

ABSTRACT
Severe fever with thrombocytopenia syndrome virus (SFTSV) is an emerging negatively stranded enveloped RNA bunyavirus that causes SFTS with a high case fatality rate of up to 30%. Macroautophagy/autophagy is an evolutionarily conserved process involved in the maintenance of host homeostasis, which exhibits anti-viral or pro-viral responses in reaction to different viral challenges. However, the interaction between the bunyavirus SFTSV and the autophagic process is still largely unclear. By establishing various autophagy-deficient cell lines, we found that SFTSV triggered RB1CC1/FIP200-BECN1-ATG5-dependent classical autophagy flux. SFTSV nucleoprotein induced BECN1-dependent autophagy by disrupting the BECN1-BCL2 association. Importantly, SFTSV utilized autophagy for the viral life cycle, which not only assembled in autophagosomes derived from the ERGIC and Golgi complex, but also utilized autophagic vesicles for exocytosis. Taken together, our results suggest a novel virus–autophagy interaction model in which bunyavirus SFTSV induces classical autophagy flux for viral assembly and egress processes, suggesting that autophagy inhibition may be a novel therapy for treating or releasing SFTS.
KEYWORDS: Autophagy, bunyavirus, sftsv, viral assembly, viral egress
Introduction
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging tick-borne hemorrhagic fever caused by SFTS virus (SFTSV). The main clinical manifestations of SFTS include fever, thrombocytopenia, leukocytopenia, vomiting, diarrhea, and even multiorgan failure with a high fatality rate of up to 30% [1,2]. SFTSV is a negatively stranded enveloped RNA virus that belongs to the genus Bandavirus in the family Phenuiviridae of the order Bunyavirales [3]. SFTSV genomes contain three segments designated as large (L), medium (M), and small (S) segment, respectively. The L segment encodes an RNA-dependent RNA polymerase (RdRp) that mediates transcription and replication of the viral genome. The M segment encodes a glycoprotein precursor (Gp) that can be cleaved by cellular proteases during translation and processed into two subunits: glycoprotein N (Gn) and glycoprotein C (Gc), which mediate assembly and entry of the virion. The S segment encodes a nucleoprotein (NP) that mediates the formation of the viral ribonucleoprotein (RNP) complex, and a nonstructural protein (NSs) that serves as an important virulence factor [4]. Like other bunyaviruses, SFTSV is a cytosolic virus that replicates in the cytoplasm, assembles in the Golgi complex, and egresses via trans-Golgi network-mediated secretory vesicles [5,6].
Autophagy is a highly conserved process responsible for maintaining cellular homeostasis [7]. Initiation of autophagy begins with the induction of a cup-shaped double-membrane structure termed the phagophore, which might originate from various cellular membrane compartments, including the endoplasmic reticulum (ER), mitochondria, ER-mitochondrial contact sites/mitochondria-associated membranes, endosomes, ER-Golgi intermediate compartments (ERGIC), Golgi complexes, and plasma membranes [8,9]. Typically, autophagy is manipulated by different classical autophagy signaling complexes, including the ULK1 complex (ULK1, ATG13, RB1CC1, and ATG101) for autophagy initiation, the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (BECN1, AMBRA1, PIK3C3/VPS34, PIK3R4/VPS15, NRBF2, and ATG14) for phagophore nucleation, the ATG12–ATG5-ATG16 L1 complex for phagophore expansion, and the STX17-SNAP29-VAMP7-VAMP8 complex for autophagosome-lysosome fusion.
Autophagy is not a passive process, typically exhibiting an anti-viral role by degrading viral particles, enhancing viral antigen presentation, or modulating the inflammatory response [10–12]. However, some studies have indicated that autophagy is sometimes subverted or hijacked at various stages by certain viruses that escape autophagy surveillance or use the autophagic process for viral maturation, assembly, replication, or exocytosis. The cleavage of Dengue virus protein prM into pr and M by cellular FURIN is required for the viral maturation [13], and inhibition of autophagy blocks the FURIN cleavage resulting in noninfectious and heat-sensitive virus particles [14]. The capsid maturation of the poliovirus is mediated by the autophagy pathway [15]. Cox-sackievirus B3 can subvert autophagy by disrupting the autophagosome-lysosome fusion process by cleaving SNAP29 [16]. Packaged picornaviruses are clustered in phosphatidylserine-enriched autophagosome-like vesicles for viral transmission [17]. Enterovirus D68, a medically important respiratory virus, remodels autophagic transport and uses the amphisome for viral egress [18]. More recently, β-coronaviruses exploit lysosomal trafficking for egress rather than the biosynthetic secretory pathway [19].
To date, few studies have discussed the role of autophagy in the life cycle of bunyaviruses [20]. Rift Valley fever virus (RVFV) is a well-known mosquito-borne bunyavirus. The TLR7-MYD88 axis, but not other canonical TLRs, is responsible for mediating the autophagy flux under RVFV infection by directly interacting with the glycoprotein of RVFV [21]. Several canonical autophagy processes, from preinitiation to elongation, are essential for limiting RVFV infection. In addition, Sin Nombre hantavirus and Hantaan virus have been shown to interact with autophagy for viral replication [22,23]. Overall, the role of autophagy under bunyavirus infection seems inconsistent and remains largely unclear. Here, to further explore the contribution of autophagy under bunyavirus infection, we aim to use SFTSV as a model and investigate its underlying interaction with autophagy.
Results
Autophagy is induced under SFTSV infection
To characterize the interplay of autophagy under SFTSV infection, the conversion of LC3, as one of the hallmarks of autophagy, was detected in Vero and HeLa cells. We observed that the level of LC3-II was accumulated during SFTSV infection (Figure 1A, B). Prolonged infection times including 48 h and 72 h were also performed, and we observed that the LC3-II level was indeed induced by SFTSV infection and peaked at 24 h and 36 h in Vero and HeLa cells, respectively (Fig. S1A). In addition, confocal microscopy assay showed that SFTSV infection resulted in accumulation of endogenous LC3 puncta in Vero and HeLa cells (Figure 1C, D). We performed immunofluorescence assays at earlier time points after SFTSV infection (3 h and 6 h) (Fig. S1B). We found that the formation of LC3 puncta appeared at 3 h after SFTSV infection. Traditionally, transmission electron microscopy is the “gold standard” for detecting the induction of autophagy. To directly visualize autophagosome formation, transmission electron microscopy was used to observe the autophagic ultrastructure under SFTSV infection. Interestingly, a number of large, single membrane autophagic vacuoles were observed (Figure 1E). Moreover, accumulated LC3 puncta were found in mouse spleen after SFTSV infection (Figure 1F). Taken together, these results clearly demonstrate that SFTSV infection is able to induce autophagy.
Figure 1.
