

Summary
Background
The microbiome is emerging as a crucial player of the immune checkpoint in cancer. Melanoma is a highly immunogenic tumour, and the composition of the gut microbiome has been correlated to prognosis and evolution of advanced melanoma and proposed as a biomarker for immune checkpoint therapy.
Objectives
We investigated the gut fungal and bacterial compositions in early‐stage melanoma and correlated microbial profiles with histopathological features.
Methods
Sequencing of bacterial 16S rRNA and the fungal internal transcribed spacer region was performed on faecal samples of patients with stage I and II melanoma, and healthy controls. A meta‐analysis with gut microbiota data from patients with metastatic melanoma was also carried out.
Results
We found a combination of gut fungal and bacterial profiles significantly discriminating patients with melanoma from controls. In patients with melanoma, we observed an abundance of Prevotella copri and yeasts belonging to the order Saccharomycetales. We found that the bacterial and fungal community correlated to melanoma invasiveness, whereas the specific fungal profile correlated to melanoma regression. Bacteroides was identified as general marker of immunogenicity, being shared by regressive and invasive melanoma. In addition, the bacterial communities in patients with stage I and II melanoma were different in structure and richer than those from patients with metastatic melanoma.
Conclusions
The composition of the gut microbiota in early‐stage melanoma changes along the gradient from in situ to invasive (and metastatic) melanoma. Changes in the microbiota and mycobiota are correlated to the histological features of early‐stage melanoma, and to the clinical course and response to immune therapies of advanced‐stage melanoma, through direct or indirect immunomodulation.
Short abstract
What is already known about this topic?
The microbial community living in symbiosis with our human body (i.e. the microbiota) is fundamental for health.
In the cancer field, and in particular in melanoma, the gut microbiota is drawing attention due to its ability to control immune checkpoints and to determine the success of immune therapies.
What does this study add?
This study evaluates for the first time the gut microbial community of patients with early melanoma (stage I and II), including bacteria, yeasts and fungi.
Promising associations between gut microbial profiles and histopathological features of melanoma along the in situ–invasive (and metastatic) axis were found.
Gut microbiota and the degree of immunogenicity of early‐stage melanoma were linked.
What is the translational message?
Microbial profiles might contribute to better prognosis of early‐stage melanoma through direct or indirect immunomodulation.
Linked Comment: N.O.S. Camara. Br J Dermatol 2022; 186:12–13.
Plain language summary available online
Melanoma is a malignant tumour arising from melanocytes. 1 It causes the greatest number of skin‐cancer‐related deaths worldwide, and despite the recent advances in therapeutic strategies, its prognosis in advanced stages remains poor. 1 The environmental determinants of melanoma remain elusive, although a well‐known risk factor is sun exposure. 2 , 3
In the past 10 years, it has become increasingly evident that in addition to our genome, health status depends on complex interactions with our symbiotic microbial community (the microbiota) and the genes and functions associated with it (the microbiome). The human microbiota is emerging as a crucial player in several types of cancer, and is an excellent predictor of the outcome of cancer immune checkpoint therapy. 4 , 5 , 6 Patients with melanoma under nivolumab or pembrolizumab therapy showed a significantly higher alpha diversity and enrichment of selected microbial species positively associated with enhanced systemic or local antitumour immune reaction. 6 A specific gut bacterial composition was found to be a predictive biomarker for the clinical outcome and the possible development of adverse gastrointestinal effects to ipilimumab treatment, 6 , 7 , 8 , 9 , 10 suggesting that the gut microbiota is able to modulate both anticancer mechanisms and immune surveillance. 11 Nevertheless, none of these studies investigated the gut fungal community. Fungi are well known for their immunogenicity traits, as recently observed in autoimmune diseases, such as Crohn disease. 12 Moreover, neither gut bacterial nor fungal communities have yet been investigated in patients with early‐stage (nonmetastatic) melanoma.
This study aimed to assess the gut bacterial and fungal community composition in a group of patients with stage I and II melanoma and to compare it with that of healthy controls. We correlated clinical and histopathological features of early‐stage melanoma with the identified microbial profiles in order to discover putative microbial biomarkers. Finally, a comparison with patients with advanced‐stage metastatic melanoma was achieved with a meta‐analysis.
Patients and methods
Study participants
Twenty patients with a histopathological diagnosis of melanoma, and 16 age‐ and sex‐matched healthy controls living in the province of Florence (Tuscany, Italy) were recruited. They were compliant to the inclusion and exclusion criteria (Appendix S1; see Supporting Information) and were screened among patients referring to the university‐based outpatient service for melanoma prevention and follow‐up of the Section of Dermatology, Department of Health Sciences, University of Florence (Florence, Italy). For each enrolled patient and control, we collected demographic, personal and clinical data (Tables S1 and S2; see Supporting Information).
The study was carried out in compliance with the Declaration of Helsinki principles for medical research involving human subjects and was reviewed and approved by the local institutional ethics review committee (Comitato Etico Regionale per la Sperimentazione Clinica della Regione Toscana‐Sezione: Area Vasta Centro; study ID: 10578_BIO).
Evaluation of clinical and histopathological parameters of patients with melanoma
Clinical data and histological parameters of melanoma 13 were assessed, including body site, histopathological subtype, Breslow thickness (recorded in millimetres and measured from the granular layer or, when present, the ulcer base, to the deepest extent of invasion by tumour cells), ulceration, mitotic index (number of mitoses per mm2), lymphovascular invasion, microsatellite or in transit metastasis, perineural invasion or neurotrophism, growth phase, tumour‐infiltrating lymphocytes and type (brisk or not brisk), regression and Clark level. 13 , 14 Melanoma staging was defined according to American Joint Committee on Cancer 8th edition pathological staging of cutaneous malignant melanoma, regional lymph nodes and metastasis 15 (Table S3; see Supporting Information).
