

SUMMARY
Type 1 diabetes is a disorder of immune tolerance that leads to death of insulin-producing islet β cells. We hypothesize that inflammatory signaling within β cells promotes progression of autoimmunity within the islet microenvironment. To test this hypothesis, we deleted the proinflammatory gene encoding 12/15-lipoxygenase (Alox15) in β cells of non-obese diabetic mice at a pre-diabetic time point when islet inflammation is a feature. Deletion of Alox15 leads to preservation of β cell mass, reduces populations of infiltrating T cells, and protects against spontaneous autoimmune diabetes in both sexes. Mice lacking Alox15 in β cells exhibit an increase in a population of β cells expressing the gene encoding the protein programmed death ligand 1 (PD-L1), which engages receptors on immune cells to suppress autoimmunity. Delivery of a monoclonal antibody against PD-L1 recovers the diabetes phenotype in knockout animals. Our results support the contention that inflammatory signaling in β cells promotes autoimmunity during type 1 diabetes progression.
In brief
Inflammation in pancreatic islets occurs early during the pathogenesis of type 1 diabetes. Piñeros et al. show that deletion of the proinflammatory gene encoding the enzyme 12/15-lipoxygenase in islet β cells of type 1 diabetes-prone mice reduces inflammatory signaling, suppresses the influx of immune cells, and prevents development of diabetes.
Graphical abstract
INTRODUCTION
Type 1 diabetes (T1D) is an autoimmune disease that results in a loss of insulin-secreting β cells in pancreatic islets. In individuals with T1D, reduction of the β cell population leads to an inability to maintain glucose homeostasis and results in the need for lifelong insulin administration (DiMeglio et al., 2018). The traditional perspective has held that T1D results from the loss of immune tolerance to β cell proteins and that susceptibility to disease largely emanates from features ascribed to the immune system. This perspective arose from studies in the non-obese diabetic (NOD) mouse model of autoimmune diabetes, wherein intervention with immune-suppressant therapies resulted in the prevention or reversal of diabetes (Chaparro and DiLorenzo, 2010). Some of these interventions have also held true in human T1D, although no study of immune suppressants or tolerance-inducing agents has completely prevented or reversed the disease (Atkinson et al., 2019; Matthews et al., 2010). Whereas these observations might call for additional trials with longer-term therapy or combination therapies, they have also served as catalysts to reevaluate the interplay of immune cells and β cells with the goal of identifying alternative therapeutic approaches (Atkinson et al., 2019).
In recent years, the pathogenesis of T1D has been the subject of intense speculation with respect to the interplay between different cell types in the islet microenvironment. A picture is emerging in which β cells may be center of a dialogue with immune cells, such that proteins expressed or secreted by β cells ultimately determine the pathogenicity of immune cells. For example, neoantigens produced by β cells under conditions of oxidative and endoplasmic reticulum (ER) stress may be triggers for selection and activation of clonal T cells (Itoh et al., 1993; Marré et al., 2016; Marro et al., 2019; Thomas et al., 1998). Additionally, the expression of cell-surface molecules on β cells may promote (e.g., major histocompatibility complex [MHC] class I) (Hamilton-Williams et al., 2003; Savinov et al., 2003) or suppress (e.g., programmed death ligand 1 [PD-L1]) (Paterson et al., 2011; Yoshihara et al., 2020) cytotoxic T cell responses. Finally, the production of chemokines (e.g., CXCL9, CXCL10, CCR2) by β cells may be permissive to the influx of immune cells, thereby inviting insulitis (islet inflammation) (Collier et al., 2017; Frigerio et al., 2002). Studies in NOD mice have been supportive of the notion that autonomous characteristics of β cells might allow for their evasion from immune attack (Fenske et al., 2017; Lee et al., 2020; Rui et al., 2017; Thompson et al., 2019). Recently, inflammatory signaling by Tet2 within β cells has been proposed as a modifier of the immune response in NOD mice (Rui et al., 2021). Because Tet2 is expressed in a host of immune cells, the definitive role of inflammatory signaling within β cells and its mechanism in mediating cellular crosstalk, however, remains to be elucidated.
12/15-lipoxygenase (12/15-LOX) is an enzyme encoded by the Alox15 gene, which is situated on the insulin-dependent diabetes locus 4 (Idd4) T1D susceptibility locus on chromosome 11 in the mouse (Todd et al., 1991). 12/15-LOX catalyzes the oxygenation of phospholipid-derived fatty acids (including arachidonic acid, linoleic acid, and docosahexaenoic acid) to diffusible lipid inflammatory eicosanoids such as 12(S)-hydroperoxyeicosatetraenoic acid (12-HPETE) and 12(S)-hydroxyeicosatetraenoic acid (12-HETE) (Kulkarni et al., 2021a). The potentially damaging effects of these eicosanoids, particularly 12-HETE, on β cells have been documented to include the development of oxidative and ER stress (Chen et al., 2005; Hernandez-Perez et al., 2017; Ma et al., 2017; Tersey et al., 2014). Global deletion of this gene in NOD mice during embryonic development leads to reduced entry and/or propagation of macrophages in the islet and to the subsequent development of diabetes (McDuffie et al., 2008). Relatively high expression of Alox15 in macrophages has led to the recognition that 12/15-LOX may be a regulator of inflammatory macrophages that initiates the innate immune insults on β cells in the setting of T1D (Green-Mitchell et al., 2013; Kulkarni et al., 2021b). Nevertheless, Alox15 is also expressed in β cells and is characteristically elevated in islets of humans at risk for T1D and in residual insulin-containing islets of individuals with established T1D (Grzesik et al., 2015). These prior observations led us to hypothesize that 12/15-LOX in islets might govern an inflammatory cascade that propagates crosstalk between β cells and immune cells. In this study, we tested this hypothesis using spatially and temporally manipulable mouse models congenic on the NOD background.
RESULTS
Deletion of Alox15 in islets alters islet-immune cell crosstalk in vitro
To assess if 12/15-LOX signaling in the islet might impact immune cell phenotypes, we leveraged a co-culture activation system in vitro wherein wild-type or Alox15−/− islets were incubated with a proinflammatory cytokine cocktail (interferon gamma [IFN-γ], interleukin 1β [IL-1β], and tumor necrosis factor alpha [TNF-α]) to stimulate inflammatory signaling. After stimulation, the islets were co-cultured with mouse macrophages (as antigen-presenting cells) and naive T cells (Figure 1A). Compared with wild-type islets, loss of Alox15 led to a decreased production of effector Th1 (CD4+IFN-γ+) and Th17 (CD4+IL-17+) cells and increased production of immune-suppressive regulatory T (Treg) (CD4+Foxp3+) cells (Figure 1B). These studies suggest the potential for inflammation-induced 12/15-LOX signaling in the islet to trigger an effector T cell response that initiates or propagates autoimmune diabetes.
Figure 1. Alox15 deletion in the β cell enhances expression of β cell genes and reduces expression of proinflammatory genes.
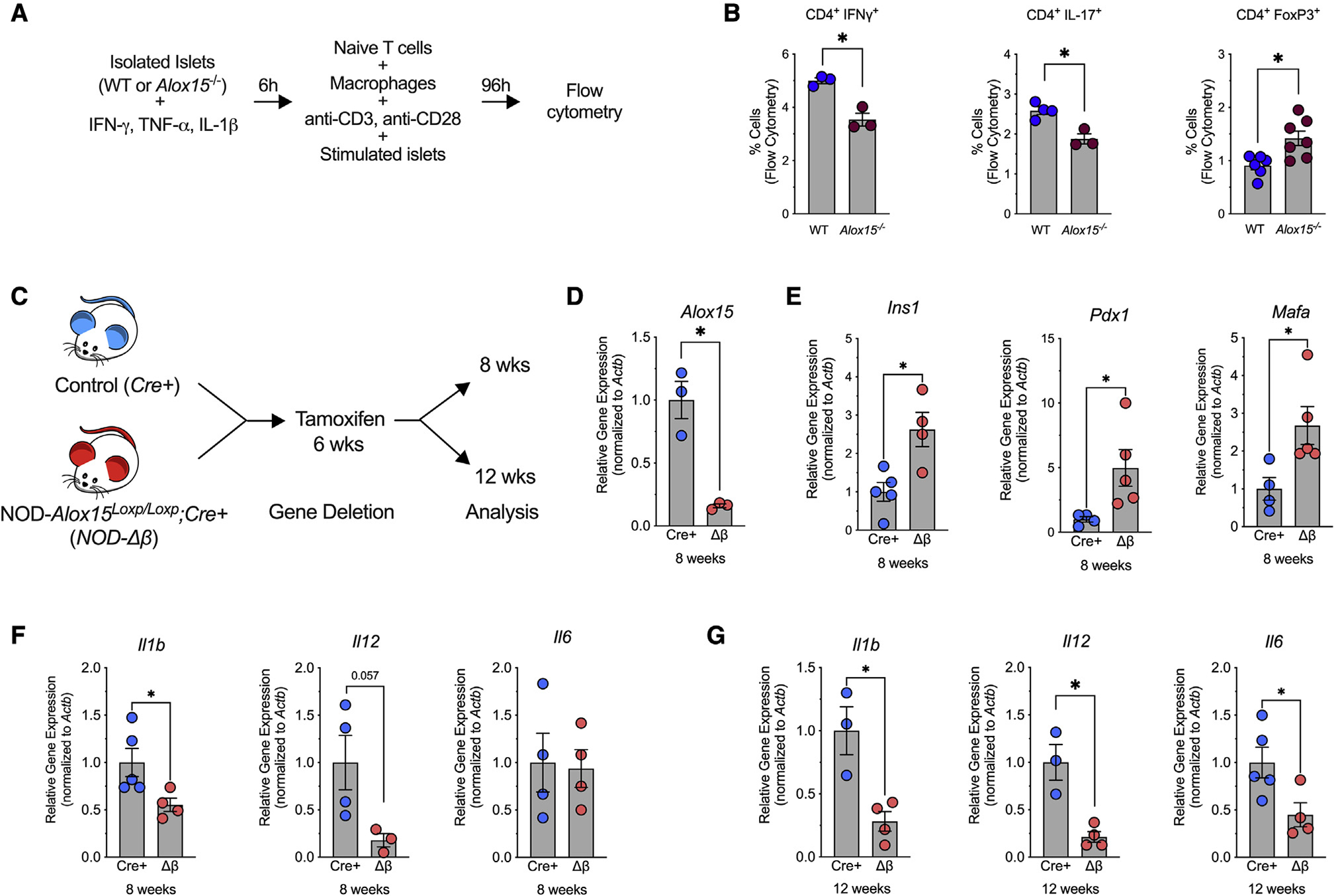
(A and B) Wild-type (WT) and Alox15−/− islets were stimulated with IFN-γ, IL-1β, and TNF-α for 6 h. After stimulation, the islets were co-cultured with WT macrophages and WT naive T cells for 96 h.
(A) Experimental schematic.
(B) T cell activation was quantified by flow cytometry for CD4+IFN-γ+, CD4+IL-17+, and CD4+FoxP3+ T cells. N = 3–7 biological replicates.
(C–G) NOD-Alox15loxP/+ mice were crossed with NOD-Pdx1PB-CreER™ mice to generate NOD-Cre+ and NOD-Δβ mice. At 6 weeks of age, the mice were administered 3 daily intraperitoneal injections of 2.5 mg of tamoxifen.
(C) Experimental design.
(D–F) Quantitative RT-PCR from isolated islets at 8 weeks of age for the indicated genes.
(G) Quantitative RT-PCR from isolated islets at 12 weeks of age for indicated genes. N = 3–5 biological replicates.
Data are expressed as the mean ± SEM. *p < 0.05.
Deletion of Alox15 in β cells of NOD mice preserves β cell identity and reduces β cell inflammation and stress
To interrogate the role of islet 12/15-LOX in propagating T1D in vivo, we introgressed Alox15Loxp/+ mice (Tersey et al., 2014) and β cell-enriched Cre driver PdxPB-CreERTM mice (Zhang et al., 2005) onto the NOD background to generate NOD-Alox15Loxp/+ mice and NOD-Pdx1PB-CreER™ mice with >99% congenicity based on microsatellite marker analysis (Table S1). These mice were then intercrossed to generate control genotypes (NOD-Alox15Loxp/Loxp and NOD-PdxPB-CreERTM) and a conditional genotype (NOD-Alox15Loxp/Loxp;NOD-PdxPB-CreERTM). To induce deletion of Alox15 in islets, we treated conditional mice (and their control littermates) with tamoxifen at 6 weeks of age, an age at which islets exhibit insulitis but that is before onset of dysglycemia/diabetes (Tersey et al., 2012), and then performed analyses of islet inflammation and immune cell phenotyping at 8 and 12 weeks of age (Figure 1C). Tamoxifen treatment led to ~80% decrease of Alox15 mRNA expression in islets isolated from mice with the conditional knockout mice (referred to henceforth as NOD-Δβ mice) compared with NOD-PdxPB-CreERTM control littermates (referred to henceforth as NOD-Cre+ mice) (Figure 1D). No change in Alox15 expression was observed in other tissues (Figure S1A), confirming specific islet deletion.
To determine how reduction in a single proinflammatory gene in the islet impacts β cell identity and inflammation, we first evaluated gene expression in islets from 8-week-old mice. Notably, islets of NOD-Δβ mice showed increased expression of Ins1, Pdx1, and Mafa (genes associated with insulin production, β cell function, and identity) compared with islets of NOD-Cre+ littermates (Figure 1E). Analysis of genes encoding proinflammatory cytokines showed decreased islet expression of Il1b and Il12, but not Il6, in NOD-Δβ mice (Figure 1F). After 12 weeks of age, islets of NOD-Δβ mice showed decreased expression of Il1b, Il12, and Il6 compared with islets of NOD-Cre+ littermates (Figure 1G). It has been shown that islet inflammation in T1D is characterized by activation of the IRE-1α pathway of ER stress (Lee et al., 2020); consistent with this observation, islets of NOD-Δβ mice showed significantly decreased expression of the target of IRE-1α, sXbp1, at 12 weeks of age (Figure S1B). Collectively, these data suggest that the loss of Alox15 in islets of NOD mice results in reduced islet inflammation and ER stress.
