

Abstract
Peptidase neurolysin (Nln) is an enzyme that functions to cleave various neuropeptides. Upregulation of Nln after stroke has identified the enzyme as a critical endogenous cerebroprotective mechanism and validated target for the treatment of ischemic stroke. Overexpression of Nln in a mouse model of stroke results in dramatic improvement of stroke outcomes, while pharmacological inhibition aggravates them. Activation of Nln has therefore emerged as an intriguing target for drug discovery efforts for ischemic stroke. Herein, we report the discovery and hit-to-lead optimization of first-in-class Nln activators based on histidine-containing dipeptide hits identified from a virtual screen. Adopting a peptidomimetic approach provided lead compounds that retain the pharmacophoric histidine moiety and possess single-digit micromolar potency over 40-fold greater than the hit scaffolds. These compounds exhibit 5-fold increased brain penetration, significant selectivity over highly homologous peptidases, greater than 65-fold increase in mouse brain stability, and ‘drug-like’ fraction unbound in the brain.
Graphical Abstract
INTRODUCTION
Ischemic stroke is caused when a vessel that supplies oxygen and nutrients to the brain is blocked, thereby reducing blood flow and resulting in death of brain cells and sudden loss of function.1,2 Globally, stroke is the second leading cause of death, with a mortality rate of approximately 5.5 million people in 2016.3 A high morbidity rate in stroke survivors resulted in 116.4 million disability-adjusted life years in 2016.4 The pharmacological treatment of stroke is based on one approved biological agent, tissue plasminogen activator, which is the gold standard and highly effective but only if administered within 3–5 h of stroke onset.5 Despite significant drug discovery efforts, no new therapeutics have been approved to treat stroke in recent years.6 Recent advances in understanding signaling pathways relevant to the self-protective mechanisms of the brain have identified novel targets for further study.7
Peptidase neurolysin (Nln), a zinc metallopeptidase, has been recognized as a key constituent of an endogenous cerebroprotective mechanism functioning to protect the brain in acute neurodegenerative disorders.8,9 This notion is based on a series of recent in vitro and in vivo experimental studies which revealed upregulation of Nln following cerebral ischemia10,11 and suggested its potential protective role in response of the brain to stroke. The functional significance of Nln in the poststroke brain has been previously established using a mouse ischemic stroke model which documented aggravation of disease outcomes following small molecule inhibition of Nln after stroke.12 In a reverse experiment, which utilized viral vector-driven overexpression of Nln in the brain, a substantial attenuation of stroke outcomes, including brain infarction, neuroinflammation, vascular permeability, brain edema, and neurological impairment, was documented in animals.12 Notably, the observed cerebroprotective role of Nln has been linked to its fundamental function as a peptidase, suggesting that in the poststroke brain, Nln inactivates several cerebrotoxic neuropeptides [bradykinin (BK), neurotensin (NT), substance P, and angiotensin II] and generates at least three [angiotensin-(1–7), Met-, and Leu-enkephalins] cerebroprotective neuropeptides.12,13
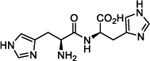
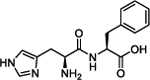
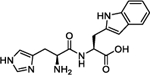
To harness this discovery and evaluate the potential of Nln as a therapeutic target for stroke and other acute neurodegenerative disorders, a structure-based discovery approach was used for rational identification of small molecules which can enhance the catalytic efficiency of Nln. While a small number of Nln inhibitors are known, no reported activators of Nln have been disclosed to the best of our knowledge. A virtual screen (VS), docking 139,725 ‘drug-like’ compounds from the NCI Developmental Therapeutics Program (DTP) Library into the hinge region of the full-length rat Nln (PDB ID: 1I1I), was performed. The top scoring 40 compounds were obtained from the DTP and screened in our developed assay. These experiments identified a number of compounds with a common histidine-containing dipeptide core as hits which enhance the activity of Nln.14–16 The top hits from this screen were purchased and rescreened in our developed isolated enzyme assay to confirm activity. Four aromatic-containing dipeptide compounds (Figure 1) possessed the ability to activate Nln based on half maximal activation concentration (A50) and maximum activation percentage (Amax). The His–Tyr dipeptide (1) possessed an A50 = 85.5 μM and Amax = 465%, the His–His compound (2) A50 = 126.5 μM and Amax = 498%, His–Phe (3) A50 = 74.5 μM and Amax = 442%, and His–Trp (4) A50 = 39.4 μM and Amax = 430%. Thorough pharmacological and biochemical/physical experiments have established the ability of these dipeptides to increase the activity of recombinant rat and human and mouse-brain isolated Nln but do not affect the activity of other peptidases closely related to Nln. These data identify valid hit compounds for further optimization as Nln activators to overcome their initial high micromolar potency and low stability.
Figure 1.
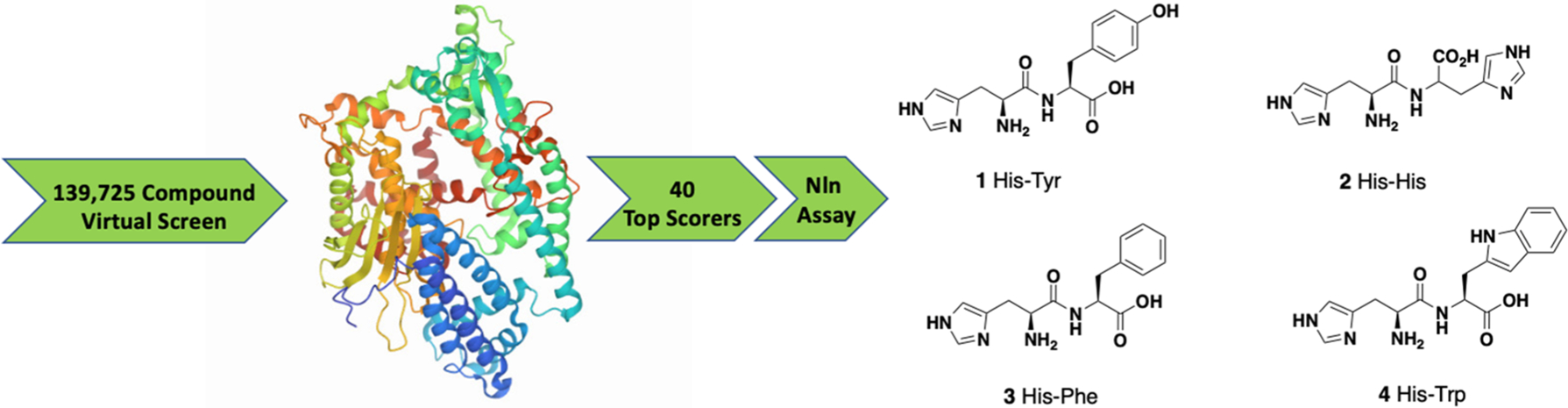
Illustrative workflow of a VS of 139,725 ‘drug-like’ compounds from the NCI Developmental Therapeutics Program (DTP) Library employing the crystal structure of Nln (PDB ID: 1I1I) identified 40 top scorers that were tested in an isolated Nln activation assay to obtain the four hit compounds depicted.
We report herein the initial structure–activity relationship (SAR) of the dipeptide hits and detail a peptidomimetic approach which affords first-in-class potent, stable, selective, brain penetrant and ‘drug-like’ small-molecule activators of Nln. These compounds represent advanced hit compounds for further study as neuroprotectants17–19 for stroke and wider neurodegenerative diseases, including Alzheimer’s disease, given the reported effects of Nln to degrade Aβ.20
RESULTS AND DISCUSSION
Chemistry.
Initial optimization of the hit dipeptides involved iterative excision of amino acid functionality to identify the minimum pharmacophore of active peptidomimetics with enhanced blood–brain barrier (BBB) penetration. A number of precautions to avoid the potential for racemization in the amide bond coupling step were employed.21 Racemization is reported to be more apparent with the use of N-acyl protecting groups at the α-amine compared with N-carbamate protection.22,23 Furthermore, the combination of the BOP coupling agent24 and Boc protection has been reported to suppress racemization in a number of syntheses.22,25 To this end, commercially available Bocprotected l- or d-histidine was coupled with an appropriately substituted primary amine in the presence of BOP coupling reagent and a base, to afford the respective amide intermediates (Scheme 1). Subsequent deprotection employing trifluoroacetic acid (TFA) yielded the targeted compounds in moderate-to-good yield. Specific rotation data obtained for selected derivatives confirmed retention of stereochemistry throughout this synthetic route when TFA concentration is limited to 20% and a 3 h reaction time is observed.
Scheme 1. Synthesis of Peptidomimeticsa.
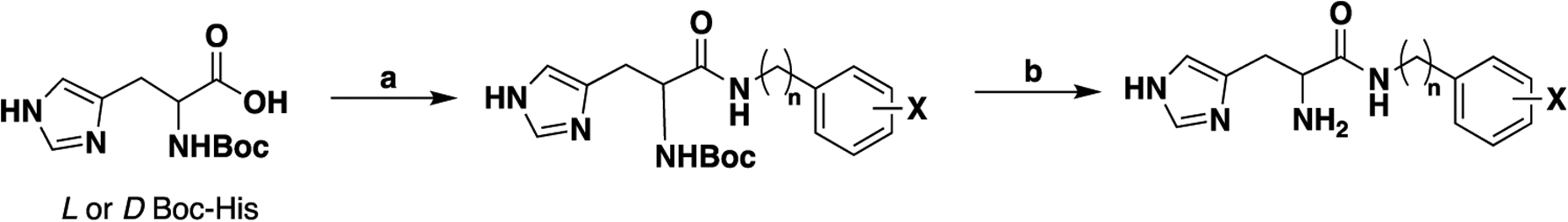
aReagents and conditions: (a) BOP, DIPEA, appropriate amine, DMF, 50 °C; (b) 20% TFA, DCM, r.t.
Structure–Activity Relationship.
Our initial studies aimed to validate the four hit dipeptides identified by our screening experiments. When these compounds were obtained from commercial sources and their structure independently confirmed by NMR, His–His compound 2 was shown to be substantially less active than the His–Tyr (1) and His–Phe (3) compounds, while His–Trp (4) was the most active. All possessed high micromolar activity (Table 1). Thus, compounds 1, 3, and 4 represented viable hit compounds for SAR studies. Characterization of each compound was performed by assignment of A50 and Amax, with 100% representing normal enzyme turnover in the latter. While these two parameters of biological activity are complementary, we have focused SAR discussions and compound development based on A50 values as these represent unambiguous evaluation criteria for drug discovery. While a 700% Amax is excellent activity alone, when combined with an A50 = 227 μM (see compound 5c), an obviously unachievable concentration in the human brain, such a compound is of little utility.
Table 1.
Structure, Nln Activation Activity, and In Silico Physicochemical Property Predictions of Monocyclic Aromatic-Containing Histidine Peptidomimetics
| Compound | Structure | A50a (μM; 95% Cl) | Amaxb {%; 95% CI) | MPOc Score | LLEd |
|---|---|---|---|---|---|
| 1 |
|
85.5 (65.1 to 112.2) |
465.2 (440.2 to 492.3) |
4 | 7.39 |
| 2 |
|
126.5 (108 to 148.3) |
498.1 (481 to 516.5) |
4 | 8.95 |
| 3 |
|
74.5 (56.2 to 98.8) |
442.3 (418.8 to 467.6) |
4.1 | 6.78 |
| 4 |
|
39.4 (27 to 57.4) |
430.3 (402.9 to 459.6) |
4.0 | 7.07 |
| 4a |
|
Inactive | Inactive | N/A | N/Ad |
| 4b |
|
Inactive | Inactive | N/A | N/A |
| 4c |
|
20.7 (10.5 to 40.8) |
337.2 (302 to 378.6) |
5 | 4.86 |
| 4d |
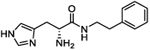
|
24.6 (17.3 to 35) |
294.6 (278.0 to 313) |
5 | 4.79 |
| 4e |
|
46 (31.5 to 68) |
339.4 (314.2 to 369.3) |
5 | 4.60 |
| 4f |
|
66.4 (48.1 to 92.7) |
378.3 (350.5 to 411.9) |
5 | 4.44 |
| 4g |
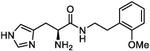
|
31.8 (21 to 48.7) |
319 (296.1 to 345.4) |
5 | 4.76 |
| 4h |
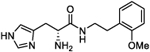
|
30 (24 to 37.5) |
328 (315.4 to 342) |
5 | 4.78 |
| 4i |
|
29.6 (22.7 to 38.8) |
367 (349.7 to 387.1) |
5 | 4.79 |
| 4j |
|
19.8 (9,5 to 40.6) |
255.3 (230.5 to 284.8) |
5 | 4.97 |
| 4k |
|
15.7 (11.6 to 22.4) |
299.4 (283.9 to 316.2) |
5 | 4.10 |
| 4l |
|
24 (17.9 to 32.2) |
330.1 (313.9 to 347.7) |
5 | 3.92 |
| 4m |
|
11.6 (5.7 to 23) |
253.8 (231.2 to 278.9) |
5 | 4.97 |
| 4n |
|
35.7 (29.3 to 43.6) |
353.8 (340.6 to 368.1) |
5 | 4.49 |
| 4o |
|
9.8 (6.3 to 15.15) |
282.4 (266.5 to 299.5) |
4 | 5.45 |
| 4p |
|
25.5 (16.7 to 38.8) |
267.5 (250.1 to 287.1) |
4 | 5.03 |
| 4q |
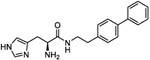
|
9.5 (6.12 to 14.6) |
258.2 (244 to 273.4) |
5 | 3.32 |
| 4r |
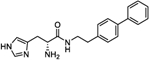
|
11.7 (8.26 to 16.7) |
269.7 (256.8 to 283.4) |
5 | 3.22 |
| 5a |
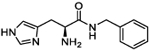
|
24.25 (7.85 to 71.4) |
152.2 (137.3 to 171.5) |
5 | 5.01 |
| 5b |
|
221 (149 to 354) |
428.8 (373 to 523.5) |
5 | 4.13 |
| 5c |
|
226.8 (146 to 392.4) |
703.5 (588.9 to 915.6) |
5 | 4.12 |
| 5d |
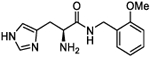
|
42.31 (32.1 to 55.6) |
385.8 (363.8 to 410.5) |
5 | 4.85 |
| 5e |
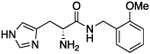
|
43.4 (30.9 to 61.5) |
324.9 (304 to 349) |
5 | 4.84 |
| 5f |
|
66.8 (51.6 to 87.1) |
327.5 (309.7 to 348.2) |
5 | 4.65 |
| 5g |
|
61.83 (46.5 to 83) |
477.1 (444 to 516) |
5 | 4.68 |
| 5h |
|
25.8 (19.5 to 34.19) |
287.7 (274.7 to 301.8) |
5 | 4.10 |
| 5i |
|
29.3 (17.1 to 48.9) |
319.1 (296.3 to 344.3) |
5 | 4.04 |
| 5j |
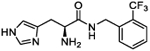
|
48.6 (31.4 to 76.7) |
362.2 (331.6 to 400.1) |
5 | 3.83 |
| 5k |
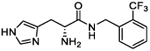
|
27.1 (17.1 to 43.3) |
345.8 (317.5 to 378.3) |
5 | 4.08 |
| 5l |
|
8.6 (4.9 to 14.9) |
270.2 (251.4 to 290.7) |
5 | 4.58 |
| 5m |
|
19.95 (11.1 to 35.4) |
282 (258.4 to 309.1) |
5 | 4.21 |
| 5n |
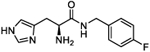
|
14.9 (10–22) |
293 (276.5–310.7) |
5 | 5.08 |
| 5o |
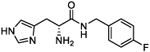
|
29.8 (20.8 to 42.9) |
320.5 (299.7 to 343.9) |
5 | 4.78 |
| 5p |
|
27.4 (17.6 to 42.81) |
310 (287.2 to 336.1) |
5 | 4.07 |
| 5q |
|
26.7 (14.9 to 46.4) |
337.1 (312 to 364.9) |
5 | 4.09 |
| 5r |
|
28.16 (22.67 to 35.03) |
287.5 (277.4 to 298.4) |
5 | 3.28 |
| 5s |
|
16.1 (9.5 to 27.1) |
251.5 (233.7 to 271.3) |
5 | 3.52 |
| 6a |
|
252 (170.6 to 406.6) |
466.6 (402.3 to 579.7) |
5 | 4.02 |
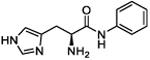
| 6b |
|
113.6 (92.8 to 140.5) |
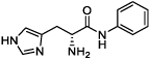
440.3 (414.9 to 470.4) |
5 | 4.37 |
| 7a |
|
20.6 (14.91 to 28.4) |
288.1 (273.6 to 303.8) |
5 | 4.49 |
| 7b |
|
21 (16 to 27.7) |
281.8 (269.9 to 294.5) |
5 | 4.48 |
| 8a |
|
126 (101 to 159) |
328 (309 to 351) |
5 | 3.17 |
| 8b |
|
27.4 (16.9 to 46.8) |
233.8 (215.9 to 254.8) |
5 | 3.84 |
Half maximal activation concentration. Values are presented as the mean of n = 4 experiments with 95% confidence intervals (CIs) in parentheses.
