

Abstract
Metal‐catalyzed C−H activations are environmentally and economically attractive synthetic strategies for the construction of functional molecules as they obviate the need for pre‐functionalized substrates and minimize waste generation. Great challenges reside in the control of selectivities, the utilization of unbiased hydrocarbons, and the operation of atom‐economical dehydrocoupling mechanisms. An especially mild borylation of benzylic CH bonds was developed with the ligand‐free pre‐catalyst Co[N(SiMe3)2]2 and the bench‐stable and inexpensive borylation reagent B2pin2 that produces H2 as the only by‐product. A full set of kinetic, spectroscopic, and preparative mechanistic studies are indicative of a tandem catalysis mechanism of CH‐borylation and dehydrocoupling via molecular CoI catalysts.
Keywords: boranes, C−H activation, cobalt, dehydrocoupling, X-ray absorption spectroscopy
An inexpensive cobalt catalyst enables the combination of benzylic CH‐borylation and dehydrocoupling to an overall acceptorless dehydrogenative functionalization of hydrocarbons at mild conditions. Mechanistic studies indicate a tandem catalysis via molecular CoI catalysts.
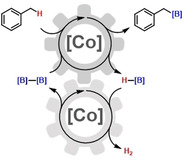
Introduction
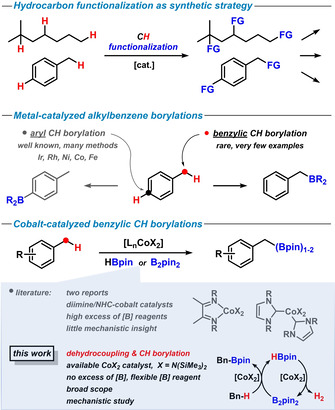
Catalytic methods of CH‐bond functionalization of easily available hydrocarbons constitute key strategic steps in the generation of synthetically valuable molecular building blocks (Scheme 1, top). [1] Among metal‐catalyzed C−H functionalizations, borylation protocols are especially attractive by virtue of the availability of various borane reagents and the diverse reactivity of the resultant polarized C−B bond in onward reactions, for example, oxidation, cross‐coupling, insertion, metallation, rearrangement.[ 2 , 3 ] Major progress was made in the development of transition metal‐catalyzed arene borylations that provide arylboron derivatives (Scheme 1, middle).[ 2 , 3 , 4 , 5 ] Iridium catalysts have most successfully been employed in these reactions that operate via oxidative addition of the aryl‐H substrates with high tolerance of functional groups. Alkyl‐H moieties are typically much less reactive under these conditions. CH‐borylations of alkyl‐H moieties are far less advanced and mostly require pre‐functionalized substrates, directing groups, and/or forcing conditions.[ 4b , 6 ] Very few protocols of undirected Csp3−H borylations in benzylic positions have been reported with Ir, Rh, Pd, Co and Ni catalysts.[ 5c , 7 , 8 , 9 , 10 ] Recently, benzylic CH‐borylations were observed with two distinct L2CoX2 pre‐catalysts: An α‐diimine cobalt(II) bis(carboxylate) enabled geminal diborylation of alkyl‐arenes using combinations of B2pin2 and HBpin as borylation reagents in excess (pin=pinacolato). [8] A cobalt(II) t‐butoxide bearing N‐heterocyclic carbene ligands afforded mixtures of mono/diboronates in the presence of excess amounts of HBpin. [9] Despite recent progress, the development of efficient methods of selective Csp3−H borylations with first‐row transition metals is still in its infancy. Reactions that operate with simple, inexpensive catalysts under mild conditions and display broad substrate scope, high product selectivity, and effective use of the employed borane reagents are being sought. Further, there is very little knowledge of the underlying mechanisms. Here, we report an efficient cobalt‐catalyzed CH‐borylation protocol with cobalt(II) bis(hexamethyldisilazide), Co(hmds)2, under mild conditions. The inexpensive catalyst fulfills dual roles: i) catalytic borylation of benzyl Csp3−H positions with B2pin2, and ii) catalytic dehydrocoupling of HBpin. This tandem cobalt catalysis allows the flexible use of borylation reagents (HBpin or B2pin2) and the complete transfer of all boron atoms in the hydrocarbon borylation (Scheme 1, bottom).
Scheme 1.
Catalytic borylations as strategic hydrocarbon functionalization (top). Cobalt‐catalyzed benzylic CH borylations (bottom).
