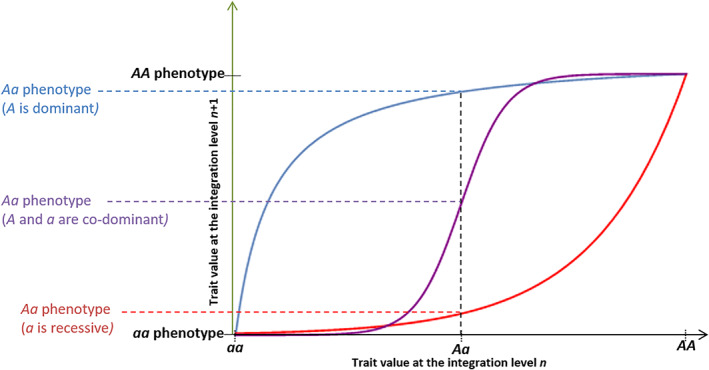
Non‐linear relationships between integration levels and consequences on dominance relationships. Here, additivity is observed at integration level n (for example RNA expression level or protein concentration), and depending on the genotype/phenotype map, different dominance levels can be observed at the integration level n + 1. (1) Assuming a concave shape (blue line), Aa and AA genotypes display similar phenotypes, therefore A is dominant over a. (2) Assuming a convex shape (red line) Aa and aa genotypes display similar phenotypes, therefore a is dominant over A. (3) Assuming a sigmoid shape (purple line): if homozygotes are located on either side of the inflexion point (as shown on the plot), then the phenotype Aa will be intermediate (additivity or semi‐dominance). By contrast, if both homozygotes are located on the concave or convex part, then the allele A is dominant or recessive respectively, similarly to cases 1 and 2.
This view was challenged by several authors (e.g. Savageau, 1992; Omholt et al., 2000; Gilchrist & Nijhout, 2001; Bagheri‐Chaichian et al., 2003), who showed that the Kacser & Burns (1981) model, by focusing on enzymatic flux, does not necessarily apply to all regulatory networks (for an extended review, see Bagheri, 2006). For instance, Omholt et al. (2000) showed that biochemical reactions implying feedback loops and regulation in a three‐locus system can lead to a large variety of results, including additivity, dominance, recessivity and even overdominance. Gilchrist & Nijhout (2001) showed that a model of spatial diffusion of gene products can give rise to non‐linearity in the genotype‐to‐phenotype map, and therefore generates various dominance patterns. More recently, Veitia and collaborators (Veitia, 2003; Bost & Veitia, 2014; Bottani & Veitia, 2017) used biochemical reaction models to predict the level of expression of a gene as a function of the rate at which transcription factors bind to its promoters. Their model allows the possibility of multiple binding sites and assumes that the transcriptional response depends on the number of sites occupied. In these conditions, the genotype‐to‐phenotype map is a sigmoid curve, whose steepness depends on the number of binding sites. As a consequence, whether a mutant is recessive, dominant or has an additive effect depends on the arbitrary position of the homozygote on the map (see Fig. 2). Along the same lines, Porter, Johnson & Tulchinsky (2017) explicitly modelled biochemical reactions underlying the level of expression of a gene assuming two or three interacting genes in a sequential manner, with genes located upstream in the reaction chain regulating the expression of genes located downstream. In line with Omholt et al. (2000), they modelled gene promoters explicitly, so that alleles can vary in their coding region, in their promoters, or both, and the expression level of a gene depends on the binding affinity of its promoter with the protein produced by the gene located directly upstream in the cascade (effectively functioning as a transcription factor). In this model, gene products compete to bind to the promoters of the downstream genes, which introduces a non‐linearity between concentration of the transcription factors and expression of the genes they regulate. By explicitly modelling the effect of variations in either transcription factors, transcription factor promoter regions, or in the promoter region of the gene targeted by the transcription factor, Porter et al. (2017) predicted that the location at which mutations emerge should lead to contrasting levels of dominance. While variations in the transcription factor itself are likely to generate dominance because of competitive binding to their target sites, variations in the promoter regions of transcription factors are expected to be associated with simple changes in levels of allele‐specific expression, and should therefore more likely lead to additivity. The molecular nature of the mutations and their effects on the binding properties of the transcription factor are therefore important biophysical properties shaping dominance relationships.
Epistatic interactions among alleles at different genes thus play an important role in dominance variation. Nevertheless, the effect of natural selection on these changes is scarcely explored. Population genetics models evaluating the invasion propensity of mutants with given epistatic effects may allow this question to be tackled. Bagheri & Wagner (2004) analysed a population genetics model with an explicit genotype‐to‐phenotype‐to‐fitness map, where the evolution of dominance emerges from the selective pressure applied to an entire gene expression network. In their model, fitness was assumed to be proportional to the flux of a simple biochemical chain comprising two enzymes. The enzyme concentration and their catalytic activities can be modified by four different loci (two for each enzyme) at which mutation can occur. The modifier alleles affecting the concentration of enzymes can be interpreted as modifiers of gene expression. As expected, natural selection on the introduced mutations led to an increase of the biochemical flux, in particular through an increase in the concentration of enzymes. More importantly, they showed that dominance is expected to evolve as a side effect of the increase of the flux rate, because of the concave relationship between enzyme concentration and flux rate: as the flux rate increased, the heterozygotes became closer to the wild‐type homozygotes (Fig. 2). Even though it was not the primary aim of the authors, this is to our knowledge the first model explicitly to link selection on modifiers of gene expression and dominance evolution. Their result can likely be generalized to other cases as long as a concave relationship exists between genotype and phenotype: if a modifier making this relationship steeper invades and goes to fixation, a necessary side‐effect is the increase of dominance (Fig. 2). Further theory is clearly needed to explore in detail such models of dominance linking gene expression to fitness, but they are still crucially lacking from the literature (Bagheri & Wagner, 2004; Bürger et al., 2008).
(2). Empirical evidence for non‐linear relationships between levels of gene expression and phenotypes
Overall, it is clear from theory that a comprehensive understanding of the patterns of dominance in a genetic system requires access to general properties of the genotype‐to‐phenotype map. Recent evidence shows that the genotype‐to‐phenotype map can indeed be non‐linear (Kemble, Nghe & Tenaillon, 2019). In the following section, we review a series of empirical studies illustrating the diversity of molecular processes by which non‐linearity can arise in genetic networks and along developmental processes, leading to different dominance relationships.
(a). Threshold effect in gene expression induces dominance
In the peppered moth Biston betularia, the melanic phenotype carbonaria was promoted by industrial activities darkening tree trunks (Haldane, 1956), and stems from a transposable element (TE) insertion in an intron of the ‘melanization’ gene cortex. The TE insertion is associated with an increase in cortex transcript numbers (van't Hof et al., 2016), and the level of expression of cortex in heterozygote developing larvae is intermediate as compared to the respective homozygotes (i.e. the effect is additive at the transcriptional level). However, at the phenotypic level, a single copy of the TE is sufficient to trigger full expression of the melanic morph, illustrating that additive transcription of the causal genes can still result in strong phenotypic dominance. This example of a strongly selected variant has been used to illustrate Haldane's sieve effect, and demographic inference suggests that this variant arose from a single recent mutation that rapidly spread across natural populations (van't Hof et al., 2016).
In another emblematic example of natural selection, lactase persistence allowing human adults to digest milk in pastoralist populations, cis‐regulatory elements acting on the LCT gene encoding lactase have been described. Lactose tolerance evolved in parallel across pastoralist populations from Europe and Africa through distinct genetic variants causing different modifications of LCT expression. The different identified SNPs are associated with different levels of blood glucose after milk ingestion in homozygote and heterozygote individuals (Tishkoff et al., 2007). However, intermediate efficiency in lactose digestion seems to be sufficient to permit full lactose tolerance. Dominance of lactose tolerance thus stems from the fact that intermediate levels of lactase confer full tolerance, pointing to a non‐linear relationship between lactase expression and the tolerance phenotype. Overall, similar threshold effects seem to be common.
(b). Non‐additive relationships between alleles: loss of function and protein heterodimers
Genetic variations leading to a loss of function at the biochemical level are often expected to be recessive because the presence of the alternative allele is generally sufficient for the gene function to be ensured at the organismal level. However, loss of function does not necessarily lead to recessivity, and a number of ‘dominant‐negative’ mutations have been described (Veitia, Caburet & Birchler, 2018). For instance, mutations in the PSEN1 gene are associated with elevated risk of Alzheimer's disease in humans. Loss‐of‐function mutations of this gene are dominant because they are able to suppress the wild‐type γ‐secretase activity (Heilig et al., 2013). Zhou et al. (2017) further revealed that this loss of function is due to hetero‐oligomerization of the mutant with the wild‐type protein, forming a non‐functional duplex.
