

Summary
DNA methylation plays key roles in transposable element (TE) silencing and gene expression regulation. DNA methylation occurs at CG, CHG and CHH sequence contexts in plants. However, the synergistic and redundant roles of CG and non‐CG methylation are poorly understood.
By introducing CRISPR/Cas9‐induced met1 mutation into the ddcc (drm1 drm2 cmt2 cmt3) mutant, we attempted to knock out all five DNA methyltransferases in Arabidopsis and then investigate the synergistic and redundant roles of CG and non‐CG DNA methylation.
We found that the homozygous ddcc met1 quintuple mutants are embryonically lethal, although met1 and ddcc mutants only display some developmental abnormalities. Unexpectedly, the ddcc met1 quintuple mutations only reduce transmission through the male gametophytes. The ddcc met1+/− mutants show apparent size divergence, which is not associated with difference in DNA methylation patterns, but associated with the difference in the levels of DNA damage. Finally, we show that a group of TEs are specifically activated in the ddcc met1+/− mutants.
This work reveals that CG and non‐CG DNA methylation synergistically and redundantly regulate plant reproductive development, vegetative development and TE silencing in Arabidopsis. Our findings provide insights into the roles of DNA methylation in plant development.
Keywords: Arabidopsis, DNA damage, DNA methylation, DNA methyltransferase, pollen development
Introduction
DNA methylation (5‐methylcytosine, 5mC) is an important epigenetic mark that is involved in many biological processes, including transposable element (TE) silencing, gene imprinting and the regulation of gene expression. In mammals, DNA methylation is primarily restricted to CG dinucleotides in somatic cells and non‐CG methylation is detected in oocytes, pluripotent embryonic stem cells, and mature neurons (Wu & Zhang, 2014). In human somatic cells, 60–80% of CG dinucleotides are methylated (Smith & Meissner, 2013). In plants, DNA methylation occurs in all sequence contexts: CG, CHG and CHH (H represents A, T or C), in all types of cells and tissues. Transposable elements and repetitive sequences are highly methylated in all sequence contexts, which closely correlates with transcriptional silencing of TEs and their neighbouring genes (Zhang et al., 2006; Zhang & Zhu, 2011). Methylation at CG context only is observed in the bodies of actively transcribed genes, but the roles of these methylation events are still unclear (Zhang et al., 2006).
In mammals, CG DNA methylation is established de novo using DNA methyltransferase 3 (Dnmt3) and maintained using Dnmt1 during DNA replication (Wu & Zhang, 2014). In plants, DNA methylation in all sequence contexts is established by Domains Rearranged Methyltransferase 2 (DRM2, homologous to mammalian Dnmt3) through an RNA‐directed DNA methylation (RdDM) pathway (Cao & Jacobsen, 2002; Law & Jacobsen, 2010; Zhong et al., 2014). Once established, the symmetric CG and CHG methylation are maintained by Methyltransferase 1 (MET1, homologous to mammalian Dnmt1) and Chromomethylase 3 (CMT3), respectively, during DNA replication. However, the asymmetric CHH methylation needs to be established de novo in a new cell cycle through the RdDM pathway (Law & Jacobsen, 2010; Matzke & Mosher, 2014) or maintained by Chromomethylase 2 (CMT2) through a pathway dependent on the chromatin remodelling protein DDM1 (Zemach et al., 2013; Stroud et al., 2014).
Dysfunction of mammalian Dnmt3 or Dnmt1 leads to embryonic lethality (Li et al., 1992; Okano et al., 1999). The Arabidopsis met1 mutants, which lack CG methylation, are viable and fertile, although they have some developmental defects, including the malformation of the apical–basal axis, shoot and root meristems and other organs in embryos due to misregulation of genes that specify embryo cell identify and disruption of auxin gradients (Xiao et al., 2006), late flowering due to transcriptional activation of the floral repressor FWA, and size divergence in self crossed progenies presumably due to epigenome divergence (Saze et al., 2003; Mathieu et al., 2007). Interestingly, some met1 individuals develop additional developmental phenotypes after continued inbreeding, which may be caused by transposition of a copia‐type retrotransposon EVD (Mirouze et al., 2009). The ddcc (drm1 drm2 cmt2 cmt3) quadruple mutant, which lacks non‐CG methylation, only shows a leaf curling phenotype due to transcriptional derepression of a single imprinted gene SDC (Henderson & Jacobsen, 2008; Johnson et al., 2008; Stroud et al., 2014). However, the simultaneous disruption of CG and non‐CG methylation in Arabidopsis causes severe developmental defects. A met1 drm2 mutant shows severely retarded growth and reduced fertility (Mathieu et al., 2007). The met1 cmt3 mutant and the drm1 drm2 cmt3 met1 mutant plants are rarely viable. The survivors have very short stature and are infertile (Xiao et al., 2006; Zhang & Jacobsen, 2006). These differences suggest that CG and non‐CG methylation compensate for each other in regulating plant development.
To better understand the functions of DNA methylation in Arabidopsis, especially to understand the overlapping functions of CG and non‐CG DNA methylation, we attempted to knock out all five DNA methyltransferases in Arabidopsis by introducing met1 mutation into the ddcc mutant using the CRISPR/Cas9 technology (Chen et al., 2019). While the homozygous quintuple mutants are embryonically lethal, we obtained the ddcc met1+/− mutants, which show severe developmental phenotypes. Phenotypic, DNA methylome and RNA transcriptome analyses of different methyltransferase mutants revealed that CG and non‐CG DNA methylation synergistically and redundantly regulate plant reproductive development, vegetative development and TE silencing. Our findings provide insights into the functions of CG and non‐CG DNA methylation in plant development.
Materials and Methods
Plant materials and growth conditions
The ddcc quadruple mutant has been described previously (Stroud et al., 2014). Plants were grown on half‐strength Murashige & Skoog (½MS) nutrient agar plates in a growth chamber under a 16 h : 8 h, light : dark cycle (Philips; TLD, 36 W/865) at 22°C for 2 wk. The seedlings were then transferred to soil and grown in a glasshouse under the same conditions. To generate the met1 mutant alleles using the CRISPR/Cas9 system, 20‐bp sgRNAs (Supporting Information Table S1) targeting MET1 were cloned into YAO promoter‐driven CRISPR/Cas9 system (Yan et al., 2015). The constructs were transformed into the wild‐type Col‐0 or ddcc mutant using Agrobacterium tumefaciens GV3101 using the standard floral dip method (Clough & Bent, 1998). Homozygous or heterozygous mutant plants with the Cas9 transgene out‐crossed were used for further experiments (Fig. S1).
Microscopy analysis
Anther and pollen morphology was observed using cryogenic scanning electron microscopy (cryo‐SEM) as described previously (Esch et al., 2004). The anthers or pollens were fast‐frozen in liquid nitrogen and transferred under vacuum to the cold stage of the chamber, where sublimation (−90°C, 5 min) and sputter coating (10 mA, 30 s) with platinum were conducted. Finally, the samples were transferred to another cold stage in the scanning electron microscope and imaged.
Embryo and ovule morphology was observed under a spinning disc confocal microscope (Zeiss Cell Observer SD; Zeiss, Oberkochen, Germany).
Alexander dye and 4′,6‐diamidino‐2‐phenylindole staining
Dehiscent anthers were placed on glass slides and the pollen were released using a dissecting needle. The pollen grains were quickly suspended in Alexander dye or 4′,6‐diamidino‐2‐phenylindole (DAPI). DAPI staining was carried out in the dark. After staining, the pollen were observed under a fluorescence microscope (Olympus BX53).
In vivo pollen germination assay
For pollen germination in vivo, the stamens of a flower bud were removed and the pistils were allowed to grow for another 2 d and then pollinated with pollen grains. After 2 d the pistils were collected and fixed in a fixing solution (acetic acid : ethanol, 1 : 3) for 2 h. After that, the pistils were washed sequentially with 70%, 50%, 30% ethanol and ddH2O for 10 min. The pistils were then softened in 8 M NaOH overnight and washed with ddH2O. The pistils were stained with 0.1% decolorised aniline blue (pH 9–11, in 108 mM K3PO4) for more than 2 h in the dark. The pollen tubes in the pistils were observed under a fluorescence microscope (Olympus BX51) equipped with an ultraviolet filter set.
Nuclei isolation and microscopy
Rosette leaves were fixed in 4% (v/v) paraformaldehyde for 20 min under vacuum at room temperature. The leaves were rinsed twice in 1× PBS and chopped with a razor blade in extraction buffer 1 (10 mM Tris‐HCl, pH 9.5, 10 mM KCl, 10 mM spermine, 4 mM spermidine, 500 mM sucrose, 0.1% mercaptoethanol, 0.1% Triton X‐100) in a Petri dish. The fine homogenate was filtered through a 30‐μm filter and then centrifuged at 600 g for 3 min at 4°C. The pellet was resuspended in 300 μl of extraction buffer 2 (125 mM sucrose in extraction buffer 1), and the suspension was gently laid on top of 300 μl of extraction buffer 3 (850 mM sucrose in extraction buffer 1) in a 1.5 ml centrifuge tube. The assembly was centrifuged at 1600 g for 30 min at 4°C. The supernatant was removed and the precipitate was resuspended with 30 μl of extraction buffer 1. The nuclei were spread onto the slides, dried, stained with DAPI and observed under a confocal microscope (LSM800; Zeiss).
