

Abstract
DNA methylation analysis by sequencing is becoming increasingly popular, yielding methylomes at single-base pair and single-molecule resolution. It has tremendous potential for cell-type heterogeneity analysis using intrinsic read-level information. Although diverse deconvolution methods were developed to infer cell-type composition based on bulk sequencing-based methylomes, systematic evaluation has not been performed yet. Here, we thoroughly benchmark six previously published methods: Bayesian epiallele detection, DXM, PRISM, csmFinder+coMethy, ClubCpG and MethylPurify, together with two array-based methods, MeDeCom and Houseman, as a comparison group. Sequencing-based deconvolution methods consist of two main steps, informative region selection and cell-type composition estimation, thus each was individually assessed. With this elaborate evaluation, we aimed to establish which method achieves the highest performance in different scenarios of synthetic bulk samples. We found that cell-type deconvolution performance is influenced by different factors depending on the number of cell types within the mixture. Finally, we propose a best-practice deconvolution strategy for sequencing data and point out limitations that need to be handled. Array-based methods—both reference-based and reference-free—generally outperformed sequencing-based methods, despite the absence of read-level information. This implies that the current sequencing-based methods still struggle with correctly identifying cell-type-specific signals and eliminating confounding methylation patterns, which needs to be handled in future studies.
Keywords: DNA methylomes, deconvolution, heterogeneity, sequencing, computational epigenetics
Introduction
Although single-cell analyses have been spearheading the progress in biomedicine lately, profiling of bulk samples is still in high demand for analyses that cannot be readily accomplished using single-cell methods due to technical or cost-related reasons. For instance, single-cell methods are still too laborious and costly [1] for profiling of long-time biobanked samples or large tumor patient cohorts [2–4]. The bulk analysis of epigenetic modifications can demonstrate epigenetic variations in large cohorts and relate those with genomic and phenotypic characteristics [5, 6].
DNA methylation, particularly occurring at cytosines in CpG context in mammals, carries highly distinguishable cell-type-specific signals [6, 7]. The inheritability over cell divisions and the chemical stability make it significantly more accessible for profiling. These features of DNA methylation motivated research associating diseases with cell-type-specific DNA methylation signals. Such disease-associated methylation differences are identified as differentially methylated regions (DMRs). To this end, it was shown that tumor subtypes can be successfully classified based on bulk DNA methylation patterns of patient biopsies [8–10]. Nevertheless, cell-type-specific DNA methylation signals can suffer from confounding factors, because DNA methylation is also associated with gender, age, various environmental influences, etc. [11, 12]. These confounding factors make cell subpopulation analysis difficult by increasing the methylation pattern complexity and obscuring cell-type-specific signals.
Even though there are in vitro techniques to purify individual cell types from bulk samples, such as cell sorting or cell enrichment, other spurious source of variation can be introduced into the samples during the experimental process and eventually further confound cell-type-specific methylation signals. As an alternative, cell-type deconvolution, a computational approach to infer the cell-type composition of bulk samples, is applicable for cell subpopulation analysis post-experimentally. Cell-type deconvolution has been extensively utilized to dissect array-based DNA methylation bulk data, e.g. Infinium 450K/EPIC microarrays [13, 14], which generates matrices of average methylation levels at specific loci in multiple samples (we refer to them as ’array-shaped data’). ’Reference-based’ deconvolution methods infer cell-type proportions based on reference methylomes of purified cell populations. Various statistical methods and algorithms were employed to establish and fit reference-based deconvolution models including different forms of regression [15–17], Expectation–Maximization (EM) algorithm and deep neural networks [18, 19]. In contrast, ’reference-free’ deconvolution methods do not require reference data to infer the proportions of underlying cell types from methylomes. Numerous reference-free methods have been proposed by us and others [20–24], also broadly reviewed [25] and compared elsewhere [26]. Overall, reference-based and reference-free methods for deconvolution of bulk DNA methylomes are well established and have been used routinely, in particular for the analysis of cellular composition in complex tumors [16, 27–30].
More recently, DNA methylation sequencing approaches, such as reduced representation bisulfite sequencing (RRBS) [31] or whole genome bisulfite sequencing (WGBS) [32], have become increasingly popular owing to dropping sequencing costs, providing much broader genome coverage [33–35] and single-molecule resolution in the form of read-level methylation calls. Furthermore, single-cell bisulfite sequencing (scBS-seq) is being used ever more frequently [36, 37], particularly in conjunction with multi-omics analyses [38, 39]. However, few issues still have to be carefully managed in sequencing data. Since it has high genome coverage, the multiple methylation states from reads covering respective sites has to be refined and well summarized. In addition, sequencing data consist of read-level methylomes so that each CpG site includes multiple methylation state values from all reads covering the site. This abundance of information makes sequencing data analysis more intricate by concealing some cell-type-specific signals. Consequently, dealing with sequencing data highly depends on which genomic regions are analyzed and how to interpret read-level information altogether.
Read-level DNA methylomes from sequencing data, in theory, should be more suitable and information-rich for cell composition inference in bulk samples, as compared with array-shaped data [28]. Diverse methods have been developed for cell-type deconvolution of sequencing-based methylome data, making use of the aforementioned advantages, foremost read-level resolution [40–44]. However, the methods published so far were evaluated based on different data sets and variable standards. Additionally, each method requires specific preprocessing procedures and generates different forms of output that might confuse less experienced users. Moreover, previous studies mainly assessed array-based cell-type deconvolution methods [26, 45]. Considering the distinctive advantages and the rapidly growing demand of sequencing data analysis, a comprehensive, standardized and unbiased assessment of deconvolution methods targeting sequencing data has become necessary.
In order to bridge this gap, we thoroughly compare and evaluate six previously published sequencing-based deconvolution algorithms: Bayesian epiallele detection (BED) [40], DXM [46], PRISM [42], MethylPurify [43], csmFinder + coMethy [41] and ClubCpG [44]. We evaluate their performance with respect to informative feature selection results and deconvolution accuracy under various experimental scenarios, using in silico mixtures of single-nucleus methylomes as well as realistic mixtures of tumor and normal WGBS data. Based on our analysis results, we finally propose efficient and trustworthy pipelines to deconvolve complex cell-mixture samples in different scenarios. To our knowledge, this benchmarking study is the first attempt to systematize and comprehensively evaluate the methodological developments in sequencing-based methylome deconvolution.
Materials and methods
Benchmarking datasets
In order to evaluate the sequencing-based deconvolution methods, we generated synthetic cell-mixture samples that we refer to as pseudo-bulk. For the pseudo-bulk generation, we have chosen two different datasets, mouse brain single-cell methylC-seq data [47] and B-cell tumor WGBS data [48].
Single-cell methylome data processing
In total, 3377 single-nucleus methylomes derived from 8-week-old mouse cortex tissue were downloaded from the Gene Expressiong Omnibus (GEO) with the accession number GSE97179. This dataset was created through high-throughput single-nucleus methylome sequencing (snmC-seq). We firstly trimmed reads twice to remove sequencing adaptors, random primer index sequence and C/T tail attached, using Adaptase with Cutadapt 2.6 [49]. Trimmed reads were aligned to the mm10 reference genome using Bismark 0.22.3 [50]. After the alignment, we sorted the resulting BAM files using samtools 1.9 [51] and removed duplicated reads using picard MarkDuplicates 1.141. Finally, the reads whose mapping quality is lower than 30 were filtered out with samtools 1.9 again. Parameters used in each step and details about the preprocessing are clarified in Supplementary Table 1.
Tumor WGBS data processing
To generate realistic tumor–normal cell mixtures, we downloaded diffuse large B-cell lymphoma and normal non-cancer B-cell WGBS data from one subject each (GEO accession number GSE137880) [52]. Trimming was conducted using Trim Galore 0.6.6, then reads were aligned by Bismark 0.22.3 [50] in paired-end mode. Only reads not aligned in paired-end mode were realigned in single-end mode, and all reads were merged and sorted through samtools 1.9 [51]. At the end, duplicated reads were removed by picard Mark Duplicates 1.141. Detailed pipeline is clarified in Supplementary Table 1.