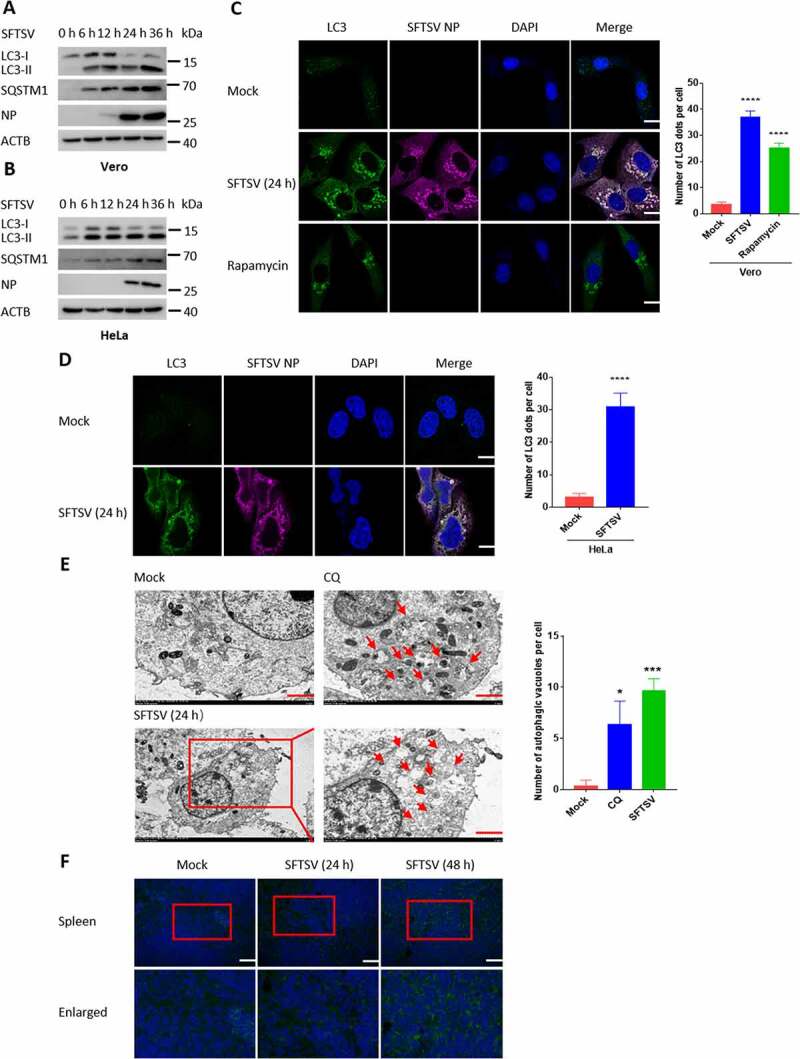
Autophagy is induced under SFTSV infection. (A and B) Vero and HeLa cells were infected with SFTSV at an MOI of 5 at different time points. Cell lysates were evaluated via western blot (WB). (C and D) Vero and HeLa cells were infected with SFTSV at an MOI of 5. Nuclear DNA, endogenous LC3, and SFTSV NP were stained as blue, green, and violet, respectively. Rapamycin served as the positive control of autophagy. For rapamycin (100 nM), cells were treated for 6 h without SFTSV infection before harvest. The number of LC3 puncta in each cell was counted, and at least 20 cells were included for each group. Scale bar: 20 μm. (E) Mock-infected, CQ-treated, or SFTSV-infected Vero cells at 24 h were processed and analyzed for the formation of autophagosomes or autolysosomes via electron transmission microscopy. For CQ (100 µM), cells were treated for 6 h without SFTSV infection before harvest. Black arrows indicate autophagic vacuoles. Scale bar: 2 μm. (F) WT C57BL/6 J mice were intraperitoneal injected with SFTSV at 106 TCID50/animal for 24 h and 48 h. The spleen sections underwent immunofluorescence assays (IFA) with LC3 (green) and DAPI (blue). Scale bar: 50 μm. Error bars, mean ± SD of three experiments. Student’s t test; *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
SFTSV-induced autophagy is a complete process
Our immunoelectron microscopy results suggest that SFTSV-induced autophagy might be a complete process (Figure 1E). To clarify this, we used two autophagy inhibitors, chloroquine (CQ) and bafilomycin A1 (Baf-A1), which can suppress autophagy by inhibiting autophagosome-lysosome fusion or the acidification of H+-ATPase, respectively. Treatment with CQ or Baf-A1 significantly increased the accumulation of LC3-II in SFTSV-infected Vero and HeLa cells (Figure 2A, B). To further confirm the induction of autophagy flux during SFTSV infection, we used a tandem reporter construct, mCherry-GFP-LC3; the GFP of this tandem reporter construct is sensitive and attenuated in a lysosomal acidic environment, whereas the mCherry is not. Therefore, fusion of autophagosomes with lysosomes results in the loss of green fluorescence and the appearance of red fluorescence. CQ and Earle’s Balanced Salt Solution (EBSS) treatments were utilized for negative and positive control, respectively. As a positive control, in EBSS-starved cells, which induces complete autophagy, GFP puncta were reduced compared with mCherry puncta (Figure 2C). While in CQ-treated cells, which induce incomplete autophagy, GFP puncta were equal to mCherry puncta (Figure 2C). Of note, GFP puncta were reduced or invisible in SFTSV-infected Vero cells, suggesting that autophagosomes fused with lysosomes during SFTSV infection (Figure 2C). Furthermore, we tracked autolysosomes with LAMP1, which is a traditional autolysosome/lysosome marker. We found that endogenous LC3 puncta were colocalized with LAMP1 in SFTSV-infected Vero cells (Figure 2D). Taken together, our results demonstrate that a complete autophagy flux is indeed induced under SFTSV infection.
Figure 2.
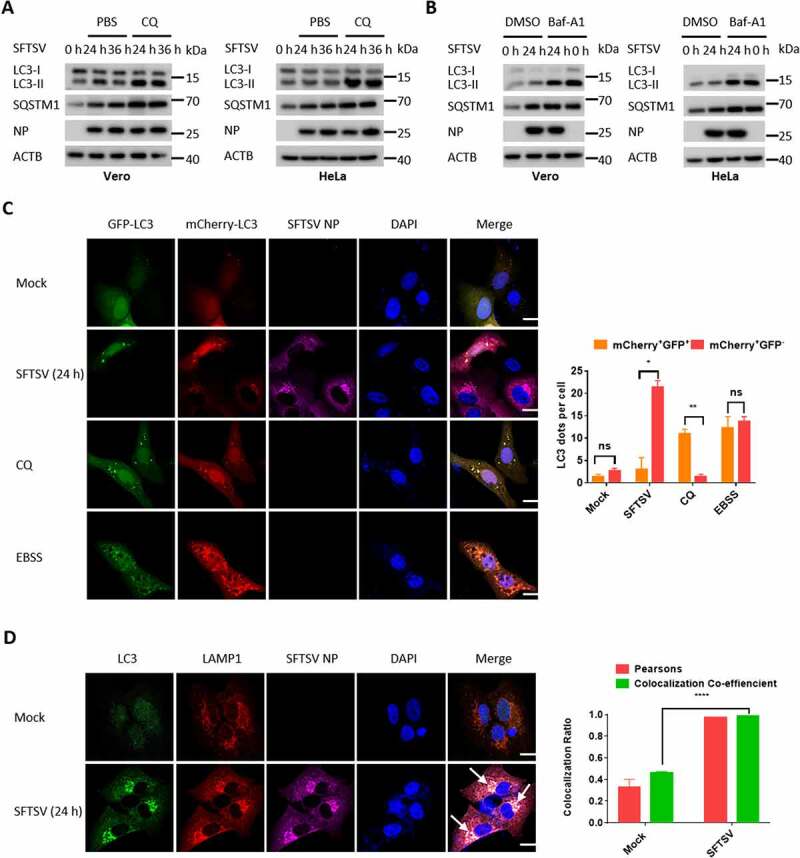
SFTSV-induced autophagy is a complete process. (A) Vero or HeLa cells were mock infected or infected with SFTSV at MOI of 5 for 24 h and 36 h and then treated with CQ for 6 h. Cell lysates were evaluated via WB. (B) Vero or HeLa cells were mock infected or infected with SFTSV for 24 h and then treated with Baf-A1 for 6 h. Cell lysates were evaluated via WB. (C) Vero cells were transfected with mCherry-GFP-LC3 for 12 h and then were mock infected or infected with SFTSV, or treated with CQ (100 µM), or starved in EBSS medium for 6 h. The number of mcherry-GFP-LC3 dots in each cell was counted, and at least 20 cells were included for each group. Single plane type of images was present. Scale bar: 20 μm. (D) Vero cells were infected with SFTSV for 24 h for analyzing the colocalization of endogenous LC3 and LAMP1. Single plane type of images was present. Scale bar: 20 μm.
Autophagy is essential for the replication of SFTSV
Inhibition of the amino-acid responsive MTOR kinase complex is a key signal for autophagosome biogenesis, primarily through the ULK1 and PtdIns3K complexes [24]. To elucidate how SFTSV induces autophagy, we determined the protein levels of ULK1, ATG101, BECN1, and ATG5 by western blot and observed that these proteins were increased during SFTSV infection (Fig. S2A). In addition, we found that the phosphorylation of MTOR (Ser2448) and ULK1 (Ser757) was also decreased after SFTSV infection (Fig. S2B), suggesting that SFTSV could induce autophagy by dephosphorylation of MTOR (Ser2448) and ULK1 (Ser757). To further demonstrate whether SFTSV-induced autophagy is dependent on these classical autophagy proteins, various autophagy gene-deficient cells were used. The LC3-II level was decreased in RB1CC1, BECN1, atg5, atg7, and ATG16L1 knockout cells (Figure 3A-E). Moreover, confocal microscopy showed that LC3 puncta significantly decreased in SFTSV infected RB1CC1, BECN1, atg5, atg7, or ATG16L1 knockout cells compared to WT cells (Figure 3F-G). These results indicate that SFTSV-triggered autophagy is classically dependent on the RB1CC1-BECN1-ATG5 axis.