Maximum % activation achieved. Values are presented as the mean of n = 4 experiments with 95% confidence intervals (CIs) in parentheses.
Multiparameter optimization.
Ligand-lipophilicity efficiency.
Not applicable.
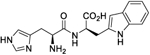
Analysis of the screening data showed that only aromatic-containing dipeptides possess activity to activate Nln. To discover potent and selective Nln activators that do not suffer from the characteristic metabolic lability of dipeptides,26–28 we adopted a peptidomimetic approach. Given the apparently conserved histidine moiety present as the Western fragment in all four hit compounds, we retained this moiety, hypothesizing it to be pharmacophoric, and focused modification on the Eastern aromatic amino acid moieties to attenuate peptide character. The tyrosine-containing compound (1) (A50 = 85.5 μM) was slightly more active than the phenylalanine-containing hit (3) (A50 = 74.5 μM) providing for the enticing prospect that phenyl ring substitution may enhance potency. The tryptophan-containing derivative (4) was the most active hit identified (A50 = 40 μM) with the indole ring suggesting that the heteroatom and/or greater aromatic character may result in higher activity.
Targeting the neuropeptidase Nln necessitates penetration of designed activators into the brain. Thus, we approximated blood–brain barrier penetration of analogues with the multiparameter optimization (MPO)29 score augmented with ligand-lipophilicity efficiency (LLE).17,30 Analysis of the MPO scores of hit compounds 1–4 (4, 4, 4.1, and 4, respectively) predict poor, or low, BBB penetrance as expected from dipeptides.31 Thus, our initial SAR strategy focused on removing the amino acid functionality that classically impedes BBB penetration, increasing lipophilicity, varying terminal aromatic ring substitution/heteroatom, and establishment of the importance of the stereocenter of the histidine amine group to determine the eutomer. Synthesized derivatives were broadly separated into two categories: substituted monocyclic aromatics (Table 1; compounds 4a–r, 5a–s, 6a–b, 7a–b, and 8a–b) and bulkier bicyclic aromatic/heteroaromatics (Table 2; compounds 9a–g, 10a–d, and 11a–b). Most derivatives possessed an MPO score of ≥4 (Tables 1, 2, and S1), validating our synthetic approach. It is worth noting that an MPO value of 5 is obtained for many derivatives due to the modifications being introduced not largely varying the underlying physicochemical properties of the target compounds. We also use LLE as a predictor of ‘drug-likeness’ (Tables 1 and 2).32 A molecule is often considered ‘drug-like’ if its LLE score exceeds five in combination with a lipophilic log P value.33,34
Table 2.
Structure, Activation Activity, and In Silico Physicochemical Property Predictions of Bicyclic Aromatic-Containing Histidine Peptidomimetics
| Compound | Structure | Asoa (μM; 95% CI) | Amaxb (%; 95% CI) | MPOc | LLEd |
|---|---|---|---|---|---|
| 4 |
|
39.4 (27 to 57.4) |
430.3 (402.9 to 459.6) |
4 | 7.07 |
| (rac)-9a |
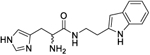
|
5.1 (1.7 to 14.2) |
197 (177.5 to 222.8) |
4.9 | N/A |
| 9b |
|
Inactive | Inactive | N/A | N/A |
| 9c |
|
Inactive | Inactive | N/A | N/A |
| 9d |
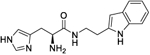
|
6.3 (4.3 to 9.2) |
252 (238 to 267) |
4.9 | 5.41 |
| 9e |
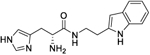
|
1.0 (0.16 to 4.4) |
132 (122.5 to 143.6) |
4.9 | 6.19 |
| 9f |
|
13.6 (10.1 to 18.2) |
323.7 (310.3 to 337.9) |
5 | 3.87 |
| 9g |
|
25.2 (19.6 to 32.4) |
339.8 (325.4 to 355.4) |
5 | 3.61 |
| 10a |
|
4.4 (1.7 to 11) |
210.9 (191.7 to 231.9) |
5 | 4.58 |
| 10b |
|
6.3 (3.8 to 10.7) |
197.5 (184.9 to 214.8) |
5 | 4.42 |
| 10c |
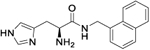
|
4.2 (3.17 to 5.6) |
264 (256.3 to 272.6) |
5 | 4.60 |
| 10d |
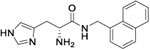
|
15.37 (10.9 to 21.5) |
343 (325.2 to 363.5) |
5 | 4.04 |
| (rac)-10c/d |
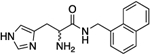
|
29.9 (23.5 to 38.1) |
370.6 (354.6 to 388.0) |
5 | N/A |
| 11a |
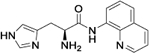
|
7.0 (4.9 to 9.9) |
281.2 (266.7 to 297.1) |
4.9 | 5.07 |
| 11b |
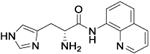
|
6.2 (4.5 to 8.5) |
288.3 (274.3 to 303.3) |
4.9 | 5.12 |
| (rac)-11a/b |
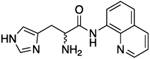
|
5.8 (3.3 to 10.1) |
349 (322.8 to 377.2) |
4.9 | N/A |
Half maximal activation concentration. Values are presented as the mean of n = 4 experiments with 95% confidence intervals (CIs) in parentheses.
Maximum % activation achieved. Values are presented as the mean of n = 4 experiments with 95% confidence intervals (CIs) in parentheses.
Multiparameter optimization.
Ligand-lipophilicity efficiency.
Not applicable.
To better understand the pharmacophoric requirements for Nln binding, compound 4a (H-Tyr-His-OH), the reverse analogue of 1, was purchased and its structure was confirmed by NMR. This reversal led to the complete amelioration of Nln activation, suggesting that histidine linked via its C-terminus is crucial for the activation of Nln. The pharmacophoric role of the primary amine of the histidine moiety was further confirmed when the derivative containing a Boc-protected carbamate at this position (4b) (Table 1) was found to be inactive. Excision of this amine, illustrated by tryptophan derivative (9c, Table 2), also results in complete amelioration of activity. Thus, a primary amine or at least a hydrogen-bond acceptor (given the elimination of hydrogen-bond acceptance ability due to the carbamate resonance structure) is required at this position of the compound for Nln activation. These observations support a critical pharmacophoric role for the terminal histidine moiety.
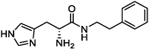
Excision of the carboxylic acid moiety from the His–Phe hit (3) to afford derivative 4c significantly improved A50 from 74.5 to 20.7 μM while reducing Amax from 442 to 337%. Moreover, the MPO score increased to 5, indicating BBB penetration, albeit with a decreased LLE value of 4.86. This observation indicated that it was possible to obtain more active compounds from this hit series (based on A50) and that further SAR studies were warranted. Given the apparent pharmacophoric nature of the free amine of the histidine, we next examined the impact of stereochemistry at this position of the molecule. The unnatural dipeptide 4d containing d-histidine, opposite to the l-histidine present in 4c, was synthesized and found to be equipotent within 95% confidence limits with an A50 of 24.6 μM and 20.7 μM and an Amax of 295 and 337%, respectively. This trend was repeated with most of the monocyclic derivatives with no eutomer identifiable for A50 activity. However, the p-nitro compounds l-4o and d-4p showed approximately twofold difference in A50 values (9.8 and 25.5 μM, respectively) but with equipotent Amax. The MPO score of both of these compounds (4o and 4p) is much lower than that compared to other derivatives. Compound d-5o possessed approximately twofold greater activity than l-5n with A50 values of 30 and 15 μM, respectively, but with similar Amax values. Within the aniline derivatives, l-6a possessed much lower activity compared with d-6b (A50 = 252 and 114 μM, respectively) but comparable Amax (467 and 440%, respectively). The trend of no correlation between activity and stereochemistry was held in the bicyclic derivatives with the exception of the naphthyl compounds l-10c and d-10d possessing A50 values of 4.2 and 15 μM and Amax values of 264 and 343%, respectively. Indeed, a racemic mixture (rac)-10c/d exhibited an A50 = 29.9 μM and an Amax of 370.6%, midline values largely within confidence intervals of the two enantiomers. Thus, while no eutomer has been identified, we deemed it prudent in these initial SAR studies to continue to synthesize both enantiomers of these compounds given the potential fourfold difference in activity identified.
We next conducted a substituent scan around the terminal phenyl ring employing an electron-donating methoxy group. The para position (l-4e and d-4f) showed the least amount of activity with A50 values of 46 and 66 μM, respectively. When the methoxy was moved to the ortho position (l-4g and d-4h), activity increased to afford equipotent compounds with a mean A50 of 31 μM. A similar activity was seen when substitution was moved to the meta position (l-4i and d-4j) with A50 values of 30 and 20 μM, respectively. When the substituent was switched to a para CF3 electron-withdrawing group (4k and 4l), activity was substantially increased with 4k possessing an A50 = 16 μM and an Amax of 299%, with the opposite enantiomer 4l being equipotent within confidence limits. A less electron-withdrawing fluorine substituent at the para position of the terminal phenyl ring (4m and 4n) affords a similar activity with 4m possessing an A50 = 12 μM and an Amax of 254%. Introduction of a para nitro group (4o and 4p) increased activity with l-4o possessing an A50 = 10 μM and an Amax of 282%, approximately double the activity of its enantiomer. Notably, both 4o and 4p possessed LLE values greater than 5 but with lower MPO scores of 4. Finally, for this series, a second phenyl ring was added to the para position of the terminal phenyl to afford compounds 4q and 4r which possessed some of the most potent A50 values identified to date, 9.5 and 11.7 μM, respectively, with equipotent Amax values of 258 and 269%. However, the lipophilicity of the biphenyl moiety reduces the LLE of these compounds to 3.3 and 3.2, respectively.
Cognizant of the potential metabolic lability of the amide bond within this scaffold, we next investigated a truncated homologation series wherein the ethyl linker to the terminal aromatic ring was reduced to a single methylene, thus increasing steric hindrance around the amide bond. Gratifyingly, benzyl derivative 5a (A50 = 24 μM, Amax = 152%) possessed a similar activity to its phenethyl counterpart 4c (A50 = 20.7 μM, Amax = 337%), albeit with lower Amax. Performing a substituent scan again with a methoxy group, activity increased in the order of ortho > meta ≫ para. Compound 5e (o-OMe) possesses an A50 = 43.4 μM and Amax = 325%, compound 5g (m-OMe) an A50 = 62 μM and Amax = 477%, and compound 5c (p-OMe) an A50 = 227 μM and Amax = 704%. This pattern highlights a significant difference from the phenethyl series wherein all positions were largely equipotent. The electron-withdrawing CF3 group afforded greater activity in the phenethyl series and so we conducted a substituent scan with this group in the benzyl series (compounds 5h–5m). Here, activity increased in the order of meta > para > ortho. Compound 5l (m-CF3) possesses an A50 = 8.6 μM and Amax = 270%, compound l-5h (p-CF3) an A50 = 26 μM and Amax = 288%, and compound 5j (o-CF3) an A50 = 49 μM and Amax = 362%. Fluorine substitution at the para position (l-5n) is equipotent, within confidence limits, with CF3 substitution, as is the p-OCF3 derivative l-5p. Introduction of a p-SCF3 to afford l-5r retains a similar activity to l-5h; however, in this case, the opposite enantiomer (d-5s) shows slightly improved activity with A50 = 16 μM and Amax = 251%.
Further homologation to completely excise the linker afforded arylamide l-6a possessing an A50 = 252 μM and Amax = 467% with an MPO score predictive of BBB penetration (5) and an LLE of 4 (Table 1). It would appear that stereochemistry has a larger role to play in activity as d-6b possesses an A50 = 114 μM and Amax = 440%, more than twofold more potent than l-6a. Extension of the linker to a propyl chain (7a) had no effect on activity compared with phenethyl (4c) and benzyl (5a) linkers. Introduction of a butyl linker (8a) significantly reduced activity (A50 = 126 μM and Amax = 328%) with the l enantiomer, but the opposite enantiomer (d-8b, A50 = 27.4 μM) retained equipotent activity to 4c and 5a. Thus, a linker length of between 1 and 3 carbons is optimal for activity, suggesting that the compounds occupy a binding pocket within Nln of limited space.
The tryptophan-containing compound (4) was the most active of the four hits and bicyclic compounds further increase steric hindrance around the amide bond, potentially affording greater stability. Derivative synthesis (Table 2) confirmed the observations from the monocyclic series; excision of the carboxylic acid moiety to afford racemate ((rac)-9a) enhances activity (A50 = 5.1 μM) and carbamate protection of the free amine (9b) ameliorates activity, as does excision of the free amine (9c), further supporting a pharmacophoric role for the histidine moiety in Nln activation. As the carboxylic acid-excised compound was initially obtained as a racemic mixture, we wished to explore the effect of stereochemistry on activity and thus, enantioselective synthesis resolved the two stereo-isomers to afford l-9d (A50 = 6.3 μM and Amax = 252%) and d-9e (A50 = 1.0 μM and Amax = 132%), significantly more potent than the parent hit and possessing ‘drug-like’ LLE scores and predictive BBB penetration by MPO scores of 4.9. Notably, the racemate was equipotent within the 95% confidence intervals to the two single enantiomers, suggesting that metabolic racemization would not significantly affect activity.
Based on these data, we next looked to excise the heteroatom in the bicyclic ring and synthesized the 2-ethylnaphthalene derivatives l-9f and d-9g, but this modification resulted in attenuation of activity with A50 values of 13.6 and 25.2 μM, respectively, suggesting a role in binding the target protein for the indole nitrogen. Following the observation that truncation of the linker chain results in retained activity, we synthesized 2-methylnaphthalene derivatives l-10a and d-10b which possessed A50 values of 4.4 and 6.3 μM, respectively. A positional switch to the 1-methylnaphthalene derivatives l-10c and d-10d did not appreciably alter activity (A50 = 4.2 and 15.4 μM, respectively). Excision of the linker and reintroduction of a nitrogen to afford 8-quinoline derivatives l-11a and d-11b afforded two of the most active derivatives to date (A50 = 7.0 and 6.2 μM, respectively). Furthermore, these compounds possess mean Amax values of 285% combined with predicted BBB penetration by an MPO score, ‘drug-like’ LLE values above 5, and enhanced stability of the amide bond given the steric congestion they afford. Likewise, the racemate (rac)-11a/b possessed equipotent A50 values to both single enantiomers within confidence intervals.
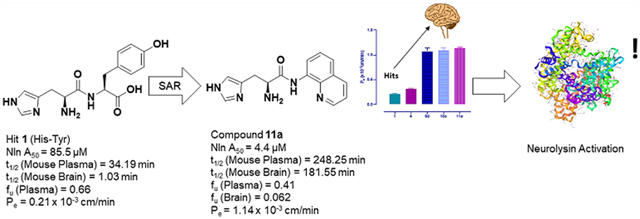
In summary, a histidine amino acid forming an amide bond with its C-terminus, possessing a free amine, forms part of the pharmacophore of this scaffold. However, stereochemistry at the amine is unimportant. Excision of the carboxylic acid from the hit scaffolds retains activity, while enhancing BBB penetration and truncation of the linker chain from the amide to the terminal aromatic moiety improves activity (Figure 2). We have identified numerous compounds exemplified by 4o, 4q, 5l, 9d, 10c, and 11a–b, with low micromolar A50 values to activate neurolysin, combined with high Amax values, ‘drug-like’ LLE values, and enhanced stability (see below).
Figure 2.
