

Abstract
Aim
To investigate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tirzepatide in Japanese participants with type 2 diabetes (T2D).
Methods
This phase 1, double‐blind, placebo‐controlled, parallel‐dose, multiple‐ascending dose study randomized participants to once‐weekly subcutaneous tirzepatide or placebo. The tirzepatide treatment groups were: 5 mg (5 mg, weeks 1‐8), 10 mg (2.5 mg, weeks 1‐2; 5 mg, weeks 3‐4; 10 mg, weeks 5‐8), and 15 mg (5 mg, weeks 1‐2; 10 mg, weeks 3‐6; 15 mg, weeks 7‐8). The primary outcome was tirzepatide safety and tolerability.
Results
Forty‐eight participants were randomized. The most frequently reported treatment‐emergent adverse events (AEs) were decreased appetite and gastrointestinal AEs, which were generally dose‐dependent and mild in severity. The plasma tirzepatide concentration half‐life was approximately 5 days. After 8 weeks of treatment, fasting plasma glucose decreased from baseline with tirzepatide versus placebo; the least squares (LS) mean decrease compared with placebo (95% confidence interval [CI]) was 52.7 (35.9‐69.6), 69.1 (52.3‐85.9), and 68.9 (53.2‐84.6) mg/dL in the 5‐, 10‐, and 15‐mg treatment groups, respectively (P < .0001 for all treatment groups). Tirzepatide also resulted in LS mean decreases from baseline versus placebo at 8 weeks in HbA1c up to 1.6% (95% CI 1.2%‐1.9%; P < .0001 for all treatment groups) and body weight up to 6.6 kg (95% CI 5.3‐7.9; P < .0001 for all treatment groups).
Conclusions
All tirzepatide doses were well tolerated. The safety, tolerability, PK, and PD profiles of tirzepatide support further evaluation of once‐weekly dosing in Japanese people with T2D.
Keywords: antidiabetic drug, GIP, GLP‐1, phase I‐II study
1. INTRODUCTION
It is estimated that 7.4 million people in Japan have diabetes, with a national prevalence of 7.9%. 1 A further 12.1 million people in Japan have impaired glucose tolerance. 1 Among Japanese people with diabetes, there has been a documented increase in the proportion of those who are overweight and obese. 2
In people with type 2 diabetes (T2D), glucagon‐like peptide‐1 (GLP‐1) receptor agonists have several therapeutic benefits, including improving glucose‐dependent insulin secretion, reducing glucagon secretion, and decreasing the rate of gastric emptying. 3 , 4 , 5 , 6 , 7 GLP‐1 receptor agonists are also associated with reduced appetite and subsequent weight loss. 3 , 4 , 5 , 6 , 7 However, not all people treated with GLP‐1 receptor agonists and/or other blood glucose‐lowering agents reach their glycemic or body weight reduction targets. 8 , 9 One potential therapeutic approach to address this limitation is a dual receptor agonist of GLP‐1 and glucose‐dependent insulinotropic peptide (GIP), which allows simultaneous targeting of additional pathways implicated in nutrient and energy metabolism. 10 , 11
Tirzepatide is a 39‐amino acid synthetic peptide dual GIP and GLP‐1 receptor agonist. Its peptide component is engineered from the GIP amino acid sequence and includes a C20 fatty di‐acid moiety, enabling albumin binding and allowing once‐weekly dosing. 12 In a global 26‐week, phase 2 study in participants with T2D, tirzepatide resulted in significant dose‐dependent reductions in HbA1c at all doses (1, 5, 10, and 15 mg) versus placebo, and for the 5‐, 10‐, and 15‐mg doses versus 1.5‐mg dulaglutide. 11 There were also significant, dose‐dependent reductions in body weight for the 5‐, 10‐, and 15‐mg doses of tirzepatide versus placebo, and for the 5‐, 10‐, and 15‐mg doses versus 1.5‐mg dulaglutide. 11 A post hoc analysis of this study showed that tirzepatide improved several markers of insulin sensitivity and beta‐cell function compared with 1.5‐mg dulaglutide. 13 Further post hoc analyses showed that tirzepatide improved participants' atherogenic lipoprotein profile and biomarkers of non‐alcoholic steatohepatitis. 14 , 15 The effects of tirzepatide on insulin sensitivity were found to be partly attributable to weight loss, supporting the hypothesis that dual receptor agonism promotes distinct mechanisms of glycemic control. 13 Another 12‐week, phase 2 study investigating several dose‐escalation regimens showed clinically significant reductions in HbA1c among participants receiving tirzepatide, and suggested that lower starting doses and smaller dose increments were better tolerated. 16
This multiple‐ascending dose phase 1 study investigated the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tirzepatide administered subcutaneously (SC) once‐weekly for 8 weeks in Japanese participants with T2D. The data from this study will support identification of an appropriate dose range for subsequent clinical studies in Japan.