Results and Discussion
In continuation of recent studies of catalytic hydrogenations and hydrofunctionalizations with non‐rare earth metal catalysts (Li, Mn, Fe) in our laboratories, [11] we have considered the related mechanistic scenarios of the reverse reactions involving metal‐catalyzed dehydrocoupling events. [12] The activation of borane reagents R2B‐X by a Lewis basic metal catalyst (such as metal amides) in the presence of activated CH bonds (such as benzyl‐H) may serve as an entry into a CH borylation reaction.[ 2 , 13 ] We initiated our investigations with the model reaction between toluene and the inexpensive and crystalline boronate source bis(pinacolato) diboron, B2pin2 (Scheme 2, top).[ 7 , 8 , 10 ] With 5 mol % Co(hmds)2, excellent conversion and near‐perfect chemo‐specificity toward benzylic CH‐borylation were obtained. The reaction resulted in the clean formation of the double borylation product, benzyl diboronate, at 80 °C in n‐hexane. Fe(hmds)2 gave moderate yields; cobalt catalysts without the basic, nucleophilic, and lipophilic hmds ligand were not active. No boronate products were detected in the absence of catalyst. Interestingly, the reaction exhibited much higher atom economy than related literature reports[ 8 , 9 ] as only 1 equiv B2pin2 sufficed for near‐quantitative diboronate formation. With B2pin2 as starting material, the cobalt‐catalyzed dehydroborylation of toluene is believed to produce low concentrations of HBpin as a primary by‐product which itself is most likely engaging in another dehydrocoupling reaction. The latter event can involve toluene or another molecule of HBpin as reaction partner, respectively, to give benzyl boronate or B2pin2 upon release of 1 equiv H2. In full accord with these hypotheses, both dehydrocoupling reactions operated under very similar conditions with the Co(hmds)2 catalyst (Scheme 2, middle and bottom). The CH‐borylation of toluene with B2pin2 (1 equiv) is about twice as fast as with HBpin (2 equiv) under identical conditions. Both reactions do not require excess amounts of the borylation reagents and produce H2 as the only by‐product (vide infra). The cobalt‐catalyzed toluene borylation protocol appears to be highly selective toward the benzylic Csp3−H position: no reaction of the arene moiety and the solvent n‐hexane was observed. The choice of B2pin2 as the more attractive substrate is governed by its lower price, easier handling, and thermal stability.
Scheme 2.
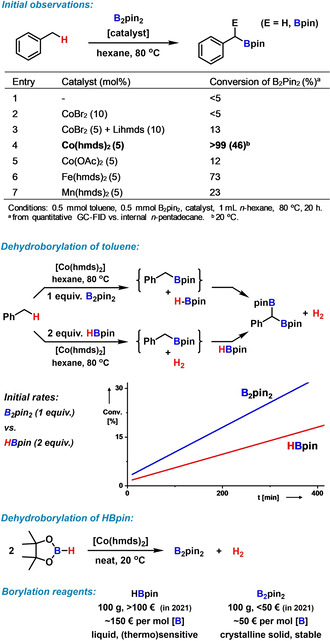
Cobalt‐catalyzed dehydroborylations: Optimization of toluene borylation (top). Dehydroborylations of toluene and HBpin under similar conditions (middle). Choice of borylation reagents (bottom). hmds=hexa‐methyldisilazide, N(SiMe3)2; pin=pinacolato, ‐OCMe2‐CMe2O‐.
The optimized conditions were then applied to a diverse set of substituted alkylarenes (Scheme 3). Substituted toluene derivatives were converted to the benzyl diboronates in moderate to good yields (mostly 50–90 %), while the benzyl monoboronates were only minor products (<5 % for toluene; 10–20 % for other methylarenes). The benzyl diboronates were separated from the monoboronates by SiO2 column chromatography (isolated yields are given). Silyl, Bpin, amine, ether, and alkyl substituents were tolerated at the arene. Reaction of 4‐methoxytoluene gave minor amounts of C−OMe cleavage product (27 %). [14] Very electron‐rich benzyl diboronates were sensitive to protodeborylation and oxidation. The crude mixture of benzyl diboronates and benzyl monoboronates (95/5 to 80/20 ratio) could be converted to the stable benzyl monoboronates in high yields by protolysis with NEt3/SiO2 (isolated yields given). Ortho‐, meta‐, and para‐xylenes gave good yields (65–77 %) of mixtures of α,α‐diboronates, α,α′‐tri‐ and tetra‐boronates with 2.1 equiv B2pin2 (Scheme 3, middle). Geminal α,α‐diborylation was favoured over α,α′‐diborylation. Notably, benzyl diboronates have potential applications in enantioselective reactions [15a] and complex molecule syntheses. [15b] Alkylbenzenes with longer side chains exhibited lower reactivity and afforded the benzyl monoboronates as major products, possibly due to steric repulsion (Scheme 3, bottom). Only 10–20 % of α,α‐diborylation were observed for n‐alkyl benzenes; only traces were formed with the bulkier i‐butyl and SiMe3 substituents. Diphenylmethane showed very low reactivity.