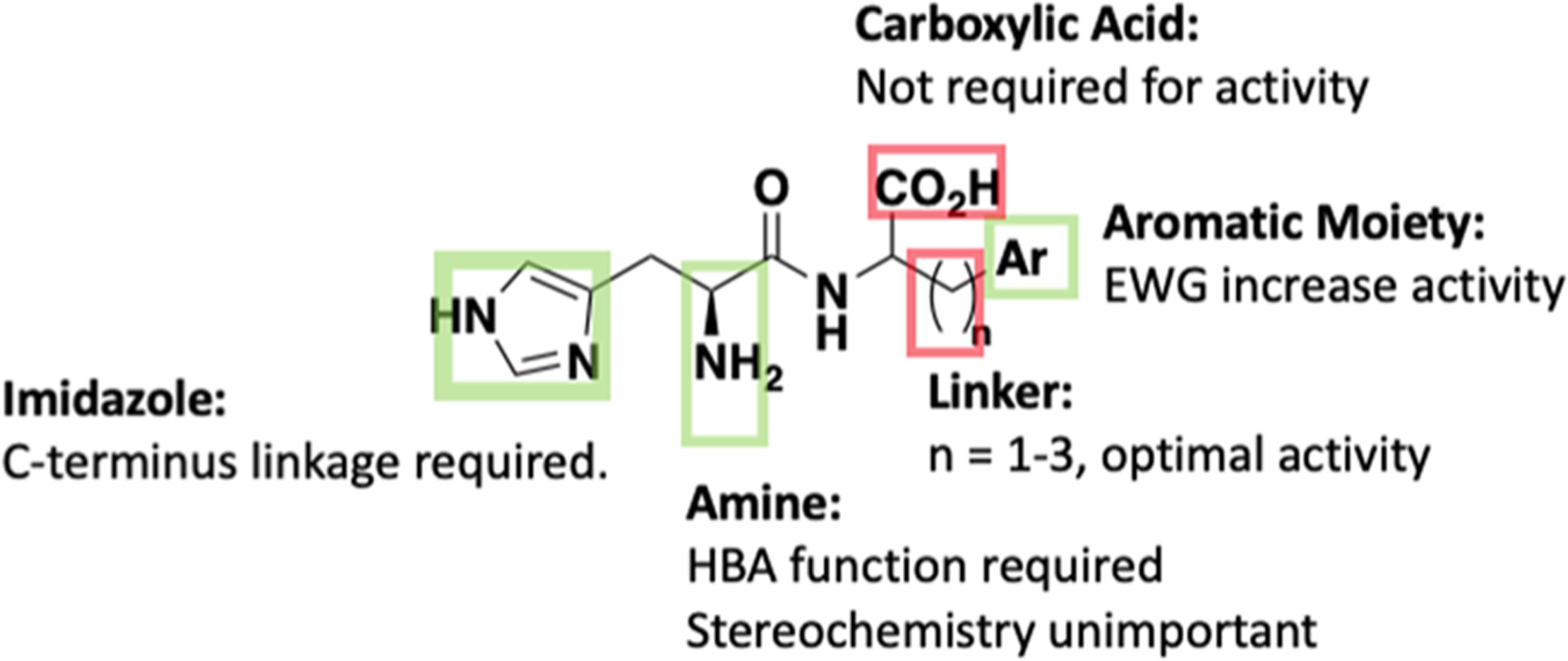
Overview of established SAR for Nln activation. Green box, histidine including the free amine (but not the stereocenter) and an aromatic terminus are pharmacophoric. Red box, the carboxylic acid and ethyl linker between the amide and terminal aromatic moiety are not required for activity.
In Vitro Metabolic Stability.
The hit His–Tyr (1) and His–Trp (4) scaffolds, along with improved derivatives 9d, 10c, and 11a, were selected to undergo in vitro plasma and brain homogenate stability determination due to their favorable combination of low micromolar A50 values and Amax values of >250% (Table 3). In vitro half-life (t1/2) values, defined as the time needed for 50% degradation of the compound, were calculated by assuming pseudo-first-order degradation. Dipeptide compound 1 possessed a half-life of 34 min in mouse plasma as expected for the labile amide bond. However, the more sterically hindered amide bond within the His–Trp hit compound 4 possessed a half-life of >300 min in mouse plasma, validating our SAR strategy to introduce increased steric hindrance around the amide bond through bicyclic aromatic moieties and truncating the linker chain. Gratifyingly, the carboxylic acid-excised derivative, 9d, and the 1-methylnaphthalene derivative, 10c, show significantly enhanced half-lives in mouse plasma of >1000 min. The quinoline derivative, 11a, showed a more moderate mouse plasma half-life of 248 min, superior to 1 and similar to 4, perhaps indicating a role for the quinoline nitrogen in intramolecular hydrogen-bonding. Both dipeptide hits showed particular instability in mouse brain homogenate with half-lives below 1.6 min. Derivatives 9d, 10c, and 11a possessed significantly improved stability in mouse brain homogenate by at least 41-fold and, in the case of 11a, up to 117-fold, with half-lives of 66, 80, and 182 min, respectively.
Table 3.
Half-Lives of Selected Nln Activators in Mouse Blood Plasma and Brain Homogenate Illustrate Increased Stability for Peptidomimetic Compounds 9d, 10c, and 11aa
| compound | A50 (μM; 95% CI) | t1/2 (min) mouse plasma | t1/2 (min) mouse brain |
|---|---|---|---|
| 1 (His–Tyr) | 85.5 (65.1 to 112.2) | 34.19 ± 1.97 | 1.03 ± 0.4 |
| 4 (His–Trp) | 39.4 (27 to 57.4) | >300 | 1.55 ± 0.37 |
| 9d | 6.3 (4.3 to 9.2) | >1000 | 65.5 ± 4.2 |
| 10c | 4.2 (3.17 to 5.6) | >1000 | 80.3 ± 2 |
| 11a | 7.0 (4.9 to 9.9) | 248.25 ± 28.1 | 181.55 ± 14.9 |
Data expressed as the mean of three experiments ± SD.
Plasma and Brain Protein Binding.
The rapid equilibrium dialysis (RED) device was used to investigate the extent of plasma and brain protein binding and calculate the fraction unbound of selected Nln activators (Table 4). His–Trp hit compound 4 has the highest unbound fraction in mouse plasma, whereas hit compound 1 has more protein binding affinity (less fu) compared with 4. Indole compound 9d showed a similar fraction unbound to hit compound 1 in plasma; however, derivatives 10c and 11a have lower unbound fractions in plasma.
Table 4.
Fraction Unbound ( f u ) in Mouse Plasma and Brain Homogenate of the Selected Nln Activators a
| compound | A50 (μM; 95% CI) | fu in plasma | fu in brain |
|---|---|---|---|
| 1 (His–Tyr) | 85.5 (65.1 to 112.2) | 0.66 ± 0.08 | N.D.b |
| 4 (His–Trp) | 39.4 (27 to 57.4) | 0.93 ± 0.13 | N.D. |
| 9d | 6.3 (4.3 to 9.2) | 0.63 ± 0.076 | 0.14 ± 0.012 |
| 10c | 4.2 (3.17 to 5.6) | 0.45 ± 0.013 | 0.052 ± 0.003 |
| 11a | 7.0 (4.9 to 9.9) | 0.41 ± 0.081 | 0.062 ± 0.011 |
Data expressed as the mean of three experiments ± SD.
Not determined.
Due to the metabolic instability of the hit compounds 1 and 4 in brain homogenate (Table 3), brain protein binding assay was not performed for these compounds. Similar to plasma, derivative 9d showed a higher fraction unbound in brain tissue compared with the other two derivatives, which have almost the same values for fu in brain tissue.
In Vitro Blood–Brain Barrier Permeability.
To determine blood–brain barrier permeability, compounds 1, 4, 9d, 10c, and 11a were added to the luminal compartment of an established co-culture system and their progressive transfer through the cells was monitored. As seen in Figure 3, a significant increase in the apical to basolateral transport of peptidomimetic activators of Nln compared to that of the dipeptide hit compounds 1 and 4 was observed (Figure 3). The Pe values for peptidomimetic derivatives 9d, 10c, and 11a (1.07 ± 0.12 × 10−3, 1.09 ± 0.09 × 10−3, and 1.14 ± 0.02 × 10−3 cm/min) were 4–5-fold higher than permeability values of hit compounds 1 and 4 (0.21 ± 0.01 × 10−3 and 0.32 ± 0.03 × 10−3 cm/min) indicating substantially increased BBB penetration.
Figure 3.
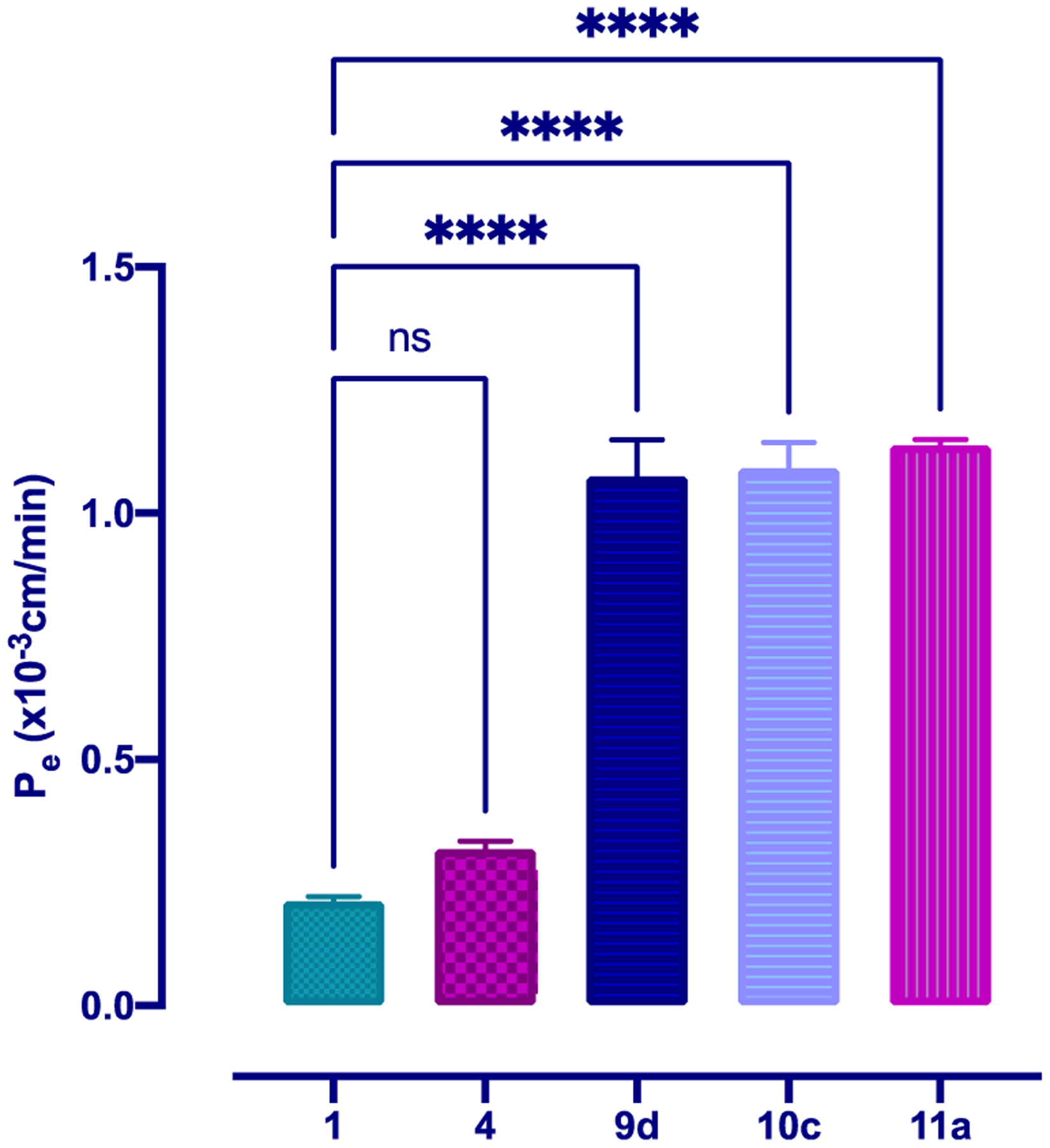
Apical to basolateral transport of Nln activators at 10 μg/mL across an in vitro co-culture model of the BBB at 37 °C illustrates the increased brain permeability of peptidomimetic compounds 9d, 10c, and 11a. The permeability coefficient (Pe) was calculated from the cleared volume of each compound versus time. Values represent the mean ± SD of 3–4 measurements. Statistics: One-way ANOVA followed by Dunnett’s test for comparison of multiple groups with compound 1, p < 0.05 was used as the statistical significance level.
Peptidase Selectivity.
The effect of selected derivatives 9d, 10c, and 11a to inhibit the highly related peptidases thimet oligopeptidase (TOP), neprilysin (NEP), angiotensin-converting enzyme 2 (ACE2), and ACE, which together with Nln belong to the same family of enzymes, was determined in an eight-point concentration–response experiment (Figure 4). No appreciable inhibition of TOP was encountered for 11a and 10c, while 9d only exhibited inhibition at concentrations approaching 300 μM (Figure 4, panels a–c). None of the three compounds showed appreciable inhibition of NEP (Figure 4, panels d–f). Compounds 9d and 11a showed inhibition of ACE2 at high concentrations, while 10c showed no inhibition up to 300 μM (Figure 4, panel h). Similarly, 9d and 11a showed inhibition of ACE at high concentration, while 10c showed no inhibition at the maximum concentration tested (Figure 4, panels j–l). These data demonstrate the high selectivity of our developed peptidomimetic neurolysin activators, providing no detectable activation of TOP, NEP, ACE, or ACE2 and inhibition activity only at high micromolar concentration.
Figure 4.
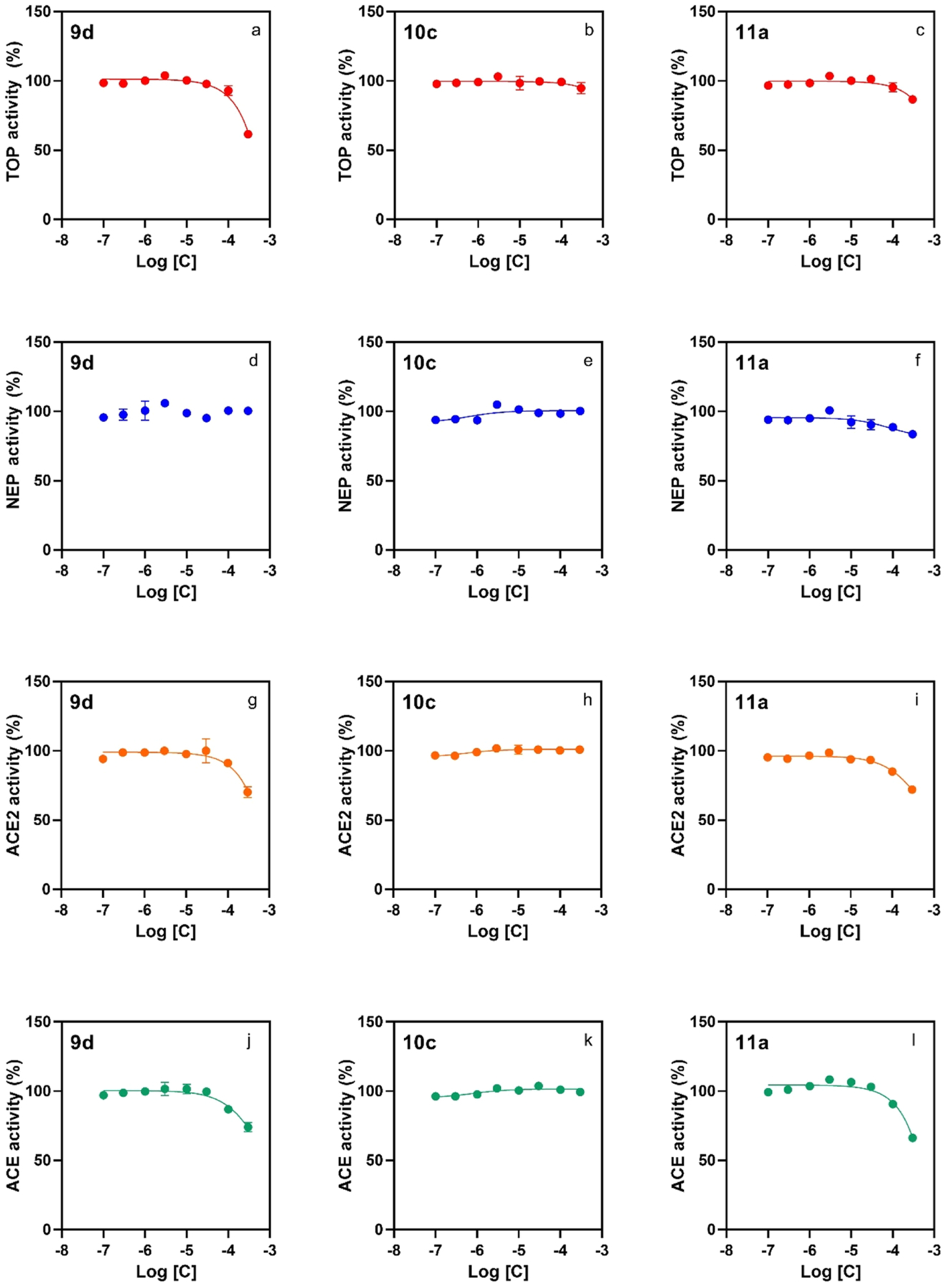
Peptidomimetic compounds 9d, 10c, and 11a have a low effect on the catalytic activity of human recombinant peptidases, illustrating selectivity toward activation of Nln over the related peptidases TOP, NEP, ACE2, and ACE. All panels document concentration-dependent effect of the indicated compounds on hydrolysis of a respective quenched fluorescent substrate (n = 4, mean ± SD): Mca-Pro-Leu-Gly-Pro-D-Lys(DNP)-OH at 15 μM for TOP (panels a–c), Mca-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys(Dnp)-OH at 10 μM for NEP (panels d–f), Mca-Ala-Pro-Lys-(Dnp)-OH at 10 μM for ACE2 (panels g–i), and angiotensin-converting enzyme (ACE; panels j–l). In all panels, the initial velocity of the hydrolysis in the absence of either compound corresponds to 100% on the vertical axis.