2. METHODS
2.1. Study design and participants
This phase 1, multiple‐site, participant‐ and investigator‐blind, placebo‐controlled, randomized, parallel dose‐group, 8‐week, multiple‐ascending dose study was conducted in Japanese participants with T2D. Eligibility criteria included age 20‐70 years at screening, T2D for 1 year or longer prior to enrolment, T2D controlled with diet and exercise alone, or stable on metformin or a dipeptidyl peptidase‐4 (DPP‐4) inhibitor (except for omarigliptin, trelagliptin, and linagliptin, which require a washout period longer than that used in this study) for 3 months or longer, and a body mass index (BMI) of 20‐35 kg/m2. Patients who had been treated with metformin or DPP‐4 inhibitors were required to withdraw their treatment and have at least a 28‐day washout period before dosing with tirzepatide or placebo. The study protocol was approved by local ethics committees and was conducted in accordance with the principles of the Declaration of Helsinki, Council of International Organizations of Medical Sciences International Ethical Guidelines, and Good Clinical Practice guidelines. All participants gave written informed consent before participation in the study. This study is registered with ClinicalTrials.gov, number NCT03322631.
2.2. Procedures
Participants who met the enrolment criteria were randomly allocated to receive either once‐weekly tirzepatide SC or placebo for 8 weeks. Randomization tables for allocation of tirzepatide or placebo were prepared by the study statistician and provided to pharmacy staff, who prepared and dispensed the study medication. The dosing regimen was participant‐ and investigator‐blinded. The tirzepatide dose groups and dose‐escalation regimens were: 5 mg (participants received 5‐mg tirzepatide weeks 1‐8), 10 mg (participants received 2.5‐mg tirzepatide, weeks 1‐2; 5 mg, weeks 3‐4; 10 mg, weeks 5‐8), and 15 mg (participants received 5‐mg tirzepatide, weeks 1‐2; 10 mg, weeks 3‐6; 15 mg, weeks 7‐8). Full details of the treatment regimens are shown in Figure S1.
All injections were administered SC into the tissue of the abdominal wall. Injection sites were alternated weekly between four sites on the abdominal wall (right and left upper quadrants, and right and left lower quadrants).
2.3. Outcomes
The primary outcome was the safety and tolerability of tirzepatide after multiple SC doses. The safety variables assessed included laboratory values, vital signs, electrocardiogram (ECG) variables, injection‐site reactions, and hypoglycemia. Hypoglycemic events were defined as plasma glucose of 70 mg/dL or less (documented glucose alert level 1). Plasma tirzepatide concentration─QT analyses were performed to assess the changes from baseline and placebo in the double delta QT interval calculated with Fridericia's correction interval relative to plasma tirzepatide concentrations. Study investigators assessed the safety of patients by monitoring for and recording any suspected adverse event (AE). The occurrence and nature of each AE was documented by the investigator, including whether there was a reasonable possibility of being related to study treatment or study procedure, considering the disease, concomitant treatment, and existing underlying pathologies and conditions at the start of the study period. A serious AE was defined as any AE that resulted in death, initial or prolonged inpatient hospitalization, a life‐threatening experience, persistent or significant disability/incapacity, congenital anomaly/birth defect, or any important medical event that could jeopardize the participant or require intervention to prevent one of the above listed outcomes.
Secondary outcomes included characterization of PK and the PD effect of tirzepatide, as measured by plasma tirzepatide concentrations and fasting plasma glucose levels. Exploratory outcomes included the PD effects of tirzepatide as measured by body weight, oral glucose tolerance test (0 to 2 hours), seven‐point self‐monitored plasma glucose (SMPG), HbA1c, and subjective appetite sensation. To assess subjective appetite sensation, participants were provided standardized lunch and dinner meals. Meal intake was recorded, and subjective rating of appetite sensations was measured by a 100‐mm visual analog scale with variables of hunger, fullness, satiety, and prospective food consumption before and 4‐5 hours after the standardized meal. Overall appetite score was calculated as the average of the four individual scores: satiety; fullness; (100 minus prospective food consumption); and (100 minus hunger). A higher overall appetite score indicated less appetite.
2.4. Statistical analysis
The sample size for the study was chosen to provide sufficient data to evaluate the primary outcome and was not intended to achieve any a priori statistical requirements. The study aimed to enrol approximately 49 participants so that approximately 32 participants would complete the study, in compliance with the defined dosing regimen.