Comet assay
The comet assays were performed as described previously (Wang & Liu, 2006) with minor modifications using a comet assay reagent kit (Trevigen, Gaithersburg, MD, USA). Briefly, 4‐wk‐old Arabidopsis leaves were chopped with a razor in 1× PBS plus 20 mM EDTA on ice. The mixture was filtered through a 60‐μm nylon mesh. Then 50 μl of the nuclei suspension was combined with 500 μl of LMAgarose at 37°C and 50 μl of the mixture were immediately pipetted into CometSlide™ wells. After incubation at 4°C in the dark for 10 min, the slides were immersed in prechilled lysis solution and incubated at 4°C for 1 h. Then the slides were immersed in alkaline unwinding solution (200 mM NaOH, 1 mM EDTA) for 40 min at 4°C for DNA unwinding. The slides were placed in an electrophoresis tray in 1× (Tris‐borate/EDTA (TBE) and electrophoresis was run at 1 V cm−1 for 10 min. After air drying, the slides were stained with SYBR® Green (1 : 10 000 dilution) and observed under a fluorescence microscope (Olympus BX53). Comets was identified and scored using Comet Score software (http://www.autocomet.com). Two hundred comets on each slide were scored. The average of the percentages of DNA in tail from three slides was calculated and presented.
RNA extraction and RT‐qPCR analysis
Total RNA was extracted from rosette leaves (30 d old) or pollen grains (stage 13 flowers) using the RNeasy Plant Mini Kit (Qiagen). Here, c. 4 μg of total RNA was used for first‐strand cDNA synthesis using the SuperScript III first‐strand synthesis system (Invitrogen) for RT‐PCR following the manufacturer's instructions. The cDNA reaction mixture was diluted 10 times, and a 1‐µl aliquot was used as the template in a 25‐μl PCR reaction. PCR was carried out using the iQ SYBR Green Supermix. The expression levels of selected genes were normalised to that of ACTIN2. The primers used for RT‐qPCR are listed in Table S1.
RNA sequencing and data analysis
PolyA RNA‐Seq library preparation and high‐throughput sequencing were performed by Beijing Novogene Co. Ltd. The NEBNext® Ultra™ RNA library prep kit for Illumina® (New England Biolabs, Ipswich, MA, USA) was used to generate sequencing libraries following the manufacturer’s recommendations. The libraries were sequenced on the Illumina HiSeq 4000 platform and paired‐end 150‐bp reads were generated.
Adapter sequences and poor‐quality reads were removed using trim_galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with flags ‐‐paired and ‐‐length 70 (minimum read length after trimming). Then the clean reads were mapped to the Arabidopsis reference genome using TopHat22 (Kim et al., 2013) with default parameters. Reads were then sorted, indexed and compressed using SAMtools3 (Li et al., 2009). Only uniquely mapped reads were kept for further analysis using SAMtools3. Bigwig files were generated using bam2wig.py (Wang et al., 2012). featurecounts was used to count reads mapped to meta‐features genes and TEs with the option –p (fragments rather than reads were counted). For TE expression analysis, if more than 20% of a TE overlapped with genes, the TE was excluded from the analysis because accurate read assignment was difficult. Differential gene expression and TE expression were analysed using deseq2 (Love et al., 2014).
Whole‐genome bisulfite sequencing and data analysis
Genomic DNA was extracted from 30‐d‐old seedlings using the DNeasy Plant Mini Kit (Qiagen) and sent to BGI (Shenzhen) for bisulfite treatment, library preparation and sequencing. Sequencing reads were filtered using Fastp (Chen et al., 2018) with the option ‐‐length_required 70. Sequencing read mapping, methylation information extraction and wiggle file conversion were performed all in methylpy (https://github.com/yupenghe/methylpy). The weighted average methylation levels of activated TEs were calculated by add‐methylation‐level from methylpy and further visualised by complexheatmap (Gu et al., 2016) in R script. Cytosines with a depth < 4 were excluded from the analysis.
Differentially methylated regions (DMRs) were called by dmrfind in methylpy for CG context with the option ‐‐min‐num‐dms 5 (at least five differentially methylated sites). Only DMRs with significant difference (P ≤ 0.01) and absolute difference greater than 0.3 were considered.
Results
DNA methylation is essential for proper plant embryo development
To better understand the overlapping functions of CG and non‐CG DNA methylation in Arabidopsis, we set out to create DNA methylation‐null mutants by disrupting all of the five DNA methyltransferases in Arabidopsis. We designed two sgRNAs that targeted the first and sixth exon of MET1 (Fig. S1a). These sgRNAs were used to introduce CRISPR/Cas9‐induced met1 mutations in the ddcc mutant. For comparison, we also generated met1 mutations in wild‐type Col‐0 using the same sgRNAs. We identified met1 chimera plants from T1 transgenic plants, and then backcrossed them to Col‐0 and ddcc, to obtain met1+/− and ddcc met1+/− heterozygote plants, respectively, with the Cas9 transgene out‐crossed (Fig. S1b). Finally, we obtained two homozygous met1 mutants in Col‐0 background from the self‐cross progenies of met1+/− heterozygote plants, named met1‐10 and met1‐11. Both mutants harboured frameshift mutations, with the mutation in met1‐10 caused by a five‐nucleotide deletion and the mutation in met1‐11 caused by a one‐nucleotide insertion (Fig. S1a). The met1 mutations in ddcc background were identical to those in Col‐0, and the corresponding heterozygous plants were named ddcc met1‐10+/− and ddcc met1‐11+/− . However, we were unable to obtain homozygous quintuple mutants from the ddcc met1+/− self‐cross progenies (Table 1), suggesting that the homozygous quintuple mutants were lethal. In ddcc met1‐10+/− and ddcc met1‐11+/− siliques, 27.0% and 22.2% of seeds, respectively, underwent early abortion; 20.8% and 22.1% of seeds, respectively, underwent late abortion at 9 d after pollination (DAP) (Fig. 1a,b). To understand why these seeds underwent abortion, we carried out an observation at 2 DAP. We found that, among 196 seeds in the siliques of ddcc met1‐11+/− plants, 21.4% were unfertilised ovules; 9.7% were single‐fertilised ovules, within which only the central cell was fertilised; and 8.7% were double‐fertilised ovules with embryo development delayed (Fig. 1c). Single‐fertilised ovules, within which only the egg cell was fertilised, could not be found (Fig. 1c). The percentage of unfertilised ovules was similar to that of early‐aborted seeds (P = 0.9275, Fisher’s exact test), suggesting that early‐aborted seeds may mainly develop from unfertilised ovules. We also observed the endosperm development in seeds from ddcc met1‐11+/− at 3 DAP. We found that 20.5% had unfertilised central cells; and 32.3% had much less endosperm nuclei (16–32) than normal (> 64), which indicated delayed endosperm development (Fig. S2b). The percentages were much higher than those for wild‐type (3.9% and 0.7%, respectively, n = 153; P < 0.01, Fisher’s exact test). To further explore the possible reasons for late abortion in ddcc met1+/− , we closely examined late‐aborted seeds in ddcc met1‐11+/− at 9 DAP (n = 278). We found that 67.5% of the late‐aborted seeds in ddcc met1+/− siliques had embryos that had arrested at the globular stage, 16.8% had embryos arrested at the heart stage and 15. 7% had no discernible embryos (Fig. 1d). To test whether the late‐aborted seeds were ddcc met1 homozygotes, we collected several late‐aborted embryos in ddcc met1‐11+/− siliques and performed genotyping. Our results revealed that both wild‐type and mutant allele of MET1 could be detected (Fig. S2a), indicating that the late‐aborted seeds could be homozygous as well as heterozygous for met1‐11. Our results suggested that CG and non‐CG DNA methylation are redundant in regulating the developmental events required for seed development (Fig. 1b,c).
Table 1.
Effect of DNA methylation loss on gamete transmission rate in Arabidopsis.
| Parental genotype (female × male) | Progeny genotype (MET1) | Total | TEF | TEM | Ratio | Expected ratio | Chi‐squared | Confidence | ||
|---|---|---|---|---|---|---|---|---|---|---|
| +/+ | +/− | −/− | ||||||||
| ddcc met1‐11+/− self‐cross | 109 | 155 | 0 | 264 | NA | NA | 1 : 1.42 : 0 | 1 : 2 : 1 | 78.41 | < 2.2e‐16 |
| Col‐0 × met1‐11+/− | 120 | 77 | NA | 197 | NA | 64.2% | 1 : 0.64 | 1 : 1 | 9.39 | 0.0021 |
| Col‐0 × ddcc met1‐11+/− | 193 | 71 | NA | 264 | NA | 36.8% | 1 : 0.37 | 1 : 1 | 56.38 | 5.977e‐14 |
| ddcc met1‐11+/− × Col‐0 | 100 | 104 | NA | 204 | 104% | NA | 1 : 1.04 | 1 : 1 | 0.08 | 0.7794 |
NA, not available; TEF, female transmission efficiency; TEM, male transmission efficiency.
Fig. 1.
