

Significance
p73 is expressed as multiple C-terminal isoforms, but their expression and activity are largely unknown. Here, we identified p73α1 as a p73 C-terminal isoform that results from exon 12 (E12) exclusion. We showed that E12 deficiency in mice leads to systemic inflammation but not spontaneous tumors. We also showed that Notch1 is regulated by p73α1 and plays a critical role in p73-dependent tumor suppression and systemic inflammation.
Keywords: p53 family, p73, p73 C-terminal isoforms, Notch1 pathway, tumor suppressor
Abstract
p73, a p53 family member, undergoes alternative splicing at the 3′ end to produce multiple isoforms, but their expression and activity are largely unknown. Thus, CRISPR was used to knock out exon 12 (E12) in human cancer cell lines and mice, leading to isoform switch from p73α to isoform p73α1. We found that p73α1 is naturally expressed and induced by DNA damage. We also found that knockout of E12 suppresses cell growth and migration in H1299 and MIA PaCa-2 cells and promotes cellular senescence in mouse embryonic fibroblasts. Similarly, ectopic expression of p73α1 suppresses cell proliferation, whereas knockdown of p73α1 restores the cell proliferative and migratory capacities of E12−/− cells. Consistently, we found that E12+/− mice are not prone to spontaneous tumors. Instead, E12+/− mice are prone to systemic inflammation and exhibit elevated TNFα expression in inflamed tissues. Moreover, we found that Notch1, a master regulator of the inflammatory response, is regulated by p73α1 and highly expressed in E12−/− cells and inflamed E12+/− mouse tissues. Furthermore, through knockdown of p73α1 and/or Notch1 in E12−/− cells, we found that Notch1 is necessary for p73α1-mediated growth suppression. Together, these data suggest that p73α1 plays a critical role in tumor suppression and the inflammatory response via Notch1.
p53 is a master transcriptional regulator and plays an integral role in tumor suppression. This feat is achieved through the ability of p53 to tightly regulate a multitude of activities, such as the cell cycle and apoptosis, in response to various genomic stressors (1). Over two decades ago, two homologs of TP53 were discovered, and termed TP63 (2–4) and TP73 (5, 6). These three proteins, better known as the p53 family, exhibit significant homology in their transactivation domain (TAD), DNA-binding domain (DBD), and oligomerization domain (OD). The discovery of p73 fueled efforts to determine whether this protein has similar functions to that of p53. Total p73-knockout (p73-KO) mice were not prone to spontaneous tumors but rather had major developmental abnormalities (7). p73-KO mice exhibited reproductive impairments due to pheromone-sensing defects (7) and profound neurological malformations due to abrogated p73 signaling in Cajal–Retzius cells (7–9). These mice also displayed signs of chronic inflammation and infection in the respiratory tract, which were later explained by the indispensable role for p73 in regulating multiciliogenesis of airway epithelia (10, 11).
TP73 is expressed as two N-terminal isoforms through the use of two promoters. Promoter 1 gives rise to the TA (transactivation) isoforms that contain the conventional TAD (12), and Promoter 2 produces the N-terminally truncated ΔN isoforms (7). While ΔNp73 was originally thought to be transcriptionally inactive, it was later found to contain a group of amino acids in the N terminus that function as a unique TAD (13). Specific TAp73- or ΔNp73-KO mouse models revealed that the N terminus of p73 is important for regulating tumor suppression and oncogenesis. Like p53-KO mice, TAp73-KO mice were prone to spontaneous tumors (14), indicating that TAp73 functions as a tumor suppressor. As such, TAp73 was found to induce cell cycle arrest and apoptosis through p21 (15) and PUMA (16), two common p53 targets. Moreover, TAp73 was shown to maintain genome stability through the Bub family proteins (17). On the other hand, ΔNp73-KO mice exhibited neurological defects but were not prone to spontaneous tumors (18). Later studies demonstrated that ΔNp73 functions as an oncogene by forming hetero-oligomers with TAp73 and wild-type (WT) p53 to inhibit their transactivation function (19). Furthermore, ΔNp73 was shown to promote metastasis and invasion (20) and is up-regulated in a variety of cancers (19, 21, 22).
While the p73 N-terminal isoforms are well studied, the C-terminal isoforms remain largely unexplored. Alternative pre-messenger RNA (mRNA) splicing of exons 11 through 13 gives rise to seven known C-terminal isoforms (α, β, γ, δ, ε, ζ, η) (7, 23, 24). Notably, the TAD, DBD, and OD are not contained in exons 11, 12, or 13, but overexpression analyses showed that the C terminus modulates p73 transcriptional activity (25). Studies suggest the differential transactivation strengths are attributed to the presence of the sterile alpha motif (SAM) (26–28) and C-inhibitory domain (TID) (29) in exons 12 through 14. These domains are present only in full-length p73α, although a few shorter isoforms contain truncated versions of the SAM and/or TID. In normal tissues, p73α and p73β are ubiquitously expressed, but p73α is the predominant isoform (26, 27). The other isoforms appear to have a more tissue-specific expression pattern (26, 27). Recently, it was discovered that p73α is necessary for proper hippocampal development in mice, and a loss of p73α produces a neurodegenerative phenotype that is highly similar to the one observed in p73-KO mice (28). On the other hand, the same group showed that an isoform switch from p73α to p73β did not impair multiciliogenesis in airway epithelia (30), suggesting that some C-terminal isoforms have overlapping functions. While p73 is rarely mutated in cancer, overexpression is common (31–34). Interestingly, cancers with p73 overexpression exhibit a shift in isoform expression, from p73α to the less abundant isoforms (35). However, the function of these C-terminal isoforms in tumor suppression or oncogenesis remains uncertain.
To explore the implication of the p73 C-terminal isoforms in cancer, we generated exon 12 knockout (E12-KO) cancer cell lines and an E12-deficient mouse model. Loss of E12 leads to the expression of two isoforms, namely, p73α1 and p73β1, from p73α and p73β, respectively. Because p73α is overwhelmingly the major isoform, we focused on characterizing the function of p73α1. We found that p73α1 is expressed in multiple cancer cell lines and normal human and tumor prostate tissue. We also found that E12-KO leads to decreased cell proliferation and migration in H1299 and MIA PaCa-2 cells but increased cellular senescence in E12-deficient mouse embryonic fibroblasts (MEFs) and mouse tissues. Moreover, we found that Exon 12+/− (E12+/−) mice were prone to systemic inflammation but not spontaneous tumors. Furthermore, we found that Notch1 is highly expressed in E12-KO cells and inflamed E12+/− mouse tissues, regulated by p73α1, and plays an important role in p73α1-mediated growth suppression. Collectively, we identify p73α1 as a naturally occurring isoform that plays a critical role in tumor suppression and the inflammatory response via Notch1.
Results
Knockout of E12 Leads to Isoform Switch from p73α to p73α1, Decreased Colony Formation and Cell Migration, and Increased Cellular Senescence.