The formation of multimeric proteins is indeed frequently associated with non‐additive dominance relationships, and the effect of various protein interactions such as dimerization, often has been described to lead to ‘dominant‐negative’ mutations (Herskowitz, 1987). If the subunits of a dimeric protein are produced by two highly differentiated alleles in a heterozygote, then the phenotype will depend on the biochemical activity of the heterodimer composed of different subunits. Assuming equal protein dosage, the heterodimer is expected to represent half of the population of proteins in the cell, so any deviation from additivity in the activity of the heterodimer would result in dominance. For instance, a non‐functional protein might suffice to render the heterodimer non‐functional by ‘poisoning’ the complex, so that 75% of the population of dimers would be non‐functional (Veitia, 2006).
This situation may also apply to binding specificity of the polymorphic positive regulatory domain zinc finger protein 9 (PRDM9) locus of mice, controlling the localization of recombination hotspots in mammals by binding to specific DNA sites, whose identity differs among PRDM9 alleles. Importantly, the PRDM9 proteins form dimers, and heterozygous individuals are expected to form 50% heterodimers, whose binding activity may be dominated by one of the two alleles. The majority of PRDM9 proteins (~75%) will thus be targeted towards DNA sites of the dominant allele, resulting in limited recombination on the sites targeted by the recessive PRDM9 allele. Moreover, protein variants compete for the recombination machinery, further altering the distribution of recombination hotspots in heterozygotes (Baker et al., 2015).
A similar situation may occur at the SRK gene, controlling female self‐incompatibility specificity in plants of the Brassicacea family. Most individuals at this gene are heterozygotes, and in many cases both alleles are expressed at the phenotypic level. In a number of cases, however, heterozygotes express only one of their two alleles at the phenotypic level, resulting in dominance interactions (Llaurens et al., 2008). This dominance phenomenon does not seem to involve transcriptional silencing for the female phenotype, since transcripts of both SRK alleles are typically detected in heterozygote genotypes (Kusaba et al., 2002; Burghgraeve et al., 2020). Interestingly, the SRK proteins form dimers, and Naithani et al. (2007) used yeast assays to suggest that affinity towards their cognate pollen proteins may depend on the binding properties of heterodimers. Direct measurement of the binding affinity of the heterodimers in a large number of combinations will now be necessary to evaluate the generality of this mechanism.
A final example has been described in disease‐resistance genes in wheat (Triticum aestivum), where the combination of multiple alleles of the nucleotide‐binding leucine‐rich repeat (LRR) resistance protein (Pm3) in the same plant by genetic transformation can result in suppression of Pm3‐based resistance. The suppression effect is post‐transcriptional, as both transcript and protein levels remain unchanged, and involve the LRR domain of the protein (Stirnweis et al., 2014). This case provides evidence that alleles of a single gene (and even partial copies of them) can block the activity of other alleles of the same gene, providing molecular evidence for interallelic interactions that can underlie the mechanism of dominance. The generality of such post‐transcriptional mechanisms remains to be determined.
(c). Duplications leading to variations in dominance
Because of complex interactions between alleles or copies of the same genes, gene duplications may interfere with dominance relationships. Haldane (1933) and Fisher (1935) already proposed that gene duplications could act as dominance modifiers: fixation of a duplicated copy of the wild‐type allele of a dosage‐sensitive gene would buffer the difference between homozygous and heterozygous genotypes at the original copy. Accordingly, haplo‐insufficient genes have on average more paralogs that haplo‐sufficient genes, suggesting that gene dosage may be an important factor for the initial fixation of duplicated copies. The evolution of gene duplication might thus be linked to the dominance relationships at the duplicated loci. In turn, dominance at one locus might be tuned by the presence of additional gene copies. Similar processes are believed to play a crucial role in the retention of gene duplicates over time (Kaltenegger & Ober, 2015; Diss et al., 2017). Our understanding of the evolution of dominance may thus benefit from the sharing of a common theoretical framework with that of genes with multiple copies (Kondrashov & Koonin, 2004).
A striking example of the link between selection on dominance and on duplication is illustrated by the insecticide‐resistance gene ace‐1 in mosquitoes, where heterozygotes carrying a susceptible (S) and a resistant (R) allele benefit from increased fitness. RR homozygotes have increased survival in treated areas, but also suffer from several fitness costs (e.g. decreased fertility and reproductive success) as compared to SS homozygotes. As the result of this trade‐off, RS heterozygotes have the greatest overall fitness. This overdominant selection has favoured the emergence of a duplicated copy of the gene, leading to an haplotype D combining the susceptible and resistant alleles (Milesi et al., 2017). Over the long term, favourable combinations of alleles can thus become fixed on a single haplotype, avoiding the constant reshuffling implied by their initial allelic status.
(d). Non‐linear protein activity
Different mutations targeting the same gene may induce similar phenotypes, but with different dominance levels, depending on how the encoded protein is affected. For instance, independently derived mutations in the coding sequence of the gene MC1R involved in skin colour variation have emerged in three lizard species (Sceloporus undulatus, Aspidoscelis inornata and Holbrookia maculata), all leading to an adaptive light phenotype in a sandy environment (Rosenblum et al., 2010). The parallel mutations in S. undulatus and A. inordata led to a reduction of pigmentation by distinct mechanisms (reduction in melanocyte membrane integration efficacy versus disruption of receptor signalling, respectively), with opposite consequences on dominance (the light allele is dominant in S. undulatus but is recessive in A. inordata). This shows that inferring dominance mechanisms from genetic variation alone is not straightforward, even in genes for which genetic pathways towards the phenotype have been well studied, and requires specific functional studies.
In some cases, different alleles of a given gene may activate distinct downstream pathways, so predicting dominance in heterozygotes requires a detailed understanding of these pathways. Some of the best‐studied examples of this phenomenon are the human leukocyte antigen (HLA) alleles in humans, which are commonly associated with elevated risks of developing autoimmune diseases. Some HLA alleles are known to overshoot non‐self targets and recognize self‐epitopes, therefore triggering an inappropriate inflammatory response. In some cases, this risk can be masked in the heterozygote state. For instance, carriers of the HLA‐DR15 allele are at greater risk of developing Goodpasture Disease, and Ooi et al. (2017) showed that the risk of developing an autoimmune response in HLA‐DR15/HLA‐DR1 heterozygotes is as low as that of HLA‐DR1 homozygotes, suggesting a dominant protective effect of HLA‐DR1. The HLA‐DR15 and HLA‐DR1 alleles exhibit distinct peptide repertoires and binding preferences, and they present the collagen epitope in different ways. This leads to the recruitment of a different population of T‐cells (‘conventional’ versus ‘regulatory’ T‐cells). In heterozygotes, the regulatory T‐cells recruited by HLA‐DR1 seem to be sufficient to prevent the inflammatory response by the conventional T‐cells recruited by HLA‐DR15, hence providing a molecular explanation for the dominant protective phenotype of HLA‐DR1 over the HLA‐DR15 allele. While important molecular details of this phenomenon and the broader related phenomenon of ‘immunodominance’ (Akram & Inman, 2012) remain to be determined, this example provides a compelling illustration that even equal production of different proteins in heterozygotes can lead to dominance at the phenotypic level, because of complex interactions with the downstream pathways.
(e). A significant role of protein interactions in dominance
Overall, various interactions involving the expressed proteins can lead to dominance relationships at the organismal level. Dosage sensitivity in different functional categories of genes may explain their contrasting levels of dominance/recessivity, with enzymes more commonly found among haplo‐sufficient genes, whereas transcription factors are more common among haplo‐insufficient genes (Jimenez‐Sanchez, Childs & Valle, 2001). Accordingly, multiprotein complexes tend to be encoded by haplo‐insufficient genes, which are generally sensitive to dosage (Papp, Pal & Hurst, 2003). The type of protein therefore strongly impacts the non‐linearity between levels of protein concentrations and heterozygote phenotype, and therefore the dominance relationships between alleles. The effect of natural selection on dominance might thus differ depending on the molecular properties of the targeted proteins.
IV. DOMINANCE EVOLUTION TRIGGERED BY CHANGES IN ALLELE‐SPECIFIC EXPRESSION
Accumulating theoretical and empirical evidence is now showing that dominance can arise from the effect of modifiers that cause changes in the relative abundance of the gene products of the two alleles in a diploid genotype.
(1). Specificity and linkage of the modifiers towards the targeted gene matter
In many metabolic models (Kacser & Burns, 1981; Gilchrist & Nijhout, 2001; Bagheri‐Chaichian et al., 2003; Veitia, 2003; Bagheri & Wagner, 2004; Bottani & Veitia, 2017), biochemical reactions occurring in heterozygotes are hypothesized to rely on the simple additive effect of the two alleles, such that their genotypic values (or enzyme/transcription factor concentration) are intermediate between the two respective homozygotes. Empirical data on allele‐specific expression (see Section IV.2), suggest that this assumption does not apply in a number of situations. Models by Omholt et al. (2000), Gjuvsland et al. (2007) and Porter et al. (2017) are the only ones where biochemical reactions are derived separately for homozygote and heterozygote genotypes. Among these, only Porter et al. (2017) considers the possibility of allele‐specific interactions. Metabolic models also generally assume that interaction rates between molecules are the product of their concentrations. However, genes are in limited number of copies within each cell and competition between transcription factors for binding sites may be high, such that stochastic effects can considerably impact allelic dominance at the cellular level [see the case of olfactory receptor (OR) genes in Section IV.2].