Sample collection and total DNA extraction
To collect faecal samples, all enrolled patients received a photographic booklet to instruct them. Stool samples were stored with 8 mL of RNAlater stabilization solution (Thermo Fisher Scientific, Waltham, MA, USA) at −20 °C until DNA extraction. Total DNA was extracted from 250 mg (wet weight) of each faecal sample using the DNeasy PowerSoil Kit (Qiagen, Venlo, the Netherlands). DNA integrity and quality were checked on 1% agarose gel and quantified using the Qubit 4 Fluorometer (Thermo Fisher Scientific).
Sample collection was performed after excision of the tumour and subsequent histopathological diagnosis of melanoma, within 12 months. We assumed that the microbiota composition should have remained stable before and after diagnosis, as patients using medication reported an intake of several years before melanoma diagnosis, and no further medication was prescribed after melanoma excision, as such a procedure is considered minor surgery. In addition, all enrolled patients had early‐stage melanoma, which does not need any treatment except for wide surgical excision and regular noninvasive follow‐up.
Microbiota and mycobiota sequencing
For each DNA sample, the bacterial 16S rRNA gene was amplified using a primer set specific for the V3–V4 hypervariable regions (341f: 5′‐CCTACGGGNGGCWGCAG‐3′ and 805r: 5′‐GACTACNVGGGTWTCTAATCC‐3′), 16 while the fungal internal transcribed spacer (ITS) was amplified using a primer set specific for the ITS1 rDNA region (ITS1f: 5′‐ CTTGGTCATTTAGAGGAAGTAA‐3′ and ITS2r: 5′‐GCTGCGTTCTTCATCGATGC‐3′). 17 Sequencing was performed on the Illumina MiSeq (Illumina, San Diego, CA, USA) with the V3 chemistry 600 cycle PE300 protocol, at the Fondazione Edmund Mach, Trento, Italy, following their internal protocol.
Sequencing data processing and statistical analysis
The sequencing data analyses are described in full in Appendix S1. Briefly, amplification primers and any sequencing adapter were removed with Cutadapt, 18 and low‐quality bases at the 5′‐end of the reads were filtered with Sickle. 19 MICCA v1.7.2 20 was used to remove sequences containing N bases, to merge forward and reverse reads (allowing a maximum 8 mismatch in a minimum overlap of 32 bp), for picking of zero‐radius operational taxonomic units (zOTUs) with the UNOISE3 21 picking algorithm (zOTUs defined by the UNOISE algorithm author), and for taxonomic assignation using the RDP classifier and database v2.11 22 for bacteria, while using the RDP classifier v2.11 and the UNITE database (release 4 July 2014) 23 for fungi. Processed reads data are available at the European Nucleotide Archive under accession number PRJEB35665.
To perform a meta‐analysis to compare bacterial communities from our study against the cohort of patients with metastatic melanoma from Matson et al., 6 16S‐targeted metagenomics raw data were firstly retrieved from the Sequence Read Archive (accession SRP116709). Given that in Matson et al. the sole V4 hypervariable region was targeted, we firstly had to make the two datasets comparable by selecting the portion corresponding to the V4 hypervariable region from our data. To do so, we used Cutadapt to trim and discard the portion of our reads before the forward primer used in Matson et al. (i.e. primer 515f; in Cutadapt we used the ‐g option for the primer sequence and the option ‐discard untrimmed). After trimming, the two datasets were joined and then processed in MICCA for OTU picking using the ‘greedy denovo’ algorithm with a similarity threshold of 97%, and for taxonomy assignment. We preferred the use of the similarity‐based OTU picking algorithm to avoid any assumptions or considerations connected to the more recent zOTU picking method (UNOISE3) that we used for our data.
For the analysis of both our original data and those from the meta‐analysis, subsequent analyses were performed in R v3.42 (R Foundation, Vienna, Austria), similarly to the methods previously reported. 24 , 25 Prior to any analysis, count data were scaled with cumulative sum scaling (CSS transform, followed by log2 scaling), as implemented in metagenomeSeq v1.26.3. 26 An extended description of the methods used can be found in Appendix S1.
Results
Patient enrolment
The mean age of the patients with melanoma was significantly higher than that of the healthy controls (66 vs. 41 years, P < 0·001), while the sex distribution and body mass index (BMI) were comparable between the two groups: eight of 20 female in the melanoma group vs. 10 of 16 female controls, P = 0·18; and mean (SD) BMI values of 26.3 (4·6) in the melanoma group vs. 24·2 (3·2) in the controls, P = 0·14.
We characterized the gut microbiota and mycobiota composition of the 20 patients with melanoma compared with the 16 healthy controls. The clinical data of both groups are reported in Tables S1 and S2. Considering the histological parameters (Table S3), in the melanoma group seven of 20 patients (35%) had in situ melanoma, 13 of 20 (65%) had invasive melanoma (Breslow thickness ranging from 0.2 to 4.1 mm) and 10 of 20 (50%) had regression.
Different gut microbial communities between patients with melanoma and healthy controls
The diversity of gut microbial communities was investigated by alpha diversity (Figure 1a, b) and beta diversity ordination (Figure 1c–e). No significant differences in alpha diversity between patients with melanoma and healthy controls were found for bacterial community (Figure 1a), while a significantly higher richness of fungi was observed in patients with melanoma (Figure 1b).
Figure 1.