Scheme 3.
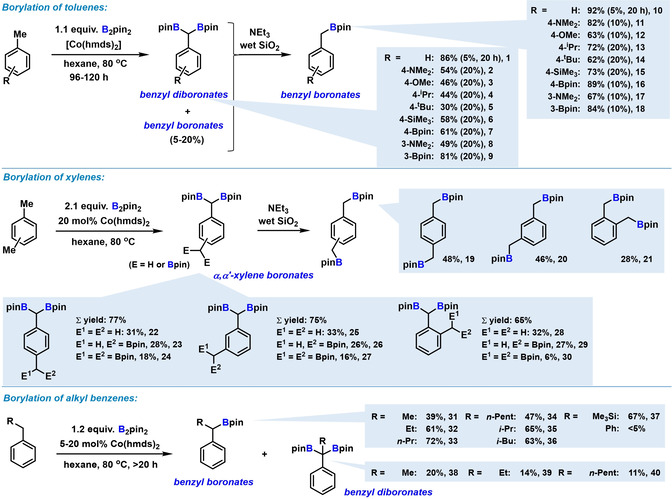
Borylation of methylarenes (top), xylenes (middle), and alkylarenes (bottom). [a] Isolated yields. Catalyst loading in parentheses. See ESI for details.
Mechanistic studies
We have performed several insightful mechanistic experiments to identify the nature of the operating catalyst which has remained ambiguous in previous reports. [9] Key observations were made in stoichiometric reactions between the pre‐catalyst and reagents, successful trapping reactions of intermediate species, operando X‐ray absorption spectroscopy, and kinetic studies of reaction progress, reaction orders, and isotope effects.
a) Kinetic studies: The borylation of toluene with 5 mol % Co(hmds)2 and B2pin2 (1.0 equiv) follows a pseudo‐1st order rate behaviour. From a van't Hoff plot ln k obs vs. ln[cat] an order in catalyst concentration of ≈1.2 was determined which indicates a monometallic mechanism. The individual order in B2pin2 was 0 (zero), that of toluene ≈1 (Table S1). Our studies of a kinetic isotopic effect (KIE) [16] documented identical rates of toluene and d5 ‐toluene and identical rates of reactions of d3 ‐toluene and [D8]toluene, respectively, under identical conditions (Figure S1). The observed k H/k D ratio of ≈2.8 suggests a 1° KIE and rate‐determining benzylic C−H cleavage by the catalyst, which is in full accord with 1st order rate dependence on toluene concentration.
b) Dehydrocoupling reactions: The borylation of toluene is much slower with HBpin as boronate source than with B2pin2 under identical conditions (Scheme 2). In borylations with B2pin2, the first CH borylation forms HBpin as by‐product which engages subsequently in another CH borylation to give benzyl diboronate. Alternatively, HBpin can undergo acceptorless dehydrocoupling to give B2pin2 and H2 (Scheme 2). The high (primary) KIE makes these reactions very slow for DBpin so that it accumulates as intermediate and can be detected in considerable amounts (Figure S2). Consistent with these observations, the formation of DBpin and D2 was observed under catalytic conditions with [D8]toluene as substrate (1H, 2H, 11B NMR). The formed D2 was used in the Pd/C‐catalyzed deuterogenation of styrene to give d2 ‐ethyl‐benzene (Figure S3). The dehydrogenative coupling of HBpin to B2pin2 in the presence of Co(hmds)2 gives low yields (<10 %), possibly due to the rapid activation of B2pin2 by the catalyst. An indirect proof of this dehydrocoupling is the reaction of the formed B2pin2 with 4,4′‐bipyridine which is known to react in a formal 1,8‐addition. [17] Consistently, we isolated N,N′‐diboryl‐4,4′‐bipyridin‐ylidene in 73 % yield when reacting HBpin (2 equiv) and Co(hmds)2 (5 mol %) in toluene solution in the presence of 4,4′‐bipyridine. There is no reaction in the absence of cobalt catalyst.