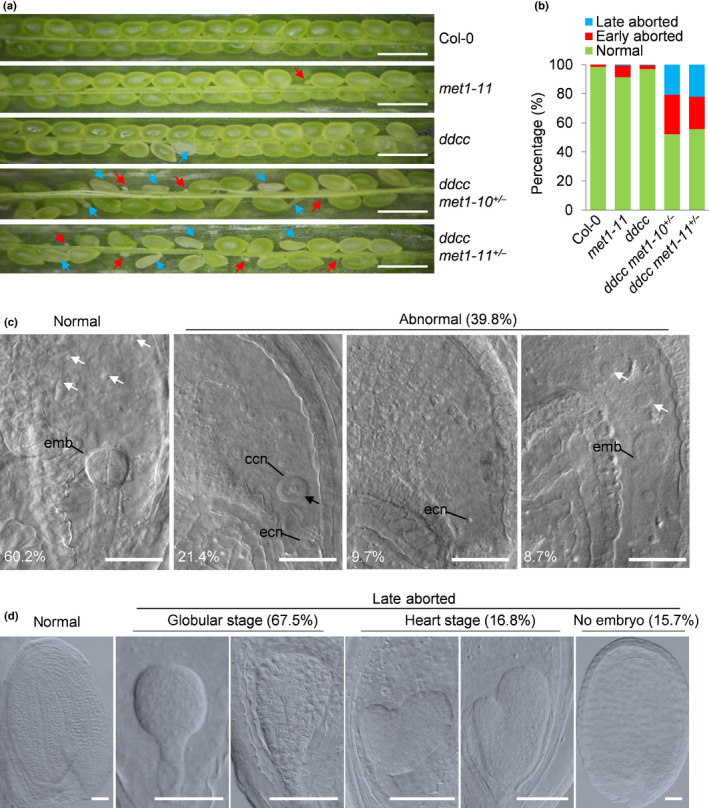
DNA methylation is essential for embryogenesis in Arabidopsis. (a) Seed set of Col‐0, met1‐11, ddcc, ddcc met1‐10+/− and ddcc met1‐11+/− plants at 9 d after pollination (DAP). Red and blue arrows indicate early‐aborted and late‐aborted seeds, respectively. Bars, 1 mm. (b) Seed abortion rates of Col‐0 (n = 438), met1‐11 (n = 442), ddcc (n = 374), ddcc met1‐10+/− (n = 1148) and ddcc met1‐11+/− (n = 1088) plants in (a). (c) Seeds in ddcc met1‐11+/− siliques (n = 196, 2 DAP). The percentages of normal and three types of aborted embryos are shown. ccn, central cell nucleus; ecn, egg cell nucleus; emb, embryo. White arrows indicate endosperm nuclei. Black arrow indicates an unfertilised central cell. Bars, 100 μm. (d) Differential interference contrast microscopy of embryos in normal and late‐aborted seeds in ddcc met1‐11+/− siliques (n = 278, 9 DAP). Bars, 50 μm.
The ddcc met1 quintuple mutations specifically impair male gametophyte transmission
Half of the gametes of ddcc met1+/− plants are expected to have a ddcc met1 genotype and a hypomethylated genome. They are an ideal system to explore the overlapping functions of CG and non‐CG DNA methylation in plant reproductive development. The only drawback is that some of the ddcc met1+/− plants have stunted growth as described below and poor vegetative growth often has adverse effects on gamete quality. To overcome this drawback, we used the ddcc met1‐11+/− plants that were not small in size (referred to as ‘big’) for the evaluation of reproductive abnormalities. Genetic analysis of ddcc met1‐11+/− self‐fertilised plants revealed a reduced transmission of the ddcc met1‐11 allele to the progeny (Table 1), suggesting that the ddcc met1‐11 mutations caused defects in male or female gametophytes. To determine whether transmission of the ddcc met1‐11 allele through the male or female gametophytes was impaired, reciprocal crosses of ddcc met1‐11+/− and wild‐type Col‐0 plants were performed. Our results showed that the female transmission was normal (Table 1). However, transmission of the ddcc met1‐11 allele through male gametophytes was significantly reduced (Table 1). To determine whether transmission of the met1‐11 allele through the male gametophytes was impaired, reciprocal crosses of met1‐11+/− and wild‐type Col‐0 plants were performed. Consistent with results of a previous study using the met1‐3 allele (Saze et al., 2003), our results showed that transmission of the met1‐11 allele through male gametophytes was reduced (Table 1). Next, wild‐type Col‐0 pistils were pollinated with Col‐0 and ddcc met1+/− pollen, respectively. Aborted seeds could rarely be found in siliques when pollinated with Col‐0 pollen. However, c. 19.5% of seeds aborted (n = 480) in siliques at 6 DAP when pollinated with ddcc met1+/− pollens (Fig. S2c). These results together suggested that the ddcc met1 quintuple mutations specifically affected male gametophyte development and/or function.
The ddcc met1 quintuple mutations impair pollen development
The reduction in male transmission could be caused by defects in pollen development or fertilisation in the ddcc met1+/− plants. To test the possibilities, we first examined anther development in wild‐type Col‐0 and the ddcc met1+/− mutant plants using cryo‐SEM. We found that the ddcc met1+/− plants had no obvious defects in anther and pollen morphology (Fig. S3a). Then we detected pollen viability using Alexander staining and found that abolition of DNA methylation produced minor effects on the viability of pollen grains (Fig. S3b; P > 0.2, Fisher’s exact test). To further assess the effects of ddcc met1 mutations on pollen development, mature pollen from Col‐0, met1‐11+/− , met1‐11, ddcc, and ddcc met1+/− plants were stained with DAPI and then observed using fluorescence microscopy (Fig. 2a–e). The results showed that, compared with wild‐type Col‐0 (95.5%), met1‐11+/− (95.6%), met1‐11 (91.34%), and ddcc (96%) plants, the ddcc met1‐10+/− and ddcc met1‐11+/− plants produced significantly lower percentages (82.1% and 82.5%, respectively; P < 0.01, Fisher's exact test) of normal trinucleated mature pollen (containing two sperm nuclei and one vegetative nucleus) but higher percentages of abnormal binucleated pollen (containing one germ cell nucleus and one vegetative nucleus), mononucleated pollen (containing one nucleus) and non‐nucleated pollen (containing no nucleus) (Fig. 2a–f; P < 0.01, Fisher's exact test). It is worth noting that the percentage of binucleated pollens (11.1%) in ddcc met1+/− is similar to that of the single‐fertilised ovules (9.7%) observed at 2 DAP (P = 0.6851, Fisher's exact test), suggesting that the single‐fertilised ovules may mainly develop from ovules fertilised by binucleated pollens. Together, our results suggested that cell divisions during pollen development were impaired, and the pollen defects contributed to seed abortion in the ddcc met1+/− plants.
Fig. 2.
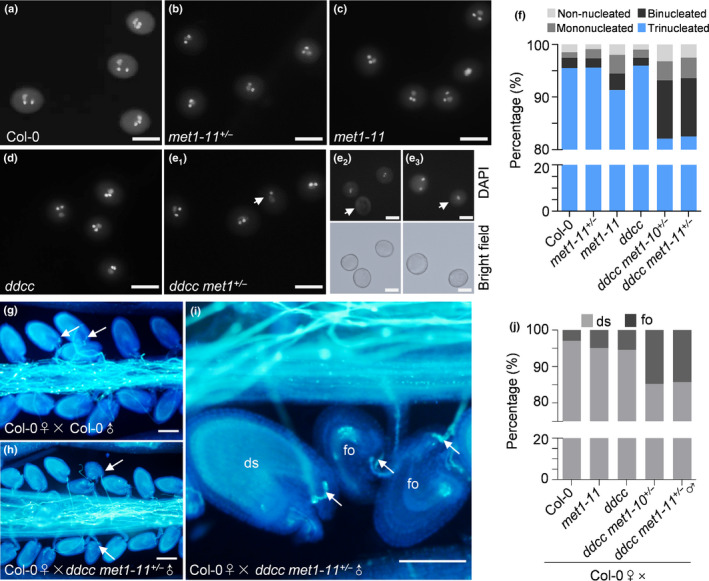
Loss of CG and non‐CG DNA methylation impairs pollen development and fertilisation in Arabidopsis. (a–e) 4′,6‐Diamidino‐2‐phenylindole (DAPI) staining of mature pollen grains from Col‐0, met1‐11+/− , met1‐11, ddcc and ddcc met1+/− mutant plants. (e1–e3) White arrows indicate pollen grains that are abnormally binucleated (e1), non‐nucleated (e2), and mononucleated (e3) in ddcc met1+/− . To confirm pollen identity for non‐nucleated (e2) and mononucleated (e3) pollen in ddcc met1+/− , images under bright field are showed. Bars, 100 μm. (f) Percentages of phenotypically abnormal pollen grains in Col‐0, met1‐11+/− , met1‐11, ddcc and ddcc met1+/− mutant plants (n > 500, for each genotype). (g–i) Aniline blue staining showing pollen tube access to each ovule at 2 d after pollination in Col‐0 ♀× Col‐0 ♂ (g) and Col‐0 ♀× ddcc met1+/− ♂ (h) siliques. Ovules that have normal pollen tube acceptance but cannot expand (failed ovules, fo) are frequently found in the Col‐0 ♀×ddcc met1+/− ♂ siliques (i). White arrows indicate normal pollen tube acceptance in a failed ovule and a developing seed (ds). Bars, 100 μm. (j) Percentages of developing seeds (ds) and failed ovules (fo) in Col‐0 ♀× Col‐0 ♂, Col‐0 ♀×met1‐11 ♂, Col‐0 ♀×ddcc ♂, and Col‐0 ♀× ddcc met1+/ − ♂ siliques (n > 200, for each genotype).