In models of variation in gene expression triggered by modifiers (Bagheri & Wagner, 2004; Nuismer & Otto, 2005), specific relationships between modifiers and alleles of the regulated gene often have been ignored, so that the modifier affects gene expression of the two heterozygous alleles indiscriminately. More recently, the evolution of dominance has been studied in models where the mode of action of modifiers of gene expression was explicit. Fyon, Cailleau & Lenormand (2015) investigated the invasion probability of modifier alleles affecting the expression of a partially linked locus, assuming cis interactions. These expression modifiers change the affinity with the transcription factors and therefore modify the level of expression of the gene copy linked in cis. The partially linked regulated locus is subject to purifying selection, and is hit by recurrent deleterious mutations. In addition, a concave relationship between gene expression and fitness is assumed. The promoter regions compete for access to the transcription factors, so that modifiers increasing the expression of one allele automatically decrease expression of the alternative allele. They showed that modifier alleles increasing gene expression are favoured when they are tightly linked to the regulated locus under purifying selection. This outcome results from the balance between two consequences of expressing deleterious alleles. A modifier increasing the expression of a deleterious allele is expected to be lost more often because it has a greater chance of being hitch‐hiked during the purging process. However, because purging is more efficient when deleterious mutations are more expressed, the modifier allele can also be found more often in association with a purged background than with a deleterious allele. The model therefore predicts that expression enhancers tightly linked to regulated genes are recurrently promoted by a runaway process, in which binding affinity of the promotor regions of the regulated gene steadily increases over the course of evolution. Nevertheless, the dominance relationship between a pair of alleles remains identical after the fixation of the expression modifier since the modifier becomes associated with the different alleles at the gene. The overall level of gene expression in the heterozygotes thus remains the same as in the ancestral state, because both alleles attract equivalent amounts of the transcription factor. However, as soon as regulatory genetic regions are polymorphic, one allele at the selected locus can be expressed more than the other, translating into different fitnesses in the heterozygotes.
In addition to this general model of evolution of gene expression modifying the fitness of heterozygotes, other models have studied the effect of modifiers of expression on specific traits. For instance, Llaurens, Joron & Billiard (2015) investigated the evolution of expression at the locus controlling mimetic colour patterns in species where individuals have chemical defences deterring predators. In this case, the colour pattern polymorphism is maintained in structured populations connected by dispersal because predators exert different selective pressures among demes. The different colour patterns are assumed to be controlled by a single bi‐allelic locus, where homozygotes have optimal colour patterns. Fitness depends on the distance to the optimal colour pattern in a given deme. As a consequence, when heterozygotes show intermediate colour patterns, they are counter‐selected across the whole population. Llaurens et al. (2015) looked at the fate of a modifier of gene expression, partially linked to the colour pattern locus, with different modes of action: the modifier can repress or enhance expression, in cis‐, in trans‐ or through both types of interactions, and it can show allelic specificity or not (i.e. the modifier can modify only one allele of colour pattern, or it can affect both). They showed that the evolutionary fate of the modifier depends strongly on its mode of action. In particular, only a modifier enhancing gene expression with an allele‐specific mode of action can invade the population, with no significant effect of the recombination rate. This model thus predicts that if dominance indeed evolved because of selection on colour pattern gene expression, then it should be due to allele‐specific enhancing modifiers.
Overall, these models by Fyon et al. (2015) and Llaurens et al. (2015) suggest that the way genetic elements control gene expression can affect the outcome of the evolutionary process. It should be noted that these models have explored only a small fraction of all possible ecological and genetic contexts, and the analysis of other models will now be necessary in order to reach a broader understanding of the link between gene expression and dominance evolution. We believe that this research avenue holds great potential to provide novel testable predictions to guide experiments and empirical discoveries.
(2). Biased allele‐specific expression can be caused by trans‐acting factors
The models presented above make specific assumptions about the way allelic expressions are controlled by molecular interactions that take place either between alleles, or within or among genes (Fig. 3). But how common are these phenomena, and what do we know about their molecular mechanisms? High‐throughput transcriptomic approaches are now allowing the routine comparison of allele‐specific transcript levels [‘the allelome’; e.g. Lappalainen et al. (2013) in humans; Crowley et al. (2015) in the mouse], and have revealed that differences in transcript levels of the two alleles in a diploid genotype are common. Identifying the general molecular mechanisms by which this bias is determined and established remains an active area of research (Gaur et al., 2013). Below, we review a series of genetic phenomena in which genetic interactions result from trans‐acting factors acting at the same locus, but interallelically (cases ④ and ⑤ in Fig. 3). The genetics literature does not have a specific name for this kind of interactions, but they are essential to understanding how the rewiring of gene regulatory networks can result in a dominance phenotype upon which natural selection can act.
Fig 3.
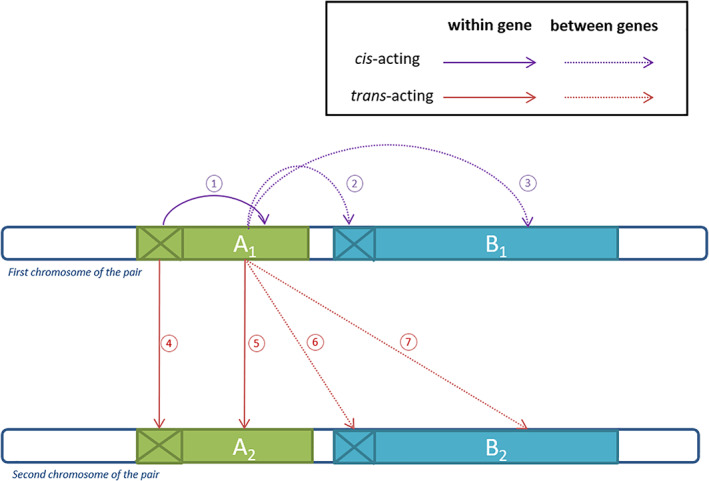
Interactions between genes (A and B) and alleles (1 and 2) in heterozygotes in a diploid individual. Two genes (A and B) located on the same chromosome pair with their respective regulatory regions (crossed boxes) are exhibited here, all at a heterozygous state (haplotypes 1 and 2). Arrows describe the influence of a genomic region on another one: cis‐ and trans‐ acting factors are shown, acting either between genes (dotted lines) or between alleles (solid lines). Enclosed numbers involve different molecular mechanisms described in the text. Depending on the different enhancing and repressing effects and specificity of the target, various departures from additivity in expression levels can be observed in heterozygotes.
Some of these phenomena have been revealed most clearly in interspecific hybrids. An example of interallelic interaction in trans‐ is the Sc locus of rice. This gene occurs as a single copy in Oryza japonica (the Sc‐j allele), while O. indica contains three duplicated copies of Sc (the Sc‐i allele). In Sc‐i/Sc‐j hybrids, high expression of Sc‐i results in suppression of the Sc‐j allele in the (diploid) sporophytic cells. This suppression is then stably maintained throughout development, leading to selective abortion of the (haploid) pollen grains carrying the Sc‐j allele, due to the lack of expression of this essential gene. This results in drastic segregation distortion (Shen et al., 2017). Overall, the Sc‐i/Sc‐j heterozygotes thus express only Sc proteins from their Sc‐i allele, which amounts to full dominance of Sc‐i alleles caused by a within‐gene trans‐acting repressive effect (case ⑤ in Fig. 3).
Another example is nucleolar dominance, a widespread phenomenon initially documented in Crepis hybrids (Navashin, 1934). In diploids, individuals inherit ribosomal RNA (rRNA) genes from their two parents, but in a number of hybrids, only one of the two sets of rRNA genes is actively expressed. In Arabidopsis, nucleolar dominance is controlled by reversible, chromatin‐mediated alterations in gene expression (Pontes et al., 2003). Preuss et al. (2008) suggest that small non‐coding RNAs may be the agents of the trans‐ modification of expression observed in the interspecific hybrid Arabidopis suecica (cases ④, ⑤, ⑥ or ⑦ in Fig. 3). However, Mohannath, Pontvianne & Pikaard (2016) suggested instead a role of chromosome positional effects in Arabidopis (see also Durica & Krider, 1978). In fact, two clusters of rRNA genes exist in A. thaliana, with typically only one being transcribed, and there is variation among A. thaliana accessions in the identity of the cluster that is expressed. Nucleolar dominance was shown to evolve rapidly, with only two generations being necessary for one of the two sets of rRNA genes to become silenced in synthetic A. suecica lines newly obtained by experimental crosses in the laboratory (Chen, Comai & Pikaard, 1998). However, Joly et al. (2004) did not observed such rapid evolution of nucleolar dominance in Glycine hybrids. Overall, it is clear that nucleolar dominance arises through both allelic (in cis, case ② or ③ in Fig. 3) and epistatic interactions (in trans, cases ④, ⑤, ⑥ or ⑦ in Fig. 3) (Rabanal et al., 2017).
