

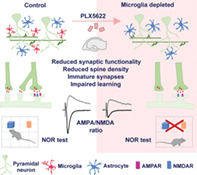
Abstract
Microglia cells are active players in regulating synaptic development and plasticity in the brain. However, how they influence the normal functioning of synapses is largely unknown. In this study, we characterized the effects of pharmacological microglia depletion, achieved by administration of PLX5622, on hippocampal CA3‐CA1 synapses of adult wild type mice. Following microglial depletion, we observed a reduction of spontaneous and evoked glutamatergic activity associated with a decrease of dendritic spine density. We also observed the appearance of immature synaptic features and higher levels of plasticity. Microglia depleted mice showed a deficit in the acquisition of the Novel Object Recognition task. These events were accompanied by hippocampal astrogliosis, although in the absence ofneuroinflammatory condition. PLX‐induced synaptic changes were absent in Cx3cr1 −/− mice, highlighting the role of CX3CL1/CX3CR1 axis in microglia control of synaptic functioning. Remarkably, microglia repopulation after PLX5622 withdrawal was associated with the recovery of hippocampal synapses and learning functions. Altogether, these data demonstrate that microglia contribute to normal synaptic functioning in the adult brain and that their removal induces reversible changes in organization and activity of glutamatergic synapses.
Keywords: glutamatergic transmission, hippocampus, learning, microglia, neuron–microglia interaction, synapses
Main Points
Microglia depletion causes weakening and functional regression of hippocampal glutamatergic synapses in adult mice; impairs learning in the NOR task.
Microglia repopulation rescues the structural, functional, and behavioral deficits.
1. INTRODUCTION
Microglia are the resident macrophage cells of the central nervous system (CNS) and their inflammatory role is well characterized (Rock et al., 2004). Beyond this, microglia perform a variety of other homeostatic functions and in particular support the development and maturation of neural circuits (Paolicelli et al., 2011; Schafer et al., 2012; Tremblay et al., 2010).
Studies in defective microglia models, such as the Cx3cr1 knockout (KO) mice (Paolicelli et al., 2014), have shown that the disruption of neuron–microglia signaling causes alterations in brain circuit development, resulting in reduced connectivity between brain areas (Y. Zhan et al., 2014) and consequent impairment of cognitive functions (Rogers et al., 2011). These alterations associated with a delay in synaptic maturation (Hoshiko et al., 2012; Paolicelli et al., 2011) have permanent consequences on synaptic properties (Basilico et al., 2019; Y. Zhan et al., 2014) and plasticity (Maggi et al., 2011; Rogers et al., 2011). Notably, defective neuron–microglia signaling in the hippocampus profoundly affects the strength and reliability of glutamatergic synapses (Basilico et al., 2019). All these effects are ascribed to the ability of microglia to foster synaptic pruning during development (Paolicelli et al., 2011), likely by contacting and phagocyting synaptic elements (Schafer et al., 2012; Tremblay et al., 2010; Weinhard et al., 2018).
In the last decade, several studies started to unravel the constitutive role of microglia in regulating synaptic plasticity and remodeling of neuronal circuits during adulthood. Loss‐of‐function mutation or deletion studies of microglial DAP12, CD200R, or CX3CR1 resulted in reduced or enhanced hippocampal long‐term potentiation (LTP) (Maggi et al., 2011; Rogers et al., 2011; Roumier et al., 2004, 2008). These, along with other evidence (reviewed in Paolicelli et al., 2014), suggest that microglia might play a critical role in learning and memory processes during adulthood. Indeed, signaling deficiency in Cx3cr1 knockout mice, microglial‐BDNF deletion or microglial depletion resulted in impaired motor learning and deficits in fear conditioning, Morris water maze and Novel Object Recognition (NOR) tasks (Maggi et al., 2011; Parkhurst et al., 2013; Rogers et al., 2011).
In this context, and given the increasing evidence of microglia role in pathological processes (Marinelli et al., 2019), it is pivotal to understand how microglial activity regulates learning processes and synaptic functions in physiological conditions. To this aim, using chronic pharmacological inhibition of colony‐stimulating factor 1 receptor (CSF1R) by systemic PLX5622 treatment (Dagher et al., 2015), we investigated the impact of microglial depletion and repopulation on structural and functional neural plasticity in the hippocampus, and its consequences on the acquisition of the NOR Task.
2. MATERIAL AND METHODS
2.1. Animals
Wild type (WT) C57BL6/J mice were used for the morphological, electrophysiological, and behavioral assessments. Thy1::GFP; Cx3cr1::cre ERT2; R26CAG‐tdtomato mice were used for dendritic spine analysis. Cx3cr1 −/− mice were used for electrophysiological analysis. All experiments were performed on male mice starting treatments at 5 weeks of age. Mice were maintained under 12‐h light/dark cycle (light on at 7 a.m.) with food and water ad libitum. All the procedures followed the guidelines of the national law (DL 26/2014) on the use of animals for research based on the European Communities Council Directive (2010/63/UE), and approved by the ethics committee of the Italian Ministry of Health.
2.2. Microglia depletion
Microglia were pharmacologically depleted by PLX5622, a selective inhibitor of CSF1R, essential for microglial proliferation, differentiation, and survival (Elmore et al., 2014). PLX5622 was kindly provided by Plexxikon Inc. (Berkeley, USA) and formulated in standard chow at 1200 PPM by Research Diets (refer to the Specific Experiments section for details on diet administration). Drug‐free standard chow (Research Diets) was used in control experiments and during PLX5622 withdrawal.
2.3. Experimental overview
2.3.1. Experiment 1. Effect of microglia depletion on astrogliosis and neuroinflammation in the hippocampal CA1
In experiment 1 (Exp. 1) we investigated whether microglia depletion causes an alteration of the brain inflammatory state. We treated three different cohorts of adult C57BL6/J mice (5 weeks old) with either control diet or PLX5622 supplemented chow for 7 consecutive days. One cohort was used for the immunofluorescence analysis of microglial (Iba1+) and astrocytic (GFAP+) markers. The second cohort was used for the ultrastructural analysis of the neuropil, microglia, and astrocytes by scanning electron microscopy. The third cohort was used to perform a gene expression profiling of hippocampi with Nanostring analysis. A different cohort of C57BL6/J mice was treated with either control diet or with PLX5622 for a period of 3–4 days for immunofluorescence analysis of Iba1 and GFAP signals.
2.3.2. Experiment 2. Effect of microglia depletion on glutamatergic synaptic activity in the hippocampal CA1
In experiment 2 (Exp. 2) we evaluated the excitatory postsynaptic currents recorded from CA1 pyramidal neurons in adult C57BL6/J mice treated with control or PLX diet as described in Exp. 1 (refer to whole‐cell patch clamp recordings section for details).
The whole‐cell patch clamp recording experiments were also performed in a group Cx3cr1 −/− mice (mice lacking the microglial fractalkine receptor), treated with control or PLX diet as described in Exp. 1 and in C53BL/6 J mice treated for 3–4 days.
2.3.3. Experiment 3. Effect of microglia depletion on hippocampal CA1 plasticity
In experiment 3 (Exp. 3) we evaluated synaptic plasticity in the hippocampal CA1 region by inducing LTP in adult C57BL6/J mice treated with control or PLX diet as described in Exp. 1 (refer to extracellular field recordings section for details).
2.3.4. Experiment 4. Effect of microglia depletion on structural plasticity in the hippocampal CA1
In experiment 4 (Exp. 4) we investigated dendritic spine density/morphology and the distribution of synapses in the hippocampal CA1 of mice treated with control or PLX diet as described in Exp. 1. We took advantage of the Thy1::GFP; Cx3cr1::cre ERT2; R26CAG‐tdtomato mouse line (triple transgenic mice expressing green fluorescent protein (GFP) in sparse excitatory neurons (Thy1::EGFP) and tdTomato in microglia (Cx3cr1::CreER16; RC::LSL‐tdTomato) (Weinhard et al., 2018) for the analysis of the spines (refer to the analysis of spine density and morphology section). For the quantification of presynaptic and postsynaptic sites we used C57BL6/J (refer to presynaptic and postsynaptic signals section).
2.3.5. Experiment 5. Effect of discontinuation of PLX5622 treatment on synaptic function, spine density and microglial morphology in the hippocampal CA1
PLX‐induced microglia depletion is reversible after treatment discontinuation (Elmore et al., 2014; Zhan et al., 2019). In experiment 5 (Exp. 5) we evaluated synaptic function and plasticity, spine density, and microglial morphology after two (Rec2W) and four (Rec4W) weeks after the discontinuation of PLX treatment. We treated four cohorts of C57BL6/J mice (5 weeks old) as described in Exp. 1, and then switched the mice to a regular diet for two or four weeks (Figure 1). The first cohort was used for the analysis of microglial density and gross morphology by performing immunofluorescence against the Iba1 marker. The second cohort was used for the ultrastructural analysis by scanning electron microscopy. The third cohort was used for extracellular field recordings. The fourth cohort was used for single‐cell recordings of excitatory postsynaptic currents in CA1 pyramidal neurons. To this last cohort, we added two extra control groups (mice treated with PLX for 3 and 5 weeks) in order to exclude that the recovery in the synaptic function after PLX5622 withdrawal was due to the mere passage of time.
FIGURE 1.
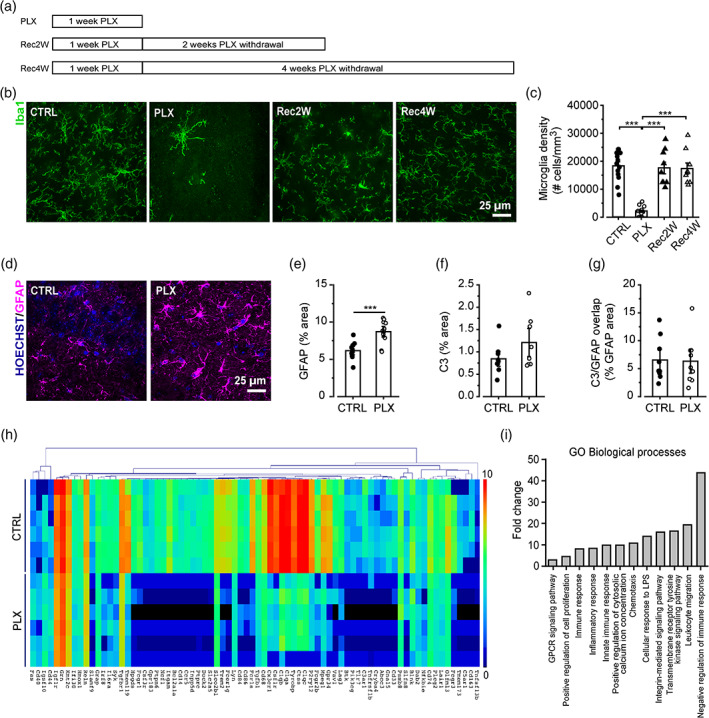
CSF1R inhibition leads to microglia depletion without causing neuroinflammation. (a) Experimental design to deplete microglia. C57BL6/J mice are fed for 7 days with 1200 PPM PLX5622. The mice are then returned to standard chow and microglial density is assessed after 2 (Rec2W) and 4 weeks (Rec4W). (b) Representative confocal images of Iba1+ microglial cells in the hippocampal CA1 stratum radiatum. (c) Quantification of microglial density in the hippocampal CA1 stratum radiatum in control condition (n = 13 fields/3 mice), after PLX5622 treatment (n = 14 fields/4 mice), 2 weeks (n = 9 fields/3 mice), and 4 weeks (n = 9 fields/3 mice) after PLX5622 withdrawal. One‐way ANOVA: F(3,44) = 33.96, p < .001; Holm‐Sidak post hoc: ***p < .001. (d) Representative images of astrocytes identified in hippocampal slices from control and PLX‐treated mice immunolabeled with anti‐GFAP antibody (magenta) and Hoechst for nuclei visualization (blue). ×60 objective (scale bar = 25 μm), acquisition from the stratum radiatum. (e) The quantification of GFAP signal, expressed as the percentual area occupied by fluorescent GFAP+ cells versus total field of view, revealed astrogliosis in PLX‐treated mice (n = 34/9/2 fields/slices/mice) versus control (n = 54/14/3). t test: t = −3.08, ***p < .001. (f) Histogram representing the quantification of the area occupied by the C3 signal in control (n = 31/9/4 FOV/slices/mice) and PLX‐treated mice (n = 26/8/4). t test t = 1.59065, p = .13254. (g) Analysis of C3 and GFAP signals overlap expressed as percentage of GFAP signal area occupied by C3 signal in control (n = 31/9/4 FOV/slices/mice) and PLX‐treated mice (n = 26/8/4). t test, t = −0.16827, p = .86862. (h) Heat map of unsupervised hierarchical clustering of the 76 differentially expressed genes in control and PLX hippocampal samples (n = 6) analyzed by Nanostring (72 genes downregulated and 4 upregulated in PLX samples). Colors in the heatmap indicate log2 counts normalized to housekeeping genes. (i) Gene ontology enrichment analysis in “Biological Process” categories for the 72 genes downregulated in PLX samples and identified according to Database for Annotation, Visualization and Integrated Discovery (DAVID) functional annotation. Data presented as Mean ± SEM (c,e) or in Log2 scale (h)
2.3.6. Experiment 6. Effect of microglia depletion on the NOR Task
In experiment 6 (Exp. 6) we determined whether the depletion of microglia in healthy adult mice is associated with cognitive deficits. We treated C57BL6/J mice (5 weeks old) with either control diet or PLX5622‐ supplemented chow for 10 consecutive days. After seven days of treatment, we initiated the NOR task. To determine whether PLX induced long‐lasting effects, a subset of animals from the PLX‐treated group were returned to the control diet over a six weeks period, and then re‐trained in the NOR Task using the same behavioral procedure with a different set of objects.
2.4. Electrophysiology
2.4.1. Extracellular field recordings
Acute hippocampal slices were obtained from adult mice (6–10 weeks old). Animals were decapitated under halothane anesthesia (Sigma‐Aldrich, Milan, Italy). Brains were rapidly extracted and immersed in ice‐cold ACSF solution continuously oxygenated with 95% O2 and 5% CO2 and containing (in mM): NaCl 125, KCl 4, CaCl2 2.5, MgSO4 1.5, NaH2PO4 1, NaHCO3 26, and glucose 10. Horizontal 350 μm thick slices were cut at +4°C using a vibratome (Thermo Scientific). Slices were transferred into an incubation chamber containing oxygenated ACSF and allowed to recover for 1 h at 30°C. Recordings were performed at 30–32°C under constant perfusion of oxygenated ACSF at 2 ml/min rate within a time window of 1–6 h after slice preparation. We placed a concentric bipolar stimulating electrode (SNE‐100 × 50 mm long, Elektronik–Harvard Apparatus GmbH) in the hippocampal stratum radiatum to stimulate Schaffer collateral fibers. Stimuli consisted of 100 μs constant current pulses of variable intensity, applied at 0.05 Hz. The recording glass micropipette (0.5–1 MΩ) was filled with ACSF and placed in the CA1 hippocampal region, at 200–600 μm from the stimulating electrode, in order to measure orthodromically‐evoked field extracellular postsynaptic potentials (fEPSP). The stimulus intensity was adjusted to evoke fEPSPs of amplitude about 50% of maximal amplitude; to ensure minimal errors in amplitude determination, we excluded from the analysis fEPSP that were contaminated by a population spike at their peak. We monitored evoked responses online, and stable baseline responses were recorded for at least 10 min. Only slices that showed stable fEPSP amplitudes (within 95%–105% of the mean of the first six evoked responses, over a 10 min period) were included in the analysis. LTP was induced by high‐frequency stimulation (HFS, 1 train of stimuli at 100 Hz, of 1 s duration). To analyze the time course of fEPSP amplitude, the recorded fEPSP was routinely averaged over 1 min (n = 3 traces). We calculated fEPSP amplitude changes following the LTP induction protocol with respect to the baseline (30 min after vs. 1 min before LTP induction). fEPSPs were recorded and filtered (low pass at 1 kHz) with an Axopatch 200A amplifier (Molecular Devices, LLC) and digitized at 10 kHz with an A/D converter (Digidata 1322A, Axon Instruments). Data were analyzed offline with Clampfit 10 software (Molecular Devices, LLC).
