

Abstract
Antibody-drug conjugates (ADCs) hold great promise for targeted cancer cell killing. Site-specific antibody-drug conjugation is highly desirable for synthesizing homogeneous ADCs with optimal safety profiles and high efficacy. We have recently reported that azide-functionalized disaccharide oxazolines of the Manβ1,4GlcNAc core were an efficient substrate of wild-type endoglycosidase Endo-S2 for Fc glycan remodeling and conjugation. In this paper, we report the synthesis and evaluation of new disaccharide oxazolines as enzyme substrates for examining the scope of the site-specific conjugation. Thus, azide-functionalized disaccharide oxazolines derived from Manβ1,4GlcNAc, Glcβ1,4GlcNAc, and Galβ1,4GlcNAc (LacNAc) were synthesized. Enzymatic evaluation revealed that wild-type Endo-S2 demonstrated highly relaxed substrate specificity and could accommodate all the three types of disaccharide derivatives for transglycosylation to provide site-specific azide-tagged antibodies, which were readily clicked with a payload to generate homogeneous ADCs. Moreover, we also found that Endo-S2 was able to accommodate drug-preloaded minimal disaccharide oxazolines as donor substrates for efficient glycan transfer, enabling a single-step and site-specific antibody-drug conjugation without the need of an antibody click reaction. The ability of Endo-S2 to accommodate simpler and more easily synthesized disaccharide oxazoline derivatives for Fc glycan remodeling further expanded the scope of this bioconjugation method for constructing homogeneous antibody-drug conjugates in a single-step manner. Finally, cell-based assays indicated that the synthetic homogeneous ADCs demonstrated potent targeted cancer cell killing.
Graphical Abstract
INTRODUCTION
Antibody–drug conjugates (ADCs) have emerged as a novel class of anticancer agents that combines the specificity of antibodies and the high potency of cytotoxic drugs.1 So far, 12 ADCs have been approved by the US Food and Drug Administration (FDA) for the treatment of cancers,2 with dozens more at various stages of preclinical and clinical development. The first-generation ADCs are generated through non-specific conjugation,3 which usually results in heterogeneous mixtures with varied drug/antibody ratios (DARs) and different pharmacological properties. Recent studies have shown that site-specific ADCs could display improved pharmacokinetics, in vivo stability, and favorable safety profiles.4–6 Recent development in the linker technology and conjugation strategies have shown promise for a new generation of ADCs with more homogeneous structures.7,8 However, most strategies involve protein engineering of antibodies and subsequent site-selective conjugation, requiring extra steps of manipulations and purification.
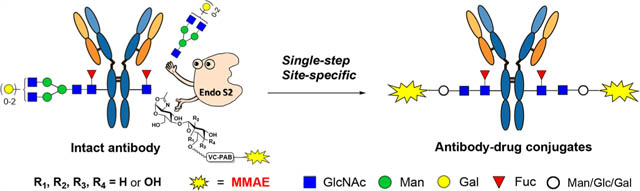
The conserved N-glycans attached to the Fc domain of IgG antibodies at Asn-297 that are spatially distant from the antigen-binding region offer an attractive conjugation site for the development of homogeneous ADCs.8–10 Significant progress has been made via the glycan-mediated chemoenzymatic bioconjugation of antibodies, which is enabled by the enzymatic transfer of azide- or other selectively modified sugars to antibodies via the catalysis of glycosyltransferases or endoglycosidase mutants.11–18 However, most of the chemoenzymatic approaches for ADC synthesis require three key steps: the deglycosylation at the Fc glycosite, the enzymatic transfer of a tagged sugar moiety, and the click conjugation with a payload. Recently, we have reported that selectively azide-tagged Man-β1,4-GlcNAc disaccharide oxazolines can act as donor substrates of several endoglycosidases, including the Endo-S and Endo-S2 from Streptococcus pyogenes, for Fc glycan remodeling with simultaneous deglycosylation and glycosylation without product hydrolysis, providing an efficient one-pot strategy for the labeling and conjugation of intact antibodies.18 The resulting azide-tagged antibodies can be efficiently converted to homogeneous ADCs with different antibody/drug ratios by subsequent click reactions.18 To further examine the scope of this method, we report herein the synthesis and evaluation of selectively modified new disaccharide oxazolines, including the Glc-β1,4-GlcNAc and Gal-β1,4-GlcNAc (LacNAc) disaccharides, as substrates for enzymatic Fc-glycan remodeling of antibodies. We found that wild-type Endo-S2 had a remarkable flexibility to accommodate the “unnatural core disaccharides” for transglycosylation to provide azide-functionalized antibodies (Figure 1). Moreover, we discovered that the wild-type Endo-S2 could perform a simultaneous deglycosylation and glycosylation of an antibody with the drug-loaded disaccharide oxazoline substrates to give homogeneous ADCs in a single step. The resulting ADCs showed high selectivity for the target cells as indicated in the cytotoxicity studies. This method opens a new avenue to producing homogeneous antibody-drug conjugates, which represents a truly single-step and site-specific bioconjugation of a payload to intact antibodies. During the preparation of this manuscript, Shi and co-workers have reported a nice and independent work on Endo-S2 catalyzed transfer of drug-modified Gal-β1,4-GlcNAc oxazolines to antibody to form antibody-drug conjugates, in which the drug and antibody are conjugated through an oxime linkage.19
Figure 1.
Design of the single-step synthesis of ADCs from intact antibodies.
RESULTS AND DISCUSSION
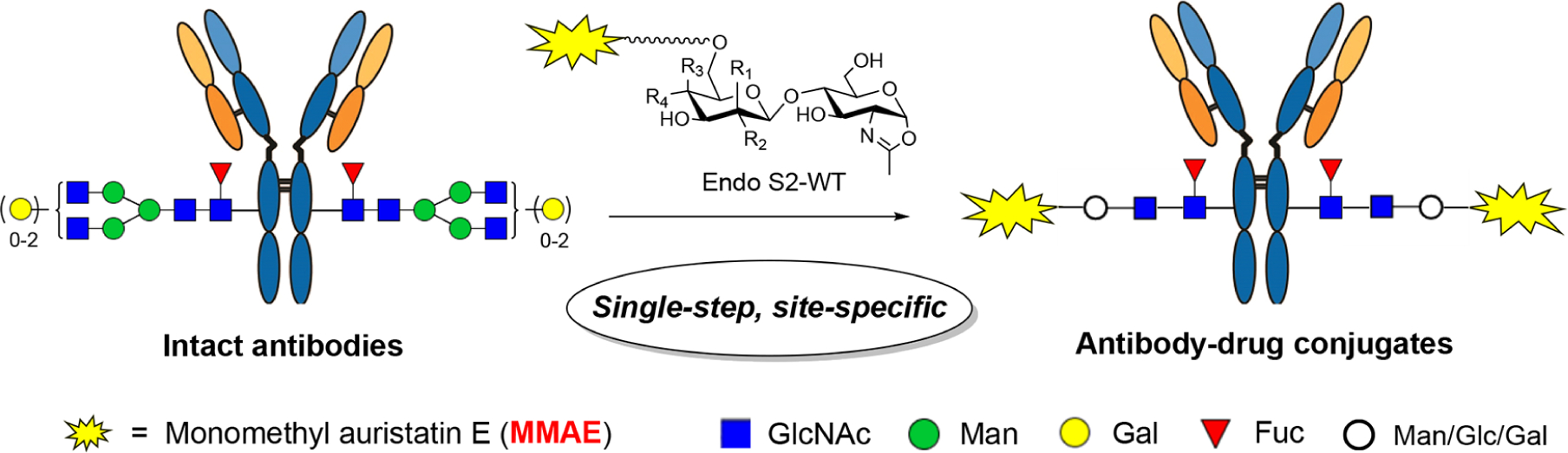
Our recent study has shown that wild-type Endo-S2 could accommodate selectively modified disaccharide oxazolines corresponding to the natural disaccharide (Manβ1,4GlcNAc) core for transglycosylation without product hydrolysis.18 However, whether this enzyme could recognize and transfer unnatural core disaccharide structures to antibodies remains to be tested. We sought to test simpler disaccharide derivatives, such as Glcβ1,4GlcNAc and Galβ1,4GlcNAc (LacNAc) oxazolines, which are much easier to synthesize than the Manβ1,4GlcNAc core. To explore the substrate specificity of Endo-S2 and to identify simple functionalized disaccharide oxazoline substrates for antibody glycan remodeling, we designed and synthesized three selectively modified disaccharide cores (Manβ1,4GlcNAc, Glcβ1,4GlcNAc, and Galβ1,4GlcNAc) with an amine or azide functional group at the 6′ position for further derivatization (Scheme 1). First, global deprotection of the known compound (4)18 afforded the Man-GlcNAc disaccharide (5) in almost quantitative yield. Under this condition, the azido group was simultaneously reduced to an amino group, which could be used for the introduction of functional groups. For the synthesis of the glucose-based disaccharide (Glc-GlcNAc), the benzoyl group in 720 was converted to benzyl group in two steps, giving 8 in 87% overall yield. Next, regioselective opening of the benzylidene moiety-with BH3·THF/Bu2BOTf furnished compound 9 with a free OH at the C-6 position in excellent yield. After the conversion of the 2-azido group into the 2-acetamido group with AcSH,21 an azido-tagged polyethylene chain was introduced at this position by treatment of 10 with NaH and N3(CH2CH2O)3Ts to give 11 in 85% yield. Catalytic hydrogenation removed all the protecting groups and afforded free glycan 12 in excellent yield. To test the substrate specificity of Endo-S2, the amine group in 12 was transformed back to an azido group by the copper-catalyzed diazo transfer reaction,22,23 giving 13 in 81% yield, which was further transformed to oxazoline 14 by treatment of DMC and Et3N in water. Finally, the galactose-based disaccharide was constructed by coupling of 1524 and 1618 under standard glycosylation conditions. Selective protection of the C6-OH with the TBDPS group at the Gal residue followed by per-benzylation after de-acetylation afforded 18 in 68% overall yield, which was transformed to 19 with AcSH in 79% yield. Upon the deprotection of TBDPS, the azido-tagged polyethylene chain was introduced to give 21, which, after global deprotection, provided 22 quantitatively. Subsequent diazo transfer reaction and oxazoline formation gave 24 in excellent yield (Scheme 1).
Scheme 1. Chemical Synthesis of Azido-Tagged Disaccharide Oxazolinesa.
aReagents and conditions: (a) Pd/C, H2, HCl (aq), THF/H2O, RT; (b) NaBrO3, Na2S2O4, EtOAc/H2O, RT; (c) DMC, Et3N, H2O, 0 °C; (d) CH3ONa, CH3OH, 50 °C; (e) BnBr, NaH, DMF, 0 °C ~ RT; (f) BH3·THF, Bu2BOTf, CH2Cl2, 0 °C; (g) AcSH, pyridine/CHCl3, 50 °C; (h) N3(CH2CH2O)3Ts, NaH, DMF, 0 °C ~ RT; (i) TfN3, K2CO3, CuSO4, CH2Cl2/MeOH/H2O, RT; (j) TMSOTf, 4 Å MS, CH2Cl2, −40 °C; (k) TBDPSCl, imidazole, DMF, RT; (l) TBAF, THF, 40 °C.
