

Abstract
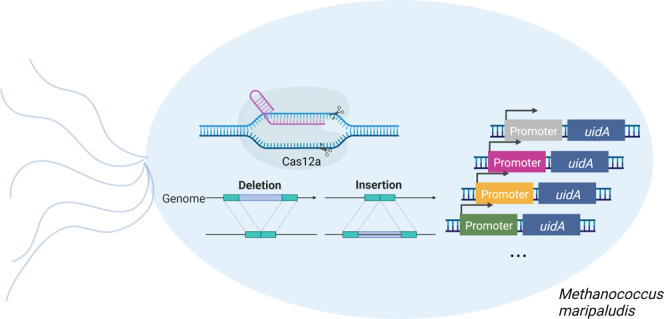
The rapid-growing and genetically tractable methanogen Methanococcus maripaludis is a promising host organism for the biotechnological conversion of carbon dioxide and renewable hydrogen to fuels and value-added products. Expansion of its product scope through metabolic engineering necessitates reliable and efficient genetic tools, particularly for genome edits that affect the primary metabolism and cell growth. Here, we have designed a genome-editing toolbox by utilizing Cas12a from Lachnospiraceae bacterium ND2006 (LbCas12a) in combination with the homology-directed repair machinery endogenously present in M. maripaludis. This toolbox can delete target genes with a success rate of up to 95%, despite the hyperpolyploidy of M. maripaludis. For the purpose of demonstrating a large deletion, the M. maripaludis flagellum operon (∼8.9 kbp) was replaced by the Escherichia coli β-glucuronidase gene. To facilitate metabolic engineering and flux balancing in M. maripaludis, the relative strength of 15 different promoters was quantified in the presence of two common growth substrates, either formate or carbon dioxide and hydrogen. This CRISPR/LbCas12a toolbox can be regarded as a reliable and quick method for genome editing in a methanogen.
Keywords: Methanococcus maripaludis, methanogens, CRISPR/Cas12a, genome editing, metabolic engineering, synthetic biology
Introduction
Methanogenic archaea are biotechnologically employed in a variety of uses, e.g., for methane production in anaerobic digestors,1 as biocatalysts in power-to-gas processes,2 and as versatile hosts for the development of synthetic pathways that convert carbon dioxide (CO2) into value-added products.3,4 Hydrogenotrophic methanogens utilize the reductive acetyl-CoA pathway for CO2 fixation,5 an energy-efficient route to synthesize organic carbon from CO2 and hydrogen (H2), which is similar to that found in acetogens.6 Subtle differences exist between the acetogenic7 and methanogenic CO2 reduction pathways in terms of ATP investment and cofactor utilization.8 Depending on the type of product that needs to be generated from CO2 as the carbon source, methanogens may be better-suited hosts than acetogens. Recently, the methanogen Methanosarcina acetivorans was re-engineered to no longer depend on methane production for its energy metabolism,9 thereby serving as an example where a methanogen could be utilized for generating an expanded repertoire of new potential products besides methane.
Methanococcus maripaludis is a promising methanogenic host organism for metabolic engineering of CO2-fixation pathways due to its advantageous growth properties, e.g., 2 h doubling time,10,11 moderate growth temperature of 38 °C, and ability to fix nitrogen.12−14 Typical electron donors for CO2 reduction in M. maripaludis include formate, H2, and bioelectrically coupled systems.15,16 Efforts to expand the product scope of M. maripaludis beyond methane are already underway. As an example, the mevalonate pathway in this methanogen was metabolically engineered to produce geraniol from CO2 and formate.4 Efficient and reliable genome-editing tools are critical for successful metabolic engineering in M. maripaludis. Marker recycling is a prerequisite for multitarget engineering. In the case of M. maripaludis, while a pop-in/pop-out markerless-based genome-editing technique has been developed,17 it tends to have a problematic low positive rate, which can sometimes be less than 5%,17 particularly for those modifications that affect cell growth. As an alternative, the CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR associated protein) system might remedy this problem because of its reputation for highly efficient genome editing.
The CRISPR/Cas9 system has already been successfully used for genome editing in a variety of organisms18−23 owing to its simplicity and high efficiency, but only a few CRISPR genome-editing toolboxes have been developed for archaea.24 The first CRISPR/Cas9-mediated genome-editing system for a methanogen was reported in 2017 using M. acetivorans as the model organism.25 This Cas9-based system recognizes and cleaves a 20-nucleotide target sequence that is flanked by a 3′-NGG protospacer adjacent motif (PAM). This contrasts with Cas12a, which instead recognizes the 5′-thymine (T)-rich PAM 5′-TTTV. This recognition site makes Cas12a the better option for developing a CRISPR toolbox in microbes with an adenine (A)- and T-rich genome. Another advantageous attribute of Cas12a lies in its ribonuclease activity, which allows the formation of multiple guide RNAs (gRNAs) from a single transcript.26,27 Since the M. maripaludis genome has a high AT content (67.1%), we decided to use the Cas12a from Lachnospiraceae bacterium ND2006 (LbCas12a) and combine it with the intrinsic homology-directed repair machinery to develop a CRISPR genome-editing toolbox. In our study, we examined how the length of the repair fragment (RF) and the distance of the RF to the double-stranded break (DSB) impact on the genome-editing efficiency. As an application of our toolbox, we deleted the M. maripaludis flagellum operon and replaced it with the Escherichia coli β-glucuronidase gene. To further expand the versatility and editing potential of this genetic toolbox, we also established a Cas9-based editing system, and we quantified the relative strength of 15 different promoters in the presence of two common growth substrates, either formate or H2 and CO2.
