

Abstract
Ovarian cancer therapy has remained fundamentally unchanged for 50 years, with surgery and chemotherapy still the frontline treatments. Typically asymptomatic until advanced stages, ovarian cancer is known as “the silent killer.” Consequently, it has one of the worst 5‐year survival rates, as low as 30%. The most frequent driver mutations are found in well‐defined tumor suppressors, such as p53 and BRCA1/2. In recent years, it has become clear that, like the majority of other cancers, many epigenetic regulators are altered in ovarian cancer, including EZH2, SMARCA2/4 and ARID1A. Disruption of epigenetic regulators often leads to loss of transcriptional control, aberrant cell fate trajectories and disruption of senescence, apoptotic and proliferation pathways. These mitotically inherited epigenetic alterations are particularly promising targets for therapy as they are largely reversible. Consequently, many drugs targeting chromatin modifiers and other epigenetic regulators are at various stages of clinical trials for other cancers. Understanding the mechanisms by which ovarian cancer‐specific epigenetic processes are disrupted in patients can allow for informed targeting of epigenetic pathways tailored for each patient. In recent years, there have been groundbreaking new advances in disease modeling through ovarian cancer organoids; these models, alongside single‐cell transcriptomic and epigenomic technologies, allow the elucidation of the epigenetic pathways deregulated in ovarian cancer. As a result, ovarian cancer therapy may finally be ready to advance to next‐generation treatments. Here, we review the major developments in ovarian cancer, including genetics, model systems and technologies available for their study and the implications of applying epigenetic therapies to ovarian cancer.
Keywords: chromatin remodeling, disease modeling, epigenetic drugs, ovarian cancer, precision oncology
Abbreviations
- ARID1A
AT‐rich interactive domain‐containing protein 1a
- CARM1, aka PRMT4
coactivator‐associated arginine methyltransferase 1
- CHD
chromodomain helicase DNA‐binding
- HDAC
histone deacetylase
- HGSOC
high‐grade serous ovarian cancer
- ISWI
imitation switch
- KDM
lysine demethylase
- KMT
lysine methyltransferase complex
- LGSOC
low‐grade serous ovarian cancer
- OCCC
ovarian clear cell carcinomas
- OEC
ovarian endometrioid carcinomas
- PARP
poly (ADP‐Ribose) polymerase
- PRC
Polycomb Repressive Complex
- PRMT
protein arginine methyltransferase
- PTMs
post‐translational modifications
- SCCOHT
small cell carcinoma of the ovary hypercalcemic type
- SWI/SNF, aka BAF
switch/sucrose non‐fermentable
1. INTRODUCTION
Ovarian cancer is one of the most lethal gynecological malignancies, with the highest incidence rates in North America, as well as Central and Eastern Europe. When diagnosed at Stage I, the 5‐year survival rate is ~90% for all subtypes. However, due to a lack of symptoms prior to metastasis throughout the abdomen, and the failure of current treatments to lead to complete remission, the vast majority of serous carcinomas are not diagnosed until Stage III (51%) or IV (29%). 1 , 2 Thus, the overall 5‐year survival rate is as low as 30% worldwide. 3 , 4 , 5 The vast majority of ovarian tumors originate from one of three cell types: epithelial (>90%), stromal (~6%) and germ cells (2%‐3%). 3 , 5 , 6
Each of these subtypes of ovarian cancer has distinct clinical features, putative cells of origin and associated driver mutations. This suggests different pathways driving the alternative cancer types, and specific weaknesses or synthetic lethalities should exist for each. As an example of inter‐subtype variation, high‐grade serous ovarian cancer (HGSOC) typically occurs in post‐menopausal women and is characterized by frequent (>90%) P53 mutations. 6 , 7 , 8 , 9 This is the most common form of ovarian cancer, accounting for 70% of all cases. 3 , 5 , 6 A highly aggressive malignancy, its cell of origin is often unclear, there has been ambiguity as to whether it has originated from ovarian surface epithelium or tubal epithelium, with recent data suggesting that it can arise from both (Figure 1). 3 , 10 Emerging technologies in cell of origin tracing, such as OriPRINT, 10 and patient‐derived organoids for normal tissue comparative analysis, 11 we are closer to tracking the specific dysregulation driving tumorigenesis.
FIGURE 1.
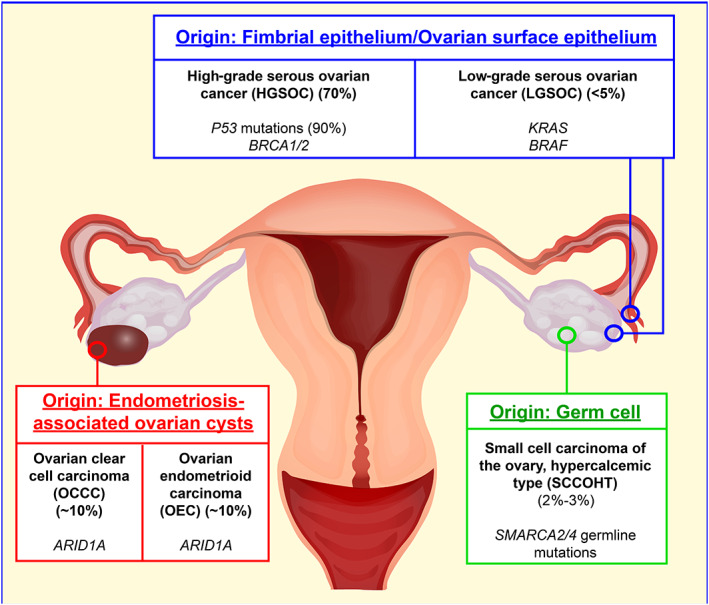
Current understanding of the putative tissues of origin for key ovarian cancer subtypes, and the most frequent mutations associated with each type
The key driver of this type of ovarian cancer, p53, is of course a common genetic lesion in many cancers, which causes genomic instability and structural variation; however, the p53 pathway has proven to be difficult to target therapeutically. 12 For these reasons, this review will focus on alternative genetic and epigenetic disruptions in the different subtypes of ovarian cancer, such as amplification and hyperactivity that may represent more viable targets for cancer therapy.
A related subtype, low‐grade serous ovarian cancer (LGSOC) typically presents in patients between 40 and 55 years of age and is associated with mutations in KRAS and BRAF. 4 , 13 On the other hand, ovarian clear cell carcinomas (OCCC) and ovarian endometrioid carcinomas (OEC) frequently feature mutations in PTEN and ARID1A. 3 , 4 OCCC and OEC are the second and third most common histological subtypes, respectively, that together account for ~20%‐25% of epithelial ovarian cancer cases. These cancers are thought to originate through endometriosis and endometriotic ovarian cysts (Figure 1). Their specific cell of origin remains unclear but related to ectopic inclusions that are suspected to be tubal or endometrial in origin. 3 , 14 , 15 , 16
The final subtype we will discuss is small cell carcinoma of the ovary. This is a very rare and highly aggressive cancer that typically occurs at a young age when compared to other ovarian cancers (~23.9 years mean). 17 Small cell carcinoma of the ovary hypercalcemic type (SCCOHT) is almost exclusively defined by germline or somatic mutations in SMARCA4, the ATPase subunit of the BAF complex, and is currently thought to originate from germ cells (Figure 1). 18 , 19 , 20
Currently, standard treatment for newly diagnosed ovarian cancer is a combination of surgical cytoreduction and platinum‐based chemotherapy. 21 At advanced stages, recurrence and platinum‐resistance of tumors is very common. 4 , 22 In recent years however, there has been significant advancement in the use of directed therapies, such as inhibition of angiogenesis and DNA repair pathways. 23 , 24 , 25 Inhibition of the DNA repair enzyme poly (ADP‐Ribose) polymerase (PARP) has shown specific activity in HGSOC with homologous recombination deficiency (~50%), with particular efficacy in BRCA‐mutated HGSOC (~20%). 8 , 21 , 24 , 26 Despite such promising advances in directed therapy, 50%‐80% of HGSOC patients do not contain the sensitive mutational profile to benefit from these therapies. This highlights the urgent need for additional therapeutics for this malignancy, targeting alternative mutational sensitivities.