To dissect the molecular causes of the pollen defects in the ddcc met1+/− plants, we performed RT‐qPCR to detect the expression levels of genes known to be involved in pollen development in mature pollens of different genotypes (Fig. S4a,b). The results showed that, compared with wild‐type Col‐0, met1‐11 and ddcc plants, the ddcc met1‐10+/− and ddcc met1‐11+/− plants had significantly reduced levels of DOU1‐ACTIVATED ATPASE1 (DAA1), DOU1‐ACTIVATED ZINC FINGER2 (DAZ2), FASCIATA 2 (FAS2), and PLANT CADMIUM RESISTANCE 11 (PCR11) in their mature pollens (Fig. S4a). DAA1, DAZ2, and PCR11 are male germline‐specific genes activated by the male germline‐specific R2R3 MYB transcription factor DUO1 POLLEN1 (DUO1) (Borg et al., 2011). FAS2 is a subunit of Chromatin Assembly Factor‐1 (CAF‐1), a histone chaperone facilitating chromatin assembly during DNA replication and DNA repair (Chen et al., 2008). The daz2 mutant and the fas2 mutant were found to have blocked generative cell division and produced high percentages of binucleated pollens (Durbarry et al., 2005; Rotman et al., 2005; Chen et al., 2008; Borg et al., 2014), suggesting that the pollen defects in the ddcc met1+/− plants could be caused by downregulation of these genes. Although the expression of genes important for germ cell development were affected, the expression of genes important for vegetative cell development, including POLYUBIQUITIN 10 (UBQ10), WPP DOMAIN INTERACTING PROTEIN 1 (WIP1), and WPP DOMAIN‐INTERACTING TAIL‐ANCHORED PROTEIN 1 (WIT1), were not affected (Fig. S4b), suggesting that DNA methylation is important for germ cell development, but may not be important for vegetative cell development.
The ddcc met1 quintuple mutations may impair fertilisation
According to our results, the transmission rate of the ddcc met1 mutations through male gametophytes was 37% (Table 1). This cannot be explained only by the development of phenotypically abnormal pollens (18%). Furthermore, the percentage of unfertilised ovules in ddcc met1+/− (21.4%, Fig. 1c) was significantly higher than that of mononucleated and non‐nucleated pollens (< 7%; P < 0.01, Fisher’s exact test), suggesting that the ddcc met1 quintuple mutations could cause other reproductive defects. To investigate this, we performed an in vivo pollen germination assay, in which Col‐0 pistils were pollinated with pollens from the ddcc met1+/− plants (Fig. 2h,i). We found that, even when pollinated with excess pollen from ddcc met1‐10+/− and ddcc met1‐11+/− , 14.7% and 14.2% of the ovules (n = 286 and 254, respectively) failed to expand. The expansion failure could not be attributed to defects in pollen tube growth and targeting because these events were normal. This type of expansion failure could rarely be seen when using met1‐11, ddcc or Col‐0 pollen (n > 200, Fig. 2g,j). These results suggested that the fertilisation process was affected in ddcc met1+/− . Next, we performed RT‐qPCR to detect the expression levels of genes known to be involved in pollen tube bursting and fertilisation using mature pollens of different genotypes (Fig. S4b). Transcript levels of genes involved in pollen tube bursting remained unaltered in mature pollen of the ddcc met1+/− plants. Interestingly, the expression level of HAP2 (also called GCS1), a gene that is expressed during late gametogenesis and is important for angiosperm fertilisation (Mori et al., 2006), was much lower in the ddcc met1+/− mutant pollens than in the met‐11 and ddcc mutant pollen (Fig. S4a). Furthermore, the hap2 mutant showed similar defects in fertilisation, suggesting that the downregulation of HAP2 may lead to the fertilisation defects in the ddcc met1+/− mutants. Collectively, these results suggested that CG and non‐CG methylation may redundantly regulate fertilisation.
The ddcc met1+/− mutants manifest severe developmental defects and size divergence
In addition to defects in siliques, the ddcc met1+/− plants showed severe developmental phenotypes during vegetative development (Fig. 3a). As previously reported, the met1 and ddcc mutants displayed a late flowering phenotype and a leaf curling phenotype, respectively (Saze et al., 2003; Stroud et al., 2014). Compared with the met1‐11 and ddcc mutants, 47.2% of the ddcc met1+/− plants (n = 144) were much smaller, and had more severe leaf curling (referred to as ‘small’) (Fig. 3a). Notably, not all of the ddcc met1+/− plants were much smaller. Some of the ddcc met1+/− plants (38.2%) were only slightly smaller than the Col‐0 plants and had sizes similar to those of met1‐11 (referred to as ‘big’) (Fig. 3a). The size divergence occurred as the ddcc met1+/− plants were generated and self‐crosses of a ‘big’ plant or a ‘small’ plant could produce both ‘big’ and ‘small’ progenies, indicating that such developmental instability was stochastic and reflected epigenetic instability (Fig. 3b). To understand whether the postembryonic size divergence was paternally, maternally or biparentally inherited, we performed more reciprocal crosses between ddcc met1+/− and ddcc. We found that the size divergence phenotype could be observed in the F1 generation of crosses between ddcc met1+/− and ddcc in either direction, suggesting that the size divergence was biparentally inherited. Furthermore, this phenotype could also be found in ddcc MET1+/+ plants segregated from the self‐cross progenies of ddcc met1+/− heterozygote plants, albeit at a lower frequency compared with the segregated ddcc MET1+/− plants, and suggesting that the size divergence was not linked to the MET1+/− genotype. Such size divergence has been observed in the self‐progenies of the met1‐3 mutant in later generations, presumably due to epigenome divergence or an increasing accumulation of TE insertions in the inbreeding met1‐3 mutant (Mathieu et al., 2007; Mirouze et al., 2009). The earlier occurrence supported a redundant role for CG and non‐CG in DNA methylation maintenance and control of development.
Fig. 3.

Arabidopsis ddcc met1+/− mutants exhibit size divergence. (a) Representative pictures of Col‐0, met1‐10, met1‐11, ddcc, ddcc met1‐10+/− and ddcc met1‐11+/− plants (24 d old). Bars, 1 cm. (b) Representative pictures of first‐generation ddcc met1‐11+/− siblings and their selfed‐progenies (30 d old). Bars, 1 cm.
Size divergence may not be caused by difference in DNA methylation
To determine whether the size divergence occurred in ddcc met1+/− plants was caused by stochastic changes to the epigenome, we investigated DNA methylomes of ‘big’ and ‘small’ ddcc met1‐11+/− plants (from this point forwards referred to as ‘big’ and ‘small’). The methylomes of wild‐type Col‐0 and met1 and ddcc mutant plants were profiled for comparison. Data analysis revealed that CG and non‐CG methylation, as expected, were eliminated in met1 and ddcc mutant plants, respectively (Fig. 4a). The ‘big’ and ‘small’ plants both lost half their CG methylation and all non‐CG methylation across the whole genome (Fig. 4a). To examine local DNA methylation changes, we identified DMRs in the CG context in ‘big’ and ‘small’. In total, 14 649 and 13 598 CG‐hypo‐DMRs were identified in ‘big’ and ‘small’, respectively, and most of the hypo‐DMRs (9641) overlapped between ‘big’ and ‘small’ (Fig. 4b). Notably, although many ‘big’‐ or ‘small’‐specific loci were not defined as hypo‐DMRs according to our criteria, the DNA methylation levels at these loci were also lower in ‘big’ and ‘small’ than in wild‐type (Fig. 4b). We next examined the CG methylation levels of CG‐hypo‐DMRs identified from met1‐11 in ‘big’ and ‘small’ and found that, overall, the CG methylation levels were comparable (Fig. 4c). Our results suggested that the size divergence occurred in ddcc met1+/− plants may not be attributed to differences in the DNA methylome. However, it was rather difficult to exclude the possibility that one or a small number of DMRs play a critical role in determining the development of plants into ‘big’ or ‘small’.
Fig. 4.
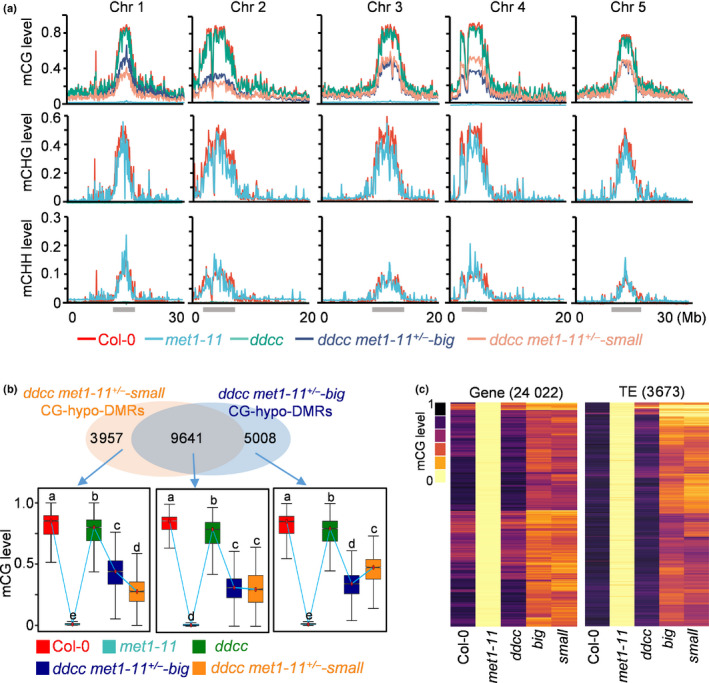
Size divergence of Arabidopsis ddcc met1+/− mutants is not caused by a difference in DNA methylation. (a) Fractional DNA methylation levels of cytosines in CG, CHG and CHH contexts across five chromosomes (Chr). The locations of pericentromeric heterochromatin are indicated with grey bars. (b) Venn diagram showing the number of CG‐hypo‐DMRs that overlap between the ‘big’ and ‘small’ plants of ddcc met1‐11+/− . Box plots show the methylation levels of CG‐hypo‐DMRs. Dark horizontal line, median; edges of boxes, 25th (bottom) and 75th (top) percentiles; whiskers, minimum and maximum percentage of DNA methylation. Significant differences between two genotypes are marked with different letters (P < 10−15, Mann–Whitney U‐test). (c) Heat map showing CG methylation levels in different genotypes at CG‐hypo‐DMRs identified in met1‐11. big: ddcc met1‐11+/−‐big; small: ddcc met1‐11+/−‐small. The number of CG‐hypo‐DMRs is shown at the top of the graph. DMR, differentially methylated region; TE, transposable element.