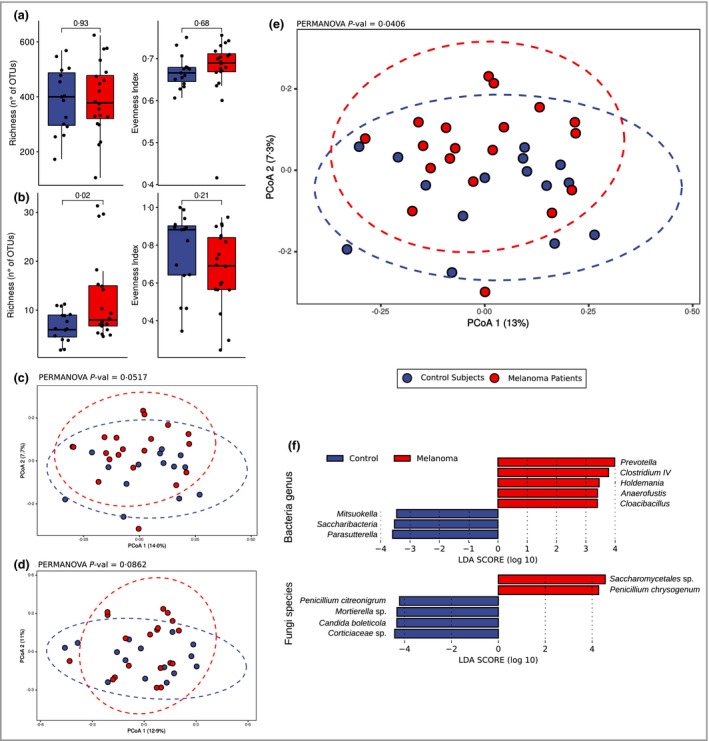
Diversity analysis of bacterial and fungal communities in patients with early‐stage melanoma. (a, b) Alpha diversity analysis using synthetic indexes of richness and evenness for (a) bacterial and (b) fungal communities. (c–e) Principal coordinates analysis (PCoA) ordination based on Bray–Curtis distance of samples from patients with melanoma and healthy controls considering bacterial community alone (c), fungal community alone (d) and combined bacterial and fungal community (e) datasets. (f) Putative differential bacterial (at the genus level) and fungal (at the species level) markers between patients with early‐stage melanoma and controls assessed by linear discriminant analysis (LDA) effect size analysis (alpha = 0.05; LDA cutoff = 2). OTU, operational taxonomic unit.
Principal coordinate analysis (PCoA) ordinations (on Bray–Curtis dissimilarity) were performed on bacterial (Figure 1c) and fungal communities (Figure 1d) separately, then the two datasets were joined and analysed together to provide a global picture of the total gut microbiota (Figure 1e). The total microbial community structure showed significant differences between controls and patients with melanoma, while marginally significant differences were found when analysing the bacterial community alone, and no significant differences were found when analysing the fungal community alone (permanova analysis P‐values in Figure 1c–e). Both the bacterial community and the total gut community showed the same differences in PCoA ordination, yet the permanova test indicated that the total gut community dataset (bacteria and fungi joined together; P = 0.041) was better able to discriminate the two cohorts. In Figures S1 and S2 (see Supporting Information) we report the composition analysis of the bacterial (at family level) and fungal communities (at order level) in all of the samples (Figure S2a) and in samples grouped by disease class (Figure S2b).
By linear discriminant analysis effect size analysis (Figure 1f and Figure S1) we discovered eight bacterial genera as candidate biomarkers of early‐stage melanoma. Among them, five were found with higher abundance and three with lower abundance in patients with melanoma compared with healthy controls. Considering the relative abundance and prevalence distribution of those markers in patients with melanoma (Figure S1), Prevotella, Clostridium IV, Holdemania and Anaerofustis were enriched in patients with melanoma, while Saccharibacteria and Parasutterella were depleted in patients with melanoma. Considering the fungal component, only the order Saccharomycetales was enriched in patients with melanoma vs. healthy controls.
Microbial profiles of patients with melanoma differ according to histopathological features
Significant correlations between the global gut biota structure and invasive melanoma (i.e. the presence or not of invasion beyond the dermoepidermal junction) were observed. In Figure 2(a, b) we show the PCoA ordination in the melanoma group with different melanoma invasion features. The ordination analyses suggested that microbial communities from patients with in situ and invasive melanoma were different, as confirmed by the significant results of permanova (Figure 2a and Table 1) and ANOSIM analysis (Figure 2c, d and Table 1). We found a significant relationship between the fungal community composition (and, to a lesser extent, the combined bacterial and fungal community) and melanoma regression (Table 1 and Figure 2e, f; fungal community alone).
Figure 2.
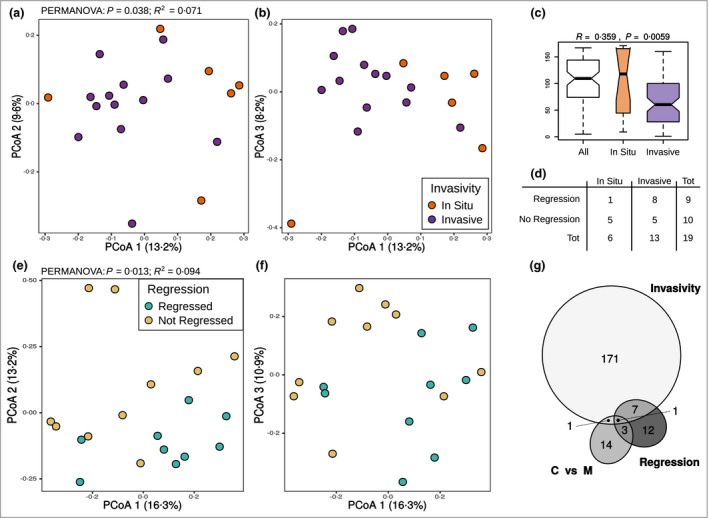
Differences in microbial communities of patients with early‐stage melanoma with respect to invasion and regression. (a, b) Principal coordinates analysis (PCoA) ordination based on Bray–Curtis distance of the combined bacterial and fungal communities in patients with melanoma. Points are coloured based on melanoma invasion features. (c) Graphical representation of the results of the ANOSIM statistical test. (d) Distribution of patients with melanoma in the different classes of melanoma invasion and regression. (e, f) PCoA ordination based on Bray–Curtis distance of the fungal community in patients with melanoma. Points are coloured based on regression features. (g) Venn diagram showing intersections between differentially abundant zero‐radius operational taxonomic units found in the comparison between (i) invasive vs. in situ melanoma (‘invasivity’), (ii) regressed vs. not regressed melanoma (‘regression’) and (iii) control vs. melanoma (C vs. M).
Table 1.
