

Abstract
Histones constitute the primary protein building blocks of the chromatin and play key roles in the dynamic control of chromatin compaction and epigenetic regulation. Histones are regulated by intricate mechanisms that alter their functionality and stability, thereby expanding the regulation of chromatin‐transacting processes. As such, histone degradation is tightly regulated to provide spatiotemporal control of cellular histone abundance. While several mechanisms have been implicated in controlling histone stability, here, we discuss proteasome‐dependent degradation of histones and the protein modifications that are associated with it. We then highlight specific cellular and physiological states that are associated with altered histone degradation by cellular proteasomes.
Keywords: histone degradation, histone modifications, proteasome, ubiquitin
Histones constitute the primary protein building blocks of the chromatin and play key roles in the dynamic control of chromatin compaction and epigenetic regulation. Histone degradation is tightly regulated to provide spatiotemporal control of histone abundance and distribution over the chromatin. Here, we discuss mechanisms of proteasome‐dependent degradation of histones. We then highlight specific cellular and physiological states which are associated with altered histone degradation by proteasomes.
Abbreviations
- CMA
chaperon‐mediated autophagy
- CP
catalytic particle
- DSB
DNA double‐strand breaks
- EGF
epidermal growth factor
- HDAC
histone deacetylases
- MAPP
MS analysis of proteolytic peptides
- Pol II
RNA polymerase II
- PROTAC
protein targeting chimeras
- PTMs
post‐translational modifications
- RP
regulatory particle
- SLE
systemic lupus erythematosus
- TSS
transcription start site
Introduction
The chromatin is a highly organized molecular complex whereby the DNA is wrapped around histones to form nucleosomes, the basic units responsible for compacting and safeguarding the DNA [1, 2, 3]. Canonical nucleosomes consist of two copies of each of the histone proteins H2A, H2B, H3, and H4, which can be further bound by the linker histone H1 that protects the internucleosomal DNA. The association of H1 with nucleosomes forms the chromatosome, which can compact the chromatin into higher‐order forms [4, 5, 6, 7]. This complex organization is key for controlling cellular processes that require chromatin modulation in a specific and temporally restricted manner, such as regulation of cell division, DNA damage response, gene expression, and cell fate [8, 9, 10, 11]. Nucleosome‐dependent chromatin modulation is also controlled by the regulation of histones’ transcriptional expression [12], mRNA stability [13], translation efficiency [13], and different post‐translational modifications (PTMs) [14]. Together, these processes endow histones with a tremendous plasticity for controlling the structure and function of the chromatin.
Another crucial layer of histone regulation is their proteolysis, which allows to control and alter histone abundance to meet the dynamic cellular demands [15, 16]. A key example is the degradation of chromatin‐unbound histones, which are extremely toxic to the cellular environment, due to their basic biochemical nature [17]. Accumulation of an unbound histone pool may occur under various conditions including hyperactive chromatin‐transacting processes that involve histone eviction like transcriptional changes, developmental transitions, response to DNA damage or damage to histones themselves, and imbalanced histone levels during S‐phase [15, 16]. Further, histone degradation is emerging as a main regulatory step in the fine‐tuned reorganization of chromatin architecture in response to developmental and environmental cues, and in response to cellular stress [18, 19, 20, 21, 22, 23, 24]. Taken together, tight regulation of the histone pool by degradation serves to both balance the changes in histone levels and chromatin binding to prevent toxicity, as well as to modulate the chromatin by tweaking histone levels.
Histone degradation has been shown to be mediated by essentially every cellular degradative machinery, including proteases, autophagy–lysosomes system, and the proteasome complex [15]. These different types of degradation underlie the versatile mechanisms required to maintain histone proteostasis in light of different functions and under distinct cellular conditions. Historically, the first observation that histones can undergo proteolysis was documented already in 1964 by Reid and Cole [25]. In the three subsequent decades, numerous papers reported the identification of different trypsin‐like and neutral proteases in the chromatin and cytoplasm that catalyze histone degradation [26], as well revealed proteasome‐dependent degradation of histones [27, 28]. Yet, these seminal studies did not fully delineate the cellular contexts and significance of such degradation events [15, 29]. These were followed by a latent era in the field until the discovery of protease‐dependent histone clipping and their potential involvement in epigenetic regulation [30, 31, 32], which reinvigorated the importance of histone proteolysis. Since then, major advances were achieved in the understanding of regulated histone degradation in cellular contexts by the two major selective degradation pathways of intracellular proteins, namely the proteasome and lysosome systems [18, 19, 20, 21, 22, 23, 24, 33, 34], which provide high specificity, selectivity, and fine‐tuning capabilities of protein abundance [35].
In this review, we will focus on proteasome‐dependent histone degradation (for a short review about autophagy‐mediated histone degradation please see Box 1). We will discuss the imperative role of distinct proteasome compositions in facilitating different types of histone degradation and highlight differences between ubiquitin‐dependent and ubiquitin‐independent processes. We will further describe mechanisms that allow for restriction and specificity in the targeting of subpopulations of histones to degradation, and the biological significance of such multilayered regulation. Finally, we will review histone degradation in the context of specific cellular and developmental conditions and human pathology.
Box 1. Histone degradation by autophagy.
While cellular autophagy is considered to target insoluble protein aggregations, long‐lived proteins, protein complexes, or organelles, to degradation, proteasomal degradation is thought to target short‐lived or soluble proteins. Histones are generally considered to be extremely stable proteins with half‐lives in the order of several months in mammalian cells [36]. However, the half‐live is representative of the contribution of chromatin‐bound histones, while non‐chromatin bound histones are rapidly degraded with a half‐life of 30–40 min in yeast cells [17, 37]. These data exemplify the need to regulate histone levels, both in their chromatin‐bound and unbound forms, in parallel by both the proteasomal and lysosomal systems [38, 39]. Chaperon‐mediated autophagy (CMA) was suggested to degrade histones to counteract histone chaperones, such as NASP which increase histone availability for immediate use in case of an unexpected demand [27]. Importantly, excess histones are rapidly degraded. Recently, it was shown that histone E3 ligases such as Hel2 and Pep5, which are localized to the cytosol, are able to degrade the excess histone in the cytoplasm [40]. A second lysosome‐dependent mechanism was shown to control the degradation of aberrant cytoplasmic chromatin fraction, a phenomena which is exacerbated in senescence [41].
Definitions:
Histone degradation/proteolysis: denotes the partial/full degradation of histones via the lysosome or proteasome.
Histone clipping: describes a process by which only part of the histone is degraded/clipped.
Histone turn‐over: this often implies turnover in chromatin‐binding or nucleosome‐binding rather than a change in total abundance of the protein.