It has become clear in recent years that disruption of the epigenetic machinery is key in development in almost all human cancers. 27 This discovery has unlocked vast potential for the field of oncology, as the plasticity of chromatin states makes epigenetic machinery attractive targets for cancer therapy. Epigenetics—the reversible modification of both DNA and DNA‐bound histones 28 —regulate the underlying genes, and disruption of the machinery results in aberrant activation/repression of key cancer‐related genes, driving oncogenesis. Therapies targeting oncogenic deregulation of the epigenome, such as EZH2 and BET‐family inhibition, have been successfully used in clinical and preclinical trials for many diverse cancers, such as non‐Hodgkins lymphoma and glioblastoma. 27 This review will focus on the key perturbations to epigenetic machinery in ovarian cancer, the sensitivities these disruptions create that can be exploited by available therapies and the advances in technologies that are bringing us closer to precision oncology for ovarian cancer.
2. EPIGENOMIC REGULATION OF TRANSCRIPTION
Over the past century, it has become clear that the organization of DNA in 3D space is one of the most fundamental controls of gene expression. Genetic information must be, in the simplest sense, physically accessible in order to be regulated. Accessibility is determined by position and information loaded in the histone‐DNA complex known as the nucleosome. The nucleosome is comprised of 147 base pairs of DNA wrapped around an octamer of histones H2A, H2B, H3 and H4. 29 , 30 Nucleosome remodeling is the alteration of the histone‐DNA interface by a dedicated set of chromatin remodeling enzymes. There is a large and complex interplay between remodelers responsible for positive and negative regulation of genetic regulatory elements. Four main subfamilies of chromatin remodelers exist, based on domain structure and organization of the catalytic ATPase translocase domain; chromodomain helicase DNA‐binding (CHD), INO80, imitation switch (ISWI) and switch/sucrose non‐fermentable (SWI/SNF, aka BAF) are the main subgroups, each of which can have distinct targets activities based on associated accessory proteins. BAF alters chromatin access through nucleosome repositioning/eviction, whereas members of the INO80 family can edit the composition of nucleosomes through incorporation of histone variants. 30 , 31 Fundamentally, chromatin remodeling enzymes (remodelers) control transcription through mobilization and organization of nucleosomes to make genetic information more or less accessible to transcriptional machinery. 31
The octamer of histones comprising the nucleosome contains post‐translational modifications (PTMs) with specific information on how the associated DNA should be organized. PTMs to the N‐ and C‐terminal tails of histones are added and removed by enzymes commonly referred to as “writers” and “erasers,” ushering in both physical changes (compaction/relaxation) and/or relay signals to be executed upon the associated regions of DNA via the actions of effector proteins (“readers”). The compaction state of a DNA region—influenced by its histone PTM status—is the central control to transcriptional output, either allowing or restricting its access to downstream transcription factors. 32 , 33 Some of the most common histone modifications include methylation, acetylation, ubiquitination and phosphorylation. Designated enzymes catalyze the transfer (writers) or removal (erasers) of these marks to specific amino acid residues. Regulators (readers) recognize these marks, and through this recognition recruit other regulators, enzymes or remodelers conferring downstream changes in chromatin compaction and accessibility. 32 , 33
For example, mono‐, di‐, or trimethylation of histone lysines is catalyzed by six major groups of lysine methyltransferase complexes (KMT1‐6), the lysine methyl “writers.” Key examples are EZH1 and EZH2 of the KMT6 family, which are the catalytic subunits of Polycomb Repressive Complex (PRC) 2, designating H3K27 mono‐, di‐ and trimethylation. 34 , 35 , 36 These “writing” enzymes catalyze the transfer of methyl groups to specific lysine residues, but the plasticity of chromatin requires the changing of states during development, transitioning from pluripotent transcriptional profiles to differentiated expression profiles through dynamic writing and erasing of key genomic sites. Thus, several families of “erasers” counteract the activity of the writer enzymes. Key examples are histone lysine demethylases (KDM) such as UTX, which removes methyl groups from H3K27. 37 , 38
The balanced writing and erasing modification states ultimately serves as information understood and acted upon by “reader” enzymes. For instance, H3K27me3 is specifically recognized and bound by the Cbx components of the PRC1 complex and is involved in chromatin compaction. 39 , 40 Thus, the complex interplay and balance of these writers, erasers and readers is key to transcriptional regulation and cellular identity.
As chromatin modification and remodeling has such a critical role in transcription, it is unsurprising that mutations in fundamental transcriptional regulators are commonly disrupted in cancers. Genomic analysis of various cancers indicates that chromatin remodelers are some of the most frequently mutated genes in human malignancies. 31 , 41 Of specific interest are the antagonistic relationships between BAF and Polycomb complexes, as well as the arginine methyltransferase CARM1. Here we discuss some of the most important chromatin regulatory complexes, their role in ovarian cancer and the therapeutic opportunities these lesions create.
3. POLYCOMB REPRESSIVE COMPLEXES 1 AND 2
Polycomb group proteins are highly conserved chromatin repressors responsible for cell fate gene regulation during development and play a fundamental role in cellular identity. Mammals contain two main multi‐subunit PRC, PRC1 and PRC2. 34 PRC1 catalyzes the ubiquitination of H2AK119ub1, a modification essential for repression at Polycomb target promoters. 42 , 43 It can be broadly divided into two subcomplexes (canonical and variant) that contribute to repression in alternative fashions. vPRC1 (variant) catalyzes the majority of H2AK119ub1, which promotes repression at promoters, at least partially, through PRC2 recruitment, 42 , 44 while cPRC1 (canonical) promotes compaction and long‐range repressive Polycomb body formation through subunits that confer structural changes on chromatin. 40 , 45 , 46
PRC2 is responsible for mono‐, di‐ and tri‐methylation of H3K27. Tri‐methylation is typically found at repressed promoters, where it is essential for maintaining transcriptional repression of non‐lineage expressed genes. Di‐methylation is found across the genome, where it has been suggested to maintain repression of tissue‐specific enhancers 47 while mono‐methylation is found at active gene bodies, though the function, if any, of this has not yet been established. The PRC2 core contains three subunits EED, SUZ12 and a catalytic methyltransferase component, either EZH1 or EZH2. 48 It has become clear that mutations in Polycomb complexes and consequent aberrant H3K27 methylation patterns have a key role in cancer development. 34 In these cases, failure to undergo cellular differentiation is a consequence of aberrant methylation patterns of H3K27. 49 , 50
3.1. EZH2
EZH2 is widely mutated in various cancer types and can act in both an oncogenic or tumor suppressive capacity. 34 “Change of function” mutations are observed in diffuse large B‐cell and follicular lymphomas, and to a lesser degree in melanoma, which cause global hyper tri‐methylation of H3K27, replacing the diffuse intergenic H3K27me2 levels. 34 , 51 Conversely, EZH2, SUZ12 and EED deletions, which cause reductions in H3K27me3 levels, are also observed in leukemias and other myeloid disorders. 52 , 53 This is an example of the importance of mutational context in cancer development and reinforces the need for patient‐specific profiling in order to choose appropriate therapeutics, as will be discussed later in this review.