β cell deletion of Alox15 protects against spontaneous diabetes development and reverses insulitis in NOD mice
To determine if deletion of Alox15 in β cells impacts progression to T1D, we monitored mice for spontaneous diabetes development through 25 weeks of age. In female mice, ~80% of control littermates (NOD-Cre+, NOD-Alox15Loxp/Loxp, and NOD-Alox15Loxp/+;Cre+) developed diabetes by 25 weeks of age, whereas only ~10% of NOD-Δβ mice developed diabetes (Figure 2A). In male mice, ~50% of control littermates (both NOD-Cre+ and NOD-Alox15Loxp/Loxp and NOD-Alox15Loxp/+;Cre+) developed diabetes, whereas none of the NOD-Δβ mice developed diabetes (Figure 2B). We performed a glucose tolerance test at 8 weeks of age (2 weeks following tamoxifen delivery) and found that NOD-Δβ mice had significantly improved glucose tolerance compared with control mice (Figure 2C), as well as increased serum insulin levels (Figure 2D). We next examined both the severity of cellular infiltration and β cell mass in NOD-Δβ mice. At 8 weeks of age, no changes were observed in β cell mass, average insulitis score, or insulitis score distribution (Figures S1C–S1F) in NOD-Δβ mice compared with control mice. However, by 12 weeks of age, pre-diabetic NOD-Δβ mice exhibited significantly greater β cell mass (Figures 2E and 2F), reduced cellular infiltration (including reductions in of T and B cells), reduced average insulitis score (Figures 2G and 2H), and an insulitis score distribution that skewed toward a lower score range (Figure S1G) compared with NOD-Cre+ littermates. Prior studies have suggested that Alox15 in β cells promotes survival from cytokine-induced apoptosis (Tersey et al., 2014). We confirmed that whereas exposure of control islets to a mix of proinflammatory cytokines (IFN-γ, TNF-α, IL-1β) enhances islet death in vitro, Alox15 islets were protected from cytokine-induced cell death (Figure S2).
Figure 2. Deletion of Alox15 in β cells protects against development of T1D in NOD mice.
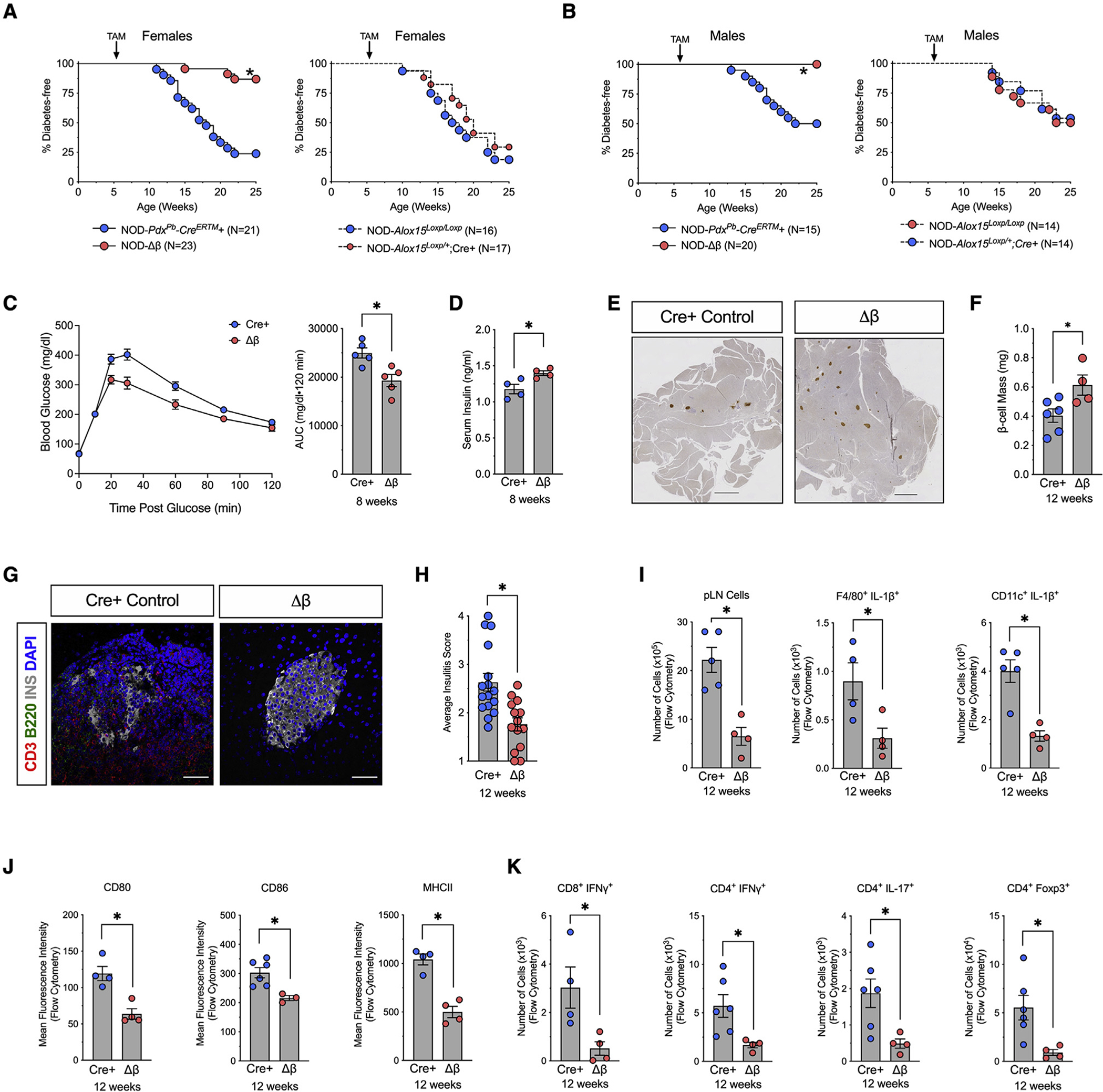
NOD-Alox15loxP/+ mice were crossed with NOD-Pdx1PB-CreER™ mice to generate NOD-Cre+ and NOD-Δβ mice. At 6 weeks of age, all mice were administered 3 daily intraperitoneal injections of 2.5 mg of tamoxifen.
(A) Kaplan-Meier curves showing diabetes incidence by genotype in females. N = 16–23 biological replicates.
(B) Kaplan-Meier curves showing diabetes incidence by genotype in males. N = 14–20 biological replicates.
(C) Glucose-tolerance test and area under the curve calculation at 8 weeks of age. N = 5 biological replicates.
(D) Serum insulin levels at 8 weeks of age. N = 5 biological replicates.
(E) Images of whole pancreatic sections from representative mice at 12 weeks of age, immunostained for β cells (insulin, brown), and counterstained with hematoxylin. Scale bars, 500 μm.
(F) Quantification of β cell mass. N = 6 mice, 5 slides per mouse.
(G) Representative images of pancreata from mice at 12 weeks of age stained for T cells (CD3, red), Β cells (B220, green), β cells (insulin, white), and nuclei (DAPI, blue). Scale bars, 50 μm.
(H) Average insulitis score in NOD-Cre+ controls and NOD-Δβ mice at 12 weeks of age. N = 3 mice, 5 slides per mouse.
(I–K) Pancreatic lymph nodes (pLNs) were isolated from NOD-Cre+ and NOD-Δβ mice at 8 weeks of age and analyzed by flow cytometry.
(I) Quantification of total cells and the number of proinflammatory macrophages (F4/80+IL-1β+) and dendritic cells (CD11c+IL-1β+).
(J) Mean fluorescent intensity of co-stimulatory molecules (CD80, CD86) and MHC class II by flow cytometry.
(K) Quantification of the number of CD4+ T cells; subclasses as indicated. N = 3–5 biological replicates.
Data are expressed as the mean ± SEM. *p < 0.05 for the comparisons indicated.
Immune cell reprogramming in pancreatic lymph nodes of NOD-Δβ mice
The striking reductions in diabetes incidence and insulitis resulting from deletion of an enzyme controlling inflammation in islets led us to interrogate further the relationship between islet inflammation and immune cells. Considering that draining pancreatic lymph nodes (pLNs) are key sites in the initial priming of autoreactive T cells (Gagnerault et al., 2002), we next investigated the activation of the immune response in the pLNs. By 12 weeks of age, when insulitis is largely diminished or absent in NOD-Δβ mice, analysis of pLNs was consistent with reduced myeloid and lymphoid populations. As shown in Figure 2I, there was a significant reduction of the total cell number in the pLNs of NOD-Δβ mice compared with NOD-Cre+ controls by conventional flow cytometry. There were striking reductions in myeloid population subtypes in NOD-Δβ mice, including reductions in populations of macrophages (F4/80+) and dendritic cells (CD11c+) that produce the proinflammatory cytokine IL-1β (Figure 2I) and a reduced population of dendritic cells that express co-stimulatory molecules (CD86 and CD80) and MHC class II (Figure 2J). Accordingly, these reductions in myeloid cell populations translated to a decrease in activated T cell populations, including CD8+IFN-γ+, CD4+IFN-γ+, and CD4+IL17+ cells (Figure 2K). At this age, CD4+Foxp3+ (Treg) cells were also reduced in pLNs of NOD-Δβ mice (Figure 2K), presumably reflecting a state of suppressed autoimmunity. Notably, these populations of T cells and innate immune cells account for only a fraction of the total cells that make up the pLNs at this age, indicating that other cell types not accounted for in our limited analysis also contribute to the total cell population.
Next, we performed an unbiased profiling of the myeloid cells and other immune cells present in the pLNs at the early time of 8 weeks of age in NOD-Cre+ and NOD-Δβ mice using cytometry by time of flight (CyTOF). The cell suspensions isolated from the pLNs were incubated with defined surface markers for myeloid cells and lymphocytes and utilized in an unsupervised algorithm for clustering and dimensionality reduction to objectively delineate and quantify immune cell populations, as shown in the t-distributed stochastic neighbor embedding (tSNE) plots in Figure 3A. We identified 7 clusters of immune cells: monocytes, two clusters of dendritic cells (DC-1 and DC-2), macrophages, CD4+ T cells, CD8+ T cells, and B cells (Figure 3A), and heatmaps were leveraged to identify the phenotypic profile of each cluster (Figure 3B). Notably, no significant differences were found in the number of general myeloid or lymphoid cell populations in the pLNs of NOD-Δβ mice compared with the NOD-Cre+ controls (Figure 3C). Upon examining immune cell subpopulations by CyTOF, however, a reduction specifically in the accumulation of proinflammatory myeloid cells producing IFN-γ (CD11b+IFN-γ+) was observed along with an increase in Treg cells (CD4+CD25+) in NOD-Δβ mice (Figures 3D and 3E). We confirmed these findings by flow cytometry of pLNs isolated at 8 weeks of age and found that although NOD-Δβ mice had no statistical changes in total cells in the pLNs compared with control mice, they exhibited decreased proinflammatory T cells (CD8+IFN-γ+) and increased Treg cells (CD4+FoxP3+) compared with control mice (Figure 3F). Collectively, these data are consistent with the persistent insulitis in NOD-Δβ mice at 8 weeks of age but still suggest subtle changes in myeloid and T cell subpopulations that are skewed toward an anti-inflammatory/regulatory phenotype. Taken together, the data in Figures 2 and 3 are consistent with a time-dependent reprogramming of immune cells from a proimmune to immune-suppressive posture upon deletion of Alox15 in the β cell.
Figure 3. Alteration in immune cell subtypes following deletion of Alox15 in β cells of NOD mice.
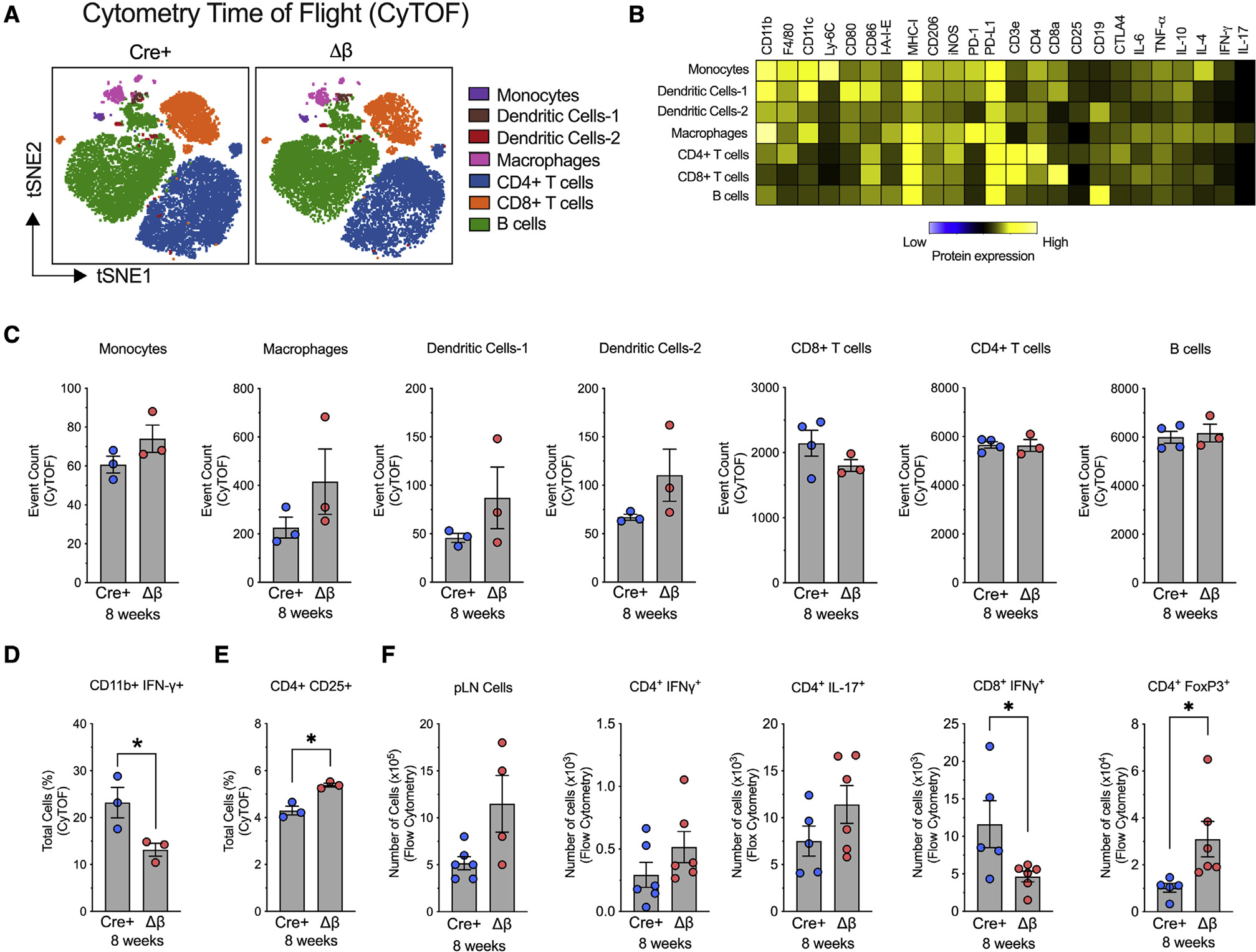
pLNs were isolated from NOD-Cre+ and NOD-Δβ mice at 8 weeks of age, dissociated into single cells, and analyzed by cytometry by time of flight (CyTOF) or flow cytometry. N = 3–4 individual animal replicates.