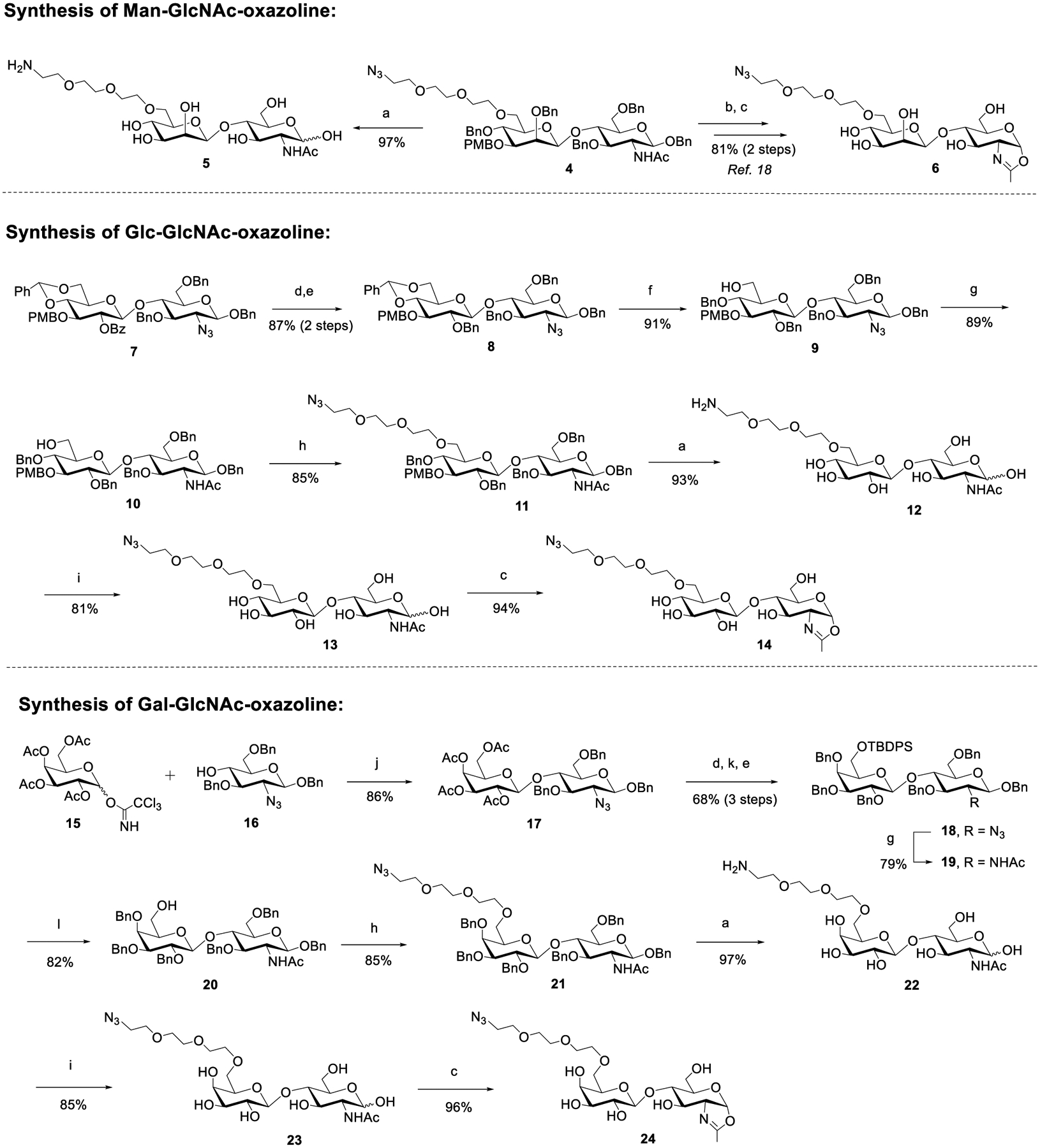
To test if the Glc- or Gal-containing disaccharide oxazolines (14 and 24) could still be recognized by wild-type Endo-S2 for Fc glycan remodeling of antibodies, we tried the one-pot transglycosylation with trastuzumab as the model antibody (Scheme 2). We found that disaccharide oxazolines 14 and 24 both could act as donor substrates for Endo-S2 catalyzed transglycosylation, but the enzymatic reactions were slower than that of the Man-β1,4-GlcNAc oxazoline (6) corresponding to the natural disaccharide core (Supporting Information, Figure S1). This result suggests that replacing the “natural” β-d-Man moiety with β-d-Glc or β-d-Gal residue reduced their substrate activity toward Endo-S2 to some extent, but Endo-S2 could still recognize them as donor substrates. Thus, more enzyme and/or prolonged reaction time were required to drive the transglycosylation to completion. We also observed that the Man-β1,4-GlcNAc oxazoline (6) could slowly be hydrolyzed by Endo-S2, while the azide-tagged Glc- or Gal-containing disaccharide oxazolines (14 and 24) were more resistant to Endo-S2 catalyzed hydrolysis. Nevertheless, the Endo-S2 catalyzed transglycosylation was much faster than the hydrolysis of the Man-β1,4-GlcNAc oxazoline (6). Thus, when an excess amount (20 equiv) of the disaccharide oxazoline (6) was used, a complete conversion of the antibody to the azide-tagged antibody product could be achieved within 30 min with 0.1% (w/w) of the enzyme under the reaction conditions, while the complete conversion with the azide-tagged Glc- or Gal-containing disaccharide oxazolines (14 and 24) would need over 2 h in the presence of 0.3 and 0.5% enzyme, respectively (Scheme 2). The ability of Endo-S2 to accept the Glcβ1,4GlcNAc and LacNAc oxazolines for transglycosylation is significant, as synthesis of these disaccharide derivatives are more efficient and straightforward than the Manβ1,4GlcNAc derivative as we have previously reported.18 The transglycosylation products, 26 and 27, were purified by a protein A affinity column, and their identity was confirmed by LC-ESI-MS analysis (Figures S2–S4).
Scheme 2. Transglycosylation of Azido-Tagged Disaccharide Oxazolinesa.
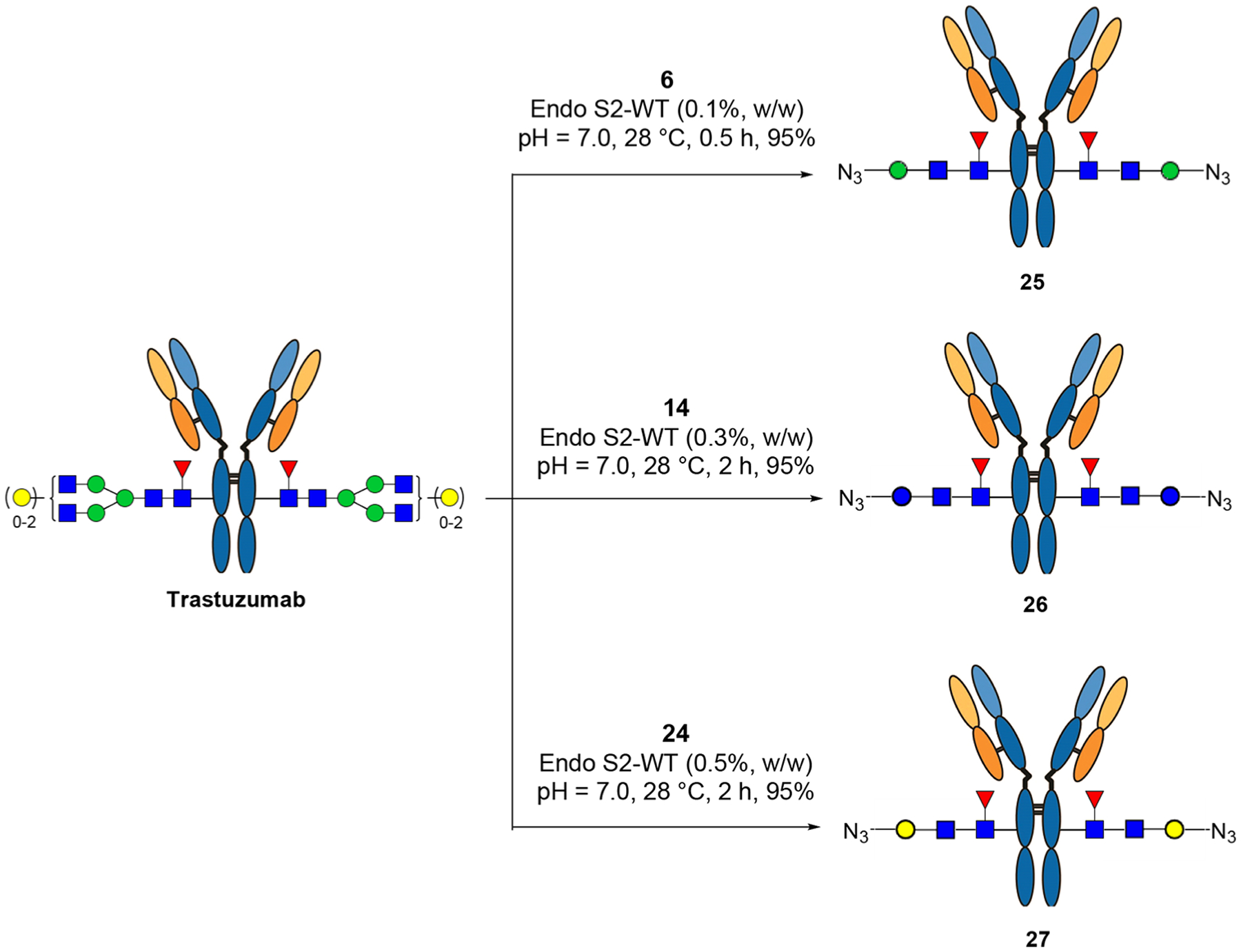
aThe reactions were performed in PBS buffer with an antibody concentration of 20 mg/mL, and 20 equiv of oxazoline was used (for each reaction site); the yields were based on the LC-MS peak intensity.
Encouraged by the excellent transglycosylation activities of the azido-tagged disaccharide oxazolines, we sought to pre-introduce the cytotoxic drug to the disaccharide oxazolines and to test the feasibility of a single-step glycan remodeling for constructing ADCs. To achieve this strategy, several factors need to be considered. First, the drug should tolerate the DMC/Et3N treatment for sugar oxazoline formation. Second, it needs to be stable under alkaline conditions. Lastly, the drug-disaccharide oxazoline should have sufficient solubility in aqueous solution. We chose monomethyl auristatin E (MMAE) as the cytotoxic drug to test the strategy. Thus, compound 2817 bearing a cleavable dipeptide linker (valine-citrulline) was reacted with bis-NHS-PEG5 to give the NHS-activated payload 29, which was further conjugated with the disaccharides via the amine-coupling reaction followed by oxazoline formation in one-pot, providing the drug-oxazoline conjugates 1–3 in good yields after RP-HPLC purification (Scheme 3).
Scheme 3. Synthesis of Drug-Oxazoline Conjugates (1–3)a.