Results of permanova and ANOSIM tests for evaluation of the relationship between microbial community structure and histopathological parameters of melanoma
| Invasivity | Breslow | Inflammation (yes/no) | Regression (yes/no) | Mitosis | ||
|---|---|---|---|---|---|---|
| Bacteria | ||||||
| permanova | R 2 | 0.069 | 0.044 | 0.053 | 0.062 | 0.042 |
| P‐value | 0.074 | 0.95 | 0.56 | 0.18 | 0.99 | |
| ANOSIM | R | 0.33 | – | 0.071 | 0.073 | – |
| P‐value | 0.008 | – | 0.13 | 0.12 | – | |
| Fungi | ||||||
| permanova | R 2 | 0.083 | 0.054 | 0.064 | 0.094 | 0.058 |
| P‐value | 0.042 | 0.5 | 0.25 | 0.013 | 0.38 | |
| ANOSIM | R | 0.36 | – | 0.078 | 0.094 | – |
| P‐value | 0.006 | – | 0.1 | 0.074 | – | |
| Bacteria + fungi | ||||||
| permanova | R 2 | 0.071 | 0.045 | 0.055 | 0.066 | 0.043 |
| P‐value | 0.039 | 0.94 | 0.48 | 0.095 | 0.98 | |
| ANOSIM | R | 0.36 | – | 0.078 | 0.094 | – |
| P‐value | 0.006 | – | 0.1 | 0.074 | – | |
Statistically significant P‐values are shown in bold.
In order to uncover significantly enriched or depleted zOTUs associated with melanoma at early stage or of different severity, we compared patients with melanoma with the control group, and subgroups of patients with melanoma according to the main histopathological features (invasiveness and regression; Figure S2). Those comparisons suggested that community rearrangement at the single zOTU level occurred both between patients and healthy controls and in relation to pivotal histopathological features of disease (Figure 2g; and Tables S4–S6; see Supporting Information).
A higher number of differentially abundant zOTUs was found for the comparison between patients with in situ and invasive melanoma (n =180; 117 bacterial zOTUs and 63 fungal zOTUs), with most of those enriched in in situ melanoma (162 of 180, 90%). In total 23 zOTUs (16 bacterial zOTUs and seven fungal zOTUs) were found to be differentially abundant between patients with regressed and not regressed melanoma. Most of those were enriched in patients with melanoma with regression (16 of 23, 70%). Finally, a total of 19 zOTUs (10 bacterial zOTUs and nine fungal zOTUs) were found to be differentially abundant in comparison between patients with melanoma and healthy controls (10 of 19 enriched in healthy controls, nine of 19 in patients with melanoma; Tables 2 and 3).
Table 2.
Zero‐radius operational taxonomic units (zOTUs) found at significantly different abundance in multiple comparisons
| zOTU | Intersection | Identification | Associated with |
|---|---|---|---|
| DENOVO40|ITS | All | Cryptococcus_fuscescens|SH215218.06FU | In situ, no regression, melanoma |
| DENOVO314|16S | Invasivity ∩ regression | Phylum Firmicutes | In situ, no regression |
| DENOVO55|16S | Invasivity ∩ regression | Genus Bacteroides | Invasive, regression |
| DENOVO256|16S | Invasivity ∩ regression | Genus Prevotella | In situ, no regression |
| DENOVO590|16S | Invasivity ∩ regression | Genus Victivallis | In situ, no regression |
| DENOVO124|ITS | Invasivity ∩ regression | Mortierella_sp|SH218045.06FU | In situ, no regression |
| DENOVO31|ITS | Invasivity ∩ regression | Mortierella_sp|SH217983.06FU | In situ, no regression |
| DENOVO16|ITS | Invasivity ∩ regression | Malasseziales_sp|SH206221.06FU | Invasive, no regression |
| DENOVO27|ITS | Invasivity ∩ melanoma control | Davidiella_tassiana|SH196750.06FU | In situ, control |
| DENOVO62|16S | Regression ∩ melanoma control | Family Clostridiales | Regression, melanoma |
| DENOVO42|ITS | Regression ∩ melanoma control | Sordariomycetes_sp|SH219629.06FU | No regression, melanoma |
| DENOVO14|ITS | Regression ∩ melanoma control | Tremellomycetes_sp|SH219359.06FU | No regression, melanoma |
Table 3.
BLAST identification of the bacterial zero‐radius operational taxonomic units (zOTUs) found at significantly different abundance in multiple comparisons
| zOTU | Hit | Identity | Query cover | E‐value | Max score | Total score | Associated with |
|---|---|---|---|---|---|---|---|
| DENOVO314|16S | Christensenella massiliensis | 88.6 | 100 | 2 × 10−139 | 494 | 494 | In situ, no regression |
| DENOVO55|16S | Bacteroides ovatus | 100 | 100 | 0.0 | 780 | 780 | Invasive, regression |
| DENOVO256|16S | Prevotella copri | 97.9 | 100 | 0.0 | 732 | 732 | In situ, no regression |
| DENOVO590|16S | Victivallis vadensis | 93.6 | 100 | 4 × 10−171 | 599 | 599 | In situ, no regression |
| DENOVO62|16S | Anaerobacterium chartisolvens | 88.6 | 99 | 3 × 10−137 | 486 | 486 | Regression, melanoma |
Of note, five zOTUs (three bacterial and two fungal) were more abundant in in situ melanoma without regression (potential poor immunogenic melanoma). On the other hand, a zOTU from the genus Bacteroides (identified in BLAST as Bacteroides ovatus) was the only zOTU enriched in samples of patients with invasive melanoma and regressing melanoma (potentially the most immunogenic melanoma).
Differences between in situ and invasive melanoma were further explored with random forest analysis, allowing the identification of the most important zOTUs in the two a priori subgroups of patients with melanoma (Figure 3a, b). Among those, a higher number of zOTUs were enriched in in situ melanoma (eight of 10), while only two zOTUs were enriched in invasive melanoma (Figure 3c and Table 4). All of the identified zOTUs belonged to the order Clostridiales, including several butyrate‐producing bacteria. Moreover, lower richness (Figure 3d) and diversity (Figure 3f) in invasive melanoma were found for bacteria, but not for fungi (Figure 3e, g).
Figure 3.