The ‘small’ plants accumulate a high level of DNA damage
To further explore why the ddcc met1+/ plants exhibited size divergence, we performed a transcriptome analysis of wild‐type Col‐0, met1‐11, ddcc, ‘big’, and ‘small’ plants. We identified 1667, 256, 1811 and 3305 upregulated genes, 2312, 488, 1566 and 1050 downregulated genes, 1388, 340, 1941 and 2042 upregulated TEs, 28, 13, 42 and 35 downregulated TEs in met1‐11, ddcc, ‘big’ and ‘small’ plants, respectively (Tables S2, S3; Fig. S5a, fold change ≥ 2, q ≤ 0.05). The ‘big’ and ‘small’ plants had comparable amounts of upregulated TEs and the upregulated TEs in ‘big’ and ‘small’ plants overlapped to a great extent (Fig. S5b). However, compared with the ‘big’ plants, the ‘small’ plants had a higher number of upregulated genes but a lower number of downregulated genes. Among 1050 downregulated genes in ‘small’, 746 (71%) were also downregulated in ‘big’ and 304 (29%) were specifically downregulated in ‘small’. Among 3305 upregulated genes in ‘small’, 1580 (48%) were also upregulated in ‘big’ and 1725 (52%) were specifically upregulated in ‘small’ (Fig. S5b).
We next pooled all of the upregulated genes in the four mutants together and performed cluster analysis. These upregulated genes were divided into five groups according to their expression levels (Fig. 5a). Group I genes were slightly upregulated in met1‐11, but their expression levels were further elevated in ddcc met1‐11+/− . Group II genes were upregulated in ddcc and ddcc met1‐11+/− . Groups III and IV genes were dramatically upregulated in met1‐11, but barely upregulated in ddcc met1‐11+/− . Group V genes were specifically upregulated in ddcc met1‐11+/− . We found that Group I and Group V genes, especially Group V genes, had much higher expression levels in ‘small’ than in ‘big’ (Fig. 5a). Gene Ontology (GO) analysis revealed that the Group I genes showed enrichment for GO terms related to DNA metabolism, DNA replication, cell cycle, chromosome organisation, DNA repair and nuclear division. Group V genes showed enrichment for GO terms related to cell cycle and cell division (Fig. 5a; Table S4). Further analysis of Group I and Group V genes revealed that there were 41, 47 and 167 genes known to be involved in DNA replication, DNA repair and cell cycle, respectively (Table S5). These genes were induced to a greater extent in ‘small’ rather than in met1‐11 and ‘big’ (Table S5). To confirm the transcriptome data, we randomly selected 12 genes among these genes for RT‐qPCR validation. The results showed that the 12 genes were generally induced in met1‐11, ‘big’ and ‘small’, but were induced mostly in ‘small’ (Fig. 5b).
Fig. 5.
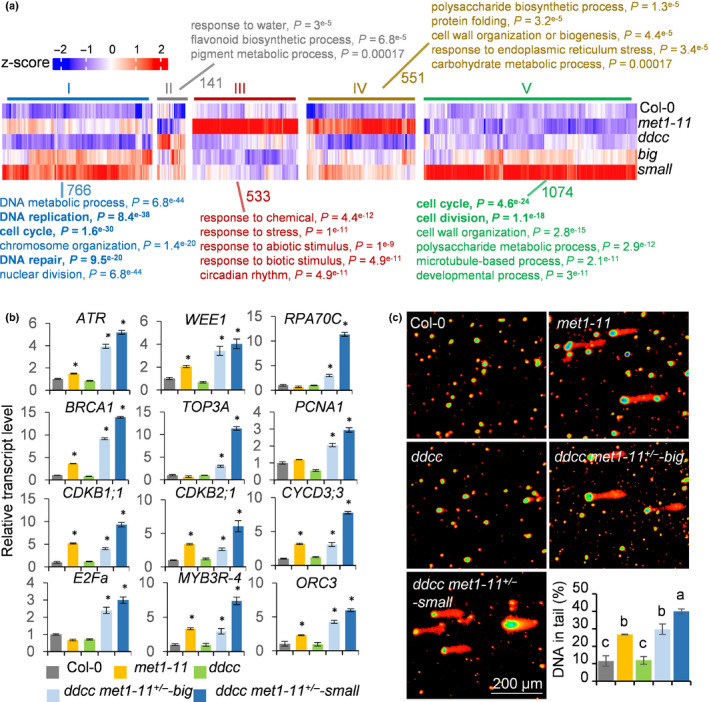
The ‘small’ plants of Arabidopsis ddcc met1‐11+/− accumulate a high level of DNA damage. (a) Heatmap and Gene Ontology analysis of the upregulated genes in met1‐11, ddcc and ddcc met1‐11+/− plants. These upregulated genes are divided into five groups based on their expression levels in the indicated mutants. The number of genes in each group is shown. (b) RT‐qPCR analysis of the expression levels of DNA repair‐, DNA replication‐ and cell cycle‐related genes in seedlings of different genotypes. Data represent the mean ± SD of three biological replicates. Asterisks indicate significant differences between Col‐0 and the indicated mutants (P < 0.05, two‐tailed Student’s t‐test). (c) Tailing of damaged DNA in different mutants as determined using the comet assay. The right‐hand bottom panel shows percentages of DNA in comet tails. The error bars represent the SD of three independent experiments. Different letters indicate significant differences (P < 0.01, one‐way analysis of variance (ANOVA)).
The differential upregulation of a large number of cell cycle, DNA replication and DNA repair genes in met1‐11, ‘big’ and ‘small’ plants could be caused by difference in DNA methylation in the promoters of these genes. Data analysis revealed that, compared with Col‐0, the ‘big’ and ‘small’ plants, both had decreased DNA methylation levels in the promoters (2 kb upstream of transcription start sites) of these genes. However, the extent of decrease in ‘big’ and ‘small’ was identical (Fig. S6). Therefore, differences in DNA methylation do not account for the differential upregulation of cell cycle, DNA replication and DNA repair‐related genes in met1‐11, ‘big’ and ‘small’ plants.
The differential upregulation of a large number of cell cycle, DNA replication and DNA repair‐related genes in met1‐11, ‘big’ and ‘small’ plants may reflect that these mutants have different levels of DNA damage (Hu et al., 2016). To test this, we performed a comet assay to detect DNA‐stranded breaks. We found that DNA‐stranded breaks were increased in met1‐11 and ddcc met1‐11+/− . Importantly, the ‘small’ plants accumulated significant higher levels of DNA‐stranded breaks than ‘big’ plants (Fig. 5c). Together, these results suggested that the met1‐11, ‘big’, and ‘small’ have different levels of DNA damage and the greater reduction in plant size in ‘small’ plants could be the result of a high level of DNA damage.
A group of TEs is specifically activated in the ddcc met1+/− mutants
DNA methylation plays an important role in TE silencing. We next examined the extent of redundancy of CG and non‐CG DNA methylation in TE silencing. As described above, we identified 1388 and 340 upregulated TEs in met1‐11 and ddcc, respectively (Fig. S5a; Table S3). The numbers of upregulated TEs in the ‘big’ and ‘small’ plants were increased to 1941 and 2042, respectively (Fig. S5a; Table S3). We identified 1498 upregulated TEs in ddm1 using previous published data with the same criteria (Osakabe et al., 2021). We compared the upregulated TEs in ddcc, ddm1, met1‐11, and ddcc met1‐11+/− (activated TEs in the ‘big’ and ‘small’ plants combined), and found that most of the activated TEs in ddcc met1‐11+/− overlapped with activated TEs in ddm1 and met1‐11 (Fig. S7a). Although a substantial amount of CG methylation was detected in the ddcc met1‐11+/− (Fig. 4), most of the TEs activated in met1‐11 (˜ 93%) were also activated in ddcc met1‐11+/− (Fig. S7a). One possibility is that reducing CG methylation to the level in ddcc met1‐11+/− was sufficient to activate these TEs. Another possibility is that, although a reduction in CG methylation was not sufficient, complete loss of non‐CG methylation adds to the effect of a reduction in CG methylation, leading to the activation of these TEs. Interestingly, 536 TEs were specifically activated in ddcc met1‐11+/− (Fig. S7a). We analysed the signature of TEs specifically activated in ddcc met1‐11+/− . We found that retrotransposons, such as long terminal repeat retrotransposons in the Gypsy superfamilies, were significantly overrepresented in TEs specifically activated in ddcc met1‐11+/− , indicating that DNA methylation preferentially silenced retrotransposons (Fig. S7b,c). Further TE family enrichment analysis revealed that VANDAL2, VANDAL5, META1, ATCOPIA95, ATHILA4B_LTR and ATGP7 TE families were specifically activated in ddcc met1‐11+/− (Fig. S7d–h).
