

ABSTRACT
Macroautophagy/autophagy is a highly conserved nutrient-recycling pathway that eukaryotes utilize to combat diverse stresses including nutrient depletion. Dysregulation of autophagy disrupts cellular homeostasis leading to starvation susceptibility in yeast and disease development in humans. In yeast, the robust autophagy response to starvation is controlled by the upregulation of ATG genes, via regulatory processes involving multiple levels of gene expression. Despite the identification of several regulators through genetic studies, the predominant mechanism of regulation modulating the autophagy response to subtle differences in nutrient status remains undefined. Here, we report the unexpected finding that subtle changes in nutrient availability can cause large differences in autophagy flux, governed by hitherto unknown post-transcriptional regulatory mechanisms affecting the expression of the key autophagyinducing kinase Atg1 (ULK1/ULK2 in mammals). We have identified two novel post-transcriptional regulators of ATG1 expression, the kinase Rad53 and the RNA-binding protein Ded1 (DDX3 in mammals). Furthermore, we show that DDX3 regulates ULK1 expression post-transcriptionally, establishing mechanistic conservation and highlighting the power of yeast biology in uncovering regulatory mechanisms that can inform therapeutic approaches.
KEYWORDS: Amino acid starvation, ATG1, autophagosome, autophagy, DDX3, DED1, RAD53, ULK1
Introduction
Macroautophagy (hereafter, autophagy) is a nutrient-recycling pathway conserved among eukaryotes [1]. The hallmark of autophagy is the de novo synthesis of a transient membranous structure which expands to form the double-membrane autophagosome [2,3]. Autophagy occurs basally to maintain homeostasis but is induced in response to various cues, including nutrient-depletion; this type of stress promotes the nonselective sequestration of cytoplasm leading to its subsequent engulfment within the lumen of autophagosomes [4,5]. Autophagosomes subsequently fuse with the lysosomes or vacuole, to promote cargo degradation leading to the generation of simple metabolites that, upon efflux back into the cytosol, act as an alternative source of nutrients [6–8]. The ability to provide nutrients makes autophagy a critical survival pathway in cancer cells [9,10]. Mutant KRAS-driven pancreatic cancers require autophagy-derived nutrients for survival [11–13]. In pancreatic ductal adenocarcinoma, pancreatic stellate cells, present in the tumor microenvironment, upregulate autophagy to generate alanine, which is supplied to the tumor cells to meet their metabolic requirements [14,15]. The identification of autophagy inhibitors has, therefore, gained importance as a therapeutic tool [13,16].
Autophagy inhibition for therapeutic purposes needs to be nuanced because a complete block of autophagy compromises survival [17,18]. This necessitates the need to understand the subtle aspects of autophagy regulation. Even in a simple eukaryote – the budding yeast Saccharomyces cerevisiae – the autophagy pathway is complex and requires the concerted activity of several Atg (autophagy-related) proteins [19,20]. Because autophagy is initiated in response to stresses such as nutrient depletion, the induction of ATG gene expression contrasts with that of most other genes. In yeast and mammalian cells, starvation leads to the activation of several pathways that suppress general transcription and translation but promotes that of ATG genes [21–23]. Furthermore, because it is primarily a degradative process, the cell needs to fine-tune autophagy to meet cellular requirements while preventing unnecessary breakdown of the cytoplasm. The expression of ATG genes is, therefore, subject to a complex regulatory network that acts at transcriptional, post-transcriptional and translational levels [24–26]. Additionally, Atg protein function is extensively regulated by post-translational modifications allowing for the exquisite regulation of autophagy in response to starvation [27,28].
Previous investigations, focused on genetically modulating the transcription of individual ATG genes, demonstrated that ATG8 [29] and ATG9 [30] expression levels are correlated with the size and frequency of autophagosome formation respectively. However, whether these mechanisms are the predominant physiological response to different nutritional challenges remains untested. Here, we study how autophagy is modulated in response to subtle differences in nutrient availability. We do so by comparing autophagy flux during nitrogen and amino acid starvation – two related but distinct starvation conditions – and show that modulation of autophagy under these conditions occurs primarily via the post-transcriptional regulation of ATG gene expression, particularly that of ATG1. Atg1 (ULK1 in mammals) is a Ser/Thr kinase that is critical for the initiation of autophagy and the activation of Atg9 through phosphorylation. We explore the regulation of ATG1 expression under these conditions and identify the kinase Rad53 as a post-transcriptional regulator of starvation-induced autophagy. Furthermore, we have identified a second novel regulator of ATG1 expression, the RNA-helicase Ded1. We show that Ded1 directly binds to the 5ʹ-UTR of ATG1 preferentially during nitrogen starvation, where it likely functions to resolve secondary structures in the ATG1 mRNA to facilitate efficient translation [31,32]. In agreement with this, we show that the loss of Ded1 leads to a greater reduction in Atg1 expression and autophagy during nitrogen starvation relative to amino acid starvation. Crucially, this mode of regulation is conserved – DDX3 (the mammalian homolog of Ded1) positively regulates ULK1 expression post-transcriptionally to promote autophagy in mammalian cells. Consistently, knockdown of DDX3 leads to a reduction in, but not a complete block of, autophagy, thereby making this protein with previously characterized pro-tumorigenic functions [33–35] an attractive candidate for therapeutic exploration.
Results
The difference in autophagy flux between nitrogen and amino acid starvation is not due to differential ATG transcription
In yeast, autophagy is initiated in response to loss of nutrient availability [36]. However, how different starvation stresses differentially modulate regulators to influence autophagy flux is unclear. To shed light on these mechanisms, we investigated the effect of differential nutrient availability on autophagy regulation by comparing complete nitrogen starvation with amino acid starvation [37]. Yeast cells subjected to nitrogen starvation were starved of organic nitrogen and limited for inorganic nitrogen, whereas amino-acid starved cells were deprived only of amino acids. We chose these two conditions for our study because despite being similar stresses overall, they represent the subtle differences that are frequently associated with divergence from homeostasis that promotes physiological responses. Additionally, while these two conditions have been studied in yeast [38], this is, to the best of our knowledge, the first large-scale comparison of the autophagy response between these conditions, thus providing the potential for novel discoveries. The transcription factor Gcn4 is a master regulator of gene expression in response to general amino acid deprivation [39]. Cytoplasmic dearth of amino acids activates the eIF2 kinase Gcn2 which promotes the efficient translation of Gcn4 [40]. Because both nitrogen and amino acid starvation lead to an amino acid deficit, the expression of Gcn4 was increased to very similar levels in cells subjected to either nitrogen starvation (“–N”) or amino acid starvation (“–A”) treatment compared to those grown in the nutrient-rich YPD medium (“+”) (Figures 1A and 1B), highlighting the similar nature of both conditions. To compare autophagy flux between these starvation treatments, we utilized the GFP-Atg8 processing assay as an end-point measurement. We found that nitrogen starvation led to the robust activation of autophagy with the autophagy response being significantly lower with amino acid starvation (Figures 1C and 1D). This finding was corroborated by the prApe1 maturation assay for autophagy flux [41], which measures the autophagy-dependent maturation of precursor aminopeptidase I (prApe1; Figures S1A and S1B). To assess the long-term effect of both starvation treatments, we carried out a longer time-course analysis using the Pho8Δ60 activity assay as an end-point measurement [42]. Extended nitrogen starvation elicited a significantly stronger autophagy response compared to extended amino acid starvation (Figure 1E) while both starvation treatments showed increased autophagy relative to nutrient-rich conditions.
Figure 1.
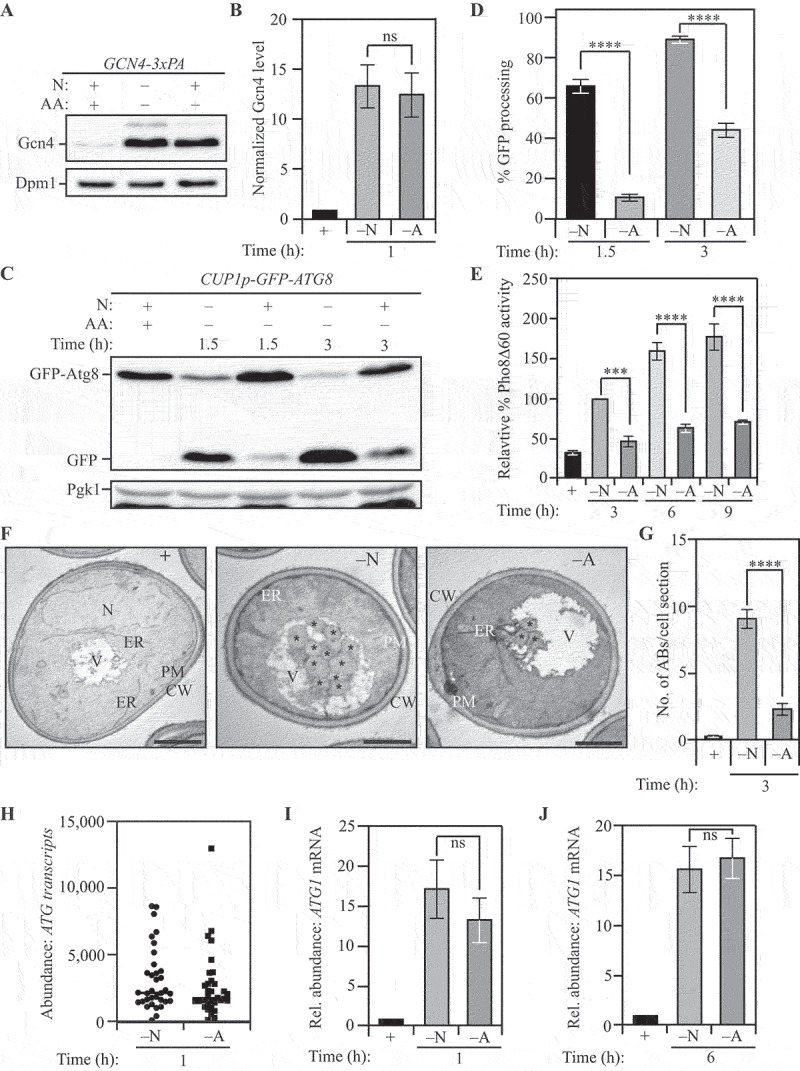
Differential autophagy flux during distinct nutrient stresses is not determined by ATG transcription. (A) Gcn4 expression is upregulated during both nitrogen and amino acid starvation: WT (SEY6210) cells with C-terminally 3x-PA tagged Gcn4 were harvested in nutrient-replete conditions or after starvation for the indicated time and examined by western blot. Gcn4 was detected using the anti-PA antibody and Dpm1 was used as a loading control. (B) Densitometric analysis for (A) from three independent biological replicates. (C) The GFP-Atg8 processing assay demonstrates increased autophagy flux during nitrogen starvation relative to amino acid starvation: WT (WLY176) cells with integrated CUP1p-GFP-ATG8 were harvested in nutrient-replete conditions or after starvation for the indicated times and assessed by western blot. The appearance of free GFP indicates autophagy flux. Pgk1 was used as a loading control. (D) Densitometric analysis of (C) from three independent biological replicates. (E) Autophagy flux is higher during nitrogen starvation compared to amino acid starvation as assessed by the Pho8Δ60 assay: WT (WLY176) cells were harvested in nutrient-replete conditions or after starvation for the indicated times and Pho8Δ60 enzyme activity was measured by colorimetry. An increase in Pho8Δ60 activity indicates increased autophagic flux. Data from three independent biological replicates. (F) Autophagosome formation is more frequent during nitrogen starvation compared to amino acid starvation: WT (SEY6210) pep4∆ vps4∆ cells were harvested in nutrient-replete conditions or after starvation for 3 h. The cells were fixed, stained and ultrastructural analysis was used to visualize the number of ABs. Scale bar: 1 µm. (G) Quantification of the number of ABs from 100 randomly selected cell profiles from two independent biological replicates. (H) RNA-Sequencing reveals similar abundance of ATG transcripts during nitrogen and amino acid starvation: DESeq2 analysis of the ATG transcriptome during nitrogen and amino acid starvation. The plot represents the mean of three independent biological replicates from WT (SEY6210) cells. (I) and (J) There is a similar abundance of ATG1 transcript in cells subjected to nitrogen or amino acid starvation: qRT-PCR detection of ATG1 mRNA in WT (SEY6210) cells after 1 h (I) or 6 h (J) of starvation. ALG9 was used as a reference gene for normalization. Data from three independent biological replicates. Data in (B), (D), (E) and (G-J) represent mean ± SEM from the indicated number of replicates. Statistical analysis for (B), (G), (I) and (J) was carried out using unpaired Student’s t-test whereas (D) and (E) were analyzed using one-way analysis of variance (ANOVA). Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
We then sought to directly examine the characteristics – frequency and size – of autophagosome formation under these starvation conditions [43]. Autophagosomes were monitored by the accumulation of autophagic bodies (ABs; the single-membrane vesicle generated by fusion of an autophagosome with the vacuole) within the vacuole lumen of yeast cells lacking the major vacuolar protease Pep4 (to prevent autophagic body degradation) and Vps4 (to block the accumulation of multivesicular bodies). Consistent with biochemical assays, transmission electron microscopy (TEM) analyses revealed that ABs were more numerous in cells subjected to nitrogen starvation compared to amino acid starvation (Figures 1F and 1G). In addition, ABs in nitrogen-starved cells were significantly larger than in amino-acid starved cells (Figure 1F and S1C). Because the SEY6210 strain, used as the parent for constructing strains for autophagy flux analysis, is an auxotroph, we confirmed the autophagy responses to nitrogen and amino acid starvation using the prototrophic strain CEN.PK [44]. Consistent with the autophagy phenotype in strain SEY6210, we found that nitrogen starvation led to increased Atg8-lipidation. Furthermore, this difference was aggravated upon treatment with the protease inhibitor PMSF, confirming that reduced flux was not responsible for the increased abundance of lipidated Atg8 (Figure S1D).
Autophagy in yeast is robustly regulated by transcriptional control [26,30,45–47], so we hypothesized a differential ATG transcriptome under these starvation conditions. We tested our hypothesis by carrying out RNA-sequencing analysis for both sets of treatments. For high-confidence identification of DEGs (differentially expressed genes), we used the following significance parameters: 2-fold enrichment with an associated p < 0.05 cutoff. Contrary to our expectations, the core ATG genes were not identified among the DEGs (Figure 1H and Data file S1 and S2), with the majority of DEGs involved in translation and metabolism (Figure S1E). ATG31 was the only core autophagy gene along with ATG32 and ATG39, involved in mitophagy [48] and reticulophagy [49], respectively, differentially expressed with higher expression in nitrogen starvation (Figures S1F and S1G, and data not shown). We confirmed that the transcriptional response did not vary with time by measuring the transcriptional upregulation of two genes crucial to the induction of autophagy: ATG1 [50–53] and ATG9 [54–57]. At both 1 h and 6 h post-starvation, nitrogen and amino acid starvation elicited similar levels of transcriptional response for both ATG1 (Figures 1I and 1J) and ATG9 (Figures S1H and S1I) consistent with our findings from the RNA-sequencing experiments. Taken together, these data suggest that differential autophagy flux during nitrogen and amino acid starvation is not due to differential transcriptional activation of ATG genes.