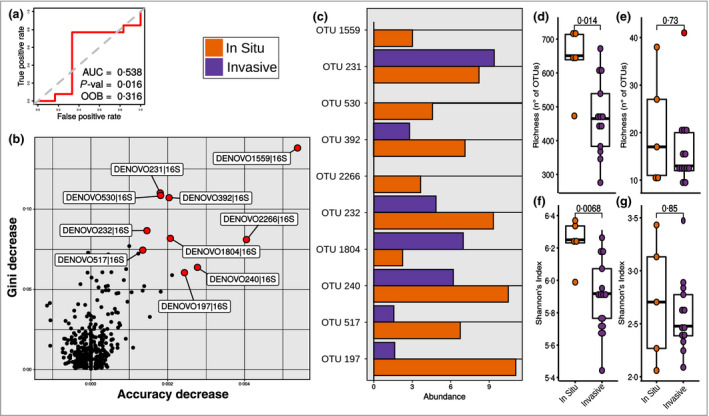
Random forest analysis for the classification of patients with early‐stage melanomas with an in situ or invasive tumour. (a) Receiver operating characteristic curve, P‐value, area under the curve (AUC) and out of bag (OOB) value showing the significance of the random forest model for the classification of patients with in situ and invasive melanoma. (b) Identification of the most important zero‐radius operational taxonomic units (zOTUs) in the random forest classification model, denoted by a red point and a label. (c) Abundance of the most important zOTUs in the random forest classification model patients with between in situ and invasive melanoma. (d–g) Alpha diversity analysis using synthetic indexes of richness (d, e) and Shannon’s index (f, g) for bacterial (d, f) and fungal (e, g) communities, in patients with in situ and invasive melanoma. Differences were assessed using the Wilcoxon test; P‐values for the comparisons are shown on the plot.
Table 4.
BLAST identification of the 10 most important zero‐radius operational taxonomic units (zOTUs) in explaining the random forest classification model of patients with in situ and invasive melanoma
| Hit | Identity | Query cover | E‐value | Max score | Total score | Enriched in | |
|---|---|---|---|---|---|---|---|
| zOTU 1559 | Hungateiclostridium cellulolyticum | 90.1 | 100 | 3 × 10−147 | 520 | 520 | In situ |
| zOTU 231 | Agathobaculum butyriciproducens | 99.8 | 100 | 0.0 | 743 | 743 | Invasive |
| zOTU 530 | Kineothrix alysoides | 97.0 | 100 | 0.0 | 678 | 678 | In situ |
| zOTU 392 | Kineothrix alysoides | 98.3 | 100 | 0.0 | 704 | 704 | In situ |
| zOTU 2266 | Desulfosporosinus fructosivorans | 89.0 | 99 | 6 × 10−150 | 529 | 529 | In situ |
| zOTU 232 | Christensenella massiliensis | 90.4 | 100 | 4 × 10−151 | 532 | 532 | In situ |
| zOTU 1804 | [Eubacterium] rectale | 99.7 | 96 | 0.0 | 710 | 710 | Invasive |
| zOTU 240 | Intestinimonas butyriciproducens | 93.1 | 100 | 2 × 10−169 | 593 | 593 | In situ |
| zOTU 517 | Sporobacter termitidis | 93.3 | 100 | 3 × 10−172 | 603 | 603 | In situ |
| zOTU 197 | Saccharofermentans acetigenes | 88.9 | 100 | 4 × 10−141 | 499 | 499 | In situ |
Evidence of different gut microbiota composition between patients with metastatic and nonmetastatic melanoma: a meta‐analysis
Finally, we compared our cohorts of patients with early‐stage melanoma against patients with metastatic melanoma from a previous study who did or did not respond to therapy. 6 PCoA ordinations (Figure 4a) showed changes in community structure between tumour stages (from early to metastatic melanoma). We did not find a clear separation of samples between patients with early melanoma and healthy controls; on the contrary, samples of patients with metastatic melanoma (both responders and nonresponders) clustered separately.
Figure 4.
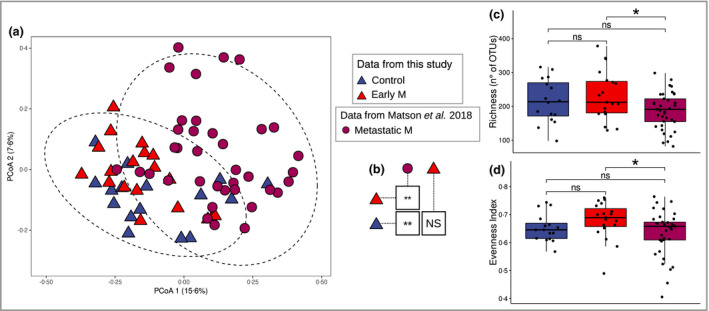
Meta‐analysis between the results of this study and that of Matson et al. 6 (a) Principal coordinates analysis (PCoA) ordination based on Bray–Curtis distance of the bacterial community in the meta‐analysis. (b) Results of pairwise permanova between the sample classes of the meta‐analysis. (c‐d) Alpha diversity analysis of bacterial communities using synthetic indexes of richness and evenness, respectively. M, melanoma; NS, not significant; OTU, operational taxonomic unit; *P < 0.05, **P < 0.01.
Pairwise permanova analysis confirmed significant differences between the bacterial communities of patients with metastatic melanoma and both patients with early melanoma and healthy controls (Figure 4b). However, no significant differences were found between patients with early melanoma and controls. Intriguingly, bacterial richness (Figure 4c) and evenness (Figure 4d) showed a significant decline in community diversity in advanced metastatic melanoma vs. early melanoma. A similar trend was also observed in invasive melanoma with respect to in situ melanoma (Figure 3).
Discussion
To date, the composition of the gut microbiota has been analysed only in patients with metastatic melanoma undergoing targeted immune therapies, 5 , 6 , 7 , 8 , 9 , 10 highlighting specific gut bacterial components related to response to immunotherapies and to side‐effects, such as colitis. Here, we focused on identification of gut microbiota profiles associated with early melanoma, speculating that a specific gut microbial community might be associated with local or systemic antimelanoma immune response and/or to invasive behaviour, given the highly immunogenic nature of this tumour.