To determine the biological function of the p73 C-terminal isoforms, CRISPR was used to generate multiple E12-KO H1299 and MIA PaCa-2 cell lines (Fig. 1A and SI Appendix, Fig. S1 A and B). Two E12-KO clones from each cell line were chosen for subsequent experiments. DNA sequencing (DNA-seq) showed a 105-nucleotide (nt) deletion in intron 11 and 45-nt deletion in E12 in H1299 E12-KO #15, and 105-nt deletion in intron 11 and 48-nt deletion in E12 in H1299 E12-KO #83. Thus, E12 would be excluded due to the lack of the E12 splice acceptor site. RT-PCR showed that p73α and a low level of p73α1 were detected in isogenic control H1299 cells (Fig. 1B). In contrast, p73α1, but not p73α, was detected in E12-KO H1299 cells (Fig. 1B), suggesting that the isoform switch from p73α to p73α1 was initiated through the deletion of the splice acceptor site. Similarly, knockout of E12 in MIA PaCa-2 cells led to a p73α-to-p73α1 isoform switch (SI Appendix, Fig. S2A). DNA-seq showed a 105-nt deletion in intron 11 and 45-nt deletion in E12 in both E12-KO MIA PaCa-2 #47 and #61. To detect p73α1 protein expression, isogenic control and E12-KO H1299 cells were treated with doxorubicin (DOX), which is known to enhance p73 protein stability for easy detection (36). p73α was detected in isogenic control cells and stabilized by DOX (Fig. 1C). However, p73α1, but not p73α, was detected and stabilized by DOX in E12-KO cells (Fig. 1C).
Fig. 1.

Isoform switch from p73α to p73α1 inhibits cell growth and migration in H1299 cells. (A) Schematic representation of TP73 E12 exclusion that leads to isoform switch from p73α to p73α1. (B) The level of p73α, p73α1, and HPRT1 transcripts was measured in isogenic control and E12−/− H1299 cells. (C) The level of TAp73α, TAp73α1, and actin proteins was measured in isogenic control and E12−/− H1299 cells treated with (+) or without (−) 0.25 µM DOX. (D) The level of p73α1 and HPRT1 transcripts was measured in H1299, HCT116, MCF7, and RKO cells treated with (+) or without (−) 0.25 µM DOX. (E) The level of p73α, p73α1, and HPRT1 transcripts was measured in normal human and tumor prostate tissues. (F) Colony formation assay was performed with isogenic control, TAp73−/−, ΔNp73−/−, and E12−/− H1299 cells. (G) Quantification of colony formation assay shown in F using relative colony area. The relative colony area in isogenic control cells was arbitrarily set as 1.0. Data are presented as the mean ± SEM of three independent experiments. One-way ANOVA was used to calculate P values. *P < 0.05; ***P < 0.001; ****P < 0.0001. (H) Isogenic control, TAp73−/−, ΔNp73−/−, and E12−/− H1299 cells were used for scratch assays. Microscopic images were taken immediately after scratch (0 h) and 24 h later to monitor cell migration. Wound closure percentages were quantified and presented below. Ctrl, control; Rel., relative.
Because aberrant splicing is a hallmark of cancer, we speculated that p73α1 would be expressed in tumors. To test this, RT-PCR was used to specifically amplify p73α1, which contains a unique DNA sequence at the junction of E11 and E13 (SI Appendix, Fig. S2B). We found that various levels of p73α1 were expressed in nine different tumor cell lines, including H1299 and MIA PaCa-2 cells (Fig. 1D and SI Appendix, Fig. S2C). Additionally, we found that p73α1 expression was increased following DOX treatment in H1299, HCT116, MCF7, and RKO cells (Fig. 1D), consistent with previous studies in which p73 transcription was induced by DNA damage (37, 38). Moreover, DNA-seq showed that the amplified complementary DNA (cDNA) was indeed p73α1. To confirm these findings, H1299 and MCF7 cells were treated with small interfering RNA (siRNA) against p73α1 to specifically knock down the p73α1 transcript (SI Appendix, Fig. S2E, Left). We found that upon treatment with si-p73α1, the p73α1 transcript was markedly decreased in these cells (SI Appendix, Fig. S2F). These data indicate that the isoform p73α1 is naturally expressed in multiple cancer cell lines.
We then wanted to determine whether p73α1 is expressed in normal human and tumor tissues. First, we analyzed p73α1 expression in normal and tumor prostate samples. We found that p73α1 was present in both normal and tumor samples (Fig. 1E and SI Appendix, Fig. S2D). Next, GTEx Portal was used to analyze RNA sequencing (RNA-seq) data mapping to TP73 in 54 different normal human tissues. Interestingly, it appeared that E12 was not transcribed in amygdala tissue, suggesting that TAp73α1 and/or ΔNp73α1 are highly expressed in normal tissues in a tissue-dependent manner.
Given that p73α1 was detected in cancer cell lines, normal tissues, and prostate tumors, we wanted to explore why a p73α-to-p73α1 isoform switch might occur. First, RNA-seq was performed to compare the gene expression pattern between isogenic control and E12-KO H1299 cells. We found that the loss of E12 led to down-regulation of numerous heterogeneous nuclear ribonucleoprotein (hnRNP) genes, which are important splicing factors (39) (SI Appendix, Fig. S3A). Second, we cross interrogated our RNA-seq data with that of prostate cancer in the The Cancer Genome Atlas (TCGA) database. We found that SNRNP200, HNRNPL, and HNRNPAB were significantly up-regulated in prostate tumors compared to normal prostate tissue (SI Appendix, Fig. S3B). SNRNP200 is associated with increased prostate cancer severity and metastasis (40), and HNRNPL is critical for prostate cancer cell growth (41). Third, the acceptor site at the junction of intron 11 and E12 for p73α expression was compared to the one at intron 12 and E13 for p73α1 expression. We found that the nucleotide sequence in the beginning of E12 (GCCCCG) was completely different from the one in the beginning of E13 (TTTTTT), although the consensus AG nucleotides were conserved in introns 11 and 12 (SI Appendix, Fig. S3C). Fourth, we found that the number and composition of intronic splicing enhancer motifs in introns 11 and 12 were quite different. Enhancer motifs are bound by splicing factors and dictate the outcome of alterative splicing (42). Since some splicing factors are up- or down-regulated in tumors, a p73α-to-p73α1 isoform switch is likely induced by preferential usage of one acceptor site over the other.
To determine whether the p73α-to-p73α1 isoform switch has an effect on growth suppression, colony formation and wound healing assays were performed with E12-KO H1299 and MIA PaCa-2 cells along with the respective isogenic controls, namely, TAp73-KO and ΔNp73-KO cells. We showed that TAp73-KO promoted, whereas ΔNp73-KO inhibited, cell growth and migration (Fig. 1 F–H and SI Appendix, Fig. S4 A–C), consistent with previous observations that TAp73 is a tumor suppressor and ΔNp73 is necessary for cell survival (14, 22). Interestingly, we found that E12-KO suppressed cell proliferation and migration in both H1299 and MIA PaCa-2 cells (Fig. 1 F–H and SI Appendix, Fig. S4 A–C), suggesting that p73α1 not only compensates for the loss of p73α but also may have stronger growth-suppressive effects than p73α. To directly demonstrate the growth-suppressive activity of p73α1, we generated multiple H1299 cell lines that inducibly express TAp73α1 under the control of a tetracycline-inducible promoter (SI Appendix, Fig. S4D). We showed that upon induction of TAp73α1, cell proliferation was markedly decreased (SI Appendix, Fig. S4E).
To determine whether the loss of E12 in murine Trp73 leads to a p73α-to-p73α1 isoform switch and subsequently inhibits cell growth, CRISPR was used to generate an E12-deficient mouse model (SI Appendix, Fig. S5 A and B and Table S3). By intercrossing E12+/− mice, a cohort of WT, E12+/-, and E12-/- MEFs were produced. We found that deletion of E12 led to an isoform switch from p73α to p73α1 in mice (Fig. 2A). We also found that E12-deficicency in MEFs increased cellular senescence evidenced by an elevated expression of senescence-associated (SA)-β-galactosidase and senescent markers (PML, p130, and p21) (Fig. 2 B and C). Moreover, we showed that E12-KO suppressed MEF cell proliferation (Fig. 2D). We then examined the effect of E12 deficiency on cellular senescence in vivo and found that the loss of E12 led to an increased expression of senescent markers p21 and p16 in liver and kidney tissues and increased SA-β-galactosidase staining in kidney tissue as compared to age- and sex-matched WT tissues (Fig. 2 E–G). These results indicate that the p73α-to-p73α1 isoform switch and the accompanying growth-suppressive effects are conserved between human and mouse.
