

ABSTRACT
WDR45-related neurodevelopmental disorder (NDD) is a clinically-heterogenous congenital disorder of macroautophagy/autophagy. The natural history of this ultra-orphan disease remains incompletely understood, leading to delays in diagnosis and lack of quantifiable outcome measures. In this cross-sectional study, we model quantitative natural history data for WDR45-related NDD using a standardized analysis of 160 published cases, representing the largest cohort to date. The primary outcome of this study was survival. Age at disease onset, diagnostic delay and geographic distribution were quantified as secondary endpoints. Our tertiary aim was to explore and quantify the spectrum of WDR45-related phenotypes. Survival estimations showed low mortality until 39 years of age. Median age at onset was 10 months, with a median diagnostic delay of 6.2 years. Geographic distribution appeared worldwide with clusters in North America, East Asia, Western Europe and the Middle East. The clinical spectrum was highly variable with a bi-phasic evolution characterized by early-onset developmental and epileptic encephalopathy during childhood followed by a progressive dystonia-parkinsonism syndrome along with cognitive decline during early adulthood. Female individuals showed milder disease severity. The majority of pathogenic WDR45 variants were predicted to result in a loss of WDR45 expression, without clear genotype-phenotype associations. Our results provide clinical and epidemiological data that may facilitate an earlier diagnosis, enable anticipatory guidance and counseling of affected families and provide the foundation for endpoints for future interventional trials.
Abbreviations: BPAN: beta-propeller protein-associated neurodegeneration; CNS: central nervous system; DEE: developmental and epileptic encephalopathy; MRI: magnetic resonance imaging; NBIA: neurodegeneration with brain iron accumulation; NDD: neurodevelopmental disorder; NGS: next-generation sequencing; WDR45/WIPI4: WD repeat domain 45
KEYWORDS: Beta-propeller protein-associated neurodegeneration, BPAN, congenital disorder of autophagy, DEE, NBIA, NBIA5, NDD, WDR45, WIPI4
Introduction
Developmental and epileptic encephalopathy (DEE) and neurodegeneration with brain iron accumulation (NBIA) both comprise a heterogeneous group of monogenic disorders. NBIA type 5, also termed beta-propeller protein-associated neurodegeneration (BPAN), is a distinct, X-linked subtype within the NBIA spectrum and a congenital disorder of autophagy. Only 68 published cases and families are on record in the latest Orphanet Report Series on the Prevalence and incidence of rare diseases [1]. Underlying pathogenic variants in the WDR45/WIPI4 (WD repeat domain 45) gene, located on chromosome Xp11.23, were first described in 2012 in individuals with NBIA type 5 [2]. WDR45 holds a core function in the autophagy pathway, notably expansion and closure of the phagophore and autophagosome maturation [3–5]. The majority of published individuals with heterozygous de novo variants in WDR45 are female. Phenotypic variability has been observed for both sexes without supporting evidence for genotype-phenotype correlations [6]. Due to the nonspecific initial clinical presentation in childhood before the occurrence of typical neuroimaging findings, genetic testing for WDR45 variants is often delayed [7]. With regard to the growing availability of targeted therapies in pediatric neurology, early diagnosis and rapid initiation of specific treatments are crucial for therapeutic success. Quantitative natural history data for ultra-orphan diseases are necessary for raising disease awareness, facilitating early diagnosis, counseling of afflicted families and the design of future clinical trials.
Using quantitative retrospective natural history modeling [8], we here provide quantifiable natural history data for WDR45-related neurodevelopmental disorder (NDD). Survival was assessed as primary endpoint of this study. Age at disease onset, diagnostic delay and geographic distribution of reported cases were quantified as secondary endpoints. The tertiary aim of this study was to explore and quantify the spectrum of WDR45-related phenotypes using Human Phenotype Ontology-based phenotyping, with a focus on subgroup analyses for sex, age-groups and genetic background.
Results
Demographic data and definition of endpoints
A total of 160 individuals harboring 109 different WDR45 variants with a median age at last follow-up of 13 years (IQR 25.2) were identified from 87 published case reports and cohort studies (Table S4 and Figure S4). Demographic data are summarized in Table 1. Most WDR45 variants occurred de novo (n = 110/160). Reported variants included frameshift variants (n = 45/160), premature stop codons (n = 44/160), splicing variants (n = 33/160), missense variants (n = 18/160), small (n = 9/160) and larger (n = 4/160) deletions, a start loss variant (n = 1/160) and variants of unknown consequence (n = 2/160). Testing for mosaicism was reported in nine individuals confirming a mosaic state in three male and two female cases. Due to the small number of cases, subgroup analysis for these individuals was not possible in a meaningful way. Skewed X–inactivation in affected females accounted for 6/8 tested cases, with X–inactivation ratios toward the mutated allele ranging from 84 to 98%. Diagnosis of WDR45-related NDD was made by next-generation sequencing (NGS) in most cases (62.2% whole-exome sequencing, 2% whole-genome sequencing and 18.2% multigene panel), followed by Sanger sequencing (16.9%) and, in one case, array-CGH (0.7%).
Table 1.
Demographic data of the WDR45 cohort.
| Total | Male | Female | |
|---|---|---|---|
| Cases: n | 160* | 21 | 126 |
| Sex: M:F | 1:6 | ||
| Median age at data collection: Years | 13 (IQR = 26.5) | 7 (IQR = 23.4) | 14.2 (IQR = 27) |
| Median age at disease onset: Years | 0.8 (IQR = 1) | 0.3 (IQR = 0.3) | 1 (IQR = 1.1) |
| Median age at diagnosis: Years | 13 (IQR = 27.7) | 7 (IQR = 23.5) | 14 (IQR = 27) |
| Median diagnostic delay: Years | 6.2 (IQR = 19.5) | 2.7 (IQR = 4) | 7.5 (IQR = 20.9) |
| Median age at last follow-up: Year | 13.0 (IQR = 25.2) | 6.0 (IQR = 20.6) | 14.0 (IQR = 27.0) |
| Outcome: | |||
| Alive: n | 155 (96.9%) | 20 (100%) | 120 (96%) |
| Deceased: n | 5 (3.1%) | 0 | 5 (4%) |
| Genetic testing method (n = 148) | |||
| Whole-exome-sequencing: n | 92 (62.2%) | ||
| Panel diagnostics: n | 27 (18.2%) | ||
| Sanger sequencing: n | 25 (16.9%) | ||
| Whole-genome sequencing: n | 3 (2%) | ||
| array-CGH: n | 1 (0.7%) | ||
| Segregation | |||
| de novo: n | 110 (68.8%) | 12 (57.1%) | 87 (69%) |
| putative parental germline mosaic: n | 5 (3.1%) | 3 (14.3%) | 2 (1.6%) |
| NA: n | 46 (28.8%) | 7 (33.3%) | 37 (29.4%) |
| Variant type | |||
| Frameshift variant: n | 45 (28.1%) | 6 (28.6%) | 38 (30.2%) |
| Premature stop codon: n | 44 (27.5%) | 3 (14.3%) | 34 (27.0%) |
| Splicing variant: n | 33 (20.6%) | 6 (28.6%) | 26 (20.6%) |
| Missense variant: n | 18 (11.2%) | 2 (9.5%) | 14 (11.1%) |
| Small deletion: n | 9 (5.6%) | 3 (14.3%) | 6 (4.8%) |
| Gross deletion: n | 4 (2.5%) | 1 (4.8%) | 1 (0.8%) |
| Start loss: n | 1 (0.6%) | 0 | 1 (0.8%) |
| other: n | 2 (1.2%) | 0 | 2 (1.6%) |
| NA: n | 4 (2.5%) | 0 | 4 (3.2%) |
*no information on sex was available in 13 cases. CGH, comparative genomic hybridization; F, female; IQR, interquartile range; M, male; n, number; NA, not available.