For the first time, we assessed the composition of the gut mycobiota, which to the best of our knowledge has never been assessed in melanoma, regardless of stage. Evidence showed that fungal cell wall components (such as chitin) can interact with the host immune system triggering proinflammatory responses, 27 , 28 , 29 or acting as a double‐edged sword, inducing both T helper (Th)1‐derived Th17 responses and Treg responses. 29 , 30 , 31 , 32 As inflammation is considered a crucial step towards cancer, as reported for several visceral cancers, 33 , 34 it is conceivable that also for skin melanoma an intestinal inflammatory status linked to the presence of altered microbial profiles 35 could contribute to maintain a systemic proinflammatory and pro‐oncogenic status. 36 In fact, increased levels of circulating proinflammatory cytokines stimulate immune cells to move towards skin, leading to local inflammation, which is considered a crucial step for neoplastic differentiation of melanocytes. 37 , 38
Our results support this evidence, and additionally suggest that gut fungal profiles could be specifically associated with melanoma regression. Regression is a phenomenon involving T lymphocytes recognizing specific melanoma antigens 39 that are capable of destroying melanoma cells, leading to the progressive (partial or complete) disappearance of the tumour and replacement with immune or fibrotic infiltration. The prognostic role of regression is still under debate 38 , 39 as the occurrence of immune or fibrotic infiltration might lead to an underestimation of the Breslow thickness. Fungi could pass across the gut barrier and induce systemic immune responses. 40 , 41 This event could help to restore the cancer immune checkpoint and induce cancer regression.
Melanoma progression from in situ to invasive showed a pauperization of the gut biota, and a lowering of alpha diversity. Species belonging to the order Clostridiales and producers of butyrate were enriched in invasive melanoma. It has been demonstrated that butyrate, which can reach circulation and tissues when produced in the gut, induces the expression of annexin A1, involved in tumoral invasion through the activation of the epithelial‐to‐mesenchymal transition signalling pathway in human melanoma cells. 42 We could speculate that an imbalance in butyrate‐producing bacteria might influence melanoma growth through increased systemic butyrate levels. However, butyrate producers were also found associated with in situ melanoma. In accordance with the controversial role in modulating colorectal cancer biology 43 , 44 this suggests that enrichment of selected butyrate producers might promote melanoma evolution towards an invasive phenotype, or towards a noninvasive phenotype.
We hypothesized that a highly immunogenic melanoma might be characterized by invasion and regression together, and connected to a worse prognosis. Immunogenicity in fact increases in the invasive advanced stage, and is related to a high mutational load. 45 , 46 Our results showed that a peculiar zOTU belonging to the genus Bacteroides was shared by regressive and invasive melanoma, and hence Bacteroides could be connected to melanoma immunogenicity. Interestingly, selected strains of B. ovatus capable of eliciting IgM‐ and IgG‐mediated responses in vitro towards human acute myelogenous leukaemia cells have been isolated from the human gut. 47 Furthermore, our result seems to be in accordance with previous studies on metastatic melanoma, in which enrichment of the phylum Bacteroidetes, to which B. ovatus belongs, has an unfavourable impact on melanoma prognosis, limiting responsiveness to immune therapy. 48
Taken together, our findings suggest that the composition of the gut microbiota and the degree of immunogenicity of early‐stage melanoma might change along the gradient from in situ to invasive and, finally, to metastatic melanoma. The observed trend of deterioration in gut microbiota from in situ to invasive also likely accompanies the transition from an early and local melanoma phenotype, to the advanced and metastatic one. In this study we cannot make any further assumptions or formulate more detailed hypotheses, as it was not among our aims, but it would certainly be interesting and promising to direct future research efforts specifically in evaluating this transition. Those microbiota transitions might implicate immunological changes, possibly involving systemic immune cell activation and circulating cytokine release, able to modulate melanoma progression, to which gut microbial changes might contribute through direct or indirect modulation of the host immune system. A growing body of evidence supports the presence of a gut–skin axis, and further investigations will be needed to demonstrate how gut fungal or bacterial communities might exert biological effects at the skin level.
The present study has a few limitations. Firstly, our findings should be tested further in larger and better balanced cohorts, as the significantly different average age between the healthy control and patients with melanoma suggests caution in interpreting the results of gut microbiota features in such groups. At the same time, the comparable age and body mass index of the invasive and in situ melanoma groups strengthens our observations on the connections between melanoma progression and gut microbiota. Secondly, even if the results of the meta‐analysis are promising, the general caution that applies to any meta‐analysis of targeted metagenomic sequencing data should be used, as possible technical bias and differences between the two studies could have influenced the results. Nevertheless, the partial overlap in ordination analysis between our samples and those of Matson et al. is reassuring for the soundness of the analysis and its correspondence with the real biology of the system. Finally, the difficulty in cultivating the microbial communities we have highlighted leads to the impossibility of proving their causal role in the pathogenesis of melanoma and in its progression, leveraging, for example, in vivo experimental models.
We can conclude that our results suggest promising future developments in early melanoma research, where the microbiota could represent a valuable tool for early patient classification, even if causal relationships were not demonstrated. It is conceivable that modulation of the microbial community through interventions on lifestyle habits, diet or treatments might help to recover a beneficial gut microbiota in which specific bacterial and fungal components might elicit a prognostic favourable immune response against melanoma cells.
Author Contribution
Francesco Vitali: Data curation (equal); Formal analysis (lead); Investigation (equal); Resources (equal); Visualization (lead); Writing‐original draft (equal); Writing‐review & editing (equal). Roberta Colucci: Conceptualization (equal); Data curation (equal); Investigation (supporting); Resources (equal); Writing‐original draft (lead); Writing‐review & editing (equal). Monica Di Paola: Conceptualization (equal); Investigation (equal); Resources (equal); Writing‐review & editing (equal). Massimo Pindo: Resources (equal); Writing‐review & editing (supporting). Carlotta De Filippo: Conceptualization (lead); Supervision (lead); Writing‐original draft (equal); Writing‐review & editing (equal). Silvia Moretti: Conceptualization (lead); Project administration (lead); Supervision (lead); Writing‐original draft (equal); Writing‐review & editing (equal). Duccio Cavalieri: Conceptualization (lead); Project administration (lead); Supervision (lead); Writing‐original draft (equal); Writing‐review & editing (equal).