Fig. 2.
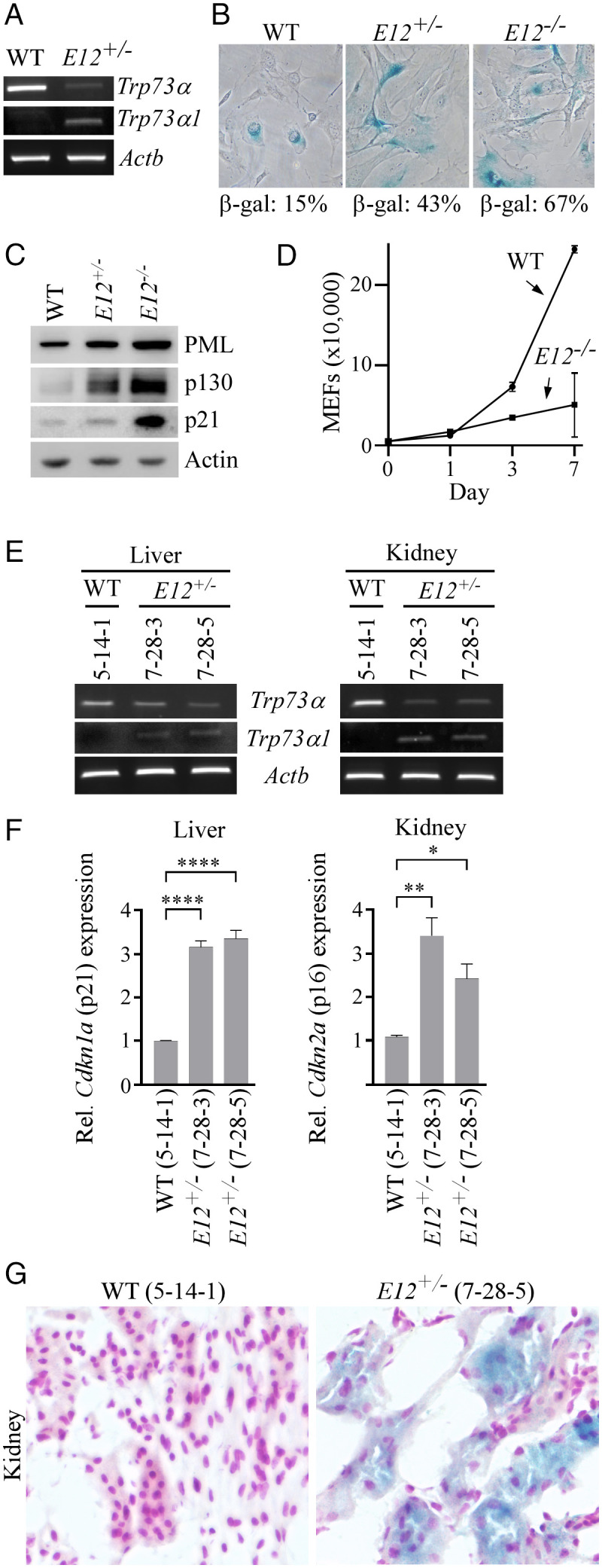
The loss of E12 in MEFs and mouse tissues leads to increased cellular senescence. (A) The level of Trp73α, Trp73α1, and Actb transcripts was measured in WT and E12+/− MEFs. (B) SA-β-galactosidase staining was performed with WT, E12+/-, and E12−/− MEFs. The percentage of SA-β-gal-positive cells was shown in each panel. (C) The level of PML, p130, p21, and actin proteins was measured in WT, E12+/-, and E12−/− MEFs. (D) Growth curves were performed with WT and E12−/− MEFs over 7 d. (E) The level of Trp73α, Trp73α1, and Actb transcripts was measured in liver and kidney tissues from age- and sex-matched WT and E12+/− mice (100 wk; Female). (F) qPCR was used to analyze relative Cdkn1a (p21) and Cdkn2a (p16) expression in liver and kidney tissues from age- and sex-matched WT and E12+/− mice (100 wk; Female). One-way ANOVA was used to calculate P values. *P < 0.05; **P < 0.01; ****P < 0.0001. (G) SA-β-galactosidase staining was performed with kidney tissues from age- and sex-matched WT and E12+/− mice (100 wk; Female).
p73α1 Is Responsible for Growth Suppression in E12-KO Cells.
To demonstrate whether the growth-suppressive effects by E12-KO are due to p73α1 activity, si-p73α1 was designed and used to specifically knock down p73α1, whereas si-E11 #1 and #2 were designed and used to knock down both p73α and p73α1 (SI Appendix, Fig. S2E). As expected, treatment with si-E11 #2, but not si-p73α1, led to a decreased expression of p73α in isogenic control H1299 cells (Fig. 3 A and B, Left column). Consistently, cell viability and migration were increased in isogenic control H1299 cells treated with si-E11 #2, but not si-p73α1 (Fig. 3 C–H, Left column). We would like to note that the low level of p73α1 in isogenic control H1299 cells was further decreased by treatment with si-E11 #2 (Fig. 3B). We also showed that p73α1 was knocked down in E12-KO H1299 cells upon treatment with either si-E11 #2 or si-p73α1 (Fig. 3 A and B, Central and Right columns). Consistently, cell viability and migration were restored in E12-KO H1299 cells upon knockdown of p73α1 (Fig. 3 C–H, Central and Right columns), suggesting that p73α1 is responsible for the growth-suppressive effect in E12-KO cells.
Fig. 3.

p73α1 inhibits cell growth and migration in E12−/− H1299 cells. (A and B) The level of p73α, p73α1, and HPRT1 transcripts was measured in isogenic control and E12−/− H1299 cells transiently transfected with (A and B) scrambled siRNA (Scr), (A) si-p73α1, or (B) si-E11#2 for 3 d. (C and D) Cells were treated as in A and used to perform a three-dimensional spheroid assay, followed by (C) microscopic images of spheroids and (D) measurement of cell viability using CellTiter-Glo 2.0 kit. The relative cell viability in isogenic control cells was arbitrarily set as 1.0. Data are presented as the mean ± SEM of three independent experiments. One-way ANOVA was used to calculate P values. ***P < 0.001; ****P < 0.0001. (E and F) The experiments were performed the same as in C and D except that si-E11#2 was used. One-way ANOVA was used to calculate P values. **P < 0.01. (G) Isogenic control and E12−/− H1299 cells were transiently transfected with Scr or si-p73α1 for 3 d and used for scratch assays. Microscopic images were taken immediately after scratch (0 h) and 18 h later to monitor cell migration. Wound closure percentages were calculated and shown to the right. (H) The experiments were performed same as in G except that si-E11#2 was used.
E12+/− Mice Are Prone to Systemic Inflammation But Not Spontaneous Tumors.
To determine how the isoform switch from p73α to p73α1 alters p73 biological activity, a cohort of E12+/−, E12−/−, and Trp73+/− mice were generated and monitored throughout their lifespan. To reduce the number of animals used, 55 WT and 26 Trp73+/− mice, which were generated and used for our earlier studies (SI Appendix, Tables S1 and S2) (43, 44), were used as controls. All previously generated mice were of the same genetic background and housed in the same facility as E12+/− mice. Initial observations revealed that E12−/− mice were runty and had a substantially shortened lifespan (SI Appendix, Fig. S5C). As such, E12+/− mice were characterized to determine p73α1 activity.