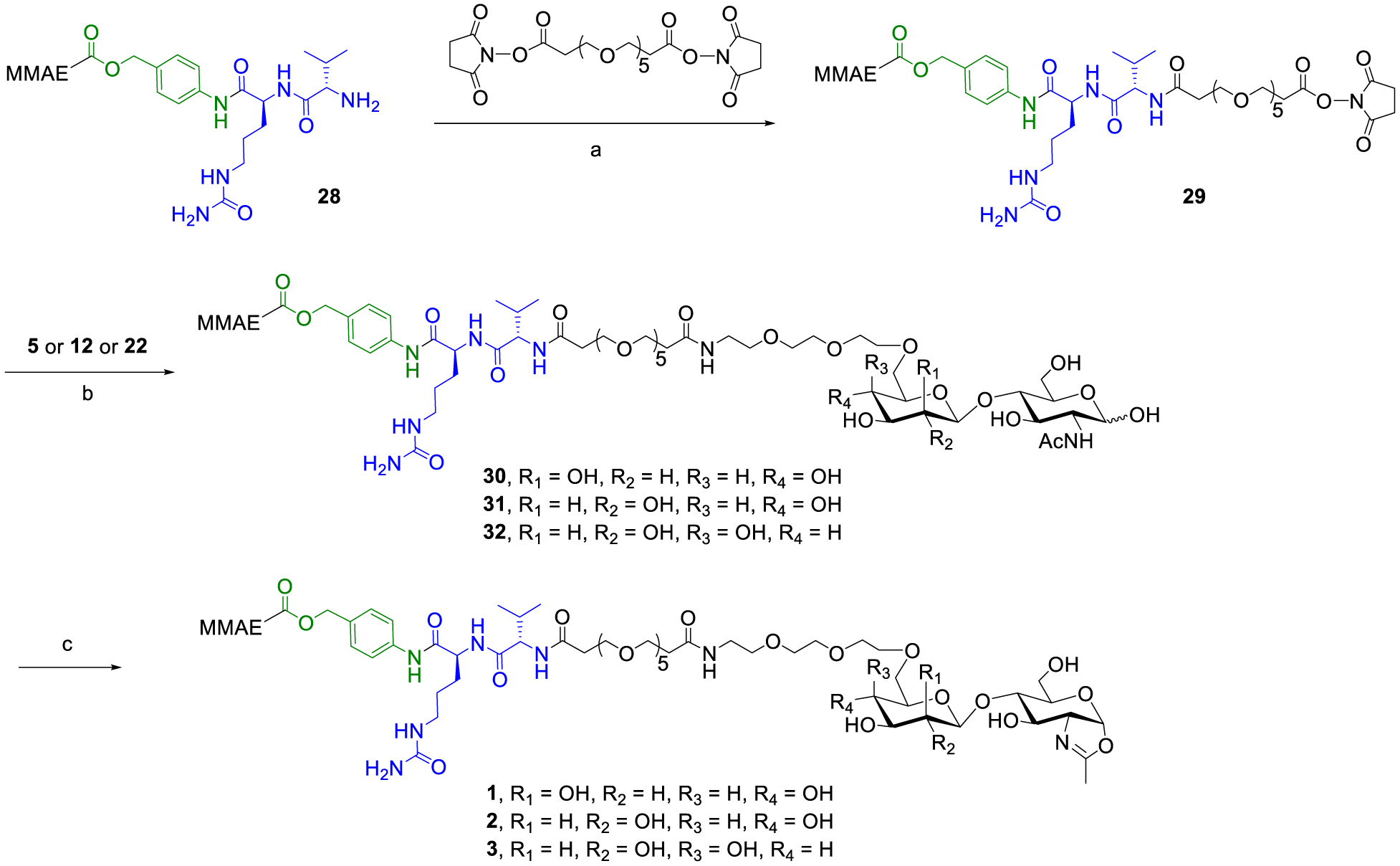
aReagents and conditions: (a) Et3N, DMSO, RT, 82%; (b) Et3N, DMSO, RT; (c) DMC, Et3N, H2O/DMSO, 0 °C, 1, 73% for 2 steps, 2, 80% for 2 steps, 3, 70% for 2 steps.
With the drug-sugar oxazoline conjugates in hand, next we investigated the one-step synthesis of ADCs via the Endo-S2-catalyzed transglycosylation (Scheme 4). Notably, the oxazolines 1–3 could not be completely dissolved in aqueous buffer solution due to the hydrophobicity of MMAE and the cleavable linker; thus, 5% DMSO was added to help dissolve the oxazolines in the reaction solution. It was found that the Man-derived drug-oxazoline conjugate (1) acted as an excellent substrate of wild-type Endo-S2, and the reaction reached completion within 1 h with a catalytic amount of enzyme (0.2%, enzyme to antibody, w/w) and 15 equiv of the sugar oxazoline donor (1). Once formed, the product was largely resistant to hydrolysis under the reaction conditions, thus offering a practical one-step method for the preparation of homogeneous ADCs with a precise control of DAR. As for Glc- and Gal-derived drug-oxazolines (2 and 3, respectively), a relatively larger amount of enzyme and oxazoline donors and longer reaction time were required to complete the transformation (Figure S5). In all the cases, the corresponding antibody-drug conjugates (33, 34, and 35) were obtained. To verify that the linker-payloads were specifically attached to the Fc domain, the ADCs (33, 34, and 35) were digested with protease IdeS, which cleaves the antibody to yield the monomeric Fc domain followed by LC-ESI-MS analysis (Figures S6–S8). The results showed high homogeneity of the products. The LC-ESI-MS analysis of the intact ADCs (33–35) showed that the shifts of molecular weight were consistent with the attached payloads (for the drug-conjugated Fc domain, calculated, M = 26,056; observed, 26,057, deconvoluted data), thus further confirming the structure of the products (Figure 2). The findings that Endo-S2 can perform simultaneous antibody deglycosylation and transfer of a payload-conjugated disaccharide oxazoline further expands the scope of the chemoenzymatic Fc glycan remodeling method for synthesizing homogeneous antibody-drug conjugates.
Scheme 4. Single-Step Transglycosylation of Drug-Oxazoline Conjugates to Make ADCs (33–35)a.
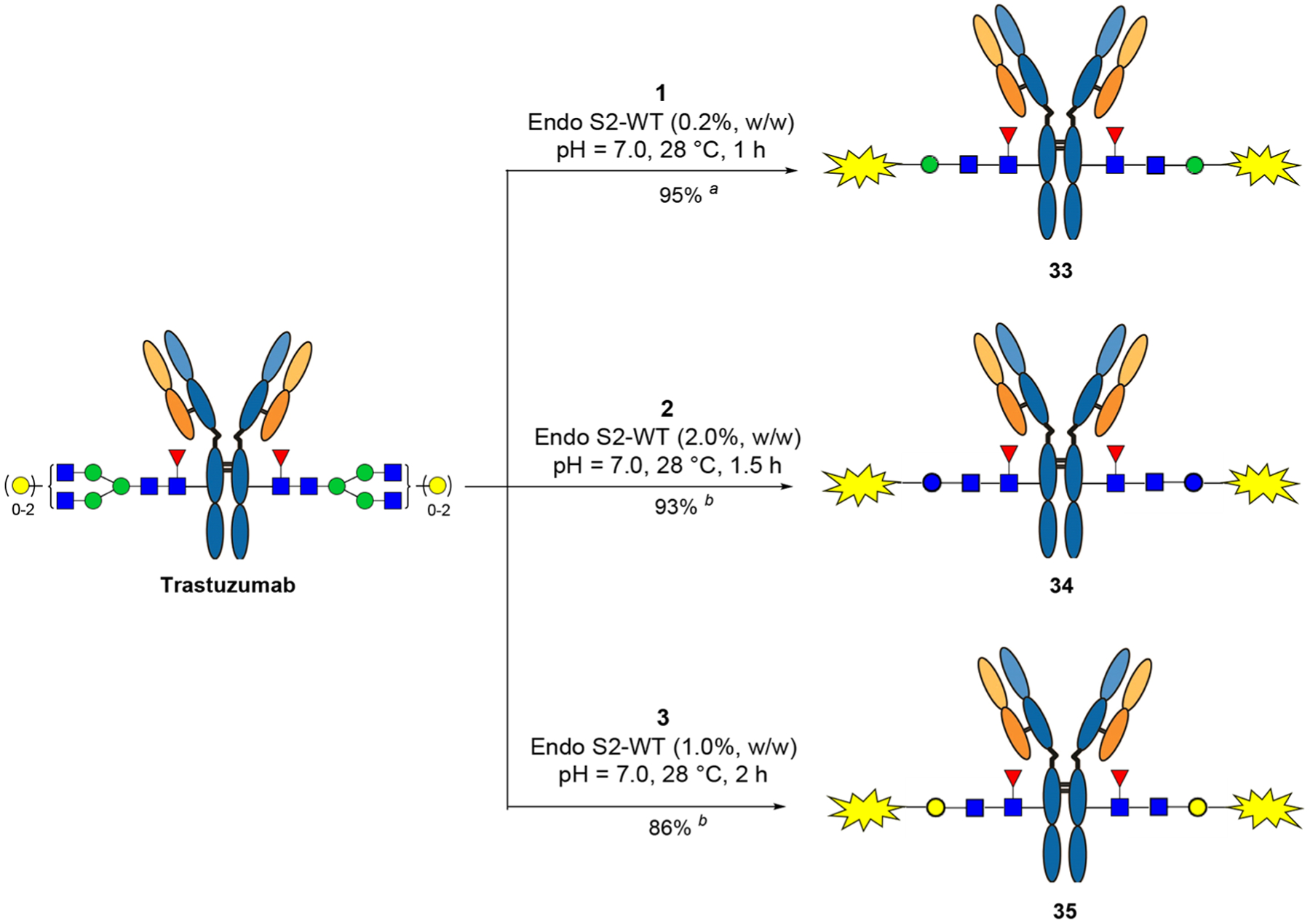
aThe reactions were performed in a PBS buffer with 5% DMSO, and the antibody concentration was 20 mg/mL (isolated yields after protein A purification). For each reaction site: (a) 15 equiv of oxazoline was used; (b) 25 equiv of oxazoline was added in two portions.
Figure 2.
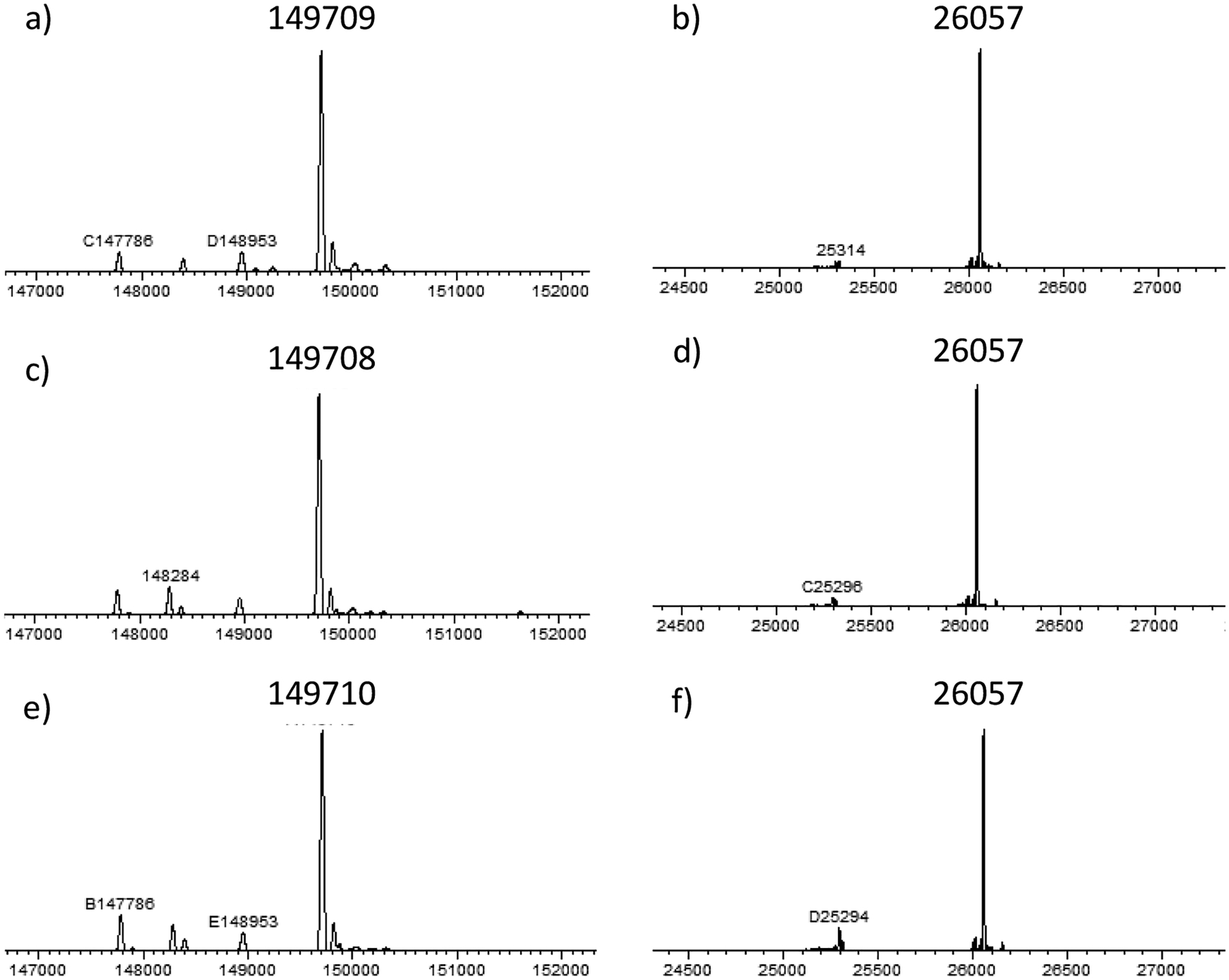
LC-ESI-MS analysis of the intact ADCs (33–35) and the Fc domains released by IdeS treatment (deconvoluted data). (a) Intact ADC 33; (b) Fc domain of 33; (c) intact ADC 34; (d) Fc domain of 34; (e) intact ADC 35; (f) Fc domain of 35.