We next determined the likely interference of 9d, 10c, and 11a on fluorescence intensity of the hydrolytic product of QFS used in the Nln activation assays to rule out false-positive/negative data. The results of these experiments indicated that compounds 10c and 11a did not interfere with the fluorescence signal under identical experimental conditions used in our assays; however, compound 9d possessed fluorescence properties (Figure S1). Notably, this property of compound 9d was taken into consideration during our data analysis to calculate A50 and Amax values.
Hydrolysis of the Endogenous Substrate.
To verify if our observations with the synthetic substrate also translate to endogenous substrates of Nln, we evaluated the effect of 9d, 10c, and 11a on hydrolysis of BK and NT [i.e., formation of BK-(1–5) and NT-(1–10)] by Nln. The results of these experiments show the concentration-dependent effect of all compounds on the formation of BK-(1–5) confirming enhanced hydrolysis of BK in the presence of these activators (Figure 5A). Similarly, hydrolysis of NT by Nln was also enhanced in the presence of all three compounds in a concentration-dependent manner (Figure 5B).
Figure 5.
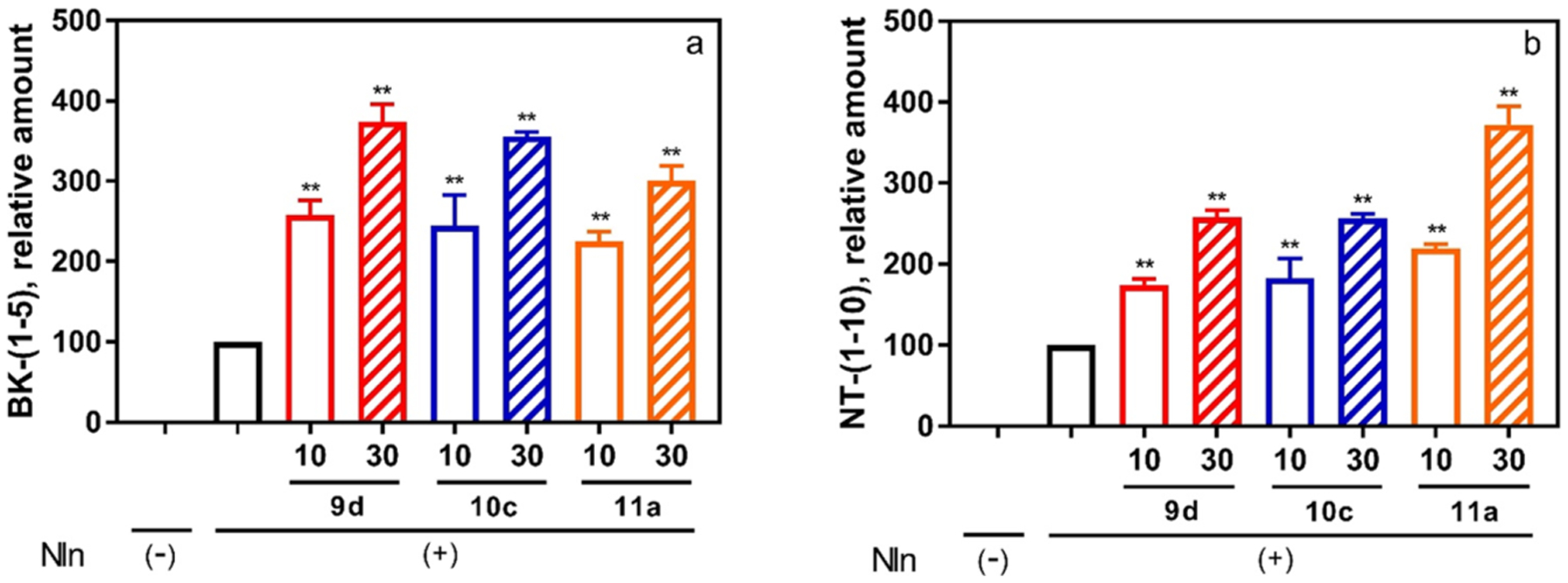
Effect of peptidomimetic compounds 9d, 10c, and 11a on the hydrolysis of Nln substrates BK and NT illustrates that activation of Nln is not an artifact of the synthetic substrate and translates to the natural system. Rat recombinant neurolysin was incubated with one of the endogenous peptides (20 μM) in the absence or presence of the test compounds (10 or 30 μM). The formation of (A) bradykinin-(1–5) (BK-(1–5)) and (B) neurotensin-(1–10) (NT-(1–10)) was documented by mass spectrometric analysis (n = 3, mean ± SD are presented; **, p < 0.001 compared to Nln-alone condition). In both panels, Nln(−) corresponds to a condition where the respective peptide substrate was incubated in assay buffer alone (i.e., no Nln, hence no product formation). Likewise, Nln(+) corresponds to a condition where the recombinant peptidase was present in the assay buffer.
CONCLUSIONS
Herein, we performed SAR studies around the aromatic side chain of four dipeptide hit compounds identified from a VS as the first scaffolds to possess Nln activation activity, albeit at a high micromolar level. The histidine moiety was found to be pharmacophoric, particularly around the free amine. Excision or carbamate protection of the amine results in complete loss of activity suggesting that a hydrogen-bond acceptor functionality is required at this position. No eutomer was identified with R-, S-, and racemic compounds affording a similar activity within 95% confidence intervals. A 1–3 carbon linker between the histidine C-terminus and aromatic moiety is optimal for activity, while a terminal aromatic moiety is pharmacophoric and tolerant of both EWGs and EDGs at varied positions along with heteroatom incorporation.
The presented SAR studies resulted in identification of peptidomimetic compounds 9d, 10c, and 11a which possess >10-fold increase in potency over the most active hit and >30-fold over the least active hit, greater than 65-fold increase in mouse brain stability, significant selectivity for activation of Nln over the four highly homologous peptidases TOP, NEP, ACE2, and ACE, 5-fold increased brain penetration, and ‘drug-like’ fraction unbound in the brain. Furthermore, these compounds were confirmed to enhance the activity of Nln to the hydrolysis of the endogenous substrates BK and NT, illustrating that activation was not an artifact of, or limited to, the synthetic substrate.
These compounds further validate the hits identified from the VS and our strategy to pursue first-in-class small-molecule activators of Nln. We are working to further determine SAR around the histidine fraction and the amide bond. The presented data strongly support this promising scaffold for further optimization to develop possible therapeutics for a range of neurological disorders.
EXPERIMENTAL SECTION
General Synthetic Procedures.
Solvents and reagents of commercial grade were purchased from Fisher Scientific, VWR, or Sigma-Aldrich and were used without additional purification. All reactions were performed in oven-dried flasks under a nitrogen atmosphere. Reaction progress was monitored using thin-layer chromatography (TLC) on aluminum-backed 20 μm silica plates supplied by Silicycle (TLA-R10011B-323) and visualized by UV (254 nm) or staining agent (ninhydrin solution, phosphomolybdic acid or iodine vapor). Flash column chromatography was performed on silica gel (40–63 μm, 60 Å) with the indicated mobile phase. Specific rotations of enantiomers were measured at 589 nm with a LAXCO polarimeter model Pol-301. The volume of the cell was 11 mL, and the path length was 1.0 dm. NMR spectrometric analysis was carried out using the indicated solvent on a Bruker AVANCE III HD spectrometer at 400 or 500 MHz for proton (1H) and 100 or 126 MHz for carbon (13C), respectively. Chemical shifts (δ) are recorded in parts per million (ppm) and reported relative to solvents; coupling constants (J) are reported in hertz (Hz). Splitting of signal peaks is indicated by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). High-resolution mass spectrometry (HRMS) was carried out on an Agilent 1200 time-of-flight mass spectrometer equipped with an electrospray ionization source. High-performance liquid chromatography (HPLC) was performed on an Agilent 1220, equipped with a 254 nm UV detector (VWD), employing a Phenomenex C18, Polar-RP column (4 μm, 250 × 4.6 mm), or RP column (5 μm, 250 × 4.6 mm). Purifications were performed using methanol: water (0.05% TFA) as the mobile phase. The purity of all final compounds was determined as ≥95%, unless otherwise specified.
General Procedure A (Synthesis of Boc-Protected Intermediates).
Under an inert atmosphere, Boc-L or d-histidine (1.1 mmol), BOP (1 mmol), and DIPEA (2 mmol) were suspended in DMF (6 mL), and the mixture was heated to 50 °C and stirred for 1 h. In a separate vessel, the respective amine (1 mmol) was dissolved in DMF (4 mL) by stirring at room temperature for 30 min. The amine solution was then added to the Boc-histidine solution and stirred overnight at 50 °C. The reaction mixture was cooled, water was added and then the mixture was extracted with ethyl acetate and washed with saturated aqueous sodium bicarbonate and brine. The organic phase was collected, dried over anhydrous Na2SO4, and concentrated in vacuo. The desired Boc-protected intermediate was then isolated from the crude extract by flash column chromatography (mobile phase: 0–20% MeOH in DCM).
General Procedure B (Boc Deprotection).
The Boc-protected intermediate was dissolved in 20% TFA in DCM (10 mL) and stirred at room temperature for 3 h. The solvent was evaporated in vacuo. DCM (20 mL) was added to the residue, perturbed, and then evaporated (×3). The residue was dissolved in ethyl acetate (50 mL) and basified (NaOH/NaHCO3 aqueous solution) to pH 7, and the organic layer was separated, dried over anhydrous Na2SO4, concentrated in vacuo, and purified by flash column chromatography using MeOH/DCM as the mobile phase.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-phenethylpropanamide (4c).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (71%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.74–2.80 (m, 3H), 2.92–2.97 (m, 1H), 3.37–3.47 (m, 2H), 3.55–3.59 (m, 1H), 6.86 (s, 1H), 7.18–7.21 (m, 3H), 7.26–7.30 (m, 2H), 7.62 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 32.10, 35.09, 40.43, 54.82, 116.91, 125.99, 128.12, 128.38, 133.55, 134.99, 138.99, 174.53. HRMS (ESI): calcd for C14H19N4O [M + H]+, 259.1553; found, 259.1560.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-phenethylpropanamide (4d).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (62%). Rf = 0.12 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.78–2.83 (m, 2H), 3.19–3.30 (m, 2H), 3.41–3.59 (m, 2H), 4.18 (t, J = 6.8 Hz, 1H), 7.21–7.31 (m, 6H), 8.79 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.45, 34.75, 40.66, 52.00, 117.95, 126.13, 127.15, 128.18, 128.34, 134.44, 138.67, 167.16. HRMS (ESI): calcd for C14H19N4O [M + H]+, 259.1553; found, 259.1560.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-methoxyphenethyl)-propanamide (4e).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (59%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.71–2.76 (m, 2H), 3.19–3.29 (m, 2H), 3.35–3.38 (m, 2H), 3.76 (s, 3H), 4.17 (t, J = 6.8 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 7.31 (s, 1H), 8.80 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.48, 33.93, 40.90, 51.97, 54.25, 113.58, 117.93, 127.19, 129.29, 130.55, 134.47, 158.45, 167.10. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1665.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-methoxyphenethyl)-propanamide (4f).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (60%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.74–2.78 (m, 2H), 3.23–3.32 (m, 2H), 3.42–3.54 (m, 2H), 3.79 (s, 3H), 4.17–4.20 (m, 1H), 6.86–6.88 (m, 2H),7.15 (d, J = 8.23 Hz, 2H), 7.36 (s, 1H), 8.89 (d, J = 1.28 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 24.65, 32.24, 39.22, 50.23, 52.57, 111.90, 116.40, 125.19, 127.61, 128.87, 132.69, 156.77, 165.37. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1659.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-methoxyphenethyl)-propanamide (4g).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (61%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.74–2.85 (m, 2H), 3.19–3.31 (m, 2H), 3.38–3.45 (m, 1H), 3.50–3.57 (m, 1H), 3.84 (s, 3H), 4.15 (t, J = 6.87 Hz, 1H), 6.84–6.89 (m, 1H), 6.95 (d, J = 8.05 Hz, 1H), 7.11 (dd, J = 1.65, 7.39 Hz, 1H), 7.21 (td, J = 1.50, 7.83 Hz, 1H), 7.31 (d, J = 0.90 Hz, 1H), 8.82 (d, J = 1.31 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.43, 29.81, 39.21, 51.97, 54.39, 110.19, 117.95, 120.12, 126.53, 127.10, 127.71, 129.98, 134.45, 157.68, 166.98. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1655.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-methoxyphenethyl)-propanamide (4h).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (63%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.74–2.85 (m, 2H), 3.20–3.29 (m, 2H), 3.37–3.45 (m, 1H), 3.50–3.57 (m, 1H), 3.84 (s, 3H), 4.16 (t, J = 6.87 Hz, 1H), 6.86 (td, J = 0.88, 7.40 Hz, 1H), 6.94 (d, J = 8.02 Hz, 1H), 7.11 (dd, J = 1.60, 740 Hz, 1H), 7.21 (td, J = 1.45, 7.83 Hz, 1H), 7.32 (s, 1H), 8.85 (d, J = 1.22 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.35, 29.80, 39.20, 51.95, 54.39, 110.19, 118.04, 120.12, 126.55, 126.94, 127.71, 130.00, 134.40, 157.69, 166.99. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1660.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-methoxyphenethyl)-propanamide (4i).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (65%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.75–2.81 (m, 2 H), 3.19–3.28 (m, 2 H), 3.41–3.48 (m, 1 H), 3.56 (dt, J = 7.55, 13.44 Hz, 1H), 3.78 (s, 3 H), 4.16 (t, J = 6.85 Hz, 1 H), 6.77–6.80 (m, 3 H), 7.21 (dd, J = 7.32, 8.89 Hz, 1H), 7.31 (d, J = 0.69 Hz, 1H), 8.81 (d, J = 1.28 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.48, 34.79, 40.57, 51.96, 54.21, 111.43, 114.12, 117.88, 120.64, 127.18, 129.19, 134.50, 140.17, 159.94, 167.08. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1659.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-methoxyphenethyl)-propanamide (4j).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (63%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.76–2.81 (m, 2H), 3.19–3.29 (m, 2H), 3.41–3.48 (m, 1H), 3.52–3.57 (m, 1H), 3.78 (s, 3H), 4.16 (t, J = 6.85 Hz, 1H), 6.77–6.80 (m, 3H), 7.19–7.23 (m, 1H), 7.31 (d, J = 0.68 Hz, 1H), 8.81 (d, J = 1.23 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.42, 34.79, 40.57, 51.91, 54.19, 111.42, 114.09, 117.94, 120.63, 127.03, 129.19, 134.49, 140.17, 159.93, 167.07. HRMS (ESI): calcd for C15H21N4O2 [M + H]+, 289.1659; found, 289.1661.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethyl)-phenethyl)propanamide (4k).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (49%). Rf = 0.17 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.91 (t, J = 7.2 Hz, 2H), 3.21–3.27 (m, 2H), 3.54 (t, J = 7.4 Hz, 2H), 4.18 (t, J = 7.0 Hz, 1H), 7.40–7.44 (m, 3H), 7.61 (d, J = 8.0 Hz, 2H), 8.88 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.34, 34.59, 40.29, 51.88, 118.11, 125.00, 125.04, 126.90, 129.07, 134.51, 143.39, 167.15. HRMS (ESI): calcd for C15H18F3N4O [M + H]+, 327.1427; found, 327.1595.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethyl)-phenethyl)propanamide (4l).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (46%). Rf = 0.16 (MeOH/DCM = 1:9). 1H NMR (400 MHz, DMSO-d6): δH 2.77–2.81 (m, 2H), 3.03–3.16 (m, 2H), 3.32–3.39 (m, 1H), 3.40–3.45 (m, 1H), 4.04 (t, J = 6.6 Hz, 1H), 7.37 (s, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.65 (d, J = 8.0 Hz, 2H), 8.58 (t, J = 5.6 Hz, 1H), 8.90 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δC: 27.11, 34.92, 51.88, 118.10, 125.60, 125.64, 129.95, 135.01, 144.48, 167.65. HRMS (ESI): calcd for C15H18F3N4O [M + H]+, 327.1427; found, 327.1433.