Figure 3.
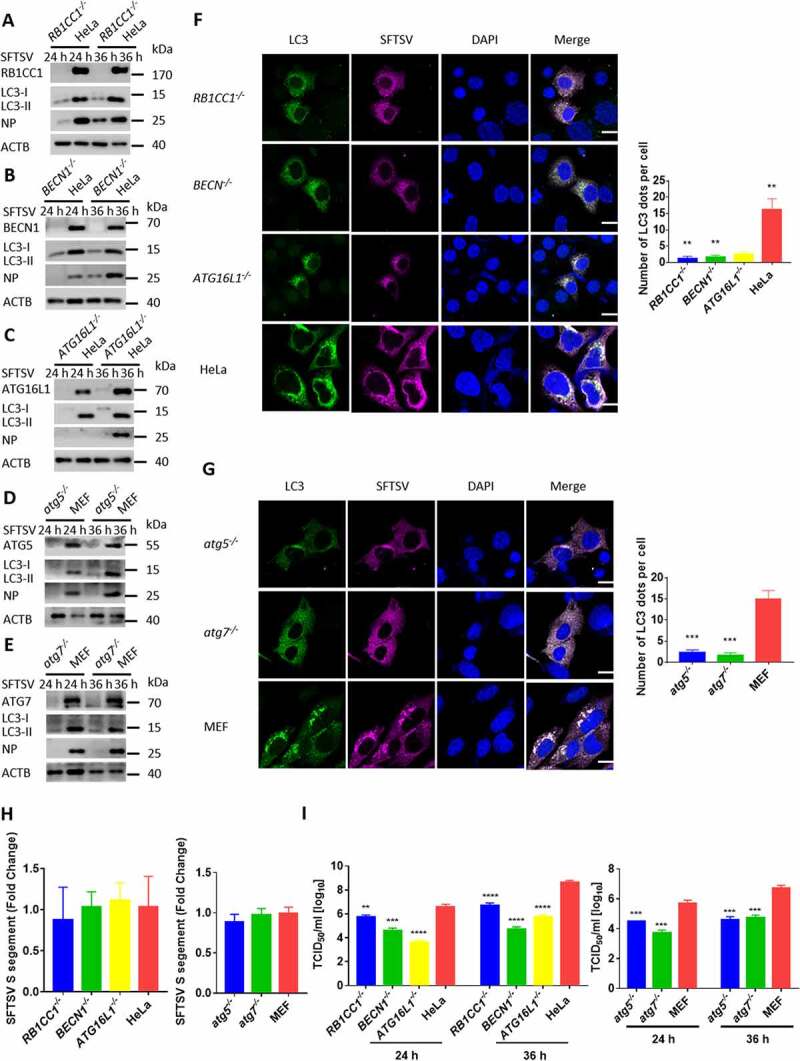
Autophagy is essential for the replication of SFTSV. (A-E) BECN1, atg5, atg7, RB1CC1, ATG16L1 knockout MEF or HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 24 h and 36 h. Cell lysates were evaluated via WB. (F-G) BECN1, atg5, atg7, RB1CC1, ATG16L1 knockout MEF or HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 24 h. Nuclear DNA, endogenous LC3, and SFTSV NP were stained as blue, green, and violet respectively. Single plane type of images was present. Scale bar: 20 μm. (H) BECN1, atg5, atg7, RB1CC1, ATG16L1 knockout MEF or HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 2 h. Then cells were washed once by PBS for three times and internalized SFTSV were detected via RT-qPCR. (I) BECN1, atg5, atg7, RB1CC1, ATG16L1 knockout MEF or HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 24 h. Endpoint 10-fold dilutions of an SFTSV stock were titrated. Values presented in the graph are calculated and expressed as the log10 of TCID50 units per ml of supernatant. Error bars, mean ± SD of three experiments. Student’s t test; *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
We next sought to determine whether the autophagy machinery can modulate SFTSV replication. HeLa cells were pretreated with 3-methyladenine (3-MA), which is a PtdIns3K inhibitor of autophagy. Viral titers in cell culture were analyzed by RT-qPCR for detecting intracellular and extracellular viruses. We observed that with the increase in the concentration of 3-MA, both intracellular and extracellular SFTSV particles were greatly reduced (Fig. S3A-C). We detected the intracellular and extracellular SFTSV S segments via RT-qPCR inRB1CC1, atg5, atg7, and ATG16L1 knockout cells (Fig. S3D-G) and found that SFTSV particles were sharply decreased in these knockout cells. Furthermore, we used TCID50 to determine the exocellular virus titers in various autophagy gene knockout cells. Similar results were obtained in RB1CC1, BECN1, atg5, atg7, and ATG16L1 knockout cells (Figure 3I). We also found that viral entry was not impaired in these autophagy genes knockout cells and viral particles could reach the endosome (Figure 3H). Moreover, the protein level of SFTSV NP was significantly reduced in 3-MA treated HeLa cells or these autophagy deficient cells (Figure 3A-E and S3A). Taken together, these results suggest that SFTSV replication is dependent on the cellular autophagy pathway.
SFTSV NP triggers BECN1-dependent autophagy
It is known that NP is the most abundant bunyavirus polypeptide. However, it is largely unclear about the role of NP in SFTSV-induced immunological studies. Here, to investigate the role of NP in the induction of autophagy, SFTSV NP was transiently transfected and expressed in Vero cells. Interestingly, we observed that overexpression of NP resulted in a significant increase in endogenous LC3-II (Figure 4A) and GFP-LC3 puncta (Figure 4B). BECN1, originally identified as a BCL2-binding protein, plays a central role in the autophagy initiation [25]. We next investigated whether NP-induced autophagy is dependent on BECN1. We found that NP-induced autophagy was significantly decreased in BECN1 knockout HeLa cells (Figure 4C), while the protein levels of BECN1 and other autophagy-related proteins were not altered by overexpression of SFTSV NP (Figure 4A). To further detect the underlying interaction between NP and BECN1, a confocal microscopy assay was used to detect colocalization between NP and BECN1. We found that both SFTSV NP and exogenously expressed NP could colocalize with endogenous BECN1 and exogenous BECN1 (Figure 4D and Fig. S4). Moreover, the coimmunoprecipitation assay showed that SFTSV NP could physically interact with endogenous BECN1 and overexpressed NP-HA could also physically interact with overexpressed BECN1-Flag (Figure 4E, G). To further evaluate the protein domain of BECN1 responsible for the interaction with SFTSV NP, six BECN1 truncations were constructed based on the functional domains of BECN1. The schematic diagram of BECN1 truncation mutants was showed in (Figure 4F). Confocal microscopy showed that BECN1 only containing coiled-coil domain (CCD) colocalized with NP-HA (Fig. S4). The coimmunoprecipitation assay also showed that BECN1 CCD interacted with NP-HA (Figure 4G). It has been reported that BCL2 functions as a negative regulator of PIK3C3-BECN1 complex-dependent autophagy by binding it to BECN1 [26]. Therefore, the effect of SFTSV NP on the interaction between BECN1 and BCL2 was further investigated. The results showed that the BECN1–BCL2 interaction was decreased during overexpression of NP (Figure 4H). Taken together, our data suggest that SFTSV NP can promote autophagy via inhibiting the association of BECN1 and BCL2.
Figure 4.