2.4.2. Whole‐cell patch‐clamp recordings
Acute hippocampal slices were obtained from adult mice (6–10 weeks old). Animals were decapitated under halothane anesthesia (Sigma‐Aldrich, Milan, Italy). Brains were rapidly extracted and immersed in ice‐cold ACSF, containing (in mM): KCl 2.5, CaCl2 2.4, MgCl2 1.2, NaHPO4 1.2, glucose 11, NaHCO3 26, glycerol 250, continuously oxygenated with 95% O2 and 5% CO2 to maintain the physiological pH. Horizontal 250 μm thick slices were cut at 4°C, using a vibratome (DSK, Dosaka EM, Kyoto, Japan) and placed in a chamber filled with oxygenated ACSF containing (in mM): NaCl 125, KCl 2.5, CaCl2 2, MgCl2 1, NaHPO4 1.2, NaHCO3 26 and glucose 10. Before use, slices were allowed to recover for at least 1 h at room temperature (24 ± 1°C). All recordings were performed on slices submerged in ACSF and perfused with the same solution in the recording chamber. In order to increase the release probability, where indicated, the extracellular Ca2+/Mg2+ concentration ratio of ACSF was increased to eight (Ca2+ = 4 mM, Mg2+ = 0.5 mM).
We recorded spontaneous and evoked excitatory postsynaptic currents (EPSCs) from hippocampal CA1 pyramidal neurons held at −70 mV, using a patch clamp amplifier (Axopatch 200A, Molecular Devices, LLC). Data were filtered at 2 kHz, digitized (10 kHz), acquired using pClamp 10.0 software (Molecular Devices), and analyzed offline using Clampfit 10 (Molecular Devices, LLC). Patch pipettes (3–5 MΩ) were filled with intracellular solution containing (in mM): Cs‐methanesulfonate 135, HEPES 10, MgATP 2, NaGTP 0.3, CaCl2 0.4, MgCl2 2, QX‐314 2, BAPTA 5 (pH adjusted to 7.3 with CsOH). Bicuculline methochloride (10 μM) was added to the extracellular solution to block GABAA receptors. For experiments comparing sEPCSs and mEPSCs, EPSCs were recorded from the same cell (one cell per slice). mEPSCs were recorded after 10 min of bath perfusion with tetrodotoxin (TTX, 1 μM). In experiments on evoked synaptic currents, electrical stimulation was applied by theta glass tubes (tip 15–20 μm) filled with an external solution. The stimulating electrode, connected to a stimulus isolation unit (Iso‐stim A320, WPI), was placed in the hippocampal CA1 stratum radiatum to activate the Schaffer collaterals projecting from CA3 to CA1 as previously described (Basilico et al., 2019). Synaptic responses were evoked by stimulating for 100 μs at 0.1 Hz; the stimulus intensity was adjusted accordingly to the experiment. AMPA‐mediated EPSCs were evoked by paired pulse stimulations (interval 50 ms), to determine the paired pulse ratio (PPR). For input/output curves, Schaffer collaterals were stimulated at increasing intensities (0.1–10 mA). Each pulse of a given intensity was repeated six times, to obtain an average response. The input/output curve of the AMPA component was determined at −70 mV, whereas that of the NMDA component was determined at +40 mV in presence of NBQX 10 μM. To minimize voltage clamp errors, when EPSC reversal potential was more positive than +10 mV, we corrected for the membrane potential error, by recording NMDA currents at +40 mV + ERev. To determine the AMPA/NMDA ratio, stimulus strength was adjusted to obtain at −70 mV stable AMPA‐mediated EPSC with an amplitude corresponding to the 50% of the maximum response. NMDA currents were recorded from the same neuron and using the same stimulus strength. For NMDA EPSC, we measured the EPSC reversal potential (ERev) in each cell and corrected the NMDA recording potential consequently (+40 mV + ERev), to avoid potential voltage clamp errors. NMDA peak amplitude was measured with a delay of 25 ms from the AMPA peak. The AMPA/NMDA ratio was then calculated using the equation: ratio = AMPA EPSC amplitude/NMDA EPSC amplitude.
We constructed I/V curves for EPSC, by measuring current amplitude at two different time points; at the current peak (for AMPA component) and at 25 ms from the peak (for NMDA component). Onset and offset kinetics were determined on EPSC recorded at −70 mV. We analyzed the synaptic delay in both control and PLX groups, by measuring the time between the peak of the stimulation artifact and the onset of the EPSC (Fedchyshyn & Wang, 2007). In order to determine the onset kinetics, we measured the ratio between the rise time (10%–90%) and the amplitude of the elicited current, excluding amplitudes larger than 100 pA. For the analysis of the offset kinetics, we calculated the Tau constant by fitting the decay phase with a single exponential function (Hestrin et al., 1990). In order to display onset and offset kinetics, control and PLX average EPSC curves were superimposed: traces were aligned (at EPSC onset) and scaled for comparison.
For current clamp experiments and sIPSC recordings, 350 μm‐thick hippocampal slices were prepared as described above, transferred to a submerged chamber, and perfused with oxygenated ACSF (3–4 ml/min; same composition as for sEPSCs). CA1 or CA3 pyramidal neurons were visualized using the IR‐DIC optics of an upright Leica DMLSF microscope and current‐ or voltage clamp recordings were obtained using a MultiClamp 700B amplifier and the pClamp9 suite (Molecular Devices). All recordings were performed at room temperature (24 ± 1°C). Patch pipettes (4–10 MΩ) were filled with intracellular solution containing (in mM): 110 K‐gluconate, KCl 12, Na2‐phosphocreatine 10, HEPES 10, Mg2‐ATP 4, Na2‐GTP 0.2, EGTA‐K 0.1 (pH 7.3 with KOH; osmolarity corrected with sucrose when needed to reach ~290–310 mOsm); for sIPSC recordings, the solution was the same except for substituting 110 mM K‐gluconate with 120 mM CsMetSO3; QX‐314 (5 mM) was added daily to avoid AP firing at depolarized potentials.
In current clamp experiments, the sub‐ and supra‐threshold properties of pyramidal CA3 neurons were studied within the first 3 min after patch rupture and while holding the membrane potential at approximately −80 mV by constant current injection. Current steps (Iinj), ranging in amplitude from 200 to 600 pA (50 pA increments; 1 s duration), were injected to estimate the input resistance (Rin) and to build the f–I relationship. The rheobase, the amplitude of the injected current necessary to induce the first AP, was estimated graphically (Clampfit) from the voltage response to a series of 5 pA‐incremented consecutive steps of Iinj (50 ms duration; range: 0–0.4 nA).
In voltage clamp, sIPSCs were recorded holding the membrane potential at +10 mV; estimated reversal potential (ERev) for glutamatergic synaptic currents was 0 mV, thus no blockers were necessary to isolate GABAergic currents (estimated ERev approximately −65 mV). To improve the whole‐cell voltage clamp, series resistance (Rs) was compensated (range 30%–70%) and recordings bearing excessive increase of Rs were excluded from analysis. In some voltage clamp experiments, 10 μM SR95531 (a blocker for GABAA receptors; Tocris) was used to verify the GABAergic nature of the currents recorded (data not shown). Current clamp recordings were filtered online at 10 kHz and sampled at 50 kHz; synaptic currents were filtered at 2 kHz and digitized at 10 kHz (amplifier in‐built 8‐pole low‐pass Bessel filter and Molecular Devices Digidata 1322A, respectively). Recordings used for analysis were post hoc filtered at 1 kHz.
2.5. Analysis of spine density and morphology
2.5.1. Dendritic spines imaging
Mice were transcardially perfused with phosphate buffer 0.9% saline solution (PBS) and 4% paraformaldehyde (PFA) in 0.1 M pH 7.4 phosphate buffer (PB), under halothane anesthesia (Sigma‐Aldrich, Milan, Italy). Brains were rapidly removed, postfixed overnight in PFA 4%, and incubated in PBS 30% sucrose solution for 24 h at +4°C. Before being frozen at −80°C, brains were placed for at least 2 h in chilled isopentane. We cut the frozen brains into 40 μm horizontal slices using a cryostat (Leica). Once fluorescent neurons of hippocampal CA1 were identified, images of primary dendrites that ramify in secondary and tertiary dendrites in the CA1 stratum radiatum were acquired using a confocal laser scanning microscope (iX83 FV1200) with a ×60 oil immersion objective. Fluorescence was separated by a dichroic mirror with 572–700 nm emission filter (green channel: for EGFP fluorescence detection) and detected Z‐stack images by photomultipliers (1600 × 1600 pixels, 0.044 μm per pixel, 0.1 μm z‐step).
2.5.2. Dendritic spines quantification and analysis
To increase resolution, images were deconvoluted (50 iterations, 0.01 of quality change) with the software Huygens Professional 18.04. Analysis was performed blinded to the experimental conditions using the NeuronStudio software. We isolated segments of known length from secondary or tertiary dendrites, and then reconstructed the segment with the software. Dendritic spines were automatically counted and classified into three morphological classes (thin, stubby, and mushroom) (Rodriguez et al., 2008). Not detected spines were added manually and classified based on shape features as follows: spines displaying a wide protuberance in absence of a neck were classified as stubby, thin and elongated spines with head and neck of similar diameter were classified as thin spines, and spines with a prominent head and a thin neck were classified as mushroom (Hering & Sheng, 2001; Peters & Kaiserman‐Abramof, 1970). To calculate dendritic spine density, spine number was divided by the length of the dendritic segment and expressed as number per micrometer.
2.6. Analysis of presynaptic and postsynaptic signals
2.6.1. Immunofluorescence
Under a sub‐lethal dose of carbon dioxide, mice were transcardially perfused with cold PBS and 4% PFA in 0.1 M PB. Brains were rapidly removed, postfixed overnight in PFA 4%, washed with PB and cryoprotected in PB 30% sucrose solution. We then collected 40 μm‐thick coronal sections with a cryostat microtome (Leica Microsystems) at −20°C. Sections containing the hippocampus were rinsed three times in PB and incubated with blocking solution, containing 10% Bovine serum albumin and 0.1% Normal donkey serum (Sigma Aldrich, St Louis) in PB 0.3% Triton X‐100 (Applichem, BioChemica, Darmstadt, Germany) 0.1 M, for 1 h at room temperature. Slices were then incubated with primary antibodies in blocking solution overnight at room temperature. We used as presynaptic and postsynaptic markers, respectively: Rabbit anti‐Synapsin I polyclonal antibody (1:100, Sigma Aldrich, St Louis) and guinea pig anti‐Drebrin polyclonal antibody (1:100, OriGene Technologies, Herford, Germany). The day after, free‐floating slices were washed (3 × 10‐min) in PB at room temperature and incubated with secondary antibodies for 2 h, followed again by three 10‐min rinses in PB. We applied DAPI for 5 min during the second rinse, dissolved 1:2000 in PB solution. Control background experiments were performed by incubating secondary antibodies alone.
2.6.2. Structured illumination (SIM) microscopy
Image acquisition was performed using a Nikon Eclipse Ti equipped with: X‐Light V2 spinning disk combined with a video confocal super‐resolution (VCS) module (CrestOptics) based on structured illumination; LDI laser source (89 North); Prime BSI Scientific CMOS (sCMOS) camera with 6.5 μm pixels (Photometrics). The images were acquired with Metamorph software version 7.10.2. (Molecular Devices) with a ×100 PlanApo Lambda oil objective (1.45 numerical aperture) and a z‐step size of 0.1 μm to obtain a total Z‐stack of about 1.2 μm. In order to achieve super‐resolution, we processed the raw data obtained by the VCS module with a modified version of the joint Richardson‐Lucy (jRL) algorithm (Chakrova et al., 2016; Ingaramo et al., 2014; Ströhl & Kaminski, 2015), where the out of focus contribution of the signal was explicitly added in the image formation model used the jRL algorithm and evaluated as a pixel‐wise linear “scaled subtraction” (Heintzmann & Benedetti, 2006) of the raw signal.
2.6.3. Analysis and quantification
Presynaptic (Synapsin I marker, in green) and postsynaptic (Drebrin marker, in red) signals were analyzed using the MetaMorph Microscopy Analysis Software (moleculardevices.com) with the following settings: for each image, five continuous single planes were selected and independently 3D rendered on the X‐Y axes by MetaMorph 4d viewer, at the minimum threshold for light objects. MetaMorph outputs the x and y coordinates of detected puncta in each plane and its corresponding size in voxels.
Cartesian coordinates of puncta were further analyzed with custom written MATLAB scripts. By combining all five imaging planes, the puncta were visualized on a 2D plane. In order to compute density, the entire imaging plane was divided into 400 bins of 10 μm2 each. The density for each bin was then computed by counting the number of voxels observed in each bin. This allows us to measure the volume of the signal accurately. Bins that overlapped with tissue damage were excluded from further analysis, by using a threshold of 300 voxels/bin., determined by visually inspecting confocal images. Data were analyzed in Graphpad PRISM 9.
2.7. Analysis of astrogliosis, microglial density and morphology
2.7.1. Immunofluorescence
Mice were perfused and brains sliced as previously described (refer to the analysis of spine density and morphology section). Sections were then rinsed in PBS (3 × 5 min) and we performed antigen retrieval by soaking slices for 40 min in a warm solution containing (in mM): 10 Na‐citrate, 0.05% Tween 20, pH 6.0, 90°C. Subsequently, slices were incubated for 45 min in blocking solution (3% goat serum and 0.3% Triton X‐100 in PBS). Primary antibodies diluted in blocking solution were incubated overnight at 4°C (anti‐GFAP z0334 DAKO 1:400, anti‐Iba1 019‐19741 Wako 1:400). After three washes in PBS, we incubated the sections for 45 min with secondary antibodies (Goat anti Rabbit‐Alexa Fluor 647, ThermoFisher) and Hoechst in order to visualize nuclei. We then coverslipped them with fluorescence mounting medium S3023 (Dako) and acquired the images by confocal microscopy (See following sections).
2.7.2. Structured illumination microscopy (SIM)
As for the analysis of presynaptic and postsynaptic signals, image acquisition was performed through a Nikon Eclipse Ti equipped with a X‐Light V2 spinning disk combined with a VCS module (CrestOptics) based on structured illumination and with a LDI laser source (89 North). The images were acquired by using Metamorph software version 7.10.2. (Molecular Devices) with a ×10 and ×60 PlanApo l oil objective (1.4 numerical aperture) and sectioning the slice in Z with a step size of 0.2 μm for spinning disk to obtain a total Z‐stack of about 10 μm (for astrocytes measurement) or 25–35 μm for microglia morphology analysis.