EZH2 overexpression/amplification has been widely reported in ovarian cancers. Increased EZH2 activity has been found in ~85% of epithelial ovarian carcinomas through genetic amplification and loss of antagonistic protein activity 54 , 55 , 56 , 57 and is functionally implicated in tumor development and proliferation. 56 , 58 While studies show in vitro cytotoxic effects of EZH2 inhibition against several ovarian cancer lines, 58 the efficacy of EZH2 targeting is highly dependent on the mutational status of several other key epigenetic regulators, which can be cooperative or antagonistic to EZH2 function, rather than the mutational status of EZH2 itself (Figure 2). 55 , 59 , 60 , 61 , 62 , 63 There is a balanced and specific interplay between chromatin regulators to maintain cellular identity and accurate gene expression. Loss of antagonists to PRC2, such as BAF components ARID1A and SMARCA2/4, leads to unrestricted EZH2 activity and improper repression of key tumor suppressor genes. This has been shown to be a fundamental driver in multiple subtypes of ovarian cancer. 64 , 65 , 66 , 67 To understand the action and potential of EZH2 inhibition in ovarian cancer, we must first discuss the mechanism of action of these PRC2 antagonists whose loss results in EZH2 dependency in cancer.
FIGURE 2.
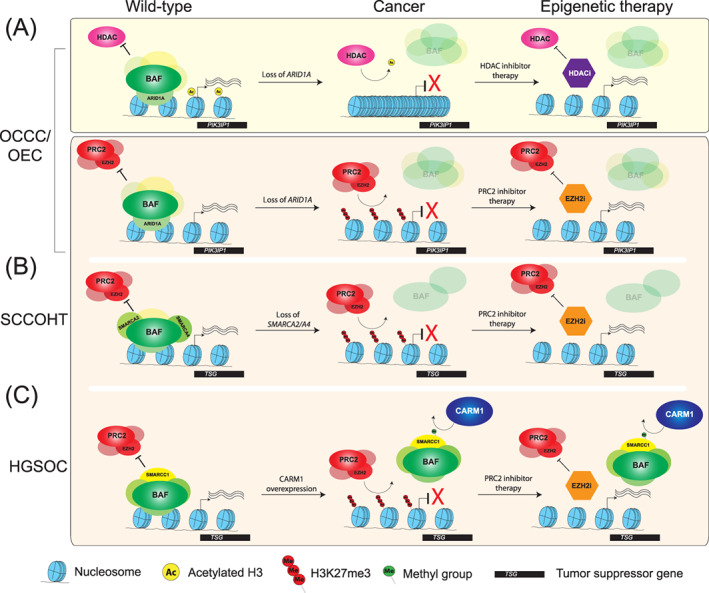
Key regulatory mechanisms in OCCC/OEC (A), SCCOHT (B), and HGSOC (C), highlighting the consequences of their disruption, the molecular sensitivities created, and how these perturbations can be exploited for therapy by epigenetic inhibitors
4. BAF
One of the best‐known nucleosome‐remodeling complexes is BAF. Its catalytic activity is important for accessibility and activation of genes, binding active promoters and enhancers through recognition and recruitment to H3K4me1 and multiple bromodomain‐containing subunits reading histone acetylation. 31 Originally identified in yeast as a major transcriptional regulating complex, it has since been found that human BAF comprises multiple configurations and compositions. 68 It contains a core set of essential components, the BAF‐core. Combinations of subunits build upon this to define three distinct compositions—BAF, PBAF and ncBAF. 31 , 69 ARID1A/B and DPF1/2/3 specify the BAF complex. PBRM1, BRD7, ARID2 and PHF10 define the PBAF complex, and ncBAF‐specific components include BRD9 and GLTSCR1/1L. 31 The presence of distinct accessory proteins specializes the complexes, directing them to primarily non‐redundant sets of genomic targets for each of these complexes, such as recruitment to active sites through the multiple acetyl lysine readers. 70
A large body of evidence has accumulated implicating a role for the BAF complex in tumor suppression. 41 , 71 In fact, components of BAF machinery are mutated in ~20% of human cancers, making it the most frequently mutated chromatin regulatory complex across human malignancies. 41 Consequently, identifying therapeutic susceptibilities of cancers carrying BAF mutations could be broadly applicable, particularly as understanding the mechanisms of specific BAF complex mutations has led to the discovery of targetable synthetic lethalities. 31 , 72 , 73 , 74 , 75 Indeed, the BAF components ARID1A and SMARCA2/4 are persistently mutated in ovarian cancer subtypes, 64 , 65 , 66 , 67 and represent promising sensitivities for epigenetic inhibition therapy, as discussed below.
4.1. ARID1A
AT‐rich interactive domain‐containing protein 1a (ARID1A) is a component of the BAF complex involved in the regulation of multiple genes. ARID1A is the most frequently mutated subunit of the BAF complex across all human malignancies. 69 ARID1A is mutated in ~57% of OCCC and ~30% of OEC, with the majority being frameshifts and nonsense, likely loss‐of‐function, mutations. 59 , 64 , 66 , 76 , 77 Its null mutations result in altered expression of several key genes such as CDKN1A and PIK3IP1, 59 , 65 , 66 both of which regulate entry into apoptosis and are aberrantly repressed in the absence of ARID1A due to changes in chromatin remodeling. 66 Mouse models with a single mutation in PIK3CA (a frequently co‐occurring mutation with ARID1A) have been shown to rapidly develop OCCC‐like tumors upon ARID1A loss. 78
The established antagonistic relationship between the Polycomb complexes and the BAF complex logically predicts that loss of function of BAF components will lead to unopposed activity of repressive Polycomb machinery. 31 , 79 Thus, repression of key apoptotic‐entry proteins in tumor cells can be expected to rely largely on EZH2 activity and suggests Polycomb complex components as promising therapeutic targets for the majority of OCCC and a significant proportion of OEC. 55 , 74 , 80 Indeed, ARID1A‐mutant ovarian cancer cell lines OVISE and TOV21G have displayed sensitivity to EZH2 inhibition, and in wild‐type ARID1A cells, its knockdown confers EZH2 sensitivity. 55 , 59 Without ARID1A, unrestrained activity of EZH2 represses ARID1A target genes, such as PIK3IP1. Once EZH2 is inhibited, PIK3IP1 repression is relieved, leading to apoptotic cell death in tumor cells (Figure 2A). Importantly, in wild‐type ARID1A cells EZH2 inhibition did not display any significant impact on cell proliferation demonstrating the specificity of this effect to ARID1A mutant ovarian cancers and underscoring the critical actionability of patients' tumor genotype in clinical practice, 59 and more specifically the way in which patient‐specific mutations and a mechanistic understanding of dysregulation in chromatin pathways can jointly inform rational therapeutic innovation.
Another prospective therapeutic target in ARID1A mutant cancers is histone deacetylase (HDAC) activity. Antagonism between the BAF complex and HDAC containing complexes such as NuRD is critical in maintaining chromatin state and cellular identity, and failure to maintain this balance contributes to cancer development. 31 , 81 In the absence of functional ARID1A, the overactivity of multiple HDACs results in aberrant repression of pro‐apoptotic genes, including p53 and PIK3IP1. 82 , 83 It has been reported that ARID1A mutant cells become dependent on HDAC6 activity, as HDAC inhibition results in accumulation of acetylated p53 Lys‐120—a pro‐apoptotic modification (Figure 2A). 82 In ARID1A mutant cells, PIK3IP1 is repressed through cooperative activity of EZH2 and HDAC2, and upon treatment with HDAC inhibitors, PIK3IP1 is derepressed and promotes cell death. Mouse models of OCCC lacking ARID1A activity display increased survival and reduced ascites accumulation and tumor progression when treated with the pan‐HDAC inhibitor SAHA, 83 making HDAC inhibition a very promising strategy for mutant ARID1A OCCC and OEC. In fact, the HDAC6 inhibitor ACY‐1215 is already in clinical trials for lymphoma, multiple myeloma and breast cancer (https://clinicaltrials.gov/) and may potentially be repurposed for OCCC or OEC. Growing evidence suggests a synergistic anti‐cancer action of the combination of EZH2 and HDAC inhibition in several cancers. 84 , 85 There is an increasing number of clinical trials implementing precision medicine and biomarker‐directed therapy, as well as combination treatments epigenetic drugs and established anti‐cancer therapies. 86 Given the evidence for epigenetic intervention as a strategy for ovarian cancer, this could be a very promising approach in the clinical setting.