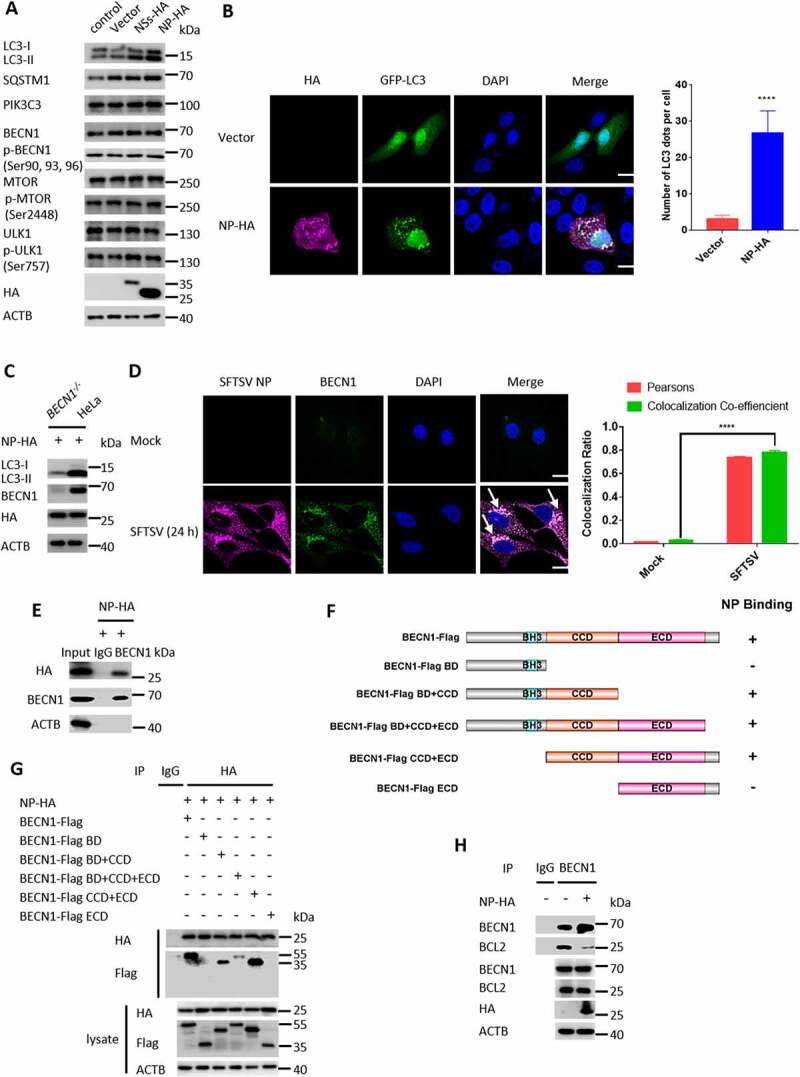
SFTSV NP triggers BECN1-dependent autophagy. (A) Vero cells were transfected with pVAX1, pVAX1-NSs-HA, or pVAX1-NP-HA for 24 h. The cell lysates were evaluated via WB.(B) Representative images of Vero cells co-transfected with EGFP-LC3 and pVAX1 or pVAX1-NP-HA for 24 h respectively. Single plane type of images was present. Scale bar: 20 μm. (C) BECN1 knockout cells or WT HeLa cells were transfected with pVAX1-NP-HA for 24 h. The cell lysates were evaluated via WB. (D) Representative images of Vero cells infected with SFTSV at an MOI of 5 for 24 h and stained for SFTSV NP and endogenous BECN1. Nuclei were stained with DAPI. Single plane type of images was present. Scale bar: 20 μm. (E) HEK293T cells were transfected with pVAX1-NP-HA for 24 h. The cell lysates were immunoprecipitated with anti-BECN1 antibody and then immunoblotted with the indicated antibody. (F) The schematic diagram of BECN1 truncation mutants and the BECN1-NP interacting domain. (G) HEK293T cells were co-transfected with pVAX1-NP-HA and Flag-BECN1, Flag-BECN1 BD, Flag-BECN1 BD+CCD, Flag-BECN1 BD+CCD+ECD, Flag-BECN1 CCD+ECD, or Flag-BECN1 ECD. The cell lysates were immunoprecipitated with anti-HA antibody and then immunoblotted with the indicated antibody. (H) Vero cells were transfected with pVAX1 or pVAX1-NP-HA for 24 h. The cell lysates were immunoprecipitated with anti-BECN1 antibody and then immunoblotted with the indicated antibody.
The autophagosome serves as SFTSV assembly platform
Given that SFTSV replication was severely limited in autophagy deficient cells (Figure 3I), we hypothesized that autophagy could be exploited for the life cycle of SFTSV. To clarify this, SFTSV particles were enriched from supernatants of SFTSV-infected HeLa cells. SFTSV NP, autophagic proteins, and membranous markers such as GOLGA2/GM130 and LAMP1 were analyzed by western blot in purified SFTSV pellets. Supernatants from mock-infected cells enriched under the same conditions served as negative controls. Interestingly, we observed high levels of NP, LC3-II, PtdIns3K complex (BECN1, ATG14, PIK3C3), ATG7-ATG12–ATG5-ATG16L1 complex in purified SFTSV pellets (Figure 5A), while ULK1 and RB1CC1 were not found in purified SFTSV particles, indicating that the SFTSV viral envelope might originate from autophagosomes. Previous studies propose that bunyavirus particles are assembled and matured in the Golgi compartment and then bud to extracellular matrix through Golgi-mediated secretory vesicles [27]. We then detected the endoplasmic reticulum-Golgi intermediate compartment marker LMAN1/ERGIC53, the cis Golgi network marker GOLGA2, and the trans-Golgi network marker TGOLN2/TGN38 in purified SFTSV pellets, and found that LMAN1, GOLGA2, and TGOLN2 were enriched in SFTSV pellets, while the endoplasmic reticulum marker CANX (calnexin) was not (Figure 5A), suggesting that the SFTSV viral envelope originated from the ERGIC and Golgi complex. To identify the purity of PEG8000 purified SFTSV particles, we observed the virus under transmission electron microscopy. The result showed that the high purity of PEG8000 purified SFTSV particles (Figure 5B). To further understand the role of autophagosomes in SFTSV assembly, we performed confocal microscopy analysis of SFTSV particles with endogenous LC3B. We found that SFTSV NP and Gn fully colocalized with LC3B (Figure 5C), suggesting the possible formation of SFTSV in autophagosomes. Moreover, by performing immunoelectron microscopy, we found SFTSV virions in LC3-positive autophagic vacuoles with a diameter of 1000 nm, and the size of autophagic vacuoles usually varies in the range of 500–1500 nm in mammalian cells [28], demonstrating the localization of SFTSV particles in autophagic vacuoles (Figure 5D). Moreover, our confocal microscopy analysis showed that Gn and autolysosome marker LAMP1 colocalized with LMAN1, GOLGA2, and TGOLN2, but not with CANX, suggesting that SFTSV was indeed assembled in the ERGIC and Golgi complex (Figure 5E). Taken together, our results strongly suggest that SFTSV assembly occurs in ERGIC- and Golgi complex-originated autophagosomes.
Figure 5.