Post-transcriptional mechanisms promote ATG gene expression during nitrogen starvation
The induction of autophagy upon starvation depends on the synthesis of key Atg proteins. For example, Atg1, which is critical for the initiation of autophagy, is robustly synthesized in response to starvation [58]. Because ATG transcription was not differentially affected we investigated the differential expression of Atg proteins that could contribute to differential autophagy flux. To this end, we compared the proteome of cells subjected to nitrogen starvation and amino acid starvation using stable isotope labeling by amino acids in culture (SILAC) (Figure S2A) [53,59]. SILAC analysis revealed that several Atg proteins were differentially expressed, with increased expression in nitrogen starvation (Figure 2A and Data file S3). The proteins with the largest and most consistent differential expression were Atg1 and Atg9 (Figure 2A). The expression of Atg9, a protein responsible for lipid delivery and transfer for phagophore formation [54], was ~45% lower in amino acid starvation compared to nitrogen starvation. We confirmed this observation with immunoblotting for endogenous Atg9 and, consistent with our SILAC analysis, found an ~ 50% reduction in amino acid starvation relative to nitrogen starvation (Figure S2B and S2C). This outcome is consistent with previous findings that suggest Atg9 levels are directly correlated with the frequency of autophagosome formation [30].
Figure 2.
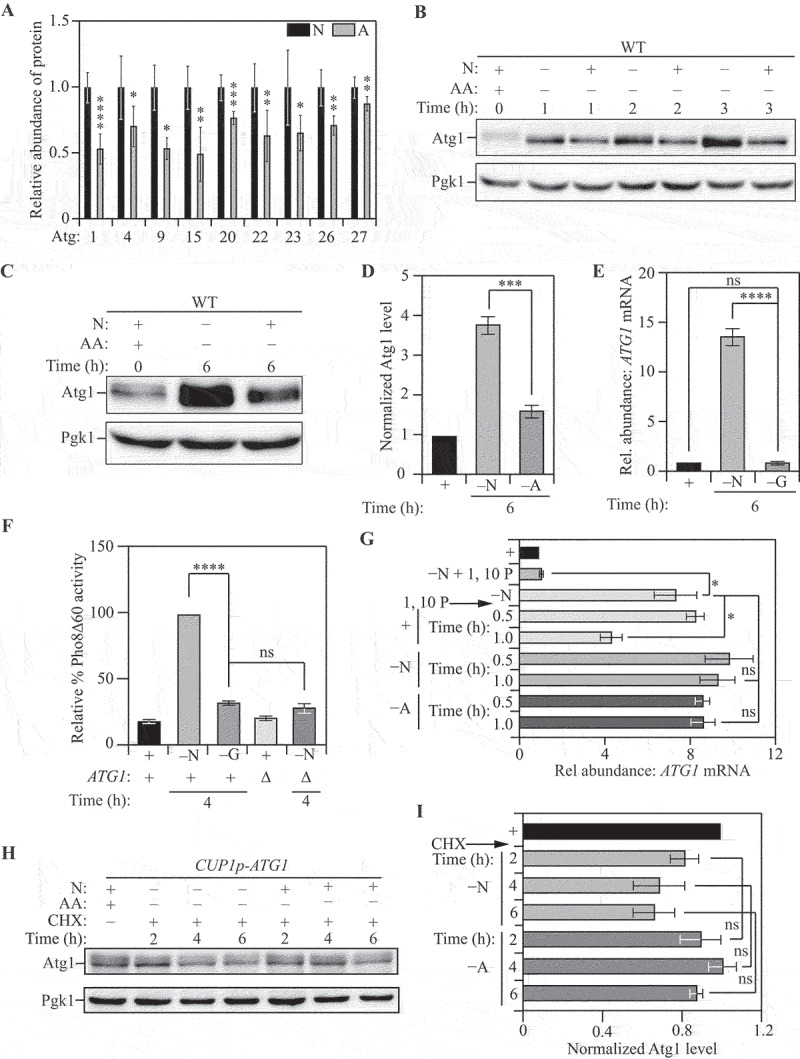
Post-transcriptional activation of ATG gene expression is a critical node determining autophagy during nitrogen starvation. (A) The Atg proteome is significantly different during nitrogen starvation compared to amino acid starvation: Triplex-SILAC labeling was used to compare the Atg protein abundance between nutrient-replete, nitrogen-starvation and amino-acid starvation conditions in SEY6210 arg4∆ cells using LC-MS/MS. The plot shows the levels of differentially expressed Atg proteins during amino acid starvation (gray bars) relative to nitrogen starvation (black bars). Individual proteins were normalized to the total protein input. Data from at least three independent biological replicates. Significant differences are highlighted. (B) and (C) Atg1 levels increase substantially more during nitrogen starvation compared to amino acid starvation. WT (SEY6210) cells were harvested in nutrient-replete conditions or after starvation for the indicated times and protein levels analyzed by western blot. Pgk1 was used as a loading control. (D) Densitometric analysis of (C) from three independent biological replicates. (E) Transcriptional upregulation of ATG1 occurs during nitrogen, but not glucose, starvation: qRT-PCR detection of ATG1 mRNA in WT (SEY6210) cells after 1 h of starvation. ALG9 was used as a reference gene for normalization. Data from three independent biological replicates. (F) Autophagy flux is upregulated during nitrogen, but not glucose, starvation: WT (WLY176) cells were harvested in nutrient-replete conditions or after starvation for the indicated times and Pho8Δ60 enzyme activity was measured by colorimetry. An increase in Pho8Δ60 activity indicates increased autophagic flux. Negative control: SEY6210 atg1∆ cells. Data from three independent biological replicates. (G) ATG1 mRNA is similarly stable under conditions of nitrogen and amino acid starvation: WT (SEY6210) cells were pulsed with nitrogen starvation to induce ATG1 transcription and/or treated with the transcriptional inhibitor 1,10-phenanthroline (1,10 P) to stop transcription. Cells were then kept in nitrogen-starvation medium or transferred to nutrient-replete medium or amino acid starvation for the indicated times. At each time point ATG1 mRNA abundance was measured by qRT-PCR. ALG9 was used as the reference gene for normalization. Data are from three independent biological replicates. (H) and (I) Atg1 protein is not preferentially degraded during amino acid starvation relative to nitrogen starvation: WT (SEY6210) cells harboring a centromeric CUP1p-ATG1 (constitutive Atg1 expression) plasmid were grown in nutrient-replete conditions and treated with cycloheximide (CHX). Following treatment, cells were transferred to nitrogen- or amino-acid starvation medium and harvested at the indicated time points. Atg1 abundance was measured by western blot. Pgk1 was used as a loading control (H). Data from three independent biological replicates (I). Data in (A), (D-G) and (I) represent mean ± SEM from indicated number of replicates. Statistical analysis for (A) and (D) was carried out using unpaired Student’s t-test whereas (E-G) and (I) were analyzed using one-way analysis of variance (ANOVA). Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
In our SILAC analysis, amino-acid starved cells showed an ~50% reduction in Atg1 expression compared to nitrogen-starved cells. In contrast, other components of the Atg1 complex such as Atg13 and Atg17 did not exhibit significant differential expression, prompting us to focus on Atg1. We confirmed differential Atg1 expression by examining endogenous Atg1 levels by immunoblotting. In agreement with our SILAC data, Atg1 levels were found to be 50% lower during amino acid starvation compared to nitrogen starvation at 2 h (Figure 2B and S2D) and ~65% lower at 6 h post-starvation (Figures 2C and 2D). A similar response was observed in the prototrophic CEN.PK strain where nitrogen starvation led to elevated Atg1 expression relative to amino acid starvation (Figure S2E). Taken together, these data suggest that post-transcriptional control is a critical node in the regulation of ATG gene expression that contributes to differential responses in autophagy flux. To rule out the possibility that ATG mRNA transcription is generally increased in response to any type of nutrient depletion, we compared the transcriptional induction of ATG1 and ATG9 during glucose starvation, which fails to significantly stimulate autophagy [7,60]. Expectedly, we found no transcriptional response for either ATG1 (Figure 2E) or ATG9 (Figure S2F), consistent with autophagy flux not being significantly induced, as assessed by the Pho8Δ60 assay (Figure 2F).
To investigate the mechanism of differential regulation further, we focused on ATG1, because Atg1 is responsible for autophagy initiation and regulating Atg9 activity through phosphorylation. We compared the stability of ATG1 mRNA in nitrogen starvation to that in amino acid starvation, to confirm that reduced protein expression during amino acid starvation is not due to mRNA instability. We induced ATG1 transcription with a pulse of nitrogen starvation, following which we treated the cells with the transcriptional inhibitor 1,10-phenanthroline [61]. Cells were then either allowed to recover in rich medium (YPD) or starved in nitrogen-starvation or amino-acid starvation medium to monitor ATG1 mRNA stability (Figure S2G). While recovery in YPD (“+” in Figure 2G) led to a significant reduction in the levels of ATG1 mRNA, there was no decrease in either nitrogen-starvation (“–N”) or amino-acid starvation (“–A”) media, highlighting that ATG1 mRNA was similarly stable under both conditions. Furthermore, to rule out the possibility that the difference in Atg1 levels is due to post-translational instability of the corresponding proteins during amino acid starvation, we used a cycloheximide chase assay. Because Atg1 levels are low during growing conditions and Atg1 is synthesized in response to starvation, we took advantage of a constitutive Atg1 expression system when measuring Atg1 stability. A strain expressing Atg1 from a CUP1 promoter was treated with cycloheximide and Atg1 protein level was followed by immunoblotting after 2, 4 and 6 h of treatment (Figure S2H). We found no significant difference in the stability of Atg1 protein between nitrogen and amino acid starvation (Figures 2H and 2I). Taken together, these findings further suggest that a post-transcriptional mechanism promotes the translation of ATG1 mRNA during nitrogen starvation.
Post-transcriptional regulation of ATG1 expression by Rad53 facilitates nitrogen starvation-induced autophagy
Higher Atg1 levels are correlated with increased autophagy flux. To identify the molecular basis for increased Atg1 expression, we sought to identify regulators that specifically promote autophagy and Atg1 expression during nitrogen starvation. Because kinases are known to be involved in autophagy regulation [62,63], we performed a screen to identify kinases that affected autophagy during nitrogen starvation. A Saccharomyces cerevisiae kinase deletion library, constructed in the BY4742 strain background, was utilized for this screen. Kinase deletion mutants were compared to wild-type BY4742 for identification of differences in autophagy flux. Autophagy flux was monitored by measuring the relative Atg8 degradation rate in the absence or presence of the serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF). In brief, PMSF blocks the vacuolar degradation of Atg8 causing an increased accumulation of Atg8–PE when autophagy flux is high [64]. From this preliminary analysis we determined that the DNA damage response-related kinase Rad53 [65–67] is a potential regulator of autophagy and that the loss of Rad53 led to a 40% decrease in autophagy flux during nitrogen starvation (Figure S3A and S3B; data for other kinases not shown). Whereas Rad53 has been previously identified as a regulator of genotoxic-stress induced autophagy, its role in starvation-induced autophagy is unexplored [68]. Consistent with this finding, compared to wild-type (WT) cells, rad53∆ sml1∆ cells (deletion of SML1 is essential for the viability of the rad53∆ strain) exhibited ~50% lower levels of Atg1 after nitrogen starvation, while the expression of Atg1 during amino acid starvation was not significantly affected (Figures 3A and 3B). During genotoxic stress, the regulation of autophagy by Rad53 occurs at the transcriptional level [68]. To determine if the effect on Atg1 expression was post-transcriptional, we probed the level of ATG1 mRNA in WT and rad53∆ sml1∆ cells using qRT-PCR and found that the steady-state levels of ATG1 transcript was not affected by the deletion of RAD53 during nitrogen starvation (Figure 3C). To investigate the effect of rad53∆ sml1∆ on autophagy flux during the two starvation conditions, we used the Pho8Δ60 assay and found that while the loss of Rad53 led to a 25% reduction in autophagy during nitrogen starvation, it had no effect on autophagy during amino acid starvation (Figure S3C and S3D). Next, we utilized the accumulation of free GFP resulting from the starvation-induced degradation of Pgi1-GFP as a marker for autophagy activity upon prolonged nitrogen starvation [69]. Pgi1-GFP has a longer half-life as an autophagy substrate during starvation relative to GFP-Atg8, preventing substrate exhaustion. Compared to WT cells or sml1∆ cells, rad53∆ sml1∆ cells showed 40% lower Pgi1-GFP processing activity (Figures 3D and 3E) after starvation, confirming that the autophagy phenotype is strictly due to the deletion of RAD53 and not SML1.
Figure 3.