The clinical endpoints assessed in this study, as well as the associated research questions and research strategies employed are summarized in Table 2.
Table 2.
Definition of clinical endpoints, associated research questions and research strategies employed in this study.
| Endpoint | Research Question/Hypothesis | Research strategy |
|---|---|---|
| Primary | Survival is affected in the WDR45 population | Kaplan-Meier method |
| Secondary | Time between onset of disease and diagnosis is delayed | Quantification of age at onset and diagnostic delay |
| Secondary | WDR45-related NDD is a disorder with worldwide distribution | Assessment of geographic distribution of individuals’ countries of origin |
| Tertiary | Diagnosis by clinical pattern recognition in infancy and childhood is challenging | Exploration of the phenotypic spectrum stratified by age groups using HPO-based phenotyping |
| Tertiary | WDR45-related NDD takes a characteristic bi-phasic disease course | Exploration of the phenotypic spectrum stratified by age groups using HPO-based phenotyping |
| Tertiary | Male sex is associated with more severe disease | Quantification of sex-specific symptom load |
| Exploration of the phenotypic spectrum stratified by sex using HPO-based phenotyping | ||
| Tertiary | Exploration of genotype-phenotype associations | Search for genetic clusters and hotspot mutational regions |
| Exploration of the phenotypic spectrum stratified by predicted mutational effects using HPO-based phenotyping |
HPO, Human Phenotype Ontology.
Individuals with WDR45 variants show low mortality within the reported follow-up period
Survival in the overall population was 96.9% (155/160) on last follow-up. All male individuals were alive at the time of publication with the oldest individual reported being 44 years old. In the female population, 95.8% were alive, while 4.2% were deceased. Probability of survival for females was estimated with 97.6% at the age of 27 years and 91.9% at 39 years (Figure 1). Probability estimations for survival above 39 years of age could not be assessed due to the low number of reported older cases.
Figure 1.
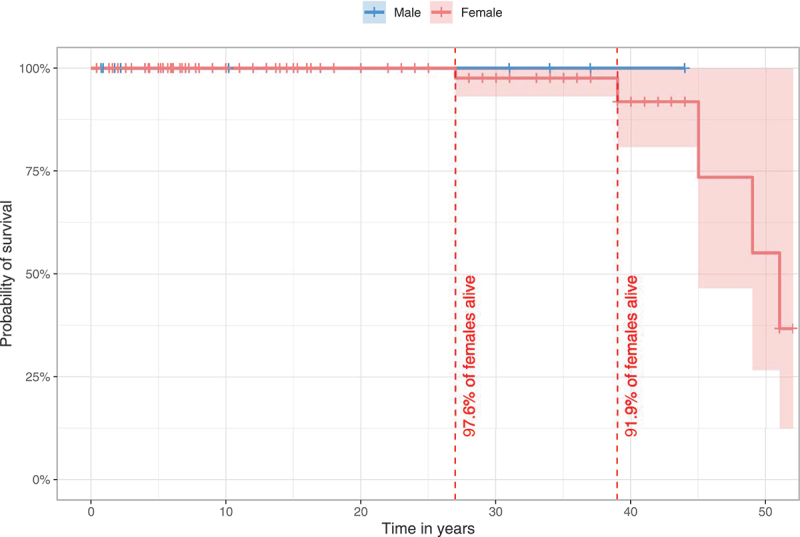
Estimated survival for WDR45-related NDD using the Kaplan-Meier method. 140/160 individuals (nmale = 20, nfemale = 120) were assessed for survival estimations. 20/160 individuals were excluded due to missing data for age at last follow-up (n = 7), sex (n = 3) or both (n = 10). Data were censored at the time of last follow-up if the individual was not reported as deceased. 95% confidence intervals are illustrated as shaded background. No significant differences were detected. Statistic testing was done using the log-rank test comparing sex.
Diagnosis of WDR45-related disease is significantly delayed
The median reported age at disease onset was within the first year of life, with a significantly earlier onset of first symptoms in males (4 months) compared to females (12 months) (Mann-Whitney U test, P ≤ 0.01). The median delay from onset of clinical signs to diagnosis was 6.2 years (IQR = 19.5) in the overall cohort, with males showing a significantly shorter diagnostic delay (2.7 years) than females (7.5 years) (Mann-Whitney U test, P ≤ 0.01) (Figure 2).
Figure 2.
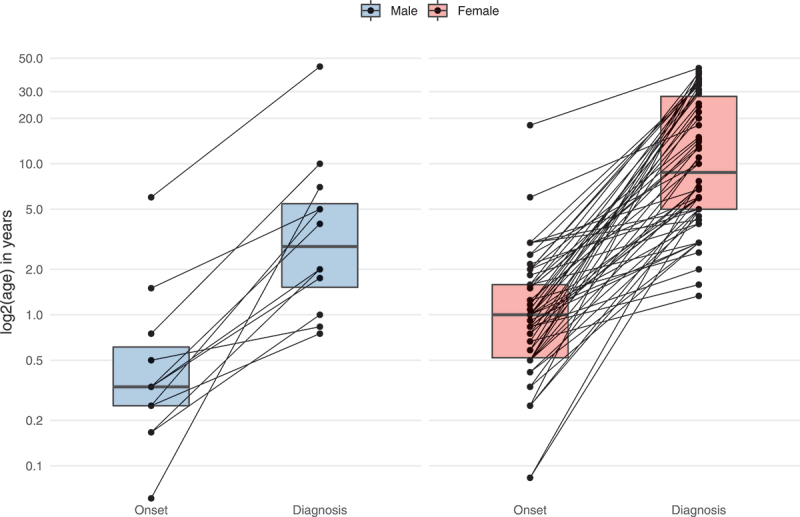
Age at onset and diagnostic delay of WDR45-related NDD. Data were assessed in 78/160 cases (nmale = 12, nfemale = 66). 82/160 individuals were excluded from analysis due to missing age (n = 69), sex (n = 1) or both (N = 12). Connecting lines represent data derived from the same individuals. Statistical testing was done using the Mann-Whitney U test.
Geographic origins of described cases involved North America, East Asia, Western Europe and, to a lesser extent, the Middle East (Figure S1).
WDR45-related NDD shows a variable and progressive CNS-specific phenotypic spectrum
Symptom burden, defined as the number of phenotypic features, represented by unique Human Phenotype Ontology terms per individual differed widely from 2 to 28, with a median number of 11 Human Phenotype Ontology terms for males (IQR = 6.75) and 10 for females (IQR = 7) in the infancy/childhood group (Figure 3A). With age, both males and females showed a progressive accumulation of clinical manifestations reaching a median symptom burden of 13 for males (IQR = 1) and 13 for females (IQR = 3.5). This increase over time was significant in the female group (Mann-Whitney U test, P ≤ 0.001). Differences in the male cohort did not reach significance.