These biases in relative expression of alleles observed in hybrids reflect the evolution of regulatory networks that diverged among species or lineages. However, direct selection on dominance in hybrids is unlikely to be common in the wild, where hybrids are typically rare. Selection on dominance is more likely to take place within species, where heterozygotes can be formed more frequently. In fact, similar interallelic interactions have also been observed within species. In Arabidopsis, comparison of the methylome of F1 hybrids obtained by crossing different natural accessions with that of their parental lines revealed a large number of non‐additive methylation patterns brought about by the processes of trans‐chromosomal methylation and demethylation, whereby the methylation state of one allele is altered to resemble that of the other allele. The majority of these loci are associated with transposable element‐derived sequences, and a proportion of these alterations of the cytosine methylation state are stably inherited to the F2 generation (Greaves et al., 2014). In the majority of cases, these altered methylation states are associated with the production of 24‐nucleotide small interfering RNA (siRNA) molecules and involve the RNA‐directed DNA methylation pathway (Greaves et al., 2016). In most cases, the ‘methylated’ state seems to be dominant over the ‘unmethylated’ state. This phenomenon represents hundreds of loci across the genome, although it remains unclear how often they lead to transcriptional changes in the nearby genes. In these situations, it is clear that the epigenetic state of an allele on one chromosome is not independent from the presence and epigenetic state of the allele on the other chromosome. These paramutation‐like phenomena remain incompletely understood, but are also common for natural epi‐alleles in the tomato Solanum lycopersicum (Gouil & Baulcombe, 2018) and also have been found in mice (Herman et al., 2003), suggesting that they could play a role in some instances of dominance modification in plants and animals.
Transvection, or trans‐sensing effects (Henikoff & Comai, 1998), has been observed in plants, fungi and mammals, and is another genetic phenomenon that can lead to trans‐ activation or trans‐ inhibition of alleles. In some cases, transvection seems to require direct physical interaction or at least proximity between the homologous chromosomes carrying the two alleles, and occurs when the two homologous chromosomes are paired during meiosis. A possible mechanism in this case is transcriptional activation of the promotor of a gene from one chromatid through recruitment of the transcriptional machinery by enhancers from the other homologous chromatid (hence implying that enhancers are acting not exclusively in cis‐; see Liu et al., 2008). Data in human, mouse and Drosophila seem to be consistent with this first model (but see Rodriguez et al., 2017). For instance, expression of the human cyclin D1 gene is controlled by trans‐allelic regulatory effects, whereby the presence of a translocated copy of the gene can alter the methylation and expression status of the other allele. The two interacting allelic sequences may be tethered at a peripheral region of the nucleolus, where transcriptional activity is low. The prevalence of transvection remains unclear, but insertion of a number of transgenes in different genomic locations in Drosophila revealed that about 30–52% of gene regulatory sequences may be associated with transvection (Mellert & Truman, 2012). Given the phylogenetic span over which epigenetic trans‐ repression was identified (including mammals, insects and plants), this may be an underappreciated phenomenon of gene regulation, with a substantial impact for our understanding of dominance.
Another fascinating example of trans‐ interaction is provided by olfactory receptor (OR) genes in mammals. Monoallelic expression of OR genes is observed at the level of an entire gene family, enabling high olfactive discrimination by individual cells. In the mouse for instance, coordinated expression of the 1,075 OR intact paralogs, each binding to specific chemical compounds, contributes to a broad spectrum of smell perception. These genes are expressed in neuronal cells of the main olfactory epithelium located in the nose in a monogenic and monoallelic fashion, whereby individual neuronal cells express a single allele of a single OR gene (Monahan & Lomvardas, 2015), therefore involving regulation both within (cases ④ and ⑤ in Fig. 3) and among (cases ②, ③, ⑥ and ⑦ in Fig. 3) a large repertoire of OR genes. This is accomplished at the single‐cell level by a complex process starting with collective epigenetic silencing of all OR genes of the genome by repressive histone marks such as H3K9me3 and H4K20me3. Expression of the lysine‐specific demethylase 1 protein (LSD1) results in slow local demethylation, de‐condensing the chromatin structure, progressively making a set of specific sequence motifs in the promotor available for transcription factor binding and contact with long‐range DNA enhancers (Lyons et al., 2013). The first randomly targeted OR sequence then becomes associated with the H3K4me3 histone mark typical of transcriptional initiation and ultimately becomes transcriptionally active. The produced OR protein then initiates a negative feedback loop that results in LSD1 downregulation, ensuring that other OR sequences can no longer become demethylated and access the active state. Ultimately, the silenced sequences become sequestered in repressive nuclear foci from which the long‐distance DNA enhancers are apparently excluded, further preventing transcription. Overall, the targeting of the expressed OR sequence corresponds to a ‘winner‐takes‐all’ process, whereby the first OR sequence to be transcribed and translated at a sufficient level induces inhibition of the other OR sequences in the genotype, a phenomenon known as ‘allelic exclusion’. The inhibition process not only concerns the other OR genes in the cluster but also the other allele of the chosen OR gene, hence in a ‘cis–trans’ manner akin to that defined herein. Any factor modifying the probability of expression of one given allele (and silencing of the others) would directly affect the dominance relationship in heterozygote individuals. At this point, however, how LSD1 is targeted to the chosen OR and whether LSD1 has different affinity for the different OR genes or different allelic variants and thus affects their propensity to become the chosen sequence is not known. Strikingly, inactivating components of the silencing feedback loop results in strong and reproducible bias towards expression of a small number of ORs (Lyons et al., 2014), suggesting a co‐evolutionary process between the strength of the promotor of the OR genes and the strength of the silencing phenomenon, resulting in approximately equal opportunity for all OR genes to be expressed in wild‐type genotypes. Hence, the ability to activate the silencing feedback loop rather than the ability to become demethylated in the first place might be the key to biasing transcriptional activity in heterozygotes. The evolutionary significance of such phenomena remains to be investigated, but they are clearly of interest in the context of how dominance could be controlled at the transcriptional level.
Several of the best‐documented cases of monoallelic expression involve self‐recognition genes. An early example concerns the gene encoding Toll‐like receptor 4 in the mouse, a situation that resembles allelic exclusion (Pereira et al., 2003). Mammalian T‐cells also show a deterministic developmental switch from monoallelic to biallelic transcription of the Gata3 (a transcription factor specifically recognizing GATA sequences) in about half of the developing T‐cell progenitors. This mono‐allelic versus bi‐allelic transcription is stably established in a parent‐of‐origin‐independent manner and the identity of the transcribed allele does not correlate with the classical repressive H3K4me3 and activating H3K27me3 epigenetic marks (Ku et al., 2015). How the switch is controlled (how the gene on the silent chromosome transitions from a silent to an expressed state) is currently under study but is clearly of interest to understand dominance between alleles: any factor modifying the expression threshold would directly affect the dominance relationship in heterozygote individuals.
Finally, a recently uncovered mechanism of mono‐allelic expression stems from dominance at the gene controlling self‐incompatibility in pollen of Brassicaceae plants (SCR), in which the interallelic interaction is mediated by small RNAs (sRNAs) produced by the dominant SCR allele targeting specific sequence motifs in the promotor of the recessive allele, leading to transcriptional repression possibly through DNA methylation (Tarutani et al., 2010; Durand et al., 2014). Specific interactions between the regulatory regions of the different SCR alleles therefore generate a dominance hierarchy between alleles (case ④ in Fig. 3), preventing expression of co‐dominant phenotypes that would be characterized by limited mating success. The evolution of these sRNAs seems complex with several emergences and losses (Durand et al., 2014), revealing a high potential for dominance evolution at the SCR gene, probably driven by strong positive selection on dominance at the SCR gene enhancing pollen mating success (Llaurens et al., 2009; Schoen & Busch, 2009).
Overall, it is clear that a variety of mechanisms can cause allele‐specific expression, as described in Fig. 3, such that transcription of the two alleles in heterozygote individuals does not need to be purely additive, as is assumed in many models of gene expression. Instead, the two alleles in a diploid genotype can interact to various degrees, potentially leading to differences in transcript levels ranging from subtle allele‐specific biases to massive mono‐allelic expression. In other words, there is a number of ways by which the relative doses of the two alleles in a heterozygote can depart from additivity, and these variations can result in dominance at the phenotypic level. While the molecular mechanisms underlying these phenomena are still not fully characterized, recent studies have highlighted the role of small RNAs and DNA methylation on modifications of allele expression, although the importance of such mechanisms at the genome scale is still unclear.
V. DISCUSSION
(1). Dominance as an evolved property of the genotype‐to‐phenotype map
The importance of dominance on the evolutionary fate of alleles, as well as the possibility for dominance to evolve have been highlighted by population geneticists since the early 20th century. The molecular mechanisms involved in dominance relationships and their modifications have been investigated by functional geneticists, sometimes using a drastically different vocabulary (Table 1). Results obtained in these research fields have rarely been combined, preventing drawing general conclusions on the evolutionary significance of dominance interactions between alleles. Dominance can be viewed as a specific genetic interaction occurring between alleles within a locus, and is as such sometimes covered by general models of gene expression based on molecular networks. The important theoretical literature on the evolution of gene regulatory networks might thus complement population genetics models focusing on dominance modifiers. Models of evolution of regulatory networks usually assumed an interaction graph between genes so that mutants at a given locus can affect the expression of another gene, depending on the property of the graph.