Pseudo-bulk generation
We created pseudo-bulks by merging reads randomly sampled from the aligned mouse brain single-nucleus methylome data and B-cell WGBS data. The cell-type proportion for bulks were decided based on Dirichlet distribution which can mimic diverse biological scenarios. For mouse neuron pseudo-bulk, five cell types including both excitatory (mDL-2, mL2-3, mL5-1 and mL6-2) and inhibitory (mPv) neuron classes were chosen. These cell types are distinctly clustered in the two-dimensional t-SNE visualization calculated by Luo et al. [47] and secure sufficient single-cell samples. For tumor pseudo-bulk, diffuse large B-cell lymphoma and normal B-cell cell types were mixed. We used generateExample function from R package MeDeCom (https://rdrr.io/github/lutsik/MeDeCom/src/R/utilities.R) to generate the cell-type proportions for pseudo-bulk samples. We followed the default parameter setup with the exception of 10 for proportion.var.factor and 1 million for number of genomic features. The numerical proportions of cell types in each pseudo-bulk sample is shown in Supplementary Table 2. This pipelines is available at https://github.com/CompEpigen/SeqDeconv_Pipeline.git.
Differentially methylated regions (DMRs)
As gold-standard to be compared with informative region selection results showing cell-type-specific features, we extracted DMRs. DMRs refer to genomic regions whose methylation states are consistently different between given groups of samples. They are also known to be associated with cell development and cell differentiation stages [53].
For mouse neuronal cell types, we compared each of 11 pure cell-type bulks (mL4, mL6-1, mL6-2, mDL-2, mL5-1, mL5-2, mL2-3, mSst-1, mNdnf-2, mPv and mVip) against all other bulks and identified respective cell type DMRs (ctDMRs). For tumor analysis, we called ctDMRs between normal B-cell non-cancer and diffuse large B-cell lymphoma. All DMRs were called using DSS package 2.34.0 [54] with the following parameters: 0.2 for delta, 0.05 for threshold of P-values, 4 for minimum number of CpG sites and default values for minimum length and the distance to merge, which are 50 bps each.
CpG selection scheme for array-based deconvolution methods
Array-shaped methylome data comes in the matrix form, where each row represents a CpG site and each column a sample. To apply array-based deconvolution methods, Houseman and MeDeCom, as a comparison group, we transformed our sequencing data to array shape using methrix [55]. When converting the sequencing data, we specified a set of CpG sites to comprise the array shape. During data conversion for MeDeCom, we chose top 20 000 CpGs with the highest beta-value variance. CpGs overlapping ctDMRs were taken to generate array-shaped data for Houseman’s method to adhere to the reference-based scenario.
Parameter values and procedures for each deconvolution method
We describe the detailed algorithm and parameter setting of each deconvolution method in this section. The pipelines are also available at https://github.com/CompEpigen/SeqDeconv_Pipeline.git.
ClubCpG
ClubCpG [44] clusters reads fully covering regions satisfying given informative region selection conditions, using density-based spatial clustering of applications with noise (DBSCAN). For our experiments, we applied informative region selection conditions as given in Table 1 that suited our dataset better than the default values suggested by authors. Even though ClubCpG package itself does not have cell-type composition estimation function, we followed the cell-type deconvolution strategy proposed by the authors [44]. Firstly, we created ClubCpG clustering results from another 100 pseudo-bulk samples as training data and extracted 20 principal components (PCs) using Principal Component Analysis algorithm from the result. Then, a multivariate linear regression model for cell-type proportion was fitted on the extracted 20 PCs. Finally, the cell-type composition was estimated using the trained multivariate linear regression model.
Table 1.
Comparison of benchmarked deconvolution methods. Sequencing-based methods have three common criteria in the informative region selection: number of CpG, region size and read coverage. Some criteria were altered to be more suitable for the dataset we used in our analyses. For array-based methods, we specifically designated CpG sites based on methylation variance or ctDMRs. Furthermore, we described differences in cell-type composition estimation step based on three criteria again: reference-requirements, number of detectable subpopulations and estimation scope
| Method | Main data type | Informative region selection | Cell-type composition estimation | ||||
|---|---|---|---|---|---|---|---|
| # CpG | Region size (bp) | Coverage | Class | # Components | Estimation Scope | ||
| BED | RRBS | >4 |
NA
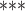
|
>20 | ref-based | 2 | local |
| ClubCpG | WGBS | >4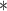
|
100 | >20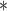
|
ref-based | 2 or more | global |
| csmFinder + coMethy | WGBS | >4 |
NA
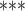
|
>10 | ref-free | 2 or more | global |
| DXM | Any kinds of BS-seq | Promoter and CpG island regions | >4 | ref-free | 2 or more | global | |
| MethylPurify | WGBS | >10 | 300/200
|
>10 | ref-free | 2 | local |
| Prism | RRBS | >4 |
NA
|
>20 | ref-free | 2 or more | local |
| Houseman | Methylation microarrays | CpGs overlapping with ctDMRs | ref-based | 2 or more | global | ||
| MeDeCom | Methylation microarrays | CpGs showing high methylation variance across pseudo-bulks | ref-free | 2 or more | global | ||
PRISM
PRISM [42] infers the composition of epigenetically distinct subpopulations in tumor bulk samples based on methylation patterns. It mainly improves the accuracy by correcting erroneous methylation patterns using Hidden Markov Model (HMM). After the correction, the method retains only loci comprised of fully methylated and unmethylated patterns, then cell-type proportions are estimated locus-specifically using EM algorithm. We applied PRISM with default setup as given in Table 1 and yielded cell-type proportions in respective samples by calculating the ratio of inferred subclones.
MethylPurify
MethylPurify [43] adopts EM algorithm to estimate tumor purity in bisulfite sequencing data. The EM algorithm in MethylPurify not only estimates methylation levels of subpopulations, but also decides which subpopulation each read would be assigned to over iterations. We used the same parameter values in both mouse neuronal and tumor pseudo-bulks; 10 for read coverage and 50 for sampling time. However, bin size parameter was set to 300 bp for mouse neuronal pseudo-bulks and to 200 bp for tumor pseudo-bulks. Although the original code is designed to estimate cell-type compositions only within CpG islands, we altered it to conduct the estimation over all informative regions including non-CpG islands. This is because the method initially failed in selecting a high-enough number of informative regions for statistically significant cell-type composition estimation. After removing the CpG island filtering, we could obtain a high-enough number of selected informative regions for cell-type deconvolution.
csmFinder + coMethy
Yin et al. [41] developed two distinct computational tools called csmFinder and coMethy. csmFinder determines genomic regions showing cell-type-specific signals denoted as putative cell-type-specific methylated (pCSM) loci. coMethy can decompose methylation-level matrix of pCSM loci by samples, using non-negative matrix factorization (NMF) approach similar to MeDeCom. For the specific input file format of csmFinder, we extracted methylation call results from our pseudo-bulk samples via bismark_methylation_extractor with comprehensive, gzip and cytosine_report options. CpG site coordinates of hg19 and mm10 reference genomes were used for corresponding samples. Running csmFinder, we followed the default parameters, minimum methylation difference of hypo- and hyper-methylation patterns 0.3 and P-value of the difference 0.05. Since coMethy works on a matrix that consists of methylation patterns at the same CpG sites across multiple samples, we collected pCSM loci detected from all samples in each experiment and filtered out only loci involving missing methylation values in any other sample than the sample that the locus was detected.