Histone degradation in cellular homeostasis
Ubiquitin‐dependent proteasomal degradation of histones
Ubiquitination is the most prevalent way by which proteins are targeted for degradation by the proteasome. Ubiquitin‐dependent degradation involves an E1‐activating, E2‐conjugating, and E3‐ligating enzymes that work in concert to modify substrates [42]. Subsequent ubiquitination events can occur repetitively on the same ubiquitination site to form polyubiquitin chains (i.e., polyubiquitination), or on different acceptor lysine residues (i.e., multi mono‐ubiquitination), both can serve for the binding of the substrate to the ubiquitin receptors on the proteasome [43, 44, 45, 46]. Canonically, a ubiquitinated target protein is recognized and unfolded, in an ATP‐dependent manner, by a proteasome regulatory particle (RP; [47]). Then, the target protein gets translocated into the catalytic particle (CP) where it gets degraded into small peptide fragments [47]. The CP, also known as the 20S core of the proteasome, consists of four stacked rings. The outer rings contain seven α‐subunits while the inner rings contain seven β‐subunits, among them are the catalytic subunits, β1, β2, and β5 [47]. The 20S core of the proteasome may be further capped by different types of RPs that control substrate recognition delivery to the catalytic core, and include the 19S subunits [48], the 11S formed by PA28α, PA28β, or PA28γ, ECM29, PI31 [49], and PA200 [50], the latter is mostly associated with ubiquitin‐independent degradation and is discussed in the relevant section below. The 19S cap is the most common activator, and together with the 20S core, they form the 26S proteasome (reviewed extensively in [51, 52]). Interestingly, the different proteasome RPs vary in their intracellular localization. Whereas PA28γ and PA200 have been mostly associated with the nucleus, PA28α and β are mainly localized to the cytosol, and the 19S was shown to be abundant in all cellular compartments, including the nucleus [53, 54, 55]. Therefore, by assembling with different regulatory subunits, proteasomes may form compartment‐specific complexes that via distinct compositions may affect function and substrate selectivity (e.g., ubiquitination‐dependency, localization, specific protein preferences).
Notably, however, not all ubiquitination events result in degradation of proteins, as numerous other signaling‐related functions have been associated with ubiquitination (see [56] for extensive review on ubiquitination code). In fact, ubiquitination of histone H2A, which was the first ubiquitination event to be reported in the literature [57], is not associated with degradation but rather is associated with transcriptional repression through the polycomb‐repressive complexes PRC1&2 [56]. Due to the scope of this review, we will focus on degradation‐related events in regulation of histones (relevant information on histone ubiquitination activities which are not covered herein may be found here [58]).
While all canonical histones were shown to be regulated by ubiquitin, mechanistic understanding regarding the effect of ubiquitination on histone stability was largely limited to H3 and more recently was expanded to H2B [19, 20, 21, 22, 33, 40] (Fig. 1, Table 1). In a seminal study describing regulated degradation of histones, surplus nonchromatin‐bound histone H3 was shown to undergo polyubiquitination that leads to its degradation by the proteasome [33]. Importantly, it was shown that this ubiquitination is partly mediated by the E2 ubiquitin‐conjugating enzymes Ubc4/5 and the HECT domain‐containing E3 ligase, Tom1. Notably, yeast strains lacking those enzymes were sensitive to histone overexpression and inhibition of replication due to the resultant massive excess in histones levels [33]. Few years after this study, four additional E3 ligases that are involved in histone H3 degradation were identified in yeast [40]. These were Pep5, Snt2, and Hel1 and Hel2. Yeast strains mutant in these enzymes were also sensitive to replication inhibitors and ectopic overexpression of histones.
Fig. 1.
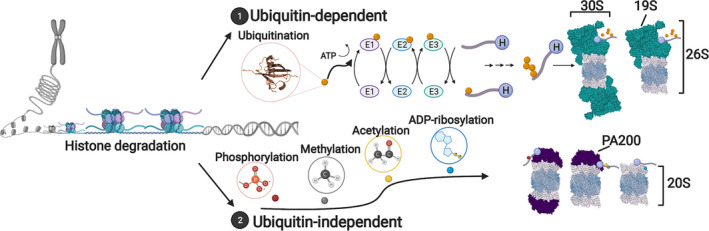
Ubiquitin‐dependent and independent degradation of histones. Schematic of histone degradation via (1) ubiquitin‐dependent or (2) ubiquitin‐independent pathways. PDB solved structures were used to generate this figure; 6KWY (PA200), 6RGQ (20S), 6FVW (19S) and 1AAR (Ubiquitin). Created with BioRender.com.
Table 1.
Summary of known and validated PTM events on histone proteins, which leads to ubiquitin‐dependent and ubiquitin‐independent degradation.
| Histone | Pre‐event | Site | PTM | Regulatory subunit | References | |
|---|---|---|---|---|---|---|
| Ubiquitin‐dependent | Canonical H3 | Phosphorylation of Y99 | N/A | PolyUb | N/A | [33, 40] |
| Canonical H3 | Phosphorylation of T11 | K4 | PolyUb | N/A | [21] | |
| H3.3 | N/A | N/A | PolyUb | N/A | [22] | |
| H2A.Z | Deacetylation of K15 | K115 | PolyUb | N/A | [64, 65] | |
| H2A.Z | Deacetylation of K15 | K121 | PolyUb | N/A | [64, 65] | |
| H2B | Multiple sites | PolyUb | [19] | |||
| Ubiquitin‐independent | H4 | R3 | De‐methylation by PRMT1 | PA200 | [23, 75] | |
| H4 | K16 | Acetylation | PA200 | [69, 71] | ||
| H3 (H3.3, H3.1) | K16 | Acetylation | PA200 | [69, 71] | ||
| H (all) | N‐terminal tail | Acetylation | N/A | [72] | ||
| H (all) | Oxidized histones | ADP‐ribosylated 20S | [113] |
A recent study provided an elegant description of ubiquitin‐dependent proteasome‐mediated degradation of histones in multicellular organisms and demonstrated how this directly affects cell programming during differentiation [20]. Specifically, the authors demonstrated that polyubiquitination and degradation of H3 are mediated by the ubiquitin‐conjugating enzyme Ube2k in both human embryonic stem cells and Caenorhabditis elegans germ cells [20]. Knockdown of Ube2k was shown to result in a global increase in the levels of histone H3, as well as of critical histone marks, including the repressive marks H3K9me3. Finally, aberrant H3K9me3 following Ube2k knockdown in embryonic stem cells was shown to revoke the upregulation of neuronal genes during differentiation. Further exploration might allude to whether beyond the change in H3K9me3, loss of Ube2k drives additional mechanisms that affect the transcriptional cellular program, and the nature by which H3 levels affect the histone modifications or vice versa.
Protein ubiquitination is often preceded by a priming removal or addition of other modifications on the same protein, such as phosphorylation, acetylation, methylation, and SUMOylation [59, 60, 61]. Phosphorylation provides substantial specificity for targeting distinct pools of histones to degradation. For example, phosphorylation of tyrosine 99 (Y99) in H3 by Rad53, which is required for efficient degradation of the H3 protein, serves to target only the nonchromatin‐bound excess H3 for degradation, while the nucleosomal H3 is spared from any inadvertent degradation [33]. This is achieved as Y99 is buried in the nucleosome and is thus not accessible for phosphorylation in a nucleosomal context [62]. In fact, the H3 Y99 residue is located in the binding site between the H3–H4 dimer and it stabilizes the heterodimer via a ring stacking interaction with the F61 residue of histone H4. This strongly implies that phosphorylation of Y99 not only primes unbound H3 for ubiquitin‐dependent degradation, but further facilitates this process by disrupting the ring stacking interaction and precluding H3–H4 dimer formation, leaving excess H3 susceptible for phosphorylation. Phosphorylation of Y99 then recruits the E2 and E3 enzymes (Ubc3, Ubc5, and Tom1) to H3, thereby inducing its ubiquitination and subsequent degradation [33, 63].