4.2. SMARCA2/4
The human BAF complex has two separate ATPases, SMARCA2 and SMARCA4 (also known as BRM and BRG1, respectively). Each individually can combine with approximately eight other core subunits to form the BAF core, which in turn assemble into BAF, PBAF or ncBAF. SMARCA2/4 act as the molecular motor for the complex, using ATP hydrolysis to power the movement of the complex over DNA resulting in the local repositioning of nucleosomes. Mutations in SMARCA4 occur in a wide range of cancers, including lung cancers, 87 melanomas 88 and lymphomas. 89 Critically, almost every case of SCCOHT is characterized by inactivating mutations in SMARCA2 and/or SMARCA4. 67 , 74 , 90 , 91
A promising clinical development has also been the sensitivity of SMARCA4 null cells to EZH2 inhibition (Figure 2B). SCCOHT cell lines display synthetic lethality with core PRC2 complex subunits, which is supported by experiments showing that SCCOHT cells are acutely sensitive to EZH2 inhibition, inducing cell cycle arrest and apoptosis. 60 , 61 Considering the frequency of SMARCA2/4 loss in SCCOHT, EZH2 inhibition, or other PRC2 inhibitors (EZH1/2 dual or EEDi), would appear to be an excellent and highly effective choice, as has been suggested in several studies. 74 However, what has been overlooked is that cases of SCCOHT are frequently familial, predisposed by germline mutations in SMARCA4. 67 While EZH2 inhibitors are tolerated in SMARCA4 wild‐type patients in clinical trials to date, patients with germline SMARCA4 mutations instead may be inherently sensitive to EZH2 inhibition. To date, studies have been carried out using cell lines with homozygous deletions of SMARCA4, displaying sensitivity to EZH2, 60 or xenografting SMARCA4+/+ mice with cell lines bi‐allelic for loss of SMARCA4. 59 , 92 A key experiment that, to our knowledge, has not yet been carried out would be testing the toxicity of EZH2 inhibition on SMARCA4 heterozygous cell lines and mice where side‐effects compared to previous trials may be greater. In addition, the capacity to generate iPSCs from patients with SMARCA4 germline mutations could be a critical paradigm for defining patient‐specific efficacy and toxicity of treatments, well beyond the predictive capability of the mouse model.
5. CARM1
Protein arginine methyltransferases (PRMTs) catalyze the methylation of arginine side chains, a fundamental process in the regulation of mRNA splicing, signal transduction, DNA repair and gene expression. 93 , 94 In addition, arginine methylation modulates the activity of many cancer‐associated proteins. 94 , 95 Coactivator‐associated arginine methyltransferase 1 (CARM1, also known as PRMT4) is an arginine methyltransferase essential during mammalian development. 96 It catalyzes asymmetric dimethylation of arginines on a small set of distinct substrates, including histone 3. 97 Its recruitment to promoters results in increased levels of H3R17 and H3R26 methylation, which are associated with active transcription.
CARM1 is overexpressed in multiple cancer types, including breast, 98 , 99 colorectal 100 and HGSOC. 62 , 94 , 101 It is involved in the activation of several cancer‐related genes, including cyclins and beta‐catenin. 94 , 98 , 100 Other than its histone substrates, CARM1 also methylates substrates involved in epigenetic chromatin remodeling such as SMARCC1 (BAF155). CARM1 forms a complex with ATP‐remodeling (BAF) factors, and its methylation of SMARCC1 results in the eviction of the BAF complex from target loci (Figure 2C). 99 The precise mechanism of how this methylation redirects BAF targeting is not entirely clear. However, EZH2 and SMARCC1 antagonistically regulate several key tumor suppressors, such as the apoptosis‐promoting gene NOXA. 62 Therefore, akin to ARID1A‐mutant OCCC, CARM1 overexpression and its consequent loss of SMARCC1 activity impairs BAF's counteraction of Polycomb repression, leading to replacement by EZH2 (Figure 2C). 62 , 63 , 99 Indeed, EZH2 inhibition is selectively effective against CARM1‐overexpressing HGSOC, 62 , 63 and thus represents a promising therapeutic strategy in this setting as well (Figure 2C).
6. BROMODOMAIN PROTEINS
Multiple bromodomain‐containing proteins may be effective targets in ovarian cancer. One such bromodomain protein, BRD4, is a chromatin reader protein crucial during embryogenesis and cell differentiation. It, like other bromodomain‐containing proteins, binds acetylated histones at transcription start sites and active enhancers, recruiting transcriptional machinery to chromatin. 102 , 103 , 104 , 105 Many cancerous cells aberrantly hyperactivate oncogenes, such as MYC, through co‐option of non‐native enhancers and BRD4 activity. 106 BRD4 inhibition therefore may be selectively toxic to cancers dependent on such enhancer or super‐enhancer activity. BRD4‐occupied enhancers function within phase separated punctae, within which drugs such as cisplatin accumulate and show specific and greater activity. The manipulation or disturbance of such particles may therefore greatly impact the efficacy of current cancer therapies. 107 In fact, recent evidence implicates disruption of condensate‐forming mechanisms by cancer cells as a mechanism of chemo‐resistance, 107 one of the major issues in ovarian cancer long‐term therapy. 22 Therefore, the nature of BRD4 activity makes it a promising broad spectrum target, especially in the instance of chemo‐resistance to cisplatin.
The expression of BRD4 target genes is frequently altered in oncogenesis. 102 , 103 Its overexpression and consequent oncogenic transcriptional profiles have been reported in HGSOC and are associated with poor overall survival. 108 , 109 However, the enhancer landscape of ovarian carcinomas remains largely unexplored. Multiple inhibitors have been developed for targeting BRD4, which are already in clinical and pre‐clinical trials 27 , 110 that may be effectively applied to HGSOC. Indeed, HGSOCs displaying increased BRD4 expression and its transcription profile exhibit sensitivity to BET inhibition. 111 Considering the current poor survival rates and intractability of HGSOC, it is promising that there may already be multiple avenues of effective epigenetic treatment approved and available for repurposing.
Another bromodomain‐containing protein of significance to ovarian cancer is BRD9. Implicated in oncogenic profiles of several cancers, it is a key component of the GLTSCR1‐BAF (ncBAF or GBAF), a remodeling complex targeting multiple loci that are involved in pluripotency. 112 Perturbations in canonical BAF components have been shown to cause a dependence on ncBAF activity 75 and BRD9 inhibition with a small‐molecule degrader successfully reverses of the oncogenic transcription pattern and significantly impedes cell proliferation in cancers associated with perturbations in the BAF‐core. 73 , 75 Due to the frequent mutation of BAF components in ovarian cancer discussed here, targeting key ncBAF components may be of great potential. A recent study demonstrated that BRD9 levels are in fact elevated in ovarian cancer, and that inhibition sensitizes ovarian cancer to PARP inhibition and cisplatin therapy through regulation of the DNA damage response machinery—a frequently disrupted pathway in ovarian cancer. 113 , 114 The success of the BRD9 degrader in clinical trials will be of great significance for future ovarian cancer therapy.