We showed that like Trp73+/− mice, E12+/− mice had a shorter lifespan than WT mice (Fig. 4A). However, unlike Trp73+/− mice, E12+/− mice were not prone to spontaneous tumors (Fig. 4B). These findings were likely due in part to increased p21 and p16 expression and increased cellular senescence in E12+/− mice (Fig. 2 F and G). Previous studies have found that a loss of Trp73 leads to systemic inflammation (7), and our data yielded similar results wherein Trp73+/− mice exhibited inflammation in three or more organs as compared to WT mice (SI Appendix, Fig. S6 A and B). Interestingly, E12+/− mice had widespread systemic inflammation, as evidenced by a majority of mice displaying inflammation in five or more organs (Fig. 4 C and D and SI Appendix, Fig. S6C). Due to the prevalence of widespread systemic inflammation in the E12+/− cohort, TNFα, a common proinflammatory cytokine, was measured and compared in age- and sex-matched WT and E12+/− liver and salivary gland tissues. We found that TNFα was significantly increased in E12+/− tissues compared to both WT and Trp73+/− liver tissues (Fig. 4E and SI Appendix, Fig. S6F). Consistently, TNFα expression was significantly increased in E12-KO H1299 cells (Fig. 4F). Additional abnormalities observed in E12+/− mice included splenic hyperplasia and extramedullary hematopoiesis (EMH) (SI Appendix, Fig. S6 D and E). These data demonstrate that a loss of one E12 allele in vivo leads to increased systemic inflammation but decreased tumor formation, the latter of which is consistent with the observation that E12-KO leads to growth suppression (Figs. 1 and 2).
Fig. 4.

E12+/− mice are prone to systemic inflammation, but not spontaneous tumors. (A) Kaplan–Meier survival curves of WT (n = 56), Trp73+/− (n = 27), and E12+/− (n = 33) mice. Median survival time was 117 wk for WT mice, 86 wk for Trp73+/− mice, and 93.5 wk for E12+/− mice. The log-rank test was used to calculate P values. (B) Tumor spectra in WT, Trp73+/−, and E12+/− mice. The asterisk (*) indicates that found dead/hydrocephalus mice were excluded. Fisher’s exact test was used to calculate P values. DLBCL, diffuse large B cell lymphoma; T-LBL, T-lymphoblastic lymphoma; LM, lymphoma. (C) Representative images of hematoxylin and eosin-stained kidney, lung, and pancreas tissues from sex-matched WT and E12+/− mice. Arrows indicate the inflammation site. (D) Proportion of WT, Trp73+/−, and E12+/− mice with chronic inflammation in five or more organs. Fisher’s exact test was used to calculate P values. *P < 0.05; ***P < 0.001. (E) qPCR was used to analyze relative Tnf (TNFα) expression in liver tissue from age- and sex-matched WT, Trp73+/−, and E12+/− mice (100 wk; Female). One-way ANOVA was used to calculate P values. **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) qPCR was used to analyze relative TNF (TNFα) expression in isogenic control, TAp73−/−, ΔNp73−/−, and E12−/− H1299 cell lines. One-way ANOVA was used to calculate P values. ***P < 0.001; ****P < 0.0001.
Notch1 Is a Target of p73α1 and Plays a Critical Role in p73α1-Dependent Growth Suppression and Inflammation.
To identify potential targets of p73α1 responsible for tumor suppression and inflammation in E12-deficient mice, RNA-seq data were analyzed to compare the differential gene expression profiles between isogenic control and E12-KO H1299 cells. Knockout of E12 was found to modulate multiple genes involved in cell growth (CDKN1A, PERP, MAP2K1, and ERK) (SI Appendix, Fig. S7A) and the inflammatory response (NF-κB2, IL1R1, SOCS3, and NFKRIB) (SI Appendix, Fig. S7B). Notably, we found that E12-KO led to up-regulation of NOTCH1, along with its ligands (JAG1 and JAG2) and targets (HES1 and HEY1) (SI Appendix, Fig. S7 A and B). Since Notch1 is known to mediate both cell growth (45) and inflammation (46), the genes associated with the Notch1 pathway were chosen for further investigation.
Notch1, a transmembrane receptor, is cleaved at both the extracellular and intracellular regions when bound to a ligand (47). The Notch1 Intracellular Domain (NICD) is then translocated to the nucleus where it cooperates with transcription factor CSL to regulate gene expression (47). To confirm the RNA-seq data, NOTCH1 and HES1 mRNAs were measured and shown to be highly induced in E12-KO H1299 and MIA PaCa-2 cells compared to TAp73-KO cells and, to a lesser extent, isogenic control cells (Fig. 5A and SI Appendix, Fig. S8A). This suggests that Notch1 can be induced by TAp73α1, and to a lesser extent by TAp73α (SI Appendix, Fig. S8C). We would like to note that NOTCH1 and HES1 mRNAs were slightly induced in ΔNp73-KO cells (Fig. 5A and SI Appendix, Fig. S8A). Since ΔNp73 is known to suppress TAp73 activity through hetero-oligomerization (19), a loss of ΔNp73 would increase TAp73 transcriptional activity, leading to an increased expression of NOTCH1 and HES1. Similarly, we found that Notch1 NTM (transmembrane/intracellular region), NICD, and Hes1 proteins were increased in E12-KO H1299 and MIA PaCa-2 cells (Fig. 5B and SI Appendix, Fig. S8B). To validate that p73α1 was responsible for Notch1 induction, NOTCH1 and HES1 mRNAs were measured in E12−/− cells wherein p73α1 was knocked down by siRNA. We found that knockdown of p73α1 led to a decreased expression of NOTCH1 and HES1 (Fig. 5C).
Fig. 5.

Notch1 is a direct target of p73α1 and highly expressed in inflamed E12+/− mouse tissues. (A) The level of NOTCH1, HES1, and HPRT1 transcripts was measured in isogenic control, TAp73−/−, ΔNp73−/−, and E12−/− H1299 cells. (Right) qPCR was used to analyze relative NOTCH1 and HES1 expression. One-way ANOVA was used to calculate P values. **P < 0.01; ****P < 0.0001. (B) The level of TAp73α, Notch1 NTM, NICD, Hes1, and vinculin proteins was measured in isogenic control, TAp73−/−, ΔNp73−/−, and E12−/− H1299 cells. (C) The level of p73α, p73α1, NOTCH1, HES1, and HPRT1 transcripts was measured in isogenic control and E12−/− H1299 cells transiently transfected with Scr or si-E11#1 for 3 d. (Right) qPCR was used to analyze relative NOTCH1 and HES1 expression. One-way ANOVA was used to calculate P values. ***P < 0.001; ****P < 0.0001. (D) ChIP analysis was performed with isogenic control and E12−/− H1299 cells. Cell lysates were immunoprecipitated with control IgG or anti-TAp73 overnight to bring down the DNA-protein immunocomplex, and the DNA fragments were visualized by PCR with primers for NOTCH1, CDKN1A, and GAPDH promoters. (E) The levels of Notch1, Hes1, and Actb transcripts were measured in liver tissue from age- and sex-matched WT and E12+/− mice (100 wk; Female). (Right) qPCR was used to analyze relative Notch1 and Hes1 expression. One-way ANOVA was used to calculate P values. **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) The level of Trp73α, Notch1, Hes1, and Actb transcripts was measured in salivary gland tissue from age- and sex-matched WT and E12+/− mice (100 wk; Female). (Right) qPCR was used to analyze relative Notch1 and Hes1 expression. **P < 0.01; ***P < 0.001; ****P < 0.0001. One-way ANOVA was used to calculate P values. (G) The level of Trp73α, Trp73α1, Notch1, Hes1, and Actb transcripts was measured in spleen tissue from age- and sex-matched WT and E12+/− mice (100 wk; Male). (Right) qPCR was used to analyze relative Notch1 and Hes1 expression. One-way ANOVA was used to calculate P values. *P < 0.05; ***P < 0.001.