Phosphorylation is a hallmark of signal transduction pathways, translating extracellular signals to gene expression changes. A noteworthy example demonstrates how phosphorylation may induce transcriptional changes in response to growth factors by priming for histone degradation. Specifically, epidermal growth factor (EGF) was shown to induce the phosphorylation of H3 on T11 by PKM [21]. Phospho‐T11 on H3 serves as a docking site for the E3 ligase RNF8, which was shown to ubiquitinate K4 of H3. This ubiquitination event of H3 was shown to induce its proteasomal degradation. While a direct effect of RNF8 was demonstrated on H3 only, RNF8 activity also led to the proteasome‐dependent destabilization of H2A, H2B, and H4, further promoting the disintegration of nucleosomes. Notably, this cascade of events was suggested to occur on the promoters of MYC and CCND1 (Cyclin D1), leading to a transcriptional activation of their genes. By altering the expression of c‐MYC and Cyclin D1, RNF8 promoted glycolysis and tumorigenesis in brain tumor models [21]. These findings provide substantial insights connecting signaling by extracellular growth factors to nuclear changes in chromosome structure and gene expression, which are mediated via the ubiquitin‐proteasome system. Specifically, this work suggested that the ubiquitin‐proteasome system can directly upregulate transcription in a locus‐localized fashion by facilitating histone degradation, thus allowing the binding of transcriptional machinery, including RNA polymerase II (Pol II) [21]. Indeed, a recent work by Rape et al. [19] has provided further evidence linking chromatin‐associated histone ubiquitination that leads to degradation by the proteasome, to transcriptional upregulation. In this elegant work, it was shown that histones undergo K48/K11‐branched ubiquitination by APC/C‐WRD5 at transcription start sites (TSSs) in human embryonic stem cells throughout mitosis, where transcription is largely halted. This ubiquitination was suggested to facilitate histone degradation at TSSs of pluripotency genes, thus leading to a rapid a faithful reactivation of their genes and cellular identity upon mitosis exit. This locus‐specific histone degradation was proposed to render the locus more accessible to the transcriptional machinery, which is otherwise isolated by nucleosome. Notably, this holistic regulatory mechanism was shown to occur specifically for H2B, and it remains to be determined if it occurs on other histones, like H3, and which sites are involved. Further, it remains to be examined whether these degradation events occur on the loci themselves, or alternatively they happen only after ubiquitinated histones are evicted from the locus.
Another example where a priming event leads to ubiquitin‐dependent proteasome degradation of histones is the deacetylation of the histone variant H2A.Z [64], which is targeted for ubiquitin‐dependent proteasomal degradation by a yet unknown E3 ligase [64, 65]. Deacetylation of K15 in H2A.Z by the deacetylase Sirtuin 1 (SIRT1/ Sir2α) triggers subsequent ubiquitination of the K115 site as well as K121 in H2A.Z and its subsequent degradation. Indeed, downregulation of SIRT1 leads to increased levels of H2A.Z. Notably, abnormally high levels of H2A.Z were suggested to result in cardiac hypertrophy in a human cell line model [64], while SIRT1 was suggested to have a dual role, depending on the interaction with other factors [66, 67]. It remains to be determined whether the effect of SIRT1 on cardiomyocytes is mediated by H2A.Z to confer protection from hypertrophy. However, we still lack a complete understanding on how these two modifications are related to each other in space and time. Likewise, it remains to be determined whether and how other modifications function in concert with ubiquitination to regulate histone degradation.
The examples described herein highlight the intricate ubiquitin‐dependent regulation that is involved in targeting specific histones, on demand, to degradation. Notably, while some of the works argued that the histone ubiquitination events occur on the chromatin, other works showed that ubiquitination occurs only after histones are evicted from the chromatin. It seems that this key difference in the localization of ubiquitination could underlie differential mechanisms of regulation. While the chromatin‐localized ubiquitination of histones may expedite direct and specific epigenetic alterations in a locus‐dependent manner, the chromatin‐unbound ubiquitination of histone might be only passive yet, an important event, in preserving low levels of unbound harmful histones.
Ubiquitin‐independent proteasomal degradation of histones
Numerous examples of ubiquitin‐independent degradation have been described to date (Fig. 1, Table 1) [68]. Such degradation is usually linked to noncanonical proteasome complexes that do not harbor ubiquitin receptors (i.e., not 26S proteasome), but are rather regulated by other types of modifications. Indeed, Qiu and colleagues showed that a special type of proteasomes, containing the PA200 activator bound to the 20S particle, is inefficient at degrading polyubiquitinated proteins [18, 50]. Interestingly, PA200‐containing proteasomes were shown to be localized to the nucleus and degrade acetylated core histones during DNA repair and replication stress, in a ubiquitin‐independent manner [18, 50, 69, 70]. Accordingly, cells depleted of PA200 are more sensitive to DNA damage [69, 71]. Acetylation of histone N‐terminal tails facilitates transcriptional activation by neutralizing the charge of the tails, thereby disrupting the interaction with DNA, or by forming a binding site for bromodomain‐containing transcription factors, some of which can remodel nucleosomes [8, 72]. Such association of acetylated histones with open regions of the chromatin can perhaps account for their susceptibility to degradation. Indeed, the degradation of acetylated histones by PA200‐capped proteasomes was suggested to be coupled to transcription and to affect the stability of the total populations of histone variant H3.3 and core histone H4, probably during histone exchange [23]. Yet, it is still unclear what initiates this process and to what extent PA200 plays an active part in it. Finally, despite the fact that other proteasome RPs such as PA28 (α/β and γ) have been reported to promote a ubiquitin‐independent degradation [73, 74], these were not shown thus far to participate in histone degradation.
In addition to acetylation, methylation has been implicated in determining the stability of histones. For example, under normal conditions, the arginine methyl transferase PRMT1 mediates symmetrical dimethylation of H4 on arginine in position 3 (H4R3me2as), which maintains histone H4 stability. Exposure of cells to oxidative stress, DNA damage, or hyperoncogenic signaling pressure (associated with senescence) leads to a reduction in PRMT1‐mediated H4R3me2, promoting the ubiquitin‐independent degradation of H4 by PA200‐capped proteasomes [23, 75]. Degradation of H4 thereby reduces nucleosomal occupancy and affects cell proliferation by promoting the transcription of cell cycle inhibitors, senescence‐associated genes, and regulators of apoptosis [75]. Interestingly, methylated histones, which are also often associated with open chromatin [76, 77], were shown to be further modified by nondegradative ubiquitination. It would be intriguing to explore whether methylated histones may also be targeted to degradation, in a ubiquitin‐dependent or ubiquitin‐independent manner, and compare the turnover rates of histones modified by different modifications in vitro and in cellular contexts.