7. FUTURE STEPS FOR THERAPEUTIC ADVANCES
What is clear from the evidence above is that the efficacy of EZH2 inhibition in multiple ovarian cancers (and indeed many other malignancies) is dependent on the mutational context, copy number or activity of other genes, that is, ARID1A, SMARCA4 and CARM1 rather than on Polycomb complex components themselves. This clearly indicates the importance of identifying individual patient mutational profiles for effective and directed therapy. Broad‐spectrum cancer therapies have limited utility, even within cancer subtypes. Ovarian cancer itself is subdivided into distinct categories, based on cell of origin, histology and pathogenesis. But, as discussed above, within these categories the underlying mutational and epigenetic landscapes vary greatly, and treatment that may be successful in one individual may be ineffective or even potentially harmful to another. In short, the key step forward in ovarian cancer treatment, and indeed cancer treatment in general, is to profile and define the specific mutational background of cells within individual tumors. Additionally, fully characterizing the cell‐of‐origin of the various subtypes, through techniques such as OriPRINT, 10 will allow proper molecular characterization and identification of subtype‐specific vulnerabilities. The burgeoning fields of organoid modeling, single‐cell technologies and epigenetic characterization can guide us through this complex area (Figure 3).
FIGURE 3.
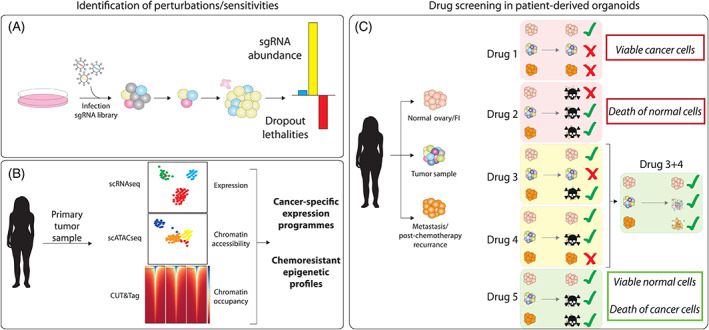
Emerging technologies for identifying improved patient‐specific treatments, illustrations of (A) CRISPR screening for identification of synthetic lethalities, (B) single‐cell technologies to identify cancer‐specific epigenetic/transcriptional changes and (C) patient‐specific organoid modeling as a basis for personalized drug screening and therapeutic discovery
7.1. Organoid modeling
Organoids mimic the fundamental characteristics of organs as multicellular, 3D in vitro cultures, derived from primary cells. 115 , 116 Originally, it was revealed that single intestinal stem cells could give rise to self‐organizing differentiated structures of the gut when provided with key factors for the in vivo intestinal stem cell niche. 115 , 116 With this paradigm, through modification of the cocktail of factors to the specific organ, there are now organoids developed that faithfully recapitulate key features of multiple vital organs, including breast, kidney and stomach. 117 , 118 , 119
Critically, 3D tumor organoids have been successfully developed for many malignancies. 116 , 120 , 121 They are already demonstrating promising results in preclinical prediction of treatment response to both targeted therapies and chemotherapeutics, 122 to improve and inform therapeutic options for broadly treatable tumors 123 and also as models to understand and characterize the mechanism of pathogenesis of intractable malignancies, such as pancreatic ductal adenocarcinoma. 124
Patient‐derived organoid models have recently been developed for ovarian cancer, through several approaches that recapitulate the genomic landscape and tumor heterogeneity of primary ovarian cancer tumors. 11 , 125 One of the key challenges in ovarian cancer is the capacity of a small subpopulation of quiescent cells to evade treatment, and results in post‐chemotherapy relapse. 126 It is becoming clear that a combination of therapies targeting individual susceptibilities of cancer subpopulations may lead to greater disease‐free survival rates. In combination with single‐cell omics technologies, organoids represent a transformative approach for making tumor heterogeneity experimentally tractable in vitro and profiling patients as to the mutational susceptibilities of their tumors (Figure 3C).
In addition, and perhaps most critically, organoid technology can be used for comparative analysis of potential treatments in tumor vs normal tissues (Figure 3C). For example, if we consider the case of the variable success of EZH2 inhibitors, characterizing ARID1A and SMARCA4 mutational status will provide valuable data as to the likely efficacy of EZH2, as mutants in both of these genes display dependency on EZH2 activity. 55 , 59 , 60 , 61 However, the question of whether the mutation is germline or somatic remains to be answered, with particular reference to germline SCCOHT SMARCA4 mutations. The burgeoning field of organoid modeling could be extremely beneficial to tackle this question. We can utilize patient‐derived normal and cancer organoids to comparatively assess the relative cytotoxicity of inhibitors to normal tissues that are specific to the patient, not just to the tumor itself.
7.2. Chromatin profiling and single‐cell technologies—deconvoluting tumor heterogeneity
The application of single‐cell transcriptomics to cancer has been illuminating in the deconvolution of tumor heterogeneity and has already been successful in identifying druggable pathways 127 and understanding sub‐populations resistant to drug treatment (Figure 3B). 128 Analysis of tumor cell populations pre‐ and post‐chemotherapy could be a fundamental in improving long‐term survival in ovarian cancer patients.
Disruption of chromatin remodeling is clearly a fundamental property in ovarian cancer. Understanding the impact of such mutations on activity and chromatin accessibility in cell lines can be readily profiled using bulk techniques, such as standard ChIP and ATAC‐seq. This is particularly relevant for BAF mutant cancers as they are a key regulator of accessibility at enhancers and promoters. 74 However, for organoid technology and patient‐specific profiling, obtaining the required cell number is a major hindrance. Primary samples have limited utility due to their propensity to undergo senescence, limiting the expansion capability and reducing input. The application of new technologies with reduced requirements for cell input overcomes such issues, such as CUT&Tag—an alternative approach to ChIP for detection of chromatin‐bound proteins using a tethered Tn5 transposase fusion protein. An antibody to the protein of interest is recognized and bound by the Tn5, which then specifically tagments adjacent DNA. 129 This technology reduces the required cell number from millions (standard ChIP‐seq) to several thousands, as well as allowing single‐cell applications 130 with the potential to increase resolution, throughput and elucidate mechanisms previously masked through bulk epigenomic analyses.
7.3. CRISPR screening
CRISPR‐based screening has been successfully used to identify synthetic lethalities in multiple cancers. 73 , 131 Identified hits often translate to logical sensitive targets to inhibition or degradation therapies. Applying this paradigm in ovarian cancer cell lines and organoids is a major step toward identifying druggable sensitivities (Figure 3A). Further classification of the intra‐tumor cell subpopulations, and the combination therapies most effective for complete cancer clearance, relies on integrative screening of organoids, CRISPR/Cas perturbation technology and single‐cell applications to fully understand the process of ovarian cancer tumorigenesis and optimize therapeutics screening and strategies (Figure 3).
The ever‐improving technologies in the field of CRISPR screening, such as knockout, activation, Perturb‐seq and domain tiling screens, have rapidly increased our ability to probe specific weaknesses in cancer cells. 131 , 132 , 133 Databases such as Depmap allow for simple identification of synthetic lethalities providing new and druggable options for targeting. 134 Focused use of such screening technologies in ovarian cancer cell models such as 2D cell lines and organoids will enhance drug target identification in a patient and mutation specific context. The coming decade will see these technologies cumulatively harnessed in order to advance ovarian cancer treatment.