Finally, we tested the cytotoxicity of the synthetic ADCs (33, 34, and 35) in breast cancer cell lines SK-BR-3 and BT474 that have high levels of HER2 expression, and T47D that has low level expression of HER2 antigen (Figure 3). The trastuzumab-MMAE conjugate (36) (with a DAR of 2) that we have synthesized previously using a two-step approach18 was used as a reference for comparison. We found that all these ADCs achieved significant killing of the high antigen-expressed cell lines (SK-BR-3 and BT474) with almost the same potency, as indicated by the EC50 values. As for the T47D line that expresses the low level of HER2 antigen, no apparent killing was observed under the same concentrations, indicating the high selectivity of the synthetic ADCs for the target cells. The anti-cancer potency of the ADCs (33, 34, and 35) synthesized by this disaccharide conjugation method was quite comparable to those ADCs produced through the full N-glycan conjugation method that we have reported previously.17 For example, for the killing of the SK-BR-3 cell line, the present ADCs (33–36) gave an EC50 of 41–71 pM, while the ADCs made from the full-glycan conjugation method gave an EC50 of 88–162 pM. It should be noted that the full glycan conjugation ADCs have a drug-to-antibody ratio (DAR) of 4, while the disaccharide-conjugated ADCs presented here have a DAR of 2. The cell-based assay data indicate the comparable efficiency of the two types of ADCs. Further evaluation and comparison of these ADCs in animal models are in progress, and the results will be reported in due course.
Figure 3.
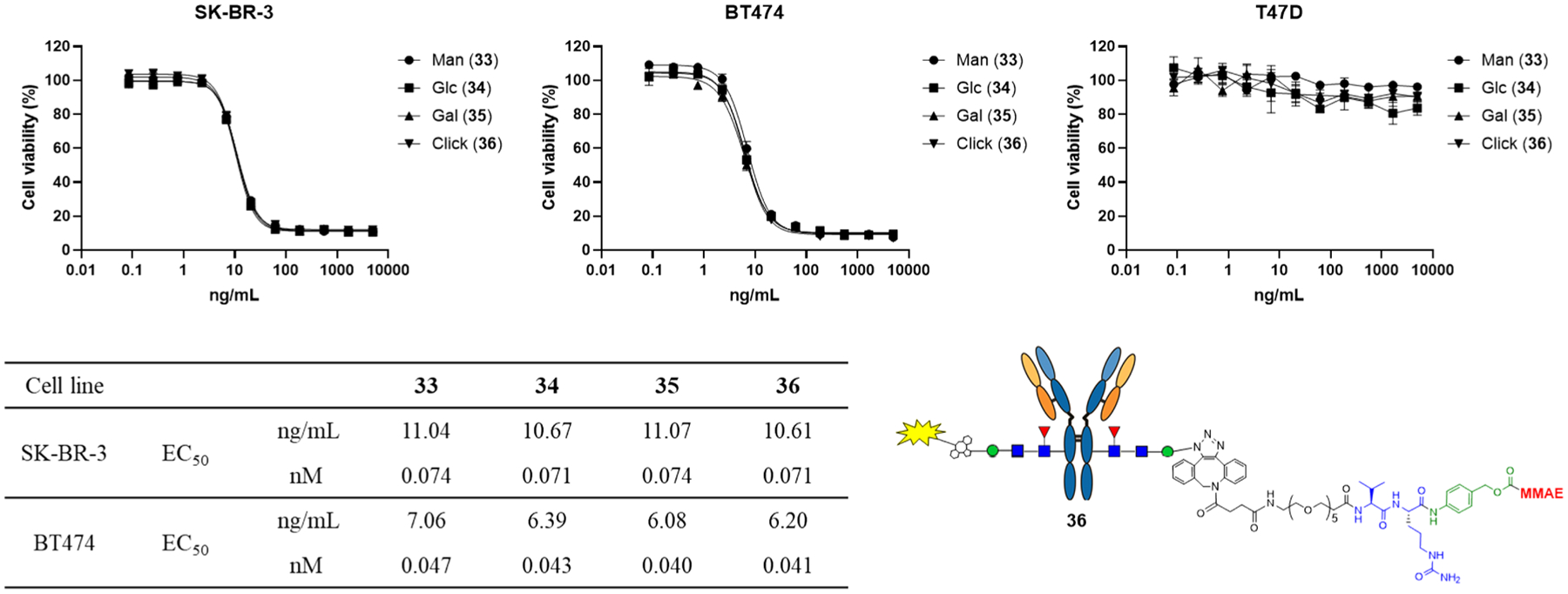
Cytotoxicity assays with breast cancer cell line SK-BR-3, BT474 (HER2 high-expression) and T47D (HER2 low-expression). All assays were performed in triplicate.
CONCLUSIONS
A highly efficient method for a single-step and site-specific chemoenzymatic synthesis of homogeneous antibody-drug conjugates is established. The findings that Endo-S2 can accept different disaccharide oxazolines, including the simpler and more easily synthesized cellobiose and N-acetyllactosamine derivatives for Fc glycan remodeling, further expands the scope of the chemoenzymatic method for antibody bioconjugation. In addition, the ability of Endo-S2 to perform deglycosylation of native antibodies and simultaneously transfer drug-preloaded disaccharide oxazolines enables a truly single-step protocol to construct homogeneous antibody-drug conjugates.
EXPERIMENTAL SECTION
General Procedure.
Chemicals, solvents, and reagents were purchased from TCI and Sigma-Aldrich and were used without further purifications unless specially noted. Silica gel (230–400 mesh) used for flash chromatography was purchased from SILICYCLE. The reactions were monitored by TLC on glass plates (Sigma-Aldrich), and spots were detected under UV-254 nm then charring with 5% (v/v) sulfuric acid in ethanol or cerium molybdate stain (CAM) followed by heating with a heat gun. NMR spectra were recorded on a Bruker-400 MHz spectrometer with CDCl3 or D2O as the solvent. Coupling constants (J) are reported in Hertz. The chemical shifts were assigned in ppm, and multiplicities are indicated by s (singlet), d (doublet), t (triplet), and m (multiplet). MALDI-TOF analysis was performed on a Bruker Autoflex Speed mass spectrometer with DHB (MeCN/H2O = 1:1) as the matrix (positive reflectron mode). HRMS was performed on an Exactive Plus Orbitrap mass spectrometer (Thermo Scientific) equipped with a C18 column. Preparative HPLC was performed with a Waters 600 HPLC instrument and Waters C18 columns (5.0 μm, 10 × 250 mm). The column was eluted with a gradient of MeCN–H2O containing 0.1% TFA or 0.1% FA at a flow rate of 4 mL/min. LC–MS analysis was performed on an Ultimate 3000 HPLC system coupled to an Exactive Plus Orbitrap mass spectrometer (Thermo Fischer Scientific) with C4 (whole antibody, gradient 5–95% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) or C8 (IdeS digestion, gradient 25–35% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) column. Deconvolution data was transformed by MagTran software.
6-O-2-[2-(2-Aminoethoxy)ethoxy]ethyl-β-d-mannopyranosyl-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (5).
A mixture of 418 (15.0 mg, 0.014 mmol) and Pd/C (10 wt.% loading, 10 mg) in THF/H2O (1.5 mL/0.5 mL) was added 3 M HCl (aq, 9 μL, 2 equiv) and then stirred under a H2 atmosphere overnight. After LC–MS analysis showed the complete conversion to free amine, the reaction mixture was filtered through a Celite pad and then concentrated and directly purified by Sephadex LH-20 (H2O) to give 5 (7.2 mg, 97%) as hydrochloride salt. 1H NMR (400 MHz, D2O) δ 5.07 (0.77H, m), 4.61−4.59 (1.31H, m), 3.96−3.95 (1.00H, m), 3.85−3.80 (0.81H, m), 3.78−3.76 (1.90H, m), 3.75−3.73 (1.04H, m), 3.69−3.67 (0.99H, m), 3.66−3.63 (3.34H, m), 3.63−3.56 (11.1H, m), 3.54−3.49 (1.90H, m), 3.48−3.43 (2.39H, m), 3.10 (2H, t, J = 4.6 Hz), 1.92 (3H, s); 13C NMR (100 MHz, D2O) δ 174.47, 174.17, 100.19, 94.81, 90.50, 79.54, 79.25, 74.68, 74.49, 72.64, 72.36, 70.44, 70.40, 69.92, 69.88, 69.73, 69.58, 69.40, 69.16, 66.58, 66.29, 63.81, 60.16, 60.03, 56.06, 53.63, 39.06, 22.13, 21.84; HRMS: [M + H]+ calcd for C20H39N2O13+, 515.2447; found, 515.2442.
Benzyl 2-O-benzyl-4,6-O-benzylidene-3-O-p-methoxybenzyl-β-d-glucopyranosyl-(1 → 4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (8).
To a solution of 720 (290 mg, 0.31 mmol) in MeOH (2.0 mL) was added sodium methoxide to maintain a pH of 10; the solution was heated to 50 °C and stirred overnight. After the complete disappearance of the starting material, the solution was diluted with CH2Cl2, successively washed with H2O and brine, and then concentrated to dryness. The residue was dissolved in dry N,N-dimethylformamide (2.0 mL) and cooled to 0 °C; sodium hydride (24 mg) and benzyl bromide (100 μL) were added successively, and the mixture was allowed to warm to room temperature. After the completion of the reaction as indicated by TLC, MeOH was added to quench the excess sodium hydride. The reaction was diluted with CH2Cl2 and successively washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4. After concentration, the residue was purified by flash column chromatography (hexanes/EtOAc = 10:1−5:1) to afford 8 (248 mg, 87% for 2 steps) as a white solid. Rf = 0.45 (hexanes/EtOAc = 5:1); 1H NMR (400 MHz, CDCl3) δ 7.53−7.51, 7.45−7.26 (27H, m, Ar-H), 6.85−6.83 (2H, m, Ar-H), 5.52 (1H, s, PhCH), 4.97−4.93 (2H, m, PhCH2), 4.88−4.83 (2H, m, PhCH2), 4.78−4.68 (4H, m, PhCH2), 4.63 (1H, d, J = 12.1 Hz, PhCH2), 4.56 (1H, d, J = 7.8 Hz), 4.43 (1H, d, J = 12.1 Hz, PhCH2), 4.31 (1H, d, J = 8.1 Hz), 4.21 (1H, dd, J = 5.0 Hz, J = 10.5 Hz), 4.06 (1H, t, J = 9.3 Hz), 3.88 (1H, dd, J = 3.7 Hz, J = 11.1 Hz), 3.82 (3H, s, OCH3), 3.70−3.58 (3H, m), 3.54−3.45 (2H, m), 3.40−3.33 (2H, m), 3.30−3.26 (1H, m), 3.22−3.15 (1H, m); 13C NMR (100 MHz, CDCl3) δ 159.26, 138.37, 138.31, 138.05, 137.42, 136.89, 130.62, 129.70, 129.00, 128.48, 128.45, 128.32, 128.27, 128.25, 128.07, 127.97, 127.95, 127.82, 127.73, 127.68, 126.07, 113.75, 102.79, 101.14, 100.38, 82.54, 81.78, 81.33, 80.83, 76.46, 75.51, 75.25, 75.18, 74.75, 73.30, 70.79, 68.71, 67.62, 65.85, 65.73, 55.29; MALDI-TOF: [M + Na]+ calcd for C55H57N3NaO11+, 958.39; found, 958.05.
Benzyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-β-d-glucopyranosyl-(1 → 4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (9).
To a solution of 8 (110 mg, 0.118 mmol) in BH3·THF (1 M, 2.0 mL) was added a solution of Bu2BOTf in CH2Cl2 (1 M, 200 μL) under an argon atmosphere at 0 °C, and the mixture was stirred at 0 °C for 40 min when TLC indicated the completion of the reaction. Et3N (150 μL) was added to the mixture followed by careful addition of MeOH (500 μL). The mixture was co-evaporated with MeOH for three times, and the residue was purified by silica gel flash chromatography (hexanes/EtOAc = 6:1−2:1) to afford 9 (100 mg, 91%) as a white solid. Rf = 0.30 (hexanes/EtOAc = 3:1); 1H NMR (400 MHz, CDCl3) δ 7.45−7.32, 7.25−7.23 (27H, m, Ar-H), 6.88−6.86 (2H, m, Ar-H), 5.00−4.96 (2H, m, PhCH2), 4.88−4.84 (3H, m, PhCH2), 4.83−4.78 (3H, m, PhCH2), 4.73 (1H, d, J = 12.0 Hz, PhCH2), 4.67−4.62 (2H, m), 4.51−4.48 (2H, m), 4.34 (1H, d, J = 8.1 Hz), 4.02 (1H, t, J = 9.4 Hz), 3.87 (1H, dd, J = 3.7 Hz, J = 11.1 Hz), 3.83 (3H, s, OCH3), 3.73−3.66 (2H, m), 3.60−3.52 (2H, m), 3.48 (1H, t, J = 9.1 Hz), 3.41−3.31 (4H, m), 3.22−3.17 (1H, m), 1.68 (1H, s); 13C NMR (100 MHz, CDCl3) δ 159.28, 138.42, 138.36, 138.13, 137.96, 136.91, 130.65, 129.56, 128.51, 128.42, 127.99, 127.91, 127.89, 127.81, 127.74, 127.71, 127.41, 113.87, 102.51, 100.41, 84.48, 82.86, 81.35, 77.97, 76.46, 75.50, 75.19, 75.16, 75.08, 75.04, 74.87, 73.40, 70.81, 67.60, 65.91, 61.84, 55.32; MALDI-TOF: [M + Na]+ calcd for C55H59N3NaO11+, 960.40; found, 959.99.
Benzyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-β-d-glucopyranosyl-(1 → 4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (10).
A solution of 9 (35.0 mg, 0.037 mmol) in a mixture of AcSH/pyridine/CHCl3 (0.6 mL/0.4 mL/0.6 mL) was stirred at 50 °C for 15 h. After the completion of the reaction as monitored by TLC, the resulting mixture was concentrated and the residue was subjected to flash chromatography on silica gel (hexanes/Acetone = 4:1−2:1) to afford 10 (31.6 mg, 89%) as a white solid. Rf = 0.20 (hexanes/acetone = 3:1); 1H NMR (400 MHz, CDCl3) δ 7.40−7.28, 7.23−7.21 (27H, m, Ar-H), 6.86−6.84 (2H, m, Ar-H), 5.65 (1H, d, J = 7.6 Hz), 5.00 (1H, d, J = 7.6 Hz), 4.94−4.92 (2H, m), 4.89−4.87 (1H, m), 4.86 (1H, m), 4.84−4.75 (3H, m), 4.67−4.58 (4H, m), 4.53−4.46 (2H, m), 4.19 (1H, t, J = 9.0 Hz), 3.89 (1H, t, J = 8.6 Hz), 3.87 (1H, dd, J = 3.9 Hz, J = 11.0 Hz), 3.82 (3H, s, OCH3), 3.75−3.69 (2H, m), 3.59−3.54 (2H, m), 3.48−3.34 (4H, m), 3.23−3.20 (1H, m), 1.85 (3H, s); 13C NMR (100 MHz, CDCl3) δ 170.47, 159.23, 138.83, 138.37, 138.12, 138.08, 137.58, 130.68, 129.51, 128.47, 128.44, 128.41, 128.40, 127.98, 127.90, 127.87, 127.81, 127.78, 127.70, 127.51, 113.83, 102.63, 98.91, 84.41, 82.81, 77.95, 77.76, 77.34, 75.43, 74.99, 74.85, 74.28, 73.32, 70.86, 68.11, 61.96, 56.87, 55.29, 23.61; MALDI-TOF: [M + Na]+ calcd for C57H63NNaO12+, 976.42; found, 976.25.
Benzyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-6-O-2-[2(2-azidoethoxy)ethoxy]ethyl-β-d-glucopyranosyl-(1 → 4)-2acetamido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (11).
To a solution of 10 (23.0 mg, 0.024 mmol) and the tosylate linker25 (23.8 mg, 0.073 mmol) in anhydrous DMF (0.6 mL) was added 60% sodium hydride (5.0 mg, 0.125 mmol) at 0 °C. After stirring for 0.5 h at 0 °C then 6 h at room temperature, MeOH (50 μL) and AcOH (10 μL) were added at 0 °C, and the reaction mixture was concentrated to dryness. The residue was purified by column chromatography on silica-gel (hexanes/acetone = 5:1 ~ 3:2) to give 11 (22.7 mg, 85%) as a white solid. Rf = 0.30 (hexanes/Acetone = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.39−7.26 7.22−7.20 (27H, m, Ar-H), 6.85−6.83 (2H, m, Ar-H), 5.67 (1H, d, NH, J = 7.8 Hz), 4.94−4.82 (6H, m), 4.79−4.74 (2H, m), 4.68−4.63 (2H, m), 4.61−4.56 (2H, m), 4.51−4.45 (2H, m), 4.05−4.03 (2H, m), 3.88 (1H, dd, J = 4.1 Hz, J = 10.7 Hz), 3.81 (3H, s, OCH3), 3.77−3.65 (3H, m), 3.61−3.48 (14H, m), 3.39 (1H, t, J = 8.1 Hz), 3.34−3.28 (3H, m), 1.82 (3H, s); 13C NMR (100 MHz, CDCl3) δ 170.14, 159.19, 139.09, 138.43, 138.40, 138.17, 137.66, 130.77, 129.49, 128.43, 128.41, 128.40, 128.33, 128.26, 128.13, 128.06, 127.96, 127.88, 127.85, 127.80, 127.74, 127.69, 127.66, 127.63, 127.47, 113.80, 102.82, 99.13, 84.50, 82.76, 77.86, 77.73, 76.92, 75.39, 75.06, 75.02, 74.99, 74.81, 73.61, 73.25, 70.87, 70.69, 70.59, 70.55, 69.93, 69.85, 68.38, 55.50, 55.29, 50.60, 23.50; MALDI-TOF MS: [M + Na]+ calcd for C63H74N4NaO14+, 1133.51; found, 1133.27.
6-O-2-[2-(2-Aminoethoxy)ethoxy]ethyl-β-d-glucopyranosyl-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (12).
A mixture of 11 (20.0 mg, 0.018 mmol) and Pd/C (10 wt.% loading, 10 mg) in THF/H2O (1.5 mL/0.5 mL) was added 3 M HCl (aq, 12 μL, 2 equiv) and then stirred under a H2 atmosphere overnight. After LC–MS analysis showed the complete deprotection and conversion of the azide to free amine, the reaction mixture was filtered through a Celite pad. The filtrate was concentrated, and the residue was purified by Sephadex LH-20 (H2O) to give 12 (9.2 mg, 93%) as hydrochloride salt. 1H NMR (400 MHz, D2O) δ 5.09 (0.52H, m), 4.62 (0.43H, d, J = 7.9 Hz), 4.43−4.39 (1.04H, m), 4.26−4.22 (0.22H, m), 4.04−4.01 (0.26H, m), 3.89−3.82 (1.40H, m), 3.80−3.73 (3.73H, m), 3.67−3.55 (13.8H, m), 3.55−3.51 (2.34H, m), 3.43−3.38 (1.57H, m), 3.36−3.31 (1.26H, m), 3.24−3.18 (1.31H, m), 3.11 (1.18, t, J = 4.9 Hz), 1.94 (3H, s); 13C NMR (100 MHz, D2O) δ 174.53, 174.29, 102.59, 94.78, 90.51, 79.45, 79.11, 75.35, 74.69, 74.48, 73.05, 72.42, 70.10, 70.02, 69.63, 69.58, 69.46, 69.20, 69.15, 66.56, 60.34, 59.99, 59.85, 56.22, 53.74, 39.11, 22.16, 21.87; HRMS: [M + H]+ calcd for C20H39N2O13+, 515.2447; found, 515.2440.
6-O-2-[2-(2-Azidoethoxy)ethoxy]ethyl-β-d-glucopyranosyl-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (13).
To a solution of 12 (9.0 mg, 0.016 mmol) in H2O (1.0 mL) was added a freshly prepared solution of TfN326 in CH2Cl2 (0.5 mL, ~0.16 mmol) containing K2CO3 (6.8 mg) and CuSO4 (0.8 mg) at 0 °C. Then MeOH was added to make the solution homogenous, and the mixture was stirred at room temperature for 36 h. The reaction mixture was filtered, and the filtrate was concentrated to dryness. The residue was purified on a Sephadex LH-20. Fractions containing the product were pooled and lyophilized and then further purified by preparative RP-HPLC (gradient, 5–15% aq MeCN containing 0.1% FA for 30 min; flow rate, 4 mL/min) to give 13 (7.2 mg, 81%) as a white solid after lyophilization. 1H NMR (400 MHz, D2O) δ 5.09 (0.58H, d, J = 2.5 Hz), 4.61 (0.47H, d, J = 7.7 Hz), 4.42−4.40 (0.97H, m), 3.88−3.83 (1.23H, m), 3.80−3.75 (2.89H, m), 3.75−3.73 (1.01H, m), 3.65−3.56 (12.9H, m), 3.53−3.49 (1.75H, m), 3.43−3.38 (2.83H, m), 3.36−3.31 (1.18H, m), 3.24−3.19 (1.20H, m), 1.94 (3H, s); 13C NMR (100 MHz, D2O) δ 174.51, 174.27, 102.61, 94.79, 90.48, 79.57, 79.30, 75.34, 74.70, 74.50, 73.05, 72.40, 70.11, 69.58, 69.53, 69.49, 69.19, 69.14, 60.05, 59.90, 56.20, 53.74, 50.10, 22.14, 21.84; HRMS: [M + H]+ calcd for C20H37N4O13+, 541.2352; found, 541.2344.
2-Methyl-{6-O-2-[2-(2-azidoethoxy)ethoxy]ethyl-β-d-glucopyranosyl-(1 → 4)-1,2-dideoxy-α-d-glucopyrano}-[2,1-d]2-oxazoline (14).
To a solution of 13 (3.3 mg, 6.1 μmol) in H2O (150 μL) were added Et3N (30 mol equiv) and 2-chloro1,3-dimethylimidazolinium chloride (DMC, 20 mol equiv) at 0 °C. The reaction mixture was kept at this temperature for 8 h then purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford 14 (3.0 mg, 94%) as a white solid after lyophilization with aq. NaOH (0.05 mol equiv, to keep the product at a basic condition). 1H NMR (400 MHz, D2O) δ 5.96 (1H, d, J = 7.3 Hz), 4.35 (1H, d, J = 7.9 Hz), 4.26−4.23 (1H, m), 4.08−4.06 (1H, m), 3.78−3.75 (1H, m), 3.70 (1H, dd, J = 2.3 Hz, J = 12.4 Hz), 3.64−3.57 (12H, m), 3.54−3.52 (1H, m), 3.47−3.43 (1H, m), 3.40−3.37 (2H, m), 3.34−3.29 (2H, m), 3.18−3.14 (1H, m), 2.88−2.86 (1H, m), 1.94 (3H, s); 13C NMR (100 MHz, D2O) δ 168.29, 104.14, 99.72, 78.35, 75.35, 74.74, 72.98, 70.79, 70.22, 69.58, 69.48, 69.18, 65.12, 61.52, 50.07, 12.85; HRMS: [M + H]+ calcd for C20H35N4O12+, 523.2246; found, 523.2233.