Taken together, the diversity in ubiquitin‐dependent and ubiquitin‐independent mechanisms, which function in concert with additional post‐translational modifications, allow for precise regulation of histone degradation by different proteasome complexes to ensure context‐dependent action (Table 1). Notably, although several papers described the association of both 26S and PA200‐20S proteasome with the chromatin, it is still unclear whether the degradation of histones is spatially controlled on the chromatin itself or even at specific genomic regions, or whether degradation occurs only after histones are dissociated from the chromatin. Since chromatin‐bound proteasomes were shown to degrade transcriptional factors on chromatin in a localized manner [78, 79], we speculate that this will be the case for histones as well. Such mechanism might facilitate fast and precise degradation on loci that require an immediate response, for example, in response to certain cues. The heterogeneity in proteasome complexes further extends and enables the multilayered regulatory network that balances histone levels and the associated chromatin marks, to maintain homeostatic conditions or respond to stress stimuli.
Histone degradation under perturbed cellular and systemic states
Changes in cell state, be it due to developmental processes, transient stress exposure, or pathology, involve high levels of chromatin‐transacting activities that challenge cells with the need for histone eviction and the resultant excess of free histones. It is therefore critical to understand the effect of histone degradation under perturbations of cellular programs or states, to comprehend how histone destabilization is implicated in pathogenesis. While the open questions still far exceed the current understanding of histone degradation in human pathology, we will describe below examples of the involvement of histone degradation in various physiological conditions (Fig. 2) and discuss potential prospects in this regard.
Fig. 2.
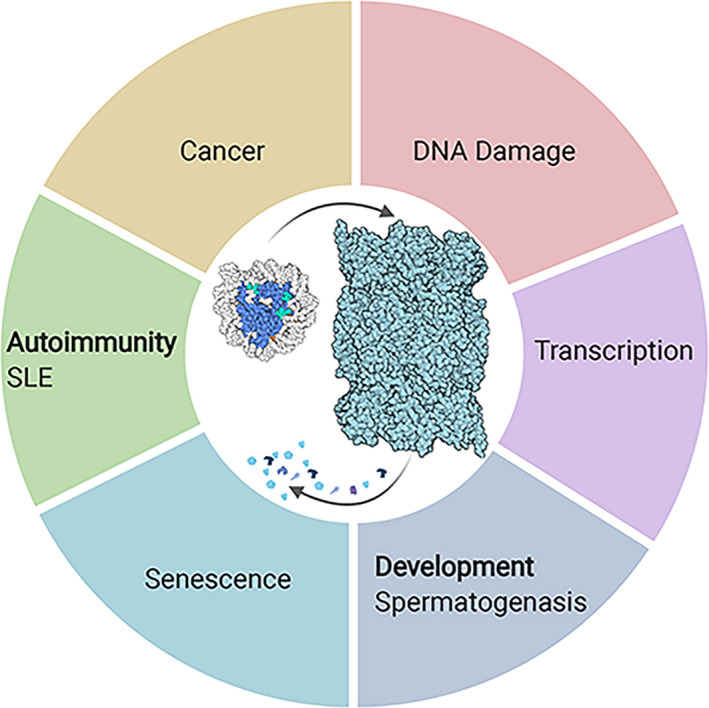
Proteasome‐mediated histone degradation under perturbed cellular and systemic states. PDB solved structures were used to generate this figure; 6RGQ (20S) and 3L3L (nucleosome). Created with BioRender.com.
Histone degradation in response to DNA damage
Cellular genomes are exposed constantly to different sources of DNA damage, requiring their repair system to disrupt and restore chromatin structure [80, 81, 82]. DNA damage challenges both genome organization and the integrity of histone proteins into the chromatin, with prominent alterations in histone variant dynamics and histone modifications. Heterochromatic chromatin has a tendency to block protein access to repair sites. Moreover, DNA double‐strand breaks (DSBs) found in heterochromatin relocate to the edge of such domains [83, 84, 85], a phenomenon that requires a certain degree of physical mobility. In response to DNA damage induced via UV radiation or zeocin, the histones are released from the chromatin and are concomitantly degraded in a proteasome‐mediated manner following phosphorylation by the DNA damage checkpoint kinase Rad53 [24, 37]. Therefore, cellular levels of histones drop by 20–40%, consequently rendering the chromatin less compacted and more flexible, which in turn enhances repair by recombination‐mediated mechanisms.
Removal of core histones is critical for efficient response to DNA damage by allowing the decompaction of the chromatin and DNA repair [24, 86]. Under DNA damaging conditions, PA200 is recruited to DNA damage sites in a manner that is dependent on the activity of DNA‐PK, a PI3‐related kinase that phosphorylates key DNA repair proteins in response to DNA damage [71]. PA200‐capped proteasomes then facilitate the degradation of acetylated histones and were implicated in enabling efficient DNA repair [50, 69, 70, 71, 87, 88]. Yet, the principles underlying the specificity of recognition of acetylated histones by PA200 are unclear, and it is not known what determines which acetylated histones will be targeted to degradation. In this regard, it is possible that the recruitment to specific DNA damage foci, for example, by histone marks that are associated with DNA damage such as phosphorylation of H3 or H2AX [89, 90], enable restriction of PA200‐dependent degradation. It will be further intriguing to explore whether PA200 facilitates the degradation of histones marked by additional modifications, such as these phosphorylated histone forms, under DNA damage.
Histone degradation in development and aging
Histone turnover plays important roles at both development and aging. In developing cells, intensive DNA replication and rapid transcriptional changes demand tight regulation of the chromatin. Indeed, during spermatogenesis, the majority of histones are transiently replaced from the chromatin of developing spermatids, by transition proteins and subsequently by protamines in postmeiotic cells, allowing flexibility of chromatin remodeling during the immense ongoing morphological changes in these cells [91]. This process was shown to be mediated by ubiquitin, which is highly abundant in the testis, with the demonstration of ubiquitination of H2A prior to its release from chromatin and replacement [92, 93]. Further, the HR6B ubiquitin‐conjugating DNA repair enzyme was shown to be required for spermatogenesis and inactivation of this protein led to male infertility in mice [94]. Moreover, the ubiquitin ligase RNF8 was shown to facilitate acetylation of histone H4 on K16, which might be an important step in nucleosome removal from the chromatin during spermatogenesis, though it is not clear whether this mechanism is facilitated through the proteasome [95].
Interestingly, PA200 is a major regulator of spermatogenesis and loss of PA200 in mice leads to delayed clearance of core histone and the formation of abnormal elongated spermatids [18]. As a result, PA200 knock‐out mice have impaired spermatogenesis and male infertility. Given the critical role of PA200‐capped proteasomes in histone degradation, these data suggest that PA200‐mediated degradation of histones may serve as an essential mechanism for sperm formation [96]. In support, other proteasome regulators such as PA28α,β or γ [97], which do not facilitate histone degradation, also do not affect spermatogenesis, highlighting a nonredundant role of PA200 in this context.