8. CONCLUSION
Ovarian cancer therapy and survival has not progressed significantly in over 30 years. Genetic and epigenetic variability within different cells in a tumor presents obvious difficulties to cancer treatment. It has become clear that therapies targeting tumors as a homogenous group of cells commonly result in drug‐resistant relapse. 135 However, with the advent of new technologies, it is becoming more evident that many of the most common ovarian cancer subtypes, such as HGSOC and OCCC, have mutational profiles that could be targeted with therapies already in clinical and preclinical trials for other malignancies. In fact, with the recent approval of the EZH2 inhibitor Tazemetostat by the FDA for treatment of follicular lymphoma, 136 improved therapy for ovarian cancer may soon be readily available. It is extremely promising that inhibitors already in use for clinical trials exhibit anti‐tumor effects for ovarian cancer. EZH2 inhibition is one of the most promising therapies discussed here for CARM1‐overexpressing HGSOC, ARID1A‐mutant OCCC/OEC and SMARCA2/4‐deficient SCCOHT. What is lacking is a more thorough genetic and molecular understanding at the patient‐specific level. Combining emerging technologies such as patient‐specific organoids, CRISPR/Cas targeting of oncogenic drivers and single‐cell technologies, we can begin to identify novel appropriate and effective therapies to eradicate the entire population of cells that comprise individual patients' cancer. We may be on the cusp of a new era of ovarian cancer therapy, as we may already have the tools at our disposal to investigate drug repositioning and combination therapies.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (to G.T.) and Fondazione Umberto Veronesi (to A.Y.C.). The authors thank Eric Conway for discussion and critical reading. Open Access Funding provided by Universita degli Studi di Milano within the CRUI‐CARE Agreement.
Coughlan AY, Testa G. Exploiting epigenetic dependencies in ovarian cancer therapy. Int. J. Cancer. 2021;149(10):1732‐1743. 10.1002/ijc.33727
Funding information Associazione Italiana per la Ricerca sul Cancro, Grant/Award Number: IG‐2017‐20016; Fondazione Umberto Veronesi
Contributor Information
Aisling Y. Coughlan, Email: aisling.coughlan@ieo.it, @aisling1751.
Giuseppe Testa, Email: giuseppe.testa@ieo.it, @gtesta72.
REFERENCES
- 1. Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284‐296. 10.3322/caac.21456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kurman RJ, Shih IM. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am J Pathol. 2016;186:733‐747. 10.1016/j.ajpath.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14:9‐32. 10.20892/j.issn.2095-3941.2016.0084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061. 10.1038/nrdp.2016.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vaughan S, Coward JI, Bast RC Jr, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719‐725. 10.1038/nrc3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bowtell DD, Böhm S, Ahmed AA, et al. Rethinking ovarian cancer II: reducing mortality from high‐grade serous ovarian cancer. Nat Rev Cancer. 2015;15:668‐679. 10.1038/nrc4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Webb PM, Jordan SJ. Epidemiology of epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2017;41:3‐14. 10.1016/j.bpobgyn.2016.08.006 [DOI] [PubMed] [Google Scholar]
- 8. Network, T. C. G. A. R . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609‐615. 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gockley A, Melamed A, Bregar AJ, et al. Outcomes of women with high‐grade and low‐grade advanced‐stage serous epithelial ovarian cancer. Obstet Gynecol. 2017;129:439‐447. 10.1097/AOG.0000000000001867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lo Riso P, Villa CE, Gasparoni G, et al. A cell‐of‐origin epigenetic tracer reveals clinically distinct subtypes of high‐grade serous ovarian cancer. Genome Med. 2020;12:94. 10.1186/s13073-020-00786-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopper O, de Witte CJ, Lohmussaar K, et al. An organoid platform for ovarian cancer captures intra‐ and interpatient heterogeneity. Nat Med. 2019;25:838‐849. 10.1038/s41591-019-0422-6 [DOI] [PubMed] [Google Scholar]
- 12. Mantovani F, Collavin L, Del Sal G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019;26:199‐212. 10.1038/s41418-018-0246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Della Pepa C, Tonini G, Santini D, et al. Low grade serous ovarian carcinoma: from the molecular characterization to the best therapeutic strategy. Cancer Treat Rev. 2015;41:136‐143. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Mang M, Wang L, Klein R, Kong B, Zheng W. Tubal origin of ovarian endometriosis and clear cell and endometrioid carcinoma. Am J Cancer Res. 2015;5:869‐879. [PMC free article] [PubMed] [Google Scholar]
- 15. Cochrane DR, Tessier‐Cloutier B, Lawrence KM, et al. Clear cell and endometrioid carcinomas: are their differences attributable to distinct cells of origin? J Pathol. 2017;243:26‐36. 10.1002/path.4934 [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi H. Ovarian cancer in endometriosis: epidemiology, natural history, and clinical diagnosis. Int J Clin Oncol. 2009;14:378‐382. 10.1007/s10147-009-0931-2 [DOI] [PubMed] [Google Scholar]
- 17. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol. 1994;18:1102‐1116. 10.1097/00000478-199411000-00004 [DOI] [PubMed] [Google Scholar]
- 18. Lu B, Shi H. An in‐depth look at small cell carcinoma of the ovary, hypercalcemic type (SCCOHT): clinical implications from recent molecular findings. J Cancer. 2019;10:223‐237. 10.7150/jca.26978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ulbright TM, Roth LM, Stehman FB, Talerman A, Senekjian EK. Poorly differentiated (small cell) carcinoma of the ovary in young women: evidence supporting a germ cell origin. Hum Pathol. 1987;18:175‐184. 10.1016/s0046-8177(87)80336-2 [DOI] [PubMed] [Google Scholar]
- 20. Testa U, Petrucci E, Pasquini L, Castelli G, Pelosi E. Ovarian cancers: genetic abnormalities, tumor heterogeneity and progression, clonal evolution and cancer stem cells. Medicines (Basel). 2018;5(1):1‐74. 10.3390/medicines5010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matulonis UA. Management of newly diagnosed or recurrent ovarian cancer. Clin Adv Hematol Oncol. 2018;16:426‐437. [PubMed] [Google Scholar]
- 22. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502‐516. 10.1038/nrc1123 [DOI] [PubMed] [Google Scholar]
- 23. Burger RA. Experience with bevacizumab in the management of epithelial ovarian cancer. J Clin Oncol. 2007;25:2902‐2908. 10.1200/JCO.2007.12.1509 [DOI] [PubMed] [Google Scholar]
- 24. George A, Kaye S, Banerjee S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat Rev Clin Oncol. 2017;14:284‐296. 10.1038/nrclinonc.2016.191 [DOI] [PubMed] [Google Scholar]
- 25. Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27 (Suppl 1):i40‐i44. 10.1093/annonc/mdw094 [DOI] [PubMed] [Google Scholar]
- 26. Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum‐sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Oncol. 2016;17:1579‐1589. 10.1016/S1470-2045(16)30376-X [DOI] [PubMed] [Google Scholar]
- 27. Brien GL, Valerio DG, Armstrong SA. Exploiting the Epigenome to control cancer‐promoting gene‐expression programs. Cancer Cell. 2016;29:464‐476. 10.1016/j.ccell.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meloni M, Testa G. Scrutinizing the epigenetics revolution. Bios. 2014;9:431‐456. 10.1057/biosoc.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature. 1997;389:251‐260. 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 30. Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP‐dependent chromatin‐remodelling complexes. Nat Rev Mol Cell Biol. 2017;18:407‐422. 10.1038/nrm.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bracken AP, Brien GL, Verrijzer CP. Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev. 2019;33:936‐959. 10.1101/gad.326066.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang YC, Peterson SE, Loring JF. Protein post‐translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24:143‐160. 10.1038/cr.2013.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259‐266. 10.1038/nsmb.2470 [DOI] [PubMed] [Google Scholar]
- 34. Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42‐48. 10.1016/j.ceb.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 35. Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343‐349. 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:1‐16. 10.1186/s13059-019-1870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Agger K, Cloos PA, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731‐734. 10.1038/nature06145 [DOI] [PubMed] [Google Scholar]