Previous studies showed that the NOTCH1 promoter contains a putative p53-response element (p53-RE) (48, 49). Since p73 is known to bind p53-RE to regulate its target gene expression, a chromatin immunoprecipitation (ChIP) assay was performed and showed that the NOTCH1 promoter DNA fragment was detected in anti-TAp73 immunoprecipitates from both isogenic control and E12-KO H1299 cells (Fig. 5D). These results suggest that like p53, both TAp73α and TAp73α1 are capable of binding to the Notch1 promoter to regulate its expression. To determine whether the induction of Notch1 is correlated with inflammation in E12+/− mouse tissues, the levels of Notch1 and Hes1 mRNAs were measured and found to be significantly up-regulated in inflamed E12+/− liver, salivary gland, and spleen tissues compared to age- and sex-matched WT counterparts (Fig. 5 E–G and SI Appendix, Fig. S8D). These findings suggest that the isoform switch from p73α to p73α1 leads to an increased expression of Notch1, thus contributing to the widespread systemic inflammation in E12+/− mice.
In addition to promoting the inflammatory response, Notch1 is a context-dependent tumor suppressor (45). Thus, p73α1 might activate Notch1 to induce growth suppression, making E12+/− mice less tumor prone. To test this, colony formation and wound healing assays were performed with isogenic control and E12-KO H1299 cells in which Notch1 was knocked down by siRNA (Fig. 6A). As expected, knockdown of Notch1 had little if any effect on cell proliferation and migration in isogenic control cells (Fig. 6B and SI Appendix, Fig. S9), suggesting that knockdown of a low basal level of NICD would not have a measurable effect on cell growth. In contrast, we found that knockdown of Notch1 restored cell proliferation and migration in E12-KO cells (Fig. 6B and SI Appendix, Fig. S9). To confirm that Notch1 is necessary for p73α1-mediated tumor suppression, p73α1 was knocked down alone or together with Notch1 in E12-KO cells (Fig. 6 C and D). We showed that knockdown of p73α1 or Notch1 led to increased cell proliferation, which was not significantly further increased by a combined knockdown of p73α1 and Notch1 in E12-KO H1299 cells (Fig. 6E). Taken together, these data provide evidence that Notch1 plays a role in p73α1-dependent tumor suppression.
Fig. 6.

Notch1 is necessary for p73α1-mediated growth suppression in H1299 cells. (A) The level of Notch1 NTM, NICD, and vinculin proteins was measured in isogenic control and E12−/− H1299 cells transiently transfected with Scr or si-Notch1 for 3 d. (B) Cells were treated as in A and used for the colony formation assay. (Right) Quantification of colony formation assay using relative colony area. The relative colony area in isogenic control cells treated with Scr was arbitrarily set as 1.0. Data are presented as the mean ± SEM of three independent experiments. One-way ANOVA was used to calculate P values. *P < 0.05; **P < 0.01; ****P < 0.0001. (C) qPCR was used to analyze relative NOTCH1 expression in E12−/− H1299 cells transiently transfected with Scr, si-Notch1, or si-p73α1 and si-Notch1 for 3 d. One-way ANOVA was used to calculate P values. ****P < 0.0001. (D) The level of p73α1 and HPRT1 transcripts was measured in E12−/− H1299 cells transiently transfected with Scr, si-p73α1, or si-p73α1 and si-Notch1 for 3 d. (E) Colony formation assay was performed with E12−/− H1299 cells transiently transfected with Scr, si-p73α1, si-Notch1, or both for 3 d. (Right) Quantification of colony formation assay using relative colony area. The relative colony area in E12−/− cells treated with Scr was arbitrarily set as 1.0. Data are presented as the mean ± SEM of three independent experiments. One-way ANOVA was used to calculate P values. ***P < 0.001; ****P < 0.0001.
Discussion
p73α is the most abundant p73 isoform in human/mouse tissues and primarily responsible for p73-associated phenotypes. TAp73α is widely recognized as a tumor suppressor, as demonstrated by p73- and TAp73-KO mouse models. The complete and partial loss of p73 in mice is associated with increased tumor formation (44, 50–52). Consistently, TAp73-KO, but not ΔNp73-KO, mice are prone to genomic instability and spontaneous tumors (14, 18). Moreover, a loss of p73 is associated with tumor dissemination and transformation in multiple types of cancer (53–57). A recent study showed that isoform switch from p73α to p73β leads to profound neurological defects, but its effect on tumorigenesis was not analyzed (28). In the present study, we showed that isoform switch from p73α to p73α1 does not increase tumor formation, suggesting that TAp73α1 compensates for the loss of TAp73α and therefore functions as a tumor suppressor. Moreover, we speculate that p73α1 has a stronger transactivation activity than p73α due to differences at the extreme C terminus. p73α has a relatively low transactivation potential (25) and is the only isoform that contains both inhibitory domains SAM (58–60) and TID (29). SAM and TID can block the binding of coactivators p300/CBP to p73 TAD, therefore attenuating the transactivation potential of p73α (29). These data indicate that despite its relatively low abundance, TAp73α1 has a strong transactivation potential and plays an important role in p73-dependent tumor suppression.
It is well defined that p73α regulates tumor suppression through both common and distinct p53 family targets, such as p21, PUMA, and HIF-1α (15, 61, 62). Similarly, RNA-seq analysis showed that p73α1 regulates several p53 family targets, such as CDKN1A and PERP, suggesting that p73α1 has a similar mode of tumor suppression as other p53 family members. However, since E12-deficient mice are prone to inflammation but not spontaneous tumors, we speculated that a p73 target may have a dual function in growth suppression and inflammation. Indeed, we found that Notch1, a master regulator of inflammatory response, is induced by p73α1 in E12-KO cells and E12-deficient mice (Fig. 5). Our data indicate that the induction of Notch1/p21/p16 and cellular senescence by p73α1 plays a role in mitigating the susceptibility of E12+/− mice to spontaneous tumors. This observation is consistent with published studies that Notch1 functions as a tumor suppressor in a p53-dependent manner (48, 49, 63) by suppressing oncogenic kinases (ROCK1/2 and MRCKα) (48) or Wnt signaling (64, 65). Additionally, studies showed that normal levels of Notch1 are necessary for maintaining malignant tumor growth, whereas overexpression of Notch1 inhibits tumor progression (66, 67). Moreover, Notch1 is decreased in late-stage cervical tumors, suggesting that excessive Notch1 signaling impairs tumor invasion and metastasis (68).