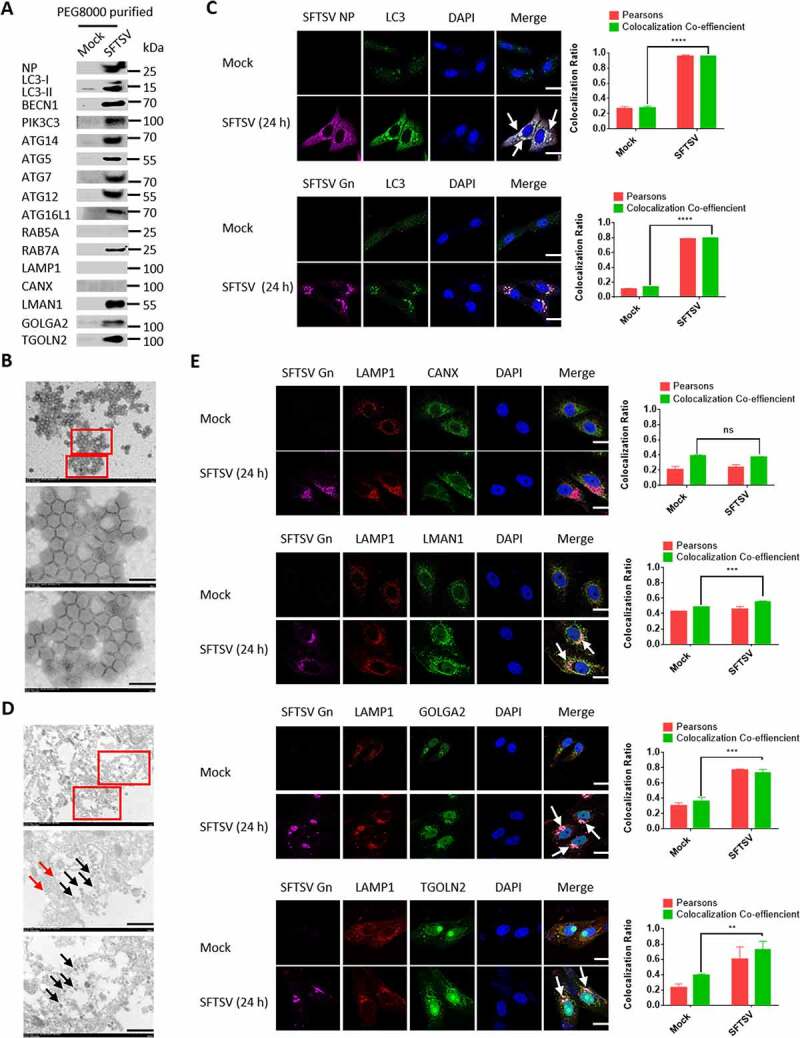
The autophagosome serves as SFTSV assembly platform. (A) HeLa cells were infected with SFTSV at an MOI of 1 for 5 days and SFTSV in supernatant were harvested and purified for WB detection. (B) PEG8000 purified SFTSV particles were analyzed via electron transmission microscopy. Scale bar: 200 nm. (C) Representative images of Vero cells infected with SFTSV at an MOI of 5 for 24 h and stained for endogenous LC3, SFTSV NP, and Gn. Nuclei were stained with DAPI. Single plane type of images was present. Scale bar: 20 μm. (D) 10-nm gold particles were used to label endogenous LC3. The red arrows refer to LC3, and the black arrows refer to SFTSV virions. Single plane type of images was present. Scale bar: 500 nm. (E) Vero cells were infected with SFTSV at an MOI of 1 for 24 h. The colocalization of Gn and LAMP1 to the ER, ERGIC and Golgi was analyzed. Single plane type of images was present. Scale bar: 20 μm.
Autophagic vacuoles is exploited by SFTSV for exocytosis
Since that SFTSV was observed to egress from autophagic vacuoles (Figure 5D), we proposed that exocytosis of SFTSV may be mediated by autophagic vacuoles. STX17 and VAMP7 are critical proteins involved in the fusion of autophagosome with lysosome [29,30]. Interestingly, we found that SFTSV NP protein levels and SFTSV titers were also inhibited in STX17 knockout cells and VAMP7 knockout cells (Figure 6A-C). We also detected the intracellular and exocellular SFTSV S segment via RT-qPCR (Fig. S5A and S5B), the result showed the same trend. To investigate the effect of lysosome on SFTSV replication, we used the E64d/pepstatin, which are inhibitors of lysosome cysteine protease and aspartic protease, respectively. We found that intracellular and extracellular SFTSV titers were not altered upon treatment with E64d/pepstatin (Fig. S5C), indicating that SFTSV replication was not affected by lysosomal/autolysosomal proteolytic enzymes or lysosomal/autolysosomal proteolytic enzymes could be suppressed by SFTSV. To investigate whether lysosomal proteolytic activity was altered under SFTSV infection, we analyzed the mature forms of CTSB and CTSD. We found that SFTSV infection reduced the levels of the mature form of CTSB and CTSD (Figure 6D). Since the persistently acidic environment of the lysosome may be essential for the maturation of pro-formed hydrolases [31], we used LysoSensor™ Green DND-189 to evaluate whether SFTSV infection affected lysosomal/autolysosomal pH. In contrast to Baf-A1 treated cells, we found that SFTSV infection did not alter lysosomal/autolysosomal pH, causing that the green fluorescence was unchanged (Figure 6E). To further validate the SFTSV trafficking process in the autolysosome, confocal microscopy analysis was performed to determine the subcellular localization of SFTSV virions. We observed that SFTSV NP and Gn colocalized with LAMP1 (Figure 6F). To directly visualize SFTSV in the autolysosome, we labeled the autolysosome with LAMP1 and found SFTSV virions in LAMP1 positive structure in SFTSV infected Vero cells (Figure 6G). We also found SFTSV virions in LC3 and LAMP1 double-labeled positive structures in SFTSV infected HeLa cells (Figure 6H). Taken together, our results indicate that SFTSV exocytosis is mediated by autolysosome.
Figure 6.
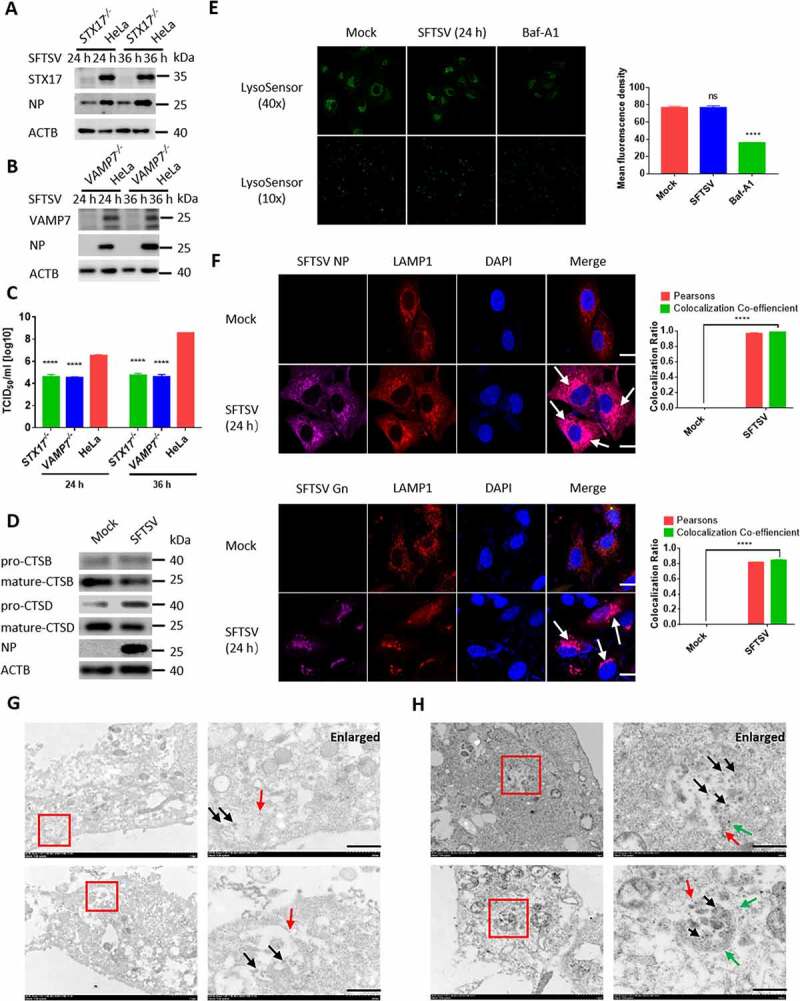
Autophagic vacuoles is exploited by SFTSV for exocytosis. (A and B) STX17 orVAMP7 knockout HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 24 h and 36 h. Cell lysates were evaluated via WB. (C) STX17 or VAMP7 knockout HeLa cells and WT cells were infected with SFTSV at an MOI of 1 for 24 h and 36 h. Viral titer was measured by TCID50. (D) Vero cells were mock infected or infected with SFTSV at MOI of 5 for 24 h. Pro-form and mature-form of CTSB and CTSD were evaluated via WB. (E) Vero cells were mock infected or infected with SFTSV at MOI of 5 for 24 h or treated with Baf-A1 for 6 h. Then autolysosome acidification was determined by LysoSensor Green DND-189 staining. Single plane type of images was present. The mean density was calculated via ImageJ. Error bars, mean ± SD of three experiments. Student’s t test; *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001. (F) Representative images of Vero cells infected with SFTSV at an MOI of 5 for 24 h and then underwent immunofluorescence assay to mark endogenous LAMP1, SFTSV NP, and Gn. Nuclei were stained with DAPI. Single plane type of images was present. Scale bar: 20 μm. (G) 10-nm gold particles were used to label endogenous LAMP1 in SFTSV infected Vero cells. The red arrows refer to LAMP1, and the black arrows refer to SFTSV virions. Scale bar: 500 nm. (H) 10-nm gold particles were used to label endogenous LC3 and 4-nm gold particles were used to label endogenous LAMP1 in SFTSV infected HeLa cells. The red arrows refer to LC3, the green arrows refer to LAMP1 and the black arrows refer to SFTSV virions. Scale bar: 500 nm.