Bayesian epiallele detection
BED algorithm [40] is based on a Bayesian model and recognizes the distribution of epialleles that indicates all possible methylation patterns at CpG sites in a specific genomic range. For the preprocessing step to deal with contiguous and missing methylation patterns, we used pipelines elucidated in bed-beta Github page (https://github.com/james-e-barrett/bed-beta). This pipeline includes all epiallele estimation processes. Barret et al. demonstrated that the proportion of reads that are not attributed to normal tissue at each loci 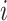 can be calculated as follows:
can be calculated as follows:
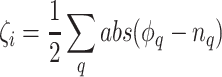 |
(1) |
This equation is calculated with respect to all epialleles 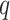 .
. 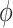 and
and 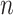 mean the distribution of epialleles at the locus and normal tissue samples each. We considered the maximum observed value in
mean the distribution of epialleles at the locus and normal tissue samples each. We considered the maximum observed value in 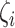 distribution to be the estimated tumor purity as this is where the local epiallele distributions and normal tissue epiallele distributions showed the biggest discrepancy.
distribution to be the estimated tumor purity as this is where the local epiallele distributions and normal tissue epiallele distributions showed the biggest discrepancy.
DXM
DXM [46] is a computational method to infer not only the number of subpopulations within bulk methylomes but also the methylation profile of estimated subpopulations based on L1-norm minimization and HMM. It first investigates the distribution of methylation beta value over the given regions and calculates L1-norm between the investigated distribution and 10 000 randomly generated distributions. The distribution with the lowest L1-norm is considered as an estimated cell-type distribution. Unlike other sequencing-based methods, DXM requires users to directly provide pre-selected specific genomic regions such as DMRs or CpG island regions. Therefore, keeping the reference-free manner and following the guidance of the authors, we gave methylation level within promoter and CpG Island regions as input of DXM after filtering the regions with minimum read coverage 4. CpG Island regions were downloaded from UCSC genome annotation database for mm10 and hg19 genomes (https://hgdownload.cse.ucsc.edu/goldenpath/mm10/database/ and https://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/). For promoter regions, we also used UCSC annotation database exposed as TxDb objects [56, 57]. HMM is used mainly for the methylation profile inference; thus, we do not include the details here.
MeDeCom
MeDeCom [20] estimates cell-type proportions in array-shaped DNA methylation data using NMF. When converting our sequencing-based data to array shape, we used methrix which is capable of loading bedGraph file format including methylation call and creates a CpG sites by samples matrix of methylation levels [55]. A bedGraph file for each pseudo-bulk sample was created through a computational tool called MethylDackel (https://github.com/dpryan79/MethylDackel). mm10 and hg19 reference genomes were used to read in the bedGraph files for mouse neuronal and B-cell tumor pseudo-bulks, respectively. As an input matrix of MeDeCom, we selected 20 000 CpG sites with the largest methylation level variance across pseudo-bulks. We ran MeDeCom with a regularization parameter 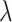 from
from 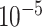 to
to 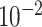 , 10 cross-validation folds, maximum iteration number 500 and random initialization number 30.
, 10 cross-validation folds, maximum iteration number 500 and random initialization number 30.
Houseman’s method
Houseman’s method [15] (or Houseman for short) was proposed to infer cell-type distribution from DNA methylation array-shaped data based on regression calibration. We used array-shaped data converted from our sequencing pseudo-bulk samples processed in the same way as for MeDeCom above. For selecting informative CpG sites, we mimicked the marker-CpG selection step of the original algorithm and chose CpG sites overlapping with ctDMRs that can provide cell-type-specific signals. We increased the number of CpG sites to 1000 in the original code considering the larger number of total CpG sites in our pseudo-bulk dataset compared with microarray data they used. For the rest, we followed up the pipeline given in the supplementary material of the original publication [15].
Performance measurement
Genomic correlation
We used genomic correlation to calculate overall genomic base-wise proximity between informative region selection results and ctDMRs. This statistic was suggested by Favorov et al. for determining the distribution of distances between two sets of genomic regions [58].
’Relative distance’ of a selected informative region 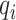 with respect to given ctDMRs is defined as below:
with respect to given ctDMRs is defined as below:
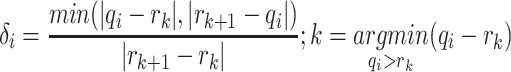 |
(2) |
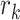 and
and 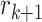 are two nearest ctDMRs from given informative region
are two nearest ctDMRs from given informative region 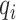 . It is calculated by dividing the distance between selected informative region
. It is calculated by dividing the distance between selected informative region 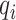 and the closest DMR by the distance between two nearest ctDMRs.
and the closest DMR by the distance between two nearest ctDMRs.
If a set of selected informative regions and ctDMRs are independent, 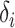 will have a uniform distribution. Hence, genomic correlation is calculated by testing if the distribution of calculated relative distances makes a uniform distribution.
will have a uniform distribution. Hence, genomic correlation is calculated by testing if the distribution of calculated relative distances makes a uniform distribution.
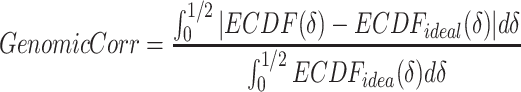 |
(3) |
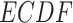 refers to empirical distribution cumulative function and
refers to empirical distribution cumulative function and 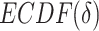 creates the distribution of observed
creates the distribution of observed 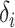 .
. 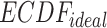 is the uniform distribution. Thus, Equation 3 yields the ratio of difference between area under
is the uniform distribution. Thus, Equation 3 yields the ratio of difference between area under 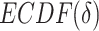 and under
and under 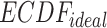 . When given selected informative regions are independent of ctDMRs,
. When given selected informative regions are independent of ctDMRs, 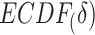 will create a uniform distribution and
will create a uniform distribution and 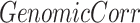 will reach 0. Conversely, if selected regions are identical to ctDMRs,
will reach 0. Conversely, if selected regions are identical to ctDMRs, 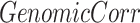 is 1.
is 1.
Absolute error between estimate and ground-truth
In cell-type composition estimation analysis, we assessed the performance by calculating absolute error between estimated proportion and ground-truth value of each pseudo-bulk sample. For coMethy and MeDeCom, we had to match cell types to estimated components by choosing the one with the lowest mean absolute error out of all possible combinations of estimate and ground-truth pair.
Mean absolute percentage error (MAPE)
MAPE is calculated by dividing the sum of individual ratios between absolute error values and the ground truth, by the number of data samples. Since this score cancels out the scale of values, we used this for the extremely low percentage of tumor cell type deconvolution evaluation.
Entropy of cell-type distribution
In information theory, entropy is a concept to describe the level of uncertainty or information in a set. We used this statistic to determine how equally cell types are distributed in the cell mixture, because cell-type composition estimation can be more intractable with extremely low or high proportion of subpopulations. Applying entropy to our cell mixtures, entropy of cell-type proportion in the cell mixture 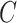 comprised of
comprised of 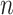 cell types,
cell types, 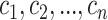 is defined as:
is defined as:
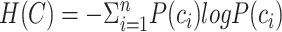 |
(4) |
 refers to the cell-type proportion of cell type
refers to the cell-type proportion of cell type 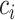 here. Thus, the entropy value is higher when all cell-type proportions are more uniformly distributed.
here. Thus, the entropy value is higher when all cell-type proportions are more uniformly distributed.
For example, let us assume we have two cell mixtures 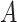 and
and 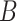 comprised of bi-components with different proportions. The dominant cell type constitutes 90% of cell mixture
comprised of bi-components with different proportions. The dominant cell type constitutes 90% of cell mixture 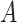 and 60% of cell mixture
and 60% of cell mixture  . According to Equation 4, the entropy of cell mixture
. According to Equation 4, the entropy of cell mixture 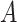 is smaller than cell mixture
is smaller than cell mixture 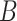 .
.