(A) tSNE maps of CD45+ immune cell populations in NOD-Cre+ and NOD-Δβ pancreata by CyTOF. Cells are colored by their FlowSOM-based cluster-assignment cell.
(B) Heatmap of protein expression levels across all populations identified by FlowSOM.
(C) Quantification of each immune cell type identified by FlowSOM-based clusters.
(D) Quantification of CD11b+IFN-γ+ cells.
(E) Quantification of CD4+CD25+ cells.
(F) Flow cytometry of pLNs of CD4+ and CD8+ T cell populations.
Data are expressed as the mean ± SEM. *p < 0.05.
Loss of Alox15 in β cells alters crosstalk with the immune system
To elucidate the mechanisms through which inflammatory signaling in β cells exerts its effect on immune cell activation in the pancreas, we analyzed the changes in the mRNA profile of islets from NOD-Δβ mice and NOD-Cre+ controls at 8 weeks of age (a time point when insulitis is present in both genotypes) using single-cell RNA sequencing (scRNA-seq). We visualized cells based on expression profiles using uniform manifold approximation and projection (UMAP) for dimension reduction plots and identified clusters representing distinct pancreatic cell types (Figure S3A): 5 β cell clusters (β1, β2, β3, β4, and β5) characterized by Ins1 and Mafa expression; 2 α cell clusters (α1 and α2) characterized by Gcg expression; 1 δ cell cluster characterized by Sst expression; 1 PP cell cluster characterized by Ppy expression; 1 acinar cell cluster characterized by Spp1 expression; and 1 duct cell cluster characterized by Reg3a expression (see Figure S3A for UMAP plots of each defining gene distribution). Additionally, we identified several clusters of immune cells (CD45/Ptprc), including macrophages (F4/80/Adgre1 and CD11b/Itgam), dendritic cells (Itgam and Cd11c/Itgax), monocytes (Ly6C), T cells (Cd4 or Cd8), and B cells (Cd19) (Figures 4A and S3A). Although immune cells are present in islets of both NOD-Δβ and NOD-Cre+ mice, phenotypes of these immune cell populations are distinctly different. Two notable changes were observed. First, Gene Ontology-gene set enrichment analysis (GO-GSEA) analysis showed that both macrophages and dendritic cells exhibit gene-expression changes in pathways associated with regulation of leukocyte activation, immune response, and cytokine production (Figures S3B and S3C), suggesting that loss of β cell Alox15 was influential in the immune phenotype of these cells. Specifically, macrophages in NOD-Δβ mice were characterized by enhanced expression of Cd274 (encoding PD-L1) and Socs1 (associated with an anti-inflammatory phenotype) (Figure 4B). PD-L1 is an immune checkpoint protein expressed on the cell surface that mediates peripheral T cell tolerance and autoimmunity via interaction with its receptor PD-1 (Karwacz et al., 2011; Qin et al., 2019). Increased PD-L1 protein was also found in myeloid cells by CyTOF analysis of whole pancreatic tissue (Figure 4C). In addition, flow cytometry also revealed an increase in PD-1 levels myeloid cells (CD11b+PD-1+) in pancreatic tissue Figure 4D). Second, scRNA-seq analysis revealed enhanced expression of Treg cell markers in the T cell cluster, with an enhanced expression of Lag3, Foxo1, Ikzf4, Ccr7, and Il10 (Figure 4E).
Figure 4. Single-cell RNA sequencing analysis of islets and CyTOF of pancreas in NOD-Cre+ and NOD-Δβ mice.
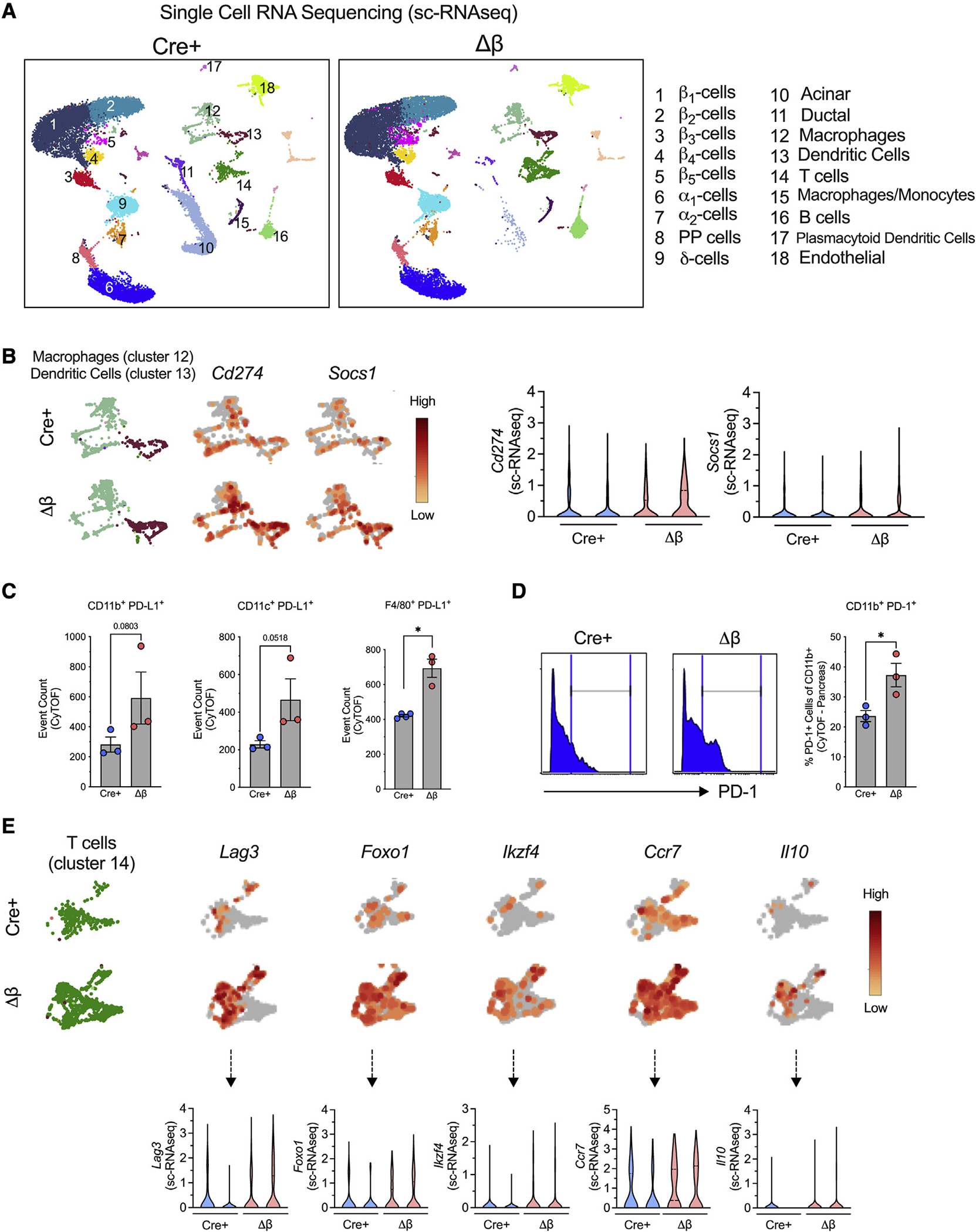
(A–E) Islets or pancreata were isolated from NOD-Cre+ and NOD-Δβ mice at 8 weeks of age, dissociated into single cells, and subjected to single-cell RNA sequencing (A, B, and E; N = 2 individual animal replicates) or CyTOF (C and D, N = 3–4 individual animal replicates), respectively.
(A) Uniform manifold approximation and projection (UMAP) embeddings of merged single-cell RNA sequencing profiles from islets colored by cell clusters that were identified.
(B) UMAP gene expression patterns and violin plots of Cd274 and Socs1 in a combined macrophage cluster (cluster 12) and a dendritic cell cluster (cluster 13) (false discovery rate [FDR] < 0.05).
(C) Number of PD-L1-positive cells expressed by CD11b+, CD11c+, and F4/80+ cells identified by CyTOF of whole pancreatic cells.
(D) Frequency of CD11b+PD-1+ cells.
(E) UMAP gene-expression patterns and violin plots of Lag3, Foxo1, Ikzf4, Ccr7, and Il-10 in the T cell cluster (cluster 14) (FDR < 0.05).
Data are expressed as the mean ± SEM. *p < 0.05.
Loss of Alox15 in β cells is linked to increased levels of PD-L1 in β cells
To clarify how these immune phenotypes are altered by loss of Alox15 in the islet, we next focused on gene expression in β cell clusters. Differential expression analysis between NOD-Δβ and NOD-Cre+ control mice at 8 weeks of age (pre-diabetic phase) using a false discovery rate <0.05 and a fold change >2 identified 429 downregulated and 262 upregulated genes in the 5 β cell clusters. Notable transcript changes in β cell clusters included those associated with enhanced cell function and survival (Esr1, Lncpint) (Zhou et al., 2018; Zhu et al., 2018) and the antioxidant response (Sod1, Nfe2l1) (Figure 5A). GO-GSEA revealed that β cell clusters in NOD-Δβ mice are significantly enriched for changes in genes associated with negative regulation of cell adhesion, regulation of cell-cell adhesion, and regulation of immune effector process and immune response (Figure 5B). This observation is consistent with the notion that loss of Alox15 in β cells led to alterations in the interactions between β cells and immune cells.
Figure 5. β cell deletion of Alox15 enhances expression of the immune checkpoint protein PD-L1.
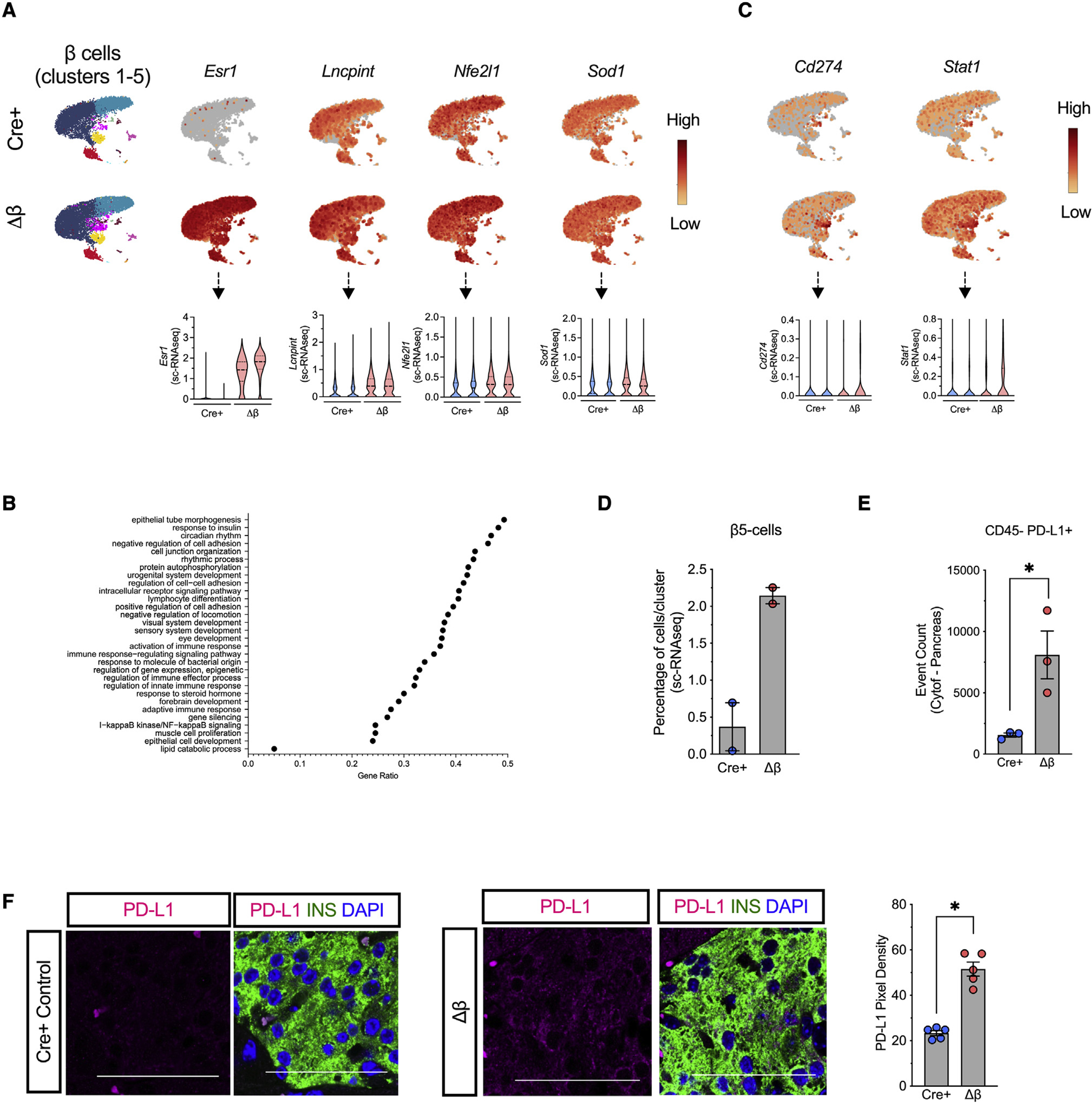
Islets were isolated from NOD-Cre+ and NOD-Δβ mice at 8 weeks of age, dissociated into single cells, and subjected to single-cell RNA sequencing. N = 2 individual animal replicates.