c) Colours and UV/Vis spectra: All successful methylarene borylation reactions with catalytic Co(hmds)2 displayed characteristic solution colours. Treatment of the pre‐catalyst Co(hmds)2 with toluene and B2pin2 in hexane resulted in an immediate colour change from green to violet/blue prior to the onset of product formation. Over the course of the reaction, the solution turned rapidly brown. The characteristic blue colour was observed with all arenes including toluene (and the other reactive methylarenes) but also with benzene and naphthalene, so that π‐arene coordination to the cobalt complex may be considered. In hexane or methylcyclohexane, the solution colour did not change. UV‐vis spectra of mixtures of Co(hmds)2, B2pin2 and arenes absorption bands in the 350–370 and 500–650 nm ranges while no absorption bands were visible in alkane solutions (Figure S4).
d) Trapping of reactive intermediates: We performed poisoning experiments of the catalyst under the reaction conditions in an effort to distinguish homotopic and heterotopic mechanisms. [18] Addition of trimethylphosphine (PMe3, 0.1–1 equiv per Co) or dibenzo[a,e]cyclooctatetraene (dct, 4 equiv per Co) led to full inhibition of catalyst turnover (Figure S5). From these observations, (equilibrating) homogeneous and hetero‐geneous catalyst species may be concluded. Addition of catalytic amounts of the radical scavenger 2,2,6,6‐tetramethylpiperidinyl‐N‐oxyl (TEMPO, 1 equiv per Co) completely inhibited the reaction (Figure S6). [19] Organic TEMPO adducts were not observed.
e) Stoichiometric reactions, byproducts, and catalyst derivatives: The operation of acid‐base reactivity in the catalytic borylation mechanism was excluded, as treatment of toluene derivatives with equimolar Co(hmds)2 at 80 °C and workup in D2O did not show any D‐incorporation. The equimolar reaction of B2pin2 and Co(hmds)2 in hexane exhibited clean formation of pinB‐N(SiMe3)2 (11B‐NMR: 25 ppm) and another boron‐containing compound (11B: 34 ppm), possibly a cobalt boryl complex (Figure S7). [20] From this crude reaction mixture, the literature‐known tetrameric cobalt cluster [CoN(SiMe3)2]4 was crystallized (while its characteristic 1H‐NMR signals were not observed in the crude reaction). [13] The formation of cobalt amido clusters from reactions containing equimolar amounts of the Lewis acidic component [Bpin] and the Lewis base [N(SiMe3)2] is especially noteworthy and may be indicative of the special role of the amido ligand hmds in the borylation catalysis mechanism. In perfect agreement with this assumption is the isolation of Co(hmds) complexes and the quantification of B2pin2 conversion: An equimolar mixture of Co(hmds)2 and B2pin2 in C6D6 turned immediately blue; 1H NMR spectra showed formation of 1 equiv (Me3Si)2N‐Bpin per Co(hmds)2 (Figure S8). The conversion of B2pin2 was ≈60 % (GC‐FID). Addition of phosphine ligands (PPh3, PCy3) to the blue solution of Co(hmds)2 and B2pin2 (2:1) in benzene enabled the isolation of cobalt(I) complexes [Co(PR3)2(hmds)] in high yields (e.g. 72 % yield, PPh3) (Figure S9). [21] This observation strongly suggests that i) the amido ligand hmds may act as a stable spectator ligand under the conditions of the borylation reaction, and ii) cobalt(I) catalysts may be operative in the mechanism. The interaction of a postulated cobalt boryl catalyst with π‐arenes (such as the substrate toluene) was corroborated by the isolation of the unusual π‐cyclohexadienyl boronate cobalt complex (dcpe)Co(η5‐C6H6‐Bpin) from the reaction of Co(hmds)2 and B2pin2 in benzene followed by addition of 1,2‐bis(dicyclohexyl‐phosphino)ethane (dcpe) (Figure S10). [22]
f) X‐ray absorption (XAS) spectroscopy provides insight into the nature of metal complexes under reaction conditions. Oxidation states, the number and type of coordinating atoms, and the nucleation of metallic species and coordination compounds can be derived from XAS experiments. [23] The XANES (X‐ray absorption near edge structure) spectra of the pre‐catalyst Co(hmds)2 in hexane and the reaction mixture of Co(hmds)2, B2pin2, and toluene showed distinct features that may be indicative of the coordination of both reactants to the cobalt center (see Figure S11). This is in perfect agreement with the formation of 1 equiv pinB‐N(SiMe3)2 and a blue Co complex from equimolar reactions of Co(hmds)2 and B2pin2 in toluene (Scheme 4, center). Time‐resolved XANES spectra also showed a continuous change of the spectral signatures over the reaction time and the formation of Co nanoparticles (NP) at the end of the reaction (Figures S11–S15). The spectral signatures of the XANES region were translated into structural parameters by analysis of the EXAFS (extended X‐ray absorption fine structure) spectra (Figure 1). Monomer and dimer species of Co(hmds)2 were observed (0.3 Co‐Co back‐scatterers at 2.61 Å distance). [24] This equilibrium shifts completely to dimeric species upon addition of B2pin2 (1.0 back‐scattering Co). The changes in the XANES spectra can be interpreted by Bpin‐coordination to cobalt. (Optimized data fits were obtained from an additional carbon coordination sphere at 2.40 Å which may result from weak interactions with the solvent; see Scheme 6 for CH‐borylation of hexane and Figure S16.) After addition of toluene to the reaction, a dimeric catalyst species with additional carbon back‐scattering contributions at (the typical π‐arene‐metal bond distance of) 2.10 Å was observed. [25] At the end of the reaction, EXAFS data documented nanoparticle formation (Co−Co coordination numbers, distances) which appear to contain (surface) light atoms from Bpin or hmds coordination (see ESI for details).