2.7.3. Astrogliosis analysis
Maximal intensity projection images (50 z‐stack planes) were analyzed using Metamorph software. After thresholding the images, astrogliosis was quantified as the area occupied by fluorescent GFAP+ cells versus total field of view.
2.7.4. Microglial morphology and density
Image processing was performed using ImageJ software (NIH). To quantify microglial density, the number of Iba1+ cells was reported as a number of somas per acquired volume. Maximal intensity projections of Iba1 confocal images were analyzed to obtain morphological indicators of cell complexity, as previously described (Basilico et al., 2019). Only cells whose body and processes were entirely contained in the image were included in the analysis. For each cell, the soma area and the arborization domain were assessed. Through skeleton analysis, we calculated the total number of microglial processes.
2.7.5. NanoString gene expression
Neuroinflammation mouse panel, containing 757 mouse neuroinflammatory genes and 13 internal reference controls, was used for control and PLX hippocampal samples. Briefly, a total of 100 ng RNA in a volume of 5 μl was hybridized to the capture and reporter probe sets at 65°C for 20 h according to the manufacturer's instructions. Data were collected using the nCounter Digital Analyzer (NanoString). RNA counts were normalized using the geometric mean of the housekeeping genes included in the panel, after validation against positive and negative controls, using the nSolver 4.0 software (NanoString). Fold changes were calculated comparing untreated (CTRL) and treated (PLX) samples.
2.7.6. Real time PCR
Total RNA was extracted from hippocampal tissue with the Quick RNA MiniPrep (Zymo Research, Freiburg, DE) and retrotranscribed with iScript Reverse Transcription Supermix for Real‐time PCR (RT‐PCR) (Bio‐Rad, Hercules, CA). RT‐PCR was carried out using Syber Green (Biorad) according to the manufacturer's instructions. The PCR protocol consisted of 40 cycles of denaturation at 95°C for 30 s and annealing/extension at 60°C for 30 s. For quantification analysis the comparative Threshold Cycle (Ct) method was used. The Ct values from each gene were normalized to the Ct value of GAPDH in the same RNA samples. Relative quantification was performed using the 2 − ∆∆Ct method (Schmittgen & Livak, 2008) and expressed as fold change in arbitrary values. Primer sequences targeted against GAPDH forw: TCG TCC CGTAGACAAAATGG, GAPDH rev: TTGAGGTCAATGAAGGGGTC; CD44 forw ACCTTGGCCACCACTCCTAA; CD44 rev GCAGTAGGCTGAAGGGTTGT; GFAP forw AGAAAGGTTGAATCGCTGGA; GFAP rev CGGCGATAGTCGTTAGCTTC; Kir 4.1 forw ATCAGAGCAGCCACTTCACC; Kir 4.1 rev GGCTCTCTGTCTGAGTCGTC; LAMP1 forw ACTGGTAACAACGGAACCTG: LAMP1 rev ACACATTGGGGTTAGGAACA; LAMP2 forw CTAGGAGCCGTTCAGTCCAA; LAMP2 rev CTTGCAGGTGAATACCCCAA.
2.8. Ultrastructural analysis of the neuropil, microglia and astrocytes
2.8.1. Immunohistochemistry for electron microscopy
Four mice per experimental group were anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and perfused with 0.1% glutaraldehyde in PFA 4%. Coronal sections containing the stratum radiatum of the hippocampal CA1 were selected based on the stereotaxic atlas Paxinos and Franklin fourth edition. The sections were washed in PBS 50 mM, (pH 7.40) to remove the cryoprotectant solution, incubated 5 min in 0.3% hydrogen peroxide and 30 min in 0.1% sodium borohydride, followed by 1 h long incubation in a blocking buffer solution (10% fetal bovine serum, 3% bovine serum albumin, 0.01% triton X‐100). The tissues were then incubated overnight with the primary antibody rabbit anti‐Iba1 (1/1000; Wako, catalogue number 019‐19741) in the blocking buffer solution at 4°C. The following day, sections were thoroughly washed before being incubated 90 min with the biotinylated secondary antibody goat‐anti rabbit (1:300; Jackson ImmunoResearch, catalogue number 111‐066‐046) at room temperature. Hippocampal sections were incubated 1 h in an avidin‐biotin complex solution (1:100, Vector Laboratories) and developed in a tris‐buffer solution (TB, 0.05 M, pH 8.0) containing 0.05% 3,3'Diaminobenzidine (DAB) with 0.015% hydrogen peroxide for 5 min. The sections were then processed for scanning electron microscopy. Briefly, sections were incubated 1 h in a solution of equal volume containing 4% osmium tetroxide and 3% potassium ferrocyanide in phosphate buffer (PB, 0.1 M, pH 7.4), 20 min in a heated solution of distilled water combined with thiocarbohydrazide and 30 min in an aqueous solution of 2% osmium tetroxide. The sections were then dehydrated with an increasing amount of ethanol (5 min each dehydrating step): 2 × 35%, 1 × 50%, 1 × 70%, 1 × 80%, 1 × 90%, 3 × 100%. Following the dehydration, the tissues were washed in propylene oxide to remove the excess ethanol and were immersed overnight in Durcupan resin. The next day, sections were placed between two ACLAR sheets (Electron Microscopy Sciences) and were left to polymerize for 3 days at 55°C.
2.8.2. Scanning electron microscopy using array tomography
The hippocampal CA1 region was then dissected from the processed biological samples using a knife and a binocular microscope. Tissues were then mounted on resin blocks using superglue and 70 nm thick ultrathin sections were acquired using a Leica UC7 ultramicrotome equipped with a diamond knife (Diatome). Floating sections were loaded onto dust‐free silicones chips (EMS) which were installed on round metal stub (EMS) inserted into a Zeiss Crossbeam 540 focused ion beam scanning electron microscope (FIB‐SEM). Once in the FIB‐SEM, the hippocampal stratum radiatum was identified using backscattered electrons (ESB) and secondary electrons (SE2) detectors at 10 kV for initial focus, then at 1.4 kV when samples were at a sufficient working distance to perform high‐resolution focus on the brain ultrastructure. These steps were controlled using SmartSEM software (Fibics). Automated imaging of whole sections was performed using ATLAS Engine 5 software (Fibics) and imaging regions of interest were drawn in order to produce 25 and 5 nm resolution mosaics for regional analysis and microglial ultrastructural analysis, respectively. Resulting mosaics were then stitched together using Atlas 5 and individually verified for precise title matching. The resulted images were exported as TIFF files using Atlas 5 as well and experimental conditions were blinded to the investigator using ImageJ.
2.8.3. Ultrastructural analyses of microglia
Four‐to‐nine microglial cell bodies were analyzed qualitatively in one animal per condition. For the qualitative analysis of microglial ultrastructure, the following parameters were evaluated: the presence of phagocytic inclusions, elongated or altered mitochondria, lysosomes, lipid bodies, lipofuscin, synaptic contacts, extracellular space and/or digestion, and the dilation of the endoplasmic reticulum (ER) and Golgi apparatus. Microglial cell bodies were identified by their immunoreactivity to Iba1, their overall shape, unique heterochromatin pattern, extracellular space pockets as well as their long and narrow ER (Peters et al., 1991; Tremblay et al., 2010). The analysis of these parameters was based on the transmission electron microscopy analysis of microglia previously published (Bisht et al., 2016; El Hajj et al., 2019; Hui et al., 2018; Savage et al., 2019; St‐Pierre et al., 2019). Briefly, we considered mitochondria elongated if their length was greater than 1000 nanometer and, altered if they appeared swollen, or if broken cristae were observed (Bisht et al., 2016; Hui et al., 2018). We identified lysosomes by the presence of granules, the association with vesicles or lipid bodies and their electron‐dense heterogeneous content (El Hajj et al., 2019). We distinguished lipofuscin from lysosomes by their distinct fingerprint‐like pattern (Peters et al., 1991). We identified lipid bodies by their round shape and homogenous content (Hui et al., 2018). We determined extracellular space and digestion by the presence of electron‐lucent content found between the microglia and the parenchyma (Tremblay et al., 2012). We characterized the synaptic contact by interaction with either a presynaptic axon terminal and/or postsynaptic dendritic spine recognized by the presence of a postsynaptic density. We determined the dilation of the ER and Golgi apparatus by the electron‐lucent pockets found inside the organelles (Hui et al., 2018; St‐Pierre et al., 2019).
2.9. NOR task
2.9.1. Equipment
NOR task was carried out in an open field arena (40 × 40 × 40 cm) made in plexiglass placed in a dimly (30–40 lx) illuminated soundproof room. Stimulus objects were made of ceramic or plastic and varied in color, size, and shape. The role (familiar or novel) and the relative position of the objects was counterbalanced and randomly permuted for each mouse. After each session, before starting the new trial, the open field arena and the stimulus objects were cleaned with a 70% ethanol solution to ensure the absence of olfactory cues. Mouse behavior was recorded by a video tracking and analysis system (ANY‐maze video‐tracking software 6.3; Stoelting Co., Wood Dale, Il).
2.9.2. Behavioral procedure
The experiments were performed blinded to the treatment condition. Mice were handled for seven consecutive days (1 min/twice a day) prior to the behavioral assessment to ensure an adequate habituation to the experimenter. On the first day of the experiment, mice were habituated to the open field arena by allowing them to freely explore for 10 min in the absence of any other behaviorally relevant stimulus (twice/60 min apart). On the second day, familiarization/training started: mice were placed in the open field arena containing two identical copies of the same object. Mice were allowed to freely explore the objects for 10 min (3 trials/60 min apart). The same procedure was repeated on the third day and during the first two trials on the fourth day. On the fourth day, 60 min after the last familiarization/training trial, mice were subjected to an acquisition test (5 min). For this purpose, one of the objects used during training was randomly replaced by a novel one. Exploration of the object was defined as active exploration with the nose pointing toward the object at a distance smaller than 1 cm. The discrimination index (D.I.) was calculated as the ratio of the difference between the exploration time of the unfamiliar (UF) minus the familiar (F) object and the total exploration time: DI = (UF − F)/Total exploration time × 100 (Broadbent et al., 2004). We also recorded the number of crossings, the total distance traveled (meters), the distance traveled in the center of the arena (meters), the average speed (meters/sec), and the time spent grooming.
2.10. Statistical analysis
All data are reported as mean ± SEM. Origin 6 and SigmaPlot 12 (Systat Software Inc., San Jose, CA) softwares were used for statistical analysis. For electrophysiological recordings, n/N refers to numbers of slices/mice (field recordings) and cells/mice (whole‐cell patch clamp). Electrophysiological data were analyzed by unpaired t test, when comparing only two groups, or one‐way analysis of variance (ANOVA), when comparing more than two groups. Kolmogorov–Smirnov test was used to compare cumulative probability curves. Two‐way ANOVA (treatment x stimulus intensity) was used to analyze the input–output curves. Post hoc comparisons were performed using the Holm–Sidak test. Dendritic spine density and morphology, microglial density and morphology, and astrogliosis were analyzed by one‐way ANOVA, followed by Holm–Sidak post hoc analysis. For Nanostring analysis, we determined significant Differential Expressed (DE) genes by t test using MeV software considering fold changes higher than 1.5 fold, lower than 0.5 fold and p ≤ .05. We performed unsupervised hierarchical clustering and heat map analysis using MeV software. Repeated measure ANOVA or Two‐way ANOVA (treatment x objects) was used, to analyze object exploration time during the familiarization/training trials in the NOR task. Post hoc comparisons between trials were performed using the Tukey's test. Unpaired t test was used to compare locomotor activity and performance in the acquisition test. Normality tests were performed with Prism 9 and nonparametric tests were used when appropriate.
3. RESULTS
3.1. PLX5622 treatment causes microglia depletion in the hippocampus without triggering neuroinflammation
In order to decipher the role of microglia in adult hippocampal synaptic functioning, 5‐weeks‐old C57BL6/J mice were fed with a chow containing 1200 PPM of PLX5622 for 7 days (Figure 1a).
As previously reported (Huang et al., 2018; Zhan et al., 2019), PLX‐treated mice showed a 90% reduction of Iba1+ cells in the hippocampal CA1 stratum radiatum, relative to control (Figure 1b,c). After discontinuing the PLX5622 diet for 2 weeks, microglia repopulated the mouse brain (Figure 1b,c).
Microglia depletion may cause profound changes in the brain parenchyma, due to a potential alteration of tissue homeostasis or to the death of microglia. To determine whether the effects of PLX on synapses result from an alteration of the brain inflammatory state, we performed molecular and cellular analysis of astrocytic and inflammatory markers. We observed an increase of GFAP+ signal density in CA1 stratum radiatum, indicative of astrogliosis in microglia‐depleted mice (Figure 1d,e).
Electron microscopy images of hippocampal astrocytes from microglia‐depleted mice showed signs of an active cytoplasm, characterized by numerous phagocytic vesicles and mitochondria, suggesting intense phagocytic activity and high‐energy need (Figure S1A). To characterize the phagocytic activity of astrocytes, in PLX‐treated mice, we measured by RT‐PCR the amount of lysosomal associated membrane proteins (LAMP1 and LAMP2) and performed immunofluorescence analysis of LAMP2 staining (Figure S1). In hippocampal extracts from control and PLX mice (6 each), we did not find significant differences in LAMP1 or LAMP2 transcripts level (Figure S1B). Consistently, in slices co‐stained with LAMP2 and GFAP antibodies, we found that LAMP2 is expressed in GFAP+ cells and that this expression is not modulated by PLX treatment (Figure S1C,D).
To explore the possibility that in the absence of microglia astrocytes acquire a reactive phenotype (Escartin et al., 2021), we analyzed the expression of the complement factor C3 in GFAP+ cells in control and PLX mice and after 4 weeks of recovery. Astrocytic C3 staining did not show alterations after PLX treatment (Figures 1f,g and S1E), not supporting the appearance of a reactive phenotype.
To further confirm the absence of neuroinflammation, we performed gene expression profiling of the hippocampus (Nanostring analysis of total hippocampal RNA extracts). We observed changes in gene expression for 76 over 757 genes within the Neuroinflammation mouse panel in microglia‐depleted mice (Figure 1h). Among these, 72 were downregulated in PLX‐treated mice, while only 4 (FAS, CD44, CD40, IGSF10) were upregulated. Real time PCR analysis of hippocampal extracts confirmed the increase in CD44 and highlighted the increase in GFAP transcripts, supporting immunofluorescence data (Figure S1F). The 72 downregulated genes belong to microglia‐mediated inflammatory function as determined by Gene ontology and Kegg‐pathway analysis, as for instance genes related to complement cascade (C1QA, C3AR1, C1QB, C1QC; p < .001), microglial G‐protein signaling pathway (P2RY12, CCR5, CX3CR1), and natural killer cell cytotoxicity (VAV1, PIK3CG, PTPN6, SYK; p < .001) (Figure 1i), corroborating the absence of an inflammatory state upon PLX treatment.
Consistently, in EM analysis, we did not observe clear ultrastructural signs of neuroinflammation in microglia‐depleted mice, as revealed by the absence of organellar disruption throughout the neuropil in both PLX‐treated and control mice (Figure S2A).