Figure 3.
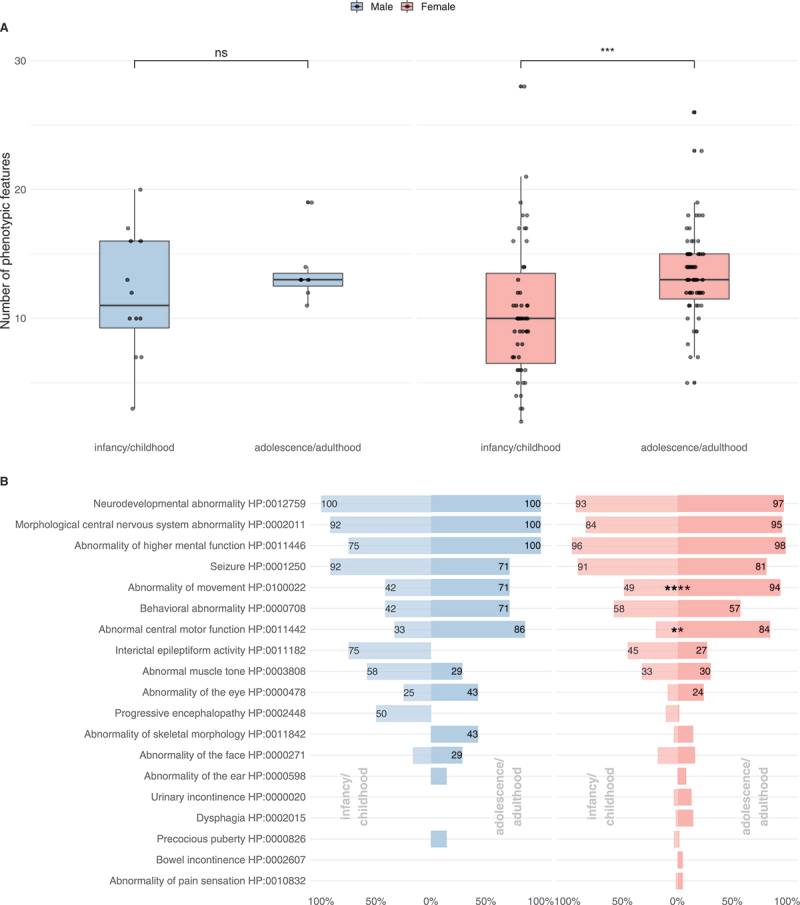
Subgroup analysis of the phenotypic spectrum of WDR45-related NDD. A total of 19 males (ninfancy/childhood = 12, nadolescence/adulthood = 7) and 118 females (ninfancy/childhood = 55, nadolescence/adulthood = 63) were examined. (A) Number of phenotypic features per individual with respect to 63 non-redundant Human Phenotype Ontology terms. Statistic testing was done using the Mann-Whitney U test. P values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg procedure. ns = not significant. (B) Frequencies of Human Phenotype Ontology ancestor terms derived phenotypic features. Frequencies greater than 20% were printed on the respective bars. Statistical testing was done using Pearson’s Chi-squared test or Fisher’s exact test (in case of less than 5 counts per subgroups). P values were adjusted for multiple hypothesis testing using the Benjamini-Hochberg procedure.
The spectrum of clinical manifestations represented by high-level Human Phenotype Ontology terms in individuals with WDR45-related NDD is shown in Figure 3B and Table S1. Both sexes showed a predominantly central nervous system (CNS) specific phenotypic spectrum. The most frequent features among all subgroups were neurodevelopmental abnormalities (mostly global developmental delay, and later, intellectual disability), morphological CNS abnormalities (in the majority of cases iron accumulation in the basal ganglia), abnormalities of higher cognitive function (such as expressive speech disorders) and seizures. Seizures occurred in 80.0% of cases and were equally distributed among males and females (Table S1). Slightly higher seizure frequencies were reported in the infancy/childhood group (91%) than in the adolescence/adulthood group (80%). A variety of seizure types were observed, including motor seizures (38%), febrile infection induced seizures (33%), generalized-onset seizures (24%), epileptic encephalopathy (18%) and focal-onset seizures (9%). In many cases, multiple seizure types existed. In infancy/childhood, the predominant seizure types for both genders were motor seizures (83% in males and 51% in females); infantile spasms, a subgroup of motor seizures, were present in 58% of males while only reported in 18% of females. Similarly, infancy/childhood onset epileptic encephalopathies as such were more frequent in males (67% vs. 27%). Along these lines, progressive encephalopathy was reported in 50% of males in comparison to 11% of females in the infancy/childhood group. By contrast, febrile infection induced seizures were comparatively more frequent in females both in infancy/childhood (38% vs. 25%) and in adolescence/adulthood (41% vs. 14%). For 49/128 individuals (38%) experiencing seizures interictal EEG abnormalities were reported while no interictal EEG abnormalities were found in two individuals (2%) with seizures; no information was available for the remaining cases. Treatments for WDR45-associated epilepsy included a variety of anti-seizure medications such as levetiracetam, valproic acid, lamotrigine, carbamazepine, clonazepam, clobazam and phenobarbital with no evidence supporting the use of a specific agent or sex-specific sensitivity to antiepileptic treatment. Out of 128 individuals with epilepsy, 23 were reported to be seizure free at the time of publication while 10 continued to experience seizures, no information on seizure outcome was available for the remaining individuals and no sex-dependent seizure outcomes were identified. Treatments for infantile spasms were anecdotally reported and included prednisolone, adrenocorticotropic hormone and the ketogenic diet.
While the overall frequency of seizures slightly decreased in adolescence/adulthood, movement disorders (abnormalities of movement and abnormal central motor function) became more prominent, in the female population reaching significance. Behavioral abnormalities were present in all subgroups, most prominent in males above 12 years of age.
WDR45-related NDD evolves from an early-onset developmental and epileptic encephalopathy to a later-onset neurodegenerative disorder
Initial symptoms were developmental delay (56.2%, n = 90/160), seizures (28.1%, n = 45/160), or both (1.2%, n = 2/160). Onset of developmental delay and seizures, particularly epileptic spasms, was usually within the first two years of life, with males experiencing a slightly earlier onset than females, yet without reaching significance (Table 3 and Figure 4A).
Table 3.
Early and late WDR45-related manifestations stratified by sex.
| Male | Female | |
|---|---|---|
| Initial symptom | ||
| Developmental delay: n | 15 (71.4%) | 75 (59.5%) |
| Seizures: n | 5 (23.8%) | 38 (30.2%) |
| Other: n | 2 (9.5%) | 9 (7.1%) |
| NA: n | 9 (7.1%) | |
| Median age at first report of developmental delay: Years | 0.3 (IQR = 0.3) | 1 (IQR = 1.8) |
| Median age at first report of seizures: Years | 0.5 (IQR = 1.1) | 1.3 (IQR = 1) |
| Median age at first report of epileptic spasms: Years | 0.4 (IQR = 0.2) | 0.9 (IQR = 0.9) |
| Median age at detection of brain iron accumulation: Years | 8.2 (IQR = 18.6) | 11 (IQR = 22.8) |
| Median age at first report of movement disorders: Years | 24 (IQR = 24) | 24 (IQR = 18.9) |
| Median age at first report of developmental regression: Years | 27 (IQR = 1) | 25.5 (IQR = 9.5) |
| Median age at first report of mental deterioration: Years | 26 (IQR = 6) | 26 (IQR = 8) |
Figure 4.