(A) UMAP gene expression patterns and violin plots of Esr1, Lncpint, Nfe2l1, Sod1, and Cd74 in the 5 β cell clusters (clusters 1–5; FDR < 0.05).
(B) UMAP gene-expression patterns and violin plots of Cd274 and Stat1 in the 5 β cell clusters (clusters 1–5; FDR < 0.05).
(C) Gene ontology-gene set enrichment analysis (GO-GSEA) in the 5 β cell clusters (clusters 1–5).
(D) Percentage of cells in β cell cluster 5.
(E) Number of non-immune cells (CD45-) expressing PD-L1 as measured by mass cytometry of the pancreas.
(F) Representative images of pancreata from 8-week-old mice immunostained for PD-L1 (magenta), β cells (insulin, green), and nuclei (DAPI, blue), and quantification of PD-L1 pixel density from islets (from N = 5 mice). Scale bars, 50 μm. Data are expressed as the mean ± SEM. *p < 0.05.
PD-L1 is expressed in leukocytes and non-hematopoietic cells including pancreatic islet cells (Fife et al., 2006, 2009; Paterson et al., 2011). Our scRNA-seq revealed that NOD-Δβ mice had enhanced expression of Cd274 (the gene encoding PD-L1) in all 5 clusters of β cells, as well as Stat1, which encodes an upstream activator of Cd274 (Garcia-Diaz et al., 2017) (Figure 5C). Of note, cluster β5 was characterized by high PD-L1 expression in both NOD-Cre+ and NOD-Δβ mice, but in NOD-Δβ mice, there was a significantly increased percentage of cells in this cluster Figure 5D). Increased numbers of non-immune cells (CD45-) exhibiting cell-surface PD-L1 protein in the pancreas of NOD-Δislet mice was also confirmed by flow cytometry (Figure 5E), and increased PD-L1 specifically in β cells of NOD-Δβ was confirmed by immunolabeling (Figure 5F).
Release of the integrated stress response underlies the production of PD-L1
To identify a mechanistic link between 12/15-LOX signaling and PD-L1 production, we leveraged primary cultured human islets. Human β cells express a functionally similar lipoxygenase (12-LOX) encoded by the ALOX12 gene (Dobrian et al., 2019), which is selectively inhibited by the small molecule ML355 (Ma et al., 2017). As shown in Figure 6A, human islets treated with proinflammatory cytokines (IFN-γ, IL-1β) to mimic conditions of T1D exhibited a previously characterized increase in PD-L1 levels (Colli et al., 2018). When islets were co-incubated with 10 μM ML355, levels of PD-L1 increased further (Figure 6A), validating a direct link between 12-LOX activity and levels of PD-L1.
Figure 6. 12/15-LOX signaling is linked to activation of the integrated stress response and relative suppression of PD-L1 expression in islets.
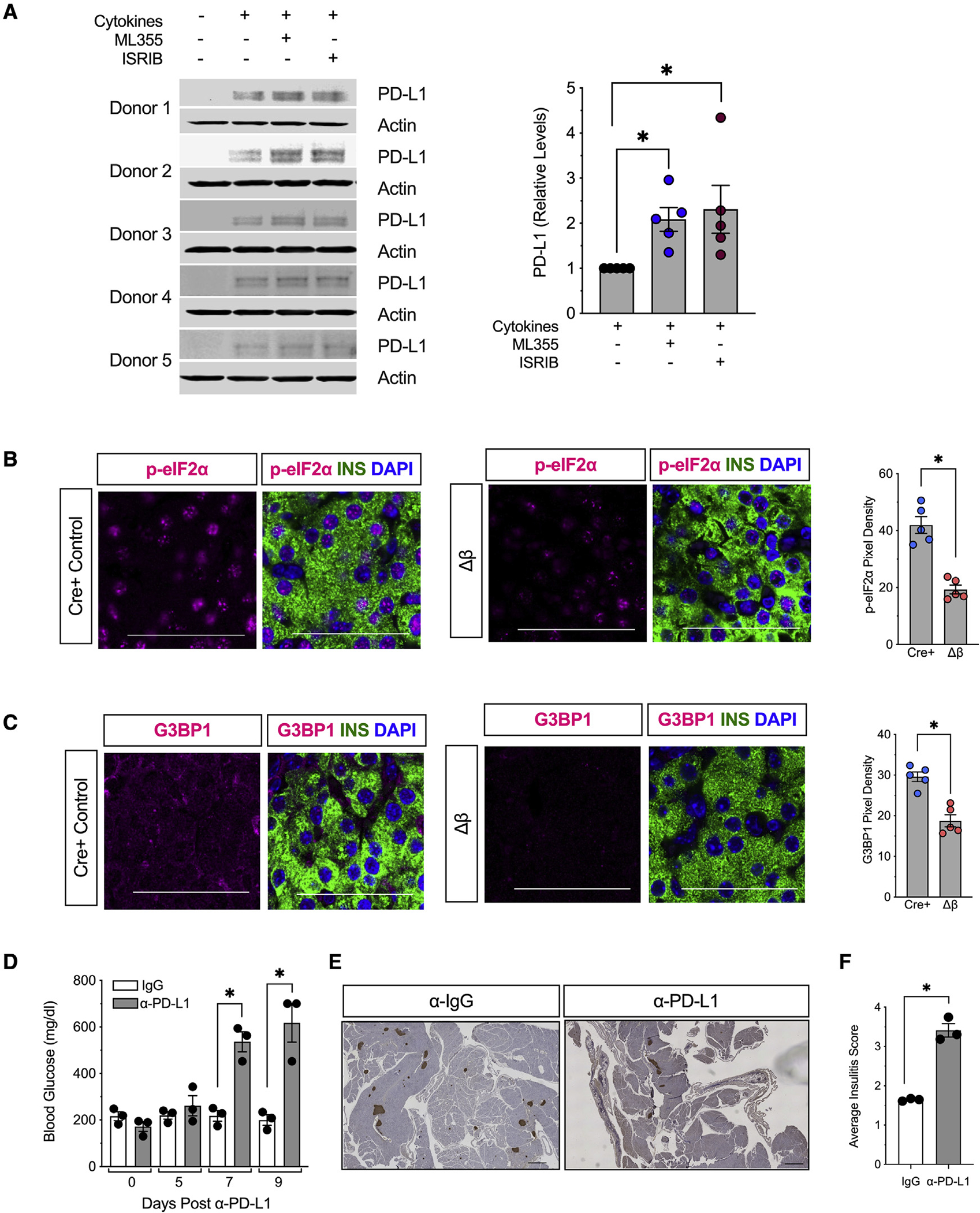
(A) Isolated human islets (N = 5 different donors) were treated with a mixture of proinflammatory cytokines (IL-1β and IFN-γ) for 24 h in the presence of the human 12-LOX inhibitor ML-355 prior to protein isolation. Individual immunoblots (left) and quantification of protein for PD-L1 (right).
(B) Representative images of pancreata from 8-week-old mice for phospho-eIF2α (magenta), β cells (insulin, green), and nuclei (DAPI, blue), and quantification of phospho-eIF2α pixel density from islets (from N = 5 mice). Scale bars, 50 μm.
(C) Representative images of pancreata from 8-week-old NOD-Cre+ and NOD-Δβ immunostained for G3BP1 (magenta), β cells (insulin, green), and nuclei (DAPI, blue), and quantification of G3BP1 pixel density from islets (from N = 5 mice). Scale bars, 50 μm.
(D–F) Female NOD-Δβ mice were administered a single dose of anti-PD-L1 monoclonal antibody (α-PD-L1) (N = 3 mice) or IgG isotype control (N = 3 mice) intraperitoneally.
(D) Non-fasting blood glucose levels.
(E) Images of whole pancreatic sections from representative mice at 9 days post α-PD-L1 or IgG administration immunostained for β cells (insulin, brown) and counterstained with hematoxylin. Scale bars, 500 μm.
(F) Average insulitis score (from N = 3 mice).
Data are expressed as the mean ± SEM. *p < 0.05.
To clarify the cell biological processes that mediate the inflammatory stress response in β cells, we interrogated our recently published RNA deep sequencing analysis of alternatively spliced RNA transcripts from human islets treated with proinflammatory cytokines (Wu et al., 2021). GO analysis of each of 5 alternatively spliced transcript modes (skipped exon, mutually exclusive exons, alternative 5′ splice site, alternative 3′ splice site, and retained intron) revealed selective pathways related to protein transport/processing, ER/golgi transit, and antigen processing and presentation (Figure S4). These findings are consistent with alterations in mRNA translational processes in β cells and with the activation of the integrated stress response (ISR), which culminates in the phosphorylation of the translation factor eIF2α and the global suppression of protein synthesis (Taniuchi et al., 2016). Downstream of the phospho-eIF2α, the ISR is characterized by the formation of G3BP1+ stress granules, which are non-membranous intracellular inclusions that sequester translationally repressed mRNAs and associated translation factors (Protter and Parker, 2016). We hypothesized that the increase in PD-L1 under circumstances where 12/15-LOX is deleted or inhibited might be enabled by alleviation of the ISR. To test this hypothesis, we first verified the reduction in the ISR in NOD-Δβ mice. As shown in Figures 6B and 6C, we observed reduced islet immunostaining of phospho-eIF2α and the stress granule marker G3BP1 in 8-week-old pre-diabetic NOD-Δβ mice compared with NOD-Cre+ littermates. Together, Figures 6B and 6C suggest a reduction in the ISR in islets of NOD-Δβ mice. To link the ISR with PD-L1 production, we treated primary human islets with proinflammatory cytokines and the ISR inhibitor ISRIB, which blocks the translational inhibitory function of phospho-eIF2α and reverses stress granule formation (Sidrauski et al., 2015). As shown in Figure 6A, treatment with ISRIB resulted in augmented PD-L1 levels by immunoblot analysis. Collectively, these findings suggest that blockade of 12-LOX activity acutely enhances PD-L1 production likely by alleviating the translationally suppressive effects of phospho-eIF2α.
PD-L1 blockade recovers immune cell infiltration and T1D development in NOD-Δβ mice
To clarify the link between islet 12/15-LOX signaling, PD-L1, and immune cell interactions, we next treated NOD-Δβ mice with a rat anti-mouse monoclonal neutralizing antibody against PD-L1 (α-PD-L1) or a rat immunoglobulin G2b (IgG2b) isotype control antibody (α-IgG). After Alox15 deletion with tamoxifen, female NOD-Δβ mice were administered a single dose of α-PD-L1 or IgG intraperitoneally. As shown Figure 6D, within 1 week of α-PD-L1 administration, NOD-Δβ animals developed diabetes, while NOD-Δβ animals treated with IgG remained normoglycemic. Histology of the pancreas of NOD-Δβ animals receiving α-PD-L1 showed evidence of severe insulitis, with an increase in average insulitis score compared with NOD-Δβ mice receiving control IgG (Figures 6E–6F).
DISCUSSION
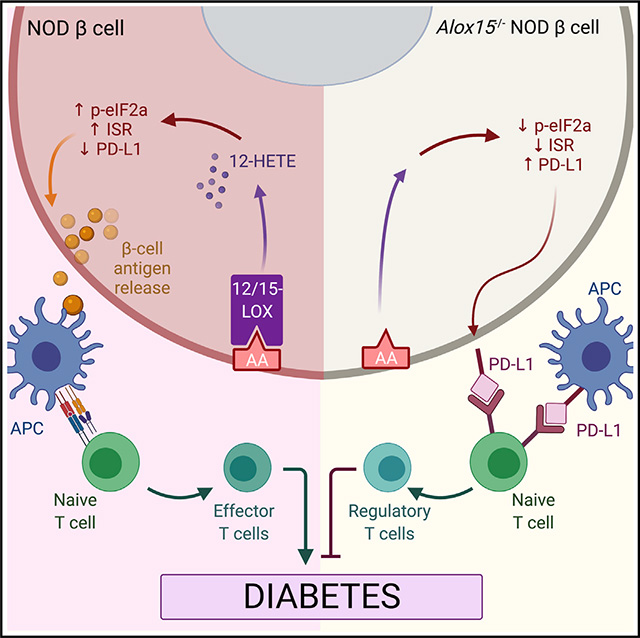
In recent years, there has been a reevaluation of the early events that culminate in autoimmunity in T1D. The emergence of new knowledge on the role of β cells, in particular, has shifted focus to the identification of mechanisms and pathways that alter the way β cells present antigens to the immune system. The concept that β cells might themselves initiate this process, which is then exacerbated by the immune system, has been reviewed recently (Roep et al., 2020). What remains unclear in the literature is whether this process, once initiated, can be reversed simply by suppressing stress responses in β cells. A challenge in directly testing this possibility is the generation of autoimmune model systems that allow for temporal and spatial alterations of β cell stress pathways. Here, we have created a NOD mouse model system that allows for the inducible deletion of a proinflammatory gene whose downstream products have been shown to generate reactive oxygen species and ER stress. Our findings show that in the absence of Alox15, there is increased generation of PD-L1 on β cells that may account, at least in part, for the cellular crosstalk that leads to suppression of insulitis and effector T cell subtypes and the protection from T1D. Our findings provide insight into how the intracellular inflammatory cascade within the β cell, in our case regulated via 12/15-LOX, might suppress an otherwise more robust protective mechanism to suppress autoimmunity. What remains a topic of speculation is how 12-LOX signaling mediates PD-L1 production and whether or not it is a direct effect of 12-LOX in response to inflammation or a secondary effect mediated by an integrated process (the ISR).