Supporting information
Figure S1 Relative abundance distribution of putative markers of bacterial and fungal communities between patients with early‐stage melanoma and controls.
Figure S2 Volcano plots representing results of DESeq analyses used to identify differentially abundant operational taxonomic units in the comparison of (i) melanoma vs. control, (ii) invasive vs. in situ melanoma and (iii) regressed vs. not regressed melanoma.
Table S1 Clinical data of the patients.
Table S2 Clinical data of the healthy controls.
Table S3 Histological parameters of patients with melanoma.
Table S4 Differentially abundant operational taxonomic units between healthy controls and patients with melanoma.
Table S5 Differentially abundant operational taxonomic units between patients with regressed vs. regressed melanoma.
Table S6 Differentially abundant operational taxonomic units between patients with invasive and in situ melanoma.
Appendix S1 Supplementary Material.
Powerpoint S1 Journal Club Slide Set.
Acknowledgments
The authors are grateful to Professor Nicola Pimpinelli and Dr Vincenzo De Giorgi for their collaboration in patient enrolment. Open Access Funding provided by Universita degli Studi di Firenze within the CRUI‐CARE Agreement.
Funding sources This work was supported by funds from ‘Fondi Ateneo’ to S.M. (Silvia Moretti Ricaten; Morinn projects), by funds from the European Community under the Horizon 2020 Framework Programme to D.C. (FNS‐Cloud project) and from the European Community under the Joint Programming Initiative a Healthy Diet for a Healthy Life (JPI‐HDHL) to C.D.F. (MeatIC project).
Conflicts of interest The authors declare they have no conflicts of interest.
Data availability The data that support the findings of this study are openly available from the European Nucleotide Archive at https://www.ebi.ac.uk/ena/browser/view/PRJEB35665. A GitHub repository (https://github.com/FrancescoVit/Supplementary_Vitali_Melanoma) was created to store the data (bacterial and fungal community counts; bacterial and fungal zero‐radius operational taxonomic unit taxonomy) and code to reproduce all of the analyses reported in the manuscript.
F.V. and R.C. contributed equally to this work.
Plain language summary available online
References
- 1. Schadendorf D, van Akkooi ACJ, Berking C et al. Melanoma. Lancet 2018; 392:971–84. [DOI] [PubMed] [Google Scholar]
- 2. Cust AE, Mishra K, Berwick M. Melanoma – role of the environment and genetics. Photochem Photobiol Sci 2018; 17:1853–60. [DOI] [PubMed] [Google Scholar]
- 3. Mancebo SE, Wang SQ. Skin cancer: role of ultraviolet radiation in carcinogenesis. Rev Environ Health 2014; 29:265–73. [DOI] [PubMed] [Google Scholar]
- 4. Christofi T, Baritaki S, Falzone L et al. Current perspectives in cancer immunotherapy. Cancers (Basel) 2019; 11:1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaikh FY, Gills JJ, Sears CL. Impact of the microbiome on checkpoint inhibitor treatment in patients with non‐small cell lung cancer and melanoma. EBioMedicine 2019; 48:642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matson V, Fessler J, Bao R et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science 2018; 359:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubin K, Callahan MK, Ren B et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint‐blockade‐induced colitis. Nat Commun 2016; 7:10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weber JS, Dummer R, de Pril V et al. Patterns of onset and resolution of immune‐related adverse events of special interest with ipilimumab: detailed safety analysis from a phase 3 trial in patients with advanced melanoma. Cancer 2013; 119:1675–82. [DOI] [PubMed] [Google Scholar]
- 9. Chaput N, Lepage P, Coutzac C et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 2017; 28:1368–79. [DOI] [PubMed] [Google Scholar]
- 10. Frankel AE, Coughlin LA, Kim J et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017; 19:848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell 2016; 165:276–87. [DOI] [PubMed] [Google Scholar]
- 12. Di Paola M, Rizzetto L, Stefanini I et al. Comparative immunophenotyping of Saccharomyces cerevisiae and Candida spp. strains from Crohn’s disease patients and their interactions with the gut microbiome. J Transl Autoimmun 2020; 3:100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slater D, Walsh M. Standards and datasets for reporting cancers. Dataset for the histological reporting of primary cutaneous malignant melanoma and regional lymph nodes. Available at: https://www.rcpath.org/resourcelibrary/dataset‐for‐the‐histological‐reporting‐of‐primary‐cutaneous‐malignant‐melanoma‐and‐regional‐lymph‐nodes.html (last accessed 12 August 2021).
- 14. Scolyer RA, Judge MJ, Evans A et al. Data set for pathology reporting of cutaneous invasive melanoma: recommendations from the International Collaboration on Cancer Reporting (ICCR). Am J Surg Pathol 2013; 37:1797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gershenwald JE, Scolyer RA, Hess KR et al. Melanoma staging: evidence‐based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin 2017; 67:472–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takahashi S, Tomita J, Nishioka K et al. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next‐generation sequencing. PLOS ONE 2014; 9:e105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. White TJ, Bruns TD, Lee SB, Taylor JW. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols: A Guide to Methods and Applications (Innis MA, Gelfand DH, Sninsky JJ, White TJ, eds). New York: Academic Press, 1990, 315–22. [Google Scholar]
- 18. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 2011; 17(1):10–12. [Google Scholar]
- 19. Joshi NA, Fass JN. Sickle: a sliding‐window, adaptive, quality‐based trimming tool for FastQ files (version 1.33) [software], 2011.