(S)-2-Amino-N-(4-fluorophenethyl)-3-(1H-imidazol-4-yl)-propanamide (4m).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (61%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.79 (t, J = 7.4 Hz, 2H), 3.14–3.25 (m, 2H), 3.41–3.54 (m, 2H), 4.19 (t, J = 7.0 Hz, 1H), 7.02 (t, J = 8.8 Hz, 2H), 7.22–7.25 (m, 2H), 7.37 (s, 1H), 8.86 (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.38, 33.97, 40.72, 51.92, 114.70, 118.05, 126.99, 129.99, 130.07, 134.5, 134.62, 161.6, 167.12. HRMS (ESI): calcd for C14H18FN4O [M + H]+, 277.1459; found, 277.1461.
(R)-2-Amino-N-(4-fluorophenethyl)-3-(1H-imidazol-4-yl)-propanamide (4n).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (58%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.69 (t, J = 7.0 Hz, 2H), 2.84–3.31 (m, 4H), 4.10 (t, J = 6.8 Hz, 1H), 6.90 (t, J = 8.8 Hz, 2H), 7.11–7.14 (t, J = 5.6 Hz, 2H), 7.29 (s, 1H), 8.76 (d, J = 0.8 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.53, 33.97, 40.73, 52.01, 114.7, 117.93, 127.32, 130.0, 134.53, 161.08, 167.21. HRMS (ESI): calcd for C14H18FN4O [M + H]+, 277.1459; found, 277.1465.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-nitrophenethyl)-propanamide (4o).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (60%). Rf = 0.06 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 2.96 (t, J = 7.21 Hz, 2H), 3.22–3.36 (m, 2H), 3.57 (t, J = 7.17 Hz, 2H), 4.19 (t, J = 6.92 Hz, 1H), 7.41 (d, J = 1.10 Hz, 1H), 7.49 (d, J = 8.72 Hz, 2H), 8.18–8.19 (m, 2H), 8.89 (d, J = 1.35 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.37, 34.60, 40.04, 51.96, 118.17, 123.21, 126.95, 129.56, 134.44, 146.75, 146.83, 167.29. HRMS (ESI): calcd for C14H18N5O3 [M + H]+, 304.1404, found 304.1407.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-nitrophenethyl)-propanamide (4p).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (61%). Rf = 0.06 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 2.96 (t, J = 7.22 Hz, 2H), 3.22–3.36 (m, 2 H), 3.56 (t, J = 7.22 Hz, 2H), 4.20 (t, J = 6.93 Hz, 1H), 7.41 (d, J = 1.13 Hz, 1H), 7.49 (d, J = 8.72 Hz, 2H), 8.17–8.19 (m, 2H), 8.89 (d, J = 1.36 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.38, 34.59, 40.05, 52.02, 118.21, 123.22, 126.96, 129.55, 134.42, 146.71, 146.86, 167.33. HRMS (ESI): calcd for C14H18N5O3 [M + H]+, 304.1404; found, 304.1414.
(S)-N-(2-([1,1′-Biphenyl]-4-yl)ethyl)-2-amino-3-(1H-imidazol-4-yl)propanamide (4q).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (63%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 2.85–2.88 (m, 2H), 3.22–3.31 (m, 2H), 3.48–3.61 (m, 2H), 4.18 (t, J = 6.00 Hz, 1H), 7.31–7.35 (m, 4H), 7.43 (t, J = 7.69 Hz, 2H), 7.56–7.60 (m, 4H), 8.85 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.39, 34.44, 40.65, 51.91, 118.04, 126.39, 126.74, 126.90, 128.47, 128.88, 134.48, 137.78, 139.41, 140.65, 167.05. HRMS (ESI): calcd for C20H23N4O [M + H]+, 335.1866; found, 335.1869.
(R)-N-(2-([1,1′-biphenyl]-4-yl)ethyl)-2-amino-3-(1H-imidazol-4-yl)propanamide (4r).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (62%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 2.84–2.88 (m, 2H), 3.22–3.31 (m, 2H), 3.48–3.61 (m, 2H), 4.19 (t, J = 6.85 Hz, 1H), 7.31–7.35 (m, 4H), 7.43 (t, J = 7.76 Hz, 2H), 7.56–7.60 (m, 4H), 8.85 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.35, 34.43, 40.65, 51.93, 118.11, 126.40, 126.73, 126.87, 128.46, 128.90, 134.39, 137.80, 139.37, 140.67, 167.12. HRMS (ESI): calcd for C20H23N4O [M + H]+, 335.1866; found, 335.1868.
(S)-2-Amino-N-benzyl-3-(1H-imidazol-4-yl)propanamide (5a).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (67%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.32–3.33 (m, 2H), 4.19–4.22 (m, 1H), 4.39 (dd, J = 14.6, 44.6 Hz, 2H), 7.24–7.36 (m, 6H), 8.80 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.23, 43.01, 52.00, 118.09, 126.80, 127.26, 127.56, 128.29, 134.32, 137.77, 166.81. HRMS (ESI): calcd for C13H17N4O [M + H]+, 245.1397; found, 245.1570.
(S)-2-Amino-3-(1H-3l4-imidazol-4-yl)-N-(4-methoxybenzyl)-propanamide (5b).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (65%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.30–3.32 (m, 2H), 3.80 (s, 3H), 4.18 (t, J = 7.06 Hz, 1H), 4.27 (d, J = 14.46 Hz, 1H), 4.38 (d, J = 14.47 Hz, 1H), 6.89 (d, J = 8.68 Hz, 2H), 7.17 (d, J = 8.64 Hz, 2H), 7.30 (d, J = 0.68 Hz, 1H), 8.75 (d, J = 1.24 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.44, 42.48, 52.09, 54.30, 113.60, 117.89, 127.22, 128.91, 129.68, 134.41, 159.28, 166.73. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1506.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-methoxybenzyl)-propanamide (5c).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (68%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.81–2.86 (m, 1H), 2.96–3.01 (m, 1H), 3.62 (t, J = 6.8 Hz, 1H), 3.78 (s, 3H), 4.23–4.35 (m, 2H), 6.85–6.87 (m, 3H), 7.12 (d, J = 6.8 Hz, 2H), 7.60 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 32.99, 42.08, 54.29, 54.97, 113.48, 117.02, 128.48, 130.26, 133.52, 134.94, 159.01, 174.56. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1439.
(S)-2-Amino-3-(1H-3l4-imidazol-4-yl)-N-(2-methoxybenzyl)-propanamide (5d).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (66%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.31 (d, J = 7.15 Hz, 2H), 3.85 (s, 3H), 4.24 (t, J = 7.10 Hz, 1H), 4.32 (d, J = 14.42 Hz, 1H), 4.46 (d, J = 14.42 Hz, 1H), 6.91 (t, J = 7.44 Hz, 1H), 6.98 (d, J = 8.17 Hz, 1H), 7.19 (dd, J = 7.43, 1.51 Hz, 1H), 7.25 (s, 1H), 7.30 (td, J = 7.85, 1.43 Hz, 1H), 8.76 (d, J = 1.28 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.26, 38.55, 51.86, 54.47, 110.20, 117.97, 120.05, 125.23, 126.82, 128.91, 129.17, 134.18, 157.44, 166.81. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1521.
(R)-2-Amino-3-(1H-3l4-imidazol-4-yl)-N-(2-methoxybenzyl)-propanamide (5e).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (62%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.30 (d, J = 7.12 Hz, 2H), 3.85 (s, 3H), 4.22 (t, J = 7.08 Hz, 1H), 4.32 (d, J = 14.43 Hz, 1H), 4.46 (d, J = 14.43 Hz, 1H), 6.92 (t, J = 7.45 Hz, 1H), 6.98 (d, J = 8.19 Hz, 1H), 7.19 (dd, J = 1.56, 7.44 Hz, 1H), 7.23 (d, J = 0.60 Hz, 1H), 7.28–7.32 (m, 1H), 8.70 (d, J = 1.22 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.39, 38.74, 52.10, 54.91, 110.59, 117.78, 120.34, 124.97, 126.83, 129.29, 129.36, 134.07, 157.32, 167.15. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1507.
(S)-2-Amino-3-(1H-3l4-imidazol-4-yl)-N-(3-methoxybenzyl)-propanamide (5f).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (60%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.30–3.39 (m, 2H), 3.80 (s, 3H), 4.22 (t, J = 7.08 Hz, 1H), 4.31 (d, J = 14.67 Hz, 1H), 4.42 (d, J = 14.66 Hz, 1H), 6.79–6.82 (m, 2H), 6.86 (dd, J = 8.20, 2.32 Hz, 1H), 7.25 (t, J = 7.89 Hz, 1H), 7.34 (s, 1H), 8.81 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.26, 42.93, 52.02, 54.29, 112.36, 113.35, 118.11, 119.65, 126.81, 129.35, 134.29, 139.24, 159.97, 166.89. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1529.
(R)-2-Amino-3-(1H-3l4-imidazol-4-yl)-N-(3-methoxybenzyl)-propanamide (5g).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (58%). Rf = 0.10 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.34–3.36 (m, 2H), 3.80 (s, 3H), 4.23 (t, J = 7.08 Hz, 1H), 4.33 (d, J = 14.67 Hz, 1H), 4.43 (d, J = 14.67 Hz, 1H), 6.79–6.88 (m, 3H), 7.25 (t, J = 7.90 Hz, 1H), 7.34 (s, 1H), 8.81 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 24.65, 41.32, 50.40, 52.67, 110.73, 111.77, 116.49, 118.04, 125.20, 127.74, 132.71, 137.64, 158.37, 165.24. HRMS (ESI): calcd for C14H19N4O2 [M + H]+, 275.1503; found, 275.1503.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethyl)benzyl)-propanamide (5h).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (55%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.33–3.44 (m, 2H), 4.28 (t, J = 7.0 Hz, 1H), 4.50 (q, J = 12.9 Hz, 2H), 7.42 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 8.86 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.30, 42.49, 52.04, 118.16, 125.09, 125.58, 126.99, 127.15, 127.94, 134.47, 142.39, 167.23. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1440.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethyl)benzyl)-propanamide (5i).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (59%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.32–3.45 (m, 2H), 4.29 (t, J = 6.8 Hz, 1H), 4.50 (q, J = 13.6 Hz, 2H), 7.45 (t, J = 8.8 Hz, 3H), 7.63 (d, J = 8.0 Hz, 2H), 8.86 (d, J = 0.8 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.33, 42.49, 52.06, 118.14, 122.89, 125.07, 125.11, 125.14, 125.58, 127.04, 127.93, 129.15, 129.47, 134.48, 142.39, 167.24. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1284.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-(trifluoromethyl)benzyl)-propanamide (5j).
Following general synthetic procedure B, the title compound was synthesized as a white solid (61%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.34–3.43 (m, 2H), 4.29 (t, J = 7.05 Hz, 1H), 4.62 (s, 2H), 7.39 (d, J = 1.27 Hz, 1H), 7.50 (t, J = 7.36 Hz, 2H), 7.63 (t, J = 7.56 Hz, 1H), 7.72 (d, J = 7.43 Hz, 1H), 8.87 (d, J = 1.35 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.27, 39.70, 39.73, 51.92, 118.15, 125.65, 125.71, 125.76, 126.89, 127.69, 127.80, 129.69, 132.22, 134.44, 135.67, 167.22. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1274.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-(trifluoromethyl)benzyl)-propanamide (5k).
Following general synthetic procedure B, the title compound was synthesized as a white solid (65%). Rf = 0.13 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.35–3.43 (m, 2H), 4.31 (t, J = 7.06 Hz, 1H), 4.62 (s, 2H), 7.39 (s, 1H), 7.49 (dd, J = 7.43, 4.84 Hz, 2H), 7.62 (t, J = 7.50 Hz, 1H), 7.72 (d, J = 7.81 Hz, 1H), 8.87 (d, J = 1.34 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.27, 39.70, 39.72, 51.92, 118.16, 125.52, 125.66, 125.70, 126.90, 127.52, 127.69, 127.77, 129.68, 132.22, 134.44, 135.68, 167.23. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1281.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-(trifluoromethyl)benzyl)-propanamide (5l).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (53%). Rf = 0.11 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.84 (dd, J = 7.6, 14.4 Hz, 1H), 3.00 (dd, J = 6.0, 14.4 Hz, 1H), 3.65 (t, J = 6.6 Hz, 1H), 4.44 (q, J = 15.2 Hz, 2H), 6.85 (s, 1H), 7.43–7.62 (m, 5H); 13C NMR (100 MHz, MeOH-d4): δC 32.37, 42.08, 54.99, 116.78, 123.51, 123.55, 123.85, 123.89, 125.60, 128.90, 130.18, 130.91, 133.66, 134.96, 139.96, 175.09. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1280.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-(trifluoromethyl)benzyl)-propanamide (5m).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (56%). Rf = 0.15 (MeOH/DCM = 1:9). 1H NMR (400 MHz, DMSO-d6): δH 3.14–3.27 (m, 2H), 4.20 (t, J = 6.6 Hz, 1H), 4.36–4.49 (m, 2H), 7.83 (s, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.54–7.64 (m, 3H), 8.92 (s, 1H), 9.13 (t, J = 5.6 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δC 27.03, 42.48, 51.97, 118.15, 124.40, 124.44, 127.81, 129.87, 131.89, 134.99, 140.31, 167.90. HRMS (ESI): calcd for C14H16F3N4O [M + H]+, 313.1271; found, 313.1278.
(S)-2-Amino-N-(4-fluorobenzyl)-3-(1H-imidazol-4-yl)-propanamide (5n).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (62%). Rf = 0.12 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.84 (dd, J = 7.2, 14.4 Hz, 1H), 2.99 (dd, J = 7.2, 14.4 Hz, 1H), 3.63 (t, J = 6.8 Hz, 1H), 4.34 (q, J = 16.4 Hz, 2H), 6.85 (s, 1H), 7.00–7.05 (m, 2H), 7.17–7.21 (m, 2H), 7.61 (s, 1H). 13C NMR (100 MHz, MeOH-d4): δC 32.34, 41.80, 55.02, 114.67, 116.96, 129.0, 133.59, 134.40, 134.95, 162.03, 174.84. HRMS (ESI): calcd for C13H16FN4O [M + H]+, 263.1303; found, 263.1305.