Scheme 4.
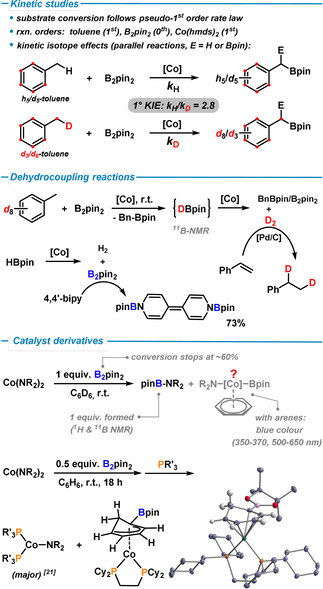
Key mechanistic experiments: Reaction orders, kinetic isotope effects (top). Products of the dehydrocoupling reactions of toluene and HBpin (center). Stoichiometric reactions toward CoI catalyst derivatives (bottom); molecular structure of (dcpe)Co(η5‐C6H6‐Bpin).
Figure 1.
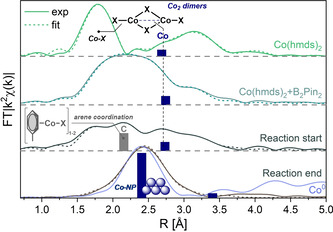
EXAFS spectra with atomic distances, selected back‐scattering atoms (Co–Co, grey bars; Co–C(toluene), blue bars), and postulated catalyst species.
Scheme 6.
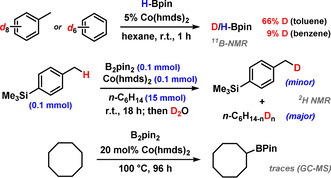
Competing pathways of aryl, benzyl, and alkyl C−H activations.
Based on the collected data from preparative, kinetic, isotope labelling, NMR, and X‐ray spectroscopy studies, we postulate a reaction mechanism involving CoI catalysts that may form through bimetallic reductive elimination of 1 equiv (Me3Si)2N‐Bpin from dimeric CoX2 pre‐catalysts (Scheme 5, top). [26] The π‐coordination of arenes to CoI complexes (A) sets the stage for benzylic CH activation that is most likely facilitated by concomitant B‐B cleavage (σ‐bond metathesis). [27] Elimination of the benzyl boronate regenerates an active CoI catalyst. The produced HBpin undergoes another benzylic CH borylation (slower than benzylation of B2pin2) or cobalt‐catalyzed dehydrocoupling to give B2pin2 and H2. It is important to note that the chemoselectivity of CH‐borylation was very high (Scheme 6). At room temperature, rapid H/D exchange of benzene (after 1 h: 9 % deuteration) and toluene (66 % deuteration, Figure S17) with HBpin was observed, whereas borylation was the major pathway at >60 °C (and with B2pin2 as borylation reagent). Aryl‐H borylation only occurred at elevated temperatures and neat conditions (PhH, full conversion at 80 °C, 72 h, by 11B‐NMR). Minor amounts of alkyl‐H activation of the solvent n‐hexane were observed in some cases. Traces of alkane borylation were detected from the reaction of cyclooctane with 20 mol % catalyst at 100 °C under neat conditions (Scheme 6, bottom).
Scheme 5.