Mice deficient in total p73 or TAp73 are prone to chronic inflammation (7, 44, 50–52). Whether or not mice deficient in ΔNp73 or E13 (p73α to p73β isoform switch) are susceptible to chronic inflammation has not been reported (18, 28, 30). Here, we found that mice deficient in E12 are prone to multiorgan inflammation along with an increased expression of TNFα in inflamed tissues. Mechanistically, our studies revealed three possible pathways by which p73α1 promotes systemic inflammation. First, the RNA-seq analysis showed that E12-KO leads to an induction of NOTCH1, its ligands (JAG1 and JAG2), and its targets (HES1 and HEY1). Previous studies showed that JAG1 and JAG2 are regulated by the p53 family proteins (69, 70). Thus, induction of NOTCH1 and its ligands by p73α1 would amplify the potency of Notch1 to promote proinflammatory cytokine production (71), leading to systemic inflammation. Second, we showed that MEFs and mouse tissues deficient in E12 are prone to cellular senescence. Despite their growth-arrested state, senescent cells are metabolically active and secrete an array of proteins that constitute the senescence-associated secretory phenotype (SASP). SASP includes growth factors and cytokines, such as IL-1β, IL-6, and IFN-γ. Thus, increased cytokine production by senescent cells may contribute to p73α1-mediated inflammation. Third, our RNA-seq analysis showed that E12-KO modulates several pro- and anti-inflammatory pathways, including NF-κB and SOCS3 (72, 73). While much work is needed to validate these RNA-seq data, it is likely that some of these alterations may directly, or together with Notch1 and SASP, promote p73α1-dependent systemic inflammation.
In summary, we identified p73α1 as a naturally expressed C-terminal isoform. Despite its relatively low abundance, p73α1 has a strong transactivation potential due to a lack of C-terminal inhibitory domains and can compensate for the loss of TAp73α in tumor suppression. We also revealed that p73α1 regulates tumor suppression and the inflammatory response in part through Notch1. Considering that the function of multiple p73 C-terminal isoforms is unclear, exon exclusion by CRISPR to induce isoform switch should be further explored to determine how p73 exerts its activity in tumor suppression.
Materials and Methods
Reagents.
Anti-Actin (catalog number [Cat#] sc-47778, 1:2,000), anti-p130 (Cat# sc-374521, 1:3,000), anti-p21 (Cat# sc-53870, 1:3,000), anti-p130 (Cat# sc-374521, 1:3,000), and anti-PML (Cat# sc-377390, 1:3,000) were purchased from Santa Cruz Biotechnology. Anti-TAp73 (Cat# A300-126A, 1:1,000) was purchased from Bethyl Laboratories, Inc. Anti-HA (Cat# 901513, 1:2,000) was purchased from BioLegend. Anti-Cleaved Notch1 (Cat# 4741, 1:1,000), anti-Notch1 (Cat# 4380T, 1:1,000), and anti-Hes1 (Cat# 11988S, 1:800) were purchased from Cell Signaling Technology. WesternBright ECL HRP substrate (Cat# K-12043-D20) was purchased from Advansta. Scrambled siRNA (5′-GGC CGA UUG UCA AAU AAU U-3′), sip73α1 siRNA (5′-ACC UGG GGC CCG UGG UUU-3′), siE11 siRNA#1 (5′-GCA CAG UUC GGC AGC UAC A-3′), siE11 siRNA#2 (5′-UCC UCU CGC CCA UGA ACA A-3′), and siNotch1 siRNA (5′-ACA AAG AUA UGC AGA ACA A-3′) were purchased from Horizon Discovery Biosciences Limited. RNAiMax (Cat# 13778150, Invitrogen), Protease Inhibitor Mixture (Cat# 78438), Magnetic Protein A/G beads (Cat# 78609), RevertAid RT Reverse Transcription Kit (Cat# K1691), and DreamTaq DNA Polymerase (Cat# EP0702) were all purchased from ThermoFisher. All reagents were used according to the manufacturer’s protocol.
Plasmid Generation.
The pSpCas9(BB)-2A-Puro expression vector was generated by the Zhang Lab (74) and purchased from Addgene. To generate a vector expressing a single guide RNA (sgRNA) that targets TAp73, ΔNp73, or E12, two 25-nt oligos were annealed and cloned into the pSpCas9(BB)-2A-Puro expression vector via BbsI. To generate the pcDNA4 vector expressing HA-tagged TAp73α1, a 620-bp cDNA fragment was amplified from H1299 E12−/− cells and then used to replace the C terminus of the previously generated HA-TAp73α vector (29) via EcoRI and XhoI. All primer sequences were listed in SI Appendix, Table S4.
Cell Culture and Cell Line Generation.
H1299 cells and their derivatives, HCT116 cells, MCF7 cells, and RKO cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat# A4766801). H1299, HCT116, MCF7, and RKO cell lines were purchased from ATCC;tested negative for mycoplasma; and used at passage 20 or lower. We did not authenticate the cell lines used in this study because ATCC has thoroughly authenticated these cell lines. To generate TAp73−/−, ΔNp73−/−, and E12−/− cell lines using CRISPR/Cas9, H1299 cells were transfected with a pSpCas9(BB)-2A-Puro vector expressing a sgRNA, and subsequently selected with puromycin (0.66 µM) for 2 to 3 wk. Individual clones were picked ,and the appropriate knockout cell lines were confirmed via Sanger DNA sequencing and/or Western blot analysis. HA-TAp73α inducible H1299 cell lines were generated previously (75). To generate HA-TAp73α1 inducible H1299 cell lines, H1299 cells expressing a tetracycline repressor were transfected with a pcDNA4-HA-TAp73α1 vector. Cells were selected with zeocin (33 µM) for 2 to 3 wk, and individual clones were picked and confirmed via Western blot analysis. To induce TAp73α or TAp73α1 expression, 1 µM tetracycline was added to the media for 24 h.
Mouse Model Generation.
WT and Trp73+/− mice were generated as described previously (53). The E12-KO strategy was outlined in SI Appendix, Fig. S4A. E12-KO mice were generated by the Mouse Biology Program at University of California Davis. All animals and use protocols were approved by University of California Davis Institutional Animal Care and Use Committee. All genotyping primers were listed in SI Appendix, Table S5.
MEF Isolation.
To generate WT, E12+/−, and E12−/− MEFs, E12+/− mice were crossbred and MEFs were isolated from mouse embryos that were 12.5 to 13.5 d postcoitum, as described previously (76). MEFs were cultured in DMEM supplemented with 10% FBS, 55 μM β-mercaptoethanol, and 1× non-essential amino acids (Gibco, Cat# 11140050).
Histological Analysis.
Mouse tissue was fixed in 10% (wt/vol) neutral buffered formalin, processed, and embedded in paraffin blocks. Tissues blocks were sectioned at 6 μm and stained with Modified Meyer’s Hematoxylin (Richard Allan Scientific, Cat# 22-110-639) and Eosin-Y (Richard Allan Scientific, Cat# 22-110-637).
RNA Isolation, RT-PCR, and qPCR.
Total RNA was isolated with the TRIzol reagent according to the manufacturer’s manual, followed by cDNA synthesis using RevertAid Reverse Transcriptase. The PCR program used for amplification was 1) 94 °C for 5 min, 2) 94 °C for 30 s, 3) 60 to 63 °C for 30 s, 4) 72 °C for 30 s, and 5) 72 °C for 10 min. Steps 2 to 4 were repeated for 25 cycles to amplify ATCB, HPRT1, and GAPDH or 28 to 35 times to amplify other genes of interest. For qPCR, PowerUp Syber Green Master Mix (Applied Biosystems, Cat# A25742) was used according to the manufacturer’s protocol. All primers used for RT-PCR and qPCR were listed in SI Appendix, Table S5.
Western Blot Analysis.
Western blot analysis was performed as previously described (77). Briefly, the whole-cell lysate was harvested with 1× sodium dodecyl-sulfate (SDS) lysis buffer. Proteins were separated in a 8 to 10% SDS polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with the indicated primary and secondary antibodies. The proteins were visualized by enhanced chemiluminescence using VisionWorksLS software.