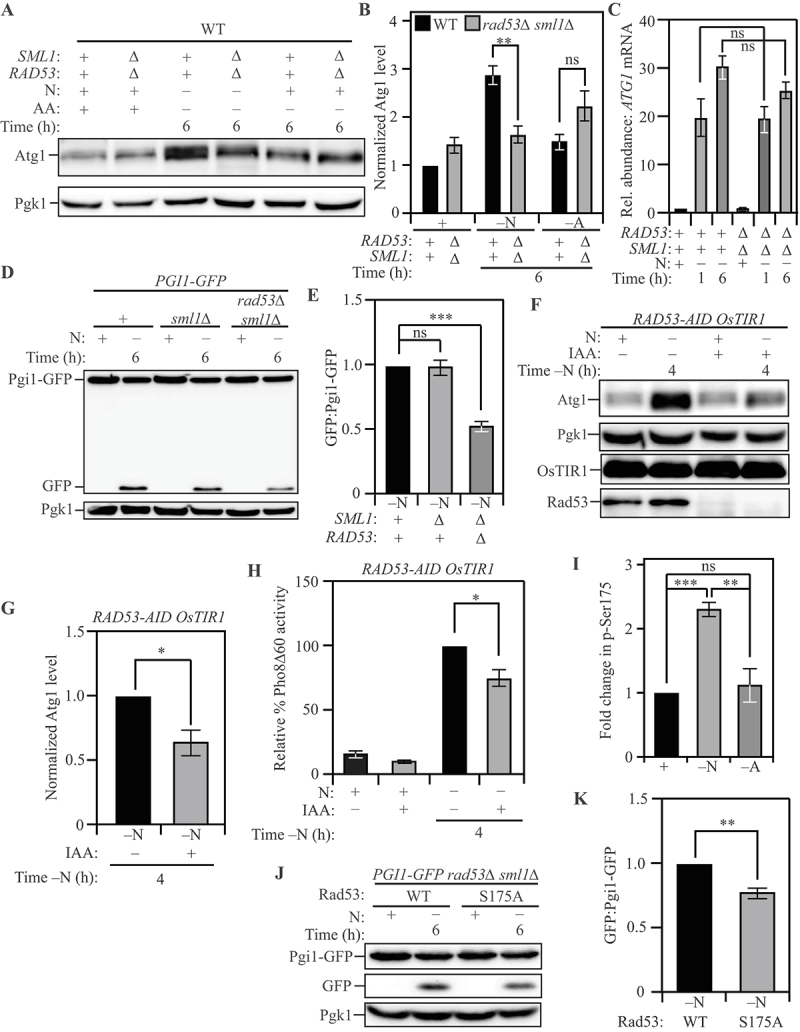
Post-transcriptional regulation of ATG1 expression by Rad53 facilitates nitrogen starvation-induced autophagy. (A) Atg1 levels exhibit a significantly greater increase in WT (SEY6210) cells relative to rad53∆ sml1∆ cells upon nitrogen starvation but not upon amino acid starvation: Cells of the indicated genotypes were harvested during nutrient-replete conditions or after nitrogen or amino acid starvation for the indicated times and protein level examined by western blot. Pgk1 was used as a loading control. (B) Densitometric analyses for (A) from three independent biological replicates. (C) A similar abundance of ATG1 transcript was detected in WT (SEY6210) and rad53∆ sml1∆ cells after nitrogen starvation: Cells of the indicated genotypes were harvested during nutrient-replete conditions or after starvation. qRT-PCR was used to determine ATG1 transcript abundance using ALG9 as the reference gene for normalization. Data from three independent biological replicates. (D) Autophagy flux during nitrogen starvation, assessed by the Pgi1-GFP processing assay, is reduced in rad53∆ sml1∆ cells compared to WT (SEY6210) and sml1∆ cells: Cells of the indicated genotypes, expressing chromosomally tagged Pgi1-GFP were harvested and examined as in (A). The appearance of free GFP indicates autophagy flux. (E) Densitometric analysis of (D) from three independent biological replicates. (F) The acute loss of Rad53 leads to a reduction in Atg1 expression during nitrogen starvation: WT (WLY176) RAD53-AID cells expressing the OsTIR1 ubiquitin ligase were treated with either IAA or DMSO and harvested during nutrient-replete conditions or after nitrogen starvation for the indicated time. IAA treatment activates the ligase activity and targets Rad53-AID for proteasomal degradation. Pgk1 was used as a loading control. (G) Densitometric analysis of (F) from three independent biological replicates. (H) The Pho8Δ60 assay reveals a reduction in autophagy flux during nitrogen starvation caused by the acute loss of Rad53: WT (WLY176) RAD53-AID OsTIR1 cells were harvested during nutrient-replete conditions or after starvation for the indicated time with or without IAA treatment, and Pho8Δ60 enzyme activity was measured by colorimetry. An increase in Pho8Δ60 activity indicates increased autophagic flux. Data are from three independent biological replicates. (I) Rad53 S175 phosphorylation levels are significantly higher in nitrogen-starvation compared to amino-acid starvation or nutrient-replete conditions: Phosphoproteome analysis of SEY6210 arg4∆ cells comparing nitrogen and amino acid starvation using triplex-SILAC labeling and LC-MS/MS analysis. The plot represents data from four independent biological replicates. (J) A non-phosphorylatable mutation of Rad53 S175 (Rad53S175A) reduces autophagy flux during nitrogen starvation, as examined by the Pgi1-GFP processing assay: WT (SEY6210) rad53∆ sml1∆ PGI1-GFP cells expressing either Rad53 or Rad53S175A were harvested during nutrient-replete conditions or after starvation for the indicated time. The appearance of free GFP indicates autophagy flux. Pgk1 used as loading control (K) Densitometric analysis of (J) from three independent biological replicates. Data in (B), (C), (E), (G-I) and (K) represent the mean ± SEM from the indicated number of replicates. Statistical analysis for (B), (C), (E) and (H) was carried out using one-way analysis of variance (ANOVA). (G) and (K) were analyzed using unpaired Student’s t-test whereas (I) was analyzed using paired Student’s t-test. Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
Next, we utilized the differential Atg8 degradation assay to demonstrate that the kinase activity of Rad53 is responsible for its stimulatory effect on autophagy. The kinase-dead Rad53DK227A,D339A mutant of Rad53 [70] exhibited a similar defect in autophagy as the rad53∆ sml1∆ strain (Figure S3E and S3F). To ensure that the autophagy phenotype of the rad53∆ sml1∆ strain is not due to chronic stress caused by the loss of Rad53, we used the auxin-inducible degron (AID) system to achieve tight temporal control of Rad53 loss [71]. Rad53-AID was degraded swiftly upon treatment with IAA (auxin) (Figure 3F; last two lanes). Compared to Rad53-AID cells treated with DMSO (vehicle), IAA-treated Rad53-AID cells showed an ~40% reduction in Atg1 expression (Figures 3F and 3G). Consistent with the results from the rad53∆ sml1∆ cells, the Pho8Δ60 activity was reduced by ~25% in Rad53-AID cells treated with IAA, compared to those treated with DMSO (Figure 3H).
The canonical activation of Rad53 downstream of DNA damage is orchestrated by the Mec1 kinase [72]. Therefore, we tested whether Mec1 has any role in autophagy during nitrogen starvation. Accordingly, we constructed a MEC1-AID strain to probe if Mec1 plays a role in starvation-induced autophagy. IAA treatment in this strain did not result in decreased Atg1 expression (Figures S4A and S4B), or reduced autophagy flux as measured by the Pho8Δ60 activity assay (Figure S4C), compared to treatment with DMSO. This contrasts with DNA damage-induced autophagy where Mec1 is involved in autophagy regulation [68], highlighting the fact that the role of Rad53 in nitrogen-starvation induced autophagy is distinct from its role in DNA-damage induced autophagy.
To probe selective Rad53 activation during nitrogen starvation we looked at differential phosphorylation of Rad53 between the two starvation conditions. The S175 site of Rad53 was previously identified by several large-scale phosphoproteome studies [73,74], but the kinase responsible for the phosphorylation remains unclear [75]. Furthermore, a recent study demonstrated that unlike Rad53 S560, which is phosphorylated extensively in response to DNA damage, Rad53 S175 is only modestly phosphorylated, suggesting a distinct regulatory function for this site [76]. Our SILAC analysis identified S175 on Rad53 as a site that was more strongly phosphorylated during nitrogen starvation relative to amino acid starvation (Figure 3I). We reasoned that if this phosphorylation is critical for the autophagy-stimulating effect of Rad53, mutation of the residue to an alanine should dampen autophagy during nitrogen starvation. Indeed, the plasmid-based re-introduction of the Rad53S175A mutant in a rad53∆ sml1∆ background revealed a partial ~25% reduction in autophagy flux, as measured by the Pgi1-GFP processing assay, compared to the re-introduction of WT Rad53 (Figures 3J and 3K). This finding indicates that the S175 site is likely an important site for Rad53 activation during nitrogen starvation but may not be the sole activation site for Rad53.
In-silico analysis suggested that the S175 residue is likely to be phosphorylated by a proline-directed kinase. Because the proline-directed kinase Cdc28 is known to regulate Rad53 phosphorylation [75,77], we examined whether Cdc28 is responsible for regulating ATG1 expression during nitrogen-starvation induced autophagy. Treatment of a CDC28-AID strain with IAA led to complete loss of Cdc28 (Figure S4D; last two lanes) but had no effect on Atg1 levels (Figures S4D and S4E) or autophagy flux (Figure S4F) indicating that Cdc28 is not involved in nitrogen starvation-induced autophagy. Taken together, these data suggest that an unconventional mode of Rad53 activation promotes Atg1 expression and autophagy during nitrogen starvation.
Ded1 binds ATG1 mRNA to promote Atg1 expression and autophagy
Our results indicated that post-transcriptional mechanisms promote the expression of Atg1 during nitrogen starvation relative to amino acid starvation. Therefore, we hypothesized that the regulation occurs via an RNA-binding protein (RBP), which binds ATG1 mRNA preferentially during nitrogen starvation and facilitates its translation. Accordingly, we carried out an unbiased preliminary screen for proteins that bind to the 5ʹ-UTR of ATG1, which identified Ded1 in addition to several previously characterized ATG1 mRNA-binding proteins (see Materials and Methods for details on the screen methodology).
Ded1 is an essential RNA-helicase involved in promoting translation initiation under nutrient-rich conditions [78] that has recently been demonstrated to be a Rad53 substrate [79]. Because Ded1 was identified from a single large-scale dataset, we used RNA-immunoprecipitation (RNA-IP) to verify that Ded1 binds ATG1 mRNA in vivo during nitrogen starvation. For this purpose, we tagged Ded1 with a 3xPA-tag and affinity isolated Ded1-PA, harvested from cells subjected to nitrogen starvation, using IgG-Sepharose beads. This affinity isolation was followed by the extraction of bound RNA and detection using qRT-PCR. As a control, we used a strain where Ded1 was not epitope tagged with PA, which served as the background to eliminate nonspecific isolates [1,69]. Using PGK1 mRNA as an internal control, and normalizing detection to the untagged strain, we found that Ded1 specifically associates with the 5ʹ-UTR of ATG1 mRNA in vivo (Figure 4A). We validated this interaction using a reciprocal approach: we synthesized 500 bp of the 5ʹ-UTR of ATG1 mRNA immediately upstream of the ORF and labeled the synthesized RNA with desthiobiotin. We incubated this RNA with nitrogen-starved yeast cell lysates. Following cross-linking and streptavidin affinity isolation, we probed the interaction between the in vitro synthesized ATG1 5ʹ-UTR fragment (ATG1 fragment) and endogenous epitope-tagged Ded1 (Ded1-PA) from nitrogen starved cell lysates by immunoblotting. Indeed, we found that Ded1-PA exhibited a 4-fold enrichment when affinity isolated with the ATG1 fragment compared to the control RNA fragment (Figures 4B and 4C), indicating specific binding to the ATG1 fragment.
Figure 4.
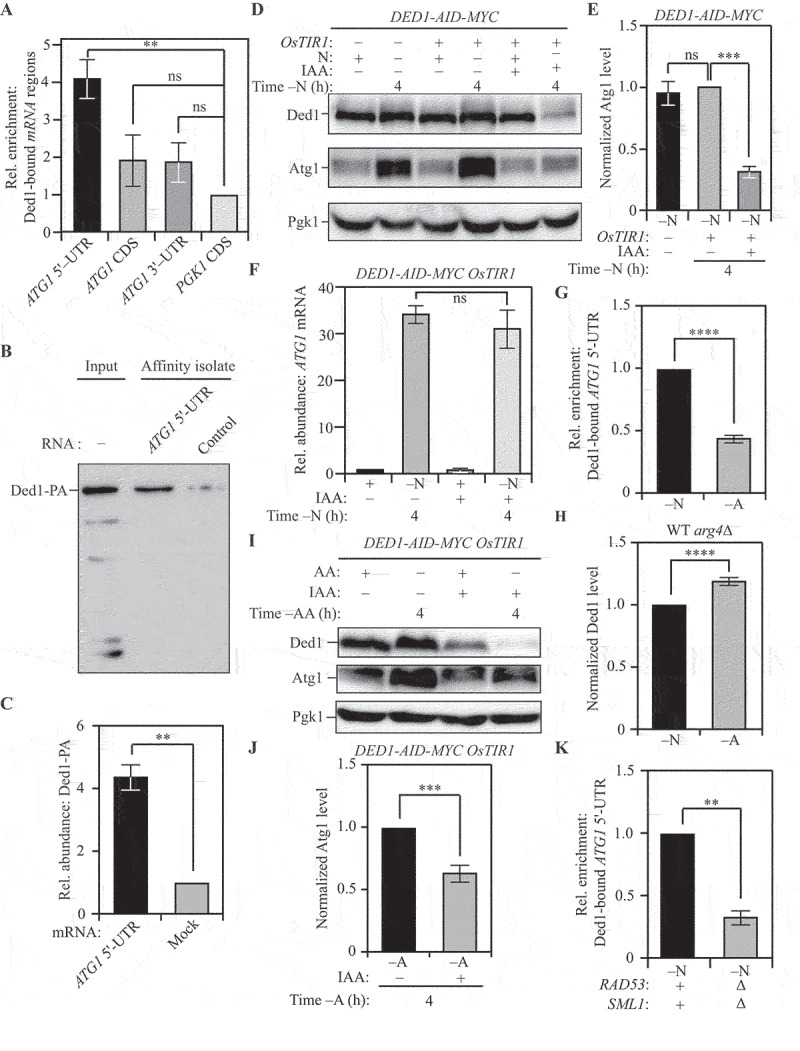
Ded1 binds ATG1 mRNA to promote Atg1 expression. (A) RNA-IP analysis demonstrates Ded1 binding to the 5ʹ-UTR of ATG1 mRNA during nitrogen starvation: PA-tagged Ded1 was immunoprecipitated using IgG-Sepharose beads and bound RNA was amplified and detected by qRT-PCR. Specific primers were used to identify the relative enrichment of the indicated regions of the ATG1 mRNA. Primers targeting the PGK1 mRNA coding sequence (CDS) were used as an internal control. A strain with untagged Ded1 was used as a control for normalization. (B) In vitro RNA affinity isolation confirms interaction between Ded1 and the 5ʹ-UTR of ATG1 mRNA during nitrogen starvation: The sequence of bases from 500 bp upstream of the ATG1 mRNA up to the coding sequence of ATG1 mRNA was synthesized in vitro and labeled with desthibiotin (ATG1 5ʹ-UTR fragment). The fragment was incubated with lysates from WT (SEY6210) Ded1-13xMYC cells. The RNA was affinity isolated using streptavidin, and Ded1 was probed by immunoblotting using anti-MYC antibody. The presence of Ded1 indicates binding to the ATG1 5ʹ-UTR fragment. Mock fragment (random sequence) used as a control (C) Data for (B) from three independent biological replicates. (D) The acute loss of Ded1 leads to reduced Atg1 expression during nitrogen starvation: WT (WLY176) CUP1p-GFP-ATG8 DED1-AID cells without OsTIR1 expression and WT (WLY176) CUP1p-GFP-ATG8 DED1-AID OsTIR1 cells were harvested during nutrient-replete conditions or after nitrogen starvation with or without IAA treatment and protein levels were examined by western blot. Pgk1 was used as a loading control. (E) Densitometric analysis of Atg1 levels from (D) from three independent biological replicates. (F) The acute loss of Ded1 has no effect on ATG1 transcription during nitrogen starvation: Total RNA was isolated from WT (WLY176) CUP1p-GFP-ATG8 DED1-AID OsTIR1 cells during nutrient-replete conditions or after nitrogen starvation with or without IAA treatment. qRT-PCR analysis was used to measure the ATG1 transcript level with ALG9 as a reference gene. Data from three independent biological replicates. (G) The interaction between Ded1 and the 5ʹ-UTR of ATG1 mRNA is stronger during nitrogen starvation compared to amino acid starvation, as demonstrated by RNA-IP analysis. Data are representative of three independent biological replicates. (H) Ded1 levels are higher during amino acid starvation relative to nitrogen starvation: Ded1 levels were measured in WT (SEY6210) arg4Δ cells using SILAC and normalized to total protein input per sample. (I) The acute loss of Ded1 leads to a partial decrease in Atg1 expression during amino acid starvation: WT (WLY176) CUP1p-GFP-ATG8 DED1-AID OsTIR1 cells were harvested during nutrient-replete conditions or after amino acid starvation with or without IAA treatment and protein levels examined by western blot. Pgk1 was used as a loading control. (J) Densitometric analysis of Atg1 levels in (I) from three independent biological replicates. (K) The interaction between Ded1 and ATG1 mRNA is weaker in rad53∆ sml1Δ cells relative to WT (SEY6210) cells during nitrogen starvation, as demonstrated by RNA-IP analysis. Data are representative of three independent biological replicates. Data in (A), (C), (E-H), (J) and (K) represent the mean ± SEM from the indicated number of replicates. Statistical analysis for (A), (E) and (F) was carried out using one-way analysis of variance (ANOVA) whereas (C), (G), (I) and (J) were analyzed using unpaired Student’s t-test. Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
We next tested whether Ded1 has a stimulatory role in Atg1 expression during nitrogen starvation. Because DED1 is an essential gene, we used a temperature sensitive ded1-95 strain (ded1ts) to investigate Atg1 expression at permissive (23°C) and non-permissive (35°C) temperatures relative to WT [80]. At both temperatures, the expression of Atg1 was significantly reduced in the ded1ts strain with a severe 80% reduction at the non-permissive temperature (Figure S5A and S5B). This reduction was post-transcriptional because the steady state levels of ATG1 mRNA were essentially unchanged between the wild-type and the ded1ts strains (Figure S5C).