Qualitative analysis of the whole neuropil of the stratum radiatum did not reveal the presence of stressed cells in PLX‐treated mice, as revealed by electron‐dense cyto‐ and nucleoplasm (Figure S2A). Consistently, we did not observe the presence of dark microglia in the stratum radiatum of both PLX and control mice, supporting the absence of inflammatory processes. In addition, we did not identify blatant ultrastructural signs of cell death in microglia‐depleted mice using scanning electron microscopy. In addition, we observed an increase in the perineuronal network, as revealed by WFA staining (Figure S2B,C). Moreover, the analysis by immunofluorescence of the postsynaptic (drebrin) and presynaptic markers (synapsin I) in microglia‐depleted mice, did not reveal major changes in synapses density (Figure S2D‐G).
3.2. Microglia depletion impairs glutamatergic synaptic activity in the hippocampus
To investigate whether microglia depletion could interfere with synaptic functional properties, we characterized hippocampal synapses in acute brain slices by patch‐clamp recordings in CA1 pyramidal neurons. The main finding was that PLX‐treated mice showed a significant decrease in the amplitude of spontaneous excitatory postsynaptic currents (sEPSC) relative to control mice (Figure 2a–d), without major effects on sEPSC frequency (Figure 2e,f). Notably, this effect was specific for glutamatergic synapses, since we did not observe differences in amplitude (Figure S3A) or frequency (Figure S3B) of spontaneous GABAergic IPSCs (Figure S3C).
FIGURE 2.
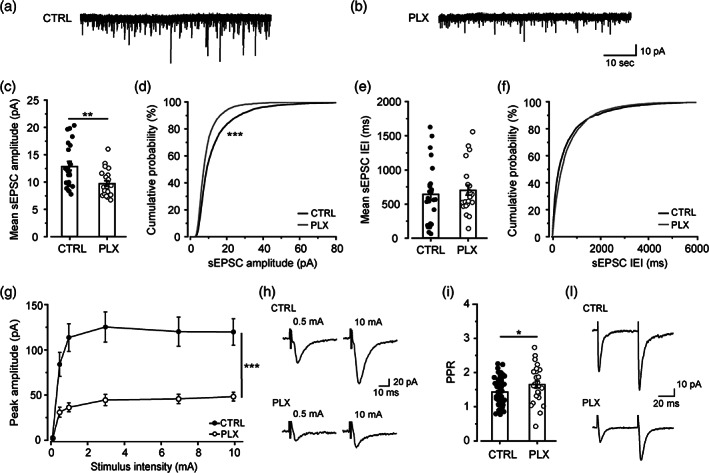
Microglia depletion impairs glutamatergic transmission in hippocampus. (a, b) Representative traces of sEPSC recorded at −70 mV from CA1 pyramidal neurons in control (CTRL) (a) and PLX‐treated mice (b). (c) Bar graph of mean sEPSC amplitudes in control (n = 25 cells/6 mice) and PLX neurons (n = 23 cells/9 mice). t test: t = 3.22, **p < .01. (d) Cumulative distributions of sEPSC amplitudes as in (c). K‐S test: D = .15, ***p < .001. (e) Bar graph of mean sEPSC interevent interval in control (n = 25 cells/6 mice) and PLX neurons (n = 23 cells/9 mice). (f) Cumulative distributions of sEPSC interevent interval as in (e). (g) Input–output curve of evoked EPSC peak amplitudes recorded at −70 mV from control (n = 24 cells/8 mice) and PLX neurons (n = 24 cells/7 mice). Note that in PLX‐treated mice, neurons show significantly lower peak amplitudes compared to control. Two‐way ANOVA: treatment F(1,275) = 105.06 ***p < .001; stimulation F = (5,275) = 20.46 ***p < .001; interaction F(5,275) = 4.44 p*** < .001. (h) Representative EPSCs were recorded at −70 mV from control and PLX neurons following Schaffer collateral stimulation at 0.5 and 10 mA, respectively. (i) Bar graph showing the PPR, determined by dividing the amplitude of the second EPSC by the first, recorded in control (n = 58 cells/20 mice) and PLX‐treated mice (n = 26 cells/10 mice). t test: t = −2.16, *p < .05. (l) Representative eEPSC induced by paired‐pulse stimulation of Schaffer collateral. Data presented as Mean ± SEM
To further investigate the impact of microglia depletion on hippocampal synaptic transmission, we analyzed evoked EPSC at CA3‐CA1 synapses (Basilico et al., 2019). As revealed by the input/output curve, EPSCs recorded at hyperpolarized potential (−70 mV; AMPA component) displayed strongly reduced amplitudes in PLX‐treated mice relative to controls (Figures 2g,h and S4A), suggesting that microglia depletion profoundly affects CA3‐CA1 synaptic functional connectivity. As shown in Figure S4, the reduction of amplitudes was not associated with changes in EPSC voltage dependency (Figure S4A,B) or kinetics (Figure S4D–G), indicating similar recording conditions. Interestingly, the reduction of the input/output curve was observed also after a short PLX treatment (3–4 days; Figure S5A,B). This was associated with microglia depletion (Figure S5C,D), but not with changes in GFAP signal (Figure S5E,F). EPSC depression was not restricted to the AMPA component. Indeed, when we analyzed the NMDA receptor (NMDAR)‐dependent component, we observed a reduction of current amplitude in PLX‐treated mice (Figure S6A,B).
Notably, PLX‐induced EPSC depression effect was not observed when experiments were repeated in Cx3cr1 −/− mice (mice lacking the microglial fractalkine receptor), indicating that PLX‐induced synaptic dysfunction is directly correlated to functional neuron–microglia crosstalk (Figure S7A,B).
To better comprehend the origin of the deficient glutamatergic synaptic transmission observed in PLX‐treated mice, we analyzed functional and structural parameters underlying synaptic efficacy (refer to next section).
3.3. Microglia depletion causes a regression in the properties of CA1 synapses, enhances long‐term potentiation and reduces spine density
Current clamp analysis of CA3 neurons (stemming excitatory inputs onto CA1 pyramidal cells) showed that both cell excitability (Figure S8A,B) and passive properties (Figure S8E–G) were unaltered in PLX‐treated mice, suggesting that microglia elimination impaired the CA3‐CA1 glutamatergic transmission at the synaptic level. Similarly, PLX treatment did not change active or passive membrane properties of CA1 pyramidal neurons (Figure S8C–G).
We thus examined more in detail the AMPAR‐mediated component of EPSCs, evoked in CA1 pyramidal neurons by stimulation of Schaffer collaterals. We observed a higher paired pulse ratio of EPSCs in slices from PLX‐treated mice, relative to controls (Figure 2i‐l), suggesting a reduction in the probability of glutamate release (Dobrunz & Stevens, 1997). Consistently, we did not observe a reduction in mEPSC amplitude (CTRL: 7.7 ± 0.4 pA; PLX: 8.6 ± 0.6; t test, p = .37) or frequency (CTRL: 1.4 ± 0.7 Hz; PLX: 1.8 ± 0.3 Hz; t test, p = .61) in slices from PLX‐treated mice (not shown), pointing to a presynaptic defect. Interestingly, when experiments were repeated in conditions of high release probability (Ca2+/Mg2+ ratio = 8) (Basilico et al., 2019), the EPSC amplitudes in the input‐output curve were significantly lower in slices from PLX‐treated animals, compared to control ones (n = 9 CTRL vs. n = 7 PLX; Two‐way ANOVA, p < .001; not shown), indicating that EPSC depression cannot be attributed simply to a reduction in the probability of glutamate release.
Further, and remarkably, in patch‐clamp recordings from PLX‐treated mice, we observed a reduction of synaptic multiplicity (indicated by the lack of difference between miniature and spontaneous EPSC amplitudes; Figure 3a–d) (Hsia et al., 1998), as well as a reduction in the AMPA/NMDA ratio (Figure 3e,f). Overall, these data suggest that microglial depletion is associated with the appearance of immature synaptic features (Basilico et al., 2019; Hoshiko et al., 2012).
FIGURE 3.
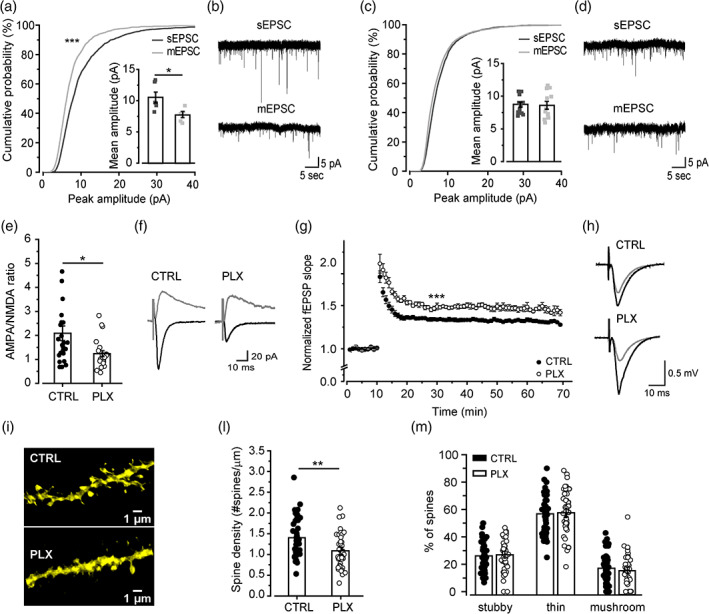
Microglia depletion causes the reappearance of immature properties and the reduction of spine density at CA3‐CA1 synapses. (a) Cumulative distribution and scatter plot showing a significant difference between mEPSC and sEPSC amplitudes in control mice (n = 6 cells/3 mice). Paired t test: t = 3.47, *p < .05. K‐S test: D = .22, ***p < .001. (b) Representative sEPSC and mEPSC traces recorded at −70 mV from pyramidal neurons of control mice. (c) In PLX‐treated mice (n = 12 cells/8 mice), the difference observed in control mice for sEPSC and mEPSC amplitudes was lost (cumulative distribution and scatter plot), indicating a defect in synaptic multiplicity. (d) Representative sEPSC and mEPSC traces recorded at −70 mV from pyramidal neurons of PLX‐treated mice. (e) The AMPA/NMDA ratio is significantly lower in PLX (n = 23 cells/10 mice) compared with control neurons (n = 26 cells/10 mice). t test: t = −2.47, *p < .05. (f) Representative traces of AMPA‐mediated currents, elicited at −70 mV, and NMDA‐mediated currents (+40 mV) evoked by Schaffer collateral stimulation. (g) Time course of fEPSP slope responses evoked at 0.05 Hz and normalized. Note that fEPSP amplitudes are higher in PLX‐treated mice after LTP induction (CTRL, n = 14 slices/6 mice; PLX, n = 8 slices/4 mice). t test: t = −6.79, ***p < .001. The absolute amplitude values of baseline fEPSP did not differ in average between control (−0.59 ± 0.04 mV, n = 19) and PLX‐treated mice (−0.55 ± 0.06 mV, n = 14; p = .6). (h) Representative field potential waveforms before (gray) and after (black) 30 min of HFS induction, for each condition as indicated. (i) Representative confocal images of dendritic segments belonging to control and PLX pyramidal neurons from CA1 stratum radiatum. (l) Bar graph showing the reduction of the overall spine density in PLX neurons (n = 46 dendrites/5 mice; control n = 46 dendrites/5 mice). t test: t = 3.28, **p < .01. (m) Quantification of the percentage of stubby, thin, and mushroom spines does not reveal any difference between the two experimental conditions. Data presented as Mean ± SEM
We then evaluated the effects of microglia depletion on synaptic plasticity in the hippocampal CA1 region, applying a 100 Hz stimulation of Schaffer collateral inputs. We observed a significant increase in LTP amplitude at CA1 synapses in PLX‐treated mice (1.5 ± 0.03) relative to controls (1.3 ± 0.01) (Figure 3g,h).
In order to investigate the structural determinants of the functional synaptic changes described above, we analyzed the density, morphology, and distribution of dendritic spines in the CA1 stratum radiatum of control and PLX‐treated mice. We observed that microglia depletion induces a significant reduction in spine density (Figure 3i,l), without changes in spine morphology (Figure 3m).
3.4. Microglia repopulation is followed by recovery of synaptic function
It is well established that microglia replenish the brain once the PLX5622 diet is discontinued (Dagher et al., 2015; Zhan et al., 2019). However, these cells may present distinct morphological features compared with the normal microglia (Elmore et al., 2015). To address this question, we performed a qualitative ultrastructural analysis of microglia two and four weeks after PLX5622 withdrawal (examples are shown in Figure 4a). Hippocampal microglia cells displayed more mitochondria and phagocytic inclusions, compared with controls, after 2 weeks of PLX withdrawal (Rec2W), recovering the typical cytoplasmic appearance after 4 weeks (Rec4W; Figure 4a). In line with these observations, our immunofluorescence analyses showed that the repopulation process was associated with a progressive return to normal microglial morphology in the hippocampus (Figure 4b). Indeed, after 2 weeks of PLX withdrawal (Rec2W) microglia density was fully restored (Figure 1b,c), but the cells showed a larger soma area and reduced ramification, relative to untreated mice (Figure 4c–e). After 4 weeks of withdrawal (Rec4W), microglial soma size returned to control levels, but the number of branches was still low, compared with untreated mice (Figure 4c–e). On the other hand, PLX‐induced astrogliosis did not recover during the time window considered, indicating a prolonged alteration of tissue homeostasis, following microglia depletion (Figure 4f).
FIGURE 4.