We next divided the upregulated TEs in the ddcc met1‐11+/− plants into five groups according to their activation levels in met1‐11 and ddcc (Fig. 6a). Group I TEs were activated by either met1‐11 or ddcc mutation, an indication that both CG and non‐CG DNA methylation are required for silencing these TEs. Group II TEs were activated in ddcc but not in met1‐11, indicating that non‐CG methylation is responsible for silencing these TEs. Group III TEs were activated in met1‐11 but not in ddcc, indicating that CG methylation is responsible for the silencing of these TEs. Group IV TEs were not activated in met1‐11 or ddcc, but were specifically activated in ddcc met1‐11+/− , indicating that CG and non‐CG DNA methylation redundantly regulated the expression of these TEs. Group V TEs did not belong to any of the above four groups. Among the 2172 upregulated TEs in the ddcc met1‐11+/− plants, only 105 (4.8%) and 49 (2.3%) TEs belonged to Groups I and II, respectively, while 990 (45.6%) TEs belonged to Group III (Fig. 6a). These results suggested that CG DNA methylation is more important than non‐CG methylation in TE silencing. Importantly, 747 (34.4%) TEs were only upregulated in ddcc met1‐11+/− , suggesting a high extent of redundancy of CG and non‐CG DNA methylation in TE silencing (Fig. 6a).
Fig. 6.
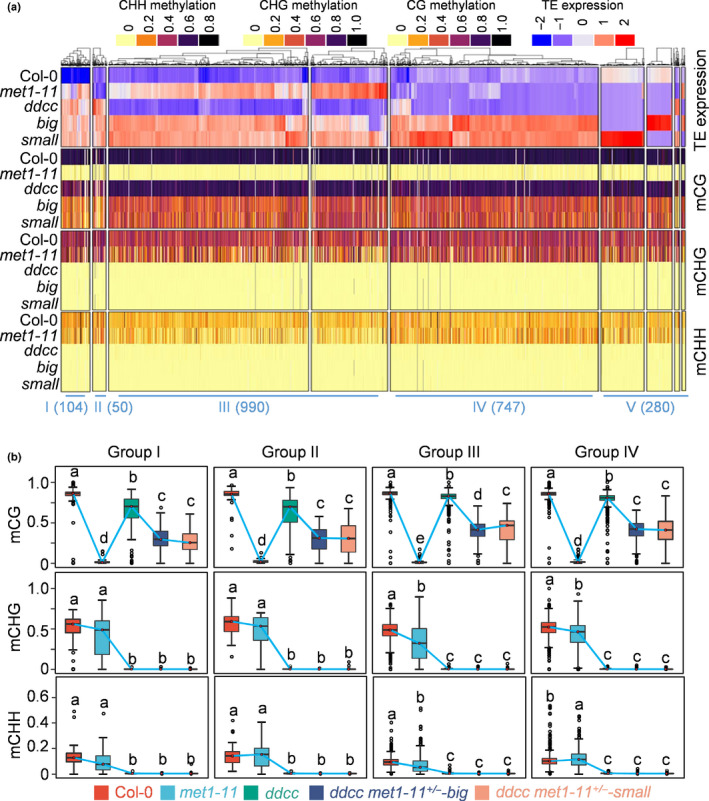
CG and non‐CG DNA methylation synergistically and redundantly control transposable element (TE) silencing in Arabidopsis. (a) Heat map of the activated TEs in the ddcc met1‐11+/− mutant plants. The DNA methylation levels of each TE are shown. These activated TEs are divided into five groups. Group I, TEs that are activated in either met1 or ddcc. Group II, TEs that are activated in ddcc but not in met1. Group III, TEs that are activated in met1 but not in ddcc. Group IV, TEs that remain silenced in either met1 or ddcc, but are activated in ddcc met1‐11+/− . Group V, others TEs. The number of TEs in each group is shown. big, ddcc met1‐11+/−‐big; small, ddcc met1‐11+/−‐small. (b) Box plots show the DNA methylation levels of the four groups of TEs in the indicated mutants. Dark horizontal line, median; edges of boxes, 25th (bottom) and 75th (top) percentiles; whiskers, minimum and maximum percentage of DNA methylation. Significant differences between two groups are marked with different letters (P < 10−15, Mann–Whitney U‐test).
To further dissect the role of DNA methylation in TE silencing, we calculated the DNA methylation levels of the four groups of TEs. We found that CG methylation decreases to a similar extent in all the four groups of TEs in ddcc met1‐11+/− , suggesting that disrupting one copy of the MET1 gene equally affected CG methylation across the four groups of TEs (Fig. 6a). Interestingly, CHG and CHH DNA methylation in Group III TEs were significantly reduced in met1‐11, suggesting that CG DNA methylation was important for the maintenance of non‐CG methylation in these TEs. Compared with Group III TEs, Group IV TEs had higher CHH DNA methylation in met1‐11 (Fig. 6a,b). This may explain why Group IV TEs were not activated in met1‐11. Group IV TEs may be only activated when CHH DNA methylation level is low enough. These results suggested that CG and non‐CG DNA methylation redundantly regulated a group of TEs in Arabidopsis.
Discussion
Studies carried out in mammals have reveal the essential roles of CG DNA methylation in normal spermatogenesis, embryonic stem cell differentiation and postnatal development (Smith & Meissner, 2013). Plants have both CG and non‐CG DNA methylation. Previous studies have suggested that CG and non‐CG DNA methylation play redundant roles in plant development. However, the exact redundant roles of CG and non‐CG DNA methylation in plant development are still unclear. In this study, we found that CG and non‐CG DNA methylation redundantly regulated plant reproductive development, vegetative development and TE silencing. We further investigated the mechanisms.
CG and non‐CG methylation play redundant roles in embryogenesis
The roles of DNA methylation in development have been investigated in different plant species. In rice, the null mutation of OsMET1b severely impairs seed development and leads to seedling lethality (Hu et al., 2014; Yamauchi et al., 2014). Moreover, loss of function of OsCMT3a induces pleiotropic developmental defects, including severe defects at the reproductive stage (Cheng et al., 2015). In maize, seeds that lack CMT3 (Zmet2/Zmet5) or DDM1 (Chr101/Chr106) could not be produced (Li et al., 2014). These finding suggested that CG or non‐CG DNA methylation is essential for development in these plants. However, their lethality makes it impossible to study whether CG and non‐CG methylation play redundant roles.
In Arabidopsis, the met1 and ddcc mutants are viable, and only show weak developmental phenotypes. A previous study has revealed that some of the embryos in met1‐6 mutant are abnormal due to misexpression of some embryo identity genes and improper formation of auxin gradients (Xiao et al., 2006). Further reduction of non‐CG methylation through generating double mutants exacerbated the embryonic developmental defects, suggesting that CG and non‐CG methylation redundantly regulated embryo development (Zhang & Jacobsen, 2006). In this work, we attempted to generate the homozygous quintuple mutant ddcc met1. However, we only obtained ddcc met1+/− . Nearly half of the embryos were aborted in the ddcc met1+/− siliques. Our results proved that CG and non‐CG methylation play redundant roles in the regulation of embryo development.
DNA methylation is only essential for transmission through the male gametophytes
It was unexpected that the ddcc met1 quintuple mutations only impaired the transmission through the male gametophytes, while female transmission was normal (Table 1). Our findings are consistent with previous findings that MET1 is expressed at a lower level in female gametophytes than in seedlings (Jullien et al., 2012) and that the transcription factor ARID1 can prevent the expression of MET1 in the egg cell and the central cell (Li et al., 2017). These results suggested that DNA methylation is not required for female gametophyte development. However, we could not exclude the possibility that other DNA methyltransferases function redundantly with MET1 in the maintenance of CG methylation in megasporocytes. There are three additional MET1 homologues (MET2a, MET2b, and MET3) in the Arabidopsis genome, although the expression levels of these genes are very low (Finnegan & Kovac, 2000; Ashapkin et al., 2016).
Consistent with our findings, other studies have also reported that DNA methylation is important for male fertility. In Capsella, knockout of NRPD1 led to the arrest of pollen development at the microspore stage (Wang et al., 2020). In mice, conditional knockout of Dnmt3a impaired spermatogenesis (Kaneda et al., 2004). These results suggested that DNA methylation plays a conserved role in male germ cell development in plants and mammals. Interestingly, the level of DNA methylation in vegetative cells is much lower than that in sperm (Calarco et al., 2012). It was found that active DNA demethylation played an important role in lowering DNA methylation levels in vegetative cells and that the removal of DNA methylation is critical for male fertility in Arabidopsis (Schoft et al., 2011; Khouider et al., 2021).
Defects in male transmission of the met1 mutant allele could be attributed to the abnormalities in pollen development and fertilisation. Further dissection of the molecular mechanism revealed that DAA1, DAZ2, FAS2 and PCR11, genes involved in germ cell development, and HAP2, a gene involved in fertilisation, were downregulated in the ddcc met1+/− mutant pollens (Fig. S4a). However, we found that there was no obvious DNA methylation enrichment at the gene body and promoter regions of DAA1, DAZ2, FAS2 and PCR11 (Fig. S4c). There was DNA methylation at the gene body and promoter regions of HAP2 (Fig. S4c). While complete loss of CG or non‐CG DNA methylation in the HAP2 promoter in met1‐11 and ddcc, respectively, had no effect on the expression of HAP2, reduction in CG methylation combined with the complete loss of non‐CG methylation in the HAP2 promoter in ddcc met1‐11+/− mutant led to a significant reduction of HAP2 expression. These results suggested that the expression of HAP2 could be redundantly controlled by CG and non‐CG methylation at its promoter and that DNA methylation promotes HAP2 expression, similar to the way that DNA methylation promotes REPRESSOR OF SILENCING1 (ROS1) expression (Johnson et al., 2015; Lei et al., 2015). Therefore, DNA methylation could directly or indirectly regulate the expression of these genes to ensure normal male transmission. In addition to regulating these genes, DNA methylation may regulate the expression of other genes involved in pollen development and double fertilisation. DNA methylation may not only affect gene transcription, but also regulate alternative splicing of mRNA precursors (Lev Maor et al., 2015). It was recently found that the pollen produced by the Arabidopsis drm1drm2 mutant contained meiotic abnormalities due to mis‐splicing of the MPS1 gene (Walker et al., 2018). Future investigations are required to further dissect the roles and mechanisms of DNA methylation in gene expression regulation and plant male fertility.