Results
Comparison of methodological designs
For our benchmarking study, we chose six published sequencing-based cell-type deconvolution methods (BED, DXM, PRISM, MethylPurify, csmFinder + coMethy and ClubCpG). As a comparison group, we added two cell-type deconvolution methods for array-shaped data (reference-based constrained projection method Houseman and our own reference-free method MeDeCom). To cope with the completely different input data formats, sequencing-based and array-based cell-type deconvolution procedures follow different pipelines in our project (Fig. 1A).
Figure 1.

Schematic overview of our cell-type deconvolution benchmarking. (A) Overall scheme of cell-type deconvolution benchmarking. We synthesized in silico cell mixtures called pseudo-bulks by mixing up reads randomly sampled from mouse neuronal scBS-seq dataset and tumor WGBS dataset, respectively. Sequencing-based cell-type deconvolution methods directly take sequencing data and select informative CpG sites (informative regions) supposedly related to cell-type heterogeneity (upper row pipeline). Then, cell-type proportions are estimated from selected CpGs. For array-based cell-type deconvolution methods, we converted sequencing data into array shape with pre-identified CpGs and the methods estimated cell-type compositions from the array (bottom row pipeline). (B) Example of an informative region showing a cell-type-specific methylation pattern in mouse neuronal data (chr1:75244319-75244379). Methylation pattern of reads (each row) overlapping with a specific region was extracted from three different samples, two pure cell-type samples (mL6-2 and mPv) and a pseudo-bulk sample comprised of these cell types. Some missing methylation patterns in the figure occurred owing to the CpG sites not covered by each read. In this region, the majority of reads from mL6-2 are fully methylated, whereas most reads from mPv show a fully unmethylated pattern.
All compared sequencing-based methods consist of two common steps: informative region selection and cell-type composition estimation. In the informative region selection step, the sequencing-based cell-type deconvolution methods filter out CpGs where the methylation patterns do not clearly demonstrate cell-type heterogeneity. Since sequencing data has significantly broader genome-wide coverage than array-shaped data, using all available CpG sites for deconvolution is not the most efficient strategy for cell-type composition estimation, associated with escalating computational complexity. In many genomic regions, methylation patterns are identical across all reads covering the CpG sites regardless of the cell type and thus non-informative. On the other hand, some of patterns are very complex because of other confounding factors. Even, at such loci where read coverage is low, local cell-type population (distribution of reads in terms of cell type at a given locus) usually does not correspond to global cell-type composition (cell-type composition in the entire bulk) due to the lack of reads. The informative region selection step alleviates these problems by clearing out these confounding methylation signals.
Initially, CpGs that are proximate to each other or overlap with a specific genomic region are grouped together. Here, we call each such group a ’region’. After that, only regions satisfying particular criteria are selected for the next step, here referred to as ’informative regions’. There are three common criteria considered in all our benchmarked methods: number of CpGs, region size and read coverage. Each method retains only regions with an abundant number of overlapping CpGs, sufficient region length and high read coverage. The detailed informative region selection parameter values of each method are described in Table 1.
For the two array-based methods, however, informative region selection is not a required step, since array-shaped data is already designed as a matrix of CpG sites by samples. Therefore, we converted our sequencing data to an array shape with predefined CpG sites. We described CpG site selection procedure for each array-based method in Materials and methods.
With the selected informative regions (sequencing-based methods) or predefined CpG sites (array-based methods), cell-type deconvolution methods predict cell-type proportions within given input cell-mixture samples. This step can be characterized by three main specifications: reference requirement, number of detectable cell types/subpopulations and estimation scope (Table 1).
Requirement of reference methylomes. Among the methods we benchmarked, BED, ClubCpG and Houseman are categorized as reference-based methods. BED and Houseman require methylome profiles of pure cell types. ClubCpG fits a regression model to training cell-mixture bulk data that is provided together with known cell-type composition.
Number of detectable components. We also categorized the benchmarked methods with respect to whether the method specifically targets tumor samples or not. The difference between tumor targeting and broadly applicable methods is their assumption about cell-type composition within given samples. Methods designed specifically for the analysis of tumor samples are generally referred as ’tumor purity estimation methods’. These assume that only two subpopulations comprise given cell mixtures, tumor and healthy stroma, whereas standard cell-type deconvolution methods do not limit the number of inferred subpopulations (components). Among the methods we considered, BED and MethylPurify were developed particularly for the analysis of tumor samples. So, we additionally tested our benchmarked methods with realistically simulated tumor-normal bulk samples, since those might be better suited to deal with abnormal methylation patterns from tumor cell types.
Estimation scope. Exploring all the methods, we have found a prevalent computational approach that summarizes methylation patterns across all of selected informative regions and compute the final estimates. BED, MethylPurify and PRISM locally calculate statistics in respective informative regions and predict compositions from the peak of region-wise estimated cell-type composition distribution. On the other hand, ClubCpG, coMethy, MeDeCom and Houseman globally calculate final cell-type proportions directly over all selected informative regions.
Benchmarking study design
To estimate the influence of each step upon the final result, we comprehensively assess all methods not only with the final cell-type composition estimation results, but also with the interim results of selected informative regions for deconvolution.
Our benchmarking analysis was performed with three different datasets to test the capability of methods in various biological scenarios, two and five cell-type mouse neuronal pseudo-bulks and tumor pseudo-bulk (Table 2). Firstly, we used single-nucleus bisulfite sequencing data of mouse neuron population from Luo et al. [47] to generate in silico pseudo-bulk read/cell mixtures for two and five cell types. Secondly, to test methods in a realistic scenario of tumor bulk deconvolution, we created in silico mixtures of WGBS methylomes from normal non-cancer B-cell and B-cell lymphoma samples from Do et al. [48].
Table 2.
Details of pseudo-bulk datasets. We created three datasets of pseudo-bulk samples for evaluating our benchmarked methods. Two datasets are comprised of two and five cell-types of mouse neuronal single-cell BS-seq data each, and one dataset was created with tumor and normal cell types. We generated 20 pseudo-bulk samples in each dataset
| Resource | # Bulks | Cell types | Biological sample | Sequencing protocol | |
|---|---|---|---|---|---|
| 2 cell-type mouse neuronal pseudo-bulk | GSE97179 [47] | 20 | mL6-2, mPv | Mouse neuron | scBS-seq (Illumina HiSeq 4000) |
| 5 cell-type mouse neuronal pseudo-bulk | GSE97179 [47] | 20 | mL6-2, mPv, mDL-2, mL2-3, mL5-1 | Mouse neuron | scBS-seq (Illumina HiSeq 4000) |
| Tumor-normal pseudo-bulk | GSE137880 [48] | 20 | Normal B-cell, B-cell lymphoma | B-cell | WGBS (Illumina NovaSeq 6000) |
In the analysis of five cell-type pseudo-bulk group, we excluded BED and MethylPurify, because tumor purity estimation methods are not capable of detecting more than two cell types. Although Prism is not a tumor purity estimation method, it failed in detecting five cell types so we also excluded it from five cell-type pseudo-bulk analysis.
Evaluation of the informative region selection methods using cell-type DMRs
We hypothesized that ideal informative region selection results should overlap with ctDMRs, i.e. genomic regions showing significant methylation pattern differences between cell types (Fig. 1B). Conversely, informative region selection results with low similarity to ctDMRs are unlikely to supply enough cell-type-specific signals. Hence, we assessed informative region selection results primarily by comparing them with ctDMRs. The ctDMRs were generated by comparing one pure cell-type bulk to all others (details in Materials and methods).
For two cell-type neuronal pseudo-bulks, BED detected a significant number of informative regions overlapping with ctDMRs, but csmFinder detected the highest number of overlaps in tumor pseudo-bulks. (Fig. 2A and B) In five cell-type mouse neuronal pseudo-bulk analysis, ClubCpG showed the highest number of overlaps with ctDMRs over all cell types (Supplementary Fig. 1).
Figure 2.