All safety data are reported in the safety population, which consisted of all randomized participants who received at least one dose of study drug. Key safety measures included laboratory variables, vital signs, ECG variables, injection‐site reactions, and hypoglycemia, which were listed and summarized using standard descriptive statistics.
All PK and PD data were reported based on data from all participants who received at least one dose of study drug and had evaluable PK and PD data. PK variables were estimated using non‐compartmental methods and summarized using descriptive statistics by dose level. PD effects of tirzepatide were summarized by treatment regimen and day. Change from baseline for each PD measure was analyzed by assigned treatment regimen using mixed‐effects models with treatment regimen received as a fixed effect and the participant as a random effect. For repeated measure variables, day and treatment regimen‐by‐day interaction terms were included in the model. For all mixed models, baseline values were used as covariates, an unstructured covariance approach was used and, if the model failed to converge, alternative structures were examined using Akaikeʼs criteria. Results from mixed models are reported as least squares (LS) means and 95% confidence intervals (CIs). Statistical significance for comparisons of PD variables was set at P less than .05. All statistical analyses were performed using SAS version 9.4 or higher.
3. RESULTS
3.1. Participant disposition and demographics
A total of 48 participants entered the study, were randomly assigned treatment, and received at least one dose of study drug (N = 39) or placebo (N = 9). This included 11 participants in the 5‐mg, 12 participants in the 10‐mg, and 16 participants in the 15‐mg treatment groups. Two participants, each receiving tirzepatide (one in the 10‐mg and one in the 15‐mg group), discontinued the study: one because of patient decision (concern about study procedures/perceived risks) and the other because of an AE of decreased appetite. Three participants, all from the 15‐mg treatment group, did not escalate to 15 mg, and remained at 10 mg through the full 8‐week treatment period: one because of an AE of infective gastroenteritis and two because of an investigator decision considering the patients' overall condition, with no AEs specified as the causal reason for not escalating the treatment dose.
Among all participants, the mean (standard deviation [SD]) age was 57.4 (8.8) years, weight was 72.3 (10.4) kg, BMI was 25.4 (3.2) kg/m2, HbA1c was 8.0% (0.8%), diabetes duration was 8.5 (4.4) years, and fasting serum glucose was 172.5 (28.9) mg/dL. Baseline demographic and clinical characteristics were similar by treatment regimen, with the exception of body weight, which was lower in participants randomized to the placebo group compared with the other treatment groups (Table 1).
TABLE 1.
Baseline demographics and clinical characteristics
| Demographics and clinical characteristics | Placebo | 5‐mg tirzepatide a | 10‐mg tirzepatide b | 15‐mg tirzepatide c | Overall |
|---|---|---|---|---|---|
| N = 9 | N = 11 | N = 12 | N = 16 | N = 48 | |
| Age, y | 57.4 (11.6) | 57.5 (7.9) | 56.9 (9.5) | 57.7 (8.0) | 57.4 (8.8) |
| Sex, n (%) | |||||
| Male | 9 (100) | 11 (100) | 12 (100) | 15 (93.8) | 47 (97.9) |
| Female | 0 | 0 | 0 | 1 (6.3) | 1 (2.1) |
| Weight, kg | 63.0 (7.8) | 75.4 (11.0) | 74.9 (9.5) | 73.3 (9.9) | 72.3 (10.4) |
| Body mass index, kg/m2 | 22.6 (2.1) | 26.7 (3.3) | 25.5 (2.8) | 26.1 (3.1) | 25.4 (3.2) |
| HbA1c, % | 7.8 (0.9) | 7.7 (0.3) | 8.1 (0.8) | 8.2 (0.9) | 8.0 (0.8) |
| Fasting serum glucose, mg/dL | 173.9 (33.1) | 164.5 (25.2) | 181.2 (23.3) | 170.7 (33.1) | 172.5 (28.9) |
| Diabetes duration, y | 9.5 (3.4) | 7.0 (5.0) | 8.4 (3.8) | 9.1 (4.8) | 8.5 (4.4) |
Note: Data are presented as mean (standard deviation) unless otherwise indicated.
Participants in the 5‐mg treatment group received 5‐mg tirzepatide weeks 1‐8.
Participants in the 10‐mg treatment group received 2.5‐mg tirzepatide weeks 1‐2, followed by 5 mg weeks 3‐4, and 10 mg weeks 5‐8.
Participants in the 15‐mg treatment group received 5‐mg tirzepatide weeks 1‐2, followed by 10 mg weeks 3‐6, and 15 mg weeks 7‐8.