- 38. Rotili D, Mai A. Targeting histone demethylases: a new avenue for the fight against cancer. Genes Cancer. 2011;2:663‐679. 10.1177/1947601911417976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl‐lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870‐1881. 10.1101/gad.1110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Isono K, Endo TA, Ku M, et al. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell. 2013;26:565‐577. 10.1016/j.devcel.2013.08.016 [DOI] [PubMed] [Google Scholar]
- 41. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and Bioinformatic analysis of mSWI/SNF (BAF) complexes reveals extensive roles in human malignancy. Nat Genet. 2013;45:592‐601. 10.1038/ng.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tamburri S, Lavarone E, Fernández‐Pérez D, et al. Histone H2AK119 mono‐ubiquitination is essential for polycomb‐mediated transcriptional repression. Mol Cell. 2020;77:840‐856.e845. 10.1016/j.molcel.2019.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cao R, Tsukada Y, Zhang Y. Role of Bmi‐1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845‐854. 10.1016/j.molcel.2005.12.002 [DOI] [PubMed] [Google Scholar]
- 44. Scelfo A, Fernández‐Pérez D, Tamburri S, et al. Functional landscape of PCGF proteins reveals both RING1A/B‐dependent‐and RING1A/B‐independent‐specific activities. Mol Cell. 2019;74:1037‐1052.e1037. 10.1016/j.molcel.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kundu S, Ji F, Sunwoo H, et al. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol Cell. 2017;65:432‐446.e435. 10.1016/j.molcel.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Plys AJ, Davis CP, Kim J, et al. Phase separation of Polycomb‐repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 2019;33:799‐813. 10.1101/gad.326488.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferrari KJ, Scelfo A, Jammula S, et al. Polycomb‐dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014;53:49‐62. 10.1016/j.molcel.2013.10.030 [DOI] [PubMed] [Google Scholar]
- 48. Deevy O, Bracken AP. PRC2 functions in development and congenital disorders. Development. 2019;146(19):1‐13. 10.1242/dev.181354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Béguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677‐692. 10.1016/j.ccr.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol. 2007;27:3769‐3779. 10.1128/MCB.01432-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Morin RD, Johnson NA, Severson TM, et al. A. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet. 2010;42:181‐185. 10.1038/ng.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665‐667. 10.1038/ng.620 [DOI] [PubMed] [Google Scholar]
- 53. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722‐726. 10.1038/ng.621 [DOI] [PubMed] [Google Scholar]
- 54. Guo J, Cai J, Yu L, Tang H, Chen C, Wang Z. EZH2 regulates expression of p57 and contributes to progression of ovarian cancer in vitro and in vivo. Cancer Sci. 2011;102:530‐539. 10.1111/j.1349-7006.2010.01836.x [DOI] [PubMed] [Google Scholar]
- 55. Alldredge JK, Eskander RN. EZH2 inhibition in ARID1A mutated clear cell and endometrioid ovarian and endometrioid endometrial cancers. Gynecol Oncol Res Pract. 2017;4:17. 10.1186/s40661-017-0052-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8:1610‐1618. 10.1158/1541-7786.MCR-10-0398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lu C, Bonome T, Li Y, et al. Gene alterations identified by expression profiling in tumor‐associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007;67:1757‐1768. 10.1158/0008-5472.CAN-06-3700 [DOI] [PubMed] [Google Scholar]
- 58. Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18:185‐197. 10.1016/j.ccr.2010.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bitler BG, Aird KM, Garipov A, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A‐mutated cancers. Nat Med. 2015;21:231‐238. 10.1038/nm.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Y, Chen SY, Karnezis AN, et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcemic type. J Pathol. 2017;242:371‐383. 10.1002/path.4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128‐134. 10.1038/nm.4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Karakashev S, Zhu H, Wu S, et al. CARM1‐expressing ovarian cancer depends on the histone methyltransferase EZH2 activity. Nat Commun. 2018;9:1‐11. 10.1038/s41467-018-03031-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karakashev S, Fukumoto T, Zhao B, et al. EZH2 inhibition sensitizes CARM1‐high, homologous recombination proficient ovarian cancers to PARP inhibition. Cancer Cell. 2020;37:157‐167.e156. 10.1016/j.ccell.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science (New York, N.Y.). 2010;330:228‐231. 10.1126/science.1196333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guan B, Wang TL, Shih IM. ARID1A, a factor that promotes formation of SWI/SNF‐mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718‐6727. 10.1158/0008-5472.CAN-11-1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takeda T, Banno K, Okawa R, et al. ARID1A gene mutation in ovarian and endometrial cancers (review). Oncol Rep. 2016;35:607‐613. 10.3892/or.2015.4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Witkowski L, Carrot‐Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438‐443. 10.1038/ng.2931 [DOI] [PubMed] [Google Scholar]
- 68. Becker PB, Workman JL. Nucleosome remodeling and epigenetics. Cold Spring Harb Perspect Biol. 2013;5:1‐19. 10.1101/cshperspect.a017905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mashtalir N, D'Avino AR, Michel BC, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 2018;175:1272‐1288.e1220. 10.1016/j.cell.2018.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Valencia AM, Kadoch C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol. 2019;21:152‐161. 10.1038/s41556-018-0258-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481‐492. 10.1038/nrc3068 [DOI] [PubMed] [Google Scholar]
- 72. McBride MJ, Mashtalir N, Winter EB, et al. The nucleosome acidic patch and H2A ubiquitination underlie mSWI/SNF recruitment in synovial sarcoma. Nat Struct Mol Biol. 2020;27:836‐845. 10.1038/s41594-020-0466-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brien GL, Remillard D, Shi J, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife. 2018;7:1‐26. 10.7554/eLife.41305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pan J, McKenzie ZM, D'Avino AR, et al. The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity‐independent genomic targeting. Nat Genet. 2019;51:618‐626. 10.1038/s41588-019-0363-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Michel BC, D'Avino AR, Cassel SH, et al. A non‐canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20:1410‐1420. 10.1038/s41556-018-0221-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Okawa R, Banno K, Iida M, et al. Aberrant chromatin remodeling in gynecological cancer. Oncol Lett. 2017;14:5107‐5113. 10.3892/ol.2017.6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1:1‐17. 10.1126/sciadv.1500447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chandler RL, Damrauer JS, Raab JR, et al. Coexistent ARID1A‐PIK3CA mutations promote ovarian clear‐cell tumorigenesis through pro‐tumorigenic inflammatory cytokine signalling. Nat Commun. 2015;6:6118. 10.1038/ncomms7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kadoch C, Williams RT, Calarco JP, et al. Dynamics of BAF‐Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2017;49:213‐222. 10.1038/ng.3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim KH, Kim W, Howard TP, et al. SWI/SNF‐mutant cancers depend on catalytic and non‐catalytic activity of EZH2. Nat Med. 2015;21:1491‐1496. 10.1038/nm.3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Mohd‐Sarip A, Teeuwssen M, Bot AG, et al. DOC1‐dependent recruitment of NURD reveals antagonism with SWI/SNF during epithelial‐mesenchymal transition in oral cancer cells. Cell Rep. 2017;20:61‐75. 10.1016/j.celrep.2017.06.020 [DOI] [PubMed] [Google Scholar]
- 82. Bitler BG, Wu S, Park PH, et al. ARID1A‐mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962‐973. 10.1038/ncb3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Fukumoto T, Park PH, Wu S, et al. Repurposing Pan‐HDAC inhibitors for ARID1A‐mutated ovarian cancer. Cell Rep. 2018;22:3393‐3400. 10.1016/j.celrep.2018.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Takashina T, Kinoshita I, Kikuchi J, et al. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non‐small‐cell lung cancer cells. Cancer Sci. 2016;107:955‐962. 10.1111/cas.12957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lue JK, Prabhu SA, Liu Y, et al. Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin Cancer Res. 2019;25:5271‐5283. 10.1158/1078-0432.CCR-18-3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Morel D, Jeffery D, Aspeslagh S, Almouzni G, Postel‐Vinay S. Combining epigenetic drugs with other therapies for solid tumours ‐ past lessons and future promise. Nat Rev Clin Oncol. 2020;17:91‐107. 10.1038/s41571-019-0267-4 [DOI] [PubMed] [Google Scholar]