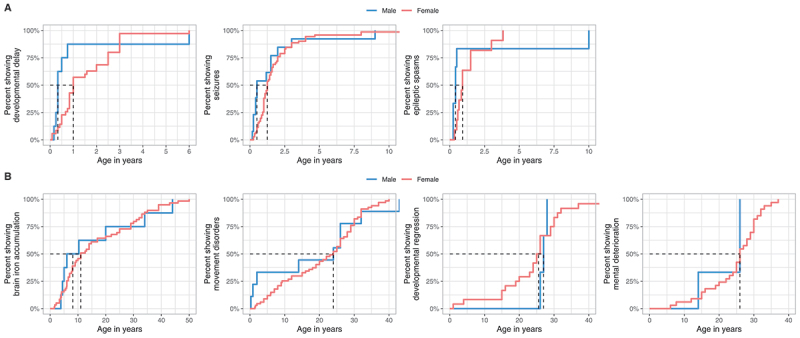
Ages at first report of early and late WDR45-related manifestations stratified by sex. (A) Age at first report of developmental delay (nmale = 8, nfemale = 35), epileptic seizures (nmale = 13, nfemale = 71) and epileptic spasms (nmale = 6, nfemale = 11) shown as time-dependent proportions. Age is depicted in years. The median age at onset is highlighted with dashed lines. (B) Age at first report of brain iron deposition (nmale = 8, nfemale = 59), movement disorders (nmale = 9, nfemale = 67), developmental regression (nmale = 3, nfemale = 24) and mental deterioration (nmale = 3, nfemale = 33) shown as time-dependent proportion. Age is depicted in years. The median age at onset is highlighted with dashed lines. No significant differences were detected between the two groups. Statistical testing was done using the Mann-Whitney U test.
Brain iron accumulation was detected in 99/160 cases at a median age of 11 years (IQR = 23), preceding the occurrence of extrapyramidal movement disorders for many years (Table 3 and Figure 4B). Median age at last normal brain MRI, available in 21/160 cases was 3 years (IQR = 2). Therapeutic approaches targeted at reducing brain iron accumulation with iron chelation therapy were reported in 2 cases (started after the onset of movement disorders) without clinical response.
Median onset of movement disorders was in the third decade for both males and females (Table 3 and Figure 4B). The predominant movement disorders were parkinsonism, dystonia, and gait disturbances, most of which were more pronounced in the female cohort. Tremors were more prevalent in males. Stereotypies were exclusively reported in females. Levodopa to alleviate extrapyramidal movement disorders was reported in 31 cases with beneficial effects in 51.6%. Developmental regression or cognitive decline, sometimes described as dementia, typically occurred during the mid-to-late 20s (Table 3 and Figure 4B).
The distribution of reported pathogenic variants along the chromosomal region of the WDR45 gene is shown in Figure 5A (for a complete list of reported variants including the respective frequencies in the cohort see Table S2). No clear mutational hotspot regions could be identified.
Figure 5.
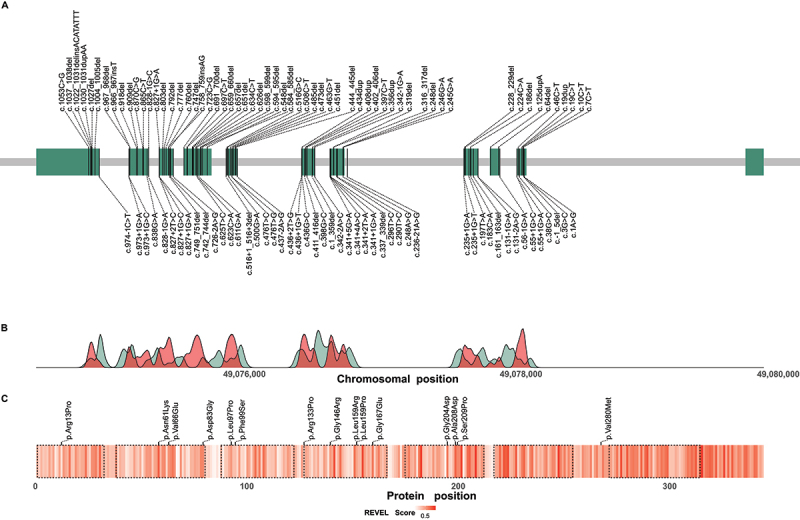
Distribution of reported disease-causing variants in WDR45. (A) Chromosomal region of WDR45 including 11 exons illustrated as green rectangles. Chromosomal positions of disease-causing variants are indicated by vertical lines. Variants resulting in frameshifts and premature stop codons are plotted above and the remaining variants below the gene. (B) Densities of pathogenic WDR45 variants from the literature (red) and benign variants from the gnomAD genomes database (green) according to their chromosomal position. Densities show mainly reciprocal distributions suggesting little tolerance in the healthy population toward variation in regions of pathogenic variant enrichment. (C) Heat map visualization of variant effect prediction score values for all possible missense variants according to their position of amino acid exchange within the WDR45 protein sequence. Arithmetic means of REVEL score values at any protein position are color-coded. WD repeat domains are highlighted by dashed lines. Reported WDR45 missense variants are shown as black ticks. Accumulation of high score values in the C-terminal section of WDR45 suggest a functional relevance of this region with low tolerance to missense variation.
The phenotypic spectrum of WDR45-related NDD stratified by predicted variant consequence (“Protein loss” vs. “Residual protein function”) is shown in Table 4 and Figure 6. Neither the frequency of single phenotypic features nor symptom burden were different between the two groups. Variants predicted to lead to a loss of WDR45 expression were associated with an earlier median onset of developmental delay and epileptic spasms, whereas movement disorders seemed to occur later (Table 4). None of these differences reached significance.
Table 4.
Demographic data of the WDR45 cohort stratified by predicted mutational effect.
| Protein loss | Residual protein function | |
|---|---|---|
| Cases: n | 129 | 18 |
| Sex: M:F | 1:9 | 1:7 |
| Initial symptom | ||
| Developmental delay: n | 74 (57.4%) | 7 (38.9%) |
| Seizures: n | 36 (27.9%) | 7 (38.9%) |
| Other: n | 10 (7.8%) | 3 (16.7%) |
| NA: n | 13 (10.1%) | 2 (11.1%) |
| Median age at disease onset: Years | 0.8 (IQR = 1.1) | 1 (IQR = 1.2) |
| Median age at first report of developmental delay: Years | 0.8 (IQR = 1.6) | 2.5 (IQR = 1.5) |
| Median age at first report of seizures: Years | 1.2 (IQR = 1.0) | 1.5 (IQR = 1.2) |
| Median age at first report of epileptic spasms: Years | 0.6 (IQR = 0.5) | 1.2 (IQR = 0.3) |
| Median age at detection of brain iron accumulation: Years | 11 (IQR = 23.1) | 12.6 (IQR = 11.5) |
| Median age at first report of movement disorders: Years | 24 (IQR = 18.9) | 15 (IQR = 23) |
| Median age at first report of developmental regression: Years | 26 (IQR = 6.0) | 21 (IQR = 13.8) |
| Median age at first report of mental deterioration: Years | 26 (IQR = 6.5) | 21.5 (IQR = 16) |
| Median symptom load (phenotypic features per individual): n | 12 (IQR = 5) | 12 (IQR = 5) |
F, female; IQR, interquartile range; M, male; n, number; NA, not available.