3.2. Safety and tolerability
No deaths or serious AEs occurred during the study. Most AEs were mild in severity, few were moderate, and none were reported as severe. All AEs resolved by the end of the study. Decreased appetite and gastrointestinal AEs (constipation [35.4% overall], diarrhoea [16.7% overall], and abdominal discomfort [12.5% overall]) were the most commonly reported AEs and occurred more frequently with increasing dose of tirzepatide (Table 2). One participant had treatment‐related transient elevations in lipase (>5× upper limit of normal [ULN]; moderate AE) and amylase (>2× ULN) with no symptoms suggesting pancreatitis. Lipase and amylase levels returned to the normal range the next day in this participant with no change in tirzepatide dose.
TABLE 2.
Treatment‐emergent adverse events reported in ≥2 participants
| Treatment‐emergent adverse events | Placebo | 5‐mg tirzepatide a | 10‐mg tirzepatide b | 15‐mg tirzepatide c |
|---|---|---|---|---|
| N = 9 | N = 11 | N = 12 | N = 16 | |
| Decreased appetite | 0 | 5 (45.5) | 6 (50.0) | 10 (62.5) |
| Constipation | 1 (11.1) | 0 | 8 (66.7) | 8 (50.0) |
| Diarrhea | 0 | 0 | 3 (25.0) | 5 (31.3) |
| Abdominal discomfort | 0 | 0 | 2 (16.7) | 4 (25.0) |
| Abdominal distension | 1 (11.1) | 3 (27.3) | 1 (8.3) | 0 |
| Headache | 0 | 0 | 3 (25.0) | 1 (6.3) |
| Vomiting | 0 | 1 (9.1) | 1 (8.3) | 2 (12.5) |
| Blood triglycerides increased | 2 (22.2) | 0 | 0 | 1 (6.3) |
| Dyspepsia | 0 | 0 | 3 (25.0) | 0 |
| Lipase increased | 0 | 0 | 1 (8.3) | 2 (12.5) |
| Nausea | 0 | 1 (9.1) | 0 | 2 (12.5) |
| Viral upper respiratory tract infection | 0 | 1 (9.1) | 1 (8.3) | 1 (6.3) |
| White blood cell count increased | 0 | 1 (9.1) | 0 | 1 (6.3) |
Note: Data are presented as n (%). Adverse events were listed by Medical Dictionary for Regulatory Activities preferred term. Adverse events with a change of severity were only counted once at the highest severity.
Participants in the 5‐mg treatment group received 5‐mg tirzepatide weeks 1‐8.
Participants in the 10‐mg treatment group received 2.5‐mg tirzepatide weeks 1‐2, followed by 5 mg weeks 3‐4, and 10 mg weeks 5‐8.
Participants in the 15‐mg treatment group received 5‐mg tirzepatide weeks 1‐2, followed by 10 mg weeks 3‐6, and 15 mg weeks 7‐8.
There were no clinically relevant changes in laboratory values, vital signs, or ECG variables, except among participants receiving tirzepatide who experienced serum tirzepatide concentration‐dependent increases in pulse rate and heart rate (Figure S2). Analysis of mean predose heart rate, pulse rate, systolic blood pressure, and diastolic blood pressure showed initial increases in heart rate and pulse rate that subsequently stabilized, and no significant changes in blood pressure, in participants treated with tirzepatide (Figure S3).
No injection‐site or hypersensitivity AEs were reported. There was no correlation between incidence of antidrug antibodies and AEs (hypersensitivity and/or injection‐site reactions).
One participant in the 5‐mg tirzepatide group and three participants in the 15‐mg tirzepatide group experienced hypoglycemic events. All events resolved either without treatment or with self‐treatment with food or drink. No severe hypoglycemia or nocturnal hypoglycemia events were reported.
3.3. Pharmacokinetics
Tirzepatide plasma concentrations for non‐compartmental analysis were available for 39 participants (Figure 1A,B and Table S1). The median tmax during weeks 1 and 8 were within the range of 24‐48 hours post‐tirzepatide dose (Table S1). The mean t1/2 was approximately 5 days and steady state was expected within 4‐5 weeks of dosing. The mean steady state apparent total body clearance of drug calculated after extra‐vascular administration (CL/F) and apparent volume of distribution during the terminal phase after extra‐vascular administration (Vz/F) were 0.0288 L/h and 5.27 L, respectively, for the 5‐mg tirzepatide group.
FIGURE 1.