Results and Discussion
CRISPR/Cas12a-Based Introduction of Double-Stranded Breaks and Transformation Efficiency
For this study, we utilized the host strain M. maripaludis JJΔupt in all experiments as well as a recently established natural transformation protocol.28 The E. coli/M. maripaludis shuttle vector pLW4029 served as the backbone for constructing the final toolbox plasmid pMM002P (Table S1). Further details about pMM002P and its construction are given in Figure 1a. The transformation efficiency of pMM002P into JJΔupt cells was calculated to be about 5 × 104 colony forming units per 2 μg DNA [cfu (2 μg DNA)−1] (Figure 1b), which was similarly obtained for pMM001 (pMM002P lacking LbCas12a) (data not shown). The high transformation efficiency suggests that LbCas12a expression is not toxic in M. maripaludis. Because the coexpression of LbCas12a with either one or two gRNA sequences resulted in only 3–18 and 0–3 transformant colonies, respectively (Figure 1b,c), we conclude that the LbCas12a–gRNA complex can cause a lethal DSB in the M. maripaludis chromosome. Since nonhomologous end-joining (NHEJ) machineries for DNA repair are rare in archaea,30 and as M. maripaludis JJ lacks a homolog of the Ku protein (which has a strong binding affinity for free DNA ends or nicks), NHEJ is not expected to provide an escape from such DSBs.
Figure 1.
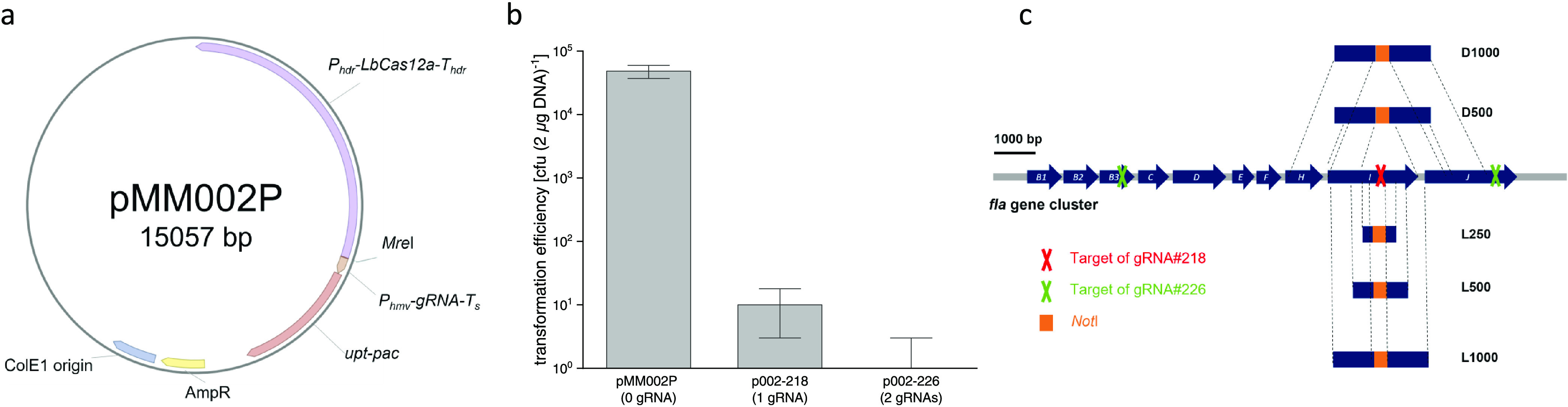
General features of the CRISPR/LbCas12a genome editing. (a) Genetic map of the CRISPR/LbCas12a pMM002P plasmid. The M. maripaludis S2 uracil phosphoribosyltransferase gene (upt), which serves as a counter-selective marker, and the codon-optimized puromycin N-acetyltransferase gene (pac) are coexpressed via the Pmcr promoter.4 LbCas12a expression is driven by the Phdr promoter from Methanococcus voltae A3. gRNA expression is driven by the M. voltae A3 Phmv histone promoter. Two PaqCI sites between the direct repeat sequence and the synthetic terminator in the opposite direction for spacer fusion is used for gRNA insertion (not displayed). The gRNA of the plasmid pMM002P that contains two PaqCI sites does not target the chromosome. An MreI restriction site assigned between the gRNA and Cas elements is used for RF insertion. (b) CRISPR/LbCas12a triggered DSBs. Shown are the transformation efficiencies [cfu (2 μg DNA)−1] for the CRISPR/LbCas12a pMM002P plasmids with one, two, or no gRNAs that were used to transform M. maripaludis. Error bars represent the standard deviation of the values obtained for the transformation efficiency (n = 3). (c) Schematic outline of the repair fragment edits. A NotI site is placed between the two homologous arms.