Discussion
Based on our knowledge, there have been few reports that have systematically addressed the interaction of autophagy and bunyavirus [21,23]. Here, using several autophagy-related gene deficient cells, we clearly demonstrated that SFTSV infection was capable of inducing complete autophagy flux in a RB1CC1-BECN1-ATG5-dependent manner. Our study showed that SQSTM1/p62 protein levels were increased under SFTSV infection status, supporting that NSs-TRIM21 interaction leads to increased stability and oligomerization of SQSTM1 [32]. It is worth noting that SQSTM1 itself is considered as a classical autophagy receptor, but SQSTM1 protein levels may not fully reflect the dynamics of autophagy [33]. Typically, autophagy restricts viral infection by directly degrading virions or viral particles via the autolysosome. In certain circumstances, autophagy is utilized for viral replication, such as picornaviruses, coronaviruses, and flaviviruses [7]. Here, we confirmed that autophagy exerted a pro-viral effect on bunyavirus SFTSV replication by using autophagy inhibitor 3-MA and several autophagy-related gene deficient cells that are critical for autophagy initiation, elongation, and fusion processes, indicating that the life cycle of SFTSV was dependent on the induction of autophagy.
In addition, our results showed that SFTSV NP-mediated dissociation of BECN1 and BCL2 was important for the induction of autophagy [34] without modulating the protein level of PIK3C3 and BECN1. To our surprise, although SFTSV-induced autophagy was dependent on the phosphorylation of MTOR (Ser2448) and ULK1 (Ser757), overexpression of SFTSV NP did not modulate the activity of MTOR. Interestingly, overexpression of SFTSV NSs resulted in the accumulation of LC3-II, while colocalization of glycoprotein Gn with autophagy marker LC3 and LAMP1 was observed under SFTSV infection. Also, the importance of glycoprotein in autophagy induction is clarified in bunyavirus RVFV and Hantaan virus [21,23,35]. These results suggest that SFTSV-induced autophagy seems more complicated and is not just dependent on SFTSV NP. Other SFTSV components could collaborate with NP in the induction of autophagy, which remains further investigation to clarify the underlying mechanisms.
To date, a number of studies have investigated the processes of internalization, replication, recruitment, and assembly of bunyaviruses [27,36]. Bunyavirus internalization into early endosomes is initiated by the interaction of viral glycoprotein and cell surface receptor. Subsequently, bunyavirus is transported into the acidic late endosome, which triggers a conformational change of the glycoprotein and membrane fusion to release the viral genome into the cytoplasm for replication. In addition, it is generally believed that the assembled bunyavirus RNP, of which NP and RdRp alone are not localized in the ERGIC and Golgi complex [37], is recruited to the lumen of the Golgi complex through interaction with the cytoplasmic tail of the glycoprotein Gn, mediating the production of infectious viral particle. Although previous studies conclude that mature bunyavirus particle buds into Golgi-mediated secretory vesicle and is released into the extracellular space [27], it remains unclear the exact mechanism of bunyavirus assembly and release and whether additional patterns exist.
In our study, the existence of lipidated LC3, PtdIns3K complex (ATG14, PIK3C3, BECN1), and ATG7-ATG12–ATG5-ATG16L1 complex was found in purified SFTSV particles, indicating that autophagosome membrane could be utilized as SFTSV envelope. Unfortunately, RB1CC1 and ULK1 were not found in virus particles. The reason for this was that SFTSV assembly might begin at a certain stage after autophagy initiation, and the specific mechanism remained to be determined. Other studies also show LC3-II is found in matured Epstein Barr virus and Varicella-Zoster virus particles [38,39]. Zimmermann et al. find that LC3-II and SQSTM1 are incorporated into extracellular human cytomegalovirus virions [40]. Taisne et al. also find that integration of parts of the autophagy machinery including ATG12–ATG5 and BECN1 into human cytomegalovirus particles [41]. We also detected RAB7A in purified SFTSV particles, indicating that late endosome or amphisome might participate in the process of SFTSV assembly.
It is known that a variety of cellular membranes could be the source of autophagosome [8,42]. Here, we found that both ERGIC marker (LMAN1) and Golgi complex marker (GOLGA2 and TGOLN2) also served as SFTSV envelope components, while ER (CANX) was not. Colocalization of the autolysosome marker LAMP1 with ERGIC and Golgi complex markers was also observed under SFTSV infection. These interesting findings suggested that SFTSV could be initially assembled in the ERGIC or Golgi complex that were then recruited as an autophagosome membrane. Notably, our finding indicated that mature SFTSV particles were localized in autophagic vacuoles and egress into extracellular space from the autolysosome once fusing with the cell membrane (Figure 7). Alternatively, SFTSV could assemble in Golgi complex and Golgi-formed secretory vesicles might mediate the egress of SFTSV via autolysosome indirectly [43]. Although lysosome/autolysosome pH was not altered, the activity of lysosome/autolysosome enzymes (CTSB and CTSD) was indeed partially decreased under SFTSV infection. Consistently, the replication of SFTSV was not affected by the treatment of lysosomal/autolysosomal hydrolase inhibitor E64/pepstatin. Moreover, the increased SQSTM1 protein levels under SFTSV infection with CQ or Baf-A1 treatments might explain that autolysosome protease activity was partially weakened.
Figure 7.
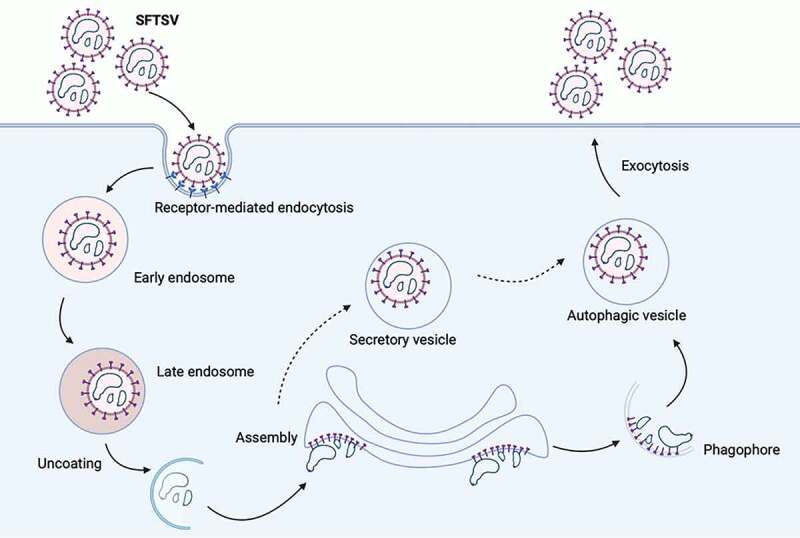
Schematic presentation of the SFTSV replication cycle. SFTSV Gp is synthesized by membrane bound ribosomes at the ER and then translocated to ERGIC and Golgi complex. Gp is expected to recruit SFTSV RNP complex, comprising SFTSV NP, RdRp, and genomic RNA, for SFTSV assembly and maturation. Traditionally, virus is considered to be formed by budding into Golgi complex lumen. Then matured virus particles are released from cell via exocytosis. Here, we provided a novel model of the exocytosis of bunyavirus SFTSV. ERGIC and Golgi complex were recruited as autophagic vesicles for SFTSV assembly and exocytosis via autolysosome. Alternatively, Golgi complex-originated secretory vesicle containing virions might fuse with autophagosome and egressed via autolysosome.
In summary, our research clearly showed a novel pattern that bunyavirus SFTSV manipulated autophagy flux for viral life cycle. SFTSV not only exploited the autophagosome as an assembly platform, but also utilized autophagy-dependent vesicles for exocytosis, of which the fusion of autophagosome and lysosome was also essential for the exocytosis of SFTSV. Currently, no approved vaccines or specific drugs are available and approved for the treatment of SFTSV. Our results would expand the knowledge of the underlying SFTSV pathogenicity mechanism that could provide a new idea for SFTSV therapy via the autophagy pathway.