Investigation of end‐of‐life processes, such as aging or age‐related diseases, revealed another level of chromatin dynamics, which includes the exchange of canonical histones with histone variants. Histone variants are expressed throughout the cell cycle and can be incorporated into chromatin in a highly dynamic manner that is independent of replication. Such substitution of histones affects chromatin organization, thereby allowing for diverse regulation of transcriptional processes. In aging, histone variant levels were shown to change whereby in some cases, variants such as H2A.1 and H3.1 are downregulated [98] and in other cases variants accumulate and replace canonical histones, such as replacement of H2A by H2A.2 and canonical H3.1/H3.2 by H3.3 by the age of 18 months in mice [99]. In fact, in mouse neuronal chromatin, H3.3 constitutes only a small fraction of the histone H3 pool in embryonic stages, whereas it accumulated to 94% of the H3 pool within old age. In humans, postmortem brain analysis revealed that accumulation of H3.3 occurs through the first decade of life, reaching near saturation by mid‐adolescence [22]. The histone variant H3.3 is polyubiquitinated by an unknown E3 ligase and is degraded by the proteasomes in mouse embryonic neurons and glia cells [22]. It has been suggested that the polyubiquitination and proteasomal degradation of H3.3 allow for its eviction from chromatin and induce transcriptional remodeling during periods of intense neuronal activity [22]. Thus, ubiquitin‐dependent regulation of H3.3 incorporation maintains the “openness” and flexibility of the chromatin even in later stages of development and life. With numerous different E3s that have been associated with nuclear localization, deciphering the regulatory principles underlying histone‐mediated neural plasticity will require further investigation.
Human diseases with aberrant histone degradation: from Systemic Lupus Erythematosus to cancer
Lupus
Recently, a novel signature of aberrant proteasomal degradation of histones was suggested in the context of systemic lupus erythematosus disease (SLE), an autoimmune disorder characterized by autoreactive T and B cells and the production of autoantibodies against self‐epitopes. Specifically, a proteasomal profiling approach of MS analysis of proteolytic peptides (MAPP) revealed marked degradation of histones in PBMCs of SLE patients [100]. This signal was almost completely absent from PBMCs of healthy individuals, suggesting a role for aberrant proteasomal degradation in the pathophysiology of SLE. Notably, autoantibodies against histone‐associated epitopes were identified in SLE [101]. As proteasome‐dependent degradation is one of the first steps in antigen presentation [102], required to generate the primary epitopes that are presented on MHC class I, the identification of increased histone degradation in SLE suggests that proteasome‐dependent presentation of histone epitopes may be an early event in the pathogenesis of SLE [100].
Targeting of histones to degradation may be driven by various signals such as the active cellular programs, chromatin remodeling, and post‐translational modifications, as discussed in this review. One intriguing example to consider is the proteasomal degradation of oxidized histones [27]. Since oxidative stress is typical in SLE patients [103], it could potentially be implicated in the observed increased histone degradation. It will be intriguing to examine other diseases that are associated with oxidative stress and their impact on histone degradation.
Cancer
Chromatin remodeling is a key feature of tumorigenesis, allowing for intensive transcriptional activity and the reshaping of the transcriptome. While alterations in the patterns of histone PTMs have been extensively linked to cancer [104, 105, 106, 107], how histone degradation is affected by oncogenic transitions, or whether dysregulation of histone degradation is involved in tumor development, is thus far not understood. Particularly, it is intriguing to speculate how a fine‐regulated destabilization of specific histones may facilitate the balance that cancer cells must maintain between extensive remodeling of the chromatin and yet retaining the original tissue’s transcriptional signatures. For example, it is known that acetylation of histones allows the decompaction of the chromatin, and accordingly, histone acetylation is typically deregulated in cancer [107, 108]. Furthermore, the acetylated form of the histone variant H2A.Z has been implicated in upregulation of proto‐oncogenes such as c‐MYC and neoplastic transformation [65]. However, whether increased acetylation of histones in cancer leads to an increase in their degradation has not been determined. Interestingly, the expression of PA200, which facilitates the degradation of acetylated histones, is observed to be upregulated in various types of cancer [109], raising the possibility that, indeed, targeted degradation of acetylated histones may contribute to nucleosome turnover in cancer. In agreement with this, inhibitors of histone deacetylases (HDACs) are being considered in clinical studies as anti‐cancer treatments. Nevertheless, acetylation and deacetylation of histones, and the function of HDACs, have pleiotropic effects on the cellular transcriptome and nonhistone substrates and may act as double‐edged swords in cancer [108]. For example, the HDAC sirtuin 1 (SIRT1) was shown to target H2A.Z to degradation in both cardiomyocytes and prostate cancer cells [64, 65]. SIRT1 is upregulated in certain cancers and downregulated in others, including prostate cancer where it is believed to act as a tumor suppressor [65, 108, 110]. Therefore, in the case of prostate cancer, and potentially other cancer types, activation of SIRT1, rather than inhibition, will be beneficial as an anti‐cancer treatment. Such studies provide evidence for the therapeutic potential of compounds, which enhance HDAC activity (such as resveratrol) [111], in combination with other chromatin‐remodeling compounds that increase H2A.Z degradation by the proteasome, in cancer.
A direct connection between proteasome‐dependent degradation of histones and cancer was demonstrated by the effect of H3 degradation on brain tumor development in mice and primary human brain cancer cells. As discussed earlier, the E3 ligase RNF8 promotes the ubiquitin‐dependent degradation of H3 in response to signaling through EGFR [21], a growth factor receptor which is overexpressed or mutated in close to 60% of human glioblastoma cases. The RNF8‐mediated degradation of H3 occurs in a phosphorylation‐dependent manner and then promotes the expression of the proto‐oncogenes c‐MYC and Cyclin D1 and increases cellular glycolysis. Strikingly, ubiquitination of H3 on Lys4 by RNF8 was shown to be required for brain tumor formation, and a mutant of either H3 lacking this lysine acceptor site or lacking the phosphorylation priming site, or a mutant of RNF8 which cannot bind H3, all abrogated the growth of brain tumors. It is intriguing to explore whether RNF8 exerts a similar effect in other types of cancer, and whether additional ubiquitin E3 ligases mediate changes in the expression of tumor suppressors or proto‐oncogenes and thereby affect tumorigenesis.
Outlook
Histones are the key elements that define chromatin structure and the accessibility of DNA to the transcriptional machinery. As such, their regulation is critical to maintain homeostasis on the one hand, yet allow for flexibility on the other hand to respond to changing cellular conditions and to alter the transcriptional programs accordingly. Post‐translational modifications of histones have been extensively studied, and great efforts are invested in modulating and controlling histone modifications for therapeutic purposes, such as cancer treatment. Yet, though the prospect of proteasomal degradation of histones as a critical determinant in shaping the cellular transcriptome is only emerging, it opens intriguing avenues for investigation in both basic and translational research aspects.