Plots for informative region selection. (A, B) Overlaps between ctDMRs for (A) two cell-type mouse neuronal pseudo-bulks and (B) tumor pseudo-bulks. The colored box plots at the top of each graph show the number of overlaps between a pair of methods connected at the middle, across all pseudo-bulks, and the gray box plots at the right side display the number of informative regions detected by each method or the number of regions in ctDMRs. Overlap sizes were calculated with respect to the number of bases. In each plot, different ctDMRs are distinguished by different colors. (C–E) Genomic correlation between ctDMRs and selected informative regions in (C) two cell-type mouse neuronal pseudo-bulks, (D) five cell-type mouse neuronal pseudo-bulks and (E) tumor pseudo-bulks. Higher genomic correlation means higher similarity between ctDMRs and selected informative regions with respect to the number of overlaps and the proximity.
Since the size of the selected informative region set differs between methods, the number of overlaps may not exactly correspond to how similar each selected informative region set is to ctDMRs. We found that the set size of selected informative regions and the number of overlaps with ctDMRs have a strong correlation in all datasets (Supplementary Fig. 4). Consequently, our results clearly showed that selecting more informative regions possibly increases the number of overlaps with ctDMRs even though the selected region set may also include many more non-overlapping regions. Thus, we conducted another statistical analysis to compare the similarity between selected informative regions and ctDMRs.
Favorov et al. [58] designed a statistic called ’genomic correlation’ that measures the distributions of distances between two sets of genomic regions(details in the Materials and methods). This statistic is [0,1] interval-bound, with lower value indicating larger distance between genomic regions from the two compared sets. We evaluated the informative region selection results based on the proximity to ctDMRs using genomic correlation score (Fig. 2C-E).
Although informative regions of BED overlapped the largest number of ctDMRs in two cell-type mouse neuronal pseudo-bulk samples, their genomic correlation with each ctDMR set was the lowest. This implies that ctDMRs comprised a very small fraction of the informative regions selected by BED. In contrast, csmFinder, Prism and MethylPurify showed high genomic correlation with ctDMRs in both two cell-type mouse neuronal and tumor pseudo-bulks, meaning that ctDMRs make up a large portion of the selected regions in spite of the small absolute total number. For five cell-type mouse neuronal pseudo-bulks, csmFinder yielded the highest genomic correlation with all ctDMRs compared with other sequencing-based methods, ClubCpG and DXM.
We further explored genome annotations of the selected informative regions (Supplementary Figs 7–9). Compared with ctDMRs, the selected informative regions included a noticeably larger amount of promoter regions in all types of pseudo-bulk analyses with the exception of BED in two cell-type mouse neuronal pseudo-bulk result. Considering that promoter methylation can regulate gene expression in a cell-type-specific manner, we expect that the informative region selection step of the benchmarked methods is well able to identify methylation patterns which contribute to cell-type identity.
We also calculated the methylation level difference between two cell types particularly with bi-component (two cell-type mouse neuronal and tumor) pseudo-bulks. Within each set of selected informative regions, we extracted methylation beta-value at CpGs from two pure cell-type methylomes and calculated the difference. The distribution of differences was calculated in each pseudo-bulk (Supplementary Figs 5 and 6). If the selected regions covers CpGs with cell-type-specific methylation patterns, the methylation level difference value must be close to either 1 or -1 depending on which of the cell types is hypomethylated at that CpG site. Cell-type DMRs, as expected, mostly cover CpGs with absolute methylation level difference of 1. Regions designated for array-based methods also showed the methylation difference distribution peaking close to -1 and 1, as ctDMRs and CpGs with the highest variance of methylation value were given to Houseman’s method and MeDeCom, respectively. However, CpGs selected by sequencing-based methods mostly had a methylation level difference 0, especially for two cell-type mouse neuronal pseudo-bulks. From tumor pseudo-bulk samples, csmFinder, MethylPurify and Prism were more successful in detecting CpGs showing high methylation difference between non-cancer and B-cell lymphoma cell types.
In conclusion, we found that sequencing-based methods can detect ctDMRs through their informative region selection step. However, despite the large number of overlaps with ctDMRs, the selected informative region sets still include some uninformative genomic regions, distant from ctDMRs or showing zero methylation level difference across different cell types. Methylation patterns from such regions may hinder accurate cell-type deconvolution, if it is not handled during the cell-type composition estimation step.
Mouse neuronal single-cell pseudo-bulk cell-type deconvolution
As explained above, we evaluated all methods with two groups of mouse neuronal single-cell pseudo-bulk samples. Absolute error value between the ground-truth and estimated value was our main performance score for the cell-type composition estimation step. Reference-based and reference-free methods were evaluated separately for a fair comparison in terms of additional prior information.
Our results showed that reference-based methods, with the exception of BED, perform better than reference-free methods in both groups (Fig. 3A and B). Furthermore, we analyzed predicted proportion of each cell type within each pseudo-bulk sample (Fig. 3C and D). We realized that ClubCpG can produce a cell-type proportion estimate lower than 0 or higher than 1, when ground-truth proportion is relatively low or high, respectively. This is because ClubCpG does not restrict its prediction value between 0 and 1. Yet, other methods successfully made all predictions in the expected range.
Figure 3.

Cell-type composition estimation for mouse neuronal single-cell pseudo-bulk scenario. (A, B) Absolute error between ground-truth and estimated cell-type proportion, calculated in each sample and each cell type, for (A) two cell-type and (B) five cell-type mouse neuronal pseudo-bulks. Black line at the middle refers to the median and the both ends of bar are the first and third quantiles. Numbers above box plots indicate the median value. (C, D) Estimated cell-type proportions and ground-truth values (black line) ordered with respect to the ground-truth proportion, in (C) two cell-type and (D) five cell-type mouse neuronal pseudo-bulks. For two cell-type pseudo-bulks, only results of mPv cell type are shown to avoid redundancy in two-component mixtures.
In the two cell-type pseudo-bulk analysis, the best accuracy was achieved by Houseman’s method requiring pure cell-type methylome profiles. Among reference-free methods, coMethy performed best with a median absolute error value of 0.044, even though MethylPurify and Prism inferred more accurate values for bulks with high proportion of mPv.
Among reference-based methods, the pseudo-bulk with five cell types showed the same results as with two cell types: the lowest median absolute error was achieved by Houseman. However, among reference-free methods, DXM performed best with the median absolute error value 0.058. In bulk-wise comparisons stratified by cell types, mL2-3 was the the most difficult cell type to estimate for coMethy, but ClubCpG and MeDeCom showed the lowest accuracy in mDL-2. Even though all other methods showed better performance with two cell-type pseudo-bulks, Houseman and DXM exhibited lower median absolute error with five cell-type pseudo-bulks than with two cell-type pseudo-bulks.
Tumor-normal cell mixture deconvolution
Tumor heterogeneity analysis is one of the most crucial and widely studied research topics in cancer biology. DNA methylation patterns can facilitate the deconvolution of cell types in tumor microenvironment and malignant clones. BED and MethylPurify were specifically designed for tumor purity estimation of tumor-normal cell mixture as described in Table 1. For this reason, we additionally assessed the benchmarked methods using pseudo-bulk samples comprised of B-cell lymphoma and normal non-cancer B-cell (Fig. 4). In order to account for the high variation that may exist within a tumor, we used WGBS data derived from expectedly homogeneous cell lines for generating pseudo-bulks rather than sparse scBS-seq data.
Figure 4.

Cell-type composition estimation results of tumor pseudo-bulk scenario. (A) Absolute error between estimated and ground-truth cell-type proportions. (B) Estimated proportions by each method and ground-truth values in order. All details of two plots are the same as in Figure 3. Again, we present the results only for normal B-cell due to the symmetric cell-type proportions in two-component mixtures.