- 87. Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543‐550. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251‐263. 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B‐cell lymphomas. Blood Cancer J. 2015;5:e361. 10.1038/bcj.2015.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jelinic P, Mueller JJ, Olvera N, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424‐426. 10.1038/ng.2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427‐429. 10.1038/ng.2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fillmore CM, Xu C, Desai PT, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumors to TopoII inhibitors. Nature. 2015;520:239‐242. 10.1038/nature14122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1‐13. 10.1016/j.molcel.2008.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37‐50. 10.1038/nrc3409 [DOI] [PubMed] [Google Scholar]
- 95. Feng J, Dang Y, Zhang W, et al. PTEN arginine methylation by PRMT6 suppresses PI3K–AKT signaling and modulates pre‐mRNA splicing. Proc Natl Acad Sci U S A. 2019;116:6868‐6877. 10.1073/pnas.1811028116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yadav N, Lee J, Kim J, et al. Specific protein methylation defects and gene expression perturbations in coactivator‐associated arginine methyltransferase 1‐deficient mice. Proc Natl Acad Sci U S A. 2003;100:6464‐6468. 10.1073/pnas.1232272100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T. Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep. 2002;3:39‐44. 10.1093/embo-reports/kvf013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. El Messaoudi S, Fabbrizio E, Rodriguez C, et al. Coactivator‐associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci U S A. 2006;103:13351‐13356. 10.1073/pnas.0605692103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wang L, Zhao Z, Meyer MB, et al. CARM1 Methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. 2014;25:21‐36. 10.1016/j.ccr.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ou CY, LaBonte MJ, Manegold PC, et al. A coactivator role of CARM1 in the dysregulation of β‐catenin activity in colorectal cancer cell growth and gene expression. Mol Cancer Res. 2011;9:660‐670. 10.1158/1541-7786.mcr-10-0223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zeng L, Zhou MM. Bromodomain: an acetyl‐lysine binding domain. FEBS Lett. 2002;513:124‐128. 10.1016/s0014-5793(01)03309-9 [DOI] [PubMed] [Google Scholar]
- 103. Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. 10.1186/s12943-018-0915-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee JE, Park YK, Park S, et al. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun. 2017;8:2217. 10.1038/s41467-017-02403-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sabari BR, Dall'Agnese A, Boija A, et al. Coactivator condensation at super‐enhancers links phase separation and gene control. Science (New York, N.Y.). 2018;361:1‐11. 10.1126/science.aar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lancho O, Herranz D. The MYC enhancer‐ome: long‐range transcriptional regulation of MYC in cancer. Trends Cancer. 2018;4:810‐822. 10.1016/j.trecan.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Klein IA, Boija A, Afeyan LK, et al. Partitioning of cancer therapeutics in nuclear condensates. Science (New York, N.Y.). 2020;368:1386‐1392. 10.1126/science.aaz4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ucar D, Lin DI. Amplification of the bromodomain‐containing protein 4 gene in ovarian high‐grade serous carcinoma is associated with worse prognosis and survival. Mol Clin Oncol. 2015;3:1291‐1294. 10.3892/mco.2015.622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rhyasen GW, Yao Y, Zhang J, et al. BRD4 amplification facilitates an oncogenic gene expression program in high‐grade serous ovarian cancer and confers sensitivity to BET inhibitors. PLoS One. 2018;13:e0200826. 10.1371/journal.pone.0200826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Alqahtani A, Choucair K, Ashraf M, et al. Bromodomain and extra‐terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA. 2019;5:FSO372. 10.4155/fsoa-2018-0115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Baratta MG, Schinzel AC, Zwang Y, et al. An in‐tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112:232‐237. 10.1073/pnas.1422165112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gatchalian J, Malik S, Ho J, et al. A non‐canonical BRD9‐containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat Commun. 2018;9:5139. 10.1038/s41467-018-07528-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nguyen L, Martens JWM, Van Hoeck A, Cuppen E. Pan‐cancer landscape of homologous recombination deficiency. Nat Commun. 2020;11:5584. 10.1038/s41467-020-19406-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhou Q, Huang J, Zhang C, et al. The bromodomain containing protein BRD‐9 orchestrates RAD51‐RAD54 complex formation and regulates homologous recombination‐mediated repair. Nat Commun. 2020;11:2639. 10.1038/s41467-020-16443-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262‐265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 116. Schutgens F, Clevers H. Human organoids: tools for understanding biology and treating diseases. Annu Rev Pathol. 2020;15:211‐234. 10.1146/annurev-pathmechdis-012419-032611 [DOI] [PubMed] [Google Scholar]
- 117. Sachs N, de Ligt J, Kopper O, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373‐386.e310. 10.1016/j.cell.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 118. Schutgens F, Rookmaaker MB, Margaritis T, et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat Biotech. 2019;37:303‐313. 10.1038/s41587-019-0048-8 [DOI] [PubMed] [Google Scholar]
- 119. Bartfeld S, Bayram T, van de Wetering M, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148:126‐136.e126. 10.1053/j.gastro.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sachs N, Clevers H. Organoid cultures for the analysis of cancer phenotypes. Curr Opin Genet Dev. 2014;24:68‐73. 10.1016/j.gde.2013.11.012 [DOI] [PubMed] [Google Scholar]
- 121. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science (New York, N.Y.). 2019;364:952‐955. 10.1126/science.aaw6985 [DOI] [PubMed] [Google Scholar]
- 122. Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science (New York, N.Y.). 2018;359:920‐926. 10.1126/science.aao2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Verissimo CS, Overmeer RM, Ponsioen B, et al. Targeting mutant RAS in patient‐derived colorectal cancer organoids by combinatorial drug screening. eLife. 2016;5:1‐26. 10.7554/eLife.18489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324‐338. 10.1016/j.cell.2014.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Velletri T, Villa CE, Lupia M, et al. Single cell derived organoids capture the self‐renewing subpopulations of metastatic ovarian cancer. bioRxiv. 2018;1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Jones BA, Varambally S, Arend RC. Histone methyltransferase EZH2: a therapeutic target for ovarian cancer. Mol Cancer Ther. 2018;17:591‐602. 10.1158/1535-7163.mct-17-0437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Izar B, Tirosh I, Stover EH, et al. A single‐cell landscape of high‐grade serous ovarian cancer. Nat Med. 2020;26:1271‐1279. 10.1038/s41591-020-0926-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rambow F, Rogiers A, Marin‐Bejar O, et al. Toward minimal residual disease‐directed therapy in melanoma. Cell. 2018;174:843‐855.e819. 10.1016/j.cell.2018.06.025 [DOI] [PubMed] [Google Scholar]
- 129. Kaya‐Okur HS, Wu SJ, Codomo CA, et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019;10:1930. 10.1038/s41467-019-09982-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bartosovic M, Kabbe M, Castelo‐Branco G. Single‐cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotech. 2021;39(7):825‐835. 10.1038/s41587-021-00869-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Boettcher M, Tian R, Blau JA, et al. Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat Biotechnol. 2018;36:170‐178. 10.1038/nbt.4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein‐tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635‐646. 10.1016/j.cell.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Dixit A, Parnas O, Li B, et al. Perturb‐Seq: dissecting molecular circuits with scalable single‐cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853‐1866.e1817. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell. 2017;170:564‐576.e516. 10.1016/j.cell.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. 10.1016/j.cell.2017.01.018 [DOI] [PubMed] [Google Scholar]
- 136. Hoy S, Tazemetostat M. First approval. Drugs. 2020;80:513‐521. 10.1007/s40265-020-01288-x [DOI] [PubMed] [Google Scholar]