Benzyl 2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1 → 4)-2-azido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (17).
A mixture of trichloroacetimidate donor 1527 (810 mg, 1.65 mmol), acceptor 1618 (570 mg, 1.2 mmol), and activated 4 Å molecular sieves (1.5 g) in anhydrous CH2Cl2 (15.0 mL) was stirred at room temperature under an argon atmosphere for 1.5 h. It was then cooled to −40 °C, and TMSOTf (27 μL, 0.15 mmol) was added. After stirring at −40 °C for 50 min, the mixture was quenched with triethylamine (50 μL). The mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel chromatography (hexanes/EtOAc = 10:1−3:2) to give 17 (832 mg, 86%) as a white foam. Rf = 0.30 (hexanes/EtOAc = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.46−7.30 (15H, m, Ar-H), 5.29 (1H, d, J = 3.3 Hz), 5.13 (1H, dd, J = 8.0 Hz, J = 10.4 Hz), 4.99−4.93 (2H, m), 4.85 (1H, dd, J = 3.5 Hz, J = 10.4 Hz), 4.81−4.74 (2H, m), 4.70 (1H, d, J = 12.0 Hz, PhCH), 4.60 (1H, d, J = 8.0 Hz), 4.50 (1H, d, J = 12.0 Hz, PhCH), 4.30 (1H, d, J = 8.1 Hz), 4.07−4.01 (2H, m), 3.87 (1H, dd, J = 6.0 Hz, J = 11.1 Hz), 3.79−3.72 (2H, m), 3.59 (1H, t, J = 7.0 Hz), 3.50 (1H, dd, J = 8.3 Hz, J = 9.8 Hz), 3.37 (1H, t, J = 9.1 Hz), 3.32−3.30 (1H, m), 2.13 (3H, s), 2.01 (3H, s), 2.00 (3H, s), 1.99 (3H, s); 13C NMR (100 MHz, CDCl3) δ 170.19, 170.07, 169.18, 138.21, 137.73, 136.76, 128.63, 128.49, 128.23, 128.13, 128.04, 128.00, 127.95, 127.93, 127.72, 100.51, 100.07, 80.95, 76.05, 75.14, 74.85, 73.70, 70.95, 70.93, 70.52, 69.58, 67.44, 66.80, 65.78, 60.60, 20.78, 20.66, 20.64, 20.59; MALDI-TOF: [M + Na]+ calcd for C41H47N3NaO14+, 828.29; found, 828.05.
Benzyl 2,3,4-Tri-O-benzyl-6-o-(tert-butyldiphenylsilyl)-β-d-galactopyranosyl-(1 → 4)-2-azido-3,6-di-O-benzyl-2deoxy-β-d-glucopyranoside (18).
To a solution of 17 (340 mg, 0.422 mmol) in MeOH (4.0 mL) was added sodium methoxide until pH = 10, the solution was heated to 50 °C and stirred overnight. After the complete disappearance of the starting material, the reaction mixture was diluted with CH2Cl2 and successively washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated to dryness to give the crude deacylated intermediate. The residue was then dissolved in dry N,N-dimethylformamide (3.0 mL); imidazole (144 mg, 2.11 mmol) and tert-butyl(chloro)diphenylsilane (312 μL, 1.2 mmol) were added successively; and the resulting mixture was stirred at room temperature until the completion of the reaction as indicated by TLC. The reaction mixture was diluted with CH2Cl2 and successively washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated to dryness. Then, the residue was dissolved in dry N,N-dimethylformamide (3.0 mL) and cooled to 0 °C, sodium hydride (135 mg, 3.38 mmol) and benzyl bromide (300 μL, 2.53 mmol) were added successively, and the mixture was slowly warmed to room temperature. After the completion of the reaction as monitored by TLC, MeOH was added to quench the excess sodium hydride. The reaction mixture was diluted with CH2Cl2 and successively washed with H2O and brine. The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrated was concentrated, and the residue was purified by flash column chromatography on silica gel (hexanes/EtOAc = 15:1−8:1) to give 18 (328 mg, 68% for 3 steps) as a colorless syrup. Rf = 0.40 (hexanes/EtOAc = 4:1); 1H NMR (400 MHz, CDCl3) δ 7.59−7.55, 7.46−7.25 (40H, m, Ar-H), 5.12 (1H, d, J = 11.4 Hz), 5.00 (1H, d, J = 10.0 Hz), 4.94 (1H, d, J = 12.1 Hz), 4.86−4.76 (4H, m), 4.71−4.62 (3H, m), 4.60−4.58 (2H, m), 4.44−4.37 (2H, m), 4.28 (1H, d, J = 8.1 Hz), 4.07 (1H, d, J = 2.3 Hz), 3.96 (1H, t, J = 9.2 Hz), 3.89−3.84 (2H, m), 3.79 (1H, t, J = 9.5 Hz), 3.73−3.70 (1H, m), 3.65 (1H, dd, J = 9.5 Hz, J = 5.0 Hz), 3.48−3.42 (2H, m), 3.33−3.23 (3H, m), 1.08 (9H, s); 13C NMR (100 MHz, CDCl3) δ 139.27, 138.88, 138.68, 138.31, 138.01, 136.99, 135.57, 134.86, 133.25, 133.19, 129.85, 129.73, 128.54, 128.48, 128.37, 128.29, 128.17, 128.10, 128.04, 127.97, 127.92, 127.88, 127.81, 127.77, 127.71, 127.64, 127.58, 127.51, 127.38, 127.21, 102.75, 100.45, 82.44, 81.38, 80.22, 76.11, 75.50, 75.40, 75.32, 74.79, 74.33, 73.76, 73.26, 72.84, 70.76, 67.85, 65.67, 61.16, 27.02, 26.96, 26.62, 19.23; MALDI-TOF: [M + Na]+ calcd for C70H75N3NaO10Si+, 1168.51; found, 1168.22.
Benzyl 2,3,4-Tri-O-benzyl-6-O-(tert-butyldiphenylsilyl)-β-d-galactopyranosyl-(1 → 4)-2-acetamido-3,6-di-O-benzyl2-deoxy-β-d-glucopyranoside (19).
A solution of 18 (300 mg, 0.262 mmol) in a mixture of AcSH/pyridine/CHCl3 (0.8 mL/0.6 mL/0.8 mL) was stirred at 50 °C for 14 h. After the completion of the reaction as monitored by TLC, the resulting mixture was concentrated and the residue was subjected to flash chromatography on silica gel (hexanes/EtOAc = 4:1−1:1) to afford 19 (240 mg, 79%) as a colorless syrup. Rf = 0.30 (hexanes/EtOAc = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.60−7.56, 7.44−7.23, 7.20−7.18 (40H, m, Ar-H), 5.78 (1H, d, J = 7.7 Hz), 5.08 (1H, d, J = 11.3 Hz), 4.97 (1H, d, J = 6.8 Hz), 4.89 (1H, d, J = 11.9 Hz), 4.86−4.77 (5H, m), 4.63−4.51 (4H, m), 4.42−4.39 (2H, m), 4.06−4.02 (2H, m), 3.94 (1H, t, J = 7.5 Hz), 3.89−3.76 (4H, m), 3.68−3.64 (2H, m), 3.55−3.49 (1H, m), 3.45 (1H, dd, J = 9.8 Hz, J = 2.8 Hz), 3.30−3.26 (1H, m), 1.81 (3H, s), 1.07 (9H, s); 13C NMR (100 MHz, CDCl3) δ 170.12, 139.12, 138.70, 138.62, 138.50, 138.37, 137.68, 135.53, 133.14, 129.87, 129.75, 128.44, 128.32, 128.30, 128.22, 128.12, 128.10, 127.99, 127.89, 127.82, 127.79, 127.76, 127.68, 127.66, 127.56, 127.53, 127.37, 127.19, 103.13, 99.06, 82.26, 80.08, 77.13, 76.66, 75.32, 75.24, 74.65, 74.31, 73.84, 73.68, 73.16, 72.89, 70.74, 68.73, 61.38, 55.02, 26.95, 23.47, 19.19; MALDI-TOF: [M + Na]+ calcd for C72H79NNaO11Si+, 1184.53; found, 1184.07.
Benzyl 2,3,4-Tri-O-benzyl-β-d-galactopyranosyl-(1 → 4)-2-acetamido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (20).
To a solution of 19 (215 mg, 0.185 mmol) in THF (2.0 mL) was added TBAF (1 M in THF, 900 μL), and the mixture was stirred at 40 °C for 2 h. After the completion of the reaction as monitored by TLC, the resulting mixture was concentrated and the residue was subjected to flash chromatography on silica gel (hexanes/EtOAc = 5:1−1:1) to afford 20 (140 mg, 82%) as a colorless syrup. Rf = 0.25 (hexanes/acetone = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.38−7.24 (30H, m, Ar-H), 5.70 (1H, d, J = 7.8 Hz), 4.99−4.93 (3H, m), 4.90 (1H, d, J = 12.1 Hz), 4.85−4.73 (4H, m), 4.64−4.54 (4H, m), 4.44−4.41 (2H, m), 4.13 (1H, t, J = 8.2 Hz), 3.96 (1H, t, J = 8.0 Hz), 3.88−3.78 (3H, m), 3.74−3.73 (1H, m), 3.69−3.62 (2H, m), 3.55−3.49 (1H, m), 3.45−3.36 (2H, m), 3.26−3.23 (1H, m), 1.85 (3H, s); 13C NMR (100 MHz, CDCl3) δ 170.42, 138.67, 138.62, 138.49, 138.39, 138.29, 137.61, 128.47, 128.38, 128.36, 128.32, 128.25, 128.19, 128.10, 127.92, 127.77, 127.74, 127.57, 127.54, 127.50, 103.19, 98.95, 82.48, 79.92, 77.48, 75.34, 75.13, 75.00, 74.41, 74.38, 73.71, 73.17, 73.11, 70.72, 68.64, 62.00, 55.71, 23.57; MALDI-TOF: [M + Na]+ calcd for C56H61NNaO11+, 946.41; found, 946.04.
Benzyl 2,3,4-Tri-O-benzyl-6-O-2-[2-(2-azidoethoxy)ethoxy]ethyl-β-d-galactopyranosyl-(1 → 4)-2-acetamido3,6-di-O-benzyl-2-deoxy-β-d-glucopyranoside (21).
To a solution of 20 (120 mg, 0.13 mmol) and the tosylate linker25 (128 mg, 0.39 mmol) in anhydrous DMF (2.5 mL) was added 60% sodium hydride (26 mg, 0.65 mmol) at 0 °C. After stirring for 0.5 h at 0 °C then 6 h at room temperature, the reaction was diluted with CH2Cl2, successively washed with H2O and brine and dried over anhydrous Na2SO4. The mixture was filtered, and the filtrate was concentrated. The residue was purified by column chromatography on silica-gel (hexanes/acetone = 6:1−2:1) to give 21 (120 mg, 85%) as a colorless syrup. Rf = 0.30 (hexanes/Acetone = 2:1); 1H NMR (400 MHz, CDCl3) δ 7.37−7.23 (30H, m, Ar-H), 5.78 (1H, d, J = 7.7 Hz), 4.99 (1H, d, J = 11.5 Hz), 4.96−4.89 (3H, m), 4.87−4.80 (2H, m), 4.76−4.74 (2H, m), 4.64−4.54 (4H, m), 4.47−4.41 (2H, m), 4.07 (1H, t, J = 8.0 Hz), 4.00 (1H, t, J = 7.5 Hz), 3.93 (1H, d, J = 2.5 Hz), 3.88−3.84 (1H, m), 3.81−3.77 (2H, m), 3.69−3.60 (8H, m), 3.57−3.51 (4H, m), 3.48−3.40 (4H, m), 3.35 (2H, t, J = 5.0 Hz), 1.86 (3H, s); 13C NMR (100 MHz, CDCl3) δ 170.12, 138.98, 138.88, 138.66, 138.50, 138.34, 137.68, 128.39, 128.34, 128.31, 128.21, 128.16, 127.99, 127.90, 127.84, 127.69, 127.59, 127.55, 127.49, 127.46, 103.15, 99.12, 82.26, 79.92, 75.34, 75.22, 74.62, 73.76, 73.60, 73.13, 72.72, 70.70, 70.61, 70.55, 70.39, 70.04, 69.05, 68.72, 55.12, 50.65, 23.53; MALDI-TOF: [M + Na]+ calcd for C62H72N4NaO13+, 1103.50; found, 1103.10.