In the reference-based setting, Houseman’s method again showed the best performance in tumor cell-type deconvolution. compared with mouse neuronal pseudo-bulk deconvolution, Houseman’s method accomplished the lowest median absolute error of 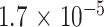 in tumor cell-type deconvolution. Among reference-free methods, MeDeCom estimated tumor pseudo-bulk cell-type compositions with the lowest median absolute error. Sample-wise performance for one cell type also showed that Houseman estimated the most accurate proportions for all samples and ClubCpG exceeded the range of 0 and 1 for extreme proportions, similarly to mouse neuronal pseudo-bulk analysis results.
in tumor cell-type deconvolution. Among reference-free methods, MeDeCom estimated tumor pseudo-bulk cell-type compositions with the lowest median absolute error. Sample-wise performance for one cell type also showed that Houseman estimated the most accurate proportions for all samples and ClubCpG exceeded the range of 0 and 1 for extreme proportions, similarly to mouse neuronal pseudo-bulk analysis results.
As in some tumor tissues, rare cell types can play a critical role [59], we performed an additional evaluation for the scenario of rare B-cell lymphoma cell type within a bulk (Supplementary Fig. 10). We further generated 10 more tumor pseudo-bulk increasing the ratio of B-cell lymphoma cell type by 0.1% from 0.1% to 1%. The cell-type composition estimation result was assessed with MAPE score considering the extremely small range of ground-truth value. Houseman’s method invariably outperformed all other reference-based methods, but, among reference-free methods, MethylPurify showed the best performance. In general, the benchmarked methods were not capable of inferring the cell-type ratio value below 0.1 aside from Houseman’s method that was able to generate estimates below 0.1. Unlike the preceding benchmarking results, MeDeCom and coMethy showed much lower performance.
Identifying factors that influence cell-type deconvolution performance
Based on the results so far, we investigated whether informative region selection results have detectable influence upon cell-type composition estimation. For this, we compared the rank of mean absolute error together with the rank of mean genomic correlation separately (Fig. 5A and Supplementary Fig. 2). Both absolute error and genomic correlation results from a method were averaged across all samples, but separately in each cell type, whereafter we ranked the methods based on the averaged values. After ranking the methods, we excluded array-based methods whose pipeline does not include informative region selection. For tumor pseudo-bulks, non-cancer B-cell and B-cell lymphoma cell types involve same ctDMRs produced by comparing only those two cell types. Consequently, in two cell-type mouse neuronal and tumor pseudo-bulk samples, the accuracy of cell-type composition estimation (complemented of the mean absolute error value) tends to be proportional to the performance of genomic correlation between selected informative regions and ctDMRs. From this result, even though all methods are designed with different algorithms, we claim that in two-component mixtures detecting CpGs overlapping with ctDMRs significantly contributes to the cell-type deconvolution performance.
Figure 5.

Influential factors in cell-type deconvolution performance. (A) Mean absolute error versus mean genomic correlation between selected informative regions and ctDMRs. The points indicate results of each cell type from each deconvolution method. (B) Mean absolute error versus entropy of cell-type proportions in each bulk sample. In both plots, we fitted dots in a linear function (the line with gray background).
We additionally discovered that all methods except DXM could infer more accurate cell-type proportions in the mixtures with more balanced cell-type composition in five cell-type pseudo-bulks (Fig. 5B). This was determined using the entropy value that measures the uniformity of given proportions or given distribution (details in Materials and methods). The entropy of cell-type distribution was negatively correlated with the mean absolute error showing the P-value lower than 0.05 in the results of ClubCpG, coMethy, Houseman and MeDeCom. We presume that, in cell-type mixtures with low entropy values where cell population is distributed in extremely biased way, minor cell types may not provide enough cell-type-specific signals. Nevertheless, DXM differed from the other methods in this regard. The lower the entropy within a given pseudo-bulk, the better its performance. We suppose that the design of DXM algorithm, searching the best fit out of randomly generated distributions rather than gradually fitting a model, becomes more powerful when applied to an extreme distribution of cell types, by disregarding regularization.
In both two cell-type deconvolution scenarios, not all methods performed better with higher entropy of samples. For instance, Houseman in the two cell-type pseudo-bulk analysis and coMethy and Prism in tumor pseudo-bulk analysis rather showed positive correlation between the two values (Supplementary Fig. 3). This might be caused by some pseudo-bulks with high entropy, where cell types are more uniformly distributed, resulting in more complex methylation patterns.
Discussion
Here we extensively reviewed and assessed five sequencing-based cell-type deconvolution methods with standardized measurements for unbiased evaluation. Two more array-based deconvolution methods, MeDeCom and Houseman, were also added to the analyses as a comparison group, to evaluate the effectiveness of the benchmarked methods to leverage the unique properties of sequencing data.
In order to reflect various biological scenarios, our analyses were done with two different datasets, mouse neuronal scBS-seq dataset and B-cell tumor WGBS dataset. We generated pseudo-bulk samples by merging randomly sampled reads from pure cell-type samples in each dataset. For mouse neuronal pseudo-bulk samples, we generate mixtures of two and five cell types, respectively, to study scenario with different numbers of subpopulations.
All sequencing-based methods we evaluated include two essential steps. First, each method selects genomic regions that are considered to show clear cell-type-specific methylation patterns (informative region selection). In the second step, final cell-type composition is inferred based on the selected regions (cell-type composition estimation). Thus, we evaluated the performance separately in respective steps and finally examined the influential factors in cell-type deconvolution performance.
While evaluating informative region selection, we regarded ctDMRs as the gold-standard cell-type-specific loci and compared them with informative regions selected by each method. Although, in mouse neuronal pseudo-bulk analyses, ClubCpG detected the largest number of overlaps with ctDMRs, csmFinder showed the best genomic correlation. This is because that large number of overlaps can be confounded by the large number of total selected informative regions, rather than the capacity to detect cell-type-specific regions (Supplementary Fig. 1).
To assess cell-type composition estimation results, we calculated the absolute error between the estimated and ground-truth proportion of each cell type in each sample. Since introducing prior knowledge from reference data naturally improves estimation performance, for the evaluation we grouped methods according to whether reference data is required. Among reference-based methods, Houseman’s method strongly outperformed other methods. In the comparison of reference-free methods, coMethy inferred the most accurate cell-type compositions in both two and five cell-type pseudo-bulks.
Cancer-associated DNA methylation patterns are particularly aberrant and the cell subpopulations in cancerous tissues containing both healthy normal and tumor cell types are often of more complex composition [60]. Therefore, we separately evaluated cell-type deconvolution results in tumor pseudo-bulk samples generated as described above. In informative region selection analysis, csmFinder not only showed large number of overlaps with the ctDMRs, but also reached the highest genomic correlation. For cell-type composition estimation, Houseman and MeDeCom outperformed all other reference-based and reference-free methods, respectively.
Lastly, we have confirmed the significance of selected informative regions for the overall cell-type deconvolution performance. As shown in Figure 5A, mean absolute error and genomic correlation of evaluated methods have a negative rank correlation in bi-component pseudo-bulks. This result highlights that the more similar informative regions to ctDMRs a method can detect, the better performance the method would achieve in cell-type composition estimation for cell mixtures comprised of two distinct subpopulations. Yet, in five cell-type mouse neuronal pseudo-bulks, entropy of cell-type distribution showed a negative correlation with the absolute error except in the case of DXM (Fig. 5B). This implies that the distribution of subpopulations can be more influential in cell-type deconvolution of complex bulk samples.
Although our benchmarked methods yielded reasonable cell-type deconvolution results in most analyses, there are still some limitations that have to be resolved in sequencing-based cell-type deconvolution. Firstly, based on our results, sequencing-based methods did not outperform array-based methods in cell-type composition estimation. Not only did Houseman’s method perform best of all reference-based methods, but also MeDeCom achieved the lowest median absolute error among reference-free methods in the tumor pseudo-bulk analysis. One complication of the sequencing-based approach is the high complexity of sequencing data itself. The complexity arises because, unlike array data consisting of summarized methylation beta-values, sequencing data captures a methylation state from a limited number of DNA molecules at each CpG site, yet with single-molecule resolution. Thus, sequencing-based methods should be capable of eliminating redundant methylation patterns and better modeling subpopulation distribution based on the remaining informative methylation patterns.