CRISPR/LbCas12a Genome Editing by Providing a Repair Fragment on the pMM002P-Derived Plasmid
For CRISPR/LbCas12a genome editing, a gRNA was expressed on pMM002P (p002-218) that targets the flaI (MMJJ_11570) gene of the M. maripaludis flagellum operon. The lethal effect of the functional gRNA could be relieved by including RFs with various lengths of the homology arms (Figures 1c and 2a). A 1000 bp homology arm on either side resulted in a transformation efficiency of 2.8 × 104 cfu (2 μg DNA)−1. We therefore used homology arms of this length for all subsequent experiments. While 250 bp homology arms were long enough to repair DSBs created by the Cas12a/gRNA complex, the transformation efficiency was 70 times lower. We also examined what effect the distance between the RF and the DSB would have on the transformation efficiency by testing three different distances (i.e., ∼25, ∼500, and ∼1000 bp) (Figures 1c and 2b). The transformation efficiency was found to be five times lower with the 500 bp distance than with the 25 bp distance (two-sided t-test, P < 0.001), but no significant difference was observed whether 500 or 1000 bp distances were used (two-sided t-test, P > 0.05). Because M. maripaludis contains an active PstI restriction modification system, cells are able to digest foreign DNA containing unmethylated PstI sites, which lowers the transformation efficiency by 1.6–3.4 fold per PstI site.31 This reduction of the transformation efficiency is exemplified by the presence of one PstI site in each of the 500 and 1000 bp homology arms, as any restriction digestion would likely be responsible for a lower number of transformants (Figure 2b). The similar transformation efficiency obtained with the 500 and 1000 bp distances to the DSB suggests that the genome-editing efficiency is unaffected within a distance length of 1000 bp. To assess the positive rate of genome editing, two sets of primers were used to amplify a DNA sequence on both sides of the homology arms on the chromosome. Each of the PCR products was then subjected to NotI digestion. Here, a NotI restriction site was engineered between the left and right RFs, so that the wild-type polyploid genome copies are distinguishable from the edited ones (Figure 1c). The PCR products were sequenced afterward to confirm that the sites were edited as expected. As a result, genome editing was highly efficient, displaying a positive rate of 89–100% (Figure 2a). All results taken together (see Figure 2), 63 out of 66 colonies had been correctly edited, which equates to an average positive rate of nearly 95%. Hence, we conclude that our CRISPR/LbCas12a toolbox can reliably perform genome editing in M. maripaludis (see the Supporting information for a detailed description of the general procedure for utilizing the CRISPR/LbCas12a genome-editing toolbox in M. maripaludis).
Figure 2.
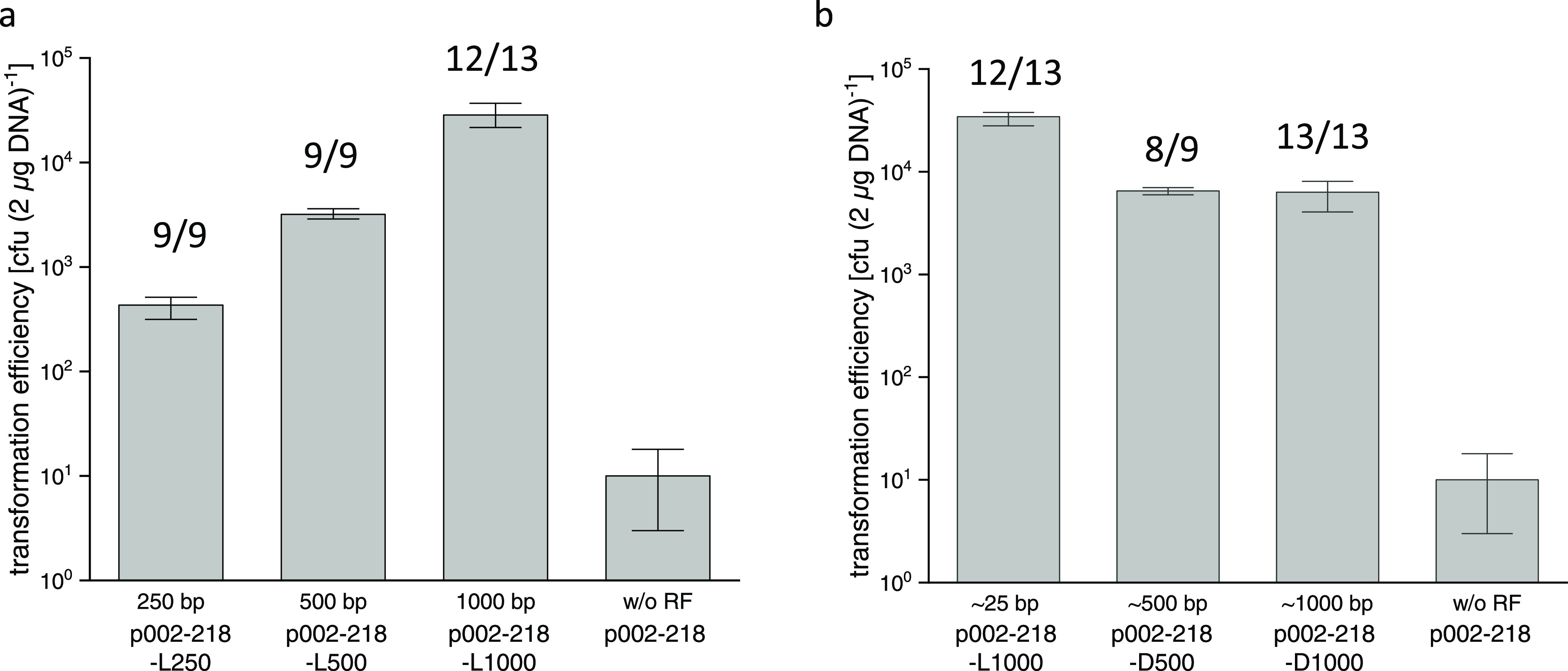
Effect on transformation and genome-editing (positive rates) efficiencies when the length and position of the RF are modified. Transformation efficiency and positive rate in relation to the length and position of the RF. Shown are the transformation efficiencies [cfu (2 μg DNA)−1] for the CRISPR/LbCas12a pMM002P-derived plasmids that were used to transform M. maripaludis. (a) CRISPR/LbCas12a plasmid p002-218, in which the lengths of the homology arms flanking the RF are 250, 500, and 1000 bp (p002-218-L250, p002-218-L500, and p002-218-L1000, respectively). The distance from the RF to DSB for all plasmids is ∼25 bp. (b) CRISPR/LbCas12a plasmid p002-218 with 1000 bp homologous arms, in which the distance between the RF and the DSB is ∼25, ∼500, and ∼1000 bp (p002-218-L1000, p002-218-D500, and p002-218-D1000, respectively). p002-218 without the RF is included as a control. Error bars represent the standard deviation of the values obtained for the transformation efficiency (n = 3). Positive rates representing the fraction of correctly edited colonies per colonies tested by PCR are shown for all plasmid transformations (numbers above bars).