Contrary to the reduction in Atg1 levels, a significant reduction was not noticed in the levels of Atg9 in the ded1ts strain, which highlights the specificity of Ded1 for ATG1 mRNA (Figures S5D and S5E). To eliminate the possibility of the defects being caused due to chronic stress in the ded1ts strain, we generated an auxin-inducible Ded1 (Ded1-AID) strain to temporally control the loss of Ded1. Treatment with IAA led to degradation of cellular Ded1 (Figure 4D, last lane). We used this strain to probe for differences in Atg1 expression upon Ded1 degradation. Relative to DMSO treatment, degradation of Ded1 by IAA treatment led to an 80% reduction in Atg1 expression (Figures 4D and 4E), consistent with the reduction observed in the ded1ts strain. Once again, loss of Ded1 by IAA treatment did not affect ATG1 mRNA levels (Figure 4F) indicating post-transcriptional regulation. To ensure that the acute loss of Ded1 did not affect general translation, we used Coomassie Brilliant Blue staining to compare total protein profiles of Ded1-AID cells treated with or without IAA after nitrogen starvation (Figure S5F). Quantification of lane profiles indicated that there was no significant decrease in the total protein content upon IAA-mediated Ded1 degradation (Figure S5G).
Next, we tested whether the strength of the interaction between Ded1 and the 5ʹ-UTR of the ATG1 mRNA differed in amino acid starvation relative to nitrogen starvation. Using epitope-tagged Ded1 (Ded1-13xMYC) for RNA-IP, we investigated this interaction in cells subjected to nitrogen starvation and amino acid starvation. Consistent with our hypothesis, Ded1 binding to the 5ʹ-UTR of the ATG1 mRNA was reduced by ~60% in amino acid starvation relative to nitrogen starvation (Figure 4G). This was not due to reduced Ded1 expression because Ded1 levels were higher during amino acid starvation relative to nitrogen starvation (Figure 4H). This finding suggests that while a basal level of Ded1-ATG1 mRNA interaction is present during amino acid starvation, increased Ded1 binding to the ATG1 mRNA promotes increased Atg1 synthesis during nitrogen starvation. Indeed, when probing the levels of Atg1 after amino acid starvation in the Ded1-AID strain, we observed an ~35% reduction in Atg1 level upon Ded1 degradation by IAA (Figures 4I and 4J), compared to the ~75% reduction observed during nitrogen starvation. This result highlights the fact that Ded1 promotes Atg1 expression preferentially during nitrogen starvation.
Finally, to investigate whether Rad53 promotes the binding of Ded1 to ATG1 mRNA, we compared the ability of epitope-tagged Ded1 (Ded1-13xMYC) to bind the 5ʹ-UTR of ATG1 mRNA in WT and rad53∆ sml1∆ cells using RNA-IP. Using PGK1 mRNA as an internal control, we determined that the ability of Ded1 to bind the 5ʹ-UTR of ATG1 mRNA was reduced by 65% in a rad53∆ sml1∆ background (Figure 4K), mirroring the reduction in binding in amino-acid starvation relative to nitrogen-starvation conditions. Taken together, these findings indicate that Ded1 binds the 5ʹ-UTR of ATG1 mRNA. Moreover, they also reveal that this binding preferentially occurs during nitrogen starvation and is mediated, at least in part, by Rad53.
Having determined that Ded1 regulates Atg1 expression, we investigated the role of Ded1 in autophagy. To measure the impact of Ded1 on autophagy flux, we transformed WT and ded1ts cells with an ATG8-promoter driven GFP-ATG8 plasmid and followed the appearance of free GFP after nitrogen starvation. Autophagy flux was reduced in the ded1ts strain at both permissive and non-permissive temperatures, relative to WT (Figures S5H and S5I). We confirmed this phenotype with biochemical assays utilizing the Ded1-AID strain chromosomally expressing a CUP1 promoter-driven GFP-Atg8, where treatment with IAA led to a 70% reduction in autophagy flux compared to treatment with DMSO (Figures 5A and 5B). This was corroborated by the Pho8Δ60 activity assay, where the loss of Ded1 led to a 60% reduction in autophagy flux (Figure 5C). Mirroring its effect on Atg1 expression during amino acid starvation relative to nitrogen starvation, loss of Ded1 by IAA treatment led to a smaller (~30%) reduction in autophagy flux during amino acid starvation, as assessed by the Pho8Δ60 activity assay (Figure 5D). Next, we used a pep4∆ vps4∆ Ded1-AID strain to directly compare autophagosome formation in the presence and absence of Ded1 (Figure 5E) after nitrogen starvation using TEM. The loss of Ded1 by IAA treatment caused a severe reduction in the number of ABs accumulated within the vacuole, indicating a lower frequency of autophagosome formation (Figures 5F and 5G). The size of the ABs was also reduced in the IAA-treated Ded1-AID cells (Figure 5H). Taken together, these findings implicate Ded1 in the regulation of autophagy flux through the regulation of Atg1 expression (Figure 5I).
Figure 5.
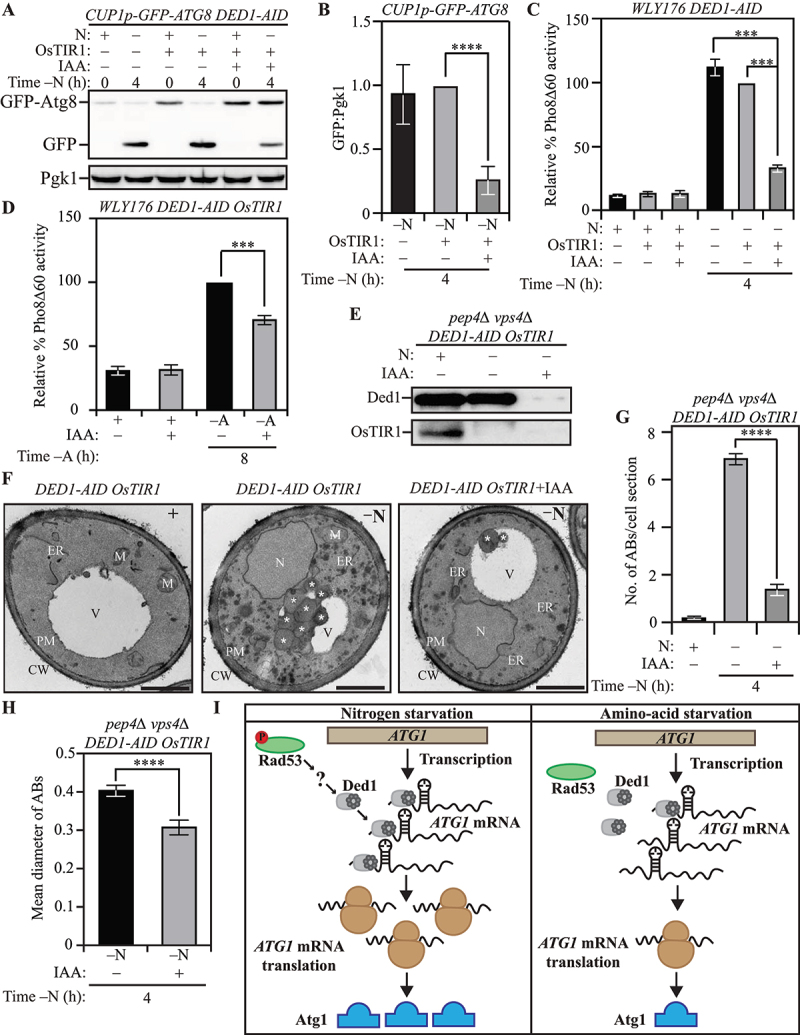
Ded1 regulates autophagy in yeast. (A) The acute loss of Ded1 causes a reduction in autophagy flux during nitrogen starvation as assessed by the GFP-Atg8 processing assay: WT (WLY176) CUP1p-GFP-ATG8 DED1-AID cells lacking OsTIR1 and WT (WLY176) CUP1p-GFP-ATG8 DED1-AID OsTIR1 cells were harvested during nutrient-replete conditions or after nitrogen starvation with or without IAA treatment and examined by western blot. The appearance of free GFP indicates autophagy flux. (B) Densitometric analysis of (A) from three independent biological replicates. (C) and (D) The acute loss of Ded1 reduces autophagy flux significantly more during nitrogen starvation than amino acid starvation as demonstrated by the Pho8Δ60 assay: WT (WLY176) CUP1p-GFP-ATG8 DED1-AID cells and WT (WLY176) CUP1p-GFP-ATG8 DED1-AID OsTIR1 cells were harvested during nutrient-replete and nitrogen-starvation (C) or amino-acid starvation (D) conditions and Pho8Δ60 enzyme activity was measured by colorimetry. An increase in Pho8Δ60 activity indicates increased autophagic flux. Data are from three independent biological replicates. (E) Degradation of Ded1-AID upon IAA treatment in SEY6210 pep4∆ vps4∆ DED1-AID OsTIR1 cells. (F-H) Defects in autophagosome formation during nitrogen starvation due to acute loss of Ded1: (F) SEY6210 pep4∆ vps4∆ DED1-AID OsTIR1 cells were harvested during nutrient-replete conditions or after 3-h nitrogen starvation with or without IAA treatment. The cells were fixed, stained and ultrastructural analysis was used to visualize the number and size of ABs. Scale bar: 1 µm. (G) Quantification of the number of ABs from 100 randomly selected cell profiles from two independent biological replicates. (H) Quantification of the diameter of ABs counted in (G). (I) Schematic depicting the proposed post-transcriptional regulation of ATG1 expression that regulates autophagy differentially between nitrogen and amino acid starvation. Data in (B-D), (G) and (H) represent mean ± SEM from the indicated number of replicates. Statistical analysis was carried out using one-way analysis of variance (ANOVA) Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
DDX3 regulates ULK1 expression and autophagy in mammalian cells
To examine whether the function of Ded1 is conserved, we investigated the ability of DDX3, the mammalian homolog of Ded1 [81], to regulate ULK1 expression. We used the pancreatic ductal cancer-derived cell line PANC-1 as well as the fibrosarcoma-derived cell line HT-1080 to probe for a role of DDX3 in autophagy regulation. We found that stable knockdown (KD) of DDX3 led to a reduction in the level of ULK1 protein (Figure 6A) in PANC-1 cells. Crucially, the reduction in ULK1 levels occurred without reduction in the level of ULK1 mRNA (Figure 6B) indicating post-transcriptional regulation. Next, PANC-1 cells with stable DDX3 KD were treated with the MTOR inhibitor rapamycin for up to 4 h to measure changes in autophagy. At all the time points tested (0.5, 2 and 4 h), DDX3 KD cells (Figures 6C and 6D) showed reduced ULK1 expression (Figures 6C and 6E) and reduced LC3-lipidation ratio (LC3-II:LC3-I; Figures 6C and 6F) relative to control cells. Additionally, co-treatment with bafilomycin A1 enhanced the LC3 lipidation ratio in both control and DDX3 KD cells, with control cells still exhibiting significantly higher levels of lipidated LC3 compared to DDX3 KD. This finding highlights the fact that the decreased LC3 lipidation ratio in DDX3 KD cells could not be attributed to accelerated autophagy flux but was caused by an overall reduction in autophagy [82] (Figures 6C and 6F). Finally, the increased accumulation of SQSTM1 in DDX3 KD cells relative to control cells confirms that autophagy flux was reduced upon loss of DDX3 [83] (Figures 6C and 6G). In contrast, the level of ATG5 was unaffected upon DDX3 KD highlighting the fact that DDX3 loss did not affect general ATG gene expression (Figure 6C and S6A). The phenotypes observed in PANC-1 cells were consistent in HT-1080 cells: stable DDX3 KD (Figures S6B and S6C) led to a reduction in ULK1 levels without alteration of ULK1 mRNA (Figures S6B, S6D and S6E), as well as a reduction in the LC3 lipidation ratio (both with and without bafilomycin A1; Figures S6B and S6F). Similarly, DDX3 KD caused an increased accumulation of SQSTM1 following rapamycin treatment, but no further increase with bafilomycin A1, indicating a reduction in autophagy rather than accelerated flux (Figures S6B and S6G) without affecting ATG5 levels (Figures S6B and S6H). Taken together, these data indicate that DDX3 plays a selective role in modulating ULK1 expression and regulating autophagy in mammalian cells.