DNA methylation is essential for genome integrity
In this study, we found extensive DNA damage and misregulation of DNA replication‐, DNA repair‐ and cell cycle‐related genes in met1‐11 and ddcc met1+/− . In mouse embryonic fibroblast cells, loss of DNA methylation upon depletion of Dnmt1 causes aberrant expression of genes involved in cell cycle control and p53‐dependent apoptosis (Jackson‐Grusby et al., 2001), suggesting that the loss of DNA methylation may also lead to DNA damage in mammalian cells. Therefore, DNA methylation may play a conserved role in the maintenance of genome integrity. An important question is why DNA methylation is important for genome integrity. It is known that DNA methylation, especially CG DNA methylation, is important for maintenance of the heterochromatin structure (Mathieu et al., 2007). In met1‐3, high proportions of nuclei have decondensed chromocenter in the 3rd and 4th generation. In the met1‐3 drm2‐2 double mutant, the compaction of centromeric 180 bp repeats and 5S rDNA is lost (Mathieu et al., 2007). Our results also confirmed that heterochromatin was decondensed in met1‐11, and that the ddcc met1‐11+/− mutant had a higher level of heterochromatin decondensation (Fig. S8). This was coincident with the finding that the ddcc met1‐11+/− mutant accumulated more DNA damage than did met1‐11 (Fig. 5c). We propose that heterochromatin decondensation may render cells susceptible to increased DNA damage. Supporting this, the ddm1 plants, which have decondensed heterochromatin, were sensitive to the DNA‐damaging reagent methyl methane sulfonate (Yao et al., 2012). Alternatively, the activated TEs in met1 and ddcc met1+/− may translocate to induce DNA damage. Previous studies have found that retrotransposons are mobile in met1 and ddm1 mutants (Mirouze et al., 2009; Tsukahara et al., 2009). Furthermore, conflicts between the replication machinery and the transcription machinery as a consequence of massive TE upregulation in the mutants could induce DNA damage (García‐Muse & Aguilera, 2016; Hamperl et al., 2017). However, we could not exclude the possibility that loss of DNA methyltransferases itself, instead of loss of DNA methylation, was responsible for the occurrence of DNA damage. In mammalian cells, Dnmt1 is located at double‐stranded breaks after DNA damage, and this is independent of its methyltransferase activity (Mortusewicz et al., 2005). The absence of Dnmt1 at the replication forks can activate replication stress checkpoints (Unterberger et al., 2006). Therefore, loss of DNA methyltransferases may cause replication fork stalling or breakage to induce DNA damage.
As the DNA methylation level and pattern, heterochromatin decompaction, and TE activation are all similar between ‘big’ and ‘small’, why ‘big’ and ‘small’ have different levels of DNA damage is still unclear. Our speculation is that epigenetic instability could lead to different levels of DNA damage in ‘big’ and ‘small’. As observed in met1‐3 (Mathieu et al., 2007), in ddcc met1‐11+/− mutants, due to reduced DNA methylation, the remaining epigenetic mechanisms (such as H3K9 methylation) may start to operate and they may operate in a highly stochastic fashion. This would lead to progressive deposition de novo of epigenetic marks transgenerationally, even at previously unmarked locations, and eventually lead to different levels of DNA damage in different ddcc met1‐11+/− populations and in different generations. Alternatively, differential TE insertions or TE upregulation could lead to different levels of DNA damage in ‘big’ and ‘small’. Future investigations are required to test these possibilities.
Author contributions
WJL, JCL and WQQ designed the research. WJL, JCL and ZJL performed the experiments. LHS and YL performed the bioinformatics analysis. WJL, JCL, LHS and WQQ analysed the data. WJL, JCL and WQQ wrote the article. WJL, JCL and LHS contributed equally to this work.
Supporting information
Fig. S1 Generation of MET1 mutants in Arabidopsis using the CRISPR/Cas9 system.
Fig. S2 Loss of DNA methylation affects endosperm development and compromises transmission through the male gametophytes in Arabidopsis.
Fig. S3 Loss of DNA methylation does not affect pollen viability in Arabidopsis.
Fig. S4 Loss of DNA methylation in Arabidopsis leads to the downregulation of some genes involved in pollen development and double fertilisation in mature pollens.
Fig. S5 Loss of DNA methylation leads to global changes of gene expression in Arabidopsis seedlings.
Fig. S6 The upregulation of cell cycle‐, DNA replication‐ and DNA repair‐related genes in Arabidopsis ddcc met1‐11 +/− seedlings is not due to loss of DNA methylation in the promoters of these genes.
Fig. S7 The Arabidopsis ddcc met1‐11 +/− mutation specifically activates a group of TEs.
Fig. S8 CG and non‐CG DNA methylation synergistically regulate chromocenter condensation in Arabidopsis.
Table S1 Primers and sgRNAs used in this study.
Table S2 List of differentially expressed genes in the indicated mutants.
Table S3 List of differentially expressed TEs in the indicated mutants.
Table S4 GO analysis of differentially expressed genes in the indicated mutants.
Table S5 List of upregulated genes involved in DNA repair, DNA replication, and cell cycle control in met1‐11 and ddcc met1‐11 +/− .
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank Steven Jacobsen for providing the ddcc quadruple mutant and Qi Xie for providing the CRISPR/Cas9 system. This study was supported by the National Key R&D Program of China (grant no. 2018YFE0204700 and 2016YFA0500800) and the National Natural Science Foundation of China (grant no. 31970614) to WQQ.
Data availability
Methylome and transcriptome data have been submitted to the NCBI Sequencing Read Archive under accession no. PRJNA686693. The DNA methylome of Col‐0 and ddcc had been previously deposited in SRR1005412 and SRR1005415, respectively (Stroud et al., 2013, 2014). RNA‐seq data of ddm1 and the corresponding Col‐0 were previously deposited in GSE150436 (Osakabe et al., 2021).
References
- Ashapkin VV, Kutueva LI, Vanyushin BF. 2016. Plant DNA methyltransferase genes: multiplicity, expression, methylation patterns. Biochemistry 81: 141–151. [DOI] [PubMed] [Google Scholar]
- Borg M, Brownfield L, Khatab H, Sidorova A, Lingaya M, Twell D. 2011. The R2R3 MYB transcription factor DUO1 activates a male germline‐specific regulon essential for sperm cell differentiation in Arabidopsis . Plant Cell 23: 534–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M, Rutley N, Kagale S, Hamamura Y, Gherghinoiu M, Kumar S, Sari U, Esparza‐Franco MA, Sakamoto W, Rozwadowski K et al. 2014. An EAR‐dependent regulatory module promotes male germ cell division and sperm fertility in Arabidopsis . Plant Cell 26: 2098–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calarco J, Borges F, Donoghue M, Van Ex F, Jullien P, Lopes T, Gardner R, Berger F, Feijó JA, Becker JD et al. 2012. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151: 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Jacobsen SE. 2002. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Current Biology 12: 1138–1144. [DOI] [PubMed] [Google Scholar]
- Chen K, Wang Y, Zhang R, Zhang H, Gao C. 2019. CRISPR/Cas genome editing and precision plant breeding in agriculture. Annual Review of Plant Biology 70: 667–697. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Tan JL, Ingouff M, Sundaresan V, Berger F. 2008. CHROMATIN ASSEMBLY FACTOR 1 regulates the cell cycle but not cell fate during male gametogenesis in Arabidopsis thaliana . Development 135: 65–73. [DOI] [PubMed] [Google Scholar]
- Cheng C, Tarutani Y, Miyao A, Ito T, Yamazaki M, Sakai H, Fukai E, Hirochika H. 2015. Loss of function mutations in the rice chromomethylase OsCMT3a cause a burst of transposition. The Plant Journal 83: 1069–1081. [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. 1998. Floral dip: a simplified method for Agrobacterium‐mediated transformation of Arabidopsis thaliana . The Plant Journal 16: 735–743. [DOI] [PubMed] [Google Scholar]
- Durbarry A, Vizir I, Twell D. 2005. Male germ line development in Arabidopsis. duo pollen mutants reveal gametophytic regulators of generative cell cycle progression. Plant Physiology 137: 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esch JJ, Chen MA, Hillestad M, Marks MD. 2004. Comparison of TRY and the closely related At1g01380 gene in controlling Arabidopsis trichome patterning. The Plant Journal 40: 860–869. [DOI] [PubMed] [Google Scholar]
- Finnegan EJ, Kovac KA. 2000. Plant DNA methyltransferases. Plant Molecular Biology 43: 189–201. [DOI] [PubMed] [Google Scholar]
- García‐Muse T, Aguilera A. 2016. Transcription‐replication conflicts: how they occur and how they are resolved. Nature Reviews Molecular Cell Biology 17: 553–563. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M. 2016. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. 2017. Transcription‐replication conflict orientation modulates R‐loop levels and activates distinct DNA damage responses. Cell 170: 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Jacobsen SE. 2008. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non‐CG DNA methylation and initiate siRNA spreading. Genes & Development 22: 1597–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Li N, Xu C, Zhong S, Lin X, Yang J, Zhou T, Yuliang A, Wu Y, Chen YR et al. 2014. Mutation of a major CG methylase in rice causes genome‐wide hypomethylation, dysregulated genome expression, and seedling lethality. Proceedings of the National Academy of Sciences, USA 111: 10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Cools T, De Veylder L. 2016. Mechanisms used by plants to cope with DNA damage. Annual Review of Plant Biology 67: 439–462. [DOI] [PubMed] [Google Scholar]