The concept that proinflammatory signaling in β cells might be conducive to immunogenicity of β cells was suggested by several studies in NOD mice in the last decade. Our group showed that NOD mice in the pre-diabetic phase display defects in prohormone processing, impairments in insulin secretion, and activation of ER stress (Tersey et al., 2012); these findings were confirmed in human studies that utilized donor pancreas tissues and blood samples from new-onset T1D subjects (Marhfour et al., 2012; Sims et al., 2016). The collective implication of these findings is that alterations in protein folding and post-translational modifications might lead to the formation of “neo-epitopes” that trigger invasion of innate and adaptive immune cells into the islet, then intitiate a vicious cycle of β cell stress, epitope spreading, and further immune cell invasion (Maganti et al., 2014). Studies in vitro and in vivo (Engin et al., 2013; Lee et al., 2020; Marre et al., 2018; Marré et al., 2016) have shown that suppression of the “endpoint” ER stress cascade might prevent the initiation of this vicious cycle. A recent elegant study (Lee et al., 2020) inducibly deleted the IRE1α arm of the ER stress pathway in β cells on the NOD mouse background and demonstrated protection from T1D, a result apparently related to β cell dedifferentiation and evasion of autoimmunity. However, in that study, β cell deletion was induced before the initiation of insulitis or any evidence of T1D, and it was not possible to ascertain the role of β cells in maintaining a pathogenic dialogue with the immune system. By contrast, in our study, the deletion of Alox15 after the development of insulitis allowed us to evaluate the role of an upstream signaling pathway that converges on stress endpoints. In particular, we show that 12/15-LOX signaling is linked not only to ER stress but, more broadly, to the ISR. Like ER stress, the ISR is characterized by the phosphorylation of Ser51 of eIF2α, a translation initiation factor, but, unlike ER stress, this phosphorylation can be triggered by a variety of kinases that respond to a diversity of stress events, including amino-acid depravation and viral infection (Taniuchi et al., 2016). Phosphorylated eIF2α sequesters the initiation factor eIF2B and leads to a block in the translational initiation of many mRNA transcripts in an attempt to divert energy expenditure toward cellular recovery processes (Muaddi et al., 2010), and these transcripts are appropriated into stress granules (Matheny et al., 2019). If the block in translational initiation is not alleviated (because of ongoing stress), the production of prosurvival proteins is suppressed, and the cell is bound for an apoptotic fate. In our NOD-Δβ mice, we observed reductions in phospho-eIF2α and stress-granule formation in islets, consistent with a reduction in the ISR. The increase in PD-L1, an immune checkpoint protein, in β cells of these mice is consistent with the alleviation of the ISR, since treatment of human islets with ISRIB, which blocks the sequestration of eIF2B by phospho-eIF2α (Sidrauski et al., 2015), similarly increased PD-L1 levels. Further studies are needed to clarify if the mRNA encoding PD-L1 (Cd274) might be translationally blocked by the ISR.
The importance of PD-L1 in protection against autoimmunity in T1D has been highlighted in recent studies. A new population of β cells resistant to immune cell attack and cellular dysfunction were identified in NOD mice that are characterized, among other features, by enhanced expression of PD-L1 (Rui et al., 2017). Moreover, residual β cells in pathologic specimens from humans with T1D show positivity for PD-L1 (Colli et al., 2018). In mice, overexpression of PD-L1 in β cells confers protection against immune cell attack (El Khatib et al., 2015; Yoshihara et al., 2020). Much of the work on PD-L1 has derived from studies of cancerous cells. Expression of PD-L1 protects cancer cells from immune cell attack by engaging with its receptor PD-1. The PD-1/PD-L1 immune checkpoint signaling inhibits T cell activation and effector function (Fife et al., 2009; Karwacz et al., 2011). Consistent with these prior studies, our work here advances the narrative that PD-L1 expression on β cells may be a critical player in β cell/immune cell crosstalk, such that its expression after initiation of insulitis alters immune cell phenotypes and suppresses T1D. Our studies offer two clear lines of evidence to support this observation: (1) the increase in β cell PD-L1 by single-cell sequencing and immunostaining in NOD-Δβ mice is co-incident with the early increase in Treg cells and anti-inflammatory myeloid cell types and the suppression of effector T cells in pLNs and pancreas, and (2) blockade of PD-L1 using a monoclonal antibody reversed the T1D protective phenotype in NOD-Δβ mice. Regarding the latter, we note that our findings show a rather rapid onset of diabetes using the monoclonal antibody, a finding that may be reflective of the aggressive blockade of PD-L1 molecules on both the β cell and innate immune cells. Whereas immune checkpoint blockade has been leveraged for cancer therapy, our studies provide an approach by which these proteins might be increased for T1D therapy, namely through inhibition of 12/15-LOX and/or the ISR.
Finally, our studies emphasize both the heterogeneity of β cells in the islet and how gene expression in subsets of β cells might broadly impact the survival of other cells. Heterogeneity of β cells in islets of NOD mice was described previously (Rui et al., 2017), but what remains unclear from these studies is how specific subpopulations of β cells might be distributed throughout the islet, such that these cells might have broader impact throughout the islet. Also, our study does not directly address if 12/15-LOX contributes to heterogeneity, though it does suggest that 12/15-LOX can alter cell numbers in these heterogeneous populations. Apart from direct cell-to-cell communication between β cells and other cell types, recent studies have now supported the concept that β cells exhibit a “secretome” that impacts phenotypes of neighboring cells. This concept is exemplified by processes such as the senescence-associated secretory phenotype (SASP) (Thompson et al., 2019) and exosome budding (Dai and Dias, 2018). In the latter case, proteins and RNA contained within exosomes can deliver transformative messages to more distant cell types that might include either antigens/neoantigens or immune checkpoint proteins (e.g., PD-L1) (Dai et al., 2017; Krishnan et al., 2019; Rahman et al., 2014; Zhou et al., 2020). In light of these intercellular communication mechanisms, it is possible that 12/15-LOX signaling in β cells links extracellular inflammatory signals (e.g., viral infections, proinflammatory cytokines) with activation of the ISR, which subsequently propagates this cellular emergency state to neighboring islet cell types and immune cells (e.g., antigen-presenting cells) to promulgate the circuit of autoimmunity. This possibility nevertheless remains to be investigated.
Limitations of the study
Some limitations of our study should be acknowledged. First, limitations exist with virtually all tissue-specific knockout approaches. Although 12/15-LOX is enriched in β cells of the islet, our experimental approach with the PdxPB-CreERTM driver line (Zhang et al., 2005) does result in a low level of deletion of Alox15 in some non-β cell islet subtypes. Therefore, we cannot know with certainty if expression of Alox15 at low levels in other islet cells might have further impacted our phenotype. Second, insulitis in NOD mice is a dynamic phenomenon whose composition changes with age. Although we inducibly deleted Alox15 after the initiation of insulitis in 6-week-old NOD mice, it remains unclear if the cellular circuit can be “shorted” at later stages of disease, where more aggressive cytotoxic CD8+ T cells prevail. Finally, our studies do not directly assess the specific product of 12/15-LOX that may confer the phenotypic effects observed, although prior studies have strongly implicated 12-HETE as the offending product (Dobrian et al., 2019). In future studies, our group will explore the roles of high- and low-affinity G protein-coupled receptors for 12-HETE, GPR31, and BLT2 (Guo et al., 2011; Yokomizo et al., 2001), in the β cell pathogenesis of T1D.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Raghavendra G. Mirmira (mirmira@uchicago.edu).
Materials availability
Unique reagents generated in this study will be made available upon reasonable request and may require completion of a materials transfer agreement. More information and requests for resources and reagents should be directed to Dr. Raghavendra Mirmira (mirmira@uchicago.edu).
Data and code availability
RNA sequencing data have been deposited in GEO. Accession numbers are listed in the key resources table. All primary data presented in this paper have been deposited as a unified Prism 9 file at Mendeley. Original microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-CD3 | BD | Cat#553057: RRID: AB_394590 |
| anti-CD28 | BD | Cat#553294: RRID:AB_394763 |
| anti-F4/80 - FITC | Biolegend | Cat# 123107: RRID:AB_893500 |
| anti-CD11c | BD | Cat# 561241: RRID:AB_10611727 |
| anti-IL-1 beta (Pro-form) monoclonal antibody | Invitrogen | Cat# 12-7114-80: RRID:AB_10736472 |
| anti-TNF-α Antibody | Biolegend | Cat# 506314: RRID:AB_493330 |
| anti-MHC-II | Biolegend | Cat# 107616: RRID:AB_493523 |
| anti-CD80 | Biolegend | Cat# 104718: RRID:AB_492824 |
| anti-CD86 | Biolegend | Cat# 105008: RRID:AB_313151 |
| anti-CD4-FITC | Biolegend | Cat# 100510: RRID:AB_312713 |
| anti-CD8 | Biolegend | Cat# 100734,:RRID:AB_2075238 |
| anti-IL17A-AlexaFluor647 | BD | Cat# 560184: RRID:AB_1645204 |
| anti-IFNg-PE | BD | Cat# 554412: RRID:AB_395376 |
| anti- FoxP3-APC | BD | Cat# 560401: RRID:AB_1645201 |
| anti-Insulin | Agilent Dako | Cat# IR002: RRID:AB_2800361 |
| anti-Insulin | ProteinTech | Cat#66198: RRID:AB_2883281 |
| anti-CD3 | Abcam | Cat# ab16669: RRID:AB_443425 |
| anti-B220 | Biolegend | Cat# 103201: RRID:AB_312986 |
| anti-phospo-eIF2a | Abcam | Cat# ab32157: RRID:AB_732117 |
| anti-G3BP1 | Novus | Cat# NBP2-16563 |
| anti-PDL1 | Abcam | Cat# ab213480: RRID:AB_2773715 |
| anti-PDL1 | Cell Signaling | Cat# 29122: RRID:AB_2798970 |
| anti-Actin | Cell Signaling | Cat# 3700: RRID:AB_2242334 |
| anti-PDL1 | BioXCell | Cat# BE0101: RRID:AB_10949073 |
| anti-IgG | BioXCell | Cat# BE0090: RRID:AB_1107780 |
| Alexa Fluor 568 donkey anti-rabbit antibody | Invitrogen | Cat# A10042: RRID:AB_2534017 |
| Alexa Fluor 488 donkey anti-guinea-pig antibody | Invitrogen | Cat# A-11055: RRID:AB_2534102 |
| anti-IL-6 | Fluidigm | 3167003C |
| anti-IL-4 | Fluidigm | 3166003C |
| anti-IL-10 | Fluidigm | 3158002C |
| anti-TNFa | Fluidigm | 3162002C |
| anti-IFNg | Fluidigm | 3165003C |
| anti-IL-17A | Fluidigm | 3174002C |
| anti-CD4 | Fluidigm | 3145002C |
| anti-CD8a | Fluidigm | 3168003C |
| anti-CD11c | Fluidigm | 3142003C |
| anti-CD206 (MMR) | Fluidigm | 3169021C |
| anti-I-A/I-E | Fluidigm | 3209006C |
| anti-CD86 | Fluidigm | 3172016C |
| anti-CD25 (IL-2R) | Fluidigm | 3151007C |
| anti-Ly-6C | Fluidigm | 3150010C |
| anti-CD19 | Fluidigm | 3149002C |
| anti-CD3e | Fluidigm | 3152004C |
| anti-CD45 | Fluidigm | 3089005C |
| anti-F4/80 | Fluidigm | 3146008C |
| anti-PDL1 | Fluidigm | 3153016C |
| anti-PD1 | Fluidigm | 3159024C |
| anti-MHC-I | Fluidigm | 3143016C |
| anti-CTLA4 | Fluidigm | 3154008C |
| anti-iNOS | Fluidigm | 3161011C |
| anti-CD11b | Fluidigm | 3143015C |
| anti-CD80 | Fluidigm | 3171008C |
| anti-IL-6 | Fluidigm | 3167003C |
| anti-IL-4 | Fluidigm | 3166003C |
| anti-IL-10 | Fluidigm | 3158002C |
| anti-TNFa | Fluidigm | 3162002C |
|
| ||
| Biological samples | ||
|
| ||
| Human Islets | IIDP; Scharp-Lacy | RRID:SAMN19470079 |
| Human Islets | IIDP; So CA Islet Cell Resource Center | RRID:SAMN19591106 |
| Human Islets | IIDP; University of Pennsylvania | RRID:SAMN19897466 |
| Human Islets | University of Alberta Islet Core | RRID:SAMN19859645 |
| Human Islets | University of Alberta Islet Core | RRID:SAMN19796386 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RPMI Medium 1640 (1X) | Gibco | 11875-093 |
| PBS | Gibco | 14190-144 |
| HEPES Buffer | Corning | 25-060-CI |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15140-122 |
| Heat inactivated FBS | Gibco | 10082-147 |
| EDTA | Thermo Fisher Scientific | BP120-500 |
| Xylene | Thermo Fisher Scientific | X5-500 |
| Methanol | Thermo Fisher Scientific | A412-500 |
| Ethanol | Thermo Fisher Scientific | A412-500 |
| Unmasking solution | Vector labs | H3300 |
| Horse blocking buffer | Vector labs | S-2012-50 |
| Immpress reagent kit peroxidase conjugated anti-rabbit Ig | Vector labs | MP-7402 |
| Vector DAB peroxidase substrate kit | Vector labs | SK-4100 |
| Hematoxilin | Sigma Aldrich | GHS216 |
| Permount | Fisher Scientific | 12-545M |
| Accutase | EMD Millipore Corporation | SCR005 |
| Collagenase | Sigma Aldrich | C7657 |
| DNAse I | ThermoFisher | AM222 |
| eBioscience™ 1X RBC Lysis Buffer | Invitrogen | 00-4333-57 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Formaldehyde | Fisher Scientific | 04042-500 |
| Tamoxifen | Sigma | T5648 |
| ISRIB | Sigma | SML0843 |
| Recombinant IFN-γ | Prospec | Cyt-358-b |
| Recombinant IL-1β | Prospec | Cyt-272-b |
| Recombinant TNF-α | Prospec | Cyt-252-b |
| Recombinant Mouse IL-2 (carrier-free) | Biolegend | 575404 |
| Recombinant Mouse IL-12 (p70) (carrier-free) | Biolegend | 577004 |
|
| ||
| Critical commercial assays | ||
|
| ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| EasySep™ Mouse Naïve CD4+ T Cell Isolation Kit | Stemcell technologies | 19765 |
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | 00-5523-01 |
| Golgi stop | BD | 512092KZ |
| Ionomycin | Sigma Aldrich | I0634 |
| Phorbol 12-myristate 13-acetate | Sigma Aldrich | P 8139 |
| Cell-IDTM Cisplatin | Fluidigm | 201064 |
| Cell-ID Intercalator-Ir — 125 μM | Fluidigm | 201192A |
| Maxpar® Cell Staining Buffer | Fluidigm | 201068 |
| Maxpar Fix and Perm Buffer | Fluidigm | 201067 |
| Maxpar Fix I Buffer (5X) | Fluidigm | 201065 |
| Maxpar Perm-S Buffer | Fluidigm | 201066 |
| Maxpar PBS | Fluidigm | 201058 |
| Maxpar Cell Acquisition Solution | Fluidigm | 201237 |
| Maxpar Water | Fluidigm | 201069 |
| Chromium Single Cell 3′ Reagent Kits V3 | 10× Genomics | PN-1000075, PN-1000073, PN-120262 |
| Live/Dead Viability/Cytotoxicity | Invitrogen | L3224 |
|
| ||
| Deposited data | ||
|
| ||
| RNA Sequencing data | GEO | Accession: GSE166572 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| B6.Cg-Alox15tm1.1Nadl/J | Jackson Laboratories | RRID:IMSR_JAX:031835 |
| Pdx1PB-CreERTM | (Zhang et al., 2005) | N/A |
| B6.129S2-Alox15/J | Jackson Laboratories | RRID:IMSR_JAX:002778 |
| NOD/ShiLtJ | Jackson Laboratories | RRID:IMSR_JAX:001976 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S3 | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Cytobank | Beckman Coulter | https://www.cytobank.org/ |
| FlowJo | FlowJo | 10.7 |
| Fiji | ImageJ | Version 2.0.0-rc-69/1.52p |
| GraphPad Prism | GraphPad software, Inc. | Version 9.0.1 |
| CV-x | Keyence | http://www.keyence.com |
| CellRanger 3.0.2 | (Butler et al., 2018; Stuart et al., 2019) | http://support.10xgenomics.com/ |
| R package Seurat | (Butler et al., 2018; Stuart et al., 2019) | Version 3.0.0.9200 |
| R package scatter | (McCarthy et al., 2017) | N/A |
| R package muscat | (Crowell et al., 2020) | Version 1.0.1 |
| EdgeR | (Robinson et al., 2010) | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse model
B6-Alox15loxp/loxp (Cole et al., 2012; Tersey et al., 2014) and Pdx1PB-CreER™ mice were backcrossed onto the NOD.ShiLt/J background using speed congenics provided by Jackson Laboratories (Table S1). After successful backcrossing, NOD-Alox15loxp/loxp mice were crossed with NOD-Pdx1PB-CreER™ mice to generate breeding colonies. The colonies were maintained at Eastern Virginia Medical School, Indiana University School of Medicine, and the University of Chicago. All mice were maintained under pathogen-free conditions according to protocols approved by the institutional animal care and use committees at each institution. Female mice were used for the studies, except where indicated in the text and figures. Diabetes outcome was studied longitudinally in mice aged from 6 weeks to 25 weeks. Mice were euthanized for studies at either 8 or 12 weeks of age.