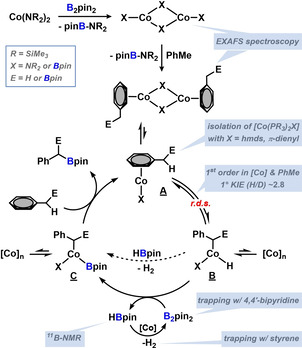
Postulated mechanism of the benzylic CH borylation involving pre‐catalyst reduction to a CoI catalyst (top), toluene borylation (center), and dehydrocoupling of HBpin to B2pin2 and H2 (bottom).
Conclusion
A new cobalt‐catalyzed benzylic CH borylation is reported that produces H2 a byproduct in an overall acceptorless dehydro‐coupling reaction. This reaction operates with Co(hmds)2 as pre‐catalyst and the bench‐stable and inexpensive B2pin2 under mild conditions to give benzyl diboronates and, after protolytic workup, benzyl boronate derivatives. No excess of borylation reagents is required. A full set of mechanistic studies was performed that support the notion of a tandem cobalt catalysis that involves benzylic borylation and borane dehydrocoupling events under identical reaction conditions with H2 being the only byproduct. Kinetic, spectroscopic, and preparative experiments were indicative of rate‐limiting CH activation, the conservation of the amido ligand during catalysis, the presence of dimeric CoI catalyst species, and π‐arene coordination motifs. The borylation reaction is highly chemospecific for benzyl‐CH bonds; the activation of aryl‐H and alkyl‐H positions were not competing under the reaction conditions. The compatibility with different borane sources and the zero‐order rate behavior in borane strongly indicate that this method can serve as a template for related CH functionalizations.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the European Research Council (CoG 683150). We acknowledge beamtime at P65, PETRA III of Deutsches Elektronensynchrotron (DESY) and thank Edmund Welter and Dr. Dieter Schaarschmidt for technical assistance. Open Access funding enabled and organized by Projekt DEAL.
P. Ghosh, R. Schoch, M. Bauer, A. Jacobi von Wangelin, Angew. Chem. Int. Ed. 2022, 61, e202110821.
Dedicated to Barry M. Trost on the occasion of his 80th birthday
References
- 1.
- 1a. Rogge T., Kaplaneris N., Chatani N., Kim J., Chang S., Punji B., Schafer L. L., Musaev D. G., Wencel-Delord J., Roberts C. A., Sarpong R., Wilson Z. E., Brimble M. A., Johansson M. J., Ackermann L., Nat. Rev. Methods Primers 2021, 1, 43; [Google Scholar]
- 1b. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]
- 1c. Roudesly F., Oble J., Poli G., J. Mol. Catal. A 2017, 426, 275–296; [Google Scholar]
- 1d. Crabtree R. H., Lei A., Chem. Rev. 2017, 117, 8481–8482; [DOI] [PubMed] [Google Scholar]
- 1e. Davies H. M. L., Morton D., J. Org. Chem. 2016, 81, 343–350; [DOI] [PubMed] [Google Scholar]
- 1f. Abrams D. J., Provencher P. A., Sorensen E. J., Chem. Soc. Rev. 2018, 47, 8925–8967; [DOI] [PubMed] [Google Scholar]
- 1g. Li B., Ali A. I. M., Ge H., Chem 2020, 6, 2591–2657; [Google Scholar]
- 1h. Moselage M., Li J., Ackermann L., ACS Catal. 2016, 6, 498–525; [Google Scholar]
- 1i. Dalton T., Faber T., Glorius F., ACS Cent. Sci. 2021, 7, 245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selected reviews:
- 2a. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 2b. Hartwig J. F., Acc. Chem. Res. 2012, 45, 864–873; [DOI] [PubMed] [Google Scholar]
- 2c. Xu L., Wang G., Zhang S., Wang H., Wang L., Liu L., Jiao J., Li P., Tetrahedron 2017, 73, 7123–7157. [Google Scholar]
- 3.
- 3a. Hall D. G., Boronic Acids. Preparation and Application in Organic Synthesis Medicine and Materials , 2nd ed., Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 4.