CRISPR/LbCas12a Genome Editing by Providing a Repair Fragment Separately
To help speed up the construction of different genome-edited mutants in parallel, we modified our CRISPR/LbCas12a toolbox by providing the RF separately as a suicide plasmid. For this alternative procedure, the CRISPR/LbCas12a cleavage plasmid was cotransformed with a suicide plasmid containing a promoter–uidA fusion expression cassette (for further details, see the section below on promoter strengths) flanked on either side by 1000 bp homology arms. While we successfully obtained transformants with this modification, the transformation efficiency was lowered by 10–50 fold, but the genome-editing efficiency was robust and remained high. As proof, when we randomly selected and examined three transformants from the 15 different genome-edited constructs made using the suicide plasmid, all of them (45/45) were found to be positive (data not shown). We also examined the possibility of cotransforming the CRISPR/LbCas12a cleavage plasmid with the RF separately by providing it as a linear PCR product. In this case, while genome editing was deemed successful, the transformation efficiency was 100–1000 times lower than that obtained using our original CRISPR/LbCas12a toolbox method with the plasmid containing the 1000 bp homology arm (p002-218-L1000).
Using CRISPR/LbCas12a Genome Editing to Replace a Large Genome Fragment with a Heterologous Gene
As an application of our CRISPR/LbCas12a method, we demonstrated its use as a tool for heterologous gene integration. We removed the entire ∼8.9 kbp flagellum operon (flaB1B2B3CDEFGHIJ MMJJ_11660 – MMJJ_11560) from the M. maripaludis chromosome and substituted it with the E. coli β-glucuronidase gene (uidA). This edit was performed using two different CRISPR/LbCas12a plasmids: p002-218-uidA, which has one gRNA that generates a single lethal DSB on the chromosome, resulting in two long distances between the DSB and the RF (i.e., 6.4 and 2.5 kbp), and p002-226-uidA, which has two different gRNAs that cleave at either side of the flagellum operon and thus shorten the distances between the DSB and the RF (i.e., 0.25 and 1.6 kbp). Both plasmids resulted in a similar transformation efficiency (Figure 3), demonstrating that our CRISPR/LbCas12a method can successfully generate a large chromosomal fragment deletion. The positive rate of genome editing was lower when only one gRNA was used, as it seems the 6.4 kbp distance (Figure 3) affects the positive rate by exceeding the 1000 bp length. A similar effect was observed for M. acetivorans, in which the positive rate was significantly reduced when the distance to the DSB went beyond 1000 bp.25 In addition, the transformation efficiency had also decreased with an increasing distance.25 Using two gRNAs to shorten the distance between the RF and the DSB might help improve the transformation and genome-editing efficiencies also in other methanogen-based CRISPR systems.
Figure 3.

CRISPR/LbCas12a genome-edited replacement of the M. maripaludis flagellum operon with the E. coli β-glucuronidase gene (uidA). Shown are the transformation efficiencies [cfu (2 μg DNA)−1] for the CRISPR/LbCas12a pMM002P-derived plasmids that were used to transform M. maripaludis. Plasmids p002-218 and p002-226 (controls) express one and two gRNAs, respectively, and do not contain the RF. Plasmids p002-218-uidA and p002-226-uidA express one and two gRNAs, respectively, but contain the RF. Error bars represent the standard deviation of the values obtained for the transformation efficiency (n = 3). Positive rates representing the fraction of correctly edited colonies per colonies tested by PCR are shown for the p002-218-uidA and p002-226-uidA transformations (numbers above bars).
To be ready for a second round of genome editing in the future, the CRISPR plasmid was removed after genome editing by being counter-selected on a plate containing 6-azauracil. The absence of the plasmid was confirmed by the inability of the cells to grow on the medium containing puromycin, and the removal rate was 9/10.
Development of the CRISPR/SpCas9 Genome-Editing System
To expand the gRNA repertoire in our CRISPR/Cas toolbox, we also constructed a Cas9-based genome-editing plasmid that uses the Streptococcus pyogenes Cas9 endonuclease. With this CRISPR/SpCas9 tool, we were able to successfully replace the 1.9 kbp fragment covering the alanine dehydrogenase–alanine racemase genes (ald-alr, MMJJ_13250 – MMJJ_13260) in the M. maripaludis chromosome with a 4.2 kbp DNA fragment containing a different heterologous gene. Here, the CRISPR/SpCas9 cleavage plasmid with one gRNA was cotransformed with a suicide plasmid containing the 4.2 kbp DNA fragment flanked on both sides by 1000 bp homology arms. The transformation efficiency was only 317 ± 123 cfu (2 μg DNA)−1, but the rate of genome editing was high (8/10 colonies were edited) (Figure 4). Since the RF contained two PstI sites, they can explain the lower transformation efficiency of this genome-editing system.
Figure 4.

CRISPR/SpCas9 genome-edited replacement of the M. maripaludis alanine dehydrogenase–alanine racemase (ald-alr 1.9 kbp) genes with a 4.2 kbp fragment. Shown are the transformation efficiencies [cfu (2 μg DNA)−1] for the CRISPR/SpCas9 plasmids that were used to transform M. maripaludis. The left bar indicates that the cells were transformed with a suicide plasmid containing the integration cassette and a CRISPR/SpCas9 plasmid carrying a gRNA targeting to the ald-alr. The right bar indicates that the cells were only transformed with a CRISPR/SpCas9 plasmid carrying a gRNA targeting to the ald-alr. Error bars represent the standard deviation of the values obtained for the transformation efficiency (n = 3). The positive rate representing the fraction of correctly edited colonies per colonies tested by PCR is shown for the transformations (numbers above the left bar).