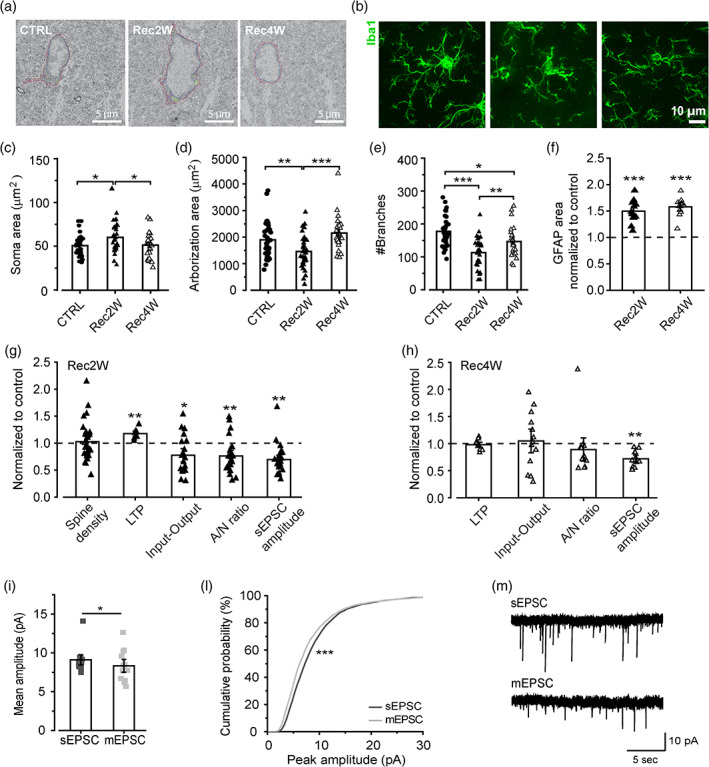
Effects of microglia repopulation on synaptic functions. (a) Electron micrograph of microglia in control (CTRL), 2 and 4 weeks recovery groups in the stratum radiatum of the hippocampal CA1. Red line = cytoplasm, blue line = nucleus, purple = mitochondria, green = phagocytic elements. Scale bar = 5 μm. (b) Representative confocal images showing morphological features of Iba1+ microglia in control, 2 and 4 weeks recovery groups in the stratum radiatum of the hippocampal CA1. Scale bar = 10 μm. (c–e) Quantitative analysis of microglial soma area (c; One‐way ANOVA: F[2,90] = 4.06, p < .05. Holm‐Sidak post hoc: *p < .05), arborization area (d; One‐way ANOVA: F[2,90] = 8.1, p < .001. Holm‐Sidak post hoc: **p < .01 ***p < .001) and a number of branches (e; One‐way ANOVA: F(2,90) = 16.06, p < .001. Holm‐Sidak post hoc: *p < .05 **p < .01 ***p < .001) in control (n = 34 cells/13 slices/3 mice), 2 weeks recovery (n = 34 cells/7 slices/3 mice) and 4 weeks recovery (n = 27 cells/7 slices/3 mice) groups. (f) Analysis of GFAP signal after 2 weeks (t test Rec2W vs. ctrl: t = −6.61, ***p < .001) and 4 weeks (t test Rec4W vs. ctrl: t = −7.92, ***p < .001) of PLX5622 withdrawal. (g) Analysis of synaptic parameters recovery after 2 weeks of PLX5622 withdrawal: dendritic spine density, LTP (t test Rec2W vs. ctrl: t = −6.19, ***p < .001), input–output (t test Rec2W vs. ctrl: t = −2.74, *p < .05), AMPA/NMDA ratio (t test Rec2W vs. ctrl: t = −3.53, **p < .01) and sEPSC amplitude during microglia repopulation. (h) Analysis of synaptic parameters recovery after 4 weeks of PLX5622 withdrawal: dendritic spine density, LTP, input output, AMPA/NMDA ratio and sEPSC amplitude (t test Rec4W vs. ctrl: t = −7.92, ***p < .001). Data in f–h presented as normalized to control values. Data shown as Mean ± SEM. (i‐l) Histogram and cumulative distribution of EPSCs amplitudes after 4 weeks PLX5622 withdrawal, showing a significant difference between mEPSC and sEPSC amplitudes (n = cells/mice). paired t test: t = 2.183 *p < .05. K‐S test: D = 0.101 ***p < .001. (m) Representative sEPSC and mEPSC traces recorded at −70 mV from pyramidal neurons of Rec4W mice. Data in (f–h) presented as normalized to control values. Data shown as Mean ± SEM
Notably, this progressive recovery of microglia density and morphology was mirrored by the recovery of the typical spine density after 2 weeks (Figure 4g) and the restoration of synaptic alterations in the hippocampal CA1 region after 4 weeks: LTP amplitude (Figure 4g,h), EPSC amplitudes (Figure 4g,h) and AMPA/NMDA ratio (Figure 4g,h), synaptic multiplicity (Figure 4i–m), except for sEPSC amplitude (Figure 4g,h).
To ascertain that synaptic recovery was specifically due to microglia repopulation and not merely time‐dependent, we measured EPSC amplitudes in conditions of continuous PLX5622 treatment at the same time points chosen for recovery (3 and 5 weeks of treatment; Figure S9A). We showed that when PLX5622 treatment was not discontinued, recovery of synaptic function was absent (Figure S9B). Similar low EPSC amplitudes were observed also in case of prolonged (8 weeks), PLX treatment (PLX, 7 cells/2 mice vs CTRL, 16/6; Two way‐ANOVA, p < .001, not shown), confirming the absence of time dependent synaptic recovery.
3.5. Microglia depletion causes a reversible impairment in the acquisition of the NOR task
In order to investigate the effects of microglia depletion on learning and memory processes, we tested mice in the NOR task (Figure 5a). We observed that PLX‐treated mice did not habituate to the exploration of the two identical objects across sessions, whereas the control group showed a progressive reduction of object exploration time across trials (Figure 5b), naturally due to a decrease in novelty after repeated exposure (ANOVA for repeated measures, F(1,182) = 11.652; p = .002; Tukey's test p < .05). During the test, PLX‐treated mice explored the familiar object as much as the novel one, showing no preference between them, whereas the control group showed a clear preference towards the novel object (Two‐way ANOVA, objects F(1,46) = 4.031; p = .05; Tukey's test p < .01; Figure 5c). Consistently, PLX‐treated mice showed a significantly lower Discrimination Index (D.I.) compared with control mice (Unpaired t test, t(27) = 4.107; p < .01; Figure 5d). Interestingly, we did not find differences between control mice and PLX‐treated mice either on the distance traveled, the velocity, the number of crossings to different areas of the arena, or on the time spent grooming and the time spent in the center and the periphery of the arena (Figure S10A–G). These results allowed us to exclude that PLX‐induced impairment in NOR task acquisition, could depend on effects on locomotor activity or anxiety‐like behavior.
FIGURE 5.
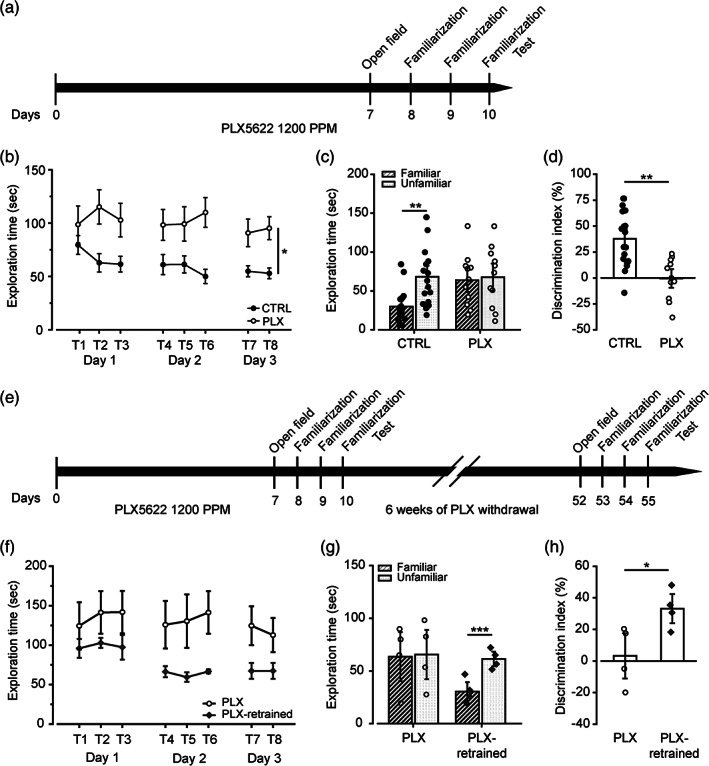
Microglia‐depleted mice show learning deficits in the NOR task.(a) Experimental design for behavioral analysis in (b–d). Thirty‐five‐days‐old mice (control n = 18, PLX‐treated n = 11) were fed with PLX5622 chow for 10 consecutive days. After 7 days of treatment, mice were placed in the open field arena to evaluate the locomotor activity. From day 8 to 10, mice were allowed to familiarize with two identical objects during repeated training sessions. On day 10, 1 h after the last training session, mice were tested in the NOR task. (b) Exploration time of the two objects across the familiarization sessions. As shown in the graph, PLX‐treated mice show increased exploratory behavior compared with control mice. ANOVA for repeated measures, F(1,182) = 11.652; p = .002; Tukey's test p < .05. (c) Bar graph representing the exploration time for the familiar and unfamiliar object during the acquisition test. (Two‐way ANOVA, objects F(1,46) = 4.031; p = .05; Tukey's test **p < .01. (d) Control mice show a higher discrimination index (D.I.) compared to PLX‐treated mice, demonstrating to distinguish between novel and familiar object. (Unpaired t test, t(27) = 4.107; **p < .01). (e) Experimental design for behavioral analysis in (f–h). (f) Exploration time of the two objects across the familiarization sessions. As shown in the graph, microglia‐repopulated mice recovered the ability to familiarize with objects across the training trials (ANOVA for repeated measures: groups F(1,42) = 3.865; p = .09; object exploration F(7,42) = 3.260; p = .007; Tukey's test p < .05). (g) Bar graph showing the exploration time for the familiar and unfamiliar object during the acquisition test. (Two‐way ANOVA, objects F(1,6) = 16.975; p = .001; Tukey's test ***p < .001). (h) Retrained mice (n = 4) show a higher discrimination index (D.I.) compared with the first training, demonstrating to distinguish between novel and familiar object. Unpaired t test, t(6) = − 2.618; *p < .05. Data presented as Mean ± SEM
To assess whether the deficit in the NOR acquisition could be rescued after microglial repopulation, we switched PLX‐treated mice (n = 4) to the control diet for six weeks and then re‐trained them in the NOR task, following the same behavioral procedure with a different set of objects (Figure 5e). Microglia‐repopulated mice recovered the ability to familiarize with objects across the training trials (ANOVA for repeated measures, F(7,42) = 3.260; p = .007; Tukey's test p < .05; Figure 5f) and to discriminate between the familiar and novel object after re‐training, as shown by the higher amount of time spent exploring the novel object (Two‐way ANOVA, objects F(1,6) = 16.975; p = .001; Tukey's test p < .001; Figure 5g) and the higher DI (Unpaired t test, t(6) = − 2.618; p = .03; Figure 5h), during the acquisition test session. In addition, we did not find differences between PLX‐treated and microglia‐repopulated mice either on the locomotor (i.e., distance traveled, average speed, number of line crossings) or on the anxiety‐related parameters (i.e., time spent grooming, time spent in the center and in the periphery of the arena; Figure S10h–p).
Altogether, our findings demonstrate that microglia depletion impaired the acquisition of the NOR task, strongly suggesting a pivotal role of microglia in regulating and modulating learning processes and hippocampal‐dependent cognitive performances.
4. DISCUSSION
In this study, we investigated the effects of PLX‐induced microglia depletion on hippocampal synaptic function in the adult brain, reporting four main findings. First, in the almost complete absence of microglia, glutamatergic synapses undergo a regression to immature properties, in association with a reduction in functional efficacy and an increase in synaptic plasticity. Second, microglial depletion reduces spine density without affecting spine morphology. Third, microglia‐depleted mice fail to acquire the NOR task. Fourth, the observed structural, functional, and behavioral deficits are rescued after microglial repopulation. We conclude that microglia are necessary for the maintenance of synaptic function in adulthood. Remarkably, our data highlight the importance of CX3CL1/CX3CR1 axis in microglia control of synaptic functioning, as synaptic depression caused by microglia depletion was absent in Cx3cr1 −/− mice.
4.1. Microglia depletion reduces the number of functional glutamatergic synapses in the hippocampus
We showed that a short microglial depletion (1 week) causes synaptic alterations in the adult mouse hippocampus. In particular, we observed in CA1 pyramidal neurons from animals treated with PLX5622, a profound depression of both evoked and spontaneous glutamatergic EPSC. The origin of such regulation is not straightforward to determine, since synaptic strength may be regulated in multiple ways, including the number of functional synapses, presynaptic or postsynaptic modifications, or neuronal excitability. Some of our results point to an alteration at the presynaptic level after microglial depletion (Dittman & Regehr, 1996). Indeed, in microglia‐depleted mice, we did not observe alterations in the postsynaptic sensitivity to the neurotransmitter (similar mEPSC amplitude in control and PLX mice) and evoked EPSC showed higher paired pulse ratio, which is typically associated with a decrease in presynaptic release probability (Basilico et al., 2019; Dobrunz & Stevens, 1997). However, recordings in high Ca2+/Mg2+ ratio did not support a presynaptic mechanism, showing reduction of EPSC amplitude also in conditions of high release probability; thus, raising the possibility of alternative or convergent mechanisms. Among these, the reduced release probability might represent a feature of synaptic “immaturity” (Basilico et al., 2019 and see below for discussion). In addition, our experiments did not reveal alterations in neuronal excitability or passive properties of CA3 or CA1 neurons after microglial removal, pointing to a reduction of functional synapse number as the most straightforward explanation of the weakening of CA3‐CA1 functional connectivity.
Although we did not highlight significant changes in the density of either presynaptic or postsynaptic immunofluorescence signals (Figure S2), the mechanisms underlying the synaptic dysfunction observed in microglia‐depleted mice may partially rely on structural changes. In fact, we observed a reduction of spine density in mice treated with PLX. These data, along with the decrease in both the AMPA and NMDA components of EPSC, are coherent with a reduction in the number of functional glutamatergic synapses in microglia‐depleted mice. Similarly, to what we observed in the hippocampus, Parkhurst and colleagues showed that microglial depletion reduces learning‐dependent formation of postsynaptic dendritic spines in the motor cortex (Parkhurst et al., 2013). In contrast, Rice and co‐workers reported no change in the number of PSD‐95 puncta in hippocampal CA1 neurons after 30 days of PLX3397‐induced microglial depletion in control mice and an increase in PSD‐95 in microglia‐depleted mice through neuronal lesion (Rice et al., 2015). In addition to differences possibly arising from the duration of the treatment, discrepancies are likely to be ascribed to the different impact of microglial removal in basal or inflammatory conditions. Recently, Ma and colleagues (Ma et al., 2020) also reported an increase in spine density in the developing visual cortex following 2 weeks of PLX3397 treatment. Most likely, this effect is related to the specific developmental window of the treatment, when spine turnover represents a major phenomenon (Cruz‐Martin et al., 2010). Our results showing a reduction of dendritic spines and a reduced AMPA/NMDA ratio in microglia‐depleted mice support the notion that microglia control synaptic functioning and stabilization in the adulthood.
A weakening of hippocampal glutamatergic transmission after microglia depletion was recently observed by Yegla et al. (2021). Consistently, it was reported that upon microglia contact with synapses increases synaptic activity and neuronal synchronization in the primary motor cortex (Akiyoshi et al., 2018). In contrast, some literature suggest that microglia negatively modulate synaptic activity. Li et al. (2012) showed that prolonged contacts between microglia and neurons cause a reduction of neuronal activity in the zebrafish larvae (Li et al., 2012). Similarly, recent evidence reported that microglia may negatively modulate neuronal activity in the striatum, upon dopamine D1 receptor agonist treatment (Badimon et al., 2020), and the local synaptic circuits in the mouse visual cortex (Liu et al., 2021). Although the experimental settings of these studies are extremely different, making the results on synaptic function hardly comparable, evidence points to a context‐ and brain region‐dependent role of microglia in regulating synaptic function. Indeed, microglia may interact with synapses using a variety of signaling mechanisms, including regulation of pruning and synaptogenesis (Paolicelli et al., 2011; Parkhurst et al., 2013), the release of substances (see Szepesi et al., 2018; Marinelli et al., 2019 for review) or physical contact (Akiyoshi et al., 2018; Weinhard et al., 2018).