(R)-2-Amino-N-(4-fluorobenzyl)-3-(1H-imidazol-4-yl)-propanamide (5o).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (68%). Rf = 0.15 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.28–3.33 (m, 2H), 4.19 (t, J = 7.2 Hz, 1H), 4. 38 (q, J = 12.2 Hz, 2H), 7.06 (t, J = 8.8 Hz, 2H), 7.26–7.35 (m, 3H), 8.83 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.33, 42.25, 52.06, 114.8, 118.06, 127.04, 129.5 133.86, 133.89, 134.41, 162.2, 166.96. HRMS (ESI): calcd for C13H16FN4O M + H]+, 263.1303; found, 263.1305.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethoxy)-benzyl)propanamide (5p).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (76%). Rf = 0.14 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.39–3.43 (m, 2H), 4.24 (t, J = 7.0 Hz, 1H), 4.43 (q, J = 12.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.38 (t, J = 8.6 Hz, 3H), 8.86 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.26, 42.24, 52.02, 118.17, 120.81, 126.92, 129.22, 134.43, 137.12, 148.42, 167.05. HRMS (ESI): calcd for C14H16F3N4O2 [M + H]+, 329.1220; found, 329.1386.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-(trifluoromethoxy)-benzyl)propanamide (5q).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (78%). Rf = 0.12 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.34–3.43 (m, 2H), 4.26 (t, J = 7.0 Hz, 1H), 4.43 (dd, J = 12.6 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.38 (t, J = 8.8 Hz, 3H), 8.85 (d, J = 0.8 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.27, 42.23, 52.04, 118.18, 120.79, 126.93, 129.19, 134.39, 137.11, 148.39, 167.11. HRMS (ESI): calcd for C14H16F3N4O2 [M + H]+, 329.1220; found, 329.1228.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-((trifluoromethyl)thio)-benzyl)propanamide (5r).
Following general synthetic procedure B, the title compound was synthesized as a white solid (62%). Rf = 0.11 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.30–3.41 (m, 2H), 4.24 (t, J = 7.02 Hz, 1H), 4.48 (q, J = 16.61 Hz, 2H), 7.40 (d, J = 8.13 Hz, 3H), 7.68 (d, J = 8.14 Hz, 2H), 8.84 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.29, 42.46, 52.12, 118.26, 122.80, 122.81, 126.94, 128.52, 128.67, 130.96, 134.40, 136.29, 141.51, 167.31. HRMS (ESI): calcd for C14H16F3N4OS [M + H]+, 345.0991; found, 345.0998.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-((trifluoromethyl)thio)-benzyl)propanamide (5s).
Following general synthetic procedure B, the title compound was synthesized as a white solid (61%). Rf = 0.11 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.31–3.43 (m, 2H), 4.27 (t, J = 7.02 Hz, 1H), 4.48 (q, J = 17.89 Hz, 2H), 7.39–7.42 (m, 3H), 7.67 (d, J = 8.17 Hz, 2H), 8.86 (d, J = 1.29 Hz, 1H);13C NMR (126 MHz, MeOH-d4): δC 26.28, 42.44, 52.08, 118.23, 122.81, 122.83, 126.93, 128.68, 134.40, 136.30, 141.53, 167.27. HRMS (ESI): calcd for C14H16F3N4OS [M + H]+, 345.0991; found, 345.0995.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-phenylpropanamide (6a).
Following general synthetic procedure B, the title compound was synthesized as a white solid (60%). Rf = 0.11 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.39–3.53 (m, 2H), 4.38 (t, J = 6.94 Hz 1H), 7.14–7.18 (m, 1H), 7.33–7.37 (m, 2H), 7.47 (s, 1H), 7.58 (dd, J = 1.00, 7.69 Hz, 2H), 8.89 (d, J = 1.19 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.31, 52.65, 118.26, 119.86, 124.71, 126.91, 128.61, 134.50, 137.33, 165.36. HRMS (ESI): calcd for C12H15N4O [M + H]+, 231.1240; found, 231.1239.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-phenylpropanamide (6b).
Following general synthetic procedure B, the title compound was synthesized as a white solid (63%). Rf = 0.11 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 3.40–3.54 (m, 2H), 4.39 (t, J = 6.94, 1H), 7.15–7.19 (m, 1H), 7.33–7.37 (m, 2H), 7.48 (s, 1H), 7.58–7.60 (m, 2H), 8.89 (d, J = 1.23 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.31, 52.66, 118.27, 119.87, 124.70, 126.92, 128.61, 134.49, 137.33, 165.38. HRMS (ESI): calcd for C12H15N4O [M + H]+, 231.1240; found, 231.1240.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-phenylpropyl)-propanamide (7a).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (52%). Rf = 0.16 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 1.79–1.82 (m, 2H), 2.61 (t, J = 7.69 Hz, 2H), 3.24–3.28 (m, 2H), 3.30–3.38 (m, 2H), 4.19 (t, J = 7.04 Hz, 1H), 7.16–7.20 (m, 3H), 7.26–7.29 (m, 2H), 7.44 (d, J = 1.23 Hz, 1H), 8.85 (d, J = 1.34 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.40, 30.50, 32.63, 38.96, 52.13, 118.17, 125.60, 127.17, 128.01, 128.06, 134.42, 141.28, 167.17. HRMS (ESI): calcd for C15H21N4O [M + H]+, 273.1710; found, 273.1713.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(3-phenylpropyl)-propanamide (7b).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (51%). Rf = 0.16 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 1.78–1.84 (m, 2H), 2.61 (t, J = 7.69 Hz, 2H), 3.24–3.26 (m, 2H), 3.28–3.40 (m, 2H), 4.22 (t, J = 7.03 Hz, 1H), 7.18 (dd, J = 13.84, 7.18 Hz, 3H), 7.27 (t, J = 7.57 Hz, 2H), 7.46 (s, 1H), 8.88 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.36, 30.48, 32.63, 38.97, 52.13, 118.22, 125.60, 127.09, 128.01, 128.05, 134.38, 141.27, 167.18. HRMS (ESI): calcd for C15H21N4O [M + H]+, 273.1710; found, 273.1715.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-phenylbutyl)-propanamide (8a).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (42%). Rf = 0.17 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 1.48–1.54 (m, 2H), 1.58–1.64 (m, 2H), 2.63 (t, J = 7.49 Hz, 2H), 3.23–3.29 (m, 2H), 3.31–3.37 (m, 2H), 4.18 (t, J = 7.02 Hz, 1H), 7.17 (dd, J = 13.89, 7.08 Hz, 3H), 7.26 (t, J = 7.53 Hz, 2H), 7.43 (s, 1H), 8.83 (d, J = 1.22 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.36, 28.31, 28.40, 34.94, 39.14, 52.01, 118.10, 125.44, 127.08, 127.95, 128.03, 134.41, 141.96, 166.96. HRMS (ESI): calcd for C16H23N4O [M + H]+, 287.1866; found, 287.1868.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(4-phenylbutyl)-propanamide (8b).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (43%). Rf = 0.17 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 1.48–1.54 (m, 2H), 1.58–1.64 (m, 2H), 2.63 (t, J = 7.49 Hz, 2H), 3.24–3.29 (m, 2H), 3.31–3.37 (m, 2H), 4.17 (t, J = 7.01 Hz, 1H), 7.17 (dd, J = 13.51, 7.14 Hz, 3H), 7.26 (t, J = 7.53 Hz, 2H), 7.44 (s, 1H), 8.84 (d, J = 1.13 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.32, 28.31, 28.39, 34.94, 39.14, 51.99, 118.12, 125.45, 127.02, 127.95, 128.03, 134.41, 141.95, 166.93. HRMS (ESI): calcd for C16H23N4O [M + H]+, 287.1866; found, 287.1868.
(rac)-N-(2-(1H-Indol-3-yl)ethyl)-2-amino-3-(1H-imidazol-4-yl)-propanamide (9a).
To a solution of Boc-protected intermediate 9b in DCM (5 mL) was added TFA (5 mL), and the reaction solution was stirred at room temperature overnight. The solvent was evaporated in vacuo. DCM (20 mL) was added to the residue, which was washed with an aqueous solution of NaHCO3, and the organic layer was basified to pH 7 (NaOH), separated, dried over anhydrous Na2SO4, and concentrated in vacuo to afford the title compound as a colorless solid (24%). 1H NMR (400 MHz, MeOH-d4): δH (major) 1.30–1.39 (m, 2H), 1.50–1.52 (m, 1H), 1.71–1.73 (m, 1H), 2.60–3.16 (m, 2H), 3.41–3.58 (m, 1H), 6.67–6.88 (m, 1H), 6.97–7.35 (m, 3H), 7.50–7.69 (m, 2H); 13C NMR (100 MHz, MeOH-d4): δC (major) 28.67, 31.13, 39.69, 54.93, 111.67, 113.01, 117.93, 118.43, 120.30, 120.94, 123.10, 129.54, 132.69, 135.37, 136.77, 174.82.
(S)-tert-Butyl(1-((2-(1H-indol-3-yl)ethyl)amino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)carbamate (9b).
Following general synthetic method A, the title compound was synthesized as a white solid (80%). Rf = 0.40 (9:1 DCM/MeOH). 1H NMR (400 MHz, MeOH-d4): δH 1.41 (s, 9H), 2.83 (dd, J = 5.2 Hz, 1H), 2.91 (t, J = 7.2 Hz, 2H), 3.02 (dd, J = 5.2 Hz, 1H), 3.43–3.56 (m, 2H), 4.27 (q, J = 5.6 Hz, 1H), 6.86 (s, 1H), 7.00–7.12 (m, 3H), 7.34 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.68 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 24.76, 27.22, 29.25, 39.83, 48.23, 54.79, 79.30, 110.83, 111.61, 117.09, 117.85, 118.23, 120.94, 122.07, 127.29, 132.94, 134.63, 136.79, 156.16, 172.61.
N-(2-(1H-Indol-3-yl)ethyl)-3-(1H-imidazol-4-yl)propanamide (9c).
To a solution of tryptamine (229 mg, 1.43 mmol) in DCM (10 mL) at 0 °C were added DMAP (0.1 mmol) and EDAC (273 mg, 1.43 mmol), and the solution was stirred for 15 min. To the reaction vessel was added a solution of deaminohistidine (200 mg, 1.43 mmol) in DCM (2 mL), and the mixture was stirred at 0 °C for 15 min and allowed to warm to room temperature with stirring continued overnight. The cream precipitate was filtered and washed with water (50 mL) and Et2O (25 mL) and allowed to air dry to afford the title compound as a cream solid (52%). 1H NMR (400 MHz, MeOH-d4): δH 2.61 (t, J = 7.36 Hz, 2H), 2.94 (t, J = 7.22 Hz, 2H), 3.13–3.16 (m, 2H), 3.24–3.27 (m, 2H), 7.05–7.09 (m, 2H), 7.13–7.17 (m, 1H), 7.20 (s, 1H), 7.39 (dt, J = 8.12 and 0.86 Hz, 1H), 7.59 (dt, J = 7.85 and 0.86 Hz, 1H), 8.16 (d, J = 1.12 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 21.23, 23.15, 34.29, 39.85, 108.85, 111.16, 115.85, 117.48, 118.65, 121.36, 122.90, 126.78, 133.66, 134.99, 136.98, 176.48.
(S)-N-(2-(1H-Indol-3-yl)ethyl)-2-amino-3-(1H-imidazol-4-yl)-propanamide (9d).
Following general synthetic procedure B, the title compound was synthesized as a white solid (72%). Rf = 0.14 (MeOH/DCM = 1:9). 21 (c 0.1, MeOH). 1H NMR (400 MHz, MeOH-d4): δH 2.93–2.98 (m, 2H), 3.16–3.28 (m, 2H), 3.51–3.56 (m, 1H), 3.61–3.66 (m, 1H), 4.16 (t, J = 6.82 Hz, 1H), 7.02 (t, J = 7.35 Hz, 1H), 7.08–7.13 (m, 2H), 7.24 (s, 1H), 7.35 (d, J = 8.09 Hz, 1H), 7.57 (d, J = 7.85 Hz, 1H), 8.72 (d, J = 1.18 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 24.59, 26.38, 40.04, 51.94, 110.96, 111.32, 117.82, 117.88, 118.31, 121.06, 122.20, 127.25, 134.29, 136.74, 167.09. HRMS (ESI): calcd for C16H20N5O [M + H]+, 298.1668; found, 298.1659.
(R)-N-(2-(1H-Indol-3-yl)ethyl)-2-amino-3-(1H-imidazol-4-yl)-propanamide (9e).
Following general synthetic procedure B, the title compound was synthesized as a white solid (70%). Rf = 0.14 (MeOH/DCM = 1:9). −21 (c 0.1, MeOH). 1H NMR (400 MHz, MeOH-d4): δH 2.95 (t, J = 7.26 Hz, 2H), 3.01–3.15 (m, 2H), 3.47–3.54 (m, 1H), 3.59–3.64 (m, 1H), 4.06 (t, J = 6.89 Hz, 1H), 7.02 (q, J = 4.90 Hz, 2H), 7.07–7.12 (m, 2H), 7.35 (d, J = 8.12 Hz, 1H), 7.57 (d, J = 7.84 Hz, 1H), 8.05 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 24.66, 28.15, 40.04, 52.84, 110.92, 111.38, 117.80, 118.27, 121.03, 122.15, 127.27, 135.13, 136.80, 167.83. HRMS (ESI): calcd for C16H20N5O [M + H]+, 298.1668; found, 298.1663.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-(naphthalen-2-yl)ethyl)-propanamide (9f).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (57%). Rf = 0.17 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.95–3.02 (m, 2H), 3.18–3.25 (m, 2H), 3.56–3.70 (m, 2H), 4.15 (t, J = 6.80 Hz, 1H), 7.24 (s, 1H), 7.40–7.46 (m, 3H), 7.69 (s, 1H), 7.80–7.82 (m, 3H), 8.70 (s, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.41, 34.91, 40.49, 51.80, 117.83, 125.16, 125.75, 126.75, 126.77, 126.87, 127.05, 127.22, 127.79, 132.34, 133.60, 134.31, 136.16, 167.12. HRMS (ESI): calcd for C18H21N4O [M + H]+; 309.1710; found, 309.1710.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(2-(naphthalen-2-yl)ethyl)-propanamide (9g).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (58%). Rf = 0.17 (MeOH/DCM = 1:9). 1H NMR (400 MHz, MeOH-d4): δH 2.95–3.02 (m, 2H), 3.15–3.26 (m, 2H), 3.53–3.60 (m, 1H), 3.64–3.69 (m, 1H), 4.17 (t, J = 6.78 Hz, 1H), 7.25 (d, J = 1.19 Hz, 1H), 7.38–7.48 (m, 3H), 7.69 (s, 1H), 7.78–7.83 (m, 3H), 8.73 (d, J = 1.37 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.33, 34.90, 40.49, 51.76, 117.91, 125.16, 125.74, 126.68, 126.75, 126.77, 127.05, 127.22, 127.79, 132.33, 133.60, 134.26, 136.16, 167.08. HRMS (ESI): calcd for C18H21N4O [M + H]+, 309.1710; found, 309.1708.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(naphthalen-2-ylmethyl)-propanamide (10a).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (68%). Rf = 0.16 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.35–3.42 (m, 2H), 4.28 (t, J = 7.04 Hz, 1H), 4.58 (q, J = 11.94 Hz, 2H), 7.36–7.38 (m, 2H), 7.48–7.50 (m, 2H), 7.75 (s, 1H), 7.82–7.86 (m, 3H), 8.76 (d, J = 1.24 Hz, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.30, 43.19, 52.07, 118.12, 125.48, 125.68, 126.00, 126.16, 126.90, 127.31, 127.35, 128.07, 132.84, 133.41, 134.28, 135.15, 167.02. HRMS (ESI): calcd for C17H19N4O [M + H]+, 295.1553; found, 295.1560.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(naphthalen-2-ylmethyl)-propanamide (10b).
Following general synthetic procedure B, the title compound was synthesized as colorless oil (65%). Rf = 0.16 (MeOH/DCM = 1:9). 1H NMR (500 MHz, MeOH-d4): δH 3.26–3.33 (m, 2H), 4.20 (t, J = 7.04 Hz, 1H), 4.49 (q, J = 11.53 Hz, 2H), 7.27–7.29 (m, 2H), 7.38–7.41 (m, 2H), 7.65 (s, 1H), 7.73–7.77 (m, 3H), 8.66 (s, 1H); 13C NMR (126 MHz, MeOH-d4): δC 26.29, 43.19, 52.09, 118.14, 125.47, 125.68, 125.99, 126.15, 126.88, 127.31, 127.35, 128.07, 132.83, 133.40, 134.26, 135.15, 167.06. HRMS (ESI): calcd for C17H19N4O [M + H]+, 295.1553; found, 295.1560.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(naphthalen-1-ylmethyl)-propanamide (10c).