Dominance has been considered as one of the simplest form of robustness, i.e. as an evolved property of the genotype‐to‐phenotype map, buffering the effect of deleterious mutations during development (Bagheri & Wagner, 2004; Bagheri, 2006). This idea has assumed several names following the debate between Wright, Haldane and Fisher about the evolution of dominance. Haldane (1930) introduced the concept of ‘factor of safety’ and considered the plateau of the enzymatic metabolic pathway as a ‘buffer’ to mutations. In the literature dealing with the evolution of genotype‐to‐phenotype maps, for instance of genes implied in development (Wagner, Booth & Bagheri‐Chaichian, 1997; Rice, 2002; Siegal & Bergman, 2002), dominance was rather referred to as a form of ‘canalization’ (Rendel, 1967) or ‘robustness’ (Bagheri & Wagner, 2004). Strikingly, however, with the exception of Bagheri & Wagner (2004), most of the recent studies interested in the evolution of canalization or robustness ignored dominance as a possible evolved by‐product. Indeed, most models either considered haploid individuals or additive effects of alleles at a given locus (e.g. Siegal & Bergman, 2002; Rünneburger & Le Rouzic, 2016). Most importantly, in such models, phenotype and fitness landscapes do not emerge from an explicit mechanistic or physiological model, which make them arbitrary to some extent, and precludes the evolution of dominance. As acknowledged by Bagheri (2006), synthesizing the physiological and evolutionary mechanisms underlying dominance is a long‐standing issue. Such a synthesis has not been achieved yet, possibly because of the still limited cross‐talk between functional biologists and evolutionary biologists (Plutynski, 2008; Billiard & Castric, 2011).
The relative independence between the modifier theory for the evolution of dominance or gene expression on the one hand, and the evolution of regulatory networks, canalization and robustness on the other hand is surprising for several reasons. First, because population genetics models dealing with the evolution of gene regulatory networks are essentially modifier models, with several interacting modifier loci. This is the approach taken by Wagner et al. (1997), Bagheri & Wagner (2004) and Fyon et al. (2015). Second, because G.P. Wagner was originally involved in both the development of the modifier theory, in particular to study the evolution of dominance (Wagner, 1981; Wagner & Bürger, 1985; Bagheri & Wagner, 2004) and of the theory dealing with the evolution of canalization and robustness (Wagner et al., 1997). Actually, Wagner explicitly links the evolution of dominance with the evolution of robustness (Bagheri & Wagner, 2004). Finally, one can see no fundamental reason why the evolutionary and population genetics of robustness, canalization, dominance and gene expression should follow separate paths. Specific models assuming competition of alleles for the transcription machinery or cooperation between alleles enhancing attraction of transcription factors could shed light on dominance at the expression level. We thus believe that a synthesis between the two theoretical frameworks is crucially needed, for instance to clarify the link between the concepts of robustness and dominance. This synthesis would aim at bringing a general answer to the question ‘how is dominance changed by the fixation of a mutant at any locus in the gene network, via the evolution of the genotype‐to‐phenotype map?’. This would require determining the conditions under which mutants affecting dominance (among other things) can invade a population in (i) a given gene network, (ii) a physiological model linking genotype to phenotype, and (iii) an ecological model linking phenotype to fitness (Fig. 1). Such a formulation would embrace all models dealing with the evolution of dominance, and would help clarify the relationship between dominance and epistasis.
(2). From models to empirical data: a necessary cross‐talk between evolutionary and functional biology
The limited cross‐talk between evolutionary and functional biology has prevented questioning the relevance of the assumptions used in the models of dominance evolution via modifiers. Three categories of assumptions can be questioned (Fig. 1): (i) how likely is the genetic architecture assumed to underlie the dominance modification; (ii) is the effect of the dominance modifier on the phenotype ecologically relevant; and (iii) how does the phenotype translate into fitness variation? Following these criteria, three main classes of model can be identified. (i) Most modifier models assume that dominance modifiers directly modify the fitness of the heterozygotes, without providing any hypothesis on the molecular mechanisms involved (e.g. Fisher, 1928; Otto & Bourguet, 1999; see Section I). These models are conceptual and only prove that dominance can evolve, but are difficult to apply to actually documented genes. (ii) Some models explicitly consider a gene network (e.g. Omholt et al., 2000; Gilchrist & Nijhout, 2001) but they do not define a fitness function, and as such, they cannot be used to analyse the invasion of dominance modifiers. Hence, while their assumptions about the processes underlying the genotype‐to‐phenotype map can be verified in experiments, their predictions for the evolution of dominance in natural populations are not straightforward. The model by Bagheri & Wagner (2004) makes specific assumptions about the links between the gene network, the phenotype and the effect of a modifier, but this model also assumes a linear relationship between the enzymatic flux and fitness. Under which ecological context this assumption is relevant remains undefined. (iii) Finally, models combining relatively precise genetic mechanisms for dominance, the effect of a modifier on the phenotype and on fitness have been developed more recently (e.g. Nuismer & Otto, 2005; Llaurens et al., 2009, 2015; Schoen & Busch, 2009; Fyon et al., 2015). In these models, the genotype–phenotype‐fitness map is clearly defined in a specific ecological context. Their specific predictions allow experimental validations of the evolution of dominance. The predictions of models on the evolution of dominance in the self‐incompatibility system in flowering plants (Llaurens et al., 2009; Schoen & Busch, 2009) have already partially been verified: the modifier genes have been detected (for a review see Billiard & Castric, 2011), their effects on the genotype‐to‐phenotype map and their tight association with the self‐incompatibility locus validated (Durand et al., 2014), as well as the asymmetry between female and male phenotypes (Llaurens et al., 2008). Hence, we hope that our review will stimulate the development of precise models about the evolution of dominance in ecologically relevant contexts, providing predictions that could be experimentally tested.
(3). Documenting the molecular basis of dominance to shed light on its evolution
In this review, we noted that dominance modification through different pathways (involving cis or trans mechanisms for instance, see Fig. 3) lead to different evolutionary outcomes, so that population genetics models investigating the evolution of dominance or of regulatory networks may be more relevant when considering known molecular mechanisms involved in changes in allele expression. Conversely, understanding the observed dominance patterns may require an evolutionary perspective: for instance, changes in allele expression often involve methylation mechanisms, allowing rapid changes in expression levels triggered by a limited number of genetic changes. Because selection on dominance acts in heterozygotes only and because polymorphism is transient for most loci, changes in expression triggered by simple variations might be favoured. Contrasting selection regimes acting on the different alleles might therefore favour different molecular mechanisms of dominance.
Furthermore, our review of molecular mechanisms highlights that dominance can arise at different levels of integration (Fig. 1), so that equal allelic expression may still lead to dominance at the protein or organismal levels through a variety of mechanisms: understanding dominance therefore requires precise dissection of the developmental mechanisms involved in trait variations. Finally, investigating the consequences of dominance variations on individual fitness is needed to predict evolutionary outcomes that might differ among environments. We hope that this review will motivate the combination of research efforts from different fields for a better understanding of dominance mechanisms and evolution.
VI. CONCLUSIONS
The literature on the evolution of dominance has been very active in recent years, and overall it is now clear that dominance may arise at different levels of integration: from biases in allele‐specific expression to organismal traits, involving a diverse array of molecular interactions and physiological and developmental properties, with contrasting consequences on dominance evolution.
Accordingly, mounting empirical evidence also shows that dominance can and has evolved within and across species. This can be an indirect consequence of the evolution of regulatory networks within or between species, but also of natural selection acting directly on the dominance of emerging or persistent alleles within populations.
In this review, we argue that it is now possible to provide an integrative view of dominance as resulting from the combination of general processes of gene regulation, and being an emerging property of these more general processes rather a property in itself.
Overall, our review highlights the diversity of processes by which dominance/recessivity can arise and evolve. We argue that these different processes can be seen as different layers of the same genetic, phenotypic and ecological integration framework (Fig. 1) rather than as mutually exclusive explanations, as the literature in this field has too often suggested.
VII. ACKNOWLEDGEMENTS
The authors would like to thank Louis Bernatchez for inspiring this literature review and Dominique de Vienne and Vincent Colot for insightful comments. We acknowledge funding by the European Research Council (NOVEL project, grant number 648321).