Figure 6.
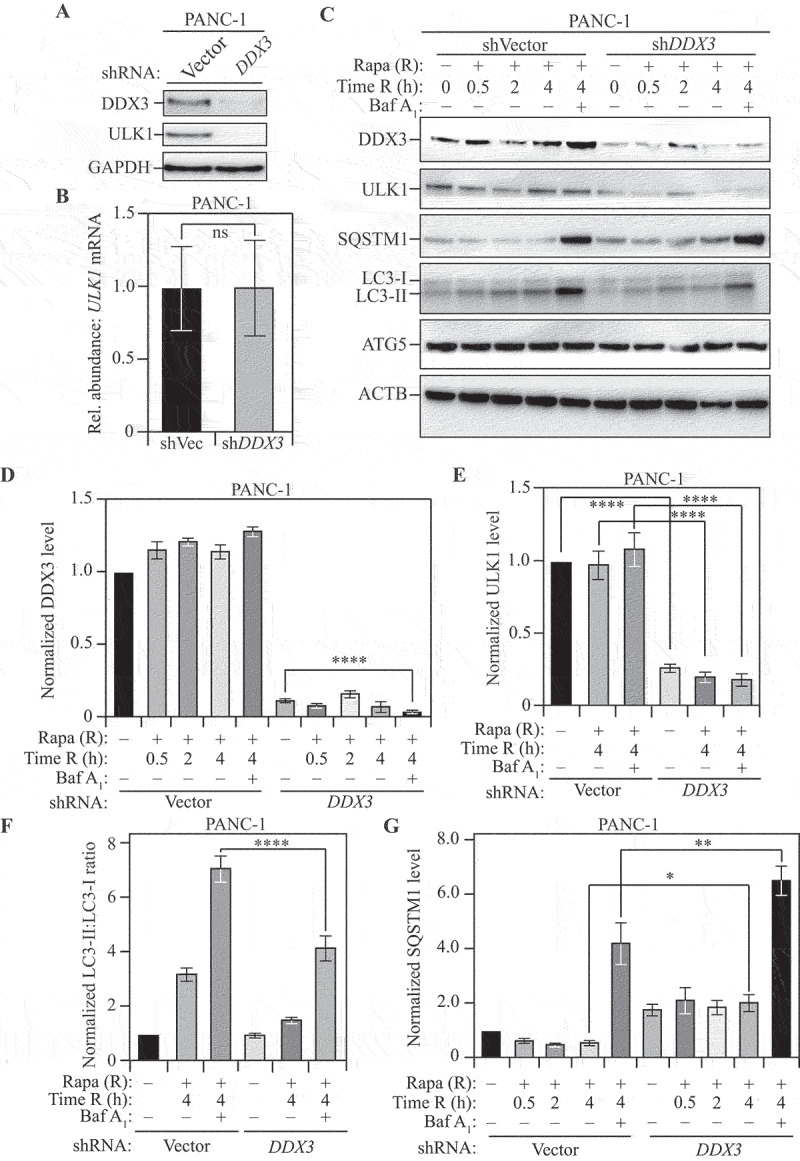
DDX3 regulates autophagy in mammalian cells. (A) Stable shRNA-mediated knockdown of DDX3 in PANC-1 cells: Western blotting to probe for DDX3 and ULK1 levels in cells transfected with shVector (control) or shDDX3. GAPDH was used as a loading control. (B) Relative abundance of ULK1 mRNA in shDDX3 cells compared to control cells. GAPDH was used as a reference gene. Data represent three independent biological replicates. (C) The loss of DDX3 leads to a reduction in ULK1 levels and autophagy in mammalian cells: PANC-1 cells, stably transfected with either vector shRNA (control) or shRNA targeting DDX3, were treated with rapamycin (Rapa; R) for the indicated times with or without co-treatment with bafilomycin A1 (Baf A1). A representative blot shows the levels of DDX3, ULK1, SQSTM1, LC3-I, LC3-II, and ATG5 with ACTB as a loading control, upon harvesting cells at the indicated time points after rapamycin treatment. (D and E) Normalized DDX3 (D) and ULK1 (E) levels at the indicated time points with the indicated treatments. Data represent three independent biological replicates. (F) Normalized LC3-II:LC3-I ratio at the indicated time points with the indicated treatments. Decreased LC3-II:LC3-I ratio in the presence of bafilomycin A1 indicates reduced autophagy flux. Data represent three independent biological replicates. (G) Normalized SQSTM1 level at the indicated time points with the indicated treatments. Increased SQSTM1 accumulation indicates reduced autophagy flux. Data represent three independent biological replicates. Data in (B) and (D-G) represent mean ± SEM from indicated number of replicates. (B) was analyzed using unpaired Student’s t-test whereas the statistical analysis for (D-G) was carried out using one-way analysis of variance (ANOVA). Multiple comparisons were carried out using Tukey’s multiple comparisons test. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001 ns: not significant.
Discussion
Autophagy is a highly complex process, and genetic studies in the model yeast system have been crucial in identifying regulators of autophagy. The expression of ATG1, which encodes the Ser/Thr kinase responsible for autophagy initiation, is subject to multiple levels of regulation. ATG1 is transcriptionally regulated by Gcn4 [45], Pho23 [30] and Rph1 [84]: Gcn4 promotes ATG1 transcription during nitrogen starvation, whereas Pho23 and Rph1 repress transcription during nutrient-replete conditions. The cytoplasmic exoribonuclease Xrn1 regulates the stability of the ATG1 mRNA, mediating its degradation during nutrient-replete conditions [85]. In contrast, the Pat1-Lsm complex prevents 3ʹ-5ʹ degradation of ATG1 mRNA by the exosome during nitrogen starvation, thereby stabilizing the ATG1 mRNA [61]. In nutrient-replete conditions, the RNA helicase Dhh1 associates with Dcp2 to facilitate the degradation of ATG1 transcripts to reduce autophagy [86], while associating with Eap1 to promote ATG1 translation and autophagy during sustained nitrogen starvation [69]. Atg1 expression is enhanced during long-term nitrogen starvation by the RGG motif-containing protein Psp2, which associates with components of the translational machinery eIF4E and eIF4G2, to promote the translation of ATG1 mRNA [58]. However, several questions remain unanswered including which of these mechanisms of regulation is physiologically critical and whether there are yet unknown regulators of ATG1 expression.
We have compared two different nutrient-starvation treatments to uncover the fact that autophagy is physiologically regulated at the level of post-transcriptional control in yeast. ATG1 undergoes transcriptional upregulation during both amino acid starvation and nitrogen starvation but post-transcriptional mechanisms that allow facile translation of the ATG1 mRNA occur simultaneously only during nitrogen starvation. While the physiological rationale driving the disconnect between ATG1 transcription and translation during amino acid starvation is unclear, an attractive hypothesis is as follows: nitrogen starvation imposes a stricter nutrient stress response that warrants swift autophagy activation, but the milder amino acid starvation initiates transcriptional priming without promoting unnecessary self-consumption.
In the process of elucidating cellular mechanisms that promote autophagy during nitrogen starvation, we have uncovered the kinase Rad53 as a post-transcriptional regulator of ATG1 expression. While Rad53 has previously been implicated in regulating autophagy transcriptionally in response to genotoxic stress [68], a role in promoting Atg1 expression post-transcriptionally during nitrogen starvation is novel. We confirmed that there was no differential DNA damage in nitrogen starvation, relative to amino acid starvation, which would be responsible for differential Rad53 activation. Results from our SILAC analysis indicated that conventional markers for DNA damage in yeast including S129 phosphorylation of Hta2 [87–90] and the expression of RNR3 (ribonucleotide reductase 3) [91] were not significantly different between nitrogen and amino acid starvation (Figure S4G and S4H). Additionally, we found no evidence of a role for Mec1 – a key mediator of the DNA-damage response pathway that activates Rad53 – in nitrogen-starvation induced autophagy. Consistent with this novel function of Rad53, we found that a previously identified but incompletely characterized phosphorylation site on Rad53, Ser175, was more abundantly phosphorylated during nitrogen starvation compared to amino acid starvation. Indeed, the Rad53S175A mutant exhibited reduced autophagy flux during nitrogen starvation, supporting selective activation of Rad53 during nitrogen starvation. Previous studies suggest that Rad53 S175 phosphorylation is independent of DNA damage or spindle checkpoint responses and is a proline-directed site likely to be phosphorylated by Cdc28. However, we found that Cdc28 was not involved in autophagy regulation during nitrogen starvation, indicating that Rad53 S175 phosphorylation during nitrogen starvation was not mediated by Cdc28. Our future analyses will focus on the identification of the kinase responsible for this phosphorylation.
We also identified the RNA-helicase Ded1 as a downstream effector that regulates the expression of ATG1 mRNA. The ATG1 mRNA is a highly structured mRNA with stem-loops in the 5ʹ-UTR and the 5ʹ-UTR proximal CDS [69]. We hypothesized that Ded1 would bind the 5ʹ-UTR of ATG1 where it would function to resolve secondary structures to promote facile translation [31,32]. Using RNA-immunoprecipitation, we confirmed that Ded1 binds to the 5ʹ-UTR of ATG1 mRNA. Consistent with its hypothesized role in promoting ATG1 translation during nitrogen starvation, the loss of Ded1 activity led to a reduction in Atg1 expression after nitrogen starvation, without affecting general translation. We also demonstrated that the Ded1-ATG1 mRNA interaction is 60% lower in amino acid starvation relative to nitrogen starvation, highlighting the fact that increased Ded1 binding likely drives increased ATG1 translation during nitrogen starvation. Ded1 has previously been identified as a Rad53 substrate [79], indicating the possibility that Ded1 is regulated differentially by Rad53 during distinct nutrient-starvation conditions. Indeed, we show that in cells lacking Rad53, the interaction between Ded1 and ATG1 mRNA during nitrogen starvation is reduced by ~65%, consistent with the difference seen between the two starvation treatments. TEM analysis revealed a reduction in number and size of autophagosomes following nitrogen starvation in cells suffering transient loss of Ded1, indicating its importance in mediating the autophagy response.
Our study demonstrates an intriguing role for Ded1 within the landscape of regulators identified to modulate ATG1 expression. Whereas negative regulators such as Xrn1 prevent unnecessary ATG1 expression during nutrient-rich conditions, positive regulators such as Pat1 and Psp2 promote ATG1 expression during starvation by stabilizing ATG1 mRNA and promoting ATG1 mRNA translation, respectively. Dhh1 switches from a negative regulatory role to a positive regulatory role as nutrient levels diminish. However, there is a temporal delay among these processes: whereas Pat1 stabilizes ATG1 mRNA shortly after cells are exposed to nitrogen starvation (1–2 h), the roles of Psp2 and Dhh1 in promoting ATG1 mRNA translation occur after extended starvation (24 h). This difference suggests that another regulator is involved in promoting ATG1 translation within this time window, and our data suggest that this regulator is Ded1. Accordingly, strong binding of Ded1 to ATG1 mRNA promotes translation during nitrogen starvation, whereas weaker binding during amino acid starvation leads to decreased Atg1 expression. What happens to the ATG1 transcripts during amino acid starvation is an intriguing question. It is probable that the ATG1 mRNAs are sequestered in specialized RNA-containing structures such as stress granules or P-bodies. Because stress granules and P-bodies have largely been studied in the context of glucose starvation, this will be a challenging but interesting subject of investigation for a subsequent analysis .
Our study also demonstrated that DDX3, the human homolog of Ded1 [81], is involved in the post-transcriptional activation of ULK1 expression, highlighting a conservation of function. DDX3 is a DEAD-box protein involved in RNA metabolism [92], influencing several cellular pathways including cell cycle regulation [93,94], WNT signaling [95–97] and apoptosis [98]. DDX3 has been implicated in stimulatory roles in the development of several cancers including breast cancer, lung cancer and colorectal cancer [33,34,97,99,100]. DDX3 knockdown reduces cell migration and metastasis highlighting the oncogenic role of DDX3 in malignant cancers [35]. Using pancreatic cancer and fibrosarcoma-derived human cell lines, we show that DDX3 is also responsible for mediating the autophagy response; DDX3 KD cells exhibited reduced ULK1 expression and autophagy flux as assessed by LC3-lipidation and SQSTM1-accumulation assays. In mammalian cells, ULK1 and ULK2 show some functional redundancy. It is possible that DDX3 may not regulate ULK2 expression, which may explain why we saw a limited reduction of LC3-II and partial block in autophagy in the knockdown cells. Indeed, a partial block in autophagy may be therapeutically more desirable than a complete block. Additionally, targeting ULK1 for autophagy inhibition is an approach that had already been adopted. For example, SBI-0206965, a small-molecule kinase inhibitor of ULK1, has shown promise in pre-clinical studies in cellular models of cancer [101–103]. Therefore, targeting DDX3 function, which would compromise both autophagy-dependent and autophagy-independent tumor survival pathways, could be an attractive therapeutic avenue for treating autophagy-addicted tumors.
Materials and Methods
Yeast growth and starvation media
Yeast cells were cultured in YPD (Bacto-yeast extract 10 g; Bacto-peptone 20 g; 2% dextrose; double-distilled H2O to 1 L) to mid-log phase (O.D = 0.8–1.0) before harvesting. For SILAC experiments yeast cells were cultured in SMD (0.67% yeast nitrogen base without amino acids; 2% D-glucose; and appropriate amino acids and nucleic acid bases) with light, medium or heavy lysine and arginine. Strains of interest carrying centromeric plasmids were grown in SMD selective medium in which the appropriate amino acids and/or nucleic acid bases were omitted. Nitrogen starvation was carried out in SD(-N) medium (0.17% yeast nitrogen base without amino acids and ammonium sulfate, with 2% glucose). Amino acid starvation was carried out in SD(-A) medium (0.67% yeast nitrogen base without amino acids; 2% D-glucose; and appropriate nucleic acid bases). If the strain of interest was a temperature-sensitive mutant, cells were grown at a permissive temperature and shifted to a nonpermissive temperature for an appropriate period before harvesting.