With the leverage of single-cell profiling, it was convincingly demonstrated that the variable composition of numerous normal cell types and tumor cell subclones underlies intratumor heterogeneity [61–63]. However, tumor purity estimation methods in our analyses could only consider tumor samples as binary mixtures of tumor and normal cell subpopulations. These results are not able to explain the actual complexity of tumor microenvironment that has been widely investigated, particularly in relation to success of various therapeutic strategies, e.g. immunotherapy [64]. Therefore, to support upcoming tumor studies with the analysis of cell-type composition variation, more advanced tumor-targeting cell-type deconvolution methods addressing multi-component intra-tumor heterogeneity need to be implemented.
Last but not least, the software availability, sustainability and deployment should be considered important when cell-type deconvolution tools are implemented and released. To this end, we found that some of our benchmarked methods have significant methodological and technical limitations. For example, PRISM and BED do not ensure its applicability to other types of data than RRBS. Furthermore, most of benchmarked methods are available only for samples processed with Bismark [50]. The limited availability, complexity of deployment and lack of input standardization eventually hinder efficient utilization of evaluated methods for real-life analyses. In addition, software maintenance is another crucial issue to develop a sustainable cell-type deconvolution tool. Importantly, to be able to execute the tools, we had to implement multiple bugfixes. Considering recent innovations in bisulfite sequencing technology [33, 36], sequencing-based cell-type deconvolution tools should be well maintained and updated to remain useful as the field evolves.
Taken together, our analysis results suggest a clear paradigm of how to conduct cell-type deconvolution for sequencing data. It will pave the way towards more accurate cell-type composition inference and more precise analysis of cell-type-specific methylation patterns to allow further method development and improvement. Because of the intrinsic benefit of read-level information, which provides detailed methylation states at each CpG, it should be possible to accomplish better accuracy in the inference of cell-type composition from sequencing-based DNA methylation data compared with summarized (array-shaped) data, something which currently available tools cannot achieve yet. Future work should be aimed towards more thoroughly designed methods for the extraction of cell-type-specific signals, while adjusting for confounding factors that affect sequencing data. Precise cell-type deconvolution of DNA methylation sequencing data can broaden the range of available cell population inference tools with diverse clinical and biomedical applications.
Key Points
Sequencing-based DNA methylomes contain highly informative read-level cell-type-specific patterns enabling cell-type deconvolution.
The majority of previously proposed deconvolution methods are comprised of two main steps: informative region selection and cell-type composition estimation.
The informative region selection step of benchmarked methods chose different genomic regions that showed a high impact upon the cell-type composition estimation step.
The benchmarked sequencing-based deconvolution methods did not significantly outperform the array-based methods with respect to cell-type composition estimation.
This evaluation study highlights the necessity for more advanced cell-type deconvolution methods taking an advantage of unique sequencing data properties.
Data availability
The single-cell mouse neuronal methylomes and tumour WGBS data are publicly available in the NCBI GEO data repository (mouse neuronal single-cell: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97179, B-cell lymphoma and non-cancer WGBS: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137880). For the detailed data preprocessing, please see Supplementary Table 1.
Code availability
All conducted analyses are reproducible following steps described in markdown files in our github repository (https://github.com/CompEpigen/SeqDeconv_Pipeline). Detailed pipelines for each method are explained in the file named METHOD_deconvolution_analysis.md. We also uploaded the bug-fixed version of some methods through git fork function. The details of fixed lines in the code are clarified in commit comments in each Github repository. The list of methods uploaded as a new bug-fixed version is below:
MethylPurify: https://github.com/CompEpigen/MethylPurify
For the methods which could be successfuly run in published form their source code is available under the following URLs:
csmFinder: https://github.com/Gavin-Yinld/csmFinder
MeDeCom: https://github.com/lutsik/MeDeCom
Houseman: Supplementary file 2 on https://doi.org/10.1186/1471-2105-13-86
The pipeline used for generating pseudo-bulk samples is available in the same github repository (https://github.com/CompEpigen/SeqDeconv_Pipeline/blob/main/Pseudo_bulk_generation_pipeline.md).
Author contributions statement
Y.J. mainly conducted all analyses and wrote the manuscript together with P.L. P.L also presented the idea, designed experiments and analyses together with Y.J. M.G. contributed to the analysis result presentation. K.B and R.T contributed to data preprocessing pipelines. L.S and D.T contributed to the analyses conducted with DXM method. C.P. provided support for the study and critically discussed results with Y.J. and P.L. All authors reviewed the manuscript and provided critical feedback.
Supplementary Material
Acknowledgements
We acknowledge guidance on their deconvolution method pipelines from James Barrett (UCL), Hehuang Xie (Virginia Tech), Robert A. Waterland (Baylor College of Medicine) and Anthony Scott (Mercury Data Science). We also appreciate Katherine Kelly’s help (DKFZ) with proof-reading the manuscript.
Yunhee Jeong is a doctorate researcher in the Division of Cancer Epigenomics at DKFZ, Germany.
Lisa Barros de Andrade e Sousa and Dominik Thalmeier are AI consultants at Helmholtz AI.
Reka Toth was a postdoctoral scientist in the Division of Cancer Epigenomics at DKFZ, Germany, and currently works as a scientist at LIH, luxembourg.
Marlene Ganslmeier and Kersten Breuer worked as a master student in the Division of Cancer Epigenomics at DKFZ, Germany.
Christoph Plass is head of the Division of Cancer Epigenomics at DKFZ, Germany.
Pavlo Lutsik is a junior group leader in the Division of Cancer Epigenomics at DKFZ, Germany.
Contributor Information
Yunhee Jeong, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany; Faculty of Mathematics and Informatics, Heidelberg University, Im Neuenheimer Feld 205, 69120, Heidelberg, Germany.
Lisa Barros de Andrade e Sousa, Helmholtz AI, Helmholtz Zentrum München, Ingolstädter Landstraβ e 1, 85764, Neuherberg, Germany.
Dominik Thalmeier, Helmholtz AI, Helmholtz Zentrum München, Ingolstädter Landstraβ e 1, 85764, Neuherberg, Germany.
Reka Toth, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany.
Marlene Ganslmeier, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany.
Kersten Breuer, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany.
Christoph Plass, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany.
Pavlo Lutsik, Division of Cancer Epigenomics, German Cancer Research (DKFZ), Im Neuenheimer Feld 280, 69120, Heidelberg, Germany.
Funding
German Network for Bioinformatics partner project de.NBI-epi [031L0101A], subproject Heidelberg [to P.L.].