- Jackson‐Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E et al. 2001. Loss of genomic methylation causes p53‐dependent apoptosis and epigenetic deregulation. Nature Genetics 27: 31–39. [DOI] [PubMed] [Google Scholar]
- Johnson LM, Law JA, Khattar A, Henderson IR, Jacobsen SE. 2008. SRA‐domain proteins required for DRM2‐mediated de novo DNA methylation. PLoS Genetics 4: e1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WC, Ordway AJ, Watada M, Pruitt JN, Williams TM, Rebeiz M. 2015. Genetic changes to a transcriptional silencer element confers phenotypic diversity within and between Drosophila species. PLoS Genetics 11: e1005279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullien PE, Susaki D, Yelagandula R, Higashiyama T, Berger F. 2012. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana . Current Biology 22: 1825–1830. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. 2004. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903. [DOI] [PubMed] [Google Scholar]
- Khouider S, Borges F, LeBlanc C, Ungru A, Schnittger A, Martienssen R, Colot V, Bouyer D. 2021. Male fertility in Arabidopsis requires active DNA demethylation of genes that control pollen tube function. Nature Communications 12: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Zhang H, Julian R, Tang K, Xie S, Zhu JK. 2015. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proceedings of the National Academy of Sciences, USA 112: 3553–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev Maor G, Yearim A, Ast G. 2015. The alternative role of DNA methylation in splicing regulation. Trends in Genetics 31: 274–280. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing S . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wu W, Zhao Y, Zheng B. 2017. A reciprocal inhibition between ARID1 and MET1 in male and female gametes in Arabidopsis . Journal of Integrative Plant Biology 59: 657–668. [DOI] [PubMed] [Google Scholar]
- Li Q, Eichten SR, Hermanson PJ, Zaunbrecher VM, Song J, Wendt J, Rosenbaum H, Madzima TF, Sloan AE, Huang J et al. 2014. Genetic perturbation of the maize methylome. Plant Cell 26: 4602–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Reinders J, Caikovski M, Smathajitt C, Paszkowski J. 2007. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130: 851–862. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA. 2014. RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nature Reviews Genetics 15: 394–408. [DOI] [PubMed] [Google Scholar]
- Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paszkowski J, Mathieu O. 2009. Selective epigenetic control of retrotransposition in Arabidopsis . Nature 461: 427–430. [DOI] [PubMed] [Google Scholar]
- Mori T, Kuroiwa H, Higashiyama T, Kuroiwa T. 2006. GENERATIVE CELL SPECIFIC 1 is essential for angiosperm fertilization. Nature Cell Biology 8: 64–71. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. 2005. Recruitment of DNA methyltransferase I to DNA repair sites. Proceedings of the National Academy of Sciences, USA 102: 8905–8909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257. [DOI] [PubMed] [Google Scholar]
- Osakabe A, Jamge B, Axelsson E, Montgomery SA, Akimcheva S, Kuehn AL, Pisupati R, Lorković ZJ, Yelagandula R, Kakutani T et al. 2021. The chromatin remodeler DDM1 prevents transposon mobility through deposition of histone variant H2A.W. Nature Cell Biology 23: 391–400. [DOI] [PubMed] [Google Scholar]
- Rotman N, Durbarry A, Wardle A, Yang WC, Chaboud A, Faure JE, Berger F, Twell D. 2005. A novel class of MYB factors controls sperm‐cell formation in plants. Current Biology 15: 244–248. [DOI] [PubMed] [Google Scholar]
- Saze H, Mittelsten Scheid O, Paszkowski J. 2003. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nature Genetics 34: 65–69. [DOI] [PubMed] [Google Scholar]
- Schoft VK, Chumak N, Choi Y, Hannon M, Garcia‐Aguilar M, Machlicova A, Slusarz L, Mosiolek M, Park JS, Park GT et al. 2011. Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proceedings of the National Academy of Sciences, USA 108: 8042–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. 2013. DNA methylation: roles in mammalian development. Nature Reviews Genetics 14: 204–220. [DOI] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. 2014. Non‐CG methylation patterns shape the epigenetic landscape in Arabidopsis . Nature Structural & Molecular Biology 21: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE. 2013. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T. 2009. Bursts of retrotransposition reproduced in Arabidopsis . Nature 461: 423–426. [DOI] [PubMed] [Google Scholar]
- Unterberger A, Andrews SD, Weaver IC, Szyf M. 2006. DNA METHYLTRANSFERASE 1 knockdown activates a replication stress checkpoint. Molecular and Cellular Biology 26: 7575–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J, Gao H, Zhang J, Aldridge B, Vickers M, Higgins JD, Feng X. 2018. Sexual‐lineage‐specific DNA methylation regulates meiosis in Arabidopsis . Nature Genetics 50: 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu Z. 2006. Arabidopsis ribonucleotide reductases are critical for cell cycle progression, DNA damage repair, and plant development. Plant Cell 18: 350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W. 2012. RSeQC: quality control of RNA‐seq experiments. Bioinformatics 28: 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang Z, Butel N, Santos‐Gonzalez J, Borges F, Yi J, Martienssen RA, Martinez G, Kohler C. 2020. Polymerase IV plays a crucial role in pollen development in Capsella . Plant Cell 32: 950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. 2014. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156: 45–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Custard KD, Brown RC, Lemmon BE, Harada JJ, Goldberg RB, Fischer RL. 2006. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18: 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi T, Johzuka‐Hisatomi Y, Terada R, Nakamura I, Iida S. 2014. The MET1b gene encoding a maintenance DNA methyltransferase is indispensable for normal development in rice. Plant Molecular Biology 85: 219–232. [DOI] [PubMed] [Google Scholar]
- Yan L, Wei S, Wu Y, Hu R, Li H, Yang W, Xie Q. 2015. High‐efficiency genome editing in Arabidopsis using YAO promoter‐driven CRISPR/Cas9 system. Molecular Plant 8: 1820–1823. [DOI] [PubMed] [Google Scholar]
- Yao Y, Bilichak A, Golubov A, Kovalchuk I. 2012. ddm1 plants are sensitive to methyl methane sulfonate and NaCl stresses and are deficient in DNA repair. Plant Cell Reports 31: 1549–1561. [DOI] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman‐Derr D, Eshed‐Williams L, Thao K, Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1‐containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhu JK. 2011. RNA‐directed DNA methylation. Current Opinion in Plant Biology 14: 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jacobsen SE. 2006. Genetic analyses of DNA methyltransferases in Arabidopsis thaliana . Cold Spring Harbor Symposia on Quantitative Biology 71: 439–447. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan S‐L, Chen H, Henderson IR, Shinn P, Pellegrini M, Jacobsen SE et al. 2006. Genome‐wide high‐resolution mapping and functional analysis of DNA methylation in Arabidopsis . Cell 126: 1189–1201. [DOI] [PubMed] [Google Scholar]
- Zhong X, Du J, Hale CJ, Gallego‐Bartolome J, Feng S, Vashisht AA, Chory J, Wohlschlegel JA, Patel DJ, Jacobsen SE. 2014. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 157: 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Generation of MET1 mutants in Arabidopsis using the CRISPR/Cas9 system.
Fig. S2 Loss of DNA methylation affects endosperm development and compromises transmission through the male gametophytes in Arabidopsis.
Fig. S3 Loss of DNA methylation does not affect pollen viability in Arabidopsis.
Fig. S4 Loss of DNA methylation in Arabidopsis leads to the downregulation of some genes involved in pollen development and double fertilisation in mature pollens.
Fig. S5 Loss of DNA methylation leads to global changes of gene expression in Arabidopsis seedlings.
Fig. S6 The upregulation of cell cycle‐, DNA replication‐ and DNA repair‐related genes in Arabidopsis ddcc met1‐11 +/− seedlings is not due to loss of DNA methylation in the promoters of these genes.
Fig. S7 The Arabidopsis ddcc met1‐11 +/− mutation specifically activates a group of TEs.
Fig. S8 CG and non‐CG DNA methylation synergistically regulate chromocenter condensation in Arabidopsis.
Table S1 Primers and sgRNAs used in this study.
Table S2 List of differentially expressed genes in the indicated mutants.
Table S3 List of differentially expressed TEs in the indicated mutants.
Table S4 GO analysis of differentially expressed genes in the indicated mutants.
Table S5 List of upregulated genes involved in DNA repair, DNA replication, and cell cycle control in met1‐11 and ddcc met1‐11 +/− .
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Methylome and transcriptome data have been submitted to the NCBI Sequencing Read Archive under accession no. PRJNA686693. The DNA methylome of Col‐0 and ddcc had been previously deposited in SRR1005412 and SRR1005415, respectively (Stroud et al., 2013, 2014). RNA‐seq data of ddm1 and the corresponding Col‐0 were previously deposited in GSE150436 (Osakabe et al., 2021).