Following general synthetic procedure B, the title compound was synthesized as yellowish oil (72%). Rf = 0.16 (MeOH/DCM = 1:9). 26 (c 0.1, MeOH). 1H NMR (400 MHz, MeOH-d4): δH 3.29–3.32 (m, 2H), 4.22 (t, J = 7.11 Hz, 1H), 4.78 (d, J = 14.87 Hz, 1H), 4.97 (d, J = 14.81 Hz, 1H), 7.20 (s, 1H), 7.45–7.46 (m, 2H), 7.54–7.56 (m, 2H), 7.85 (t, J = 4.77 Hz, 1H), 7.91–7.93 (m, 1H), 8.00 (d, J = 9.20 Hz, 1H), 8.61 (d, J = 1.22 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.26, 40.99, 51.92, 117.91, 122.92, 125.05, 125.63, 126.17, 126.58, 126.71, 128.27, 128.50, 131.14, 132.86, 133.96, 134.05, 166.75. HRMS (ESI): calcd for C17H19N4O [M + H]+, 295.1553; found, 295.1549.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(naphthalen-1-ylmethyl)-propanamide (10d).
Following general synthetic procedure B, the title compound was synthesized as yellowish oil (70%). Rf = 0.16 (MeOH/DCM = 1:9). −26 (c 0.1, MeOH). 1H NMR (400 MHz, MeOH-d4): δH 3.29–3.31 (m, 2H), 4.21 (t, J = 7.12 Hz, 1H), 4.78 (d, J = 14.64 Hz, 1H), 4.97 (d, J = 14.63 Hz, 1H), 7.19 (d, J = 1.03 Hz, 1H), 7.45–7.46 (m, 2H), 7.54–7.56 (m, 2H), 7.86 (t, J = 4.76 Hz, 1H), 7.92–7.94 (m, 1H), 8.00–8.02 (m, 1H), 8.61 (d, J = 1.35 Hz, 1H); 13C NMR (100 MHz, MeOH-d4): δC 26.32, 40.98, 51.93, 117.83, 122.92, 125.05, 125.63, 126.17, 126.59, 126.83, 128.27, 128.49, 131.13, 132.86, 133.95, 134.10, 166.76. HRMS (ESI): calcd for C17H19N4O [M + H]+, 295.1553; found, 295.1554.
(S)-2-Amino-3-(1H-imidazol-4-yl)-N-(quinolin-8-yl)propanamide (11a).
Following general synthetic procedure B, the title compound was synthesized as yellowish oil (53%). Rf = 0.14 (MeOH/DCM = 1:9). 10 (c 0.4, MeOH). 1H NMR (500 MHz, MeOH-d4): δH 3.54 (dd, J = 6.87, 4.72 Hz, 2H), 4.76 (t, J = 7.05 Hz, 1H), 7.56 (s, 1H), 7.61–7.64 (m, 2H), 7.77 (dd, J = 8.30, 1.11 Hz, 1H), 8.39 (dd, J = 8.33, 1.61 Hz, 1H), 8.57 (dd, J = 7.64, 1.01 Hz, 1H), 8.91–8.92 (m, 2H); 13C NMR (126 MHz, MeOH-d4): δC 26.41, 52.81, 118.52, 119.84, 121.93, 124.11, 126.72, 126.89, 128.48, 132.57, 134.66, 137.31, 138.72, 148.76, 165.92. HRMS (ESI): calcd for C15H16N5O [M + H]+, 282.1349; found, 282.1355.
(R)-2-Amino-3-(1H-imidazol-4-yl)-N-(quinolin-8-yl)propanamide (11b).
Following general synthetic procedure B, the title compound was synthesized as yellowish oil (51%). Rf = 0.14 (MeOH/DCM = 1:9). −10 (c 0.1, MeOH). 1H NMR (500 MHz, MeOH-d4): δH 3.54 (dd, J = 6.80, 5.20 Hz, 2H), 4.76 (t, J = 7.04, Hz, 1H), 7.55 (s, 1H), 7.60–7.64 (m, 2H), 7.77 (dd, J = 8.31, 1.08 Hz, 1H), 8.39 (dd, J = 8.33, 1.60 Hz, 1H), 8.57–8.58 (m, 1H), 8.90–8.92 (m, 2H); 13C NMR (126 MHz, MeOH-d4): δC 26.44, 52.80, 118.49, 119.18, 121.94, 123.90, 126.58, 126.90, 128.42, 132.83, 134.70, 136.80, 138.97, 148.93, 165.78. HRMS (ESI): calcd for C15H16N5O [M + H]+, 282.1349; found, 282.1355.
Biology.
All studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Texas Tech University Health Sciences Center, Lubbock, Texas (IACUC protocol # 09007).
Enzymatic Assays and Compound Screening.
The effect of synthesized compounds on activity of in-house-produced recombinant rat Nln was evaluated in a continuous assay using a quenched fluorescence substrate (QFS, MCA–Pro–Leu–Gly–Pro–D–Lys (DNP)–OH, 15 μM assay concentration) as described in our previous publications.11,12,35 In brief, the assays were performed in 100 μL volume in the artificial cerebrospinal fluid (aCSF) at 37 °C, the reaction was initiated by the addition of QFS, and the fluorescence was continuously measured for 20 min (at 320 nm excitation and 405 nm emission wavelength, SynergyMX, BioTek). All compounds were dissolved in DMSO to have 30 to 50 mM stock solutions; they were preincubated with Nln at 0.1–300 μM assay concentrations (37 °C) for 10 min before initiation of the reaction.
The fluorescence quenching or enhancing property of compounds 9d, 10c, and 11a was tested under identical conditions described above with the exception that Mca-Pro-Leu-OH (hydrolytic product of QFS, 1.5 μM assay concentration) was used in these experiments.
The effect of compounds 9d, 10c, and 11a on the activity of human NEP, human angiotensin-converting enzyme, human ACE2, and human TOP (products 1182-ZNC, 929-ZN, 933-ZN, and 3439-ZN, respectively, R&D Systems) was evaluated following the protocol described above with the exception that different fluorogenic substrates were used. In these experiments, Mca-Ala-Pro-Lys (Dnp) was used for ACE2,36 whereas Mca-Arg-Pro-Pro-Gly-Phe-Ser-Ala-Phe-Lys(Dnp)-OH was used for ACE and NEP (both substrates at 10 μM assay concentration).37,38 The activity of TOP was measured under conditions identical to Nln except that dithiothreitol, at 0.1 mM assay concentration, was present in the assay.39
Measurement of Substrate Hydrolysis Using LC–MS/MS.
Quantification of BK and NT hydrolysis by Nln was carried out using LC–MS/MS. In these experiments, Nln (0.5 nM) was preincubated with compounds 9d, 10c, or 11a (10 and 30 μM assay concentration) for 10 min at 37 °C, followed by the addition of BK or NT (20 μM assay concertation, Phoenix Peptides) and incubation for 10 additional minutes at 37 °C. The reaction was carried out in aCSF, in a total volume of 30 μL, and it was stopped by the addition of 1 μL of 1.0 N HCl and stored in −80 °C. Liquid chromatography was carried out on a Shimadzu Nexera ultra-performance liquid chromatography (UPLC) system using a Kinetex-EVO-C18 100 Å column (1.7 μm, 50 × 2.1 mm, Phenomenex), and ionization was performed on a triple quadrupole ion-trap (AB SCIEX QTRAP 5500) mass spectrometer. Solvents A (LC–MS-grade water) and B (LC–MS-grade acetonitrile) both with 0.1% formic acid as the ion-pairing agent were used as the buffer solutions and were run at 0.3 mL/min flow rate with column temperature set at 40 °C. Peptides were eluted with the following elution gradient profile: 5% solvent B over 0.1 min, sustained at 5% for 0.9 min, afterward, a 1.5 min linear gradient to 50% B, reaching 80% B over 2 min, followed by a 0.2 gradient to 95% B. By calculating the mass-to-charge ratio, as M + 1, (M + 2)/2, or (M + 3)/3, the precursor ion of the charged state was sorted. Fragment ions were produced by collision-induced dissociation gas and the collision energy applied for NT and BK and their respective hydrolytic products NT-(1–10) and BK-(1–5) is 34, 40, 50, and 45 V, respectively. Detection of the peptides was performed in the positive polarity and multiple-reaction monitoring mode. In order to obtain internal standard (IS) response ratio versus analyte peak area, synthetic opioid peptide DAMGO (Tocris Bioscience) was spiked into each sample. In multiple reaction monitoring (MRM), the selected precursor ion (Q1) and fragment ions (Q3) for the peptides are as follows: m/z 531.1 precursor ion to m/z 120.1 for BK and m/z 573.3 precursor ion to m/z 237.1 for BK-(1–5), m/z 558.4 precursor ion to m/z 136.3 for NT and m/z 642.3 precursor ion to m/z 136.3 for NT-(1–10), and m/z 514 precursor ion to m/z 134.1 for DAMGO. Analyst software (version 1.7.2) was used to calculate the ratio of peak areas of the peptides to IS.
In Vitro Metabolic Stability.
Metabolic stability was determined using mouse plasma and freshly made 10% mouse brain homogenate according to literature methods.40,41 Briefly, stock solutions of the compounds were prepared at 10 μg/mL in LC–MS-grade water. Each biological matrix was spiked with compounds to afford a final concentration of 500 ng/mL. The tubes were placed on a water bath shaker at 37 °C for an incubation period of up to 4 h. Sample aliquots (50 μL) of compound-containing matrices were removed at 0, 2, 5, 15, 30, 60, 120, 180, and 240 min. The samples were analyzed using an LC–MS/MS method developed in our laboratory.
Plasma and Brain Protein Binding.
The RED plate and single-use inserts with a membrane molecular weight cutoff = 8 kDa and sealing tapes for 96-well plates were purchased from Pierce Biotechnology, Thermo Fisher Scientific, Waltham, MA. Test compounds were dissolved in dimethyl sulfoxide and then diluted to 10 μg/mL with water for binding studies. The stock solutions (10 μg/mL) were added to mouse brain homogenates or plasma. The final concentration of the compounds for the equilibrium dialysis experiment was 500 ng/mL. The RED device was assembled according to the manufacturer’s instructions. A 200 μL aliquot of plasma or brain homogenate spiked with 500 ng/mL of the selected compound was added to the sample chamber of insert (donor), and 400 μL of Dulbecco’s phosphate-buffered saline (DPBS) was added to the buffer chamber of the dialysis membrane (receiver). Then, the base plate was covered with a sealing tape and was incubated at 37 °C on an orbital shaker at 250 rpm for 4 h to achieve equilibrium. Compounds were evaluated in triplicate for each experiment. At the end of incubation, equal volumes from both donor and receiver chambers were taken and placed in separate microcentrifuge for content analysis using LC–MS/MS.
Calculation of fu.
Fraction unbound (fu) was calculated using eqs 1 and 2 as described previously.42 For calculation of brain protein binding, the dilution factor (D = 5), which was used to prepare the brain homogenate, should be considered, whereas for plasma protein binding, no dilution was applied and only eq 1 was used to calculate fu.
| (1) |
| (2) |
In Vitro BBB Permeability.
The BBB model was a co-culture of primary mouse astrocytes and immortalized mouse endothelial cells (bEnd3). bEnd3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, St. Louis, MO) with the addition of 10% fetal bovine serum (FBS) (Atlanta biologicals, Minneapolis, MN) and 1% nonessential amino acid and 1% penicillin–streptomycin (PS) (Sigma, St. Louis, MO). Cells (passage 26–30) were maintained in a humidified cell culture incubator at 37 °C and with 5% CO2/95% air. Mouse primary astrocytes were isolated from the cerebral cortices of one day-old pups (CD-1 mice, Charles Rivers Laboratory) following a literature method.43 After extracting the brain, cortices were separated, minced, and placed in Hank’s balanced salt solution (HBSS) without calcium and magnesium, supplemented with gentamycin (10 μg/m). Then, cortices were incubated and digested with 0.25% trypsin for 15 min at 37 °C, followed by neutralizing with astrocyte media containing DMEM plus 10% FBS and 1% PS. The cell suspensions were seeded into a T75 flask and the medium was changed every 3 days for 10–14 days or until reaching confluency.
For bEnd3 and astrocyte co-culture, the Transwell filters (0.4 lm pore size, 12-well; Corning, Lowell, MA) were used. First, the Transwell filter was inverted and astrocytes at a density of 150,000 cells/filter were seeded onto the abluminal side of the filter and allowed to adhere for 4 h. Then, the Transwell filter was inverted back and the cells were grown for 2 days in astrocyte medium. At the end of 48 h, bEnd3 cells at a density of 50,000 cells/filter were seeded onto the upper/luminal side of the filter and the co-culture of astrocytes and bEnd3 cells was grown for an additional 8 days. All permeability experiments were conducted with endothelial/astrocyte cultures that had a trans-endothelial cell resistance measurement of >70 Ω·cm2. The medium for both compartments were changed every other day according to literature procedures.44
The apparent permeability coefficient (Pe, in cm/min) was calculated according to the cleared volume of each time point, as previously described.45,46
Permeability measurement for each compound was run for 120 min. The media was removed and transwells were rinsed and incubated with HBSS buffer at 37 °C for 30 min. Then, 10 μg/mL of each activator in HBSS was introduced to the apical/donor chamber. At different time points (0, 30, 60, and 120 min) following the addition of the compounds, 100 μL of assay buffer was collected from the receiver compartment of the wells in duplicate for concentration determination and the removed volume was replaced with fresh buffer to avoid the back diffusion of the tested compound. Concentration of compounds was determined by LC–MS/MS. Since the blank Transwell insert itself (without cells) provides resistance to the passage of buffer and compound from the donor to receiver chamber, we also measured the permeability of each compound in a blank insert and the final permeability coefficient results were calculated by considering the permeability of each compound in a blank insert.
LC–MS/MS Sample Preparation and Analysis.
To prepare plasma, brain, and buffer standard curves for each compound, these matrices were spiked with stock solutions of each compound to achieve final concentrations within the range of 7.8–1000 ng/mL, and then, these concentrations were subjected to the sample preparation procedure similar to unknown samples. Also, blank control solutions were prepared accordingly without adding the compounds.
To prepare a sample for LC–MS/MS analysis, aliquoted matrix samples were precipitated using an organic solvent specific for each peptide.
Sample aliquots of histidine–tryptophan were precipitated using cold TFA solution (1% v/v) in methanol/water (1:1) at a 1:3 ratio.
Sample aliquots of histidine–tyrosine were precipitated using cold formic acid (FA) solution (0.1% v/v) in acetonitrile (ACN) at a 1:3 ratio, followed by thoroughly vortexing, and then adding 1:1 ratio water.
Sample aliquots of 9d, 10c, and 11a were precipitated using cold FA solution (0.1% v/v) in ACN/water (80:20 mixture) at a 1:3 ratio.
After precipitation, the samples were vortexed thoroughly for 1 min and centrifuged at 14,000 rpm for 10 min at 4 °C. The supernatants were collected in the vials and analyzed using LC–MS/MS.
The samples were analyzed using an AB SCIEX QTRAP 5500triple quadrupole mass spectrometer attached to a Nexera UPLC system (Shimadzu Corporation). The UPLC system contained an autosampler (Sil-30AC), pumps (LC-30AD), a controller (CBM-20A), a degasser (DGA-20A5), and a column oven (CTO-30A). Analyst software was used for data acquisition and quantification. For chromatographic separation, gradient elution using specific mobile phases for each compound was used with different chromatographic columns. Detailed information for the chromatographic separation of each compound is summarized below.
Histidine–Tryptophan.
Column: BEH-C18 (2.1 mm × 50 mm, 1.7 μm; Waters, Milford, MA, USA); mobile phases: (A): water + 0.1% FA, (B): methanol + 0.1% FA; flow rate: 0.25 mL/min; gradient: 0–0:50 min; 0–5% B, 0:50–2 min; to 30% B, 2–3:50 min; to 90% B, 3:50–5 min; to 5% B.
Histidine–Tyrosine.
Column: RFP (2.1 mm × 100 mm, 2.7 μm; Raptor FluoroPhenyl, RESTEK, USA); mobile phases: (A): water + 0.1% FA, (B): ACN+ 0.1% FA; flow rate: 0.4 mL/min; gradient: 0–0:10 min; 0–95% B, 0:10–3 min; to 50% B, 3–5 min; to 95% B.
9d, 10c, and 11a.