4.2. Hippocampal CA1 glutamatergic synapses display immature features and enhanced plasticity after microglia depletion
In addition to the reduced strength, following PLX treatment, CA1 synapses display altered functional features, similar to those observed during development. In particular, we observed a reduced efficacy and immature properties in CA1 glutamatergic synapses, as showed by the lower AMPA/NMDA ratio (Hsia et al., 1998), reduced synaptic multiplicity (Hsia et al., 1998; Paolicelli et al., 2011), together with the higher paired pulse ratio described above. A possible scenario is that, in the absence of microglia, synaptic properties regress, resembling those found in immature glutamatergic CA1 synapses during early postnatal development (Basilico et al., 2019). Consistently with the increased plasticity in microglia depleted mice (see below), this “regression” may, in fact, reveal the formation of new, immature synapses, as secondary effect of increased synapse loss. An alternative explanation could be that the absence of microglia is associated with a defined pattern of synaptic properties, similar to those present during development. Consistently with this hypothesis, the properties of hippocampal CA1 synapses following microglia depletion with PLX5622 administration closely resemble those observed in a dysfunctional microglia model (Basilico et al., 2019). In this regard, it should be emphasized that PLX treatment did not produce any significant effect on synaptic properties of mice lacking the fractalkine receptor (Cx3cr1 −/− mice; Figure S7), indicating an overlapping effect between microglial removal and dysfunctional neuron–microglia signaling (Basilico et al., 2019). We interpret these results as a proof that PLX effect relies specifically on microglia. Indeed, they cannot be reproduced where microglia functions are impaired by the lack of CX3CR1 (Cardona et al., 2006). Our results also highlight CX3CL1/CX3CR1 signaling in neuron–microglia crosstalk (see Basilico et al., 2019; Gunner et al., 2019; Paolicelli et al., 2011). In particular, the coincidence between the effects observed in transgenic mouse models of defective signaling (Basilico et al., 2019) with those of the acute removal of microglia, gives great emphasis to the key role of this axis in microglia control of synaptic functioning.
We also observed a significant increase in LTP amplitude at hippocampal CA1 synapses in microglia‐depleted mice. In line with the hypothesis described above, increased LTP was also observed in Cx3cr1 −/− mice (Maggi et al., 2011) and it is consistent with the presence of immature synapses. Indeed, a higher level of synaptic plasticity is a general phenomenon in the developing brain (Feldman et al., 1999; Hogsden & Dringenberg, 2009), and higher LTP magnitude has been previously reported in developing rodent CA1 (Dumas, 2012; Dumas & Foster, 1995). At the mechanistic level, we may speculate that enhanced plasticity after microglial depletion could rely on the inclusion of “immature” NMDAR subunits at CA1 synapses (i.e., NR2B). Indeed, a decrease of NR2A/NR2B ratio lowers the threshold for LTP (Yashiro & Philpot, 2008), and alterations in the density of NMDAR subunits has been observed after LPS‐induced microglial activation (Ma et al., 2014).
Although LTP is largely accepted as a mechanism underlying the encoding and storage of memories (Martin et al., 2000), a nonlinear relationship between LTP magnitude and learning has been proposed (Barnes et al., 1994; Diógenes et al., 2011). We can speculate that the elevated LTP amplitude observed in microglia‐depleted mice (i.e., dysfunctional LTP or LTP saturation) could contribute to the impairment in the acquisition of the NOR task (see below). In line with this hypothesis, it has been previously demonstrated that inhibiting neuronal‐microglial fractalkine signaling, increases LTP amplitude leading to a lack of responsiveness to environmental stimuli (Maggi et al., 2011; Milior et al., 2016).
4.3. Microglia depletion impairs recognition learning in mice
We also report that microglia‐depleted mice show a deficit in the acquisition of object recognition memory (NOR), as revealed by the failure in discriminating between the novel and familiar objects during the test. Notably, upon microglial repopulation, after the discontinuation of the PLX treatment, mice recovered the ability to discriminate the novelty. Microglia‐depleted mice also displayed enhanced exploration of both objects and did not habituate across training sessions. Consistent with our results, one week of PLX5622 treatment impaired spatial memory acquisition in adult mice (Torres et al., 2016). Similarly, one week of microglial depletion, achieved using the tamoxifen‐inducible CX3CR1CreER to drive diphtheria toxin receptor expression in microglia, impaired learning in both fear conditioning and the NOR task (Parkhurst et al., 2013). While Torres et al., 2016 found that spatial memory alterations were not present after a longer period of microglial depletion (i.e., 21 days), Yegla and colleagues (2021), by using a novel context‐object discrimination (similar to our behavioral procedure), found behavioral deficits also after 21 days of PLX treatment. Noteworthy, we observed that alterations at CA3‐CA1 synapses were evident in mice treated with PLX for either 3, 7, 21, or 35 days, suggesting that synaptic alterations are stable, independently of the duration of microglia depletion. Moreover, it has been recently shown that microglial repopulation in aged animals has beneficial effects on cognition, by improving spatial memory and reversing age‐dependent deficits in synaptic plasticity (Elmore et al., 2018). Overall, our results strongly support the hypothesis that microglia are actively involved in the regulation of learning related processes. It should be noted that the deficit in the NOR task is not explained by alterations in spontaneous locomotor activity or anxiety, consistently with previous findings that PLX treatment does not alter these parameters in different behavioral procedures (Dagher et al., 2015; Elmore et al., 2014). However, our results and the above‐mentioned studies are at odds with findings of an intact spatial memory in healthy mice treated with PLX for longer periods (up to 3 weeks) (Dagher et al., 2015; Elmore et al., 2014) or evidence supporting the PLX‐restorative effects on spatial memory deficits in an aged mouse model of Alzheimer's disease (Dagher et al., 2015). While the restorative effects of microglial depletion in animal models of neurological disorders are likely resulting from a decreased inflammation process (Dagher et al., 2015; Rice et al., 2015), we speculate that in healthy animals the effects of microglial depletion on learning‐related processes probably depend on the duration of microglial removal or the cell‐specificity of the depletion method. It should be also noted that prolonged inhibition of CSF1R might have systemic effect on hematopoiesis and macrophage functions (Lei et al., 2020).
4.4. Recovery of synaptic function following microglia repopulation
To further understand the role of microglia on synaptic functioning, we monitored the functionality of hippocampal synapses after discontinuation of PLX5622 treatment. After 2 weeks of PLX withdrawal, we observed (i) microglial repopulation of the brain parenchyma (Elmore et al., 2014; Zhan et al., 2019) and (ii) the gradual re‐acquisition of microglia morphological features typical of physiological condition (within 4 weeks after PLX removal), following a trend similar to that previously described (Elmore et al., 2015; Zhan et al., 2019). Remarkably, we observed a functional recovery of synaptic properties in parallel with the recovery of the typical microglial morphology. In particular, 2 weeks after PLX withdrawal, when microglia re‐gained normal density (though with lower branching), the typical spine density was re‐established. At this stage, hippocampal CA1 synapses were still weak, immature, and more plastic, as demonstrated by the reduced synaptic efficacy, the lower AMPA/NMDA ratio, and the higher LTP, all indicative of an ongoing process of maturation. After 4 weeks of microglial repopulation, we observed a full functional synaptic recovery, with the only exception of the amplitude of the spontaneous EPSC that remained low. Notably, the recovery of synaptic function was specifically related to the repopulation and recovery of microglia and not simply an effect of time or age, as aged‐matched mice with prolonged PLX administration did not show functional recovery of synapses (Figure S9). Indeed, functional synaptic recovery coincided with complete morphological recovery of microglia, but not with the resolution of the astrogliosis. The lack of recovery of sEPSC amplitude in the considered time window (4 weeks after PLX withdrawal) raises the possibility that evoked and spontaneous EPSCs represent the activity of partially segregated synaptic pools, whose function is regulated by different mechanisms. Based on the above‐mentioned temporal coincidence, we could also speculate that synaptic function regulation in the absence of microglia might involve astrocytes, during depression or recovery processes. There are several experimental contexts in which astrocyte regulation of synaptic transmission is reported (see Dallérac et al., 2018 for review), although no one of them fully applies to the modulation of synaptic activity here reported. In particular, reactive astrocytes might reduce mEPSC amplitudes in culture (Liddelow et al., 2017). Although our results do not support the development of a “C3‐like” reactive astrocytic phenotype in the absence of microglia (see below for discussion), we cannot exclude that GFAP upregulation and sEPSC amplitude decrease are mechanistically connected.
4.5. Microglia are necessary for the maintenance of hippocampal synaptic function and learning in adulthood
In summary, in the absence of normal microglia–neuron interactions, hippocampal CA1 glutamatergic activity is depressed, with weak and less reliable synapses. Even if we do not know yet the primary mechanistic trigger(s) of this process, functional changes include the reduction of functional synapses and of glutamate release and the boost in plasticity mechanisms, possibly associated with a circuit rearrangement and leading to learning impairment. Of note, the recovery of synaptic function after drug withdrawal was paralleled by recovery of the NOR performance in PLX‐treated mice, similarly to what was reported by Yegla et al. (2021) for context discrimination. Indeed, the proper functioning of hippocampal CA3‐CA1 synapses is of critical importance for efficient recognition learning (Broadbent et al., 2010; Cohen et al., 2013; Cohen & Stackman, 2015). Accordingly, rats with hippocampal lesions display higher rates of object exploration time and lack of habituation during training in the NOR task, similarly to our microglia‐depleted mice (Broadbent et al., 2010). Our results suggest that the role of microglia in supporting hippocampal excitatory synaptic function is critical for learning and memory.
However, our findings should be interpreted with caution, because our microglial depletion strategy is systemic, and is therefore likely that deficits in other structures might have concurred with the performance in the NOR task. Indeed, beside the hippocampus, other brain structures relevant to the NOR task, such as the perirhinal cortex (Winters et al., 2008), might have concurred to the behavioral performance observed.
4.6. Methodological limitations
As mentioned above, when discussing our results, it has to be considered that the strategy used to deplete microglia may also have undesired side effects. Indeed, the treatment with CSF1R antagonist is systemic and not restricted to a precise brain area. However, it has been previously shown that PLX5622 effects in the CNS are limited to microglia and do not affect mice general health conditions (Elmore et al., 2014; Torres et al., 2016). Although prolonged PLX treatments may have effects on peripheral immune cells (Lei et al., 2020), PLX treatment do not cause peripheral myeloid cells infiltration in healthy conditions (Rice et al., 2015) and repopulation occurs by self‐renewal (Zhan et al., 2019). Remarkably, the peripheral effects observed by Lei and colleagues develop only after prolonged PLX administration (3 weeks). In this respect, it is important to notice that we observed a decrease in synaptic function also following a short (3–4 days) PLX treatment.
However, we considered the possibility that PLX5622‐induced microglia depletion may cause direct or indirect effects on brain homeostasis, eventually due to microglia cell death. Our analyses do not indicate the presence of neuroinflammatory processes in the hippocampus of microglia‐depleted mice, based on the reduced expression of inflammatory genes. Indeed, in PLX‐treated mice, we found the down regulation of genes related to microglia‐mediated inflammatory function, in the absence of hippocampal or systemic inflammation. The only four upregulated genes did not allow gene ontology analysis and are likely related to astrocytic functions (Bhat et al., 2017; Lee et al., 2000; Naruse et al., 2013). In addition, we did not observe ultrastructural signs of cell death (shrunken size, electron‐dense cells) or the appearance of dark microglia, previously associated with ultrastructural signs of oxidative stress (e.g., condensation of the nucleo‐ and cytoplasm, dilatation of the reticulum endoplasm, mitochondrial alterations) and found in regions where synaptic loss is exacerbated (e.g., in stratum radiatum) in Alzheimer's disease and chronic stress (Bisht et al., 2016). Interestingly, microglial elimination also resulted in the increase of the perineuronal network density in the hippocampus (Figure S2) and somatosensory cortex (see Figure S11D,E), in line with the ubiquitous accumulation of perineuronal network in the brain of PLX5622 treated mice, found by Crapser et al. (2020). This finding endorses a role of microglia as homeostatic regulators of perineuronal network formation in the healthy brain (Crapser et al., 2020).
The most striking change observed in hippocampal parenchyma following PLX treatment is astrogliosis, indicated by the increase of GFAP signal density (Figures 1 and S1), also observed in the cortex (see Figure S11B,C) of microglia–depleted mice (Figure S11A). The increase in GFAP in our experimental conditions could be regarded as a reactive astrocytic rearrangement (see Dallérac et al., 2018 for review), also supported by EM observations of high metabolic activity and phagocytosis in astrocytes. However, this does not necessarily involve a detrimental astrocytic phenotype, as highlighted by the lack of C3 overexpression (Liddelow et al., 2017). Recently, Konishi et al. (2020), reported that astrocytes readily phagocytose microglial debris after microglial ablation, leading to the speculation that dysfunctional or a decreased number of microglia may induce a compensatory phagocytic activity of astrocytes. It also remains unknown whether astrocytes act as spare phagocytes for live cellular components, such as synapses and axons; (Chung et al., 2013; Nguyen et al., 2011), thus contributing to the decrease of functional synapses. In our conditions, despite the observation of phagocytic inclusions in EM images, data on LAMP expression do not support a significant increase in astrocytic phagocytosis. It should be considered that LAMP1/2 genes are expressed in other cell types, including microglia. Therefore, ablation of microglia would be expected to lead to an overall reduction in LAMP1/2 mRNA expression. While this is plausible, our data do not support an increase in astrocytic LAMP2 expression in microglia‐depleted mice. Still, we cannot completely exclude an upregulation of LAMP1in astrocytes, which has been recently reported in spinal cord in strong inflammatory conditions (Sanmarco et al., 2021).
The potential involvement of astrocytes in the synaptic depression induced by microglia depletion has to be considered with caution, considering our observation that short PLX treatment (3 days) causes a likewise decrease in synaptic function, in the absence of evident signs of astrogliosis. Still, we speculate that in the absence of microglia, brain parenchyma homeostasis may require the involvement of astrocytes. In this regard, the lack of a neurotoxic or phagocytic phenotype does not completely exclude an involvement of these cells in the mechanisms underlying synaptic modulation in the absence of microglia. In particular, given that astrocytes, in addition to providing metabolic support to neurons (Bonvento & Bolaños, 2021), release and regulate several neuroactive molecules that can affect neuronal activity and modulate synaptic plasticity (Allen & Barres, 2005; Ota et al., 2013; Perez‐Alvarez et al., 2014), we cannot exclude their contribution to the alterations in synaptic plasticity observed in the absence of microglia.
In addition, our results should be discussed in light of previous evidence, showing that microglial depletion and repopulation may exacerbate (Rubino et al., 2018) or favor the resolution (Han et al., 2019) of different neuroinflammatory conditions. Indeed, during microglial repopulation, the brain parenchyma is going through an unnatural developmental process, where microglia although present and responsive (Elmore et al., 2015) are gradually re‐acquiring their abilities of modulating synapses. Still, the study of synaptic recovery after microglial depletion can be informative about the importance of microglia–neuron interaction in brain plasticity and brain responses after traumatic or inflammatory events.
Although further understanding is needed to resolve many of the above‐mentioned important questions, our findings clearly demonstrate that microglia are necessary to stabilize the “adult” properties of hippocampal glutamatergic synapses. We provide evidence that the synaptic changes associated with microglial depletion are mechanistically connected to the absence of microglia, as demonstrated by: (i) the temporal coincidence of microglia depletion and morphological recovery during repopulation with the impairment/recovery of synaptic functions; (ii) the absence of effect of PLX on the functionality of synapses in Cx3cr1 −/− mice; (iii) the absence of cell death and neuroinflammation.
5. CONCLUSIONS
Our study highlights the importance of microglia in regulating synaptic functioning in the adult brain. In the absence of microglia, glutamatergic synapses work less efficiently and display immature properties, ultimately affecting cognitive abilities. We conclude that microglia are central in the control of excitatory synaptic activity: besides participating in synaptic development, they are required for the maintenance of glutamatergic synaptic function in adulthood.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interests.