Materials and methods
Viruses and cells
Vero, HeLa, HEK293T, atg5 knockout MEF, atg7 knockout MEF, RB1CC1 knockout HeLa, BECN1 knockout HeLa, ATG16L1 knockout HeLa, STX17 knockout HeLa and VAMP7 knockout HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher, 11,965,092), supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2. Vero (CCL-81), Hela (CCL-2) and HEK293T (CRL-3216) cells were obtained from ATCC. atg5 and atg7 knockout MEF cells were kindly provided by Dr. Ming-Zhou Chen (Wuhan University, China). RB1CC1 and ATG16L1 knockout HeLa cells were kindly provided by Dr. Feng Shao (Peking University, China), and BECN1 knockout HeLa cells were kindly provided by Dr. Wen-Sheng Wei (Peking University, China). STX17 and VAMP7 knockout HeLa cells were kindly provided by Dr. Jian-Guo Chen (Peking University, China). SFTSV isolate JS2011-013-1 was cultured in Vero cells. Virus-infected cells were maintained in the DMEM containing 2% fetal bovine serum and penicillin-streptomycin. For the in vivo experiment, C57B6L mice were injected with SFTSV at 106 TCID50/mouse.
Antibodies and reagents
Primary antibodies were purchased from the indicated manufacturers as follows: Anti-LC3B (3868S), anti-MTOR (2983S), anti-phospho-MTOR-Ser2448 (5536S), anti-ULK1 (8054S), anti-phospho-ULK1-Ser757 (14202S), anti-RB1CC1/FIP200 (12436S), anti-ATG101 (13492S), anti-BECN1/Beclin1 (3495S), anti-ATG5 (12994S), anti-ATG7 (8558S), anti-ATG16L1 (8089S), and anti-RAB5A (3547S) were purchased from Cell Signaling Technology. Anti-SQSTM1/p62 (18,420-1-AP), anti-LAMP1 (65,051-1-Ig), anti-CANX/calnexin (10,427-2-AP), anti-LMAN1/ERGIC53 (13,364-1-AP), anti-GOLGA2/GM130 (11,308-1-AP), anti-TGOLN2/TGN38 (13,573-1-AP), anti-RAB7A (55,469-1-AP), anti-STX17 (17,815-1-AP), anti-VAMP7 (22,268-1-AP), anti-PIK3C3/VPS34 (12,452-1-AP), anti-BCL2 (60,178-1-Ig), and anti-Flag tag (80,010-1-RR) were purchased from Proteintech. Anti-LAMP1 (DF7033), anti-phospho-BECN1/Beclin1-Ser90/93/96 (AF7386), and anti-HA tag (T0008) were purchased from Affinity Biosciences. Anti-CTSD (ABP50871) and anti-CTSB (ABP50870) were purchased from Abbkine Scientific Corporation. Anti-ACTB/actin (sc-8432) were purchased from Santa Cruz Biotechnology. Anti-SFTSV NP and anti-Gn primary antibody were produced in the laboratory of the author (Institute of Pathogenic Microbiology, Jiangsu Provincial Center for Disease Prevention and Control). Fluorescence-labeled secondary antibodies including Alexa Fluor 488 goat anti-mouse IgG, IgM (H + L) (A-10680), Alexa Fluor 568 goat anti-mouse IgG (H + L) (A-11004), and Alexa Fluor 647 goat anti-human IgG (H + L) (A-21445) were purchased from Thermo Fisher. Alexa Fluor 594 goat anti-human IgG (bs-0297 G-AF594), Alexa Fluor 594 goat anti-rabbit IgG (bs-0295 G-AF594), and Alexa Fluor 488 goat anti-rabbit IgG (bs-0295 G-AF488) were purchased from BIOSS. HRP-conjugated goat anti-mouse IgG (G1214) and goat anti-rabbit IgG (G1213) secondary antibodies were purchased from Servicebio. HRP-conjugated goat anti-human IgG (bs-0297 G-HRP) was purchased from BIOSS.
Chloroquine (CQ; C6628) was purchased from Sigma Aldrich. 3-Methyladenine (3-MA; HY-19312) was purchased from MedChemExpress. Bafilomycin A1 (Baf-A1; MB5505-L) was purchased from Meilunbio. Earle’s Balanced Salt Solution (EBSS; 24,010,043) was purchased from Thermo Fisher. LysoSensor™ Green DND-189 (40767ES50) was purchased from Yeasen. For CQ, Baf-A1 and EBSS, cells were treated for 6 h before harvest.
Production of anti-SFTSV antibodies (anti-NP and anti-Gn)
Monoclonal anti-NP or anti-Gn was isolated using recombinant SFTSV NP or Gn protein as a bait from a phage-display antibody library derived from the peripheral blood mononuclear cells of a patient that recovered from SFTS disease. The variable region of anti-NP or anti-Gn antibody fused with the human heavy chain constant region gene (IgG1) by overlapping PCR and then cloned into pCAGGS eukaryotic expression vector. The recombinant expression vectors were transfected into HEK293T cells, then anti-NP or anti-Gn antibody was purified using a protein A column (Genscript, L00464) and a Superdex 200 column (Cytiva, 28,989,335).
SFTSV internalization assay
Cells were infected with SFTSV at an MOI of 1 at 37°C for 2 h to allow virus internalization. Then, cells were washed with PBS for three times to remove noninternalized viruses. Internalized SFTSV was detected using RT-qPCR.
RNA extraction and RT-qPCR
Intracellular RNA was extracted with RNA isolation kit (Servicebio, G3013), extracellular RNA was extracted with TRIzol™ LS Reagent (Thermo Fisher, 10,296,010). RT-qPCR was performed using the Hiscript III Reverse Transcriptase (Vazyme, R333-01), ChamQ SYBR Color qPCR Master Mix (Vazyme, Q411-02), and an CFX96 touch system (BIO-RAD, Hercules, CA, USA) according to the manufacturers’ protocols. The following primers were used: SFTSV S segment, forward 5′-GGGTCCCTGAAGGAGTTGTAAA-3′ and reverse 5′- TGCCTTCACCAAGACTATCAATGT-3′; human GAPDH, forward 5′- GGAGCGAGATCCCTCCAAAAT-3′ and reverse 5′- GGCTGTTGTCATACTTCTCATGG −3′.
Immunofluorescence and confocal microscopy
Cell monolayers were fixed with 4% paraformaldehyde for 10 min and then permeabilized using 0.02% Triton X-100 (Solarbio, T8200) in PBS (Solarbio, P1010). After blocking with 2% bovine serum albumin (Solarbio, A8020), cells were incubated with indicated primary antibodies overnight at 4°C and fluorescence-labeled secondary antibodies for 30 min at room temperature. Cell nuclei was stained with DAPI (Beyotime, C1005). Laser-scanning confocal microscopy was performed on a Zeiss LSM880 (Institute of Medicine, Wuhan University, Wuhan, China). All images were taken with the same acquisition condition, brightness/contrast of images adjusted only for presentation purposes. For colocalization analysis, colocalization finder plugin of ImageJ software was utilized.
Western blot and co-immunoprecipitation
For western blot analysis, cells were washed with PBS and lysed with 1x SDS loading buffer (Servicebio, G2013). Cell lysates were centrifuged at 12,000 g for 5 min at 4°C and then heated at 95°C for 5 min. Cell lysates were subjected to 12% SDS-polyacrylamide gel electrophoresis (PAGE) and then transferred to 0.22-µm polyvinylidene difluoride (PVDF) membranes (Cytiva, 10,600,021). Membranes were blocked with 5% nonfat milk in TBST (1× TBS [Solarbio, T1080], 0.05% Tween 20 [Solarbio, T8220]) and then probed with primary antibodies and HRP-labeled secondary antibodies. Protein signals were visualized by enhanced chemiluminescence kit (Thermo Fisher, 32,132) using Amersham Imager 600 system (GE Healthcare, Chicago, IL, USA).
For co-immunoprecipitation, cells were washed with PBS and lysed with 200 µl immunoprecipitation lysis buffer (Servicebio, G2038). To detect the target proteins, 80 µl of the lysates were subjected for immunoblot analysis. The rest of lysates were incubated overnight with a control IgG or the indicated primary antibodies at 4°C and then incubated with the protein A/G agarose (Beyotime, P2055) at 4°C for 2 h. The beads were washed four times with an immunoprecipitation lysis buffer and resuspend in 60 µl 1× SDS loading buffer before western blot analysis.