Promoter Strengths of 15 Different Promoter–uidA Fusions Constructed by CRISPR/LbCas12a
Fifteen different promoters delivered as a promoter–uidA fusion expression cassette were integrated into the locus of the acetyl-CoA synthetase gene (MMJJ_09370) of M. maripaludis JJ using the CRISPR/LbCas12a toolbox (for further details, see earlier section; the sequences of the 15 promoters are listed in the Supporting information). Three promoters (Pmcr_JJ, PmcrR_JJ, and Pfla_JJ) originated from M. maripaludis JJ, while the remaining twelve were from the closely related methanogen, Methanococcus vannielii SB. The relative strengths of these promoters were measured in the presence of two common growth substrates, either formate or H2 and CO2. All promoters except Pnif and PhdrC1 had successfully driven the expression of uidA using both growth substrates (Figure 5). PhdrC1 had allowed gene expression in only the formate-containing growth medium, while no expression was detected for Pnif in both growth substrates. Since Pmcr is regarded as a strong constitutive promoter in methanogens,32 then by comparison, PglnA, Pmtr, Pmcr, Pmcr_JJ, and Pfla_JJ can be judged as strong promoters in M. maripaludis. Transcription from Pnif is normally repressed by the nitrogen regulatory protein R (NrpR) but can become highly active when N2 gas serves as the sole nitrogen source or else in the absence of NrpR.33 With this in mind, we deleted the nrpR gene from the genome-edited Pnif–uidA strain and found that Pnif was no longer repressed and had instead increased in strength significantly (2670 ± 58 nmol min–1 OD600–1) (two-sided t-test, P < 0.001). It is tempting to speculate that the ΔnrpR–Pnif strain might be a useful host for target genes requiring very strong expression. For the majority of the promoters, their strengths were similar for both growth substrates.
Figure 5.

Quantification of promoter strengths for the two different growth conditions formate or H2/CO2, measured after the culture has reached OD600 = ca. 0.5. The promoters mcr_JJ, mcrR_JJ, and fla_JJ are from M. maripaludis JJ. The remaining promoters are from M. vannielii SB. Error bars represent the standard deviation (n = 3). The activity of the hdrC1 promoter in H2/CO2 medium and the nif promoter in formate and H2/CO2 medium cannot be detected. *P < 0.05; **P < 0.01; ***P < 0.001.
While Peha normally drives the expression of the energy-converting hydrogenase A gene (eha) in M. vannielii, it was observed as a weak promoter in M. maripaludis. Peha does not directly control the eha gene expression, but instead first drives the expression of a putative transcriptional factor (TF) gene that precedes the eha gene, wherein both genes are presumably part of an operon. We also examined a uidA construct that includes the TF gene after the Peha w.TF promoter sequence. The uidA expression using both growth substrates was found to be significantly higher for the Peha w.TF construct than the Peha one (two-sided t-test, P < 0.01). These results suggest that this transcriptional factor might regulate eha expression. On the other hand, PglnA from M. vannielii was unexpectedly strong in M. maripaludis, even with the presence of the NrpR repressor or ammonium in the growth medium. DNA sequencing of the integrated PglnA–uidA expression cassette eliminated a possible point mutation or other change as being responsible for this unusual promoter strength. Likewise, constructing and testing a new PglnA–uidAM. maripaludis strain still gave the same result. Thus, one can confidently conclude that PglnA from M. vannielii functions as a strong promoter in M. maripaludis. In M. maripaludis, native PglnA normally directs basal constitutive expression levels when ammonium is present.34 Although the glnA operator for PglnA is the same in M. vannielii and M. maripaludis, the PglnA–uidA strain appears to have the highest promoter strength among all others tested in this study.
Conclusions
M. maripaludis already possesses several efficient genetic tools and transformation protocols for standard applications,17,28,31 e.g., such as the classic pop-in/pop-out genome-editing technique.17 In some instances, however, this genetic editing tool is hindered by a low positive rate, with many colonies requiring to be screened to obtain a desired genotype, particularly when the targets to be engineered affect cell growth.32 As a solution, we have developed a reliable CRISPR/Cas12a toolbox that can efficiently knock-in or knock-out genes in M. maripaludis with a positive rate of at least 95%. Notably, our system requires only a single round of homologous recombination and lacks merodiploid formation, which then lowers the workload of genome editing and increases the overall success rate. The option of providing the RF separately as a suicide plasmid or PCR fragment might further speed up the genome-editing process. Since Cas12a displays ribonuclease activity that can process a single continuous multi-gRNA transcript,26,27 it might be convenient to express two gRNAs via our CRISPR/LbCas12a system, thus saving the time and cost of additional plasmid construction. Our CRISPR/LbCas12a toolbox can also allow for heterologous protein production in M. maripaludis, as it drives the stable integration of genes into the chromosome. M. maripaludis might become an attractive expression host for many proteins that are difficult to be produced in E. coli, e.g., such as formate dehydrogenase,16 methyl-coenzyme M reductase,35 and heterodisulfide reductase.36 While a variety of promoters have thus far been studied and used for synthetic biology in M. maripaludis,(14,33,37,38) a uniform system to compare their strengths has been lacking. In that context, our CRISPR/LbCas12a genome-editing toolbox now represents a versatile system for engineering and balancing metabolic fluxes in M. maripaludis strains.