AUTHOR CONTRIBUTIONS
Bernadette Basilico and Laura Ferrucci carried out and analyzed all evoked EPSC and sEPSC experiments. Patrizia Ratano designed and performed behavioral experiments with help from Ingrid Reverte. Laura Maggi, and Maria T. Golia performed LTP experiments. Massimiliano Renzi performed GABAergic and excitability recordings. Maria Rosito, Caterina Sanchini, Alfonso Grimaldi, Valentina Ferretti, Ingrid Reverte, Maria C. Marrone, Silvia Marinelli, Laura Ferrucci, Maria Giubettini, Valeria De Turris, Stefano Garofalo, Brijesh Modi, Marcello Raspa, and Ferdinando Scavizzi performed immunohistochemistry, confocal acquisition, and morphology analysis. Marie‐Ève Tremblay, Micael Carrier, and Marie‐Kim St‐Pierre performed EM acquisition and analysis. Maria Rosito designed performed and analyzed RT‐PCR, Debora Salerno performed Nanostring analysis. Bernadette Basilico, Davide Ragozzino, and Silvia Di Angelantonio designed the experiments with input from Laura Ferrucci. Davide Ragozzino, Bernadette Basilico, and Silvia Di Angelantonio wrote the article with the help of Ingrid Reverte, Daniele Caprioli, Patrizia Ratano, Cornelius T. Gross, and Cristina Limatola. Davide Ragozzino conceived the project.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGMENTS
The work was supported by a grant from MIUR (PRIN 2017HPTFFC_003) to Davide Ragozzino and in part by funds to Silvia Di Angelantonio (CrestOptics‐IIT JointLab for Advanced Microscopy) and Daniele Caprioli (Istituto Pasteur‐Fondazione Cenci Bolognetti). Bernadette Basilico, and Laura Ferrucci were supported by the PhD program in Clinical‐Experimental Neuroscience and Psychiatry, Sapienza University, Rome; Caterina Sanchini was supported by the PhD program in Life Science, Sapienza University, Rome and by the Italian Institute of Technology, Rome. The authors thank Alessandro Felici, Claudia Valeri, Arsenio Armagno, and Senthilkumar Deivasigamani for help with animal husbandry and transgenic colonies management. They also wish to thank Piotr Bregestovski and Michal Schwartz for helpful discussions and criticism. PLX5622 was provided under Materials Transfer Agreement by Plexxikon Inc. (Berkeley, CA). Open Access Funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement.
Basilico, B. , Ferrucci, L. , Ratano, P. , Golia, M. T. , Grimaldi, A. , Rosito, M. , Ferretti, V. , Reverte, I. , Sanchini, C. , Marrone, M. C. , Giubettini, M. , De Turris, V. , Salerno, D. , Garofalo, S. , St‐Pierre, M.‐K. , Carrier, M. , Renzi, M. , Pagani, F. , Modi, B. , … Ragozzino, D. (2022). Microglia control glutamatergic synapses in the adult mouse hippocampus. Glia, 70(1), 173–195. 10.1002/glia.24101
Bernadette Basilico, Laura Ferrucci, and Patrizia Ratano contributed equally to this work. Silvia Di Angelantonio and Davide Ragozzino contributed equally to this work.
Funding information Ministero dell'Istruzione, dell'Università e della Ricerca, Grant/Award Number: PRIN 2017HPTFFC_003
Contributor Information
Bernadette Basilico, Email: bernadette.basilico@ist.ac.at.
Davide Ragozzino, Email: davide.ragozzino@uniroma1.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Akiyoshi, R. , Wake, H. , Kato, D. , Horiuchi, H. , Ono, R. , Ikegami, A. , Haruwaka, K. , Omori, T. , Tachibana, Y. , Moorhouse, A. J. , & Nabekura, J. (2018). Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro, 5(5), 88–106. 10.1523/ENEURO.0088-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, N. J. , & Barres, B. A. (2005). Signaling between glia and neurons: Focus on synaptic plasticity. Current Opinion in Neurobiology, 15(5), 542–548. 10.1016/j.conb.2005.08.006 [DOI] [PubMed] [Google Scholar]
- Badimon, A. , Strasburger, H. J. , Ayata, P. , Chen, X. , Nair, A. , Ikegami, A. , Hwang, P. , Chan, A. T. , Graves, S. M. , Uweru, J. O. , Ledderose, C. , Kutlu, M. G. , Wheeler, M. A. , Kahan, A. , Ishikawa, M. , Wang, Y. C. , Loh, Y. H. E. , Jiang, J. X. , Surmeier, D. J. , … Schaefer, A. (2020). Negative feedback control of neuronal activity by microglia. Nature, 10, 1–7. 10.1038/s41586-020-2777-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, C. A. , Jung, M. W. , McNaughton, B. L. , Korol, D. L. , Andreasson, K. , & Worley, P. F. (1994). LTP saturation and spatial learning disruption: Effects of task variables and saturation levels. Journal of Neuroscience, 14(10), 5793–5806. 10.1523/jneurosci.14-10-05793.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilico, B. , Pagani, F. , Grimaldi, A. , Cortese, B. , di Angelantonio, S. , Weinhard, L. , Gross, C. , Limatola, C. , Maggi, L. , & Ragozzino, D. (2019). Microglia shape presynaptic properties at developing glutamatergic synapses. Glia, 67(1), 53–67. 10.1002/glia.23508 [DOI] [PubMed] [Google Scholar]
- Bhat, S. A. , Goel, R. , Shukla, R. , & Hanif, K. (2017). Platelet CD40L induces activation of astrocytes and microglia in hypertension. Brain, Behavior, and Immunity, 59, 173–189. 10.1016/j.bbi.2016.09.021 [DOI] [PubMed] [Google Scholar]
- Bisht, K. , Sharma, K. P. , Lecours, C. , Gabriela Sánchez, M. , el Hajj, H. , Milior, G. , Olmos‐Alonso, A. , Gómez‐Nicola, D. , Luheshi, G. , Vallières, L. , Branchi, I. , Maggi, L. , Limatola, C. , Butovsky, O. , & Tremblay, M. È. (2016). Dark microglia: A new phenotype predominantly associated with pathological states. Glia, 64(5), 826–839. 10.1002/glia.22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvento, G. , & Bolaños, J. P. (2021). Astrocyte‐neuron metabolic cooperation shapes brain activity. Cell Metabolism, 33(8), 1546–1564. 10.1016/j.cmet.2021.07.006 [DOI] [PubMed] [Google Scholar]
- Broadbent, N. J. , Gaskin, S. , Squire, L. R. , & Clark, R. E. (2010). Object recognition memory and the rodent hippocampus. Learning and Memory, 17(1), 794–800. 10.1101/lm.1650110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent, N. J. , Squire, L. R. , & Clark, R. E. (2004). Spatial memory, recognition memory, and the hippocampus. Proceedings of the National Academy of Sciences, 101, 14515–14520. 10.1073/pnas.0406344101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona, A. E. , Pioro, E. P. , Sasse, M. E. , Kostenko, V. , Cardona, S. M. , Dijkstra, I. M. , Huang, D. R. , Kidd, G. , Dombrowski, S. , Dutta, R. J. , Lee, J. C. , Cook, D. N. , Jung, S. , Lira, S. A. , Littman, D. R. , & Ransohoff, R. M. (2006). Control of microglial neurotoxicity by the fractalkine receptor. Nature Neuroscience, 9(7), 917–924. 10.1038/nn1715 [DOI] [PubMed] [Google Scholar]
- Chakrova, N. , Rieger, B. , & Stallinga, S. (2016). Deconvolution methods for structured illumination microscopy. Journal of the Optical Society of America A, 33(7), B12. 10.1364/JOSAA.33.000B12 [DOI] [PubMed] [Google Scholar]
- Chung, W.‐S. , Clarke, L. E. , Wang, G. X. , Stafford, B. K. , Sher, A. , Chakraborty, C. , Joung, J. , Foo, L. C. , Thompson, A. , Chen, C. , Smith, S. J. , & Barres, B. A. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 504, 394–400. 10.1038/nature12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, S. J. , Munchow, A. H. , Rios, L. M. , Zhang, G. , Ásgeirsdóttir, H. N. , & Stackman, R. W. (2013). The rodent hippocampus is essential for nonspatial object memory. Current Biology, 23(17), 1685–1690. 10.1016/j.cub.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, S. J. , & Stackman, R. W. (2015). Assessing rodent hippocampal involvement in the novel object recognition task. A review. Behavioural Brain Research, 285, 105–117. 10.1016/j.bbr.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapser, J. D. , Ochaba, J. , Soni, N. , Reidling, J. C. , Thompson, L. M. , & Green, K. N. (2020). Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington's disease. Brain, 143, 266–288. 10.1093/brain/awz363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz‐Martin, A. , Crespo, M. , & Portera‐Cailliau, C. (2010). Delayed stabilization of dendritic spines in fragile X mice. Journal of Neuroscience, 30(23), 7793–7803. 10.1523/JNEUROSCI.0577-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher, N. N. , Najafi, A. R. , Kayala, K. M. N. , Elmore, M. R. P. , White, T. E. , Medeiros, R. , West, B. L. , & Green, K. N. (2015). Colony‐stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg‐AD mice. Journal of Neuroinflammation, 12(1), 139. 10.1186/s12974-015-0366-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallérac, G. , Zapata, J. , & Rouach, N. (2018). Versatile control of synaptic circuits by astrocytes: Where, when and how? Nature Reviews. Neuroscience, 19, 729–743. 10.1038/s41583-018-0080-6 [DOI] [PubMed] [Google Scholar]
- Diógenes, M. J. , Costenla, A. R. , Lopes, L. V. , Jerónimo‐Santos, A. , Sousa, V. C. , Fontinha, B. M. , Ribeiro, J. A. , & Sebastião, A. M. (2011). Enhancement of LTP in aged rats is dependent on endogenous BDNF. Neuropsychopharmacology, 36(9), 1823–1836. 10.1038/npp.2011.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman, J. S. , & Regehr, W. G. (1996). Contributions of calcium‐dependent and calcium‐independent mechanisms to presynaptic inhibition at a cerebellar synapse. Journal of Neuroscience, 16(5), 1623–1633. 10.1523/jneurosci.16-05-01623.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz, L. E. , & Stevens, C. F. (1997). Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron, 18(6), 995–1008. 10.1016/S0896-6273(00)80338-4 [DOI] [PubMed] [Google Scholar]
- Dumas, T. C. , & Foster, T. C. (1995). Developmental increase in CA3‐CA1 presynaptic function in the hippocampal slice. Journal of Neurophysiology, 73(5), 1821–1828. 10.1152/jn.1995.73.5.1821 [DOI] [PubMed] [Google Scholar]
- Dumas, T. C. (2012). Postnatal alterations in induction threshold and expression magnitude of long‐term potentiation and long‐term depression at hippocampal synapses. Hippocampus, 22(2), 188–199. 10.1002/hipo.20881 [DOI] [PubMed] [Google Scholar]
- El Hajj, H. , Savage, J. C. , Bisht, K. , Parent, M. , Vallières, L. , Rivest, S. , & Tremblay, M. È. (2019). Ultrastructural evidence of microglial heterogeneity in Alzheimer's disease amyloid pathology. Journal of Neuroinflammation, 16(1), 87. 10.1186/s12974-019-1473-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore, M. R. P. , Hohsfield, L. A. , Kramár, E. A. , Soreq, L. , Lee, R. J. , Pham, S. T. , Najafi, A. R. , Spangenberg, E. E. , Wood, M. A. , West, B. L. , & Green, K. N. (2018). Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell, 17(6), e12832. 10.1111/acel.12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore, M. R. P. , Lee, R. J. , West, B. L. , & Green, K. N. (2015). Characterizing newly repopulated microglia in the adult mouse: Impacts on animal behavior, cell morphology, and Neuroinflammation. PLoS One, 10(4), e0122912. 10.1371/journal.pone.0122912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore, M. R. P. , Najafi, A. R. , Koike, M. A. , Dagher, N. N. , Spangenberg, E. E. , Rice, R. A. , Kitazawa, M. , Matusow, B. , Nguyen, H. , West, B. L. , & Green, K. N. (2014). Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82(2), 380–397. 10.1016/J.NEURON.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin, C. , Galea, E. , Lakatos, A. , O'Callaghan, J. P. , Petzold, G. C. , Serrano‐Pozo, A. , Steinhäuser, C. , Volterra, A. , Carmignoto, G. , Agarwal, A. , Allen, N. J. , Araque, A. , Barbeito, L. , Barzilai, A. , Bergles, D. E. , Bonvento, G. , Butt, A. M. , Chen, W. T. , Cohen‐Salmon, M. , … Verkhratsky, A. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nature Neuroscience, 24, 312–325. 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn, M. J. , & Wang, L.‐Y. (2007). Activity‐dependent changes in temporal components of neurotransmission at the juvenile mouse calyx of held synapse. The Journal of Physiology, 581, 581–602. 10.1113/jphysiol.2007.129833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman, D. E. , Nicoll, R. A. , & Malenka, R. C. (1999). Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. Journal of Neurobiology, 41(1), 92–101. [DOI] [PubMed] [Google Scholar]
- Gunner, G. , Cheadle, L. , Johnson, K. M. , Ayata, P. , Badimon, A. , Mondo, E. , Nagy, M. A. , Liu, L. , Bemiller, S. M. , Kim, K. W. , Lira, S. A. , Lamb, B. T. , Tapper, A. R. , Ransohoff, R. M. , Greenberg, M. E. , Schaefer, A. , & Schafer, D. P. (2019). Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nature Neuroscience, 22(7), 1075–1088. 10.1038/s41593-019-0419-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Zhu, K. , Zhang, X. M. , & Harris, R. A. (2019). Enforced microglial depletion and repopulation as a promising strategy for the treatment of neurological disorders. Glia, 67, 217–231. 10.1002/glia.23529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann, R. , & Benedetti, P. A. (2006). High‐resolution image reconstruction in fluorescence microscopy with patterned excitation. Applied Optics, 45(20), 5037–5045. 10.1364/AO.45.005037 [DOI] [PubMed] [Google Scholar]
- Hering, H. , & Sheng, M. (2001). Dentritic spines: Structure, dynamics and regulation. Nature Reviews Neuroscience, 2(12), 880–888. 10.1038/35104061 [DOI] [PubMed] [Google Scholar]
- Hestrin, S. , Nicoll, R. A. , Perkel, D. J. , & Sah, P. (1990). Analysis of excitatory synaptic action in pyramidal cells using whole‐cell recording from rat hippocampal slices. Journal of Physiology, 422, 203–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogsden, J. L. , & Dringenberg, H. C. (2009). NR2B subunit‐dependent long‐term potentiation enhancement in the rat cortical auditory system in vivo following masking of patterned auditory input by white noise exposure during early postnatal life. European Journal of Neuroscience, 30(3), 376–384. 10.1111/j.1460-9568.2009.06835.x [DOI] [PubMed] [Google Scholar]
- Hoshiko, M. , Arnoux, I. , Avignone, E. , Yamamoto, N. , & Audinat, E. (2012). Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of Thalamocortical synapses in the barrel cortex. Journal of Neuroscience, 32(43), 15106–15111. 10.1523/JNEUROSCI.1167-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia, A. Y. , Malenka, R. C. , & Nicoll, R. A. (1998). Development of excitatory circuitry in the hippocampus. Journal of Neurophysiology, 79(4), 2013–2024. 10.1152/jn.1998.79.4.2013 [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Xu, Z. , Xiong, S. , Sun, F. , Qin, G. , Hu, G. , Wang, J. , Zhao, L. , Liang, Y. X. , Wu, T. , Lu, Z. , Humayun, M. S. , So, K. F. , Pan, Y. , Li, N. , Yuan, T. F. , Rao, Y. , & Peng, B. (2018). Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nature Neuroscience, 21(4), 530–540. 10.1038/s41593-018-0090-8 [DOI] [PubMed] [Google Scholar]