Protein sample preparation and immunoblotting
For yeast samples, proteins were precipitated using 10% TCA and the cell pellet was washed with acetone and dried. Dried, precipitated cell pellets were lysed by vortexing with glass beads in MURB buffer (50 mM sodium phosphate, pH 7.0, 25 mM MES, 1% SDS [w:v], 3 M urea, 1 mM NaN3, 1% β-mercaptoethanol, 0.01% bromophenol blue) for 5 min. Lysed samples were incubated at 55°C for 15 min before being collected by centrifugation at 10,000xg for 3 min. The supernatant was used as the sample for immunoblotting. Immunoblotting was carried out with standard denaturing SDS-PAGE followed by a semi-dry transfer using a Trans-Blot® SD Semi-Dry Transfer Cell (Bio-Rad). After blocking with TBST containing 5% skim milk for 1 h, the membrane was incubated overnight at 4°C with various primary antibodies (1:1000; anti-YFP [Clontech/Takara Bio, 632381], anti-Dpm1 [Molecular Probes/Fisher, A6429] and rabbit peroxidase-anti-peroxidase [Jackson Immunoresearch, 323-005-024]). After incubation with peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG secondary antibody [Fisher, ICN55676; 1:1000]; rabbit anti-mouse IgG secondary antibody [Jackson, 315-035-003; 1:1000]) for 1 h at room temperature, the signals were visualized by chemiluminescence using Clarity and Clarity Max ECL Western Blotting Substrates (Bio-Rad, 1705061 and 1705062, respectively) on a ChemiDoc Touch Imaging System (Bio-Rad).
For mammalian samples, cells were lysed in 1× cell lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor (Roche, 11836153001) on ice for 10 min. After centrifugation at 14,000 × g for 15 min at 4°C, the supernatants were collected and quantified using the BCA assay (Thermo Fisher Scientific, 23225). Then, 30 μg of each sample were resolved on 4–12% Criterion XT Bis–Tris gels (Bio-Rad, 3450124) in XT MES running buffer (Bio-Rad, 1610789) and transferred to PVDF membranes (Bio-Rad, 1620233) using the Trans-Blot Turbo Transfer Pack and System (Bio-Rad). After blocking with TBST containing 5% skim milk for 1 h, the membrane was incubated overnight at 4°C with various primary antibodies (1:1000). After incubation with peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG secondary antibody [Cell Signaling Technology, 7074; 1:1000]; rabbit anti-goat IgG secondary antibody [Abcam, ab6741; 1:1000]) for 1 h at room temperature, the signals were visualized by chemiluminescence using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34095). We collected protein from each cell line in three biologically independent samples and mixed them together for western blot analysis. The relative intensities of the bands of western blots from three regions were automatically analyzed and normalized to a loading control using the ChemiDoc Touch Imaging System Version 1.2 (Bio-Rad).
RNA isolation, RNA-Sequencing, and qRT-PCR
The RNA extraction protocol and qPCR primers are published previously (Hu et al., 2015). In brief, RNA isolation was performed using the Macherey-Nagel Mini kit (Clontech/Takara Bio, 740955) for RNA purification. For RNA-Sequencing, isolated RNA was frozen and submitted to BGI Genomics Inc. Transcriptome profiling was carried out using the DNBSeq™ technology and bioinformatics analysis was done using three well-established workflows: DESeq2, EBSeq and NOIseq.
For qRT-PCR using yeast cells, cDNA synthesis was carried out using random primers and the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™/Fisher Scientific, 4368814). cDNA samples were analyzed using a Bio-Rad CFX Connect Real-Time System. Samples were tested in Hard-Shell 96-clear well black shell plates (Bio-Rad, HSP9661). The reaction mix (15 μl final volume) consisted of 7.5 μl Radiant Green Lo-ROX qPCR kit (Alkali Scientific, QS1020), 0.6 μl each primer (400 nM final concentration), 1.3 μl H2O, and 5 μl of a 1:5 dilution of the cDNA preparation. The thermocycling program consisted of an initial hold at 95°C for 3 min, followed by 40 cycles of 5 s at 95°C and 25 s at 62°C. After completion, a melting curve was generated to verify PCR specificity, as well as the absence of contamination and primer dimers. The transcript abundance in samples was determined using the CFX Manager Software regression method. Relative abundance of reference mRNAs and normalization for different total RNA amounts was carried out as described previously (Hu et al., 2015) .
For qRT-PCR using mammalian cells, total RNA was extracted and purified from cultured cells using the RNA extraction kit (E.Z.N.A.® HP Total RNA Kit; Omega Bio-tek, R6812) according to the manufacturer’s instructions. The RNA was quantified by determining absorbance at 260 nm. One microgram of total RNA from each sample was reverse transcribed into cDNA using the iScript cDNA synthesis kit (Bio-Rad, 1708890) in a volume of 20 μl; cDNA from cell samples was amplified. The qPCR was performed using 2X SYBR Green q-PCR master mix (Bimake, B21202) on the C1000 Touch Thermocycler CFX96 Real-Time System (Bio-Rad) according to the manufacturer’s protocol. Analysis was performed using Bio-Rad CFX Manager software 3.1 (Bio-Rad). The gene expression was calculated via the 2−ΔΔCt method and normalized to GAPDH. The relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1.
SILAC sample preparation and LC-MS/MS analysis
Samples were prepared as described previously [104]. Briefly, dried TCA-treated cell pellets (50 mg) of each labeling were mixed and lysed in urea buffer (8 M urea, 50 mM Tris-HCl, pH 8.0). Proteins were then alkylated by treatment with 5 mM iodoacetamide for 30 min and digested by Lys-C (lysyl endopeptidase; WAKO Chemicals/Fisher Scientific, NC9223464) for 4 h. The concentration of urea was diluted to 1 M and the peptides were digested with trypsin (Promega, V5113) overnight. On the following day, peptides were acidified and purified by solid-phase extraction using HR-X columns in combination with C18 cartridges (Macherey-Nagel, 731802). Buffers used were as follows: Buffer A, 0.1% formic acid in deionized water; Buffer B, 80% acetonitrile and 0.1% formic acid in deionized water. Eluates were frozen in liquid nitrogen and lyophilized overnight. On the third day, peptides were fractionated by HpH reversed-phase chromatography [105]. The dry peptide powder was suspended in 5% ammonium hydroxide and fractionated using a Waters XBridge BEH130 C18 3.5- μm 4.6- × 250-mm column on an Ultimate 3000 HPLC (Thermo Scientific). Peptides were loaded with 100% HpH buffer A containing 10 mM ammonium formate in deionized water (pH 10) and fractionated by increasing acetonitrile concentration from 1% to 40% using buffer B (10 mM ammonium formate and 90% acetonitrile; pH 10) in 25 min. Ninety-six fractions were collected in a 96-deep well plate. Fractions were mixed with an interval of 12 to yield 8 final fractions. The peptides were acidified, frozen in liquid nitrogen and lyophilized overnight. On the fourth day, the dry peptides were suspended in 200 μl 80% acetonitrile with 0.1% TFA. Phosphopeptides were enriched either by TiO2 beads (GL Sciences, GL-5020-75010) manually [106] or by Fe(III)-NTA cartridges (Agilent, G5496-60085) automatically using the Bravo Automated Liquid Handling Platform (Agilent) [107]. Samples were concentrated by vacuum concentration and resuspended in 20 μL of 0.1% formic acid for LC-MS/MS analysis. The tip flow-through was stored at −80°C for proteome analysis.
LC-MS/MS measurements were performed on a QExactive (QE) Plus and HF-X mass spectrometer coupled to an EasyLC 1000 and EasyLC 1200 nanoflow-HPLC, respectively (all Thermo Scientific). Peptides were fractionated on a fused silica HPLC-column tip (I.D. 75 μm, New Objective, self-packed with ReproSil-Pur 120 C18-AQ, 1.9 μm (Dr. Maisch, r119.ag.0001) to a length of 20 cm) using a gradient of A (0.1% formic acid in water) and B (0.1% formic acid in 80% acetonitrile in water): samples were loaded with 0% B with a flow rate of 600 nL/min; peptides were separated by 5%–30% B within 85 min with a flow rate of 250 nL/min. Spray voltage was set to 2.3 kV and the ion-transfer tube temperature to 250°C; no sheath and auxiliary gas were used. Mass spectrometers were operated in the data-dependent mode; after each MS scan (mass range m/z = 370–1750; resolution: 70,000 for QE Plus and 120,000 for HF-X) a maximum of ten, or twelve MS/MS scans were performed using a normalized collision energy of 25%, a target value of 1,000 (QE Plus)/5,000 (HF-X) and a resolution of 17,500 for QE Plus and 30,000 for HF-X. MS raw files were analyzed using MaxQuant (version 1.6.2.10) [108,109] using a Uniprot full-length S. cerevisiae database (March, 2016) and common contaminants such as keratins and enzymes used for in-gel digestion as reference. Carbamidomethylcysteine was set as fixed modification and protein amino-terminal acetylation, serine, threonine and tyrosine phosphorylation, and oxidation of methionine were set as variable modifications. The MS/MS tolerance was set to 20 ppm and three missed cleavages were allowed using trypsin/P as enzyme specificity. Peptide, site, and protein FDR based on a forward-reverse database were set to 0.01, minimum peptide length was set to 7, the minimum score for modified peptides was 40, and minimum number of peptides for identification of proteins was set to one, which must be unique. The “match-between-run” option was used with a time window of 0.7 min. MaxQuant results were analyzed using Perseus [110].
Ultrastructural analysis
The sample preparation protocol for TEM analysis was adapted from a previously described protocol [43]. SEY6210 pep4∆ vps4∆ or SEY6210 pep4∆ vps4∆ DED1-AID-MYC OsTIR1-MYC cells were cultured in YPD, with or without auxin, and 20 OD600 unit equivalents of cells in log phase were harvested by centrifugation at 3,000 g for 5 min at room temperature (RT). Cells were then washed once with 10 ml of distilled water. Cell pellets were subsequently resuspended in 1 ml of freshly prepared ice-cold 1.5% KMnO4 (Sigma Aldrich, 223468) and transferred into microcentrifuge tubes. The microcentrifuge tubes were filled completely with ice-cold 1.5% KMnO4 to exclude air and incubated on a slow-moving rotating wheel for 30 min at 4°C. Cells were then centrifuged at 3,000 x g for 3 min at 4°C and the supernatant discarded. Pellets were again resuspended in 1.5 ml of ice-cold 1.5% KMnO4 and microcentrifuge tubes incubated on a rotating wheel overnight at 4°C. After 5 washes with 1 ml of distilled water, cells were collected by centrifugation at 5,000 x g for 3 min.
Dehydration was performed by incubating the cells progressively in 1 ml of 10, 30, 50, 70, 90 and 95% acetone (acetone for analysis ENSURE®; MERCK, 1.00014.2500) with at least 20 min incubation after each step at RT. Between incubation steps, cells were collected by centrifugation at 5,000 x g for 4 min. Cells were then incubated 3 times in 1 ml of water-free acetone (dried acetone; MERCK, 1.00299.0500) for at least 20 min each time, on a slow-motion rotating wheel at RT. This was followed by incubation in 33% freshly made Spurr’s resin (11.8 g nonenyl succinic anhydride [Ted Pella, 18301], 8.2 g ERL 4221 epoxide resin [Ted Pella, 18306–4221], 1.9 g diglycidyl ether of poly [propylene glycol] 736 [Ted Pella, 18310], 0.2 g dimethylaminoethanol [Ted Pella, 18315]) on a slow-motion rotating wheel for at least 1 h at RT. Cells were collected by centrifugation at 5,000 x g for 5 min and the supernatants discarded. Following this, cells were incubated in 100% Spurr’s resin on a rotating wheel overnight at RT and collected by centrifugation at 5,000 x g for 5 min and the supernatant discarded. Incubation in 100% Spurr’s resin was repeated for 8 h at RT and the samples transferred to conic embedding capsules (BEEM embedding capsules size 00; EMS, 70010-B). The capsules were centrifuged at 5,000 x g for 5 min and the supernatant discarded. The tubes were then topped with fresh 100% Spurr’s resin and heated at 65°C for 4 days to polymerize the resin.
Ultra-thin 55-nm sections were cut using a Leica ultramicrotome (Leica Microsystems) and collected on 50-mesh copper grids (EMS, G50-Cu) that have been formvar carbon-coated. Cell sections were stained with a filtered lead-citrate solution (80 mM lead nitrate, 120 mM sodium citrate, pH 12) for 2 min at RT. Sections were viewed either in a CM100bio TEM or a Talos F200i (FEI). The average number of ABs per cell section was determined by counting 100 randomly selected cell profiles over 3 grids for each analyzed condition. The average diameter of ABs was measured using the FIJI - ImageJ software [111] and examining at least 100 ABs profiles randomly selected.
Auxin-inducible degradation
S. cerevisiae SEY6210 cells were first transformed with the plasmid pNHK53 (ADH1p-OsTIR1-9MYC). Genes of interest (DED1, RAD53, CDC28 and MEC1) was then tagged with AID-9MYC by homologous recombination. The DNA fragments used for transformation were amplified with pHIS3-AID*-9MYC (Addgene, 99524; deposited by Dr. Helle Ulrich) as the template DNA. The auxin-inducible degron refers to the 71–116 amino acids of the AT1G04250/ATIAA17 protein in plants. To deplete target protein levels, the cells were treated with 500 mM 3-indoleacetic acid (IAA/auxin; Sigma, I2886) or DMSO (vehicle) during mid-log phase growth in YPD medium for 60–90 min (depending on the protein of interest) to induce degradation of the target protein. Subsequently, samples were collected for downstream analyses: enzymatic assays, immunoblots or qRT-PCR.
RNA immunoprecipitation
The RNA immunoprecipitation protocol was modified from previously published procedures [61,69,112]. For determining Ded1-ATG1 mRNA interaction, a Ded1-PA tagged strain and an untagged (control) strain were cultured to mid-log phase and subjected to nitrogen starvation for 4 h. Cross-linking was performed by adding formaldehyde, to a final concentration of 0.8%, and shaking slowly for 10 min at room temperature. Cross-linking was halted with glycine treatment, to a final concentration of 0.2 M, with shaking for 5 min. Cultures were then harvested, washed in PBS, and resuspended in FA lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100 [Sigma, T8787], 0.1% sodium deoxycholate [Sigma, D6750], 0.1% SDS), containing 5 mM PMSF, 1 tablet of complete protease inhibitor cocktail (Roche, 1873580) and RNasin® PLUS RNase inhibitor (Promega/Fisher Scientific, PRN2615). Yeast cells were lysed by vortexing with glass beads (USA Scientific, 7400–2405) at 4°C, centrifuged (5000 x g, 1 min) and the supernatant was collected. Samples were sonicated at 4°C using three 15-s pulses of 45% amplitude, with 60-s pauses for cooling on ice. Sonicated samples were collected by centrifugation (10,000 x g, 10 min), and the supernatant was collected and divided into input and IP fractions. IP fractions were incubated with IgG Sepharose 6 Fast Flow beads (Fisher Scientific, 45000173), overnight with shaking at 4°C, while input fractions were frozen in liquid nitrogen and left at −80°C. IP fractions were washed with FA lysis buffer several times, resuspended in RIP elution buffer (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 1% SDS) and incubated at 70°C for 10 min with intermittent vortexing. IP supernatant and input samples were collected and incubated with a combination of proteinase K (Worthington Biochemical Corporation, LS004249) and RNase inhibitor at 42°C for 1 h, followed by 1 h at 65°C to ensure degradation of proteins bound to RNA.