ChIP Assay.
The ChIP assay was performed as previously described (78). Briefly, chromatin was cross-linked in 1% formaldehyde in phosphate-buffered saline (PBS). Chromatin lysates were sonicated to yield ∼200- to 1,000-base pair DNA fragments and immunoprecipitated with 1 μg of control IgG or anti-TAp73 antibody at 4 °C overnight. The protein–DNA immunocomplex was reverse crosslinked and purified using ChIP DNA Clean & Concentrator (Cat# 50-44-363) from Zymo Research. PCR was used to amplify the DNA fragments. Primers used for ChIP assays were listed in SI Appendix, Table S5.
Colony Formation Assay.
A total of 103 cells was seeded per well in a six-well plate and cultured for 15 d. At the end point, cells were fixed by methanol/glacial acetic acid (7:1, vol/vol) and then stained with 0.1% crystal violet solution. Relative colony area was quantified using ColonyArea from ImageJ (79).
Spheroid Assay.
Cells were resuspended in MammoCult (Cat# 05620) (StemCell) and mixed with Matrigel (Cat# 354234) (Corning) in a 3:4 ratio. A total of 15 µL of cell/Matrigel mixture was plated in a ring-like shape in a well of 96-well plate and incubated at 37 °C for 20 min followed by the addition of 100 µL of MammoCult to the each well. At 72 h, cells were washed with PBS and treated with 50 µL of Dispase (5 mg/mL) (Corning, Cat# 354235) at 37 °C for 1.5 h. Cell viability was measured using the CellTiter-Glo 2.0 Cell Viability Assay kit (Cat# G9241) (Promega) according to the manufacturer’s protocol. The assay was performed in triplicates to ensure proper statistical analyses.
Wound Healing Assay.
Cells were seeded at a density of 4 × 105 in a 6-well plate and allowed to grow into monolayers. The monolayered cells were scraped with a P2 micropipette tip and washed two times with PBS. Microscopic images were taken at the indicated time points with phase contrast microscopy. Wound closure percentage was quantified using the ImageJ plugin MRI Wound Healing Tool.
Senescence Assay.
The senescence assay was performed as described previously (80). Briefly, 5 × 104 primary MEFs at passage five were seeded in one well of a 6-well plate for 24 h. Cells were washed with PBS, fixed with a solution of 2% formaldehyde/0.2% glutaraldehyde for 15 min at room temperature, and then incubated with an SA-β-galactosidase staining solution (1 mg/mL 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 40 mm citric acid/sodium phosphate [pH 6.0], 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 150 mm NaCl, and 2 mm MgCl2) at 37 °C overnight. The percentage of senescent cells was calculated by dividing the number of SA-β-galactosidase-positive cells by the total number of cells counted. For SA-β-galactosidase staining on tissue, fresh-frozen tissue was embedded in Tissue Plus O.C.T. (optimal cutting temperature) Compound (Fisher Healthcare, Cat# 23-730-571), cryo-sectioned at 10 μm, stained according to the protocol mentioned above, and counter stained with Nuclear Fast Red Solution (RICCA, Cat# R5463200-500A).
Growth Rate Determination.
To determine the rate of cell growth, 5x103 WT, E12+/-, and E12-/- MEFs were seeded in 60 mm diameter plates. At the time points indicated, two plates were rinsed two times with PBS to remove dead cells and debris. Live cells on the plates were trypsinized and collected separately. Cells from each plate were counted four times with the Coulter cell counter. The average number of cells from the plates was used to determine growth rate.
Statistical Analysis.
The Log-rank test was used for Kaplan–Meier survival analysis. Fisher’s exact test was used to determine statistical significance for the proportion of mice with inflammation, splenic hyperplasia, and EMH. One-way ANOVA was used to determine statistical significance in colony formation and tumor sphere assays and qPCR analysis. P < 0.05 was considered as statistically significant.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health R01 grants (CA081237 and CA224433) and UC Davis Cancer Center Core Support Grant CA093373 to X. Chen, by TRDRP T31IP1727 to J.Z., and T32 HL007013 to K.N.L. The authors would like to graciously thank Dr. Yanhong Zhang for her technical support, and the members of the Chen Lab for their suggestions.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2123202119/-/DCSupplemental.
Data Availability
All study data are included in the article and/or SI Appendix.
References
- 1.Ko L. J., Prives C., p53: Puzzle and paradigm. Genes Dev. 10, 1054–1072 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Schmale H., Bamberger C., A novel protein with strong homology to the tumor suppressor p53. Oncogene 15, 1363–1367 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Trink B., et al. , A new human p53 homologue. Nat. Med. 4, 747–748 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Osada M., et al. , Cloning and functional analysis of human p51, which structurally and functionally resembles p53. Nat. Med. 4, 839–843 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Kaghad M., et al. , Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90, 809–819 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Jost C. A., Marin M. C., Kaelin W. G. Jr., p73 is a human p53-related protein that can induce apoptosis. Nature 389, 191–194 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Yang A., et al. , p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404, 99–103 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Meyer G., Perez-Garcia C. G., Abraham H., Caput D., Expression of p73 and Reelin in the developing human cortex. J. Neurosci. 22, 4973–4986 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abraham H., Meyer G., Reelin-expressing neurons in the postnatal and adult human hippocampal formation. Hippocampus 13, 715–727 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Marshall C. B., et al. , p73 is required for multiciliogenesis and regulates the Foxj1-associated gene network. Cell Rep. 14, 2289–2300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemajerova A., et al. , TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 30, 1300–1312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arrowsmith C. H., Structure and function in the p53 family. Cell Death Differ. 6, 1169–1173 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Liu G., Nozell S., Xiao H., Chen X., DeltaNp73β is active in transactivation and growth suppression. Mol. Cell. Biol. 24, 487–501 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomasini R., et al. , TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 22, 2677–2691 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J., Jiang J., Zhou W., Chen X., The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res. 58, 5061–5065 (1998). [PubMed] [Google Scholar]
- 16.Melino G., et al. , p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 279, 8076–8083 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Vernole P., et al. , TAp73α binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle 8, 421–429 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Wilhelm M. T., et al. , Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 24, 549–560 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaika A. I., et al. , DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J. Exp. Med. 196, 765–780 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steder M., et al. , DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell 24, 512–527 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Concin N., et al. , Transdominant DeltaTAp73 isoforms are frequently up-regulated in ovarian cancer. Evidence for their role as epigenetic p53 inhibitors in vivo. Cancer Res. 64, 2449–2460 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Müller M., et al. , TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 12, 1564–1577 (2005). [DOI] [PubMed] [Google Scholar]
- 23.De Laurenzi V., et al. , Two new p73 splice variants with different transcriptional activity. J. Exp. Med. 188, 1763–1768 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Laurenzi V. D., et al. , Additional complexity in p73: Induction by mitogens in lymphoid cells and identification of two new splicing variants ε and ζ. Cell Death Differ. 6, 389–390 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Ueda Y., Hijikata M., Takagi S., Chiba T., Shimotohno K., New p73 variants with altered C-terminal structures have varied transcriptional activities. Oncogene 18, 4993–4998 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Rufini A., et al. , p73 in cancer. Genes Cancer 2, 491–502 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grespi F., Amelio I., Tucci P., Annicchiarico-Petruzzelli M., Melino G., Tissue-specific expression of p73 C-terminal isoforms in mice. Cell Cycle 11, 4474–4483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amelio I., et al. , The C terminus of p73 is essential for hippocampal development. Proc. Natl. Acad. Sci. U.S.A. 117, 15694–15701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu G., Chen X., The C-terminal sterile α motif and the extreme C terminus regulate the transcriptional activity of the α isoform of p73. J. Biol. Chem. 280, 20111–20119 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Buckley N., et al. , P73 C-terminus is dispensable for multiciliogenesis. Cell Cycle 19, 1833–1845 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaika A. I., Kovalev S., Marchenko N. D., Moll U. M., Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res. 59, 3257–3263 (1999). [PubMed] [Google Scholar]