Figure 6.
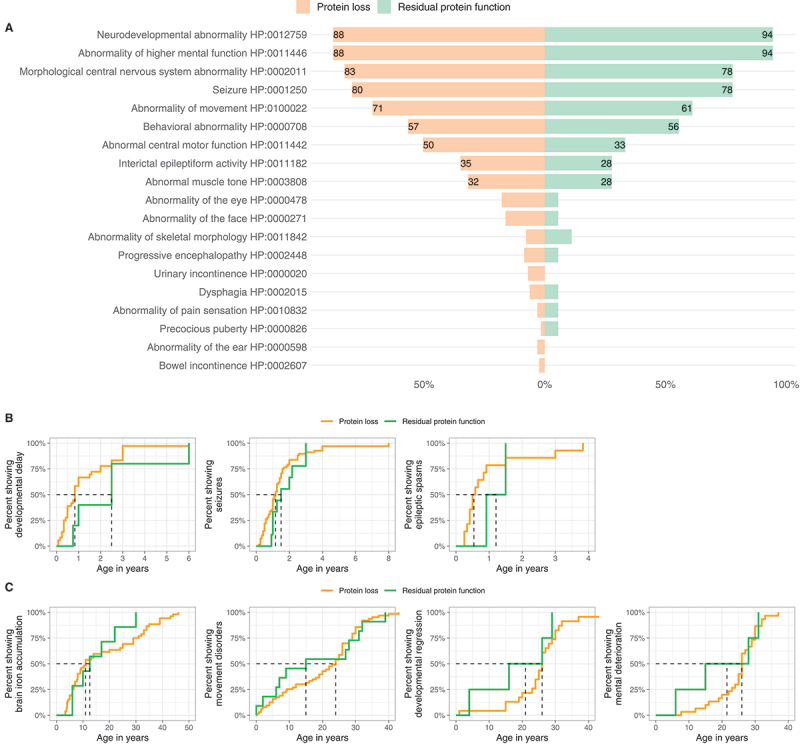
Genotype-phenotype association analysis of WDR45-related NDD. (A) Frequencies of reported Human Phenotype Ontology ancestor terms derived phenotypic features stratified for predicted variant effects (nProtein loss = 129/160, nResidual protein function = 18/116). Frequencies greater than 20% are printed on the respective bars. No significant differences were detected. Statistical testing was done using Pearson’s Chi-squared test or Fisher’s exact test (in case of less than 5 counts per subgroup). (B and C) Age at first report of early and late manifestations stratified by predicted mutational effects. (B). First report of ndevelopmental delay (nProtein loss = 30, nResidual protein function = 4), epileptic seizures (nProtein loss = 68, nResidual protein function = 9) and epileptic spasms (nProtein loss = 14, nResidual protein function = 2). (C). First report of brain iron deposition (nProtein loss = 52, nResidual protein function = 7), movement disorders (nProtein loss = 63, nResidual protein function = 11), developmental regression (nProtein loss = 23, nResidual protein function = 4) and mental deterioration (nProtein loss = 30, nResidual protein function = 4). Age is depicted in years. The median age at presence of specific symptoms is marked by dashed lines. No significant differences were detected. Statistic testing was done using the Mann-Whitney U test.
Splicing and missense variants appeared to be enriched at the C terminus of WDR45, a region that generally yielded higher variant effect prediction scores in our prediction model than the N-terminal sections of the protein (Figures 5 and S2). Accordingly, the variants c.436G>C; p.(Gly146Arg) and c.623C>A; p.(Ala208Asp) yielded the highest scores among all reported missense variants and were rated as the most damaging by 6/10 and 2/10 variant effect prediction scores, respectively, whereas the least damaging effects were predicted for the variants c.38G>C; p.(Arg13Pro), c.398G>C; p.(Arg133Pro), and c.838G>A; p.(Val280Met) (Figure S2). No phenotypic differences between individuals harboring these predicted “most damaging” and “least damaging” missense variants were detected (data not shown).
Discussion
Using a systematic cross-sectional analysis of 160 published cases, we provide quantitative data delineating the natural history of WDR45-related neurodevelopmental disorder (NDD).
As primary endpoint of this study we quantified survival, allowing predictions on life-expectancy in the WDR45 cohort. Overall mortality was low with 96.9% of individuals being alive at last follow-up. All deaths occurred in the female population with a low estimated mortality rate of 8.1% up to the age of 39 years.
As secondary endpoints, we assessed age at disease onset, diagnostic delay and geographic distribution. We showed that WDR45-related NDD typically starts in infancy or early childhood as a developmental and epileptic encephalopathy (DEE) with significantly earlier presentation and shorter time to diagnosis in male individuals, likely reflecting a more severe course at onset and more rapid progression. These findings support previous observations and may be explained by the X-linked inheritance pattern of the disease [2,7]. Median diagnostic delay in the overall population was 6.2 years. This significant diagnostic delay is likely due to the nonspecific clinical manifestations in infancy and childhood. Our study, together with previous reports, argues for the inclusion of WDR45-related NDD into the phenotypic group of DEE, which reflects the typical presentation in nearly all affected infants [9–11]. WDR45 is increasingly important in the differential diagnosis of early-onset epilepsy. Due to the broad range of differential diagnoses in this age group, we recommend the use of genome-wide screening assays, which is justified and appropriate to identify WDR45-related DEE before the occurrence of specific MRI changes. Early identification of WDR45 variants in individuals with DEE provides opportunities for anticipatory guidance and inclusion in interventional trials at an early stage before significant neurodegeneration has occurred.
With increasing and cost-effective access to genome/exome-wide sequencing expected to lead to the identification of more individuals with WDR45 variants, the standardized collection of phenotypic information becomes even more important. This can facilitate the interpretation of novel variants and builds a platform for prospective natural history studies. Unfortunately, phenotypic data on this ultra-rare disease are thus far scattered among a small number of case reports and case series using mostly non-standardized terms for disease-specific clinical features. To address this issue, we translated available clinical information into standardized Human Phenotype Ontology terminology providing a unique and novel data set on WDR45-related phenotypic features (Figure 3 and Table S1), which will facilitate further research and serve as powerful phenotype-driven genomic-diagnostic tool to prioritize genes in exome-sequencing approaches [12,13].
Using Human Phenotype Ontology terminology, we delineated the phenotypic spectrum of WDR45-related DEE as tertiary endpoint. Both sexes showed a broad range of phenotypic features, almost exclusively involving the central nervous system. An increase in the number of clinical manifestations was observed with increasing age as a surrogate of disease progression. This increase in symptom burden over time was less pronounced in males, suggesting a possibly more continuous disease course, in contrast to the rather bi-phasic evolution in females (Figure 3A).