6-O-2-[2-(2-Aminoethoxy)ethoxy]ethyl-β-d-galactopyranosyl-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (22).
To a mixture of 21 (75.0 mg, 0.069 mmol) and Pd/C (10 wt.% loading, 40 mg) in THF/H2O (4.5 mL/1.5 mL) was added 3 M HCl (aq, 46 μL, 2 equiv), and then the mixture was stirred under a H2 atmosphere overnight. After ESI-MS indicated the completion deprotection and conversion of the azide to amine, the reaction mixture was filtered through a Celite pad. The filtrate was concentrated, and the residue was purified by Sephadex LH-20 (H2O) to give 22 (37.2 mg, 97%) as hydrochloride salt. 1H NMR (400 MHz, D2O) δ 5.10 (0.56H, m), 4.62 (0.42H, d, J = 7.6 Hz), 4.37 (1.05H, d, J = 7.9 Hz), 3.90−3.82 (2.22H, m), 3.81−3.76 (3.46H, m), 3.74−3.70 (0.71H, m), 3.65−3.55 (15.61H, m), 3.47−3.42 (1.28H, m), 3.00−2.98 (1.61H, m), 1.94 (3H, s); 13C NMR (100 MHz, D2O) δ 174.51, 174.26, 102.98, 94.83, 90.51, 79.40, 79.07, 74.69, 73.33, 72.48, 72.40, 71.67, 70.80, 70.09, 70.00, 69.87, 69.62, 69.55, 69.49, 69.42, 69.21, 68.67, 67.95, 67.91, 60.35, 60.08, 59.93, 56.12, 53.70, 39.30, 22.18, 21.89; HRMS: [M + H]+ calcd for C20H39N2O13+, 515.2447; found, 515.2440.
6-O-2-[2-(2-Azidoethoxy)ethoxy]ethyl-β-d-galactopyranosyl-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (23).
To a solution of 22 (9.0 mg, 0.016 mmol) in H2O (1.0 mL) was added a freshly prepared solution of TfN326 in CH2Cl2 (0.5 mL, ~0.16 mmol) containing K2CO3 (6.8 mg) and CuSO4 (0.8 mg) at 0 °C, and then MeOH was added to make the solution homogenous. The mixture was stirred at room temperature for 36 h, and then the reaction mixture was filtered. The filtrate was concentrated to dryness, and the residue was purified on a Sephadex LH-20 column by elution with H2O. Fractions containing the product were pooled and lyophilized and then further purified by preparative RP-HPLC (gradient, 5–15% aq MeCN containing 0.1% FA for 30 min; flow rate, 4 mL/min) to give 23 (7.5 mg, 85%) as a white solid. 1H NMR (400 MHz, D2O) δ 5.09 (0.64H, m), 4.61 (0.43H, d, J = 7.6 Hz), 4.37 (1.01H, d, J = 7.8 Hz), 3.88−3.82 (2.28H, m), 3.79−3.75 (3.59H, m), 3.74−3.70 (0.62H, m), 3.67−3.54 (15.67H, m), 3.51−3.50 (0.41H, m), 3.47−3.38 (2.97H, m), 3.12−3.09 (0.23H, m), 1.94 (3H, s); 13C NMR (100 MHz, D2O) δ 174.50, 174.25, 102.98, 94.82, 90.47, 79.44, 79.17, 74.72, 73.36, 72.46, 72.40, 70.79, 70.12, 69.94, 69.57, 69.55, 69.49, 69.18, 68.64, 60.11, 59.96, 56.12, 53.71, 50.11, 22.14, 21.84; HRMS: [M + H]+ calcd for C20H37N4O13+, 541.2352; found, 541.2336.
2-Methyl-{6-O-2-[2-(2-azidoethoxy)ethoxy]ethyl-β-d-galactopyranosyl-(1 → 4)-1,2-dideoxy-α-d-glucopyrano}-[2,1-d]-2-oxazoline (24).
To a solution of compound 23 (5.3 mg, 9.8 μmol) in H2O (200 μL) were added Et3N (30 mol equiv) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC, 20 mol equiv) at 0 °C. The reaction mixture was kept at this temperature for 7 h then purified by gel filtration on a Sephadex G-10 column that was eluted with 0.1% aq Et3N to afford 24 (4.9 mg, 96%) as a white solid after lyophilization with aqueous NaOH (0.05 mol equiv). 1H NMR (400 MHz, D2O) δ 5.98 (1H, d, J = 7.3 Hz), 4.31 (1H, d, J = 7.8 Hz), 4.29−4.27 (1H, m), 3.80 (1H, d, J = 3.1 Hz), 3.74−3.69 (2H, m), 3.67−3.57 (14H, m), 3.56−3.50 (2H, m), 3.42−3.38 (3H, m), 3.36−3.32 (1H, m), 1.96 (3H, s); 13C NMR (100 MHz, D2O) δ 168.22, 104.68, 99.86, 78.52, 73.50, 72.49, 71.02, 70.79, 70.23, 70.07, 69.57, 69.55, 69.51, 69.37, 69.20, 68.85, 65.29, 61.67, 50.12, 12.90; HRMS: [M + H]+ calcd for C20H35N4O12+, 523.2246; found, 523.2236.
Compound (25).
To a solution of commercial trastuzumab (100 μg) in a PBS buffer (4 μL, 150 mM, pH = 7.0) was added oxazoline 618 (14.0 μg, 20 equiv per reaction site) and wild-type Endo-S2 (0.1 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. Within 30 min, LC–MS analysis indicated the disappearance of the starting material and the formation of the desired product 25 with complete transglycosylation at both reaction sites. LC–MS analysis: for whole antibody (m/z), calculated, M = 146,909 Da; found 146,912 (deconvoluted data); for Fc domain released by IdeS digestion (m/z), calculated, M = 24,656 Da; found 24,656 (deconvoluted data).
Compound (26).
To a solution of commercial trastuzumab (100 μg) in a PBS buffer (4 μL, 150 mM, pH = 7.0) was added oxazoline 14 (14.0 μg, 20 equiv per reaction site) and wild-type Endo-S2 (0.3 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. Within 2 h, LC–MS analysis indicated the disappearance of the starting material and the formation of the desired product 26 with complete transglycosylation at both reaction sites. LC–MS analysis: for whole antibody (m/z), calculated, M = 146,909 Da; found 146,911 (deconvoluted data); for Fc domain released by IdeS digestion (m/z), calculated, M = 24,656 Da; found 24,656 (deconvoluted data).
Compound (27).
To a solution of commercial trastuzumab (100 μg) in a PBS buffer (4 μL, 150 mM, pH = 7.0) was added oxazoline 24 (14.0 μg, 20 equiv per reaction site) and wild-type Endo-S2 (0.5 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. Within 2 h, LC–MS analysis indicated the disappearance of the starting material and the formation of the desired product 27 with complete transglycosylation at both reaction sites. LC–MS analysis: for whole antibody (m/z), calculated, M = 146,909 Da; found 146,912 (deconvoluted data); for Fc domain released by IdeS digestion (m/z), calculated, M = 24,656 Da; found 24,657 (deconvoluted data).
Compound (29).
To a solution of bis-PEG5-NHS (6.0 mg, 11.3 μmol) in anhydrous DMSO (100 μL) was added 2817 (3.2 mg, 2.8 μmol) in anhydrous DMSO (100 μL) in 5 portions every 10 min, and Et3N was added to keep the pH = 8.5. After the completion of the reaction as monitored by LC–MS, 10% TFA (aq) was added (60 μL), and the reaction mixture was directly purified by preparative-HPLC (gradient, 30–70% aq MeCN containing 0.1% TFA for 40 min, 4 mL/min) to give 29 (3.6 mg, 82%) as a white foam. RP-HPLC retention time, tR = 22.5 min (gradient, 20–70% aq MeCN containing 0.1% FA for 30 min; flow rate, 0.4 mL/min). ESI-MS [M + H]+ calcd for C76H122N11O22+, 1540.88; found, 1541.29; [M + Na]+ calcd for C76H121N11NaO22+, 1562.86; found, 1563.28.
Compound (1).
To a solution of 5 (1.4 mg, 2.55 μmol) and 29 (2.1 mg, 1.36 μmol) in anhydrous DMSO (40 μL) was added Et3N (0.6 μL) to adjust pH = 8.5. The mixture was kept at room temperature until the complete consumption of 29 to give the crude product 30 in DMSO that was directly used in next step without further purification. RP-HPLC retention time for 30, tR = 16.9 min (gradient, 20–70% aq MeCN containing 0.1% FA for 30 min; flow rate, 0.4 mL/min). HRMS: [M + H]+ calcd for C92H155N12O32+, 1941.0898; found, 1941.0847. To the residue obtained in the first step was added H2O (80 μL) and Et3N (40 mol equiv); the mixture was cooled to 0 °C; and 2-chloro-1,3-dimethylimidazolinium chloride (DMC, 30 mol equiv) was added. After 12 h at 0 °C, the reaction was purified by preparative-HPLC (gradient, 25–60% aq MeCN containing 0.1% NH3·H2O for 40 min, 4 mL/min) to give oxazoline 1 (1.9 mg, 73% for 2 steps) as a white foam. HRMS: [M + H]+ calcd for C92H153N12O31+, 1922.0759; found, 1922.0703.
Compound (2).
To a solution of 12 (1.4 mg, 2.60 μmol) and 29 (2.0 mg, 1.30 μmol) in anhydrous DMSO (40 μL) was added Et3N (0.6 μL) to adjust pH = 8.5. The mixture was kept at room temperature until the complete consumption of 29 to give the crude product (31), which was used in the next step without further purification. RP-HPLC retention time for 31, tR = 17.1 min (gradient, 20–70% aq MeCN containing 0.1% FA for 30 min; flow rate, 0.4 mL/min). HRMS: [M + H]+ calcd for C92H155N12O32+, 1941.0898; found, 1941.0851. To the residue obtained in the first step was added H2O (80 μL) and Et3N (40 mol equiv). The mixture was cooled to 0 °C, and 2-chloro-1,3-dimethylimidazolinium chloride (DMC, 30 mol equiv) was added. After 12 h at 0 °C, the reaction was purified by preparative-HPLC (gradient, 25–60% aq MeCN containing 0.1% NH3·H2O for 40 min, 4 mL/min) to give oxazoline 2 (2.0 mg, 80% for 2 steps) as a white foam. HRMS: [M + H]+ calcd for C92H153N12O31+, 1922.0759; found, 1922.0800.
Compound (3).
To a solution of 22 (2.3 mg, 4.14 μmol) and 29 (3.2 mg, 2.07 μmol) in anhydrous DMSO (60 μL) was added Et3N (0.8 μL) to adjust pH = 8.5. The mixture was kept at room temperature until the complete consumption of 29 to give the crude product (32), which was used directly for the oxazoline formation without further purification. RP-HPLC retention time for 32, tR = 17.1 min (gradient, 20–70% aq MeCN containing 0.1% FA for 30 min; flow rate, 0.4 mL/min). HRMS: [M + H]+ calcd for C92H155N12O32+, 1941.0898; found, 1941.0836. To the residue obtained in the first step was added H2O (100 μL) and Et3N (40 mol equiv), the mixture was cooled to 0 °C and 2-chloro-1,3-dimethylimidazolinium chloride (DMC, 30 mol equiv) was added. After 12 h at 0 °C, the reaction mixture was subjected to preparative-HPLC (gradient, 25–60% aq MeCN containing 0.1% NH3·H2O for 40 min, 4 mL/min) to give oxazoline 3 (2.8 mg, 70% for 2 steps) as a white foam. HRMS: [M + H]+ calcd for C92H153N12O31+, 1922.0759; found, 1922.0731.