Mounting evidence suggests that proteasome‐mediated histone degradation occurs only after histones are evicted from the chromatin. However, recent works raise the possibility that proteasomes might be to degrade histones while they are bound onto the chromatin. In such case, we further speculate that proteasomes might be qualified as bona‐fide chromatin‐remodeling factors that are directly involved in shaping the chromatin architecture. This undescribed role might be complementary to their current established role in countering excess of free histones proteins. As such, controlling stability of histones and other regulatory factors, in a localized manner, may afford a novel paradigm by which protein‐targeting chimeras (PROTACs [112]) may be coupled to induce degradation of chromatin modulators, on demand, for novel therapies in a diverse set of human diseases. Moreover, with the advent of PROTACs, revealing ubiquitin E3 ligases that function in the proximity to chromatin may offer novel means to harness endogenous ubiquitination activities for genetic modulation. Together, untangling the crosstalk of ubiquitin‐dependent and ubiquitin‐independent proteasomal degradation, with the various histone modifications, will provide a comprehensive view of how cells cope with varying conditions by dynamically controlling chromatin remodeling and gene expression.
Conflicts of interest
The authors declare no conflict of interest.
Author contributions
MDS, DS, and YM planned the manuscript’s structure and content. MDS and DS conceived, researched, and wrote the manuscript. AEL and YM provided critical review and editing. All authors critically reviewed the text and approved the final version.
Acknowledgements
We apologize to our colleagues whose work was not cited due to space constraints.
M.S.D. is supported by Marie Sklodowska‐Curie Individual Fellowship (Horizon 2020 Grant No. GAP‐845066). D.S. is supported by a fellowship granted by Israel Council for higher education (VATAT). Y.M. is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No 677748); The I‐CORE Program of the Planning and Budgeting Committee and The Israel Science Foundation (Grant No. 1775/12) and the Israeli Science Foundation (Grant No. 2109/18); The Gruber Peter & Patricia award. Y.M. is supported by the incumbent of the Leonard and Carol Berall Career Development Chair.
Merav D. Shmueli and Daoud Sheban contributed equally to this article
References
- 1. Kornberg RD & Thomas JO (1974) Chromatin structure: oligomers of the histones. Science 184, 865–868. [DOI] [PubMed] [Google Scholar]
- 2. Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. [DOI] [PubMed] [Google Scholar]
- 3. Olins AL & Olins DE (1974) Spheroid chromatin units (v bodies). Science 183, 330–332. [DOI] [PubMed] [Google Scholar]
- 4. Simpson RT (1978) Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 17, 5524–5531. [DOI] [PubMed] [Google Scholar]
- 5. Luger K, Mäder AW, Richmond RK, Sargent DF & Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 6. Campos EI & Reinberg D (2009) Histones: annotating chromatin. Annu Rev Genet 43, 559–599. [DOI] [PubMed] [Google Scholar]
- 7. Fyodorov DV, Zhou BR, Skoultchi AI & Bai Y (2018) Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 19, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allis CD & Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500. [DOI] [PubMed] [Google Scholar]
- 9. Margueron R & Reinberg D (2010) Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buschbeck M & Hake SB (2017) Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18, 299–314. [DOI] [PubMed] [Google Scholar]
- 11. Stewart‐Morgan KR, Petryk N & Groth A (2020) Chromatin replication and epigenetic cell memory. Nat Cell Biol 22, 361–371. [DOI] [PubMed] [Google Scholar]
- 12. Osley MA (1991) The regulation of histone synthesis in the cell cycle. Annu Rev Biochem 60, 827–861. [DOI] [PubMed] [Google Scholar]
- 13. Marzluff WF, Wagner EJ & Duronio RJ (2008) Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat Rev Genet 9, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jenuwein T & Allis CD (2001) Translating the histone code. Science 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 15. Dhaenens M, Glibert P, Meert P, Vossaert L & Deforce D (2015) Histone proteolysis: a proposal for categorization into “clipping” and “degradation”. BioEssays 37, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gunjan A, Paik J & Verreault A (2006) The emergence of regulated histone proteolysis. Curr Opin Genet Dev 16, 112–118. [DOI] [PubMed] [Google Scholar]
- 17. Singh RK, Liang D, Gajjalaiahvari UR, Kabbaj MHM, Paik J & Gunjan A (2010) Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 9, 4236–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y et al. (2013) XAcetylation‐mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY & Rape M (2020) Gene expression and cell identity controlled by anaphase‐promoting complex. Nature 579, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fatima A, Irmak D, Noormohammadi A, Rinschen MM, Das A, Leidecker O, Schindler C, Sánchez‐Gaya V, Wagle P, Pokrzywa W et al. (2020) The ubiquitin‐conjugating enzyme UBE2K determines neurogenic potential through histone H3 in human embryonic stem cells. Commun Biol 3, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia Y, Yang W, Fa M, Li X, Wang Y, Jiang Y, Zheng Y, Lee JH, Li J & Lu Z (2017) RNF8 mediates histone H3 ubiquitylation and promotes glycolysis and tumorigenesis. J Exp Med 214, 1843–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL et al. (2015) Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang TX, Ma S, Han X, Luo ZY, Zhu QQ, Chiba T, Xie W, Lin K & Qiu XB (2021) Proteasome activator PA200 maintains stability of histone marks during transcription and aging. Theranostics 11, 1458–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen‐Hughes T et al. (2017) Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24, 99–107. [DOI] [PubMed] [Google Scholar]
- 25. Reid BR & Cole RD (1964) Biosynthesis of a lysine‐rich histone in isolated calf thymus nuclei. Proc Natl Acad Sci USA 51, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garrels JI, Elgin SCR & Bonner J (1972) A histone protease of rat liver chromatin. Biochem Biophys Res Commun 46, 545–551. [DOI] [PubMed] [Google Scholar]
- 27. Ullrich O, Sitte N, Sommerburg O, Sandig V, Davies KJA & Grune T (1999) Influence of DNA binding on the degradation of oxidized histones by the 20S proteasome. Arch Biochem Biophys 362, 211–216. [DOI] [PubMed] [Google Scholar]
- 28. Haas A, Reback PM, Pratt G & Rechsteiner M (1990) Ubiquitin‐mediated degradation of histone H3 does not require the substrate‐binding ubiquitin protein ligase, E3, or attachment of polyubiquitin chains. J Biol Chem 265, 21664–21669. [PubMed] [Google Scholar]
- 29. Carter DB, Efird PH & Chae CB (1978) Chromatin‐bound proteases and their inhibitors. Methods Cell Biol 19, 175–190. [DOI] [PubMed] [Google Scholar]
- 30. Duncan EM, Muratore‐Schroeder TL, Cook RG, Garcia BA, Shabanowitz J, Hunt DF & Allis CD (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Osley MA (2008) Epigenetics: how to lose a tail. Nature 456, 885–886. [DOI] [PubMed] [Google Scholar]
- 32. Santos‐Rosa H, Kirmizis A, Nelson C, Bartke T, Saksouk N, Cote J & Kouzarides T (2009) Histone H3 tail clipping regulates gene expression. Nat Struct Mol Biol 16, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh RK, Kabbaj MHM, Paik J & Gunjan A (2009) Histone levels are regulated by phosphorylation and ubiquitylation‐dependent proteolysis. Nat Cell Biol 11, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cook AJL, Gurard‐Levin ZA, Vassias I & Almouzni G (2011) A specific function for the histone chaperone NASP to fine‐tune a reservoir of soluble H3–H4 in the histone supply chain. Mol Cell 44, 918–927. [DOI] [PubMed] [Google Scholar]