Human islets
Non-diabetic human islets were obtained from the Integrated Islet Distribution Program (IIDP) or the University of Alberta Diabetes Institute Islet Core. Both the IIDP and the Alberta Islet Core require consent from family members of deceased donors prior to islet procurement and distribution to qualified islet research labs. The use of primary human islets from deidentified organ donors was approved by the Institutional Review Board of the University of Chicago and is not considered human subjects research. Islets from both males and females were used in this research and characteristics of the donors are provided in Table S2.
METHOD DETAILS
Animal studies
Mice were administered 3 daily intraperitoneal injections of 2.5 mg of tamoxifen dissolved in peanut oil at 6 weeks of age. For diabetes incidence, blood glucose was monitored weekly between 9 and 10 am by tail snip using an AlphaTrak glucometer. Female NOD-Δβ mice were administered a single dose of 200 mg/kg body weight α-PD-L1 or 200 mg/kg body weight α-IgG intraperitoneally at 7 weeks of age and blood glucose values were monitored for 9 days. At the indicated experimental time points, mice were euthanized, serum was collected, and tissues were harvested. For islet isolation, collagenase was injected into the pancreatic bile duct to inflat the pancreas prior to removal. The pancreas is then dissociated in HBSS with BSA. A histopaque-HBSS gradient is the applied to the dissociated pancreas, followed by centrifugation at 900 × g for 18 min. The mouse islets are then removed from the center of the gradient and cultured in RPMI medium (Stull et al., 2012). Islets were handpicked and allowed to recover overnight before experimentation.
Islet studies
Islets from non-diabetic human donors were treated with proinflammatory cytokines (1000 IU/mL IFN-γ, 50 IU/mL IL-1β) and 10 μM ML355 for 24 h, then lysed in lysis buffer (Thermo Fisher) and subjected to electrophoresis on a 4%–20% gradient SDS–polyacrylamide gel. Antibodies included mouse monoclonal antibody directed against PD-L1 (1:1000; Cell Signaling) and mouse monoclonal antibody directed against β-actin (1:1000; Cell Signaling). Immunoblots were visualized using fluorescently labeled secondary antibodies (LI-COR Biosciences) and were quantified using LI-COR software.
Islets were obtained from wildtype and Alox15−/− mice and treated with a proinflammatory cytokine cocktail containing 25 ng/mL IL-1β (Prospec), 50 ng/mL TNF-α (Prospec), and 100 ng/mL IFN-γ (Prospec) for 18 h. Islets were then transferred to a glass-bottom culture dish (MatTek) and islet death was measured using the Live/Dead Viability/Cytotoxicity kit (Invitrogen). Images were acquired using a MVX10 microscope (Olympus) and quantification was completed using ImageJ.
Single cell isolations
Pancreatic lymph nodes (pLNs) were harvested by dissection and single cells obtained by passing through a filter. Cells were then lysed with RBC lysis buffer (Thermo Fisher Scientific) and washed with PBS. For pancreatic cells, whole pancreata were isolated and digested with 1 mg/mL of collagenase type XI (Sigma Aldrich) at 37°C for 20 min. Digested pancreata were then dissociated with a syringe and the collagenase was inactivated with 10% FBS. The leukocyte fraction was enriched by gradient centrifugation using Histopaque 1077 for 20 min at 900 × g. Cells located in the interphase were collected and stimulated with 100 ng/mL PMA (Sigma Aldrich) and 500 ng/mL Ionomycin (Sigma Aldrich).
Cytometry by time of flight (CyTOF) analysis
CyTOF was performed on both single cells isolated from the pLN or whole pancreas harvested from 8 week-old mice. All mass spectrometry reagents were purchased from Fluidigm Inc. Appropriate antibody dilution was determined by serial dilutions stained to minimize background. Cell suspensions were stained with 5 μM cisplatin in RPMI without FBS for 5min at room temperature (RT) and quenched with complete RPMI. Cells were then incubated with a cocktail of surface markers antibodies diluted in Maxpar Cell staining buffer for 30 min at RT (Table S2). For intracellular markers, cells were fixed with Maxpar Fix I buffer and permeabilized with Maxpar Perm-S buffer according to the manufacturer’s recommendations. Next, cells were incubated with a cocktail of intracellular markers (Table S2) for 30 min at RT. After intracellular staining, the cells were fixed with 4% PFA for 10 min and stained with DNA intercalator (125 μM MaxPar Intercalator-Ir, Fluidigm). Cells were subsequently washed with Maxpar Cell staining buffer followed by Maxpar water. Cells were diluted to 500,000 cells/ml in Maxpar water containing EQ calibration beads. The samples were the processed on a CyTOF2 (Fluidigm) mass cytometer at an acquisition rate of <500 events/s. The resulting FCS files were concatenated and normalized using a bead-based normalization algorithm in the CyTOF acquisition software and analyzed with Cytobank. FCS files were manually pre-gated on Ir191 DNA + vs CD45+ events, excluding dead cells, doublets, and DNA negative debris. Pre-gated cells were then analyzed using viSNE and FlowSOM algoritm.
Flow cytometry
Dissociated cells from pancreatic lymph nodes were suspended in buffer containing 2% FBS in PBS. To stain for surface antigens, cells were incubated with specific antibodies to F4/80 (BM-8, Biolegend), CD11c (HL3, BD Pharmigen), CD4 (RM4–5, Biolegend), CD45 (30- F11, BD Biosciences), CD8a (53–6.7, eBioscience), CD80 (16-10A1, Biolegend), CD274 (10F.9G2, Biolegend), CD86 (GL-1, Biolegend), and MHC-II (M5/114.15.2, Biolegend) or the appropriate isotype controls for 30 min. For the intracellular staining, cells were stimulated with 100ng/mL PMA (Sigma Aldrich), 500ng/ml Ionomycin (Sigma Aldrich), and Golgi Stop Plug (BD Pharmigen, 1/1000). After stimulation, cells were first stained for surface antigens, then fixed and permeabilized with BD Cytofix/Cytoperm™ (BD Pharmigen), according to the manufacturer’s recommendations. Cells were then incubated with specific antibodies to TNF-α (MP6-XT22, Biolegend), IL-1β (NJTEN3, Thermo Fisher Scientific), IL-17 (TC11-18H10, BD Pharmigen), IFN-γ (XMG12, BD Pharmigen), and FoxP3 (MF23, BD Pharmigen). After staining, the cells were washed, filtered, and analyzed on a FACS Canto II cytometer (BD). Data were analyzed using FlowJo software (Tree Star).
Single cell RNA sequencing (scRNAseq) processing and analysis
Islets from 8-week-old mice were isolated as described above. After isolation, islets were handpicked and digested with Accutase (EMD Millipore Corporation) containing 2 U/ml of DNAse for 5 min at 37°C with agitation (1000 rpm). The cells were then washed with PBS and 2% FBS to eliminate DNAse and filtered using a 40 μm cell strainer. Each cell suspension, a collection of 180–200 islets isolated from an individual mouse, was first inspected under a microscope for cell number, cell viability, and cell size. Samples with more than 90% viability were used for scRNAseq. Single cell 3′ RNA-seq experiments were conducted using the Chromium single cell system (10x Genomics, Inc). Cell suspensions from mouse islets were first inspected under microscope for cell number, cell viability, and cell size. Single cell capture and library preparation were carried out according to the Chromium Single cell 3′ Reagent kits V3 User Guide (10X Genomics PN-1000075, PN-1000073, PN-120262). ~17,000 cells were loaded on a multiple-channel microfluidics chip of the Chromium Single Cell Instrument (10x Genomics) with a targeted cell recovery of 10,000. Single cell gel beads in emulsion containing barcoded oligonucleotides and reverse transcriptase reagents were generated with the v3 single cell reagent kit (10X Genomics). Following cell capture and cell lysis, cDNA was synthesized and amplified. Illumina sequencing libraries were prepared with the amplified cDNA. The resulting libraries were assessed with Agilent TapeStation. The final libraries were sequenced on Illumina NovaSeq 6000. About 400 million read pairs were generated for each sample, with 28 bp of cell barcode and unique molecular identifier (UMI) reads, and 91 bp RNA reads.
For analysis, CellRanger 3.0.2 (http://support.10xgenomics.com/) was utilized to process the raw sequence data generated. The filtered gene-cell barcode matrices generated from CellRanger were used for further analysis with the R package Seurat (Seurat development version 3.0.0.9200) (Butler et al., 2018; Stuart et al., 2019) with Rstudio version 1.1.453 and R version 3.5.1. Quality control (QC) of the data was implemented as the first step in our analysis. We filtered out genes that detected in fewer than five cells and cells with fewer than 200 genes. To exclude low-quality cells in downstream analysis, we used the function isOutlier from R package Scater (McCarthy et al., 2017) together with visual inspection of the distributions of number of genes, UMIs, and mitochondrial gene content. Cells with extremely high or low number of detected genes/UMIs were excluded. In addition, cells with high percentage of mitochondrial reads were also filtered out. After removing likely multiplets and low-quality cells, the gene expression levels for each cell were normalized with the NormalizeData function in Seurat. Highly variable genes were subsequently identified.
To integrate the single cell data of the Cre + control and NOD-Δβ samples, functions FindIntegrationAnchors and IntegrateData from Seurat were applied. The integrated data was scaled and principal component analysis was performed. Clusters were identified with the Seurat functions FindNeighbors and FindClusters. The FindConservedMarkers function was subsequently used to identify cell cluster marker genes. Cell cluster identities were manually defined with the cluster-specific marker genes or known marker genes. The cell clusters were visualized using Uniform Manifold Approximation and Projection (UMAP) plots. To investigate cell cluster/type-specific responses of the deletion of Alox15, a pseudo bulk count approach was applied. The counts of each gene in all cells within a cell cluster for each biologic replicate were aggregated. The R package muscat (version 1.0.1) (Crowell et al., 2020) was employed for generating pseudo bulk counts and differential gene expression analysis between conditions with the same cluster. The Seurat object generated from the integrative analysis was first converted into a SingleCellExperiment object using the as.SingleCellExperiment function in Seurat. The SingleCellExperiment object was then used as input for muscat. Gene-cell count data was aggregated with the function aggregateData and differential gene expression analysis of the pseudo counts was carried out with edgeR (Robinson et al., 2010). Gene ontology gene set enrichment analysis (GO-GSEA) was performed using the R package clusterProfiler (Yu et al., 2012). The datasets generated and analyzed in the present study are available in the GEO repository (accession number: GSE166572).
Gene expression analysis
RNA extraction was performed using RLT Buffer, according to the manufacturer’s instructions (Qiagen). iScript cDNA synthesis Kit was used was used for cDNA synthesis (Bio-Rad), and quantitative PCR (qPCR) was performed on a QuantStudio 3 (Thermo Fisher Scientific). Relative gene expression was calculated using the comparative threshold cycle (Ct) and expressed relative to control groups (ΔΔCt method). Primers for Actb, Il6, Il12, Il10, Il1β, Alox15, Pdx1, Ins1, Xbp1s, Atf4, Cd274, and Mafa were purchased from Integrated DNA Technologies or Eurofins Genomics (Table S3).
Cocxulture of macrophages with T cells
Islets isolated from Alox15−/− or wildtype littermate were stimulated with a cytokine cocktail containing 25 ng/mL IL-1β (Prospec), 50 ng/mL TNF-α (Prospec), and 100 ng/mL IFN-γ (Prospec) for 6 h. Islets were then removed from the stimulated media, washed, and cocultured with peritoneal macrophages (5 × 105 per well) and naive T cells (1 × 105 per well). Mouse peritoneal macrophages were isolated following euthanasia by injecting ice-cold RPMI into the peritoneal cavity using a 25-gauge needle (Ray and Dittel, 2010). The injected RPMI was then removed, and the isolated cells were lysed with RBC lysis buffer (eBioscience) to remove red blood cells. NaïveT cells were isolated and purified from spleenof C57BL/6J mice using EasySep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies) and stimulated with anti-CD3 (3 μg/mL, BD Pharmigen) and anti-CD28 (5 μg/mL, BD Pharmigen) in U-bottom 96-well plates for 96 h. Cells were then stimulated with 100 ng/mL PMA (Sigma Aldrich), 500 ng/mL Ionomycin (Sigma Aldrich), and Golgi Stop Plug (BD Pharmigen, 1/1000) for 4 h and then stained for FoxP3, IL-17 and IFN-γ and analyzed by FACS cytometry.