Materials and Methods
Plasmids and Strains
All plasmids and strains used in this study are listed in Tables S1 and S2, respectively. Links to plasmid maps are listed in Table S3. M. maripaludis JJΔupt28 and plasmid pLW4029 are gifts from Prof. Kyle Costa, University of Minnesota. Plasmid pMEV44 was kindly provided by Prof. William B Whitman, University of Georgia. M. maripaludis S239 was kindly provided by Prof. John Leigh and Dr. Thomas Lie, University of Washington. E. coli NEB5α (New England Biolabs) was used for plasmid construction. The plasmids pMM002P and pMM005 were constructed by Gibson assembly.40 The construction protocol and primers for pMM002P and pMM005 are described in Tables S4 and S5, respectively. All of the cleavage plasmids were constructed in the following manner. For LbCas12a gRNA, the forward primer consisted of 5′-AGAT and 24-nucleotide guide sequence, whereas the reverse primer consisted of 5′-TATC and 24-nucleotide reverse complement guide sequence. For SpCas9 gRNA, the forward primer consisted of 5′-AGTG and 20-nucleotide guide sequence, whereas the reverse primer consisted of 5′-AAAC and 20-nucleotide reverse complement guide sequence. Both sets of forward and reverse primers containing the gRNA and a 5′ four-nucleotide overhang were annealed. The annealing product was ligated to PaqCI-digested pMM002P or pMM005 vector DNA. CRISPR guide sequences were designed using the CHOPCHOP webtool (https://chopchop.cbu.uib.no/).41 The RF was inserted into the corresponding cleavage plasmid at the MreI restriction site. Additional primers used in this study are listed in Table S6.
Growth Media and Conditions
Lysogeny broth medium (10 g L–1 tryptone, 10 g L–1 NaCl, and 5 g L–1 yeast extract) containing 50 mg L–1 ampicillin was used for plasmid construction. Liquid McC medium was used for growing M. maripaludis strains with an anoxic headspace (2.8 bar, 80% H2/20% CO2).28 Sealed culture tubes were incubated at 37 °C with 200 rpm agitation. McFC medium with an anoxic headspace (1 bar, 80% N2/20% CO2) was used when formate served as the carbon source.42 Sealed culture tubes were incubated statically at 37 °C. Puromycin (2.5 μg mL–1) or 6-azauracil (0.25 mg mL–1) was added as required.
M. Maripaludis Transformation
The natural transformation of M. maripaludis was performed using a previously described protocol.28 Briefly, a sealed tube containing a 5 mL of M. maripaludis culture was grown overnight to an OD600 between 0.7 and 1.2. Two micrograms of DNA was then added directly to the culture. This was followed by flushing the headspace with a gas mixture of 80% H2 and 20% CO2 for 30 s and adjusting the pressure to 2.8 bar. The sealed culture tube was then incubated at 37 °C with 200 rpm agitation for 4 h, after which the cells were spread-plated onto solid McC medium supplemented with 2.5 μg mL–1 puromycin and grown anaerobically at 37 °C.
Curing of the CRISPR/Cas Plasmid from M. Maripaludis Strains
M. maripaludis strains containing the CRISPR/Cas toolbox plasmid were grown in 5 mL of liquid McC medium without antibiotics to an OD600 between 0.7 and 1.2. A 100 μL aliquot of each culture was used to inoculate another 5 mL of liquid McC medium lacking antibiotics and allowed to incubate overnight. A single droplet of culture was then streaked out onto solid McC medium containing 0.25 mg mL–1 6-azauracil. After 3–5 days, several isolated colonies were selected and streaked out onto another plate of the same medium for purification of plasmid-free cells.
β-Glucuronidase Activity Measurements
4-Nitrophenyl β-d-glucuronide (4-NPG, Sigma-Aldrich) served as the substrate and was prepared as a 10 mg mL–1 stock solution in 50 mM sodium phosphate buffer, pH 7.0 (Na-PB). For measuring β-glucuronidase activity, a tube of M. maripaludis cells was first grown to an OD600 of ∼0.5 (BioPhotometer plus, Eppendorf) and then a 1 mL aliquot of culture was centrifuged at 10 000 g for 2 min. The pelleted cells were resuspended with Na-PB (500 μL) and the cell suspension was subjected to glass bead (30 μL) disruption for 5 min. Afterward, the cell-free lysate was recovered by centrifugation at 10 000 g for 2 min. To take activity measurements, the cell-free lysate was diluted appropriately to 500 μL of Na-PB and incubated for 20 min at 37 °C. A 40 μL aliquot of 4-NPG stock solution (see above) was added to the mix and allowed to react for 15 min at 37 °C. The reaction was stopped by adding a 400 μL aliquot of 200 mM sodium carbonate, and the absorbance measurement was taken at 405 nm with a UV–vis spectrophotometer. Specific activity calculations were made with E. coli K12 β-glucuronidase (Cat. no. 3 707 580 001, Sigma-Aldrich) as the standard using the conversion factor of 398 nmol min–1.
Acknowledgments
The authors thank the summer laboratory assistants Han Le, An Nguyen, and Pradhuman Jetha for their work on plasmid constructions and M. maripaludis transformations. They also kindly thank the following individuals: Prof. Kyle Costa for providing the M. maripaludis JJΔupt strain and plasmid pLW40, Prof. William B. Whitman for providing plasmids pMEV4 and pMEV4mTs, and the M. voltae A3 strain, and Prof. John Leigh and Dr. Thomas Lie for providing the M. maripaludis S2 strain. Prof. Michael Rother, Prof. Qunxin She, Prof. Dipti Nayak, and Dr. Zhe Lyu are acknowledged for their helpful discussions. They are also thankful to Dr. Ingemar von Ossowski, Dr. Norman Adlung, and Dr. Vera Jäger for providing critical and helpful comments on the manuscript. The Novo Nordisk Foundation and the Academy of Finland are acknowledged for funding this study (grant NNF19OC0054329 and grant 326020 to S.S.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.2c00137.