- 35. Ciechanover A (2005) Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 6, 79–86. [DOI] [PubMed] [Google Scholar]
- 36. Commerford SL, Carsten AL & Cronkite EP (1982) Histone turnover within nonproliferating cells. Proc Natl Acad Sci USA 79, 1163–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gunjan A & Verreault A (2003) A Rad53 kinase‐dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae . Cell 115, 537–549. [DOI] [PubMed] [Google Scholar]
- 38. Zou C & Mallampalli RK (2014) Regulation of histone modifying enzymes by the ubiquitin‐proteasome system. Biochim Biophys Acta Mol Cell Res 1843, 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kirkin V, McEwan DG, Novak I & Dikic I (2009) A role for ubiquitin in selective autophagy. Mol Cell 34, 259–269. [DOI] [PubMed] [Google Scholar]
- 40. Singh RK, Gonzalez M, Kabbaj MHM & Gunjan A (2012) Novel E3 ubiquitin ligases that regulate histone protein levels in the budding yeast saccharomyces cerevisiae. PLoS One 7, e36295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adams PD, Ivanov A, Pawlikowski J, Manoharan I, Van TJ, Nelson DM, Singh Rai T, Shah PP, Hewitt G, Korolchuk VI et al. (2013) Lysosome‐mediated processing of chromatin in senescence. J Cell Biol 202, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwartz AL & Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49, 73–96. [DOI] [PubMed] [Google Scholar]
- 43. Pickart CM & Eddins MJ (2004) Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta Mol Cell Res 1695, 55–72. [DOI] [PubMed] [Google Scholar]
- 44. Wiekowski M, Miranda M, Nothias JY & DePamphilis ML (1997) Changes in histone synthesis and modification at the beginning of mouse development correlate with the establishment of chromatin mediated repression of transcription. J Cell Sci 110, 1147–1158. [DOI] [PubMed] [Google Scholar]
- 45. Lu Y, Lee BH, King RW, Finley D & Kirschner MW (2015) Substrate degradation by the proteasome: a single‐molecule kinetic analysis. Science 348, 1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D & Gygi SP (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21, 921–926. [DOI] [PubMed] [Google Scholar]
- 47. Tanaka K (2009) The proteasome: overview of structure and functions. Proc Japan Acad Ser B Phys Biol Sci 85, 12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharon M, Taverner T, Ambroggio XI, Deshaies RJ & Robinson CV (2006) Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol 4, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rechsteiner M & Hill CP (2005) Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol 15, 27–33. [DOI] [PubMed] [Google Scholar]
- 50. Ustrell V, Hoffman L, Pratt G & Rechsteiner M (2002) Pa200, a nuclear proteasome activator involved in DNA repair. EMBO J 21, 3516–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Collins GA & Goldberg AL (2017) The logic of the 26S proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E & Martin A (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D & Dikic I (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Eisenberg‐Lerner A, Benyair R, Hizkiahou N, Nudel N, Maor R, Kramer MP, Shmueli MD, Zigdon I, Cherniavsky Lev M, Ulman A et al. (2020) Golgi organization is regulated by proteasomal degradation. Nat Commun 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. von Mikecz A (2006) The nuclear ubiquitin‐proteasome system. J Cell Sci 119, 1977–1984. [DOI] [PubMed] [Google Scholar]
- 56. Komander D & Rape M (2012) The ubiquitin code. Annu Rev Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- 57. Goldknopf IL, Taylor CW, Baum RM, Yeoman LC, Olson MO, Prestayko AW & Busch H (1975) Isolation and characterization of protein A24, a “histone like” non histone chromosomal protein. J Biol Chem 250, 7182–7187. [PubMed] [Google Scholar]
- 58. Zhang Y (2003) Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev 17, 2733–2740. [DOI] [PubMed] [Google Scholar]
- 59. Hunter T (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 28, 730–738. [DOI] [PubMed] [Google Scholar]
- 60. Caron C, Boyault C & Khochbin S (2005) Regulatory cross‐talk between lysine acetylation and ubiquitination: role in control of protein stability. BioEssays 27, 408–415. [DOI] [PubMed] [Google Scholar]
- 61. Zhao Y, Brickner JR, Majid MC & Mosammaparast N (2014) Crosstalk between ubiquitin and other post‐translational modifications on chromatin during double‐strand break repair. Trends Cell Biol 24, 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. White CL, Suto RK & Luger K (2001) Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J 20, 5207–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Freeman L, Kurumizaka H & Wolffe AP (1996) Functional domains for assembly of histones H3 and H4 into the chromatin of Xenopus embryos. Proc Natl Acad Sci USA 93, 12780–12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen IY, Lypowy J, Pain J, Sayed D, Grinberg S, Alcendor RR, Sadoshima J & Abdellatif M (2006) Histone H2A.z is essential for cardiac myocyte hypertrophy but opposed by silent information regulator 2α. J Biol Chem 281, 19369–19377. [DOI] [PubMed] [Google Scholar]
- 65. Baptista T, Graça I, Sousa EJ, Oliveira AI, Costa NR, Costa‐Pinheiro P, Amado F, Henrique R & Jerónimo C (2013) Regulation of histone H2A.Z expression is mediated by sirtuin 1 in prostate cancer. Oncotarget 4, 1673–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alcendor RR, Kirshenbaum LA, Imai SI, Vatner SF & Sadoshima J (2004) Silent information regulator 2α, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res 95, 971–980. [DOI] [PubMed] [Google Scholar]
- 67. Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF & Sadoshima J (2007) Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100, 1512–1521. [DOI] [PubMed] [Google Scholar]
- 68. Erales J & Coffino P (2014) Ubiquitin‐independent proteasomal degradation. Biochim Biophys Acta Mol Cell Res 1843, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mandemaker IK, Geijer ME, Kik I, Bezstarosti K, Rijkers E, Raams A, Janssens RC, Lans H, Hoeijmakers JH, Demmers JA et al. (2018) DNA damage‐induced replication stress results in PA 200‐proteasome‐mediated degradation of acetylated histones. EMBO Rep 19, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Blickwedehl J, McEvoy S, Wong I, Kousis P, Clements J, Elliott R, Cresswell P, Liang P & Bangia N (2007) Proteasomes and proteasome activator 200 kDa (PA200) accumulate on chromatin in response to ionizing radiation. Radiat Res 167, 663–674. [DOI] [PubMed] [Google Scholar]
- 71. Blickwedehl J, Agarwal M, Seong C, Pandita RK, Melendy T, Sung P, Pandita TK & Bangia N (2008) Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc Natl Acad Sci USA 105, 16165–16170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sterner DE & Berger SL (2000) Acetylation of histones and transcription‐related factors. Microbiol Mol Biol Rev 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ & O’Malley BW (2006) The SRC‐3/AIB1 coactivator is degraded in a ubiquitin‐ and ATP‐independent manner by the REGγ proteasome. Cell 124, 381–392. [DOI] [PubMed] [Google Scholar]
- 74. Strickland E, Hakala K, Thomas PJ & DeMartino GN (2000) Recognition of misfolding proteins by PA700, the regulatory subcomplex of the 26 S proteasome. J Biol Chem 275, 5565–5572. [DOI] [PubMed] [Google Scholar]