Immunohistochemistry, immunofluorescence, and β-cell mass
Pancreata from at least 5 different mice per group were fixed in 4% paraformaldehyde, paraffin embedded, and sectioned onto glass slides. Pancreata were stained for immunofluorescence using the following antibodies: anti-p-eIF2α (ab32157, Abcam), anti-G3BP1 (NBP2-16563, Novus), anti-PD-L1 (ab213480, Abcam), anti-CD3 (ab16669, Abcam), anti-B220 (103201, Biolegend), and anti-insulin antibody (IR002, Dako). Alexa Fluor antibodes were used as secondary antibodies (Invitrogen). All samples were stained with DAPI for nuclei identification (Thermo Fisher Scientific, D1306). Immunostainings were quantified by measuring pixel density per insulin + cell. Images were acquired using an LSM 800 (Carl Zeiss) or A1 (Nikon) confocal. For β-cell mass and insulitis scoring, pancreata from at least 5 different mice per group were fixed in 4% paraformaldehyde, paraffin embedded, and sectioned onto glass slides. Pancreata were immunostained using rabbit anti-insulin (1:1000; ProteinTech), Immpress reagent kit peroxidase conjugated anti-rabbit Ig (Vector), and DAB peroxidase substrate kit (Vector). β Cell mass was calculated as previously detailed. β-cell mass was calculated by measuring insulin positive area and pancreas area using CV-X software on a Keyence fluorescent microscope system (Keyence) (Tersey et al., 2018). Insulitis was scored with the following scores: 1) no insulitis, 2) infiltrate <50% circumference, 3) infiltrate >50% circumference, 4) infiltration within islet (McDuffie et al., 2008; Tersey et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean ± SEM. One-way ANOVA (with Dunnett’s post-test) was used for comparisons involving more than two conditions, a two-way ANOVA was used for comparisons with multiple time points, and a two-tailed Student’s t test was used for comparisons involving two conditions. For NOD mouse diabetes outcome experiments, a log rank (Mantel-Cox) test was used to determine significance between groups. Prism 9 software (GraphPad) was used for all statistical analyses. Statistical significance was assumed at p < 0.05.
Supplementary Material
Highlights.
12/15-lipoxygenase in β cells increases transcripts associated with inflammation
12/15-lipoxygenase-driven β cell inflammation promotes T cell infiltration and diabetes
12/15-lipoxygenase promotes the integrated stress response and suppresses PD-L1
PD-L1 blockade restores the diabetes phenotype in NOD mice lacking 12/15-lipoxygenase
ACKNOWLEDGMENTS
The authors thank Ms. Karishma Randhave (University of Chicago), Mr. Advaita Chakraborty (University of Chicago), and Ms. Jennifer Nelson (University of Chicago) for their technical assistance and Drs. Christopher Reissaus (Indiana University) and Marimar Hernadez-Perez (Indiana University) for their thoughtful discussions. Veralox Therapeutics kindly supplied ML355. This work was supported by National Institutes of Health grants R01 DK105588 (to R.G.M.) and U01 DK127786 (to R.G.M. and C.E.-M.) and by a gift from Joseph Romic and Kimberly Romic. This study utilized Diabetes Center core resources supported by National Institutes of Health grants P30 DK097512 (to Indiana University) and P30 DK020595 (to the University of Chicago). M.G. was supported by a Merit Review Award from the Department of Veterans Affairs (1IO1 BX0037440-01) and National Institutes of Health grant R01 DK120626.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Footnotes
DECLARATION OF INTERESTS
R.G.M. and J.L.N. serve on the scientific advisory board for Veralox Therapeutics. J.L.N. is a co-inventor on a patent for selective inhibitors of 12-lipoxygenase.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111011.
REFERENCES
- Atkinson MA, Roep BO, Posgai A, Wheeler DCS, and Peakman M (2019). The challenge of modulating β-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. 7, 52–64. 10.1016/S2213-8587(18)30112-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaparro RJ, and DiLorenzo TP (2010). An update on the use of NOD mice to study autoimmune (Type 1) diabetes. Expert Rev. Clin. Immunol. 6, 939–955. 10.1586/eci.10.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Yang ZD, Smith KM, Carter JD, and Nadler JL (2005). Activation of 12-lipoxygenase in proinflammatory cytokine-mediated beta cell toxicity. Diabetologia 48, 486–495. [DOI] [PubMed] [Google Scholar]
- Cole BK, Morris MA, Grzesik WJ, Leone KA, and Nadler JL (2012). Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediat. Inflamm. 2012, 851798. 10.1155/2012/851798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colli ML, Hill JLE, Marroquí L, Chaffey J, Dos Santos RS, Leete P, Coomans de Brachène A, Paula FMM, Op de Beeck A, Castela A, et al. (2018). PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. EBioMedicine 36, 367–375. 10.1016/j.ebiom.2018.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier JJ, Sparer TE, Karlstad MD, and Burke SJ (2017). Pancreatic islet inflammation: an emerging role for chemokines. J. Mol. Endocrinol. 59, R33–R46. 10.1530/JME-17-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell HL, Soneson C, Germain P-L, Calini D, Collin L, Raposo C, Malhotra D, and Robinson MD (2020). Muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 11, 6077. 10.1038/s41467-020-19894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai YD, and Dias P (2018). Exosomes or microvesicles, a secreted subcellular organelle contributing to inflammation and diabetes. Diabetes 67, 2154–2156. 10.2337/dbi18-0021. [DOI] [PubMed] [Google Scholar]
- Dai YD, Sheng H, Dias P, Jubayer Rahman M, Bashratyan R, Regn D, and Marquardt K (2017). Autoimmune responses to exosomes and candidate antigens contribute to type 1 diabetes in non-obese diabetic mice. Curr. Diabetes Rep. 17, 130. 10.1007/s11892-017-0962-4. [DOI] [PubMed] [Google Scholar]
- DiMeglio LA, Evans-Molina C, and Oram RA (2018). Type 1 diabetes. Lancet 391, 2449–2462. 10.1016/S0140-6736(18)31320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrian AD, Morris MA, Taylor-Fishwick DA, Holman TR, Imai Y, Mirmira RG, and Nadler JL (2019). Role of the 12-lipoxygenase pathway in diabetes pathogenesis and complications. Pharmacol. Ther. 195, 100–110. 10.1016/j.pharmthera.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khatib M, Sakuma T, Tonne JM, Mohamed MS, Holditch SJ, Lu B, Kudva YC, and Ikeda Y (2015). β-cell-targeted blockage of PD1 and CTLA4 pathways prevents development of autoimmune diabetes and acute allogeneic islets rejection. Gene Ther. 22, 430–438. 10.1038/gt.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F, Yermalovich A, Nguyen T, Ngyuen T, Hummasti S, Fu W, Eizirik DL, Mathis D, and Hotamisligil GS (2013). Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci. Transl. Med. 5, 211ra156. 10.1126/scitranslmed.3006534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenske RJ, Cadena MT, Harenda QE, Wienkes HN, Carbajal K, Schaid MD, Laundre E, Brill AL, Truchan NA, Brar H, et al. (2017). The inhibitory G protein α-subunit, gαz, promotes type 1 diabetes-like pathophysiology in NOD mice. Endocrinology 158, 1645–1658. 10.1210/en.2016-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, and Bluestone JA (2006). Insulin-induced remission in new-onset NOD mice is maintained by the PD-1–PD-L1 pathway. J. Exp. Med. 203, 2737–2747. 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, and Bluestone JA (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR–induced stop signal. Nat. Immunol. 10, 1185–1192. 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Holländer GA, and Piali L (2002). Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat. Med. 8, 1414–1420. 10.1038/nm1202-792. [DOI] [PubMed] [Google Scholar]
- Gagnerault M-C, Luan JJ, Lotton C, and Lepault F (2002). Pancreatic lymph nodes are required for priming of β cell reactive T cells in NOD mice. J. Exp. Med. 196, 369–377. 10.1084/jem.20011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green-Mitchell SM, Tersey SA, Cole BK, Ma K, Kuhn NS, Cunningham TD, Maybee NA, Chakrabarti SK, McDuffie M, Taylor-Fishwick DA, et al. (2013). Deletion of 12/15-lipoxygenase alters macrophage and islet function in NOD-Alox15(null) mice, leading to protection against type 1 diabetes development. PLoS One 8, e56763. 10.1371/journal.pone.0056763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesik WJ, Nadler JL, Machida Y, Nadler JL, Imai Y, and Morris MA (2015). Expression pattern of 12-lipoxygenase in human islets with type 1 diabetes and type 2 diabetes. J. Clin. Endocrinol. Metab. 100, E387–E395. 10.1210/jc.2014-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly A-K, Hsu A, Zhou S, Maddipati KR, Liu J, et al. (2011). Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 286, 33832–33840. 10.1074/jbc.M110.216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton-Williams EE, Palmer SE, Charlton B, and Slattery RM (2003). Beta cell MHC class I is a late requirement for diabetes. Proc. Natl. Acad. Sci.U S A 100, 6688–6693. 10.1073/pnas.1131954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Perez M, Chopra G, Fine J, Conteh AM, Anderson RM, Linnemann AK, Benjamin C, Nelson JB, Benninger KS, Nadler JL, et al. (2017). Inhibition of 12/15-lipoxygenase protects against β-cell oxidative stress and glycemic deterioration in mouse models of type 1 diabetes. Diabetes 66, 2875–2887. 10.2337/db17-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, and Inada M (1993). Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J. Clin. Invest. 92, 2313–2322. 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, and Escors D (2011). PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol. Med. 3, 581–592. 10.1002/emmm.201100165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan P, Syed F, Jiyun Kang N, Mirmira RG, and Evans-Molina C (2019). Profiling of RNAs from human islet-derived exosomes in a model of type 1 diabetes. Int. J. Mol. Sci. 20. 10.3390/ijms20235903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Nadler JL, Mirmira RG, and Casimiro I (2021a). Regulation of tissue inflammation by 12-lipoxygenases. Biomolecules 11. 10.3390/biom11050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Pineros AR, Walsh MA, Casimiro I, Ibrahim S, Hernandez-Perez M, Orr KS, Glenn L, Nadler JL, Morris MA, et al. (2021b). 12-Lipoxygenase governs the innate immune pathogenesis of islet inflammation and autoimmune diabetes. JCI Insight 6, 147812. 10.1172/jci.insight.147812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lee Y-S, Harenda Q, Pietrzak S, Oktay HZ, Schreiber S, Liao Y, Sonthalia S, Ciecko AE, Chen Y-G, et al. (2020). Beta cell dedifferentiation induced by IRE1α deletion prevents type 1 diabetes. Cell Metab. 31, 822–836.e5. 10.1016/j.cmet.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Xiao A, Park SH, Glenn L, Jackson L, Barot T, Weaver JR, Taylor-Fishwick DA, Luci DK, Maloney DJ, et al. (2017). 12-Lipoxygenase inhibitor improves functions of cytokine-treated human islets and type 2 diabetic islets. J. Clin. Endocrinol. Metab. 102, 2789–2797. 10.1210/jc.2017-00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maganti A, Evans-Molina C, and Mirmira R (2014). From immunobiology to β-cell biology: the changing perspective on type 1 diabetes. Islets 6, e28778. 10.4161/isl.28778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhfour I, Lopez XM, Lefkaditis D, Salmon I, Allagnat F, Richardson SJ, Morgan NG, and Eizirik DL (2012). Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55, 2417–2420. 10.1007/s00125-012-2604-3. [DOI] [PubMed] [Google Scholar]