Plasmids; strains and primers lists; plasmid maps; guidelines on the use of the CRISPR/Cas genome-editing toolbox in M. maripaludis;M. maripaludis natural transformation protocol; and promoter sequences (PDF)
Author Contributions
J.B. and S.S. conceived the study and wrote the manuscript. J.B. and E.D.M. performed the experiments. All authors commented and approved the final version of the manuscript. All authors declare no conflict of interest.
The authors declare no competing financial interest.
Supplementary Material
References
- Wintsche B.; Jehmlich N.; Popp D.; Harms H.; Kleinsteuber S.. Metabolic adaptation of methanogens in anaerobic digesters upon trace element limitation Front. Microbiol., 2018; Vol. 9, 1−10 10.3389/fmicb.2018.00405. [DOI] [PMC free article] [PubMed]
- Braga Nan L.; Trably E.; Santa-Catalina G.; Bernet N.; Delgenès J. P.; Escudié R. Biomethanation processes: new insights on the effect of a high H2 partial pressure on microbial communities. Biotechnol. Biofuels 2020, 13, 1–17. 10.1186/s13068-020-01776-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge J.; Carr S.; et al. Anaerobic production of isoprene by engineered Methanosarcina species Archaea. Appl. Environ. Microbiol. 2021, 87, e02417–e02420. 10.1128/AEM.02417-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu Z.; Jain R.; Smith P.; Fetchko T.; Yan Y.; Whitman W. B. Engineering the autotroph Methanococcus maripaludis for geraniol production. ACS Synth. Biol. 2016, 5, 577–581. 10.1021/acssynbio.5b00267. [DOI] [PubMed] [Google Scholar]
- Simpson P. G.; Whitman W. B.. Anabolic pathways in methanogens. In Methanogenesis; Springer, Boston: MA, 1993; 10.1007/978-1-4615-2391-8_11 pp 445–472. [Google Scholar]
- Ragsdale S. W.; Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784, 1873–1898. 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V. New Horizons in acetogenic conversion of one-carbon substrates and biological hydrogen storage. Trends Biotechnol. 2019, 37, 1344–1354. 10.1016/j.tibtech.2019.05.008. [DOI] [PubMed] [Google Scholar]
- Lemaire O. N.; Jespersen M.; Wagner T.. CO2 -fixation strategies in energy extremophiles: what can we learn from acetogens? Front. Microbiol., 2020; Vol. 11, 1−8 10.3389/fmicb.2020.00486. [DOI] [PMC free article] [PubMed]
- Schöne C.; Poehlein A.; Jehmlich N.; Adlung N.; Daniel R.; von Bergen M.; Scheller S.; Rother M. Deconstructing Methanosarcina acetivorans into an acetogenic archaeon. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2113853119 10.1073/pnas.2113853119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydock A. K.; Porat I.; Whitman W. B.; Leigh J. A. Continuous culture of Methanococcus maripaludis under defined nutrient conditions. FEMS Microbiol. Lett. 2004, 238, 85–91. 10.1016/j.femsle.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Müller A. L.; Gu W.; Patsalo V.; Deutzmann J. S.; Williamson J. R.; Spormann A. M. An alternative resource allocation strategy in the chemolithoautotrophic archaeon Methanococcus maripaludis. Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2025854118 10.1073/pnas.2025854118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler P. S.; Daniel C.; Leigh J. A. Ammonia switch-off of nitrogen fixation in the methanogenic archaeon Methanococcus maripaludis: mechanistic features and requirement for the novel GlnB Homologues, NifI1 and NifI2. J. Bacteriol. 2001, 183, 882–889. 10.1128/JB.183.3.882-889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler P. S.; Leigh J. A. Genetics of nitrogen regulation in Methanococcus maripaludis. Genetics 1999, 152, 1343–1351. 10.1093/genetics/152.4.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie T. J.; Wood G. E.; Leigh J. A. Regulation of nif expression in Methanococcus maripaludis: roles of the euryarchaeal repressor NrpR, 2-oxoglutarate, and two operators. J. Biol. Chem. 2005, 280, 5236–5241. 10.1074/jbc.M411778200. [DOI] [PubMed] [Google Scholar]
- Goyal N.; Zhou Z.; Karimi I. A. Metabolic processes of Methanococcus maripaludis and potential applications. Microb. Cell Fact. 2016, 15, 107 10.1186/s12934-016-0500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohner S. T.; Deutzmann S.; Logan B. E.; Leigh J.; Spormann A. M. Hydrogenase-independent uptake and metabolism of electrons by the archaeon Methanococcus maripaludis. ISME J. 2014, 8, 1673–1681. 10.1038/ismej.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B. C.; Leigh J. A. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. J. Bacteriol. 2005, 187, 972–979. 10.1128/JB.187.3.972-979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F. A.; Hsu P. D.; Wright J.; Agarwala V.; Scott D. A.; Zhang F. Genome engineering using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O.; Sanjana E. N.; Hartenian E.; Shi X.; Scott D. A.; Mikkelsen T. S.; Heckl D.; Ebert B. L.; Root D. E.; Doench J. G.; Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–88. 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaestecker W.; Buono R. A.; Pfeiffer M. L.; Vangheluwe N.; Jourquin J.; Karimi M.; van Isterdael G.; Beeckman T.; Nowack M. K.; Jacobs T. B. CRISPR-Tsko: A technique for efficient mutagenesis in specific cell types, tissues, or organs in Arabidopsis. Plant Cell 2019, 31, 2868–2887. 10.1105/tpc.19.