- 20. Albanese D, Fontana P, De Filippo C et al. MICCA: a complete and accurate software for taxonomic profiling of metagenomic data. Sci Rep 2015; 5:9743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edgar RC. UNOISE2: improved error‐correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016; 81257. [Google Scholar]
- 22. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007; 16:5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nilsson RH, Larsson K‐H, Taylor AFS et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 2018; 47:D259–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Butera A, Di Paola M, Vitali F et al. IL‐13 mRNA tissue content identifies two subsets of adult ulcerative colitis patients with different clinical and mucosa‐associated microbiota profiles. J Crohns Colitis 2020; 14:369–80. [DOI] [PubMed] [Google Scholar]
- 25. Meriggi N, Di Paola M, Vitali F. Saccharomyces cerevisiae induces immune enhancing and shapes gut microbiota in social wasps. Front Microbiol 2019; 10:2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paulson JN, Pop M, Bravo HC. metagenomeSeq: statistical analysis for sparse high‐throughput sequencing. Bioconductor package 1.26.3. Available at: http://cbcb.umd.edu/software/metagenomeSeq (last accessed 12 August 2021).
- 27. Luan C, Miao H, Zhu B. Gut mycobiota and adenomas. Gut Microbes 2015; 6:331–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao R, Kong C, Li H et al. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur J Clin Microbiol Infect Dis 2017; 36:2457–68. [DOI] [PubMed] [Google Scholar]
- 29. Rizzetto L, Ifrim DC, Moretti S et al. Fungal chitin induces trained immunity in human monocytes during cross‐talk of the host with Saccharomyces cerevisiae . J Biol Chem 2016; 291:7961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramazzotti M, Stefanini I, Di Paola M et al. Population genomics reveals evolution and variation of Saccharomyces cerevisiae in the human and insects gut. Environ Microbiol 2019; 21:50–71. [DOI] [PubMed] [Google Scholar]
- 31. Elinav E, Nowarski R, Thaiss CA et al. Inflammation‐induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13:759–71. [DOI] [PubMed] [Google Scholar]
- 32. Lafouresse F, Groom JR. A task force against local inflammation and cancer: lymphocyte trafficking to and within the skin. Front Immunol 2018; 9:2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Picardo SL, Coburn B, Hansen AR. The microbiome and cancer for clinicians. Crit Rev Oncol Hematol 2019; 141:1–12. [DOI] [PubMed] [Google Scholar]
- 34. Yu J, Li SM, Kong Y et al. Association of immune‐inflammation index with outcome of high‐risk acral melanoma patients treated with adjuvant high dose interferon. J Clin Oncol 2017; 10:719–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneider SL, Ross AL, Grichnik JM. Do inflammatory pathways drive melanomagenesis? Exp Dermatol 2015; 24:86–90. [DOI] [PubMed] [Google Scholar]
- 36. Neagu M, Constantin C, Caruntu C et al. Inflammation: a key process in skin tumorigenesis. Oncol Lett 2019; 17:4068–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ribero S, Moscarella E, Ferrara G et al. Regression in cutaneous melanoma: a comprehensive review from diagnosis to prognosis. J Eur Acad Dermatol Venereol 2016; 30:2020–37. [DOI] [PubMed] [Google Scholar]
- 38. Cartron AM, Aldana PC, Khachemoune A. Reporting regression in primary cutaneous melanoma. Part 1: history, histological criteria and pathogenesis. Clin Exp Dermatol 2021; 46:28–33. [DOI] [PubMed] [Google Scholar]
- 39. Aung PP, Nagarajan P, Prieto VG. Regression in primary cutaneous melanoma: etiopathogenesis and clinical significance. Lab Invest 2017; 97:657–68. [DOI] [PubMed] [Google Scholar]
- 40. Rizzetto L, De Filippo C, Cavalieri D. Mycobiota: micro‐eukaryotes inhabiting our body as commensals or opportunistic pathogens. Fungal Genom Biol 2015; 5:120. [Google Scholar]
- 41. Ko JS. The immunology of melanoma. Clin Lab Med 2017; 37:449–71. [DOI] [PubMed] [Google Scholar]
- 42. Shin J, Song IS, Pak JH, Jang SW. Upregulation of annexin A1 expression by butyrate in human melanoma cells induces invasion by inhibiting E‐cadherin expression. Tumour Biol 2016; 37:14577–84. [DOI] [PubMed] [Google Scholar]
- 43. Bach Knudsen KE, Lærke HN, Hedemann MS et al. Impact of diet‐modulated butyrate production on intestinal barrier function and inflammation. Nutrients 2018; 10:1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu X, Wu Y, He L et al. Effects of the intestinal microbial metabolite butyrate on the development of colorectal cancer. J Cancer 2018; 9:2510–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kwoonel K, Kin HS, Kim JY et al. Predicting clinical benefit of immunotherapy by antigenic or functional mutations affecting tumour immunogenicity. Nat Commun 2020; 11:951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yusko E, Vignali M, Wilson RK et al. Association of tumor microenvironment T‐cell repertoire and mutational load with clinical outcome after sequential checkpoint blockade in melanoma. Cancer Immunol Res 2019; 7:458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ulsemer P, Henderson G, Toutounian K et al. Specific humoral immune response to the Thomsen‐Friedenreich tumor antigen (CD176) in mice after vaccination with the commensal bacterium Bacteroides ovatus D‐6. Cancer Immunol Immunother 2013; 62:875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gopalakrishnan V, Spencer CN, Nezi L et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 2018; 359:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Relative abundance distribution of putative markers of bacterial and fungal communities between patients with early‐stage melanoma and controls.
Figure S2 Volcano plots representing results of DESeq analyses used to identify differentially abundant operational taxonomic units in the comparison of (i) melanoma vs. control, (ii) invasive vs. in situ melanoma and (iii) regressed vs. not regressed melanoma.
Table S1 Clinical data of the patients.
Table S2 Clinical data of the healthy controls.
Table S3 Histological parameters of patients with melanoma.
Table S4 Differentially abundant operational taxonomic units between healthy controls and patients with melanoma.
Table S5 Differentially abundant operational taxonomic units between patients with regressed vs. regressed melanoma.
Table S6 Differentially abundant operational taxonomic units between patients with invasive and in situ melanoma.
Appendix S1 Supplementary Material.
Powerpoint S1 Journal Club Slide Set.