References
- 1. Schwartzman O, Tanay A. Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet 2015;16(12):716–26. [DOI] [PubMed] [Google Scholar]
- 2. Horak P, Heining C, Kreutzfeldt S, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov 2021;11(11):2780–95. [DOI] [PubMed] [Google Scholar]
- 3. Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014;383(9933):1990–8. [DOI] [PubMed] [Google Scholar]
- 4. Lam LL, Emberly E, Fraser HB, et al. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci 2012;109(Suppl. 2):17253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci 2007;104(3):973–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wen Y, Wei Y, Zhang S, et al. Cell subpopulation deconvolution reveals breast cancer heterogeneity based on DNA methylation signature. Brief Bioinform 2017;18(3):426–40. [DOI] [PubMed] [Google Scholar]
- 7. Hui T, Cao Q, Wegrzyn-Woltosz J, et al. High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations. Stem Cell Rep 2018;11(2):578–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature 2018;555(7697):469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun 2021;12(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kozlenkov A, Wang M, Roussos P, et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res 2016;44(6):2593–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boks MP, Derks EM, Weisenberger DJ, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PloS One 2009;4(8):e6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang FF, Cardarelli R, Carroll J, et al. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011;6(5):623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CPG site resolution. Genomics 2011; 98(4):288–95. [DOI] [PubMed] [Google Scholar]
- 14. Pidsley R, Zotenko E, Peters TJ, et al. Critical evaluation of the illumina methylationepic beadchip microarray for whole-genome DNA methylation profiling. Genome Biol 2016;17(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform 2012;13(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chakravarthy A, Furness A, Joshi K, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun 2018;9(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Teschendorff AE, Breeze CE, Zheng SC, et al. A comparison of reference-based algorithms for correcting cell-type heterogeneity in epigenome-wide association studies. BMC Bioinform 2017;18(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H, Cai R, Dai J, et al. Emeth: an em algorithm for cell type decomposition based on DNA methylation data. Sci Rep 2021;11(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levy JJ, Titus AJ, Petersen CL, et al. Methylnet: an automated and modular deep learning approach for DNA methylation analysis. BMC Bioinform 2020;21(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lutsik P, Slawski M, Gasparoni G, et al. Medecom: discovery and quantification of latent components of heterogeneous methylomes. Genome Biol 2017;18(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andres Houseman E, Kile ML, Christiani DC, et al. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinform 2016;17(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Onuchic V, Hartmaier RJ, Boone DN, et al. Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell Rep 2016;17(8):2075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rahmani E, Schweiger R, Shenhav L, et al. Bayescce: a Bayesian framework for estimating cell-type composition from DNA methylation without the need for methylation reference. Genome Biol 2018;19(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rahmani E, Schweiger R, Rhead B, et al. Cell-type-specific resolution epigenetics without the need for cell sorting or single-cell biology. Nat Commun 2019;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scherer M, Schmidt F, Lazareva O, et al. Machine learning for deciphering cell heterogeneity and gene regulation. Nat Comput Sci 2021;1(3):183–91. [DOI] [PubMed] [Google Scholar]
- 26. Decamps C, Privé F, Bacher R, et al. Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software. BMC Bioinform 2020;21(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goeppert B, Toth R, Singer S, et al. Integrative analysis defines distinct prognostic subgroups of intrahepatic cholangiocarcinoma. Hepatology 2019;69(5):2091–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scherer M, Nazarov PV, Toth R, et al. Reference-free deconvolution, visualization and interpretation of complex DNA methylation data using decomppipeline, medecom and factorviz. Nat Protoc 2020;15(10):3240–63. [DOI] [PubMed] [Google Scholar]
- 29. Chen Y, Toth R, Chocarro S, et al. Diverse routes of club cell evolution in lung adenocarcinoma. bioRxiv. 2021.
- 30. Simon M, Mughal SS, Horak P, et al. Deconvolution of sarcoma methylomes reveals varying degrees of immune cell infiltrates with association to genomic aberrations. J Transl Med 2021;19(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meissner A, Gnirke A, Bell GW, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005;33(18):5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009;462(7271):315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shu C, Zhang X, Aouizerat BE, et al. Comparison of methylation capture sequencing and infinium methylationepic array in peripheral blood mononuclear cells. Epigenet Chromatin 2020;13(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou W, Dinh HQ, Ramjan Z, et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat Genet 2018;50(4):591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salhab A, Nordström K, Gasparoni G, et al. A comprehensive analysis of 195 DNA methylomes reveals shared and cell-specific features of partially methylated domains. Genome Biol 2018;19(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clark SJ, Smallwood SA, Lee HJ, et al. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scbs-seq). Nat Protoc 2017;12(3):534–47. [DOI] [PubMed] [Google Scholar]
- 37. Guo H, Zhu P, Xinglong W, et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 2013;23(12):2126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Argelaguet R, Clark SJ, Mohammed H, et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 2019;576(7787):487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shuhui Bian Y, Hou XZ, Li X, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018;362(6418):1060–3. [DOI] [PubMed] [Google Scholar]
- 40. Barrett JE, Feber A, Herrero J, et al. Quantification of tumour evolution and heterogeneity via Bayesian epiallele detection. BMC Bioinform 2017;18(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yin L, Luo Y, Xiguang X, et al. Virtual methylome dissection facilitated by single-cell analyses. Epigenet Chromatin 2019;12(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee D, Lee S, Kim S. Prism: methylation pattern-based, reference-free inference of subclonal makeup. Bioinformatics 2019;35(14):i520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zheng X, Zhao Q, Wu H-J, et al. Methylpurify: tumor purity deconvolution and differential methylation detection from single tumor DNA methylomes. Genome Biol 2014;15(7):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anthony Scott C, Duryea JD, MacKay H, et al. Identification of cell type-specific methylation signals in bulk whole genome bisulfite sequencing data. Genome Biol 2020;21(1):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Titus AJ, Gallimore RM, Salas LA, et al. Cell-type deconvolution from DNA methylation: a review of recent applications. Hum Mol Genet 2017;26(R2):R216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fong J, Gardner JR, Andrews JM, et al. Determining subpopulation methylation profiles from bisulfite sequencing data of heterogeneous samples using DXM. Nucleic Acids Res 2021;49(16):e93–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Luo C, Keown CL, Kurihara L, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 2017;357(6351):600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Do C, Dumont ELP, Salas M, et al. Allele-specific DNA methylation is increased in cancers and its dense mapping in normal plus neoplastic cells increases the yield of disease-associated regulatory SNPs. Genome Biol 2020;21(1):1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 2011;17(1):10–2. [Google Scholar]
- 50. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 2011;27(11):1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and samtools. Bioinformatics 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Do C, Lang CF, Lin J, et al. Mechanisms and disease associations of haplotype-dependent allele-specific DNA methylation. Am J Hum Genet 2016;98(5):934–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neidhart M. DNA Methylation and Complex Human Disease. Radarweg 29, 1043 NX Amsterdam, The Netherlands: Academic Press, 2015. [Google Scholar]
- 54. Hao W, Wang C, Zhijin W. A new shrinkage estimator for dispersion improves differential expression detection in RNA-seq data. Biostatistics 2013;14(2):232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mayakonda A, SchÃnung M, Hey J, et al. Methrix: an R/Bioconductor package for systematic aggregation and analysis of bisulfite sequencing data. Bioinformatics 2020;36(22–23):5524–5. [DOI] [PubMed] [Google Scholar]
- 56. Team BC, Maintainer BP. Txdb. mmusculus. ucsc. mm10. knowngene: annotation package for txdb object (s). r package version 3.10. 0. 2020.
- 57. Marc Carlson and Bioconductor Package Maintainer. Txdb. hsapiens. ucsc. hg19. knowngene. 2015.
- 58. Favorov A, Mularoni L, Cope LM, et al. Exploring massive, genome scale datasets with the genometricorr package. PLoS Comput Biol 2012;8(5):e1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Egyud M, Tejani M, Pennathur A, et al. Detection of circulating tumor DNA in plasma: a potential biomarker for esophageal adenocarcinoma. Ann Thorac Surg 2019;108(2):343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res 2009;15(12):3927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu Y, He S, Wang X-L, et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat Commun 2021;12(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dong X, Wang F, Liu C, et al. Single-cell analysis reveals the intra-tumor heterogeneity and identifies mlxipl as a biomarker in the cellular trajectory of hepatocellular carcinoma. Cell Death Discov 2021;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou Y, Yang D, Yang Q, et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun 2020;11(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Baghban R, Roshangar L, Jahanban-Esfahlan R, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal 2020;18(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single-cell mouse neuronal methylomes and tumour WGBS data are publicly available in the NCBI GEO data repository (mouse neuronal single-cell: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97179, B-cell lymphoma and non-cancer WGBS: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137880). For the detailed data preprocessing, please see Supplementary Table 1.