Purification of SFTSV pellets from supernatant
HeLa cells were infected by SFTSV at an MOI of 1 and cell culture supernatant was harvested at 120 h post-infection. The cell culture supernatant was centrifuged at 200 g for 5 min and the supernatant was transferred to a new tube and centrifuged at 3,200 g for 15 min at 4°C to remove the cell debris thoroughly. Next, the supernatant was mixed with PEG8000 solution (5×) (Abcam, ab100538) and refrigerated overnight. Finally, the supernatant was centrifuged at 4,000 g for 30 min at 4°C, and carefully removed supernatant by aspiration. The beige or white pellet containing virus was collected.
Transmission electron microscopy and immunoelectron microscopy
For transmission electron microscopy, Vero cells were infected by SFTSV for 24 h at an MOI of 5 or treated with CQ (100 μM) for 6 h, and then cells were fixed with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for 2 h. The cells were harvested and fixed with 2.5% glutaraldehyde on ice for 2 h followed by postfixation in 2% osmium tetroxide, and dehydrated with sequential washes in 50%, 70%, 90%, 95%, and 100% ethanol. Areas containing cells were block mounted and thinly sliced using Leica EM UC7. Samples were imaged using a HITACHI HT7800 transmission electron microscope at 80 kv.
For immunoelectron microscopy, Vero cells were infected with SFTSV at an MOI of 1, and then cells were fixed in 2% glutaraldehyde in a neutral phosphate buffer, postfixed in osmium tetroxide, and embedded in Epon (Sigma Aldrich, 45,345–250ML-F). Sections were cut at 70–80 nm and examined under a HITACHI HT7800 transmission electron microscope at 80 kv.
Plasmids and DNA transfections
pcDNA3.1 (+)-3× Flag-BECN1 and mutants Flag-BECN1-BD, Flag-BECN1-BD+CCD, Flag-BECN1-BD+CCD+ECD, Flag-BECN1-CCD+ECD, Flag-BECN1-ECD were kindly provided by Dr. Jian-Guo Wu (Wuhan University, China). The PCR products of NP and NSs were both cloned into pVAX1. EGFP-LC3 were purchased from Addgene (11,546; deposited by Karla Kirkegaard). pmCherry-C1-EGFP-LC3B was purchased from Bioworld Technology (PPL00191-2d). Vero cells were grown on confocal dishes to 50% confluent and transfected using Hieff TransTM Liposomal Transfection Reagent (Yeasen, 40802ES02) according to the instructions.
Statistical analysis
Data were expressed as means ± standard error of the means (SEM) and were analyzed with Prism software (GraphPad) by using Student’s t-test or one-way analysis of variance (ANOVA) test comparisons. P values less than 0.05 were considered statistically significant. Most experiments were performed three times.
Supplementary Material
Funding Statement
This study was supported by the National Natural Science Foundation of China[81971939 and 31570167] and the Fundamental Research Funds for the Central Universities [2042021kf0046]. The funders had no role in the study design, data collection and analysis, decision to publish, or the preparation of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Yu XJ, Liang MF, Zhang SY, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Li H, Lu QB, Xing B, et al. Epidemiological and clinical features of laboratory-diagnosed severe fever with thrombocytopenia syndrome in China, 2011-17: a prospective observational study. Lancet Infect Dis. 2018;18:1127–1137. [DOI] [PubMed] [Google Scholar]
- [3].Kuhn JH, Adkins S, Alioto D, et al. taxonomic update for phylum Negarnaviricota (Riboviria: orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch Virol. 2020;165:3023–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu S, Chai C, Wang C, et al. Systematic review of severe fever with thrombocytopenia syndrome: virology, epidemiology, and clinical characteristics. Rev Med Virol. 2014;24:90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Novoa RR, Calderita G, Cabezas P, et al. Key Golgi factors for structural and functional maturation of bunyamwera virus. J Virol. 2005;79:10852–10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Salanueva IJ, Novoa RR, Cabezas P, et al. Polymorphism and structural maturation of bunyamwera virus in Golgi and post-Golgi compartments. J Virol. 2003;77:1368–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Choi Y, Bowman JW, Jung JU.. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tooze SA, Yoshimori T.. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. [DOI] [PubMed] [Google Scholar]
- [9].Ge L, Wilz L, Schekman R. Biogenesis of autophagosomal precursors for LC3 lipidation from the ER-Golgi intermediate compartment. Autophagy. 2015;11:2372–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. [DOI] [PubMed] [Google Scholar]
- [11].Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013;494:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Orvedahl A, MacPherson S, Sumpter R, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hsieh SC, Wu YC, Zou G, et al. Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J Biol Chem. 2014;289:33149–33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mateo R, Nagamine CM, Spagnolo J, et al. Inhibition of cellular autophagy deranges dengue virion maturation. J Virol. 2013;87:1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Richards AL, Jackson WT. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012;8:e1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mohamud Y, Shi J, Qu J, et al. Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 2018;22:3292–3303. [DOI] [PubMed] [Google Scholar]
- [17].Chen YH, Du W, Hagemeijer MC, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015;160:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Corona AK, Saulsbery HM, Velazquez AFC, et al. Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 2018;22:3304–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghosh S, Dellibovi-Ragheb TA, Kerviel A, et al. Beta-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 2020;183:1520–35 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou CM, Yu XJ. Unraveling the underlying interaction mechanism between dabie bandavirus and innate immune response. Front Immunol. 2021;12:676861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moy RH, Gold B, Molleston JM, et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity 2014;40:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hussein IT, Cheng E, Ganaie SS, et al. Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J Virol. 2012;86:7520–7529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang K, Ma H, Liu H, et al. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 2019;27:2075–91 e5. [DOI] [PubMed] [Google Scholar]
- [24].Munson MJ, Ganley IG. MTOR, PIK3C3, and autophagy: signaling the beginning from the end. Autophagy 2015;11:2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Antonioli M, Di Rienzo M, Piacentini M, et al. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42:28–41. [DOI] [PubMed] [Google Scholar]
- [26].Maejima Y, Kyoi S, Zhai P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wichgers Schreur PJ, Kortekaas J. Single-molecule FISH reveals non-selective packaging of rift valley fever virus genome segments. PLoS Pathog. 2016;12:e1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. [DOI] [PubMed] [Google Scholar]
- [29].Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. 2019;218:757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dingjan I, Linders PTA, Verboogen DRJ, et al. Endosomal and phagosomal SNAREs. Physiol Rev. 2018;98:1465–1492. [DOI] [PubMed] [Google Scholar]
- [31].Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. [DOI] [PubMed] [Google Scholar]
- [32].Choi Y, Jiang Z, Shin WJ, et al. Severe fever with thrombocytopenia syndrome virus NSs interacts with TRIM21 to activate the p62-Keap1-Nrf2 pathway. J Virol. 2020;94(6):e01684-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Umemura A, He F, Taniguchi K, et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29:935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Menon MB, Dhamija S, Dingjan I, et al. Beclin 1 phosphorylation - at the center of autophagy regulation. Front Cell Dev Biol. 2018;6:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nakamoto M, Moy RH, Xu J, et al. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 2012;36:658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu J, Xu M, Tang B, et al. Single-Particle tracking reveals the sequential entry process of the bunyavirus severe fever with thrombocytopenia syndrome virus. Small 2019;15:e1803788. [DOI] [PubMed] [Google Scholar]
- [37].Lundu T, Tsuda Y, Ito R, et al. Targeting of severe fever with thrombocytopenia syndrome virus structural proteins to the ERGIC (endoplasmic reticulum Golgi intermediate compartment) and Golgi complex. Biomed Res. 2018;39:27–38. [DOI] [PubMed] [Google Scholar]
- [38].Nowag H, Guhl B, Thriene K, et al. Macroautophagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine 2014;1:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Buckingham EM, Jarosinski KW, Jackson W, et al. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016;90:8673–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zimmermann C, Kramer N, Krauter S, et al. Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release. Autophagy 2021;17:779–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Taisne C, Lussignol M, Hernandez E, et al. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci Rep. 2019;9:4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wei Y, Liu M, Li X, et al. Origin of the autophagosome membrane in mammals. Biol Med Res Int. 2018;2018:1012789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].De Tito S, Hervas JH, van Vliet AR, et al. The Golgi as an assembly line to the autophagosome. Trends Biochem Sci. 2020;45:484–496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.