- 4a. Fernández E., in Topics in Organometallic Chemistry, Springer, Berlin, Heidelberg, 2020, pp. 1–19; [Google Scholar]
- 4b. Ros A., Fernández R., Lassaletta J. M., Chem. Soc. Rev. 2014, 43, 3229–3243; [DOI] [PubMed] [Google Scholar]
- 4c. Wright J. S., Scott P. J. H., Steel P. G., Angew. Chem. Int. Ed. 2021, 60, 2796–2821; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 2830–2856; [Google Scholar]
- 4d. Kuroda Y., Nakao Y., Chem. Lett. 2019, 48, 1092–1100. [Google Scholar]
- 5.Selected recent examples:
- 5a. Dombray T., Werncke C. G., Jiang S., Grellier M., Vendier L., Bontemps S., Sortais J.-B., Sabo-Etienne S., Darcel C., J. Am. Chem. Soc. 2015, 137, 4062–4065; [DOI] [PubMed] [Google Scholar]
- 5b. Obligacion J. V., Semproni S., Chirik P. J., J. Am. Chem. Soc. 2014, 136, 4133–4136; [DOI] [PubMed] [Google Scholar]
- 5c. Furukawa T., Tobisu M., Chatani N., Chem. Commun. 2015, 51, 6508–6511; [DOI] [PubMed] [Google Scholar]
- 5d. Zhang H., Hagihara S., Itami K., Chem. Lett. 2015, 44, 779–781; [Google Scholar]
- 5e. Mazzacano T. J., Mankad N. P., J. Am. Chem. Soc. 2013, 135, 17258–17261. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hartwig J. F., Larsen M. A., ACS Cent. Sci. 2016, 2, 281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Hartwig J. F., Chem. Soc. Rev. 2011, 40, 1992;21336364 [Google Scholar]
- 6c. Li Q., Liskey C. W., Hartwig J. F., J. Am. Chem. Soc. 2014, 136, 8755–8876; [DOI] [PubMed] [Google Scholar]
- 6d. Larsen M. A., Cho S. H., Hartwig J., J. Am. Chem. Soc. 2016, 138, 762–765; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Reyes R. L., Iwai T., Maeda S., Sawamura M., J. Am. Chem. Soc. 2019, 141, 6817–6821; [DOI] [PubMed] [Google Scholar]
- 6f. Chen H., Schlecht S., Semple T. C., Hartwig J. F., Science 2000, 287, 1995–1997. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Shimada S., Batsanov A. S., Howard J. A. K., Marder T. B., Angew. Chem. Int. Ed. 2001, 40, 2168–2171; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2226–2229; [Google Scholar]
- 7b. Ishiyama T., Ishida K., Takagi J., Miyaura N., Chem. Lett. 2001, 30, 1082–1083; [Google Scholar]
- 7c. Larsen M. A., Wilson C. V., Hartwig J. F., J. Am. Chem. Soc. 2015, 137, 8633–8643; [DOI] [PubMed] [Google Scholar]
- 7d. Yoshii D., Yatabe T., Yabe T., Yamaguchi K., ACS Catal. 2021, 11, 2150–2155. [Google Scholar]
- 8.
- 8a. Palmer W. N., Obligacion J. V., Pappas I., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 766–769. [DOI] [PubMed] [Google Scholar]
- 9. Jayasundara C. R. K., Sabasovs D., Staples R. J., Oppenheimer J., M. R. Smith III , R. E. Maleczka, Jr. , Organometallics 2018, 37, 1567–1574. [Google Scholar]
- 10. Manna K., Ji P., Lin Z., Greene F. X., Urban A., Thacker N. C., Lin W., Nat. Commun. 2016, 7, 12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Ghosh P., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2021, 60, 16035–16043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16171–16179; [Google Scholar]
- 11b. Ghosh P., Jacobi von Wangelin A., Org. Chem. Front. 2020, 7, 960–966; [Google Scholar]
- 11c. Gieshoff T. N., Chakraborty U., Villa M., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2017, 56, 3585–3589; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3639–3643; [Google Scholar]
- 11d. Chakraborty U., Reyes-Rodriguez E., Demeshko S., Meyer F., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2018, 57, 4970–4975; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5064–5069; [Google Scholar]
- 11e. Chakraborty U., Demeshko S., Meyer F., Jacobi von Wangelin A., Angew. Chem. Int. Ed. 2019, 58, 3466–3470; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3504–3508. [Google Scholar]
- 12.
- 12a. Melen R. L., Chem. Soc. Rev. 2016, 45, 775–788; [DOI] [PubMed] [Google Scholar]
- 12b. Braunschweig H., Guethlein F., Angew. Chem. Int. Ed. 2011, 50, 12613–12616; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12821–12824; [Google Scholar]
- 12c. Arrowsmith M., Braunschweig H., Stennett T. E., Angew. Chem. Int. Ed. 2017, 56, 96–115; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 100–120; [Google Scholar]
- 12d. Lee C.-I., Zhou J., Ozerov O. V., J. Am. Chem. Soc. 2013, 135, 3560–3566. [DOI] [PubMed] [Google Scholar]
- 13. Ohki Y., Shimizu Y., Araake R., Tada M., Sameera W. M. C., Ito J.-I., Nishiyama H., Angew. Chem. Int. Ed. 2016, 55, 15821–15825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16053–16057. [Google Scholar]
- 14.