- 32.Yokomizo A., et al. , Overexpression of the wild type p73 gene in human bladder cancer. Oncogene 18, 1629–1633 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Chen C.-L., Ip S.-M., Cheng D., Wong L.-C., Ngan H. Y. S., P73 gene expression in ovarian cancer tissues and cell lines. Clin. Cancer Res. 6, 3910–3915 (2000). [PubMed] [Google Scholar]
- 34.Pan H., Liao S.-J., Lai W.-Y., Lu H.-C., Hsiao K.-M., Overexpression but lack of mutation and methylation of p73 in hepatocellular carcinoma. Acta Oncol. 41, 550–555 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Vikhreva P., Melino G., Amelio I., p73 alternative splicing: Exploring a biological role for the C-terminal isoforms. J. Mol. Biol. 430, 1829–1838 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X., Zheng Y., Zhu J., Jiang J., Wang J., p73 is transcriptionally regulated by DNA damage, p53, and p73. Oncogene 20, 769–774 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Marabese M., Vikhanskaya F., Rainelli C., Sakai T., Broggini M., DNA damage induces transcriptional activation of p73 by removing C-EBPalpha repression on E2F1. Nucleic Acids Res. 31, 6624–6632 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urist M., Tanaka T., Poyurovsky M. V., Prives C., p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 18, 3041–3054 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krecic A. M., Swanson M. S., hnRNP complexes: Composition, structure, and function. Curr. Opin. Cell Biol. 11, 363–371 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Jiménez-Vacas J. M., et al. , Dysregulation of the splicing machinery is directly associated to aggressiveness of prostate cancer. EBioMedicine 51, 102547 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fei T., et al. , Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl. Acad. Sci. U.S.A. 114, E5207–E5215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y., Ma M., Xiao X., Wang Z., Intronic splicing enhancers, cognate splicing factors and context-dependent regulation rules. Nat. Struct. Mol. Biol. 19, 1044–1052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H. J., et al. , Ninjurin 1 has two opposing functions in tumorigenesis in a p53-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 114, 11500–11505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y., et al. , Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 31, 1243–1256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dotto G. P., Notch tumor suppressor function. Oncogene 27, 5115–5123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shang Y., Smith S., Hu X., Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 7, 159–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radtke F., Raj K., The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat. Rev. Cancer 3, 756–767 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Lefort K., et al. , Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 21, 562–577 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yugawa T., et al. , Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell. Biol. 27, 3732–3742 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J., et al. , FDXR regulates TP73 tumor suppressor via IRP2 to modulate aging and tumor suppression. J. Pathol. 251, 284–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J., et al. , Mutant p53 antagonizes p63/p73-mediated tumor suppression via Notch1. Proc. Natl. Acad. Sci. U.S.A. 116, 24259–24267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flores E. R., et al. , Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7, 363–373 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Loukopoulos P., et al. , Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: Identification of genetic indicators that predict patient outcome. Cancer Sci. 98, 392–400 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nemajerova A., Petrenko O., Trümper L., Palacios G., Moll U. M., Loss of p73 promotes dissemination of Myc-induced B cell lymphomas in mice. J. Clin. Invest. 120, 2070–2080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nemajerova A., Palacios G., Nowak N. J., Matsui S., Petrenko O., Targeted deletion of p73 in mice reveals its role in T cell development and lymphomagenesis. PLoS One 4, e7784 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson J., et al. , p73 loss triggers conversion to squamous cell carcinoma reversible upon reconstitution with TAp73alpha. Cancer Res. 67, 7723–7730 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Chi S.-G., et al. , Elevated and biallelic expression of p73 is associated withprogression of human bladder cancer. Cancer Res. 59, 2791–2793 (1999). [PubMed] [Google Scholar]
- 58.Thanos C. D., Bowie J. U., p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Sci. 8, 1708–1710 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chi S. W., Ayed A., Arrowsmith C. H., Solution structure of a conserved C-terminal domain of p73 with structural homology to the SAM domain. EMBO J. 18, 4438–4445 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ozaki T., et al. , Deletion of the COOH-terminal region of p73α enhances both its transactivation function and DNA-binding activity but inhibits induction of apoptosis in mammalian cells. Cancer Res. 59, 5902–5907 (1999). [PubMed] [Google Scholar]
- 61.Bálint E., Phillips A. C., Kozlov S., Stewart C. L., Vousden K. H., Induction of p57(KIP2) expression by p73β. Proc. Natl. Acad. Sci. U.S.A. 99, 3529–3534 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stantic M., et al. , TAp73 suppresses tumor angiogenesis through repression of proangiogenic cytokines and HIF-1α activity. Proc. Natl. Acad. Sci. U.S.A. 112, 220–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alimirah F., Panchanathan R., Davis F. J., Chen J., Choubey D., Restoration of p53 expression in human cancer cell lines upregulates the expression of Notch1: Implications for cancer cell fate determination after genotoxic stress. Neoplasia 9, 427–434 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nicolas M., et al. , Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33, 416–421 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Devgan V., Mammucari C., Millar S. E., Brisken C., Dotto G. P., p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 19, 1485–1495 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao J., Duan L., Fan M., Yuan J., Wu X., Notch1 induces cell cycle arrest and apoptosis in human cervical cancer cells: Involvement of nuclear factor kappa B inhibition. Int. J. Gynecol. Cancer 17, 502–510 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Wang L., et al. , Overexpressed active Notch1 induces cell growth arrest of HeLa cervical carcinoma cells. Int. J. Gynecol. Cancer 17, 1283–1292 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Talora C., Sgroi D. C., Crum C. P., Dotto G. P., Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev. 16, 2252–2263 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sasaki Y., et al. , The p53 family member genes are involved in the Notch signal pathway. J. Biol. Chem. 277, 719–724 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Buckley N. E., et al. , BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 41, 8601–8614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Colombo M., et al. , Cancer cells exploit notch signaling to redefine a supportive cytokine milieu. Front. Immunol. 9, 1823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carow B., Rottenberg M. E., SOCS3, a major regulator of infection and inflammation. Front. Immunol. 5, 58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tak P. P., Firestein G. S., NF-kappaB: A key role in inflammatory diseases. J. Clin. Invest. 107, 7–11 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ran F. A., et al. , Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dohn M., Nozell S., Willis A., Chen X., “Tumor suppressor gene-inducible cell lines” in Methods in Molecular Biology, W. S. El-Deiry, Ed.(Humana Press, 2003), pp. 221–235. [DOI] [PubMed] [Google Scholar]
- 76.Scoumanne A., Cho S. J., Zhang J., Chen X., The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res. 39, 213–224 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen X., Bargonetti J., Prives C., p53, through p21 (WAF1/CIP1), induces cyclin D1 synthesis. Cancer Res. 55, 4257–4263 (1995). [PubMed] [Google Scholar]
- 78.Liu G., Xia T., Chen X., The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 278, 17557–17565 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Guzmán C., Bagga M., Kaur A., Westermarck J., Abankwa D., ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 9, e92444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dimri G. P., et al. , A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or SI Appendix.