REFERENCES
- Akram, A. & Inman, R. D. (2012). Immunodominance: a pivotal principle in host response to viral infections. Clinical Immunology 143(2), 99–115. [DOI] [PubMed] [Google Scholar]
- Anderson, T. M. , Candille, S. I. , Musiani, M. , Greco, C. , Stahler, D. R. , Smith, D. W. , Padhukasahasram, B. , Randi, E. , Leonard, J. A. & Bustamante, C. D. (2009). Molecular and evolutionary history of melanism in North American gray wolves. Science 323(5919), 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias, M. , Le Poul, Y. , Boisseau, R. , Chouteau, M. , Théry, M. & Llaurens, V. (2016). Crossing fitness valleys: empirical estimation of adaptive landscape associated with polymorphic mimicry. Proceedings of the Royal Society B: Biological Sciences 283, 20160391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri, H. C. (2006). Unresolved boundaries of evolutionary theory and the question of how inheritance systems evolve: 75 years of debate on the evolution of dominance. Journal of Experimental Zoology 306B, 329–359. [DOI] [PubMed] [Google Scholar]
- Bagheri, H. C. & Wagner, G. P. (2004). Evolution of dominance in metabolic pathways. Genetics 168(3), 1713–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri‐Chaichian, H. , Hermisson, J. , Vaisnys, J. R. & Wagner, G. P. (2003). Effects of epistasis on phenotypic robustness in metabolic pathways. Mathematical Biosciences 184(1), 27–51. [DOI] [PubMed] [Google Scholar]
- Baker, C. L. , Petkova, P. , Walker, M. , Flachs, P. , Mihola, O. , Trachtulec, Z. , Petkov, P. M. & Paigen, K. (2015). Multimer formation explains allelic suppression of PRDM9 recombination hotspots. PLoS Genetics 11(9), e1005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barson, N. J. , Aykanat, T. , Hindar, K. , Baranski, M. , Bolstad, G. H. , Fiske, P. , Jacq, C. , Jensen, A. J. , Johnston, S. E. & Karlsson, S. (2015). Sex‐dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 528, 405–408. [DOI] [PubMed] [Google Scholar]
- Billiard, S. & Castric, V. (2011). Evidence for Fisher's dominance theory: how many ‘special cases’? Trends in Genetics 27(11), 441–445. [DOI] [PubMed] [Google Scholar]
- Bost, B. & Veitia, R. A. (2014). Dominance and interloci interactions in transcriptional activation cascades: models explaining compensatory mutations and inheritance patterns. Bioessays 36(1), 84–92. [DOI] [PubMed] [Google Scholar]
- Bottani, S. & Veitia, R. A. (2017). Hill function‐based models of transcriptional switches: impact of specific, nonspecific, functional and nonfunctional binding. Biological Reviews 92(2), 953–963. [DOI] [PubMed] [Google Scholar]
- Bürger, R. (1983). Dynamics of the classical genetic model for the evolution of dominance. Mathematical Biosciences 67(2), 125–143. [Google Scholar]
- Bürger, R. , Bagheri, H. C. , Jorgensen, S. & Fath, B. (2008). Dominance and its evolution. In Encyclopedia of Ecology (eds Jorgensen S. E. and Fath B.), pp. 945–952. Elsevier, Amsterdam. [Google Scholar]
- Burghgraeve, N. , Simon, S. , Barral, S. , Foibis‐Loisy, I. , Holl, A.‐C. , Ponitzki, C. , Schmitt, E. , Vekemans, X. & Castric, V. (2020). Base‐pairing requirements for small RNA‐mediated gene silencing of recessive self‐incompatibility alleles in Arabidopsis halleri . Genetics 215, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D. & Charlesworth, B. (1975). Theoretical genetics of Batesian mimicry. 3. Evolution of dominance. Journal of Theoretical Biology 55(2), 325–337. [DOI] [PubMed] [Google Scholar]
- Chen, Z. J. , Comai, L. & Pikaard, C. S. (1998). Gene dosage and stochastic effects determine the severity and direction of uniparental ribosomal RNA gene silencing (nucleolar dominance) in Arabidopsis allopolyploids. Proceedings of the National Academy of Sciences of the United States of America 95(25), 14891–14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, C. A. & Sheppard, P. M. (1960). The evolution of dominance under disruptive selection. Heredity 14, 73–87. [Google Scholar]
- Crowley, J. J. , Zhabotynsky, V. , Sun, W. , Huang, S. , Pakatci, I. K. , Kim, Y. , Wang, J. R. , Morgan, A. P. , Calaway, J. D. & Aylor, D. L. (2015). Analyses of allele‐specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nature Genetics 47(4), 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debat, V. & David, P. (2001). Mapping phenotypes: canalization, plasticity and developmental stability. Trends in Ecology & Evolution 16(10), 555–561. [Google Scholar]
- Diss, G. , Gagnon‐Arsenault, I. , Dion‐Coté, A.‐M. , Vignaud, H. , Ascencio, D. I. , Berger, C. M. & Landry, C. R. (2017). Gene duplication can impart fragility, not robustness, in the yeast protein interaction network. Science 355(6325), 630–634. [DOI] [PubMed] [Google Scholar]
- Durand, E. , Meheust, R. , Soucaze, M. , Goubet, P. , Gallina, S. , Poux, C. , Fobis‐Loisy, I. , Gaude, T. , Sarrazin, A. , Figeac, M. , Prat, E. , Marande, W. , Bergès, H. , Vekemans, X. , Billiard, S. , et al. (2014). Dominance hierarchy arising from the evolution of a complex small RNA regulatory network. Science 346(6214), 1200–1205. [DOI] [PubMed] [Google Scholar]
- Durica, D. & Krider, H. (1978). Studies on the ribosomal RNA cistrons in interspecific Drosophila hybrids. II. Heterochromatic regions mediating nucleolar dominance. Genetics 89(1), 37–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre‐Walker, A. & Keightley, P. D. (2007). The distribution of fitness effects of new mutations. Nature Reviews Genetics 8(8), 610–618. [DOI] [PubMed] [Google Scholar]
- Feldman, M. W. & Karlin, S. (1971). The evolution of dominance: a direct approach through the theory of linkage and selection. Theoretical Population Biology 2(4), 482–492. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A. (1928). The possible modification of the response of the wild type to recurrent mutations. The American Naturalist 62, 115–126. [Google Scholar]
- Fisher, R. A. (1935). The sheltering of lethals. The American Naturalist 69(724), 446–455. [Google Scholar]
- Fragata, I. , Blanckaert, A. , Dias Louro, M. A. , Liberles, D. A. & Bank, C. (2019). Evolution in the light of fitness landscape theory. Trends in Ecology & Evolution 34(1), 69–82. [DOI] [PubMed] [Google Scholar]
- Fyon, F. , Cailleau, A. & Lenormand, T. (2015). Enhancer runaway and the evolution of diploid gene expression. PLoS Genetics 11(11), e1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur, U. , Li, K. , Mei, S. Q. & Liu, G. S. (2013). Research progress in allele‐specific expression and its regulatory mechanisms. Journal of Applied Genetics 54(3), 271–283. [DOI] [PubMed] [Google Scholar]
- Gilchrist, M. A. & Nijhout, H. F. (2001). Nonlinear developmental processes as sources of dominance. Genetics 159(1), 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjuvsland, A. B. , Hayes, B. J. , Meuwissen, T. H. , Plahte, E. & Omholt, S. W. (2007). Nonlinear regulation enhances the phenotypic expression of trans‐acting genetic polymorphisms. BMC Systems Biology 1(1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouil, Q. & Baulcombe, D. C. (2018). Paramutation‐like features of multiple natural epialleles in tomato. BMC Genomics 19(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves, I. K. , Eichten, S. R. , Groszmann, M. , Wang, A. , Ying, H. , Peacock, W. J. & Dennis, E. S. (2016). Twenty‐four–nucleotide siRNAs produce heritable trans‐chromosomal methylation in F1 Arabidopsis hybrids. Proceedings of the National Academy of Sciences of the United States of America 113(44), E6895–E6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves, I. K. , Groszmann, M. , Wang, A. , Peacock, W. J. & Dennis, E. S. (2014). Inheritance of trans chromosomal methylation patterns from Arabidopsis F1 hybrids. Proceedings of the National Academy of Sciences of the United States of America 111(5), 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane, J. B. S. (1927). A mathematical theory of natural and artificial selection, part V: selection and mutation. Proceedings of the Cambridge Philosophical Society 23, 838–844. [Google Scholar]