- 14a. Wang M., Shi Z., Chem. Rev. 2020, 120, 7348–7398; [DOI] [PubMed] [Google Scholar]
- 14b. Kaithal A., Kalsi D., Krishnakumar V., Pattanaik S., Bordet A., Leitner W., Gunanathan C., ACS Catal. 2020, 10, 14390–14397; [Google Scholar]
- 14c. Zarate C., Manzano R., Martin R., J. Am. Chem. Soc. 2015, 137, 6754–6757. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Sun C., Potter B., Morken J. P., J. Am. Chem. Soc. 2014, 136, 6534–6537; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Hong K., Liu X., Morken J. P., J. Am. Chem. Soc. 2014, 136, 10581–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gómez-Gallego M., Sierra M. A., Chem. Rev. 2011, 111, 4857–4963. [DOI] [PubMed] [Google Scholar]
- 17. Ohmura T., Morimasa Y., Suginome M., J. Am. Chem. Soc. 2015, 137, 2852–2855. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Gärtner D., Sandl S., Jacobi von Wangelin A., Catal. Sci. Technol. 2020, 10, 3502–3514; [Google Scholar]
- 18b. Crabtree R. H., Chem. Rev. 2012, 112, 1536–1554; [DOI] [PubMed] [Google Scholar]
- 18c. Widegren J. A., Finke R. G., J. Mol. Catal. A 2003, 198, 317–341. [Google Scholar]
- 19.
- 19a. Jahr D., Rebhan K. H., Schwarzhans K. E., Wiedemann J., Z. Naturforsch. B 1973, 28, 55–62; [Google Scholar]
- 19b. Jaitner P., Huber W., Gieren A., Betz H., J. Organomet. Chem. 1986, 311, 379–385. [Google Scholar]
- 20. Schaefer B. A., Margulieux G. W., Small B. L., Chirik P. J., Organometallics 2015, 34, 1307–1320. [Google Scholar]
- 21. Brennan M. R., Kima D., Fout A. R., Chem. Sci. 2014, 5, 4831–4839. [Google Scholar]
- 22.Deposition number 2097237. contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 23.
- 23a. Benedikter M., Musso J., Kesharwani M. K., Sterz K. L., Elser I., Ziegler F., Fischer F., Plietker B., Frey W., Kästner J., Winkler M., van Slageren J., Nowakowski M., Bauer M., Buchmeiser M. R., ACS Catal. 2020, 10, 14810–14823; [Google Scholar]
- 23b. Schoch A., Burkhardt L., Schoch R., Stührenberg K., Bauer M., Faraday Discuss. 2019, 220, 113–132; [DOI] [PubMed] [Google Scholar]
- 23c. Gregori B. J., Schwarzhuber F., Pöllath S., Zweck J., Fritsch L., Schoch R., Bauer M., Jacobi von Wangelin A., ChemSusChem 2019, 12, 3864–3870. [DOI] [PubMed] [Google Scholar]
- 24. Bryan A. M., Long G. J., Grandjean F., Power P. P., Inorg. Chem. 2013, 52, 12152–12160. [DOI] [PubMed] [Google Scholar]
- 25. Yang X.-J., Fan X., Zhao Y., Wang X., Liu B., Su J.-H., Dong Q., Xu M., Wu B., Organometallics 2013, 32, 6945–6949. [Google Scholar]
- 26.For selected examples, see:
- 26a. Powers D. C., Ritter T., Nat. Chem. 2009, 1, 302–309; [DOI] [PubMed] [Google Scholar]
- 26b. Wolf W. J., Winston M. S., Toste F. D., Nat. Chem. 2014, 6, 159–164; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c. Xu H., Diccianni J. B., Katigbak J., Hu C., Zhang Y., Diao T., J. Am. Chem. Soc. 2016, 138, 4779–4786; [DOI] [PubMed] [Google Scholar]
- 26d. Schenck T. G., Milne C. R. C., Sawyer J. F., Bosnich B., Inorg. Chem. 1985, 24, 2338–2344; [Google Scholar]
- 26e. Halpern J., Inorg. Chim. Acta 1982, 62, 31–37. [Google Scholar]
- 27. Obligacion J. V., Semproni S. P., Pappas I., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 10645–10653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information