Next, the samples were treated with an equal volume of acid phenol-chloroform, mixed by vortexing and centrifuged. The aqueous layer of each sample was recovered and treated with 25 µl 3 M sodium acetate, 20 µg glycogen (Roche/Sigma, 10901393001), and 625 µl ice-cold 100% ethanol to precipitate the RNA. Samples were incubated for 1 h to overnight at −80°C, following which they were centrifuged, washed with 70% ethanol, and dried for 15 min. Pellets were resuspended in 90 µl of nuclease-free water and treated with DNase (10 µl TURBO DNase buffer, 2 µl TURBO DNase [TURBO DNA-free kit; Invitrogen/Fisher Scientific, AM1907]) with 0.5 µl of RNasin® PLUS RNase inhibitor . Samples were incubated for 45 min at 37°C to eliminate DNA. Following incubation, DNase was inhibited using the DNase inactivation reagent (provided in the TURBO DNA-free kit). Samples were then subjected to qRT-PCR as described in the qRT-PCR method section.
For determining Ded1-ATG1 mRNA interaction in the WT or rad53∆ sml1∆ background, the procedure followed was the same as described in the earlier paragraph except for the following: 1) Ded1 was tagged C-terminally with 13xMYC; 2) IP fractions were incubated with MYC magnetic beads (Pierce™/Fisher Scientific, 88843) overnight with shaking at 4°C; and 3) magnetic separation was used for collecting beads during the incubation, washing and elution processes.
In vitro RNA interactome capture screen/mRNA IP
RNA in vitro transcription was performed as previously described [58]. The linearized pUC19-ATG1-5ʹ UTR was used as template to carry out RNA transcription using the HiScribe T7 High Yield RNA synthesis kit (NEB, E2040S). The yield of the resulting RNA was measured using a nanodrop and the 3ʹ-end was labeled with desthiobiotin according to the manufacturer’s instructions using the Pierce 3ʹ-End desthiobiotinylation kit (Thermo Fisher Scientific, 20,163). The labeled mRNA was used in the in vitro RNA immunoprecipitation.
Cell pellets were resuspended in lysis buffer (10 mM Tris-HCl, pH 7.5, 0.1 M Nacl, 30 mM MgCl2, 1% Triton X-100, RNasin® PLUS RNase inhibitor and Roche complete protease inhibitor cocktail and mechanically lysed used acid washed beads for 5 min at 4°C. The lysates were centrifuged at 12,000 xg for 10 min and the supernatant was collected. The affinity isolation was set up according to the manufacturer’s instructions (Thermo Fisher Scientific, 20164). Protein enrichment in the labeled RNA reaction was measured either using MS analysis (for the preliminary screen leading to Ded1 identification) or monitored by immunoblotting (for confirming the interaction between Ded1 and the ATG1-5ʹ UTR).
Mammalian cell culture, transfection, and infection
The PANC-1 (CRL-1469) and HT1080 (CCL-121) cell lines were obtained from the American Type Culture Collection. These cells were cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, 11995073) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, A3840001) and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070063) at 37°C, with 95% humidity, and 5% CO2. Lentiviral particles were generated by transfection of the DDX3 shRNA (Sigma, TRCN0000000002 and TRCN0000000003), and the 2nd generation lentiviral systems (viral packaging psPAX2 and viral envelope pMD2G) were collected, mixed with polybrene, and added into 293-T cells using Lipofectamine 2000 (Life Technologies, 11668019) for transfection. Culture media were harvested 48 h after transfection and filtered through 0.45-μm filters. Upon infection, the stable cell lines were established by selecting with 2–5 µg/ml puromycin.
Quantification and statistical analyses
Western blot images were quantified using Bio-Rad ImageLab software. Statistical analyses were performed using GraphPad Prism 9.1.0. Statistical significance was determined in all cases from at least 3 independent biological replicates using either Student’s t test, one-way ANOVA or two-way ANOVA followed by Tukey’s multiple comparisons test. Differences with a P value < 0.05 or lower were considered significant. *p < 0.05, **p < 0.01, ***p < 0.001. Number of independent experiments (n), statistical tests utilized, dispersion of measurements and significance are also described in the figure legends.
Supplementary Material
Acknowledgments
We would like to thank members of the Klionsky group for their constructive suggestions during the study, and BGI Americas Corp for their collaboration on the RNA-Sequencing experiment. V.L. would like to thank Rishav Mitra for insightful comments aiding manuscript preparation and Subhechha Roy for help with figure preparation.
Funding Statement
This work was supported by the National Cancer Institute [R01CA160417]; National Institute of General Medical Sciences [GM131919]; Swiss National Science Foundation [177088] and the canton and University of Fribourg; ENW KLEIN-1 [OCENW.KLEIN.118]; ZonMW TOP [91217002]; ALW Open Programme [ALWOP.355].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Gatica D, Lahiri V, Klionsky DJ.. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018Mar;20(3):233–242. PubMed PMID: 29476151; PubMed Central PMCID: PMCPMC6028034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chang C, Jensen LE, Hurley JH.. Autophagosome biogenesis comes out of the black box. Nat Cell Biol. 2021May;23(5):450–456. PubMed PMID: 33903736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020. Jun 1. 219(6). PubMed PMID: 32357219; PubMed Central PMCID: PMCPMC7265318. DOI: 10.1083/jcb.202002085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Corona Velazquez AF, Jackson WT. So many roads: The multifaceted regulation of autophagy induction. Mol Cell Biol. 2018. Nov 1. 38(21). PubMed PMID: 30126896; PubMed Central PMCID: PMCPMC6189458. DOI: 10.1128/MCB.00303-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. 2020Aug;21(8):439–458. PubMed PMID: 32372019. [DOI] [PubMed] [Google Scholar]
- [6].White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015Nov15;21(22):5037–5046. PubMed PMID: 26567363; PubMed Central PMCID: PMCPMC4646728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liu K, Sutter BM, Tu BP. Autophagy sustains glutamate and aspartate synthesis in Saccharomyces cerevisiae during nitrogen starvation. Nat Commun. 2021Jan4;12(1):57. PubMed PMID: 33397945; PubMed Central PMCID: PMCPMC7782722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].May AI, Prescott M, Ohsumi Y. Autophagy facilitates adaptation of budding yeast to respiratory growth by recycling serine for one-carbon metabolism. Nat Commun. 2020Oct7;11(1):5052. PubMed PMID: 33028817; PubMed Central PMCID: PMCPMC7542147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016Sep1;30(17):1913–1930. PubMed PMID: 27664235; PubMed Central PMCID: PMCPMC5066235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].White E. The role for autophagy in cancer. J Clin Invest. 2015Jan;125(1):42–46. PubMed PMID: 25654549; PubMed Central PMCID: PMCPMC4382247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011Jan15;22(2):165–178. PubMed PMID: 21119005; PubMed Central PMCID: PMCPMC3020913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011Mar1;25(5):460–470. PubMed PMID: 21317241; PubMed Central PMCID: PMCPMC3049287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mulcahy Levy JM, Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020Mar;27(3):843–857. PubMed PMID: 31836831; PubMed Central PMCID: PMCPMC7206017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fu Y, Liu S, Zeng S, et al. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer. 2018Feb19;17(1):62. PubMed PMID: 29458370; PubMed Central PMCID: PMCPMC5817854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016Aug25;536(7617):479–483. PubMed PMID: 27509858; PubMed Central PMCID: PMCPMC5228623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019Sep9;9:1167–1181. PubMed PMID: 31434711; PubMed Central PMCID: PMCPMC7306856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014. Aug;4(8):914–927. PubMed PMID: 24875857; PubMed Central PMCID: PMCPMC4125614. doi: 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020Oct15;383(16):1564–1576. PubMed PMID: 33053285. [DOI] [PubMed] [Google Scholar]
- [19].Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016Dec1;3(12):588–596. PubMed PMID: 28357331; PubMed Central PMCID: PMCPMC5348978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014Jan;241:24–41. PubMed PMID: 24366339; PubMed Central PMCID: PMCPMC3879710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gross AS, Graef M. Mechanisms of autophagy in metabolic stress response. J Mol Biol. 2020Jan3;432(1):28–52. PubMed PMID: 31626805. [DOI] [PubMed] [Google Scholar]
- [22].Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014Jan;24(1):42–57. PubMed PMID: 24343578; PubMed Central PMCID: PMCPMC3879708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011Feb;132:132–141. PubMed PMID: 21258367; PubMed Central PMCID: PMCPMC3987946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Abildgaard MH, Brynjolfsdottir SH, Frankel LB. The Autophagy-RNA Interplay: degradation and Beyond. Trends Biochem Sci. 2020;Oct;45(10):845–857. PubMed PMID: 32828649. [DOI] [PubMed] [Google Scholar]
- [25].Lahiri V, Hawkins WD, Klionsky DJ. Watch what you (self-) eat: Autophagic mechanisms that modulate metabolism. Cell Metab. 2019Apr2;29(4):803–826. PubMed PMID: 30943392; PubMed Central PMCID: PMCPMC6450419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Delorme-Axford E, Klionsky DJ. Transcriptional and post-transcriptional regulation of autophagy in the yeast Saccharomyces cerevisiae. J Biol Chem. 2018Apr13;293(15):5396–5403. PubMed PMID: 29371397; PubMed Central PMCID: PMCPMC5900762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45. PubMed PMID: 25484070; PubMed Central PMCID: PMCPMC4502723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McEwan DG, Dikic I. The three musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011Apr;21(4):195–201. PubMed PMID: 21277210; PubMed Central PMCID: PMCPMC3714536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008Aug;19(8):3290–3298. PubMed PMID: 18508918; PubMed Central PMCID: PMCPMC2488302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jin M, He D, Backues SK, et al. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr Biol. 2014Jun16;24(12):1314–1322. PubMed PMID: 24881874; PubMed Central PMCID: PMCPMC4169046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sen ND, Zhou F, Ingolia NT, et al. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res. 2015Aug;258:1196–1205. PubMed PMID: 26122911; PubMed Central PMCID: PMCPMC4510003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sen ND, Gupta N, Ka S, et al. Functional interplay between DEAD-box RNA helicases Ded1 and Dbp1 in preinitiation complex attachment and scanning on structured mRNAs in vivo. Nucleic Acids Res. 2019Sep19;47(16):8785–8806. PubMed PMID: 31299079; PubMed Central PMCID: PMCPMC7145680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wilky BA, Kim C, McCarty G, et al. RNA helicase DDX3: a novel therapeutic target in Ewing sarcoma. Oncogene. 2016May19;35(20):2574–2583. PubMed PMID: 26364611. [DOI] [PubMed] [Google Scholar]
- [34].Botlagunta M, Vesuna F, Mironchik Y, et al. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008Jun26;27(28):3912–3922. PubMed PMID: 18264132; PubMed Central PMCID: PMCPMC5576029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen HH, Yu HI, Cho WC, et al. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene. 2015May21;34(21):2790–2800. PubMed PMID: 25043297. [DOI] [PubMed] [Google Scholar]
- [36].Cebollero E, Reggiori F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2009Sep;1793(9):1413–1421. PubMed PMID: 19344676. [DOI] [PubMed] [Google Scholar]
- [37].Conrad M, Schothorst J, Kankipati HN, et al. Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev. 2014Mar;382:254–299. PubMed PMID: 24483210; PubMed Central PMCID: PMCPMC4238866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ecker N, Mor A, Journo D, et al. Induction of autophagic flux by amino acid deprivation is distinct from nitrogen starvation-induced macroautophagy. Autophagy. 2010. Oct;6(7):879–890. PubMed PMID: 20647741. doi: 10.4161/auto.6.7.12753 [DOI] [PubMed] [Google Scholar]
- [39].Natarajan K, Meyer MR, Jackson BM, et al. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol. 2001Jul;2113:4347–4368. PubMed PMID: 11390663; PubMed Central PMCID: PMCPMC87095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dever TE, Feng L, Wek RC, et al. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992Feb7;68(3):585–596. PubMed PMID: 1739968. [DOI] [PubMed] [Google Scholar]
- [41].Huang WP, Shintani T, Xie Z. Assays for autophagy I: the Cvt pathway and nonselective autophagy. Methods Mol Biol. 2014;1163:153–164. PubMed PMID: 24841304. [DOI] [PubMed] [Google Scholar]
- [42].Klionsky DJ. Monitoring autophagy in yeast: the Pho8Delta60 assay. Methods Mol Biol. 2007;390:363–371. PubMed PMID: 17951700. [DOI] [PubMed] [Google Scholar]
- [43].Backues SK, Chen D, Ruan J, et al. Estimating the size and number of autophagic bodies by electron microscopy. Autophagy. 2014Jan;10(1):155–164. PubMed PMID: 24270884; PubMed Central PMCID: PMCPMC4389869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kummel A, Ewald JC, Fendt SM, et al. Differential glucose repression in common yeast strains in response to HXK2 deletion. FEMS Yeast Res. 2010May;10(3):322–332. PubMed PMID: 20199578. [DOI] [PubMed] [Google Scholar]
- [45].Bernard A, Jin M, Xu Z, et al. A large-scale analysis of autophagy-related gene expression identifies new regulators of autophagy. Autophagy. 2015Nov2;11(11):2114–2122. PubMed PMID: 26649943; PubMed Central PMCID: PMCPMC4824583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Backues SK, Lynch-Day MA, Klionsky DJ. The Ume6-Sin3-Rpd3 complex regulates ATG8 transcription to control autophagosome size. Autophagy. 2012Dec8;12:1835–1836. PubMed PMID: 22960621; PubMed Central PMCID: PMCPMC3541296. DOI: 10.4161/auto.21845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jin M, Klionsky DJ. Regulation of autophagy: modulation of the size and number of autophagosomes. FEBS Lett. 2014Aug1;588(15):2457–2463. PubMed PMID: 24928445; PubMed Central PMCID: PMCPMC4118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kanki T, Wang K, Cao Y, et al. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009Jul;171:98–109. PubMed PMID: 19619495; PubMed Central PMCID: PMCPMC2746076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mochida K, Oikawa Y, Kimura Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015Jun18;522(7556):359–362. PubMed PMID: 26040717. [DOI] [PubMed] [Google Scholar]