- Hui, C. W. , St‐Pierre, A. , el Hajj, H. , Remy, Y. , Hébert, S. S. , Luheshi, G. N. , Srivastava, L. K. , & Tremblay, M. È. (2018). Prenatal immune challenge in mice leads to partly sex‐dependent behavioral, microglial, and molecular abnormalities associated with schizophrenia. Frontiers in Molecular Neuroscience, 11(13), 13. 10.3389/fnmol.2018.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingaramo, M. , York, A. G. , Hoogendoorn, E. , Postma, M. , Shroff, H. , & Patterson, G. H. (2014). Richardson‐Lucy deconvolution as a general tool for combining images with complementary strengths. Chemphyschem, 15(4), 794–800. 10.1002/cphc.201300831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi, H. , Okamoto, T. , Hara, Y. , Komine, O. , Tamada, H. , Maeda, M. , Osako, F. , Kobayashi, M. , Nishiyama, A. , Kataoka, Y. , Takai, T. , Udagawa, N. , Jung, S. , Ozato, K. , Tamura, T. , Tsuda, M. , Yamanaka, K. , Ogi, T. , Sato, K. , & Kiyama, H. (2020). Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. The EMBO Journal, 39(22), 1–18. 10.15252/embj.2020104464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. J. , Zhou, T. , Choi, C. , Wang, Z. , & Benveniste, E. N. (2000). Differential regulation and function of Fas expression on glial cells. The Journal of Immunology, 164(3), 1277–1285. 10.4049/jimmunol.164.3.1277 [DOI] [PubMed] [Google Scholar]
- Lei, F. , Cui, N. , Zhou, C. , Chodosh, J. , Vavvas, D. G. , & Paschalis, E. I. (2020). CSF1R inhibition by a small‐molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proceedings of the National Academy of Sciences of the United States of America, 117(38), 23336–23338. 10.1073/pnas.1922788117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Du, X. F. , Liu, C. S. , Wen, Z. L. , & Du, J. L. (2012). Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell, 23(6), 1189–1202. 10.1016/j.devcel.2012.10.027 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. , Guttenplan, K. , Clarke, L. E. , Bennett, F. , Bohlen, C. , Schirmer, L. , Bennett, M. L. , Münch, A. E. , Chung, W. S. , Peterson, T. C. , Wilton, D. K. , Frouin, A. , Napier, B. A. , Panicker, N. , Kumar, M. , Buckwalter, M. S. , Rowitch, D. H. , Dawson, V. L. , Dawson, T. M. , … Barres, B. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature Publishing Group, 541, 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.‐J. , Spangenberg, E. E. , Tang, B. , Holmes, T. C. , Green, K. N. , & Xu, X. (2021). Microglia elimination increases neural circuit connectivity and activity in adult mouse cortex. The Journal of Neuroscience, 41(6), 1274–1287. 10.1523/JNEUROSCI.2140-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Choi, B. R. , Chung, C. , Min, S. S. , Jeon, W. K. , & Han, J. S. (2014). Chronic brain inflammation causes a reduction in GluN2A and GluN2B subunits of NMDA receptors and an increase in the phosphorylation of mitogen‐activated protein kinases in the hippocampus. Molecular Brain, 7(1), 33. 10.1186/1756-6606-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Chen, K. , Cui, Y. , Huang, G. , Nehme, A. , Zhang, L. , Li, H. , Wei, J. , Liong, K. , Liu, Q. , Shi, L. , Wu, J. , & Qiu, S. (2020). Depletion of microglia in developing cortical circuits reveals its critical role in glutamatergic synapse development, functional connectivity, and critical period plasticity. Journal of Neuroscience Research, 98(10), 1968–1986. 10.1002/jnr.24641 [DOI] [PubMed] [Google Scholar]
- Maggi, L. , Scianni, M. , Branchi, I. , D'Andrea, I. , Lauro, C. , & Limatola, C. (2011). CX3CR1 deficiency alters hippocampal‐dependent plasticity phenomena blunting the effects of enriched environment. Frontiers in Cellular Neuroscience, 5, 1–10. 10.3389/fncel.2011.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli, S. , Basilico, B. , Marrone, M. C. , & Ragozzino, D. (2019). Microglia‐neuron crosstalk: Signaling mechanism and control of synaptic transmission. Seminars in Cell & Developmental Biology, 94, 138–151. 10.1016/j.semcdb.2019.05.017 [DOI] [PubMed] [Google Scholar]
- Martin, S. J. , Grimwood, P. D. , & Morris, R. G. M. (2000). Synaptic plasticity and memory: An evaluation of the hypothesis. Annual Review of Neuroscience, 23(1), 649–711. 10.1146/annurev.neuro.23.1.649 [DOI] [PubMed] [Google Scholar]
- Milior, G. , Lecours, C. , Samson, L. , Bisht, K. , Poggini, S. , Pagani, F. , Deflorio, C. , Lauro, C. , Alboni, S. , Limatola, C. , Branchi, I. , Tremblay, M. E. , & Maggi, L. (2016). Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain, Behavior, and Immunity, 55, 114–125. 10.1016/j.bbi.2015.07.024 [DOI] [PubMed] [Google Scholar]
- Naruse, M. , Shibasaki, K. , Yokoyama, S. , Kurachi, M. , & Ishizaki, Y. (2013). Dynamic changes of CD44 expression from progenitors to subpopulations of astrocytes and neurons in developing cerebellum. PLoS One, 8(1), e53109. 10.1371/journal.pone.0053109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, J. V. , Soto, I. , Kim, K.‐Y. , Bushong, E. A. , Oglesby, E. , Valiente‐Soriano, F. J. , Yang, Z. , Davis, C. H. O. , Bedont, J. L. , Son, J. L. , Wei, J. O. , Buchman, V. L. , Zack, D. J. , Vidal‐Sanz, M. , Ellisman, M. H. , & Marsh‐Armstrong, N. (2011). Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proceedings of the National Academy of Sciences of the United States of America, 108, 1176–1181. 10.1073/pnas.1013965108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota, Y. , Zanetti, A. T. , & Hallock, R. M. (2013). The role of astrocytes in the regulation of synaptic plasticity and memory formation. Neural Plasticity, 2013, 1–11. 10.1155/2013/185463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli, R. C. , Bisht, K. , & Tremblay, M.‐È. (2014). Fractalkine regulation of microglial physiology and consequences on the brain and behavior. Frontiers in Cellular Neuroscience, 8, 129. 10.3389/fncel.2014.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli, R. C. , Bolasco, G. , Pagani, F. , Maggi, L. , Scianni, M. , Panzanelli, P. , Giustetto, M. , Ferreira, T. A. , Guiducci, E. , Dumas, L. , Ragozzino, D. , & Gross, C. T. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science (New York, N.Y.), 333(6048), 1456–1458. 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- Parkhurst, C. N. , Yang, G. , Ninan, I. , Savas, J. N. , Yates, J. R. , Lafaille, J. J. , Hempstead, B. L. , Littman, D. R. , & Gan, W. B. (2013). Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell, 155(7), 1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Alvarez, A. , Navarrete, M. , Covelo, A. , Martin, E. D. , & Araque, A. (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. Journal of Neuroscience, 34(38), 12738–12744. 10.1523/JNEUROSCI.2401-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A. , Josephson, K. , & Vincent, S. L. (1991). Effects of aging on the neuroglial cells and pericytes within area 17 of the rhesus monkey cerebral cortex. The Anatomical Record, 229(3), 384–398. 10.1002/ar.1092290311 [DOI] [PubMed] [Google Scholar]
- Peters, A. , & Kaiserman‐Abramof, I. R. (1970). The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. The American Journal of Anatomy, 127(4), 321–355. 10.1002/aja.1001270402 [DOI] [PubMed] [Google Scholar]
- Rice, R. A. , Spangenberg, E. E. , Yamate‐Morgan, H. , Lee, R. J. , Arora, R. P. S. , Hernandez, X. M. X. , Tenner, A. J. , West, B. L. , & Green, K. N. (2015). Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. The Journal of Neuroscience, 35(27), 9977–9989. 10.1523/JNEUROSCI.0336-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock, R. B. , Gekker, G. , Hu, S. , Sheng, W. S. , Cheeran, M. , Lokensgard, J. R. , & Peterson, P. K. (2004). Role of microglia in central nervous system infections. Clinical Microbiology Reviews, 17(4), 942–964. 10.1128/CMR.17.4.942-964.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, A. , Ehlenberger, D. B. , Dickstein, D. L. , Hof, P. R. , & Wearne, S. L. (2008). Automated three‐dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One, 3(4), e1997. 10.1371/journal.pone.0001997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, J. T. , Morganti, J. M. , Bachstetter, A. D. , Hudson, C. E. , Peters, M. M. , Grimmig, B. A. , Weeber, E. J. , Bickford, P. C. , & Gemma, C. (2011). CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. Journal of Neuroscience, 31(45), 16241–16250. 10.1523/JNEUROSCI.3667-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier, A. , Béchade, C. , Poncer, J.‐C. , Smalla, K.‐H. , Tomasello, E. , Vivier, E. , Gundelfinger, E. D. , Triller, A. , & Bessis, A. (2004). Impaired synaptic function in the microglial KARAP/DAP12‐deficient mouse. The Journal of Neuroscience, 24(50), 11421–11428. 10.1523/JNEUROSCI.2251-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumier, A. , Pascual, O. , Béchade, C. , Wakselman, S. , Poncer, J.‐C. , Réal, E. , Triller, A. , & Bessis, A. (2008). Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS One, 3(7), e2595. 10.1371/journal.pone.0002595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino, S. J. , Mayo, L. , Wimmer, I. , Siedler, V. , Brunner, F. , Hametner, S. , Madi, A. , Lanser, A. , Moreira, T. , Donnelly, D. , Cox, L. , Rezende, R. M. , Butovsky, O. , Lassmann, H. , & Weiner, H. L. (2018). Acute microglia ablation induces neurodegeneration in the somatosensory system. Nature Communications, 9(1), 1–13. 10.1038/s41467-018-05929-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmarco, L. M. , Wheeler, M. A. , Gutiérrez‐Vázquez, C. , Polonio, C. M. , Linnerbauer, M. , Pinho‐Ribeiro, F. A. , Li, Z. , Giovannoni, F. , Batterman, K. V. , Scalisi, G. , Zandee, S. E. J. , Heck, E. S. , Alsuwailm, M. , Rosene, D. L. , Becher, B. , Chiu, I. M. , Prat, A. , & Quintana, F. J. (2021). Gut‐licensed IFNγ(+) NK cells drive LAMP1(+)TRAIL(+) anti‐inflammatory astrocytes. Nature, 590(7846), 473–479. 10.1038/s41586-020-03116-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage, J. C. , St‐Pierre, M.‐K. , Hui, C. W. , & Tremblay, M.‐E. (2019). Microglial ultrastructure in the hippocampus of a lipopolysaccharide‐induced sickness mouse model. Frontiers in Neuroscience, 13, 1340. 10.3389/fnins.2019.01340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, D. P. , Lehrman, E. K. , Kautzman, A. G. , Koyama, R. , Mardinly, A. R. , Yamasaki, R. , Ransohoff, R. M. , Greenberg, M. E. , Barres, B. A. , & Stevens, B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron, 74(4), 691–705. 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T. D. , & Livak, K. J. (2008). Analyzing real‐time PCR data by the comparative CT method. Nature Protocols, 3(6), 1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- St‐Pierre, M.‐K. , Bordeleau, M. , & Tremblay, M.‐È. (2019). Visualizing Dark Microglia. In Methods in molecular biology (Vol. 2034, pp. 97–110). Humana Press Inc. 10.1007/978-1-4939-9658-2_8 [DOI] [PubMed] [Google Scholar]
- Ströhl, F. , & Kaminski, C. F. (2015). A joint Richardson—Lucy deconvolution algorithm for the reconstruction of multifocal structured illumination microscopy data. Methods and Applications in Fluorescence, 3(1), 014002. 10.1088/2050-6120/3/1/014002 [DOI] [PubMed] [Google Scholar]
- Szepesi, Z. , Manouchehrian, O. , Bachiller, S. , & Deierborg, T. (2018). Bidirectional microglia–neuron communication in health and disease. Frontiers in Cellular Neuroscience, 12(September), 1–26. 10.3389/fncel.2018.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, L. , Danver, J. , Ji, K. , Miyauchi, J. T. , Chen, D. , Anderson, M. E. , West, B. L. , Robinson, J. K. , & Tsirka, S. E. (2016). Dynamic microglial modulation of spatial learning and social behavior. Brain, Behavior, and Immunity, 55, 6–16. 10.1016/j.bbi.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M.‐È. , Lowery, R. L. , & Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8(11), e1000527. 10.1371/journal.pbio.1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M. È. , Zettel, M. L. , Ison, J. R. , Allen, P. D. , & Majewska, A. K. (2012). Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia, 60(4), 541–558. 10.1002/glia.22287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard, L. , di Bartolomei, G. , Bolasco, G. , Machado, P. , Schieber, N. L. , Neniskyte, U. , Exiga, M. , Vadisiute, A. , Raggioli, A. , Schertel, A. , Schwab, Y. , & Gross, C. T. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9(1), 1228. 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters, B. D. , Saksida, L. M. , & Bussey, T. J. (2008). Object recognition memory: Neurobiological mechanisms of encoding, consolidation and retrieval. Neuroscience and Biobehavioral Reviews, 32, 1055–1070. 10.1016/j.neubiorev.2008.04.004 [DOI] [PubMed] [Google Scholar]
- Yashiro, K. , & Philpot, B. D. (2008). Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology, 55(7), 1081–1094. 10.1016/j.neuropharm.2008.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegla, B. , Boles, J. , Kumar, A. , & Foster, T. C. (2021). Partial microglial depletion is associated with impaired hippocampal synaptic and cognitive function in young and aged rats. Glia, 69, 1494–1514. 10.1002/glia.23975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, L. , Krabbe, G. , du, F. , Jones, I. , Reichert, M. C. , Telpoukhovskaia, M. , Kodama, L. , Wang, C. , Cho, S. H. , Sayed, F. , Li, Y. , le, D. , Zhou, Y. , Shen, Y. , West, B. , & Gan, L. (2019). Proximal recolonization by self‐renewing microglia re‐establishes microglial homeostasis in the adult mouse brain. PLoS Biology, 17(2), e3000134. 10.1371/journal.pbio.3000134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, Y. , Paolicelli, R. C. , Sforazzini, F. , Weinhard, L. , Bolasco, G. , Pagani, F. , Vyssotski, A. L. , Bifone, A. , Gozzi, A. , Ragozzino, D. , & Gross, C. T. (2014). Deficient neuron‐microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience, 17(3), 400–406. 10.1038/nn.3641 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.