Based on our findings, we defined a core set of clinical manifestations in WDR45-related DEE. In particular, we report early-onset global developmental delay and a spectrum of early-onset epilepsy as first clinical signs, presenting around the first birthday in females and during the first 6 months of life in males (Figure 4). In the majority of cases intellectual disability was in the severe to profound range, usually accompanied by a prominent expressive speech disorder or entirely absent speech (Figure 3).
Seizure disorders are not a prominent feature of most forms of NBIA. Our study, in line with previous reports, confirms that WDR45-related DEE is phenotypically distinct as 80% of reported cases experienced seizures, in 28.1% as an initial clinical manifestation (Figure 3B). Males seem to be more susceptible to epileptic encephalopathy underscored by the reported frequency of infantile spasms and other epileptic encephalopathies. These findings support the observation that seizure disorders are a core clinical feature of WDR45-related DEE. Cortical neurons and particularly highly metabolic active synaptic networks likely have an age-dependent selective vulnerability to disruptions in neuronal autophagy and mitochondrial dysfunction secondary to a loss of WDR45 [14,15].
Brain iron accumulation is the hallmark feature of NBIA. Iron deposits in the globus pallidus and substantia nigra in WDR45-related DEE were first detected on cerebral MRI in late childhood, usually preceding extrapyramidal motor dysfunction and cognitive decline. Based on the available data, no correlations between age at disease onset/duration of disease and age at first detection of brain iron MRI could be identified. However, data might be biased by the limited use of iron-sensitive MRI sequences which can accelerate diagnosis [16]. The exact molecular mechanisms leading to brain iron accumulation in NBIA are not entirely understood and diverse cellular pathways are associated with distinct NBIA syndromes. Emerging evidence suggests that primary or secondary metabolic defects converge in common downstream lysosomal and mitochondrial dysfunction, resulting in excessive reactive oxygen species production and upregulation of cellular iron uptake (reviewed in detail by Wildeman et al. [17]). Furthermore, excessive free iron in the basal ganglia has been shown to drive the formation of highly reactive oxygen species, causing localized toxicity, likely explaining the extrapyramidal movement disorders in the NBIA [18,19].
Iron chelation has been proposed as a treatment approach to decrease brain iron concentration. Indeed, results form a large, randomized trial using iron chelation therapy with deferiprone in individuals with pantothenate kinase-associated neurodegeneration (PKAN), the most common form of NBIA, showed a reduction of excess iron in the brain, however, without general clinical improvement [20,21]. In WDR45-related DEE, iron chelation therapy has been reported in two cases only and without clinical benefit. In both cases treatment was initiated in late stages of the disease (with 29 and 36 years of age) after the onset of an extrapyramidal movement disorder, limiting conclusions from these reports [22,23]. The estimated natural history data provided here can serve as valuable control group for future interventional trials in WDR45-related NDD, especially after early identification of WDR45 variants in individuals with DEE.
Movement disorders usually occurred in the second decade of life, characterized by a disabling dystonia-parkinsonism syndrome and progressive gait disturbances. While the progression of extrapyramidal symptoms was significant in females, no statistically significant increase in secondary motor decline was obvious in male individuals (Figure 3B). In some individuals therapy with levodopa led to improvement of movement disorders.
Thus, our findings confirm the bi-phasic evolution of WDR45-associated NDD, previously also referred to by the term static encephalopathy of childhood with neurodegeneration in adulthood/SENDA, reflecting the typical age-dependent disease progression. In females many of these age-dependent differences reached significance while in males no significant differences supporting a clear bi-phasic evolution of the disease could be detected (Figures 3, S3 and Table S3). These results have to be interpreted with caution as comparisons of the two sexes are statistically difficult due to the low frequency of reported males.
Most pathogenic WDR45 variants occurred de novo, although parental somatic mosaicism has been reported to account for familial recurrence [24,25]. Overall, 109 distinct disease-causing variants have been reported thus far. Most variants were predicted to lead to a loss of WDR45. We were unable to identify genotype-phenotype associations for different genetic variants, in line with previous studies [6]. Only mild trends were observed. Carriers of protein-truncating variants had a slightly earlier onset of symptoms (developmental delay and epileptic spasms) while individuals carrying missense variants with possible residual protein function showed a slightly earlier onset of movement disorders, yet without reaching significance. Genotype-phenotype analyses could, however, be biased by the assumption that missense variants might retain residual protein function. In the future, missense variants might be reclassified to result in a complete protein loss depending on the availability of functional genomic data.
Although, the distribution of benign variants in the healthy population compared to putative disrupting variants suggests specific regions of importance in WDR45 (notably the C-terminal region), no mutational hotspots were identified (Figure 5).
Pathogenic WDR45 variants in females are assumed to lead to haploinsufficiency [3]. While males with pathogenic germline WDR45 variants were thought to be non-viable, single male individuals have been reported and were included into our analyses [24–26]. A gene-dosage effect for males has been suggested from families with affected siblings of both sexes, where females showed milder phenotypes, suggesting at least some residual function of the seemingly silenced wild-type allele in females with damaging WDR45 variants [24,25]. In line with these observations, our results provide evidence for the hypothesis that males with WDR45-related DEE have more severe clinical manifestations underlined by an earlier onset of clinical signs, higher frequencies of epileptic encephalopathy, and likely more rapid disease progression. These findings are corroborated by wdr45 knockout mice that show increased susceptibility to induced seizures [4].
Future therapeutic interventions for WDR45-related DEE might aim to modulate the autophagy pathway. Autophagy is a key cellular quality control mechanism that removes misfolded proteins and damaged organelles, holding a crucial function in maintaining neural and axonal health. A role in different neurodegenerative diseases such as Alzheimer disease [27], Parkinson disease [28], amyotrophic lateral sclerosis [29] and Huntington disease [30], as well as in WDR45-related DEE [5] has been delineated. Activation of autophagy with the mTOR inhibitor rapamycin in WDR45-deficient cells was reported to reduce cell death, suggesting MTOR inhibition as an interesting mechanism for the treatment of WDR45-related DEE [5]. Rapamycin and its derivatives have been used in infants and children and can alleviate epilepsy and other manifestations in tuberous sclerosis, another monogenic disorder with impaired autophagy [31,32]. Further therapeutic approaches including suppression of endoplasmic reticulum stress using tauroursodeoxycholic acid/TUDCA or enhancing lysosomal pathways by inhibition of SNAP29 O-GlcNAcylation via small interfering RNA have been reported [5]. Standardized clinical data on the natural history of WDR45-related NDD will help in rating effects of future therapeutic concepts.