- Marré ML, Profozich JL, Coneybeer JT, Geng X, Bertera S, Ford MJ, Trucco M, and Piganelli JD (2016). Inherent ER stress in pancreatic islet β cells causes self-recognition by autoreactive T cells in type 1 diabetes. J. Autoimmun. 72, 33–46. 10.1016/j.jaut.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marre ML, McGinty JW, Chow I-T, DeNicola ME, Beck NW, Kent SC, Powers AC, Bottino R, Harlan DM, Greenbaum CJ, et al. (2018). Modifying enzymes are elicited by ER stress, generating epitopes that are selectively recognized by CD4+ T cells in patients with type 1 diabetes. Diabetes 67, 1356–1368. 10.2337/db17-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marro BS, Legrain S, Ware BC, and Oldstone MBA (2019). Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight 4, e125067. 10.1172/jci.insight.125067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheny T, Rao BS, and Parker R (2019). Transcriptome-wide comparison of stress granules and P-bodies reveals that translation plays a major role in RNA partitioning. Mol. Cell Biol. 39, e00313–19. 10.1128/MCB.00313-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews JB, Staeva TP, Bernstein PL, Peakman M, and von Herrath M; ITN-JDRF Type 1 diabetes combination therapy assessment group (2010). Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF type 1 diabetes combination therapy assessment group. Clin. Exp. Immunol. 160, 176–184. 10.1111/j.1365-2249.2010.04153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun ATL, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186. 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDuffie M, Maybee NA, Keller SR, Stevens BK, Garmey JC, Morris MA, Kropf E, Rival C, Ma K, Carter JD, et al. (2008). Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes 57, 199–208. 10.2337/db07-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M, and Koromilas AE (2010). Phosphorylation of eIF2α at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol. Biol. Cell 21, 3220–3231. 10.1091/mbc.E10-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AM, Brown KE, Keir ME, Vanguri VK, Riella LV, Chandraker A, Sayegh MH, Blazar BR, Freeman GJ, and Sharpe AH (2011). The programmed death-1 ligand 1:B7–1 pathway restrains diabetogenic effector T cells in vivo. J. Immunol. 187, 1097–1105. 10.4049/jimmunol.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, and Parker R (2016). Principles and properties of stress granules. Trends Cell Biol. 26, 668–679. 10.1016/j.tcb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Hu L, Zhang X, Jiang S, Li J, Zhang Z, and Wang X (2019). The diverse function of PD-1/PD-L pathway beyond cancer. Front. Immunol. 10. 10.3389/fimmu.2019.02298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MJ, Regn D, Bashratyan R, and Dai YD (2014). Exosomes released by islet-derived mesenchymal stem cells trigger autoimmune responses in NOD mice. Diabetes 63, 1008–1020. 10.2337/db13-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, and Dittel BN (2010). Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 10.3791/1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roep BO, Thomaidou S, van Tienhoven R, and Zaldumbide A (2020). Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 10.1038/s41574-020-00443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, and Herold KC (2017). β cells that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab. 25, 727–738. 10.1016/j.cmet.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui J, Deng S, Perdigoto AL, Ponath G, Kursawe R, Lawlor N, Sumida T, Levine-Ritterman M, Stitzel ML, Pitt D, et al. (2021). Tet2 controls the responses of β cells to inflammation in autoimmune diabetes. Nat. Commun. 12, 5074. 10.1038/s41467-021-25367-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinov AY, Wong FS, Stonebraker AC, and Chervonsky AV (2003). Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J. Exp. Med. 197, 643–656. 10.1084/jem.20021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, McGeachy AM, Ingolia NT, and Walter P (2015). The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 4. 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims EK, Chaudhry Z, Watkins R, Syed F, Blum J, Ouyang F, Perkins SM, Mirmira RG, Sosenko J, DiMeglio LA, et al. (2016). Elevations in the fasting serum proinsulin-to-C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care 39, 1519–1526. 10.2337/dc15-2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stull ND, Breite A, McCarthy RC, Tersey SA, and Mirmira RG (2012). Mouse islet of langerhans isolation using a combination of purified collagenase and neutral protease. J. Vis. Exp. 67, e4137. 10.3791/4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, and Oyadomari S (2016). Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 6, 32886. 10.1038/srep32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Nishiki Y, Templin AT, Cabrera SM, Stull ND, Colvin SC, Evans-Molina C, Rickus JL, Maier B, and Mirmira RG (2012). Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the non-obese diabetic mouse model. Diabetes 61, 818–827. 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Maier B, Nishiki Y, Maganti AV, Nadler JL, and Mirmira RG (2014). 12-Lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol. Cell Biol. 34, 3735–3745. 10.1128/MCB.00157-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersey SA, Levasseur EM, Syed F, Farb TB, Orr KS, Nelson JB, Shaw JL, Bokvist K, Mather KJ, and Mirmira RG (2018). Episodic β-cell death and dedifferentiation during diet-induced obesity and dysglycemia in male mice. FASEB J 32, 6150–6158. 10.1096/fj.201800150RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas HE, Parker JL, Schreiber RD, and Kay TW (1998). IFN-gamma action on pancreatic beta cells causes class I MHC upregulation but not diabetes. J. Clin. Invest. 102, 1249–1257. 10.1172/JCI2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, and Bhushan A (2019). Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. 29, 1045–1060.e10. 10.1016/j.cmet.2019.01.021. [DOI] [PubMed] [Google Scholar]
- Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, Knight AM, Love JM, McAleer MA, Prins JB, et al. (1991). Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature 351, 542–547. 10.1038/351542a0. [DOI] [PubMed] [Google Scholar]
- Wu W, Syed F, Simpson E, Lee C-C, Liu J, Chang G, Dong C, Seitz C, Eizirik DL, Mirmira RG, et al. (2021). The impact of pro-inflammatory cytokines on alternative splicing patterns in human islets. Diabetes, db200847. 10.2337/db20-0847. [DOI] [PubMed] [Google Scholar]
- Yokomizo T, Kato K, Hagiya H, Izumi T, and Shimizu T (2001). Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J. Biol. Chem. 276, 12454–12459. 10.1074/jbc.M011361200. [DOI] [PubMed] [Google Scholar]
- Yoshihara E, O’Connor C, Gasser E, Wei Z, Oh TG, Tseng TW, Wang D, Cayabyab F, Dai Y, Yu RT, et al. (2020). Immune-evasive human islet-like organoids ameliorate diabetes. Nature 586, 606–611. 10.1038/s41586-020-2631-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fujitani Y, Wright CV, and Gannon M (2005). Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis 42, 210–217. [DOI] [PubMed] [Google Scholar]
- Zhou K, Guo S, Li F, Sun Q, and Liang G (2020). Exosomal PD-L1: new insights into tumor immune escape mechanisms and therapeutic strategies. Front. Cell Dev. Biol. 8, 569219. 10.3389/fcell.2020.569219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Ribas V, Rajbhandari P, Drew BG, Moore TM, Fluitt AH, Reddish BR, Whitney KA, Georgia S, Vergnes L, et al. (2018). Estrogen receptor alpha protects pancreatic beta cells from apoptosis by preserving mitochondrial function and suppressing endoplasmic reticulum stress. J. Biol. Chem. 293, 805069, jbc.M117. 10.1074/jbc.M117.805069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Li Y, Dai C, Sun L, You L, De W, Yuan Q, Wang N, and Chen Y (2018). Inhibition of Lincpint expression affects insulin secretion and apoptosis in mouse pancreatic β cells. Int. J. Biochem. Cell Biol. 104, 171–179. 10.1016/j.biocel.2018.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data have been deposited in GEO. Accession numbers are listed in the key resources table. All primary data presented in this paper have been deposited as a unified Prism 9 file at Mendeley. Original microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-CD3 | BD | Cat#553057: RRID: AB_394590 |
| anti-CD28 | BD | Cat#553294: RRID:AB_394763 |
| anti-F4/80 - FITC | Biolegend | Cat# 123107: RRID:AB_893500 |
| anti-CD11c | BD | Cat# 561241: RRID:AB_10611727 |
| anti-IL-1 beta (Pro-form) monoclonal antibody | Invitrogen | Cat# 12-7114-80: RRID:AB_10736472 |
| anti-TNF-α Antibody | Biolegend | Cat# 506314: RRID:AB_493330 |
| anti-MHC-II | Biolegend | Cat# 107616: RRID:AB_493523 |
| anti-CD80 | Biolegend | Cat# 104718: RRID:AB_492824 |
| anti-CD86 | Biolegend | Cat# 105008: RRID:AB_313151 |
| anti-CD4-FITC | Biolegend | Cat# 100510: RRID:AB_312713 |
| anti-CD8 | Biolegend | Cat# 100734,:RRID:AB_2075238 |
| anti-IL17A-AlexaFluor647 | BD | Cat# 560184: RRID:AB_1645204 |
| anti-IFNg-PE | BD | Cat# 554412: RRID:AB_395376 |
| anti- FoxP3-APC | BD | Cat# 560401: RRID:AB_1645201 |
| anti-Insulin | Agilent Dako | Cat# IR002: RRID:AB_2800361 |
| anti-Insulin | ProteinTech | Cat#66198: RRID:AB_2883281 |
| anti-CD3 | Abcam | Cat# ab16669: RRID:AB_443425 |
| anti-B220 | Biolegend | Cat# 103201: RRID:AB_312986 |
| anti-phospo-eIF2a | Abcam | Cat# ab32157: RRID:AB_732117 |
| anti-G3BP1 | Novus | Cat# NBP2-16563 |
| anti-PDL1 | Abcam | Cat# ab213480: RRID:AB_2773715 |
| anti-PDL1 | Cell Signaling | Cat# 29122: RRID:AB_2798970 |
| anti-Actin | Cell Signaling | Cat# 3700: RRID:AB_2242334 |
| anti-PDL1 | BioXCell | Cat# BE0101: RRID:AB_10949073 |
| anti-IgG | BioXCell | Cat# BE0090: RRID:AB_1107780 |
| Alexa Fluor 568 donkey anti-rabbit antibody | Invitrogen | Cat# A10042: RRID:AB_2534017 |
| Alexa Fluor 488 donkey anti-guinea-pig antibody | Invitrogen | Cat# A-11055: RRID:AB_2534102 |
| anti-IL-6 | Fluidigm | 3167003C |
| anti-IL-4 | Fluidigm | 3166003C |
| anti-IL-10 | Fluidigm | 3158002C |
| anti-TNFa | Fluidigm | 3162002C |
| anti-IFNg | Fluidigm | 3165003C |
| anti-IL-17A | Fluidigm | 3174002C |
| anti-CD4 | Fluidigm | 3145002C |
| anti-CD8a | Fluidigm | 3168003C |
| anti-CD11c | Fluidigm | 3142003C |
| anti-CD206 (MMR) | Fluidigm | 3169021C |
| anti-I-A/I-E | Fluidigm | 3209006C |
| anti-CD86 | Fluidigm | 3172016C |
| anti-CD25 (IL-2R) | Fluidigm | 3151007C |
| anti-Ly-6C | Fluidigm | 3150010C |
| anti-CD19 | Fluidigm | 3149002C |
| anti-CD3e | Fluidigm | 3152004C |
| anti-CD45 | Fluidigm | 3089005C |
| anti-F4/80 | Fluidigm | 3146008C |
| anti-PDL1 | Fluidigm | 3153016C |
| anti-PD1 | Fluidigm | 3159024C |
| anti-MHC-I | Fluidigm | 3143016C |
| anti-CTLA4 | Fluidigm | 3154008C |
| anti-iNOS | Fluidigm | 3161011C |
| anti-CD11b | Fluidigm | 3143015C |
| anti-CD80 | Fluidigm | 3171008C |
| anti-IL-6 | Fluidigm | 3167003C |
| anti-IL-4 | Fluidigm | 3166003C |
| anti-IL-10 | Fluidigm | 3158002C |
| anti-TNFa | Fluidigm | 3162002C |
|
| ||
| Biological samples | ||
|
| ||
| Human Islets | IIDP; Scharp-Lacy | RRID:SAMN19470079 |
| Human Islets | IIDP; So CA Islet Cell Resource Center | RRID:SAMN19591106 |
| Human Islets | IIDP; University of Pennsylvania | RRID:SAMN19897466 |
| Human Islets | University of Alberta Islet Core | RRID:SAMN19859645 |
| Human Islets | University of Alberta Islet Core | RRID:SAMN19796386 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RPMI Medium 1640 (1X) | Gibco | 11875-093 |
| PBS | Gibco | 14190-144 |
| HEPES Buffer | Corning | 25-060-CI |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15140-122 |
| Heat inactivated FBS | Gibco | 10082-147 |
| EDTA | Thermo Fisher Scientific | BP120-500 |
| Xylene | Thermo Fisher Scientific | X5-500 |
| Methanol | Thermo Fisher Scientific | A412-500 |
| Ethanol | Thermo Fisher Scientific | A412-500 |
| Unmasking solution | Vector labs | H3300 |
| Horse blocking buffer | Vector labs | S-2012-50 |
| Immpress reagent kit peroxidase conjugated anti-rabbit Ig | Vector labs | MP-7402 |
| Vector DAB peroxidase substrate kit | Vector labs | SK-4100 |
| Hematoxilin | Sigma Aldrich | GHS216 |
| Permount | Fisher Scientific | 12-545M |
| Accutase | EMD Millipore Corporation | SCR005 |
| Collagenase | Sigma Aldrich | C7657 |
| DNAse I | ThermoFisher | AM222 |
| eBioscience™ 1X RBC Lysis Buffer | Invitrogen | 00-4333-57 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Formaldehyde | Fisher Scientific | 04042-500 |
| Tamoxifen | Sigma | T5648 |
| ISRIB | Sigma | SML0843 |
| Recombinant IFN-γ | Prospec | Cyt-358-b |
| Recombinant IL-1β | Prospec | Cyt-272-b |
| Recombinant TNF-α | Prospec | Cyt-252-b |
| Recombinant Mouse IL-2 (carrier-free) | Biolegend | 575404 |
| Recombinant Mouse IL-12 (p70) (carrier-free) | Biolegend | 577004 |
|
| ||
| Critical commercial assays | ||
|
| ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| EasySep™ Mouse Naïve CD4+ T Cell Isolation Kit | Stemcell technologies | 19765 |
| eBioscience™ Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | 00-5523-01 |
| Golgi stop | BD | 512092KZ |
| Ionomycin | Sigma Aldrich | I0634 |
| Phorbol 12-myristate 13-acetate | Sigma Aldrich | P 8139 |
| Cell-IDTM Cisplatin | Fluidigm | 201064 |
| Cell-ID Intercalator-Ir — 125 μM | Fluidigm | 201192A |
| Maxpar® Cell Staining Buffer | Fluidigm | 201068 |
| Maxpar Fix and Perm Buffer | Fluidigm | 201067 |
| Maxpar Fix I Buffer (5X) | Fluidigm | 201065 |
| Maxpar Perm-S Buffer | Fluidigm | 201066 |
| Maxpar PBS | Fluidigm | 201058 |
| Maxpar Cell Acquisition Solution | Fluidigm | 201237 |
| Maxpar Water | Fluidigm | 201069 |
| Chromium Single Cell 3′ Reagent Kits V3 | 10× Genomics | PN-1000075, PN-1000073, PN-120262 |
| Live/Dead Viability/Cytotoxicity | Invitrogen | L3224 |
|
| ||
| Deposited data | ||
|
| ||
| RNA Sequencing data | GEO | Accession: GSE166572 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| B6.Cg-Alox15tm1.1Nadl/J | Jackson Laboratories | RRID:IMSR_JAX:031835 |
| Pdx1PB-CreERTM | (Zhang et al., 2005) | N/A |
| B6.129S2-Alox15/J | Jackson Laboratories | RRID:IMSR_JAX:002778 |
| NOD/ShiLtJ | Jackson Laboratories | RRID:IMSR_JAX:001976 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S3 | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Cytobank | Beckman Coulter | https://www.cytobank.org/ |
| FlowJo | FlowJo | 10.7 |
| Fiji | ImageJ | Version 2.0.0-rc-69/1.52p |
| GraphPad Prism | GraphPad software, Inc. | Version 9.0.1 |
| CV-x | Keyence | http://www.keyence.com |
| CellRanger 3.0.2 | (Butler et al., 2018; Stuart et al., 2019) | http://support.10xgenomics.com/ |
| R package Seurat | (Butler et al., 2018; Stuart et al., 2019) | Version 3.0.0.9200 |
| R package scatter | (McCarthy et al., 2017) | N/A |
| R package muscat | (Crowell et al., 2020) | Version 1.0.1 |
| EdgeR | (Robinson et al., 2010) | N/A |