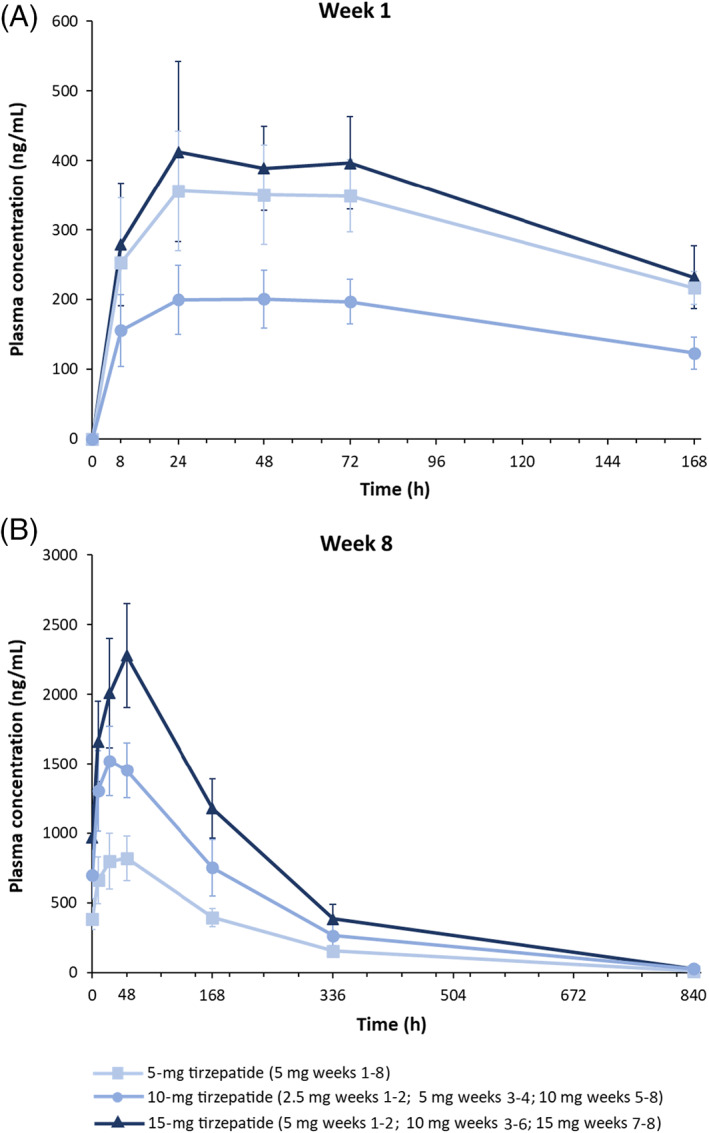
Tirzepatide mean (standard deviation) plasma concentration time profiles following A, first subcutaneous administration at week 1, and B, last subcutaneous administration at week 8. Pharmacokinetic profiles were determined for one dosing interval (168 h) after first dose on day 1 and up to 840 h after the last dose on day 50
3.4. Pharmacodynamics
Tirzepatide resulted in statistically significant decreases from baseline in fasting plasma glucose compared with placebo from week 2 to week 8. At week 8, the LS mean decreases versus placebo (95% CI) were 52.7 (35.9‐69.6), 69.1 (52.3‐85.9), and 68.9 (53.2‐84.6) mg/dL in the 5‐, 10‐, and 15 ‐mg treatment groups, respectively (Figure 2A).
FIGURE 2.
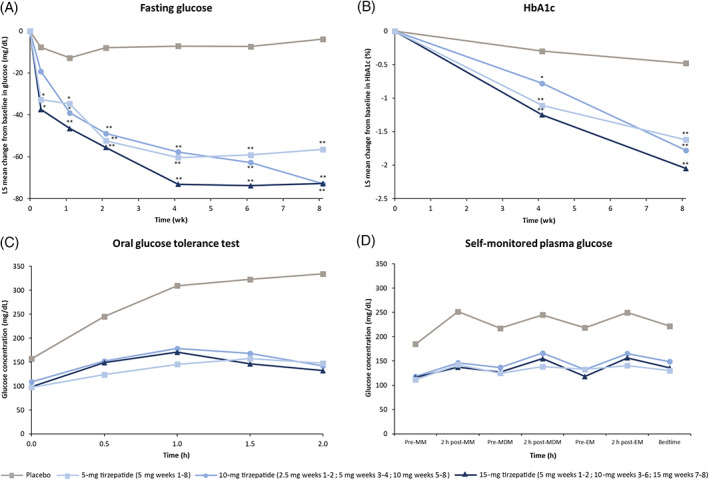
Least squares mean change from baseline in A, fasting plasma glucose, B, HbA1c, C, glucose concentrations by oral glucose tolerance test at day 51 (week 7), and D, arithmetic mean 7‐point glucose profile at day 57 (week 8). *P < .05 compared with placebo; **P < .0001 compared with placebo. EM, evening meal; LS, least squares; MDM, midday meal; MM, morning meal
Tirzepatide also resulted in statistically significant decreases from baseline in HbA1c compared with placebo. At week 8, the LS mean decreases versus placebo (95% CI) were 1.1% (0.8%‐1.5%), 1.3% (0.9%‐1.7%), and 1.6% (1.2%‐1.9%) in the 5‐, 10‐, and 15‐mg treatment groups, respectively (Figure 2B).