Compound (33).
To a solution of commercial trastuzumab (500 μg) in a PBS buffer (25 μL, 150 mM, pH = 7.0) containing 5% of DMSO was added oxazoline 1 (200 μg, 15 equiv per reaction site) and wild-type Endo-S2 (1.0 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. After 1 h, LC–MS analysis indicated the complete transglycosylation. The reaction mixture was diluted with 50 mM phosphate buffer (3.0 mL, pH = 7.2) and filtered by 0.22 μm syringe filter (remove most of the hydrophobic linker-payload) before subjecting to protein A column to give the antibody-drug conjugate 33 (470 μg, 95%). LC–MS analysis: for whole ADC (m/z), calculated, M = 149,709 Da; found 149,709 (deconvoluted data); for drug-conjugated Fc monomer released by IdeS digestion (m/z), calculated, M = 26,056 Da; found 26,057 (deconvoluted data).
Compound (34).
To a solution of commercial trastuzumab (500 μg) in 150 mM PBS buffer containing 5% of DMSO (25 μL, pH = 7.0) was added oxazoline 2 (250 μg, 20 equiv per reaction site) and wild-type Endo-S2 (10 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. After 1 h, another portion of oxazoline 2 (60 μg, 5 equiv per reaction site) was added to push the reaction. Within 1.5 h, LC–MS analysis indicated the complete transglycosylation. The reaction mixture was diluted with 50 mM phosphate buffer (3.0 mL, pH = 7.2) and filtered by a 0.22 μm syringe filter (remove most of the hydrophobic linker-payload) before subjecting to a protein A column to give the antibody-drug conjugate 34 (460 μg, 93%). LC–MS analysis: for whole ADC (m/z), calculated, M = 149,709 Da; found 149,708 (deconvoluted data); for drug-conjugated Fc monomer released by IdeS digestion (m/z), calculated, M = 26,056 Da; found 26,057 (deconvoluted data).
Compound (35).
To a solution of commercial trastuzumab (500 μg) in a PBS buffer (25 μL, 150 mM, pH = 7.0) containing 5% of DMSO was added oxazoline 3 (250 μg, 20 equiv per reaction site) and wild-type Endo-S2 (5.0 μg). The reaction was incubated at 28 °C and monitored by LC–MS aliquots. After 1 h, another portion of oxazoline 3 (60 μg, 5 equiv per reaction site) was added to push the reaction. Within 2 h, LC–MS analysis indicated the complete transglycosylation. The reaction mixture was diluted with 50 mM phosphate buffer (3.0 mL, pH = 7.2) and filtered by 0.22 μm syringe filter (remove most of the hydrophobic linker-payload) before subjecting to a protein A column to give the antibody-drug conjugate 35 (430 μg, 86%). LC–MS analysis: for whole ADC (m/z), calculated, M = 149,709 Da; found 149,710 (deconvoluted data); for drug-conjugated Fc monomer released by IdeS digestion (m/z), calculated, M = 26,056 Da; found 26,057 (deconvoluted data).
Cell Lines and Culture Conditions.
SK-BR-3 cells (ATCC HTB-30) were maintained in suspension in McCoy’s 5a Medium (ATCC 30–2007) containing 10% fetal bovine serum (FBS, not heated), 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT). BT474 cells (ATCC HTB-20) were maintained in suspension in Hybri-Care Medium (ATCC 46-X) containing 10% fetal bovine serum (FBS, pre-heated), 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT). T47D cells (ATCC HTB-133) were maintained in suspension in RPMI-1640 Medium (ATCC 30–2001) containing 10% fetal bovine serum (FBS, pre-heated), 4 mg/L insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT).
Cytotoxicity Assays.
For SK-BR-3 and T47D cell lines, the cells were planted into 96-well plates (cell number: 10,000 cells per well), and the plates were incubated for 24 h at 37 °C with 5% CO2. The ADC samples were diluted by 3-fold serial dilution with the corresponding medium from 5000 to 0.085 ng/mL (11 concentrations) and then added to the wells in triplicate (150 μL per well) for every single concentration. The cells were cultured at 37 °C with 5% CO2 for 3 days before the removal of the medium and addition of Cell Counting Kit-8 (Sigma). The absorbance of formazan released by viable cells was measured at 450 nm using a spectrophotometer after incubation at 37 °C with 5% CO2 for 2–3 h, and the background absorption was deducted by 550 nm absorbance. Finally, the cell viability curve and EC50 values were calculated by GraphPad Prism software. For the BT474 cell line, the cells were planted into 96-well plates with 4000 cells per well. The plates were incubated for 24 h at 37 °C with 5% CO2. The ADC samples were diluted by 3-fold serial dilution with the corresponding medium from 5000 to 0.085 ng/mL (11 concentrations) and then added to the wells in triplicate (200 μL per well) for every single concentration. The cells were cultured at 37 °C with 5% CO2 for 6 days before the removal of the medium and addition of Cell Counting Kit-8 (Sigma). The absorbance of formazan released by viable cells was measured at 450 nm using a spectrophotometer after incubation at 37 °C with 5% CO2 for 2–3 h, and the background absorption was deducted by 550 nm absorbance. Finally, the cell viability curve and EC50 values were calculated by GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH grant R01 AI155716).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.2c00142.
Figures S1–S8 and copies of NMR spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.2c00142
The authors declare the following competing financial interest(s): LXW is the founder and a major shareholder of GlycoT Therapeutics LLC. Other authors declare no conflict of interest. University of Maryland has filed patent applications related to this work.
Contributor Information
Xiao Zhang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Chong Ou, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Huiying Liu, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
Lai-Xi Wang, Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, United States.
REFERENCES
- (1).Drago JZ; Modi S; Chandarlapaty S Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol 2021, 18, 327–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).do Pazo C; Nawaz K; Webster RM The oncology market for antibody-drug conjugates. Nat. Rev. Drug Discovery 2021, 20, 583–584. [DOI] [PubMed] [Google Scholar]
- (3).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and challenges for the next generation of antibody−drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (4).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol 2008, 26, 925–932. [DOI] [PubMed] [Google Scholar]
- (5).Shen BQ; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu SF; Mai E; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- (6).Strop P; Delaria K; Foletti D; Witt JM; Hasa-Moreno A; Poulsen K; Casas MG; Dorywalska M; Farias S; Pios A; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol 2015, 33, 694–696. [DOI] [PubMed] [Google Scholar]
- (7).Bargh JD; Isidro-Llobet A; Parker JS; Spring DR Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev 2019, 48, 4361–4374. [DOI] [PubMed] [Google Scholar]
- (8).Walsh SJ; Bargh JD; Dannheim FM; Hanby AR; Seki H; Counsell AJ; Ou X; Fowler E; Ashman N; Takada Y; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev 2021, 50, 1305–1353. [DOI] [PubMed] [Google Scholar]
- (9).Wang LX; Tong X; Li C; Giddens JP; Li T Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem 2019, 88, 433–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Manabe S; Yamaguchi Y Antibody Glycoengineering and Homogeneous Antibody-Drug Conjugate Preparation. Chem. Rec 2021, 21, 3005–3014. [DOI] [PubMed] [Google Scholar]
- (11).Huang W; Giddens J; Fan SQ; Toonstra C; Wang LX Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 2012, 134, 12308–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Li X; Fang T; Boons GJ Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem., Int. Ed 2014, 53, 7179–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).van Geel R; Wijdeven MA; Heesbeen R; Verkade JMM; Wasiel AA; van Berkel SS; van Delft FL Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconjugate Chem. 2015, 26, 2233–2242. [DOI] [PubMed] [Google Scholar]
- (14).Parsons TB; Struwe WB; Gault J; Yamamoto K; Taylor TA; Raj R; Wals K; Mohammed S; Robinson CV; Benesch JL; Davis BG Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed 2016, 55, 2361–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tang F; Yang Y; Tang Y; Tang S; Yang L; Sun B; Jiang B; Dong J; Liu H; Huang M; et al. One-pot N-glycosylation remodeling of IgG with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody-drug conjugates. Org. Biomol. Chem 2016, 14, 9501–9518. [DOI] [PubMed] [Google Scholar]
- (16).Manabe S; Yamaguchi Y; Matsumoto K; Fuchigami H; Kawase T; Hirose K; Mitani A; Sumiyoshi W; Kinoshita T; Abe J; et al. Characterization of Antibody Products Obtained through Enzymatic and Nonenzymatic Glycosylation Reactions with a Glycan Oxazoline and Preparation of a Homogeneous Antibody-Drug Conjugate via Fc N-Glycan. Bioconjugate Chem. 2019, 30, 1343–1355. [DOI] [PubMed] [Google Scholar]
- (17).Ou C; Li C; Zhang R; Yang Q; Zong G; Dai Y; Francis RL; Bournazos S; Ravetch JV; Wang LX One-Pot Conversion of Free Sialoglycans to Functionalized Glycan Oxazolines and Efficient Synthesis of Homogeneous Antibody-Drug Conjugates through Site-Specific Chemoenzymatic Glycan Remodeling. Bioconjugate Chem. 2021, 32, 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang X; Ou C; Liu H; Prabhu SK; Li C; Yang Q; Wang LX General and Robust Chemoenzymatic Method for Glycan-Mediated Site-Specific Labeling and Conjugation of Antibodies: Facile Synthesis of Homogeneous Antibody−Drug Conjugates. ACS Chem. Biol 2021, 16, 2502–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shi W; Li W; Zhang J; Li T; Song Y; Zeng Y; Dong Q; Lin Z; Gong L; Fan S; et al. One-step synthesis of site-specific antibody-drug conjugates by reprograming IgG glycoengineering with LacNAc-based substrates. Acta Pharm. Sin. B 2021, DOI: 10.1016/j.apsb.2021.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zou G; Ochiai H; Huang W; Yang Q; Li C; Wang LX Chemoenzymatic synthesis and Fcγ receptor binding of homogeneous glycoforms of antibody Fc domain. Presence of a bisecting sugar moiety enhances the affinity of Fc to FcγIIIa receptor. J. Am. Chem. Soc 2011, 133, 18975–18991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yamaguchi T; Amin MN; Toonstra C; Wang LX Chemoenzymatic Synthesis and Receptor Binding of Mannose-6-Phosphate (M6P)-Containing Glycoprotein Ligands Reveal Unusual Structural Requirements for M6P Receptor Recognition. J. Am. Chem. Soc 2016, 138, 12472–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nyffeler PT; Liang CH; Koeller KM; Wong CH The chemistry of amine-azide interconversion: catalytic diazotransfer and regioselective azide reduction. J. Am. Chem. Soc 2002, 124, 10773–10778. [DOI] [PubMed] [Google Scholar]
- (23).Ochiai H; Huang W; Wang LX Expeditious chemoenzymatic synthesis of homogeneous N-glycoproteins carrying defined oligosaccharide ligands. J. Am. Chem. Soc 2008, 130, 13790–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang Z; Chinoy ZS; Ambre SG; Peng W; McBride R; de Vries RP; Glushka J; Paulson JC; Boons GJ A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lossouarn A; Renault K; Bailly L; Frisby A; Le Nahenec-Martel P; Renard PY; Sabot C Maleimide-based metal-free ligation with dienes: a comparative study. Org. Biomol. Chem 2020, 18, 3874–3887. [DOI] [PubMed] [Google Scholar]
- (26).Orgueira HA; Bartolozzi A; Schell P; Litjens REJN; Palmacci ER; Seeberger PH Modular synthesis of heparin oligosaccharides. Chem. – Eur. J 2003, 9, 140–169. [DOI] [PubMed] [Google Scholar]
- (27).Tiwari P; Misra AK Selective removal of anomeric O-acetate groups in carbohydrates using HClO4−SiO2. Tetrahedon Lett. 2006, 47, 3573–3576. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.