- [50].Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010Apr;22(2):132–139. PubMed PMID: 20056399. [DOI] [PubMed] [Google Scholar]
- [51].Noda T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules. 2017. Jul 7. 7(3). PubMed PMID: 28686223; PubMed Central PMCID: PMCPMC5618233. DOI: 10.3390/biom7030052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Papinski D, Schuschnig M, Reiter W, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014Feb6;53(3):471–483. PubMed PMID: 24440502; PubMed Central PMCID: PMCPMC3978657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hu Z, Raucci S, Jaquenoud M, et al. Multilayered control of protein turnover by TORC1 and Atg1. Cell Rep. 2019Sep24;28(13):3486–3496e6. PubMed PMID: 31553916. [DOI] [PubMed] [Google Scholar]
- [54].Matoba K, Kotani T, Tsutsumi A, et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol. 2020Dec;2712:1185–1193. PubMed PMID: 33106658. [DOI] [PubMed] [Google Scholar]
- [55].Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012Jul23;198(2):219–233. PubMed PMID: 22826123; PubMed Central PMCID: PMCPMC3410421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gomez-Sanchez R, Rose J, Guimaraes R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 2018Aug6;217(8):2743–2763. PubMed PMID: 29848619; PubMed Central PMCID: PMCPMC6080931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sawa-Makarska J, Baumann V, Coudevylle N, et al.Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science. 2020Sep4; 369(6508):PubMed PMID: 32883836; PubMed Central PMCID: PMCPMC7610778. doi: 10.1126/science.aaz7714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yin Z, Liu X, Ariosa A, et al. Psp2, a novel regulator of autophagy that promotes autophagy-related protein translation. Cell Res. 2019Dec;2912:994–1008. PubMed PMID: 31666677; PubMed Central PMCID: PMCPMC6951345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Deng J, Erdjument-Bromage H, Neubert TA. Quantitative comparison of proteomes using SILAC. Curr Protoc Protein Sci. 2019Feb;95(1):e74. PubMed PMID: 30238645; PubMed Central PMCID: PMCPMC6342620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lang MJ, Martinez-Marquez JY, Prosser DC, et al. Glucose starvation inhibits autophagy via vacuolar hydrolysis and induces plasma membrane internalization by down-regulating recycling. J Biol Chem. 2014Jun13;289(24):16736–16747. PubMed PMID: 24753258; PubMed Central PMCID: PMCPMC4059118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gatica D, Hu G, Liu X, et al. The Pat1-Lsm complex stabilizes ATG mRNA during nitrogen starvation-induced autophagy. Mol Cell. 2019Jan17;73(2):314–324e4. PubMed PMID: 30527663; PubMed Central PMCID: PMCPMC6338489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Licheva M, Raman B, Kraft C, et al. Phosphoregulation of the autophagy machinery by kinases and phosphatases. Autophagy. May 10 2021;1–20. PubMed PMID: 33970777. doi: 10.1080/15548627.2021.1909407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sridharan S, Jain K, Basu A. Regulation of autophagy by kinases. Cancers (Basel). 2011Jun9;3(2):2630–2654. PubMed PMID: 24212825; PubMed Central PMCID: PMCPMC3757434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Steinfeld N, Lahiri V, Morrison A, et al. Elevating PI3P drives select downstream membrane trafficking pathways. Mol Biol Cell. 2021Jan15;32(2):143–156. PubMed PMID: 33237833; PubMed Central PMCID: PMCPMC8120694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jung KW, Lee Y, Huh EY, et al.Rad53- and Chk1-dependent DNA damage response pathways cooperatively promote fungal pathogenesis and modulate antifungal drug susceptibility. mBio. 2019Jan2; 10(1):PubMed PMID: 30602579; PubMed Central PMCID: PMCPMC6315099. doi: 10.1128/mBio.01726-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Szyjka SJ, Aparicio JG, Viggiani CJ, et al. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008Jul15;22(14):1906–1920. PubMed PMID: 18628397; PubMed Central PMCID: PMCPMC2492737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cartagena-Lirola H, Guerini I, Manfrini N, et al. Role of the Saccharomyces cerevisiae Rad53 checkpoint kinase in signaling double-strand breaks during the meiotic cell cycle. Mol Cell Biol. 2008Jul;2814:4480–4493. PubMed PMID: 18505828; PubMed Central PMCID: PMCPMC2447123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Eapen VV, Waterman DP, Bernard A, et al. A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc Natl Acad Sci U S A. 2017Feb14;114(7):E1158–E1167. PubMed PMID: 28154131; PubMed Central PMCID: PMCPMC5320992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu X, Yao Z, Jin M, et al. Dhh1 promotes autophagy-related protein translation during nitrogen starvation. PLoS Biol. 2019Apr;174:e3000219. PubMed PMID: 30973873; PubMed Central PMCID: PMCPMC6459490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Holzen TM, Sclafani R. Genetic interaction of RAD53 protein kinase with histones is important for DNA replication. Cell Cycle. 2010Dec1;9(23):4735–4747. PubMed PMID: 21099362; PubMed Central PMCID: PMCPMC3048039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Morawska M, Ulrich HD. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast. 2013Sep;30(9):341–351. PubMed PMID: 23836714; PubMed Central PMCID: PMCPMC4171812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sweeney FD, Yang F, Chi A, et al. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol. 2005Aug9;15(15):1364–1375. PubMed PMID: 16085488. [DOI] [PubMed] [Google Scholar]
- [73].Chen ES, Hoch NC, Wang SC, et al. Use of quantitative mass spectrometric analysis to elucidate the mechanisms of phospho-priming and auto-activation of the checkpoint kinase Rad53 in vivo. Mol Cell Proteomics. 2014Feb;132:551–565. PubMed PMID: 33498138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Albuquerque CP, Smolka MB, Payne SH, et al. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics. 2008. Jul 7;7:1389–1396. PubMed PMID: 18407956; PubMed Central PMCID: PMCPMC2493382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Schleker T, Shimada K, Sack R, et al. Cell cycle-dependent phosphorylation of Rad53 kinase by Cdc5 and Cdc28 modulates checkpoint adaptation. Cell Cycle. 2010Jan15;9(2):350–363. PubMed PMID: 20046099. [DOI] [PubMed] [Google Scholar]
- [76].Lanz MC, Yugandhar K, Gupta S, et al. In-depth and 3-dimensional exploration of the budding yeast phosphoproteome. EMBO Rep. 2021Feb3;22(2):e51121. PubMed PMID: 33491328; PubMed Central PMCID: PMCPMC7857435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Abreu CM, Kumar R, Hamilton D, et al. Site-specific phosphorylation of the DNA damage response mediator rad9 by cyclin-dependent kinases regulates activation of checkpoint kinase 1. PLoS Genet. 2013Apr;94:e1003310. PubMed PMID: 23593009; PubMed Central PMCID: PMCPMC3616908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].De La Cruz J, Iost I, Kressler D, et al. The p20 and Ded1 proteins have antagonistic roles in eIF4E-dependent translation in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1997May13;94(10):5201–5206. PubMed PMID: 9144215; PubMed Central PMCID: PMCPMC24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lao JP, Ulrich KM, Johnson JR, et al. The yeast DNA damage checkpoint kinase Rad53 targets the exoribonuclease, Xrn1. G3 (Bethesda). 2018Dec10;8(12):3931–3944. PubMed PMID: 30377154; PubMed Central PMCID: PMCPMC6288840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Burckin T, Nagel R, Mandel-Gutfreund Y, et al. Exploring functional relationships between components of the gene expression machinery. Nat Struct Mol Biol. 2005Feb;122:175–182. PubMed PMID: 15702072. [DOI] [PubMed] [Google Scholar]
- [81].Tarn WY, Chang TH. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol. 2009Jan-Mar;6(1):17–20. PubMed PMID: 19106629. [DOI] [PubMed] [Google Scholar]
- [82].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021Jan;171:1–382. PubMed PMID: 33634751; PubMed Central PMCID: PMCPMC7996087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bjorkoy G, Lamark T, Pankiv S, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. PubMed PMID: 19200883. [DOI] [PubMed] [Google Scholar]
- [84].Bernard A, Jin M, Gonzalez-Rodriguez P, et al. Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy. Curr Biol. 2015Mar2;25(5):546–555. PubMed PMID: 25660547; PubMed Central PMCID: PMCPMC4348152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Delorme-Axford E, Abernathy E, Lennemann NJ, et al. The exoribonuclease Xrn1 is a post-transcriptional negative regulator of autophagy. Autophagy. 2018;14(5):898–912. PubMed PMID: 29465287; PubMed Central PMCID: PMCPMC6070002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hu G, McQuiston T, Bernard A, et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat Cell Biol. 2015Jul;177:930–942. PubMed PMID: 26098573; PubMed Central PMCID: PMCPMC4528364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000Dec21-28;408(6815):1001–1004. PubMed PMID: 11140636. [DOI] [PubMed] [Google Scholar]
- [88].Javaheri A, Wysocki R, Jobin-Robitaille O, et al. Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc Natl Acad Sci U S A. 2006Sep12;103(37):13771–13776. PubMed PMID: 16940359; PubMed Central PMCID: PMCPMC1564209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Redon C, Pilch DR, Rogakou EP, et al. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003. Jul;4(7):678–684. PubMed PMID: 12792653; PubMed Central PMCID: PMCPMC1326317. doi: 10.1038/sj.embor.embor871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Redon C, Pilch DR, Bonner WM. Genetic analysis of Saccharomyces cerevisiae H2A serine 129 mutant suggests a functional relationship between H2A and the sister-chromatid cohesion partners Csm3-Tof1 for the repair of topoisomerase I-induced DNA damage. Genetics. 2006Jan;172(1):67–76. PubMed PMID: 16219777; PubMed Central PMCID: PMCPMC1456192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tkach JM, Yimit A, Lee AY, et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol. 2012Sep;149:966–976. PubMed PMID: 22842922; PubMed Central PMCID: PMCPMC3434236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Soto-Rifo R, Ohlmann T. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip Rev RNA. 2013Jul-Aug;4(4):369–385. PubMed PMID: 23606618. [DOI] [PubMed] [Google Scholar]
- [93].Lai MC, Chang WC, Shieh SY, et al. DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol. 2010Nov;30(22):5444–5453. PubMed PMID: 20837705; PubMed Central PMCID: PMCPMC2976371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Heerma van Voss MR, Kammers K, Vesuna F, et al. Global effects of DDX3 Inhibition on cell cycle regulation identified by a combined phosphoproteomics and single cell tracking approach. Transl Oncol. 2018Jun;113:755–763. PubMed PMID: 29684792; PubMed Central PMCID: PMCPMC6050443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zhao L, Mao Y, Zhou J, et al. Multifunctional DDX3: dual roles in various cancer development and its related signaling pathways. Am J Cancer Res. 2016;6(2):387–402. PubMed PMID: 27186411; PubMed Central PMCID: PMCPMC4859668. [PMC free article] [PubMed] [Google Scholar]
- [96].Cruciat CM, Dolde C, de Groot RE, et al. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-beta-catenin signaling. Science. 2013Mar22;339(6126):1436–1441. PubMed PMID: 23413191. [DOI] [PubMed] [Google Scholar]
- [97].Heerma van Voss MR, Vesuna F, Trumpi K, et al. Identification of the DEAD box RNA helicase DDX3 as a therapeutic target in colorectal cancer. Oncotarget. 2015Sep29;6(29):28312–28326. PubMed PMID: 26311743; PubMed Central PMCID: PMCPMC4695062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sun M, Zhou T, Jonasch E, et al. DDX3 regulates DNA damage-induced apoptosis and p53 stabilization. Biochim Biophys Acta. 2013Jun;18336:1489–1497. PubMed PMID: 23470959; PubMed Central PMCID: PMCPMC3638797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bol GM, Xie M, Raman V. DDX3, a potential target for cancer treatment. Mol Cancer. 2015Nov5;14:188. PubMed PMID: 26541825; PubMed Central PMCID: PMCPMC4636063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ariumi Y. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front Genet. 2014;5:423. PubMed PMID: 25538732; PubMed Central PMCID: PMCPMC4257086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Egan DF, Chun MG, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015Jul16;59(2):285–297. PubMed PMID: 26118643; PubMed Central PMCID: PMCPMC4530630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zheng Y, Liu L, Wang Y, et al. Glioblastoma stem cell (GSC)-derived PD-L1-containing exosomes activates AMPK/ULK1 pathway mediated autophagy to increase temozolomide-resistance in glioblastoma. Cell Biosci. 2021Mar31;11(1):63. PubMed PMID: 33789726; PubMed Central PMCID: PMCPMC8011168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Tang F, Hu P, Yang Z, et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol Rep. 2017Jun;376:3449–3458. PubMed PMID: 28498429. [DOI] [PubMed] [Google Scholar]
- [104].Hu Z, Raucci S, Jaquenoud M, et al. Multilayered Control of Protein Turnover by TORC1 and Atg1. Cell Rep. 2019;28(13):3486–3496.e6. [DOI] [PubMed] [Google Scholar]
- [105].Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res. 2014Dec5;13(12):6176–6186. PubMed PMID: 25338131. [DOI] [PubMed] [Google Scholar]
- [106].Zarei M, Sprenger A, Rackiewicz M, et al. Fast and easy phosphopeptide fractionation by combinatorial ERLIC-SCX solid-phase extraction for in-depth phosphoproteome analysis. Nat Protoc. 2016;11(1):37–45. [DOI] [PubMed] [Google Scholar]
- [107].Post H, Penning R, Fitzpatrick MA, et al. Robust, sensitive, and automated phosphopeptide enrichment optimized for low sample amounts applied to primary hippocampal neurons. J Proteome Res. 2017;16(2):728–737. [DOI] [PubMed] [Google Scholar]
- [108].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008Dec;26(12):1367–1372. PubMed PMID: 19029910. [DOI] [PubMed] [Google Scholar]
- [109].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. [DOI] [PubMed] [Google Scholar]
- [110].Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11(12):2301–2319. [DOI] [PubMed] [Google Scholar]
- [111].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012Jul;9(7):671–675. PubMed PMID: 22930834; PubMed Central PMCID: PMCPMC5554542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Selth LA, Gilbert C, Svejstrup JQ. RNA immunoprecipitation to determine RNA-protein associations in vivo. Cold Spring Harb Protoc. 2009Jun;2009(6):pdbprot5234. PubMed PMID: 20147192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.