Oral glucose tolerance tests showed decreases in glucose area under the curve (AUC; 0‐2 h) and incremental glucose iAUC (0‐2 h) at week 8 for all doses of tirzepatide compared with placebo (Figure 2C).
The seven‐point SMPG results showed that all doses of tirzepatide resulted in improved overall daily SMPG profiles compared with placebo from week 2 to week 8. The reduction of postprandial glucose was greater than premeal glucose, resulting in a more flattened daily SMPG profile in the tirzepatide treatment groups versus the placebo group. The seven‐point SMPG profile at day 57 (week 8) is shown in Figure 2D.
Analysis of body weight showed that tirzepatide resulted in dose‐dependent, statistically significant decreases from baseline in body weight compared with placebo from week 5 to week 8. At week 8, the LS mean decreases (95% CI) versus placebo were 3.4 (2.0‐4.8), 5.0 (3.6‐6.5), and 6.6 (5.3‐7.9) kg in the 5‐, 10‐, and 15 ‐mg treatment groups, respectively (Figure 3).
FIGURE 3.
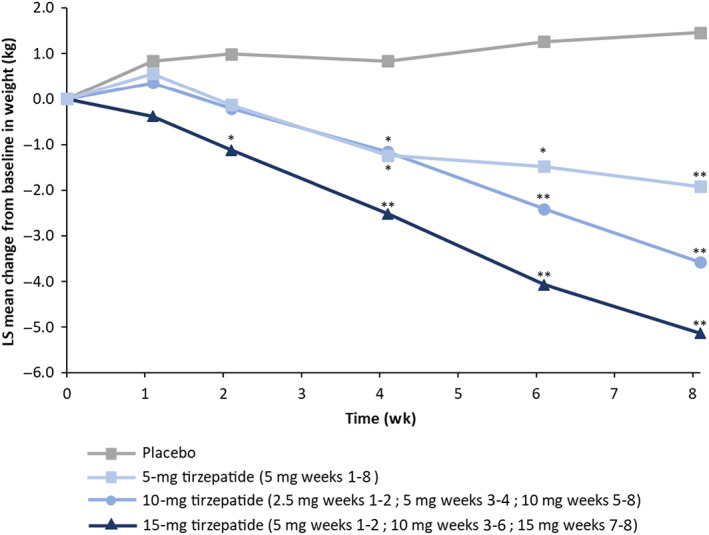
Least squares mean change from baseline in body weight. *P < .05 compared with placebo; **P < .0001 compared with placebo. LS, least squares
There were no significant differences in LDL cholesterol, HDL cholesterol, or total cholesterol concentrations at week 5 or week 8, compared with placebo, following tirzepatide administration. However, dose‐related trends suggesting reduced triglyceride levels were observed postdose at week 5 and week 8, versus at week 1 predose, for all tirzepatide treatment groups. The mean (SD) of triglyceride predose (week 1) and postdose (week 8) were: placebo, 175 (88) mg/dL and 197 (118) mg/dL, respectively; 5 mg, 204 (56) mg/dL and 161 (51) mg/dL, respectively; 10 mg, 172 (71) mg/dL and 122 (38) mg/dL, respectively; and 15 mg, 173 (121) mg/dL and 122 (55) mg/dL, respectively.
At week 8, tirzepatide resulted in a dose‐dependent decrease in the percentage of meal intake compared with placebo (Figure S4). Analysis of appetite sensations and subjective ratings of appetite sensation by visual analog scale showed numerically decreased hunger and fullness scores for both lunch and dinner, at all tirzepatide dose levels, compared with placebo (Figure S5). However, overall appetite score was not different among the treatment groups (Figure S5).