- Haldane, J. B. S. (1930). A note on Fisher's theory of the origin of dominance, and on a correlation between dominance and linkage. American Naturalist 64, 87–90. [Google Scholar]
- Haldane, J. B. S. (1933). The part played by recurrent mutation in evolution. The American Naturalist 67(708), 5–19. [Google Scholar]
- Haldane, J. B. S. (1956). The theory of selection for melanism in Lepidoptera. Proceedings of the Royal Society of London. Series B‐Biological Sciences 145(920), 303–306. [DOI] [PubMed] [Google Scholar]
- Hartfield, M. , Bataillon, T. & Glémin, S. (2017). The evolutionary interplay between adaptation and self‐fertilization. Trends in Genetics 33(6), 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama, K. , Watanabe, M. , Takasaki, T. , Ojima, K. & Hinata, K. (1998). Dominance relationships between S‐alleles in self‐incompatible Brassica campestris L. Heredity 80(2), 241–247. [Google Scholar]
- Heilig, E. A. , Gutti, U. , Tai, T. , Shen, J. & Kelleher, R. J. (2013). Trans‐dominant negative effects of pathogenic PSEN1 mutations on γ‐secretase activity and Aβ production. Journal of Neuroscience 33(28), 11606–11617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff, S. & Comai, L. (1998). Trans‐sensing effects: the ups and downs of being together. Cell 93(3), 329–332. [DOI] [PubMed] [Google Scholar]
- Herman, H. , Lu, M. , Anggraini, M. , Sikora, A. , Chang, Y. , Yoon, B. J. & Soloway, P. D. (2003). Trans allele methylation and paramutation‐like effects in mice. Nature Genetics 34(2), 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz, I. (1987). Functional inactivation of genes by dominant negative mutations. Nature 329(6136), 219–222. [DOI] [PubMed] [Google Scholar]
- Huber, C. D. , Durvasula, A. , Hancock, A. M. & Lohmueller, K. E. (2018). Gene expression drives the evolution of dominance. Nature Communications 9(1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Sanchez, G. , Childs, B. & Valle, D. (2001). Human disease genes. Nature 409(6822), 853–855. [DOI] [PubMed] [Google Scholar]
- Joly, S. , Rauscher, J. T. , Sherman‐Broyles, S. L. , Brown, A. H. D. & Doyle, J. J. (2004). Evolutionary dynamics and preferential expression of homeologous 18S‐5.8S‐26S nuclear ribosomal genes in natural and artificial Glycine allopolyploids. Molecular Biology and Evolution 21(7), 140–1421. [DOI] [PubMed] [Google Scholar]
- Kacser, H. & Burns, J. A. (1981). The molecular basis of dominance. Genetics 97, 639–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenegger, E. & Ober, D. (2015). Paralogue interference affects the dynamics after gene duplication. Trends in Plant Science 20(12), 814–821. [DOI] [PubMed] [Google Scholar]
- Karlin, S. & McGregor, J. (1974). Towards a theory of the evolution of modifier genes. Theoretical Population Biology 5(1), 59–103. [DOI] [PubMed] [Google Scholar]
- Keightley, P. D. (1996). Nature of deleterious mutation load in Drosophila . Genetics 144(4), 1993–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble, H. , Nghe, P. & Tenaillon, O. (2019). Recent insights into the genotype–phenotype relationship from massively parallel genetic assays. Evolutionary Applications 12(9), 1721–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, F. A. & Koonin, E. V. (2004). A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends in Genetics 20, 287–290. [DOI] [PubMed] [Google Scholar]
- Ku, C. J. , Lim, K. C. , Kalantry, S. , Maillard, I. , Engel, J. D. & Hosoya, T. (2015). A monoallelic‐to‐biallelic T‐cell transcriptional switch regulates GATA3 abundance. Genes & Development 29(18), 1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusaba, M. , Tung, C.‐W. , Nasrallah, M. E. & Nasrallah, J. B. (2002). Monoallelic expression and dominance interactions in anthers of self‐incompatible Arabidopsis lyrata . Plant Physiology 128, 17–20. [PMC free article] [PubMed] [Google Scholar]
- Lappalainen, T. , Sammeth, M. , Friedländer, M. R. , AC‘t Hoen, P. , Monlong, J. , Rivas, M. A. , Gonzalez‐Porta, M. , Kurbatova, N. , Griebel, T. & Ferreira, P. G. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501(7468), 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Poul, Y. , Whibley, A. , Chouteau, M. , Prunier, F. , Llaurens, V. & Joron, M. (2014). Evolution of dominance mechanisms at a butterfly mimicry supergene. Nature Communications 5(5644), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Yu, X. , Li, K. , Klejnot, J. , Yang, H. , Lisiero, D. & Lin, C. (2008). Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis . Science 322(5907), 1535–1539. [DOI] [PubMed] [Google Scholar]
- Llaurens, V. , Billiard, S. , Castric, V. & Vekemans, X. (2009). Evolution of dominance in sporophytic self‐incompatibility systems: I. Genetic load and co‐evolution of levels of dominance in pollen and pistil. Evolution 63(9), 2427–2437. [DOI] [PubMed] [Google Scholar]
- Llaurens, V. , Billiard, S. , Leducq, J.‐B. , Castric, V. & Vekemans, X. (2008). Does frequency‐dependent selection with complex dominance interactions accurately predict allelic frequencies at the self‐incompatibility locus in Arabidopsis halleri? Evolution 62(10), 2545–2557. [DOI] [PubMed] [Google Scholar]
- Llaurens, V. , Joron, M. & Billiard, S. (2015). Molecular mechanisms of dominance evolution in Müllerian mimicry. Evolution 69(12), 3097–3108. [DOI] [PubMed] [Google Scholar]
- Llaurens, V. , Whibley, A. & Joron, M. (2017). Genetic architecture and balancing selection: the life and death of differentiated variants. Molecular Ecology 26(9), 2430–2448. [DOI] [PubMed] [Google Scholar]
- Lyons, D. B. , Allen, W. E. , Goh, T. , Tsai, L. , Barnea, G. & Lomvardas, S. (2013). An epigenetic trap stabilizes singular olfactory receptor expression. Cell 154(2), 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, D. B. , Magklara, A. , Goh, T. , Sampath, S. C. , Schaefer, A. , Schotta, G. & Lomvardas, S. (2014). Heterochromatin‐mediated gene silencing facilitates the diversification of olfactory neurons. Cell Reports 9(3), 884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet, J. (1986). Hybrid zones of Heliconius butterflies in Panama and the stability and movement of warning colour clines. Heredity 56(2), 191–202. [Google Scholar]
- Mallet, J. & Barton, N. (1989). Strong natural selection in a warning‐color hybrid zone. Evolution 43(2), 421–431. [DOI] [PubMed] [Google Scholar]
- Manna, F. , Martin, G. & Lenormand, T. (2011). Fitness landscapes: an alternative theory for the dominance of mutation. Genetics 189(3), 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, G. (2014). Fisher's geometrical model emerges as a property of complex integrated phenotypic networks. Genetics 197(1), 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo, O. & Burger, R. (1997). The evolution of dominance: a theory whose time has passed? Biological Reviews 72(1), 97–110. [Google Scholar]
- Mellert, D. J. & Truman, J. W. (2012). Transvection is common throughout the Drosophila genome. Genetics 191(4), 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milesi, P. , Weill, M. , Lenormand, T. & Labbé, P. (2017). Heterogeneous gene duplications can be adaptive because they permanently associate overdominant alleles. Evolution Letters 1(3), 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohannath, G. , Pontvianne, F. & Pikaard, C. S. (2016). Selective nucleolus organizer inactivation in Arabidopsis is a chromosome position‐effect phenomenon. Proceedings of the National Academy of Sciences of the United States of America 113(47), 13426–13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan, K. & Lomvardas, S. (2015). Monoallelic expression of olfactory receptors. Annual Review of Cell and Developmental Biology 31, 721–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, H. J. (1932). Further studies on the nature and causes of gene mutations. In Proceedings of the Sixth International Congress of Genetics, Ithaca, New York, USA, pp. 213–255.