- 75. Lin C, Li H, Liu J, Hu Q, Zhang S, Zhang N, Liu L, Dai Y, Cao D, Li X et al. (2020) Arginine hypomethylation‐mediated proteasomal degradation of histone H4—an early biomarker of cellular senescence. Cell Death Differ 27, 2697–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Worden EJ, Hoffmann NA, Hicks CW & Wolberger C (2019) Mechanism of cross‐talk between H2B ubiquitination and H3 methylation by Dot1L. Cell 176, 1490–1501.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Worden EJ, Zhang X & Wolberger C (2020) Structural basis for COMPASS recognition of an H2B‐ubiquitinated nucleosome. eLife 9, e53199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Szutorisz H, Georgiou A, Tora L & Dillon N (2006) The proteasome restricts permissive transcription at tissue‐specific gene loci in embryonic stem cells. Cell 127, 1375–1388. [DOI] [PubMed] [Google Scholar]
- 79. Geng F & Tansey WP (2012) Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc Natl Acad Sci USA 109, 6060–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lukas J, Lukas C & Bartek J (2011) More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- 81. Soria G, Polo SE & Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46, 722–734. [DOI] [PubMed] [Google Scholar]
- 82. Shiloh Y (2003) ATM and related protein kinases: Safeguarding genome integrity. Nat Rev Cancer 3, 155–168. [DOI] [PubMed] [Google Scholar]
- 83. Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV & Karpen GH (2011) Double‐strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lemaître C, Grabarz A, Tsouroula K, Andronov L, Furst A, Pankotai T, Heyer V, Rogier M, Attwood KM, Kessler P et al. (2014) Nuclear position dictates DNA repair pathway choice. Genes Dev 28, 2450–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Torres‐Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert‐Boulet N, Reid R, Jentsch S, Rothstein R, Aragón L & Lisby M (2007) The Smc5‐Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9, 923–931. [DOI] [PubMed] [Google Scholar]
- 86. Hauer MH & Gasser SM (2017) Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31, 2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Blickwedehl J, Olejniczak S, Cummings R, Sarvaiya N, Mantilla A, Chanan‐Khan A, Pandita TK, Schmidt M, Thompson CB & Bangia N (2012) The proteasome activator PA200 regulates tumor cell responsiveness to glutamine and resistance to ionizing radiation. Mol Cancer Res 10, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Doherty KM, Pride LD, Lukose J, Snydsman BE, Charles R, Pramanik A, Muller EG, Botstein D & Moore CW (2012) Loss of a 20S proteasome activator in Saccharomyces cerevisiae downregulates genes important for genomic integrity, increases DNA damage, and selectively sensitizes cells to agents with diverse mechanisms of action. G3: Genes ‐ Genomes ‐ Genetics 2, 943–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rossetto D, Avvakumov N & Côté J (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7, 1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mah LJ, El‐Osta A & Karagiannis TC (2010) γh2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686. [DOI] [PubMed] [Google Scholar]
- 91. Sassone‐Corsi P (2002) Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science 296, 2176–2178. [DOI] [PubMed] [Google Scholar]
- 92. Baarends WM, Hoogerbrugge JW, Roest HP, Ooms M, Vreeburg J, Hoeijmakers JHJ & Grootegoed JA (1999) Histone ubiquitination and chromatin remodeling in mouse spermatogenesis. Dev Biol 207, 322–333. [DOI] [PubMed] [Google Scholar]
- 93. Lanneau M & Loir M (1982) An electrophoretic investigation of mammalian spermatid‐specific nuclear proteins. J Reprod Fertil 65, 163–170. [DOI] [PubMed] [Google Scholar]
- 94. Roest HP, Van Klaveren J, De Wit J, Van Gurp CG, Koken MHM, Vermey M, Van Roijen JH, Hoogerbrugge JW, Vreeburg JTM, Baarends WM et al. (1996) Inactivation of the HR6B ubiquitin‐conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell 86, 799–810. [DOI] [PubMed] [Google Scholar]
- 95. Lu LY, Wu J, Ye L, Gavrilina GB, Saunders TL & Yu X (2010) RNF8‐dependent histone modifications regulate nucleosome removal during spermatogenesis. Dev Cell 18, 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Khor B, Bredemeyer AL, Huang C‐Y, Turnbull IR, Evans R, Maggi LB, White JM, Walker LM, Carnes K, Hess RA et al. (2006) Proteasome activator PA200 is required for normal spermatogenesis. Mol Cell Biol 26, 2999–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Levy‐Barda A, Lerenthal Y, Davis AJ, Chung YM, Essers J, Shao Z, Van Vliet N, Chen DJ, Hu MCT, Kanaar R et al. (2011) Involvement of the nuclear proteasome activator PA28γ in the cellular response to DNA double‐strand breaks. Cell Cycle 10, 4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rogakou EP & Sekeri‐Pataryas KE (1999) Histone variants of H2A and H3 families are regulated during in vitro aging in the same manner as during differentiation. Exp Gerontol 34, 741–754. [DOI] [PubMed] [Google Scholar]
- 99. Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska‐Wrzesinska A & Jensen ON (2017) Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res 45, 9272–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wolf‐Levy H, Javitt A, Eisenberg‐Lerner A, Kacen A, Ulman A, Sheban D, Dassa B, Fishbain‐Yoskovitz V, Carmona‐Rivera C, Kramer MP et al. (2018) Revealing the cellular degradome by mass spectrometry analysis of proteasome‐cleaved peptides. Nat Biotechnol 36, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dieker J, Berden JH, Bakker M, Briand JP, Muller S, Voll R, Sjöwall C, Herrmann M, Hilbrands LB & van der Vlag J (2016) Autoantibodies against modified histone peptides in sle patients are associated with disease activity and lupus nephritis. PLoS One 11, e0165373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rock KL, York IA, Saric T & Goldberg AL (2002) Protein degradation and the generation of MHC class I‐presented peptides. Adv Immunol 80, 1–70. [DOI] [PubMed] [Google Scholar]
- 103. Perl A (2013) Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol 9, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M & Kurdistani SK (2005) Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- 105. Bannister AJ & Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Baylin SB & Jones PA (2016) Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 8, a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Audia JE & Campbell RM (2016) Histone modifications and cancer. Cold Spring Harb Perspect Biol 8, a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK & Patra SK (2014) Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem 62, 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F et al. (2017) A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed] [Google Scholar]
- 110. Lin Z & Fang D (2013) The roles of SIRT1 in cancer. Genes and Cancer 4, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yin Y, Zhu Q, Jiang T, Fan L & Qiu X (2019) Targeting histones for degradation in cancer cells as a novel strategy in cancer treatment. Sci China Life Sci 62, 1078–1086. [DOI] [PubMed] [Google Scholar]
- 112. Pettersson M & Crews CM (2019) PROteolysis TArgeting Chimeras (PROTACs) — past, present and future. Drug Discov Today Technol 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ullrich O & Grune T (2001) Proteasomal degradation of oxidatively damaged endogenous histones in K562 human leukemic cells. Free Radic Biol Med 31, 887–893. [DOI] [PubMed] [Google Scholar]