4. DISCUSSION
This phase 1, multiple‐ascending dose study showed that tirzepatide was well tolerated at all tested doses. The PK data reported here support a once‐weekly dosing regimen. Compared with placebo, tirzepatide treatment resulted in statistically significant reductions in fasting serum glucose, HbA1c, and body weight in Japanese participants with T2D. The overall safety and PD profiles of tirzepatide in Japanese participants were comparable with other phase 2 studies in non‐Japanese participants with T2D. 11 , 16 These findings indicate that tirzepatide has the potential to provide clinically meaningful improvements in glycemic control and body weight in people with T2D. 12
In this study, the most frequently reported AEs among Japanese participants with T2D who received tirzepatide were decreased appetite and gastrointestinal AEs (constipation, diarrhea, abdominal discomfort). All the decreased appetite and gastrointestinal AEs were mild in severity and dose‐dependent. These AEs are comparable with those observed in the 26‐ and 12‐week phase 2 studies in non‐Japanese participants, which also showed that gastrointestinal AEs (nausea, diarrhea, and vomiting) and decreased appetite were the most common AEs, were mild to moderate in severity, and were dose‐dependent. 11 , 16 Collectively, these studies suggest that most gastrointestinal AEs occur after initial doses of tirzepatide, and are likely to decrease by starting at a lower dose and gradually escalating the dose with small increments. 11 , 16
In this study, pulse rate and heart rate were increased with increasing tirzepatide doses and serum tirzepatide concentrations. An increased pulse rate among participants receiving tirzepatide was also reported in the 12‐week phase 2 clinical study. 16 GLP‐1 receptor agonists are known to increase heart rate, 15 however, a systematic review and meta‐analysis of cardiovascular outcome studies has shown that GLP‐1 receptor agonists reduce major adverse cardiovascular events and all‐cause mortality. 17 There were no other clinically relevant changes in vital signs, ECG measures, or laboratory values, which is consistent with previous phase 2 studies. In addition, as with the prior tirzepatide studies, 11 , 16 no severe hypoglycemia or nocturnal hypoglycemia events were reported. Together, these data show tirzepatide has similar AE profiles across studies with similar dosing regimens and was well tolerated among all participants, including Japanese participants with T2D.
This study showed overall benefits on glucose and weight lowering, similar to previous phase 2 studies in non‐Japanese participants. 11 , 16 In this study, there was also a dose‐dependent association between increased tirzepatide dose and reduced meal intake, suggesting decreased hunger in Japanese participants with T2D. GLP‐1 receptor agonists stimulate satiety and decrease food intake. 18 Likewise, tirzepatide has been shown to suppress food intake and body weight in preclinical studies. 12 In the current study, we found a decreasing trend in fullness (the physical sensation of how much the stomach is filled with food), and no difference in appetite scores among treatment groups. Typically, decreased food intake results in decreased fullness and hence increased hunger; however, tirzepatide appears to decrease the hunger sensation even when the stomach is not filled with food. Interestingly, satiety (the psychological satisfaction of meal intake) appears not to be changed with tirzepatide, even with decreased meal intake and the physical sensation of fullness. Our data support the hypothesis that tirzepatide has effects on hunger and/or the psychological sensation of satiety separate to that induced by physical input from the stomach for meal ingestion, which may result in long‐term effects on meal intake, as reported by the AE of decreased appetite.
There are several limitations inherent to this phase 1 study, in particular the comparatively small number of participants assessed for safety and PD. Almost all the participants were male (97.9%). The small sample size may have also limited the ability to interpret subjective appetite sensation assessments. Additionally, the comparatively short duration of treatment is a limitation for evaluating glucose lowering and weight loss. The short treatment duration also meant a short duration of dose escalation to reach the maintenance dose, which may have led to an overestimation in the incidence of gastrointestinal AEs. There was also no active comparator for other GLP‐1 receptor agonists. All data reported herein should be interpreted within the context of these limitations.
In conclusion, all tirzepatide doses were well tolerated in Japanese participants with T2D. The PK profile supports once‐weekly tirzepatide dosing. The safety, tolerability, PK, and PD profiles of tirzepatide warrant further clinical evaluation of tirzepatide in Japanese people with T2D.
CONFLICT OF INTEREST
KF has received consulting fees from Kissei Pharmaceutical. HM, TO, KO, and TI are full‐time employees of Eli Lilly Japan K. K. and own stock in Eli Lilly and Company. SU is a full‐time employee of Eli Lilly and Company, Indianapolis, Indiana, and owns stock in Eli Lilly and Company.
AUTHOR CONTRIBUTIONS
All authors contributed to designing the study, interpreting the data, and developing and writing the manuscript. All authors read and approved the final version. KF also collected data.
Supporting information
Appendix S1. Supporting information
Appendix S2. Supporting information
ACKNOWLEDGEMENTS
Medical writing support was provided by Andrew Sakko, PhD, CMPP, and editorial support was provided by Antonia Baldo, of Syneos Health, and was funded by Eli Lilly and Company in accordance with Good Publication Practice (GPP3) guidelines (www.ismpp.org/gpp3). Project management support was provided by Aki Yoshikawa from Eli Lilly Japan K. K. This study was sponsored by Eli Lilly and Company.
Furihata K, Mimura H, Urva S, Oura T, Ohwaki K, Imaoka T. A phase 1 multiple‐ascending dose study of tirzepatide in Japanese participants with type 2 diabetes. Diabetes Obes Metab. 2022;24(2):239‐246. doi: 10.1111/dom.14572
Funding information: Eli Lilly and Company.