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J.; Guo D.; Zhang J.; Huang Q.; Qin G.; Zhang X.; Wan J.; Gu H.; Qu L. J. Targeted mutagenesis in rice using CRISPR-Cas system. Cell Res. 2013, 23, 1233–1236. 10.1038/cr.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop-Fabre M. M.; Jakočiu̅nas T.; Stovicek V.; Dai Z.; Jensen M. K.; Keasling J. D.; Borodina I. EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 2016, 11, 1110–1117. 10.1002/biot.201600147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai P.; Duan X.; Wu X.; Gao L.; Ye M.; Zhou Y. J. Recombination machinery engineering facilitates metabolic engineering of the industrial yeast Pichia Pastoris. Nucleic Acids Res. 2021, 49, 7791–7805. 10.1093/nar/gkab535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gophna U.; Allers T.; Marchfelder A. Finally, archaea get their CRISPR-Cas Toolbox. Trends Microbiol. 2017, 25, 430–432. 10.1016/j.tim.2017.03.009. [DOI] [PubMed] [Google Scholar]
- Nayak D. D.; Metcalf W. W. Cas9-mediated genome editing in the methanogenic archaeon Methanosarcina acetivorans. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 2976–2981. 10.1073/pnas.1618596114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B.; Gootenberg J. S.; Abudayyeh O. O.; Slaymaker I. M.; Makarova K. S.; Essletzbichler P.; Volz S. E.; Joung J.; van der Oost J.; Regev A.; Koonin E. V.; Zhang F. Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. 10.1016/J.CELL.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B.; Heidenreich M.; Mohanraju P.; Fedorova I.; Kneppers J.; DeGennaro E. M.; Winblad N.; Choudhury S. R.; Abudayyeh O. O.; Gootenberg J. S.; Wu W. Y.; Scott D. A.; Severinov K.; van der Oost J.; Zhang F. Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca D. R.; Halim M. F. A.; Holten M. P.; Costa K. C. Type IV-like pili facilitate transformation in naturally competent archaea. J. Bacteriol. 2020, 202, 1–12. 10.1128/JB.00355-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodsworth J. A.; Leigh J. A. Regulation of nitrogenase by 2-oxoglutarate-reversible, direct binding of a PII - like nitrogen sensor protein to dinitrogenase. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9779–9784. 10.1073/pnas.0602278103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. F.; Allers T. DNA repair in the archaea - an emerging picture. FEMS Microbiol. Rev. 2018, 42, 514–526. 10.1093/femsre/fuy020. [DOI] [PubMed] [Google Scholar]
- Tumbula D. L.; Makula R. A.; Whitman W. B. Transformation of Methanococcus maripaludis and identification of a Pst I-like restriction system. FEMS Microbiol. Lett. 1994, 121, 309–314. 10.1111/j.1574-6968.1994.tb07118.x. [DOI] [Google Scholar]
- Kohler P. R. A.; Metcalf W. W. Genetic manipulation of Methanosarcina spp. Front. Microbiol. 2012, 3, 259 10.3389/fmicb.2012.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie T. J.; Leigh J. A. A novel repressor of nif and glnA expression in the methanogenic archaeon Methanococcus maripaludis. Mol. Microbiol. 2003, 47, 235–246. 10.1046/j.1365-2958.2003.03293.x. [DOI] [PubMed] [Google Scholar]
- Lie T. J.; Leigh J. A. Regulatory response of Methanococcus maripaludis to alanine, an Intermediate nitrogen source. J. Bacteriol. 2002, 184, 5301–5306. 10.1128/JB.184.19.5301-5306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu Z.; Chou C.-W.; Shi H.; Wang L.; Ghebreab R.; Phillips D.; Yan Y.; Duin E. C.; Whitman W. B. Assembly of methyl-coenzyme M reductase in the methanogenic archaeon Methanococcus maripaludis. J. Bacteriol. 2018, 200, e00746-17 10.1128/JB.00746-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienemann M.; Deutzmann J. S.; Milton R. D.; Sahin M.; Spormann A. M. Mediator-free enzymatic electrosynthesis of formate by the Methanococcus maripaludis heterodisulfide reductase supercomplex. Bioresour. Technol. 2018, 254, 278–283. 10.1016/j.biortech.2018.01.036. [DOI] [PubMed] [Google Scholar]
- Akinyemi T. S.; Shao N.; Lyu Z.; Drake I. J.; Liu Y.; Whitman W. B. Tuning gene expression by phosphate in the methanogenic archaeon Methanococcus maripaludis. ACS Synth. Biol. 2021, 10, 3028–3039. 10.1021/acssynbio.1c00322. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Berezuk A.; Khursigara C. M.; Jarrell K. F. Bypassing the need for the transcriptional activator EarA through a spontaneous deletion in the BRE portion of the fla operon promoter in Methanococcusmaripaludis. Front. Microbiol. 2017, 8, 1329 10.3389/fmicb.2017.01329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman W. B.; Shieh J.; Sohn S.; Caras D. S.; Premachandran U. Isolation and characterization of 22 mesophilic Methanococci. Syst. Appl. Microbiol. 1986, 7, 235–240. 10.1016/S0723-2020(86)80012-1. [DOI] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A.; Smith H. O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Labun K.; Montague T. G.; Krause M.; Torres Cleuren Y. N.; Cleuren Y. N. T.; Tjeldnes H.; Valen E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, w171–w174. 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F.; Wang L.; Lupa B.; Whitman W. B. A flexible system for cultivation of Methanococcus and other formate-utilizing methanogens. Archaea 2017, 2017, 7046026 10.1155/2017/7046026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.