Study limitations
This study, due to its design, has several limitations. Analysis of geographic distribution was likely biased by the availability of molecular testing, thus underestimating the prevalence of WDR45-related NDD in less developed regions. Phenotyping of affected individuals was subject to ascertainment bias and missing data. Individuals harboring WDR45 variants were often identified in the context of large cohort studies that focused on a specific phenotype, i.e. cohorts of individuals with intellectual disability or epileptic encephalopathy. In addition, respective reports only captured manifestations that were present at time of last follow-up. For instance, in infants and children, late manifestations might have been missing. Conversely, in older individuals, a history of early manifestations, such as developmental delay and seizures was often not assessed, leading to underestimated frequencies of both early and late manifestations. Along these lines, extra-neurologic manifestations were rarely reported, highlighting the need for unbiased comprehensive phenotyping of individuals harboring variants in WDR45, including peripheral and non-neurologic phenotypes.
The lack of standardized data collection and phenotyping among publications biases assessment of descriptive variables. Accordingly, we mainly focused on quantifiable clinical endpoints such as survival, disease onset and diagnostic delay. Survival estimations are based upon small sample sizes without consideration of possible confounders such as access to supportive care. As WDR45-related NDD is rare, prospective natural history studies will require a multicenter design. Quantitative natural history modeling, as presented here, may serve as a substitute until prospective studies can be completed.
Conclusions and directions for future research
In this study, we quantitatively assessed clinically relevant endpoints including survival, disease onset, diagnostic delay and geographic distribution of WDR45-related DEE. We explored the phenotypic disease spectrum based on a systematic analysis of published cases. Our results provide clinical and epidemiological data that are of interest to future interventional trials. We define a set of core clinical features that holds the potential to facilitate a diagnosis and interpretation of variants of unknown significance. Our results raise awareness for this ultra-rare disease, broaden the understanding of the clinical course and phenotypic spectrum of WDR45-related NDD, and allow better counseling of affected families.
Materials and methods
This study was designed, executed and reported in accordance with STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) criteria [33].
Literature review and definition of variables
To conduct a specific mutation-based and phenotypically unbiased literature search, we applied a conservative search strategy using the keyword “WDR45” to screen for PubMed-listed publications and entries of the Human Gene Mutation Database (HGMD). A detailed summary of the search strategy is provided in Table S4 and Figure S4. Data acquisition was completed on 12/08/2020. Epidemiological information assessed for primary and secondary endpoints included: outcome (alive or deceased), age at last follow-up, age at disease onset, age at diagnosis, sex and individuals’ country of origin. In case “country of origin” was not explicitly stated in the report, the country of the patient’s origin was attributed to the country of the first author’s institutional affiliation in the respective case description. If “age at diagnosis” or “age at last follow-up” were not explicitly stated, they were attributed to the patient’s age at data collection in the original report. Semi-quantitative age descriptions, such as “postnatal”, “postpartal” and “congenital” were defined as age 0 years. Individuals were considered alive at the time of the report if not explicitly stated otherwise.
Annotation of Human Phenotype Ontology terms
Phenotypic information was manually extracted from original reports and translated into standardized Human Phenotype Ontology vocabulary (HPO version 1.7.3; release 2020/10/12). A total of 90 unique Human Phenotype Ontology terms were generated (for a complete list of the generated Human Phenotype Ontology terms and frequencies of phenotypic features see Table S1). Disease severity was assessed by quantifying the number of 63 unique and specific Human Phenotype Ontology terms assigned per individual (symptom load). The spectrum of phenotypic features associated to WDR45-related NDD was quantified using high-level Human Phenotype Ontology terms. For a detailed description of the generation of Human Phenotype Ontology terms for the respective analyses see Supplemental Materials and Methods.
Visualization of pathogenic WDR45 variants
To visualize differences in the distribution of reported variants between affected individuals and the healthy population, all pathogenic WDR45 variants listed in the databases PubMed and the HGMD as well as all benign variants listed in the genome aggregation database (gnomAD) were collected [34].
Statistical analysis
Techniques of descriptive statistics were applied as previously reported [8]. Baseline patient demographics were summarized descriptively using patient counts and percentages of the total study population or respective subgroups, where necessary. Survival was assessed and defined as the time interval between patient birth and death. Patient data were censored at the time of last follow-up if the patient was not reported as deceased. Survival was estimated using the Kaplan–Meier method. Diagnostic delay was calculated as the time interval between age at disease onset and age at diagnosis. All analyses were performed using R version 4.0.4 (2021–02-15) and RStudio (version 1.4.1103; RStudio, Inc.). The world map was plotted using the R package “rworldmap” [35]. Missing data were not imputed. Sensitivity analyses were not conducted. Distribution of data was tested for normality by visualization with histograms and normality testing using the Shapiro–Wilk test. Since data did not follow a normal distribution, the statistic tests employed included the Mann–Whitney U test for nonparametric distributions, the Pearson’s Chi-squared test or Fisher’s exact test (in case of less than 5 counts per subgroups) for testing frequency distributions and the log-rank test for survival rates. All P values are explorative. P values reported are two-sided and adjusted for multiple hypothesis testing using the Benjamini-Hochberg procedure.
Given the X-linked inheritance and reports on males with more severe disease [2,7], subgroup analyses were done for female and male individuals separately. Moreover, considering the bi-phasic disease course, the cohort was stratified into an “infancy/childhood” group (0–12 years) and an “adolescence/adulthood” group (> 12 years), in accordance with the proposed classification of age groups by the 2017 policy statement of the American Academy of Pediatrics [36]. For genotype-phenotype association analysis, the cohort was stratified based on predicted variant consequences into a “Protein loss” and a “Residual protein function” group. All frameshift, premature stop codon and splicing variants as well as small and gross deletions were assumed to lead to a protein loss, resulting in haploinsufficiency in females and a complete loss of WDR45 in males. For missense variants without available functional data, a putative residual protein function was assumed, and pathogenicity rated using in silico prediction. In case functional data were available, variants were reclassified; for instance, the synonymous variant c.516G>C was reclassified to the “Protein loss” group after personal correspondence with the authors of the original publication confirming that this variant was demonstrated by RT-PCR to result in a 22 base pair retention of intronic sequences leading to a frameshift (p.Asp174Valfs*29). Individuals with missing information (n = 4) were excluded. Similarly, variants for which, in the absence of functional data, predictions about functional consequences were not possible, were excluded from genotype-phenotype analysis. These included in-frame deletions (n = 4), an in-frame insertion and a start loss variant, that might, with equal possibility, exert little to no effect on protein function or lead to nonsense-mediated decay resulting in loss of protein function. Furthermore, for specific subgroup analyses, individuals without quantifiable information were excluded. Sample sizes are indicated (n) for each corresponding analysis.
Supplementary Material
Acknowledgments
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SA 4171/1-1 (to AS) and the Dietmar Hopp Stiftung –1DH1813319 (to SS).
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [SA 4171/1-1]; Dietmar Hopp Stiftung [1DH1813319].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Prevalence of rare diseases: bibliographic data [Internet]. Orphanet Report Series, Rare Diseases collection, January 2021, Number 1: Diseases listed in alphabetical order; 2021. Available from: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Accessed 9 Mar 2021.