Prior presentation: These data were previously presented at the American Diabetes Associationʼs 79th Scientific Sessions, San Francisco, CA, 7‐11 June 2019 (poster 1024P) and at the 63rd Annual Meeting of the Japanese Diabetes Society, online meeting, 5‐16 October 2020 (poster II P‐75‐2).
DATA AVAILABILITY STATEMENT
Eli Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
REFERENCES
- 1. International Diabetes Foundation . Diabetes Atlas, 9th ed.; 2019. www.diabetesatlas.org. Accessed January 4, 2021.
- 2. Miyazawa I, Kadota A, Miura K, et al. Twelve‐year trends of increasing overweight and obesity in patients with diabetes: the Shiga Diabetes Clinical Survey. Endocr J. 2018;65(5):527‐536. [DOI] [PubMed] [Google Scholar]
- 3. Drucker DJ. Mechanisms of action and therapeutic application of glucagon‐like peptide‐1. Cell Metab. 2018;27(4):740‐756. [DOI] [PubMed] [Google Scholar]
- 4. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696‐1705. [DOI] [PubMed] [Google Scholar]
- 5. Holst JJ. The physiology of glucagon‐like peptide 1. Physiol Rev. 2007;87(4):1409‐1439. [DOI] [PubMed] [Google Scholar]
- 6. Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon‐like peptide 1‐induced deceleration of gastric emptying in humans. Diabetes. 2011;60:1561‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nauck MA, Meier JJ. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. 2016;4(6):525‐536. [DOI] [PubMed] [Google Scholar]
- 8. Karagiannis T, Liakos A, Bekiari E, et al. Efficacy and safety of once‐weekly glucagon‐like peptide 1 receptor agonists for the management of type 2 diabetes: a systematic review and meta‐analysis of randomized controlled trials. Diabetes Obes Metab. 2015;17(11):1065‐1074. [DOI] [PubMed] [Google Scholar]
- 9. Orme ME, Nguyen H, Lu JY, Thomas SA. Comparative effectiveness of glycemic control in patients with type 2 diabetes treated with GLP‐1 receptor agonists: a network meta‐analysis of placebo‐controlled and active‐comparator trials. Diabetes Metab Syndr Obes. 2017;10:111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Finan B, Müller TD, Clemmensen C, Perez‐Tilve D, DiMarchi RD, Tschöp MH. Reappraisal of GIP pharmacology for metabolic diseases. Trends Mol Med. 2016;22(5):359‐376. [DOI] [PubMed] [Google Scholar]
- 11. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP‐1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo‐controlled and active comparator‐controlled phase 2 trial. Lancet. 2018;392(10160):2180‐2193. [DOI] [PubMed] [Google Scholar]
- 12. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP‐1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas MK, Nikooienejad A, Bray R, et al. Dual GIP and GLP‐1 receptor agonist tirzepatide improves beta‐cell function and insulin sensitivity in type 2 diabetes. J Clin Endocrinol Metab. 2021;106(2):388‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson JM, Nikooienejad A, Robins DA, et al. The dual glucose‐dependent insulinotropic peptide and glucagon‐like peptide‐1 receptor agonist, tirzepatide, improves lipoprotein biomarkers associated with insulin resistance and cardiovascular risk in patients with type 2 diabetes. Diabetes Obes Metab. 2020;22(12):2451‐2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartman ML, Sanyal AJ, Loomba R, et al. Effects of novel dual GIP and GLP‐1 receptor agonist tirzepatide on biomarkers of nonalcoholic steatohepatitis in patients with type 2 diabetes. Diabetes Care. 2020;43(6):1352‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frias JP, Nauck MA, Van J, et al. Efficacy and tolerability of tirzepatide, a dual glucose‐dependent insulinotropic peptide and glucagon‐like peptide‐1 receptor agonist in patients with type 2 diabetes: a 12‐week, randomized, double‐blind, placebo‐controlled study to evaluate different dose‐escalation regimens. Diabetes Obes Metab. 2020;22(6):938‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kristensen SL, Rørth R, Jhund PS, et al. Cardiovascular, mortality, and kidney outcomes with GLP‐1 receptor agonists in patients with type 2 diabetes: a systematic review and meta‐analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019;7(10):776‐785. [DOI] [PubMed] [Google Scholar]
- 18. van Bloemendaal L, IJzerman RG, Ten Kulve JS, et al. GLP‐1 receptor activation modulates appetite‐ and reward‐related brain areas in humans. Diabetes. 2014;63(12):4186‐4196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information
Appendix S2. Supporting information
Data Availability Statement
Eli Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.