- Naithani, S. , Chookajorn, T. , Ripoll, D. R. & Nasrallah, J. B. (2007). Structural modules for receptor dimerization in the S‐locus receptor kinase extracellular domain. Proceedings of the National Academy of Sciences of the United States of America 104(29), 12211–12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navashin, M. (1934). Chromosome alterations caused by hybridisation and their bearing upon certain general genetic problems. Cytologia 5, 169–203. [Google Scholar]
- Nijhout, F. H. (2003). Polymorphic mimicry in Papilio dardanus: mosaic dominance, big effect and origins. Evolution and Development 5(6), 579–592. [DOI] [PubMed] [Google Scholar]
- Nowak, M. D. , Russo, G. , Schlapbach, R. , Huu, C. N. , Lenhard, M. & Conti, E. (2015). The draft genome of Primula veris yields insights into the molecular basis of heterostyly. Genome Biology 16(12), 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuismer, S. L. & Otto, S. P. (2005). Host‐parasite interactions and the evolution of gene expression. PLoS Biology 3(7), 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donald, P. & Barrett, J. A. (1973). Evolution of dominance in polymorphic Batesian mimicry. Theoretical Population Biology 4(2), 173–192. [Google Scholar]
- Omholt, S. W. , Plahte, E. , Øyehaug, L. & Xiang, K. (2000). Gene regulatory networks generating the phenomena of additivity, dominance and epistasis. Genetics 155(2), 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi, J. D. , Petersen, J. , Tan, Y. H. , Huynh, M. , Willett, Z. J. , Ramarathinam, S. H. , Eggenhuizen, P. J. , Loh, K. L. , Watson, K. A. & Gan, P. Y. (2017). Dominant protection from HLA‐linked autoimmunity by antigen‐specific regulatory T cells. Nature 545(7653), 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A. (1991). A test of Fisher theory of dominance. Proceedings of the National Academy of Sciences of the United States of America 88(24), 11413–11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A. & Betancourt, A. J. (2001). Haldane's sieve and adaptation from the standing genetic variation. Genetics 157(2), 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, S. P. & Bourguet, D. (1999). Balanced polymorphisms and the evolution of dominance. The American Naturalist 153, 561–574. [DOI] [PubMed] [Google Scholar]
- Pannell, J. (1997). The maintenance of gynodioecy and androdioecy in a metapopulation. Evolution 51(1), 10–20. [DOI] [PubMed] [Google Scholar]
- Pannell, J. R. , Dorken, M. E. & Eppley, S. M. (2005). Haldane's Sieve'in a metapopulation: sifting through plant reproductive polymorphisms. Trends in Ecology & Evolution 20(7), 374–379. [DOI] [PubMed] [Google Scholar]
- Papp, B. , Pal, C. & Hurst, L. D. (2003). Dosage sensitivity and the evolution of gene families in yeast. Nature 424(6945), 194–197. [DOI] [PubMed] [Google Scholar]
- Peischl, S. & Burger, R. (2008). Evolution of dominance under frequency‐dependent intraspecific competition. Journal of Theoretical Biology 251(2), 210–226. [DOI] [PubMed] [Google Scholar]
- Peischl, S. & Schneider, K. A. (2010). Evolution of dominance under frequency‐dependent intraspecific competition in an assortatively mating population. Evolution 64(2), 561–582. [DOI] [PubMed] [Google Scholar]
- Pereira, J. P. , Girard, R. , Chaby, R. , Cumano, A. & Vieira, P. (2003). Monoallelic expression of the murine gene encoding Toll‐like receptor 4. Nature Immunology 4(5), 464–470. [DOI] [PubMed] [Google Scholar]
- Plutynski, A. (2008). Explaining how and explaining why: developmental and evolutionary explanations of dominance. Biology and Philosophy 23(3), 363–381. [Google Scholar]
- Pontes, O. , Lawrence, R. J. , Neves, N. , Silva, M. , Lee, J. H. , Chen, Z. J. , Viegas, W. & Pikaard, C. S. (2003). Natural variation in nucleolar dominance reveals the relationship between nucleolus organizer chromatin topology and rRNA gene transcription in Arabidopsis . Proceedings of the National Academy of Sciences of the United States of America 100(20), 11418–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteous, J. W. (1996). Dominance—one hundred and fifteen years after Mendel's paper. Journal of Theoretical Biology 182(3), 223–232. [DOI] [PubMed] [Google Scholar]
- Porter, A. H. , Johnson, N. A. & Tulchinsky, A. Y. (2017). A new mechanism for mendelian dominance in regulatory genetic pathways: competitive binding by transcription factors. Genetics 205(1), 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss, S. B. , Costa‐Nunes, P. , Tucker, S. , Pontes, O. , Lawrence, R. J. , Mosher, R. , Kasschau, K. D. , Carrington, J. C. , Baulcombe, D. C. , Viegas, W. & Pikaard, C. S. (2008). Multimegabase silencing in nucleolar dominance involves siRNA‐directed DNA methylation and specific methylcytosine‐binding proteins. Molecular Cell 32(5), 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal, F. A. , Mandáková, T. , Soto‐Jiménez, L. M. , Greenhalgh, R. , Parrott, D. L. , Lutzmayer, S. , Steffen, J. G. , Nizhynska, V. , Mott, R. , Lysak, M. A. , Clark, R. M. & Nordborg, M. (2017). Epistatic and allelic interactions control expression of ribosomal RNA gene clusters in Arabidopsis thaliana . Genome Biology 18(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendel, J. M. (1967). Canalisation and Gene Control. Logos Press, London. [Google Scholar]
- Rice, S. H. (2002). A general population genetic theory for the evolution of developmental interactions. Proceedings of the National Academy of Sciences of the United States of America 99(24), 15518–15523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, J. D. , Myrick, D. A. , Falciatori, I. , Christopher, M. A. , Lee, T. W. , Hannon, G. J. & Katz, D. J. (2017). A model for epigenetic inhibition via transvection in the mouse. Genetics 207(1), 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronfort, J. & Glemin, S. (2013). Mating system, Haldane's sieve, and the domestication process. Evolution 67(5), 1518–1526. [DOI] [PubMed] [Google Scholar]
- Rosenblum, E. B. , Roempler, H. , Schoeneberg, T. & Hoekstra, H. E. (2010). Molecular and functional basis of phenotypic convergence in white lizards at White Sands. Proceedings of the National Academy of Sciences of the United States of America 107(5), 2113–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rünneburger, E. & Le Rouzic, A. (2016). Why and how genetic canalization evolves in gene regulatory networks. BMC Evolutionary Biology 16(1), 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savageau, M. A. (1992). Dominance according to metabolic control analysis: major achievement or house of cards? Journal of Theoretical Biology 154(1), 131–136. [DOI] [PubMed] [Google Scholar]
- Schoen, D. J. & Busch, J. W. (2009). The evolution of dominance in sporophytic self‐incompatibility systems. II. Mate availability and recombination. Evolution: International Journal of Organic Evolution 63(8), 2099–2113. [DOI] [PubMed] [Google Scholar]
- Shen, R. X. , Wang, L. , Liu, X. P. , Wu, J. , Jin, W. W. , Zhao, X. C. , Xie, X. R. , Zhu, Q. L. , Tang, H. W. , Li, Q. , Chen, L. T. & Liu, Y. G. (2017). Genomic structural variation‐mediated allelic suppression causes hybrid male sterility in rice. Nature Communications 8(1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal, M. L. & Bergman, A. (2002). Waddington's canalization revisited: developmental stability and evolution. Proceedings of the National Academy of Sciences of the United States of America 99(16), 10528–10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, H. G. & Priest, N. K. (2016). The evolution of sex‐specific dominance in response to sexually antagonistic selection. The American Naturalist 187(5), 658–666. [DOI] [PubMed] [Google Scholar]
- Stirnweis, D. , Milani, S. D. , Brunner, S. , Herren, G. , Buchmann, G. , Peditto, D. , Jordan, T. & Keller, B. (2014). Suppression among alleles encoding nucleotide‐binding–leucine‐rich repeat resistance proteins interferes with resistance in F1 hybrid and allele‐pyramided wheat plants. The Plant Journal 79(6), 893–903. [DOI] [PubMed] [Google Scholar]
- Tarutani, Y. , Shiba, H. , Iwano, M. , Kakizaki, T. , Suzuki, G. , Watanabe, M. , Isogai, A. & Takayama, S. (2010). Trans‐acting small RNA determines dominance relationships in Brassica self‐incompatibility. Nature 466(7309), 983–986. [DOI] [PubMed] [Google Scholar]
- Tishkoff, S. A. , Reed, F. A. , Ranciaro, A. , Voight, B. F. , Babbitt, C. C. , Silverman, J. S. , Powell, K. , Mortensen, H. M. , Hirbo, J. B. & Osman, M. (2007). Convergent adaptation of human lactase persistence in Africa and Europe. Nature Genetics 39(1), 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Hof, A. E. , Campagne, P. , Rigden, D. J. , Yung, C. J. , Lingley, J. , Quail, M. A. , Hall, N. , Darby, A. C. & Saccheri, I. J. (2016). The industrial melanism mutation in British peppered moths is a transposable element. Nature 534(7605), 102–105. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A. (2003). Nonlinear effects in macromolecular assembly and dosage sensitivity. Journal of Theoretical Biology 220(1), 19–25. [DOI] [PubMed] [Google Scholar]
- Veitia, R. A. (2006). The Biology of Genetic Dominance. Landes Bioscience, Georgetown. [Google Scholar]
- Veitia, R. A. , Caburet, S. & Birchler, J. A. (2018). Mechanisms of mendelian dominance. Clinical Genetics 93(3), 419–428. [DOI] [PubMed] [Google Scholar]
- Wagner, G. P. (1981). Feedback selection and the evolution of modifiers. Acta Biotheoretica 30(2), 79–102. [DOI] [PubMed] [Google Scholar]
- Wagner, G. P. , Booth, G. & Bagheri‐Chaichian, H. (1997). A population genetic theory of canalization. Evolution 51(2), 329–347. [DOI] [PubMed] [Google Scholar]
- Wagner, G. P. & Bürger, R. (1985). On the evolution of dominance modifiers II: a non‐equilibrium approach to the evolution of genetic systems. Journal of Theoretical Biology 113(3), 475–500. [DOI] [PubMed] [Google Scholar]
- Whibley, A. C. , Langlade, N. B. , Andalo, C. , Hanna, A. I. , Bangham, A. , Thebaud, C. & Coen, E. (2006). Evolutionary paths underlying flower color variation in Antirrhinum . Science 313(5789), 963–966. [DOI] [PubMed] [Google Scholar]
- Wilkie, A. O. (1994). The molecular basis of genetic dominance. Journal of Medical Genetics 31(2), 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. (1929). Fisher's theory of dominance. The American Naturalist 63, 274–279. [Google Scholar]
- Wright, S. (1934). Physiological and evolutionary theories of dominance. The American Naturalist 63, 25–53. [Google Scholar]
- Zhou, R. , Yang, G. H. & Shi, Y. G. (2017). Dominant negative effect of the loss‐of‐function gamma‐secretase mutants on the wild‐type enzyme through heterooligomerization. Proceedings of the National Academy of Sciences of the United States of America 114(48), 12731–12736. [DOI] [PMC free article] [PubMed] [Google Scholar]