Column: RFP (2.1 mm × 100 mm, 2.7 μm; Raptor FluoroPhenyl, RESTEK, USA); mobile phases: (A): water + 0.1% FA, (B): ACN + 0.1% FA; flow rate: 0.4 mL/min; gradient: 0–0:10 min; 0–10% B, 0:10–4 min; to 90% B, 4–5 min; to 10% B.
Data and Statistical Analysis.
The results are presented as the mean ± SD. The n-value represents the number of independent experiments. For the BBB permeability study, each experiment is from a separate cell culture preparation and comprised 3–4 Transwell inserts for each compound and data were analyzed with a one-way ANOVA, followed by Dunnett’s test for comparison of multiple groups with compound 1, p < 0.05 was used as the statistical significance level.
Half-lives are calculated based on the pseudo-first-order kinetics of the disappearance of the compounds.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by NIH grant R01NS106879 to P.C.T., V.T.K. and T.J.A. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Additional support was provided by the University of Nebraska Medical Center and Texas Tech University Health Sciences Center School of Pharmacy.
ABBREVIATIONS
- ACN
acetonitrile
- BBB
blood–brain barrier
- BK
bradykinin
- Boc
tert-butyloxycarbonyl
- BOP
benzotriazol-1-yloxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- bEnd
brain endothelial
- CNS
central nervous system
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylforma-mide
- DPBS
Dulbecco’s phosphate-buffered saline
- DTP
developmental therapeutics program
- EDG
electron-donating group
- EWG
electron-withdrawing group
- FA
formic acid
- FBS
fetal bovine serum
- f u
fraction unbound
- HBA
hydrogen-bond acceptor
- HBSS
Hank’s balanced salt solution
- His–His
histidine–histidine
- His–Phe
histidine–phenylalanine
- His–Trp
histidine–tryptophane
- His–Tyr
histidine–tyrosine
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- IACUC
Institutional Animal Care and Use Committee
- kDa
kilodalton
- IS
internal standard
- LC–MS/MS
liquid chromatography–mass spectrometry
- LLE
ligand-lipophilicity efficiency
- MHz
megahertz
- MPO
multiparameter optimization
- MRM
multiple reaction monitoring
- Nln
neurolysin
- NMR
nuclear magnetic resonance
- NT
neurotensin
- P e
permeability coefficient
- QFS
quenched fluorescence substrate
- RED
rapid equilibrium dialysis
- RP
reverse phase
- SAR
structure–activity relationship
- SD
standard deviation
- TFA
trifluoroacetic acid
- TLC
thin-layer chromatography
- UPLC
ultraperformance liquid chromatography
- UV
ultraviolet
- VS
virtual screen
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c00759.
Characterization data for all intermediates; 1H and 13C NMR spectra, and HPLC data for all final peptidomimetic compounds; and fluorescence of compounds 9d, 10c, and 11a with the Nln substrate (PDF)
Molecular formula strings (CSV)
The authors declare the following competing financial interest(s): Compounds described herein are the subject of published patent applications; PCT Int. Appl. (2020) WO2020047185 and U.S. Pat. Appl. Publ. (2021), US 20210198647 A1.
Contributor Information
Md. Shafikur Rahman, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Shikha Kumari, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Shiva Hadi Esfahani, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Saeideh Nozohouri, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Srinidhi Jayaraman, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Nihar Kinarivala, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Joanna Kocot, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Andrew Baez, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Delaney Farris, Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Thomas J. Abbruscato, Department of Pharmaceutical Sciences, School of Pharmacy and Center for Blood Brain Barrier Research, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States
Vardan T. Karamyan, Department of Pharmaceutical Sciences, School of Pharmacy and Center for Blood Brain Barrier Research, School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas 79106, United States.
Paul C. Trippier, Department of Pharmaceutical Sciences, College of Pharmacy, Fred & Pamela Buffett Cancer Center, and UNMC Center for Drug Discovery, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
REFERENCES
- (1).Mozaffarian D; Benjamin EJ; Go AS; Arnett DK; Blaha MJ; Cushman M; Das SR; de Ferranti S; Despres JP; Fullerton HJ; Howard VJ; Huffman MD; Isasi CR; Jimenez MC; Judd SE; Kissela BM; Lichtman JH; Lisabeth LD; Liu S; Mackey RH; Magid DJ; McGuire DK; Mohler ER 3rd; Moy CS; Muntner P; Mussolino ME; Nasir K; Neumar RW; Nichol G; Palaniappan L; Pandey DK; Reeves MJ; Rodriguez CJ; Rosamond W; Sorlie PD; Stein J; Towfighi A; Turan TN; Virani SS; Woo D; Yeh RW; Turner MB; Writing Group Members; American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Executive summary: Heart disease and stroke statistics–2016 update: A report from the American Heart Association. Circulation 2016, 133, 447–454. [DOI] [PubMed] [Google Scholar]
- (2).Virani SS; Alonso A; Benjamin EJ; Bittencourt MS; Callaway CW; Carson AP; Chamberlain AM; Chang AR; Cheng S; Delling FN; Djousse L; Elkind MSV; Ferguson JF; Fornage M; Khan SS; Kissela BM; Knutson KL; Kwan TW; Lackland DT; Lewis TT; Lichtman JH; Longenecker CT; Loop MS; Lutsey PL; Martin SS; Matsushita K; Moran AE; Mussolino ME; Perak AM; Rosamond WD; Roth GA; Sampson UKA; Satou GM; Schroeder EB; Shah SH; Shay CM; Spartano NL; Stokes A; Tirschwell DL; VanWagner LB; Tsao CW; American Heart Association Council on Epidemiology; Prevention Statistics Committee; Stroke Statistics Subcommittee. Heart disease and stroke statistics-2020 update: A report from the American Heart Association. Circulation 2020, 141, e139–e596. [DOI] [PubMed] [Google Scholar]
- (3).Donkor ES Stroke in the 21(st) century: A snapshot of the burden, epidemiology, and quality of life. Stroke Res. Treat 2018, 2018, 3238165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).GBD 2016 Stroke Collaborators. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019, 18, 439–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hacke W; Donnan G; Fieschi C; Kaste M; von Kummer R; Broderick JP; Brott T; Frankel M; Grotta JC; Haley EC Jr.; Kwiatkowski T; Levine SR; Lewandowski C; Lu M; Lyden P; Marler JR; Patel S; Tilley BC; Albers G; Bluhmki E; Wilhelm M; Hamilton S; Investigators AT; Investigators ET; Investigators N. r.-P. S. G. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004, 363, 768–774. [DOI] [PubMed] [Google Scholar]
- (6).Macrae IM; Allan SM Stroke: The past, present and future. Brain Neurosci. Adv 2018, 2, 2398212818810689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Iadecola C; Anrather J Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci 2011, 14, 1363–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Karamyan V The role of peptidase neurolysin in neuro-protection and neural repair after stroke. Neural Regener. Res 2021, 16, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Karamyan VT Peptidase neurolysin is an endogenous cerebroprotective mechanism in acute neurodegenerative disorders. Med. Hypotheses 2019, 131, 109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rashid M; Arumugam TV; Karamyan VT Association of the novel non-AT1, non-AT2 angiotensin binding site with neuronal cell death. J. Pharmacol. Exp. Ther 2010, 335, 754–761. [DOI] [PubMed] [Google Scholar]
- (11).Rashid M; Wangler NJ; Yang L; Shah K; Arumugam TV; Abbruscato TJ; Karamyan VT Functional up-regulation of endopeptidase neurolysin during post-acute and early recovery phases of experimental stroke in mouse brain. J. Neurochem 2014, 129, 179–189. [DOI] [PubMed] [Google Scholar]
- (12).Jayaraman S; Al Shoyaib A; Kocot J; Villalba H; Alamri FF; Rashid M; Wangler NJ; Chowdhury EA; German N; Arumugam TV; Abbruscato TJ; Karamyan VT Peptidase neurolysin functions to preserve the brain after ischemic stroke in male mice. J. Neurochem 2020, 153, 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Al-Ahmad AJ; Pervaiz I; Karamyan VT Neurolysin substrates bradykinin, neurotensin and substance P enhance brain microvascular permeability in a human in vitro model. J. Neuroendocrinol 2021, 33, No. e12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Karamyan VT; Trippier PC; Ostrov DA; Abbruscato TJ; Jayaraman S Enhancers of Neurolysin Activity. U.S. Patent 20,210,198,647 A1. 2021.
- (15).Jayaraman S; Zhu R; Wangler NJ; Mechref Y; Abbruscato TJ; Ostrov DA; Karamyan VT Allosteric potentiation of peptidase neurolysin by small molecules. Biophys. J 2016, 110, 396a–397a. [Google Scholar]
- (16).Jayaraman S; Kocot J; Esfahani SH; Wangler NJ; Uyar A; Mechref Y; Trippier PC; Abbruscato TJ; Dickson A; Aihara H; Ostrov DA; Karamyan VT Discovery of small molecule allosteric potentiators of peptidase neurolysin. J. Pharmacol. Exp. Ther DOI: 10.1124/jpet.121.000840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kinarivala N; Patel R; Boustany R-M; Al-Ahmad A; Trippier PC Discovery of aromatic carbamates that confer neuroprotective activity by enhancing autophagy and inducing the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2). J. Med. Chem 2017, 60, 9739–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kinarivala N; Suh JH; Botros M; Webb P; Trippier PC Pharmacophore elucidation of phosphoiodyn A - potent and selective peroxisome proliferator-activated receptor beta/delta agonists with neuroprotective activity. Bioorg. Med. Chem. Lett 2016, 26, 1889–1893. [DOI] [PubMed] [Google Scholar]
- (19).Kinarivala N; Morsy A; Patel R; Carmona AV; Sajib MS; Raut S; Mikelis CM; Al-Ahmad A; Trippier PC An iPSC-derived neuron model of CLN3 disease facilitates small molecule phenotypic screening. ACS. Pharmacol. Transl. Sci 2020, 3, 931–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Teixeira PF; Masuyer G; Pinho CM; Branca RMM; Kmiec B; Wallin C; Wärmländer SKTS; Berntsson RP-A; Ankarcrona M; Gräslund A; Lehtiö J; Stenmark P; Glaser E Mechanism of peptide binding and cleavage by the human mitochondrial peptidase neurolysin. J. Mol. Biol 2018, 430, 348–362. [DOI] [PubMed] [Google Scholar]
- (21).El-Faham A; Albericio F Peptide coupling reagents, more than a letter soup. Chem. Rev 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- (22).Ramu VG; Bardaji E; Heras M DEPBT as coupling reagent to avoid racemization in a solution-phase synthesis of a kyotorphin derivative. Synthesis 2014, 46, 1481–1486. [Google Scholar]
- (23).Cherkupally P; Ramesh S; de la Torre BG; Govender T; Kruger HG; Albericio F Immobilized coupling reagents: synthesis of amides/peptides. ACS Comb. Sci 2014, 16, 579–601. [DOI] [PubMed] [Google Scholar]
- (24).Castro B; Dormoy J-R; Dourtoglou B; Evin G; Selve C Peptide coupling reagents. VI. A novel, cheaper preparation of benzotriazolyloxytris[dimethylamine]phosphoniumhexafluoro-phisphate (BOP reagent). Synthesis 1976, 1976, 751–752. [Google Scholar]
- (25).Ribeiro MM; Pinto A; Pinto M; Heras M; Martins I; Correia A; Bardaji E; Tavares I; Castanho M Inhibition of nociceptive responses after systemic administration of amidated kyotorphin. Br. J. Pharmacol 2011, 163, 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yao J-F; Yang H; Zhao Y-Z; Xue M Metabolism of peptide drugs and strategies to improve their metabolic stability. Curr. Drug Metab 2018, 19, 892–901. [DOI] [PubMed] [Google Scholar]
- (27).Kumari S; Carmona AV; Tiwari AK; Trippier PC Amide bond bioisosteres: Strategies, synthesis, and successes. J. Med. Chem 2020, 63, 12290–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Muttenthaler M; King GF; Adams DJ; Alewood PF Trends in peptide drug discovery. Nat. Rev. Drug Discovery 2021, 20, 309–325. [DOI] [PubMed] [Google Scholar]
- (29).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Johnson TW; Gallego RA; Edwards MP Lipophilic efficiency as an important metric in drug design. J. Med. Chem 2018, 61, 6401–6420. [DOI] [PubMed] [Google Scholar]
- (31).Trippier PC Selecting good “drug-like” properties to optimize small molecule blood-brain barrier penetration. Curr. Med. Chem 2016, 23, 1392–1407. [DOI] [PubMed] [Google Scholar]
- (32).Wang H; Huwaimel B; Verma K; Miller J; Germain TM; Kinarivala N; Pappas D; Brookes PS; Trippier PC Synthesis and antineoplastic evaluation of mitochondrial complex II (succinate dehydrogenase) inhibitors derived from atpenin A5. ChemMedChem 2017, 12, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hopkins AL; Keseru GM; Leeson PD; Rees DC; Reynolds CH The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- (34).Leeson PD; Springthorpe B The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- (35).Wangler NJ; Jayaraman S; Zhu R; Mechref Y; Abbruscato TJ; Bickel U; Karamyan VT Preparation and preliminary characterization of recombinant neurolysin for in vivo studies. J. Biotechnol 2016, 234, 105–115. [DOI] [PubMed] [Google Scholar]
- (36).Vickers C; Hales P; Kaushik V; Dick L; Gavin J; Tang J; Godbout K; Parsons T; Baronas E; Hsieh F; Acton S; Patane M; Nichols A; Tummino P Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem 2002, 277, 14838–14843. [DOI] [PubMed] [Google Scholar]
- (37).Miners JS; Verbeek MM; Rikkert MO; Kehoe PG; Love S Immunocapture-based fluorometric assay for the measurement of neprilysin-specific enzyme activity in brain tissue homogenates and cerebrospinal fluid. J. Neurosci. Methods 2008, 167, 229–236. [DOI] [PubMed] [Google Scholar]
- (38).Joyner JC; Hocharoen L; Cowan JA Targeted catalytic inactivation of angiotensin converting enzyme by lisinopril-coupled transition-metal chelates. J. Am. Chem. Soc 2012, 134, 3396–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shrimpton CN; Glucksman MJ; Lew RA; Tullai JW; Margulies EH; Roberts JL; Smith AI Thiol activation of endopeptidase EC 3.4.24.15. A novel mechanism for the regulation of catalytic activity. J. Biol. Chem 1997, 272, 17395–17399. [DOI] [PubMed] [Google Scholar]
- (40).Li N; Han ZL; Wang ZL; Xing YH; Sun YL; Li XH; Song JJ; Zhang T; Zhang R; Zhang MN; Xu B; Fang Q; Wang R BN-9, a chimeric peptide with mixed opioid and neuropeptide FF receptor agonistic properties, produces non-tolerance-forming antinociception in mice. Br. J. Pharmacol 2016, 173, 1864–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nan D-D; Gan C-S; Wang C-W; Qiao J-P; Wang X-M; Zhou J-N 6-Methoxy-indanone derivatives as potential probes for beta-amyloid plaques in Alzheimer’s disease. Eur. J. Med. Chem 2016, 124, 117–128. [DOI] [PubMed] [Google Scholar]
- (42).Kalvass JC; Maurer TS Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dispos 2002, 23, 327–338. [DOI] [PubMed] [Google Scholar]
- (43).Du F; Qian ZM; Zhu L; Wu XM; Qian C; Chan R; Ke Y Purity, cell viability, expression of GFAP and bystin in astrocytes cultured by different procedures. J. Cell. Biochem 2010, 109, 30–37. [DOI] [PubMed] [Google Scholar]
- (44).Li G; Simon MJ; Cancel LM; Shi ZD; Ji X; Tarbell JM; Morrison B 3rd; Fu BM Permeability of endothelial and astrocyte cocultures: in vitro blood-brain barrier models for drug delivery studies. Ann. Biomed. Eng 2010, 38, 2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nozohouri S; Noorani B; Al-Ahmad A; Abbruscato TJ Estimating Brain Permeability Using In Vitro Blood-Brain Barrier Models. In Permeability Barrier; Methods in Molecular Biology; Turksen K, Ed.; Humana, New York, NY, 2020; Vol. 2367. [DOI] [PubMed] [Google Scholar]
- (46).Gaillard PJ; Voorwinden LH; Nielsen JL; Ivanov A; Atsumi R; Engman H; Ringbom C; de Boer AG; Breimer DD Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur. J. Pharm. Sci 2001, 12, 215–222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.