- [2].Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, x-linked dominant form of NBIA. Am J Hum Genet [Internet]. 2012 Dec 7;91(6):1144–1149. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23176820. Accessed 4 Dec 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet [Internet]. 2013;45(4):445–449,449e1. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23435086. Accessed 4 Dec 2020. [DOI] [PubMed] [Google Scholar]
- [4].Wan H, Wang Q, Chen X, et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy [Internet]. 2020 Mar;16(3):531–547. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31204559. Accessed 17 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ji C, Zhao H, Chen D, et al. Beta-propeller proteins WDR45 and WDR45B regulate autophagosome maturation into autolysosomes in neural cells. Curr Biol [Internet]. 2021 Feb;31(8)1666–1677.e6. Available from: https://www.ncbi.nlm.nih.gov/pubmed/33636118. Accessed 1 Mar 2021. [DOI] [PubMed] [Google Scholar]
- [6].Cong Y, So V, Tijssen MAJ, et al. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy [Internet]. 2021. Apr 12:1–16. doi: 10.1080/15548627.2021.1899669. Accessed 15 Apr 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Adang LA, Pizzino A, Malhotra A, et al. Phenotypic and imaging spectrum associated with WDR45. Pediatr Neurol [Internet]. 2020 Aug;109:56–62. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32387008. Accessed 4 Dec 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Garbade SF, Zielonka M, Komatsuzaki S, et al. Quantitative retrospective natural history modeling for orphan drug development. J Inherit Metab Dis [Internet]. 2020 Aug 26. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32845020. Accessed 4 Dec 2020 [DOI] [PubMed] [Google Scholar]
- [9].Hamdan FF, Srour M, Capo-Chichi JM, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet [Internet]. 2014 Oct;10(10):e1004772. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25356899. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carvill GL, Liu A, Mandelstam S, et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia [Internet]. 2018 Jan;59(1):e5–e13. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29171013. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Krey I, Krois-Neudenberger J, Hentschel J, et al. Genotype-phenotype correlation on 45 individuals with west syndrome. Eur J Paediatr Neurol [Internet]. 2020 Mar;25:134–138. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31791873. Accessed 23 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [12].Kohler S, Carmody L, Vasilevsky N, et al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res [Internet]. 2019 Jan 8;47(D1):D1018–D1027. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30476213. Accessed 7 Dec 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Robinson PN, Kohler S, Bauer S, et al. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet [Internet]. 2008 Nov;83(5):610–615. Available from: https://www.ncbi.nlm.nih.gov/pubmed/18950739. Accessed 7 Dec 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Seibler P, Burbulla LF, Dulovic M, et al. Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain [Internet]. 2018 Oct 1;141(10):3052–3064. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30169597. Accessed 17 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain [Internet]. 2016 Feb;139(2):317–337. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26715604. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chard M, Appendino JP, Bello-Espinosa LE, et al. Single-center experience with beta-propeller protein-associated neurodegeneration (BPAN); expanding the phenotypic spectrum. Mol Genet Metab Rep [Internet]. 2019 Sep;20:100483. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31293896. Accessed 3 Mar 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hinarejos I, Machuca-Arellano C, Sancho P, et al. Mitochondrial dysfunction, oxidative stress and neuroinflammation in neurodegeneration with brain iron accumulation (NBIA). Antioxidants (Basel) [Internet]. 2020 Oct 20;9(10). Available from: https://www.ncbi.nlm.nih.gov/pubmed/33092153. Accessed 17 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hayflick SJ, Hogarth P.. As iron goes, so goes disease? Haematologica [Internet]. 2011 Nov;96(11):1571–1572. Available from: https://www.ncbi.nlm.nih.gov/pubmed/22058278. Accessed 15 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci [Internet]. 2013 Aug;14(8):551–564. Available from: https://www.ncbi.nlm.nih.gov/pubmed/23820773. Accessed 17 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [20].Huebl J, Schneider SA. Iron chelation in pantothenate kinase-associated neurodegeneration: a possible new avenue for slowing down disease progression in neurodegeneration. Mov Disord [Internet]. 2019 Oct;34(10):1476–1477. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31483532. Accessed 15 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [21].Klopstock T, Tricta F, Neumayr L, et al. Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: a randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol [Internet]. 2019 Jul;18(7):631–642. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31202468. Accessed 15 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [22].Lim SY, Tan AH, Ahmad-Annuar A, et al. A patient with beta-propeller protein-associated neurodegeneration: treatment with iron chelation therapy. J Mov Disord [Internet]. 2018 May;11(2):89–92. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29860786. Accessed 2 Mar 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fonderico M, Laudisi M, Andreasi NG, et al. Patient affected by beta-propeller protein-associated neurodegeneration: a therapeutic attempt with iron chelation therapy. Front Neurol [Internet]. 2017;8:385. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28878728. Accessed 15 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zarate YA, Jones JR, Jones Ma, et al. Lessons from a pair of siblings with BPAN. Eur J Hum Genet [Internet]. 2016 Jul;24(7):1080–1083. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26577041. Accessed 5 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nakashima M, Takano K, Tsuyusaki Y, et al. WDR45 mutations in three male patients with west syndrome. J Hum Genet [Internet]. 2016 Jul;61(7):653–661. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27030146. Accessed 5 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [26].Abidi A, Mignon-Ravix C, Cacciagli P, et al. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur J Hum Genet [Internet]. 2016 Apr;24(4):615–618. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26173968. Accessed 4 Dec 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci [Internet]. 2017 Mar;40(3):151–166. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28190529. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Karabiyik C, Lee MJ, Rubinsztein DC. Autophagy impairment in Parkinson’s disease. Essays Biochem [Internet]. 2017 Dec 12;61(6):711–720. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29233880. Accessed 23 Feb 2021. [DOI] [PubMed] [Google Scholar]
- [29].Ramesh N, Pandey UB. Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci [Internet]. 2017;10:263. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28878620. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kaliszewski M, Knott AB, Bossy-Wetzel E. Primary cilia and autophagic dysfunction in huntington’s disease. Cell Death Differ [Internet]. 2015 Sep;22(9):1413–1424. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26160070. Accessed 23 Feb 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Impaired mitochondrial dynamics and mitophagy in neuronal models of tuberous sclerosis complex. Cell Rep [Internet]. 2016 Oct 18;17(4):1053–1070. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27760312. Accessed 22 Apr 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saffari A, Brosse I, Wiemer-Kruel A, et al. Safety and efficacy of mTOR inhibitor treatment in patients with tuberous sclerosis complex under 2 years of age - a multicenter retrospective study. Orphanet J Rare Dis [Internet]. 2019 May 3;14(1):96. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31053163. Accessed 22 Apr 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].von Elm E, Altman DG, Egger M, et al. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet [Internet]. 2007 Oct 20;370(9596):1453–1457. Available from: https://www.ncbi.nlm.nih.gov/pubmed/18064739. Accessed 7 Dec 2020. [DOI] [PubMed] [Google Scholar]
- [34].Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature [Internet]. 2020 May;581(7809):434–443. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32461654. Accessed 4 Mar 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].South A. Rworldmap: a new r package for mapping global data. R J [Internet]. 2011;3(1):35–43. Available from: https://journal.r-project.org/archive/2011-1/RJournal_2011-1_South.pdf. Accessed 20 Jan 2021. [Google Scholar]
- [36].Hardin AP, Hackell JM, Committee On P, et al. Age limit of pediatrics. Pediatrics [Internet]. 2017 Sep:140(3). Available from: https://www.ncbi.nlm.nih.gov/pubmed/28827380. Accessed 6 Dec 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.