

Abstract
We applied our computational algorithm TRUST4 to assemble immune receptor (TCR/BCR) repertoires from approximately twelve thousand RNA-seq samples from The Cancer Genome Atlas (TCGA) and seven immunotherapy studies. From over 35 million assembled complete complementary-determining region 3 (CDR3) sequences, we observed that the expression of CCL5 and MZB1 are the most positively correlated genes with T-cell clonal expansion and B-cell clonal expansion, respectively. We analyzed amino acid evolution during B-cell receptor somatic hypermutation and identified tyrosine as the preferred residue. We found that IgG1+IgG3 antibodies together with FcRn were associated with complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity or phagocytosis. In addition to B-cell infiltration, we discovered that B-cell clonal expansion and IgG1+IgG3 antibodies are also correlated with better patient outcomes. Finally, we created a website, VisualizIRR, for users to interactively explore and visualize the immune repertoires in this study.
Introduction
T cells and B cells play central roles in adaptive immunity by recognizing viral, bacterial, or parasitic pathogens and tumor antigens through their diverse receptors (TCRs and BCRs). Upon antigen recognition, B cells further undergo somatic hypermutation (SHM) and class switch recombination [1] to evolve BCRs or antibodies with better affinities to the antigen. TCR and BCR analyses have been widely adopted in studies of infectious disease [2], allergy [3], auto-immune disorders [4], tumor immunity [5] and immuno-oncology [6]. Though TCR/BCR-seq allows detailed investigation of the immune repertoire in tumors [7], these technologies are expensive and sometimes infeasible with limited tissue biopsies. In comparison, TCR and BCR sequences are contained in RNA-seq data as expressed transcripts, especially the abundant and clonally expanded receptors, but they are usually ignored in transcriptome studies. Traditional analysis methods could not process the most diverse and critical region of antigen recognition on TCR/BCRs, complementary-determining region 3 (CDR3), which is inherently different from the reference genome sequence. We and others have developed computational methods to de novo assemble immune receptor CDR3 sequences from RNA-seq data, such as TRUST [8–11], V’DJer [12], MiXCR [13], CATT [14] and ImRep [15]. Our previous work systematically characterized the TCR [8] and BCR [10] repertoires using earlier versions of the TRUST algorithm and thousands of tumor RNA-seq samples from The Cancer Genome Atlas (TCGA). These studies reveal a correlation between the immune repertoire and other tumor molecular features, such as immunogenetic somatic mutations and antibody-dependent cellular cytotoxicity.
We previously published TRUST4 [11] with significantly improved performance over previous versions of TRUST in terms of computational efficiency and the number of complete CDR3s recovered. This not only led to more accurate estimations of receptor diversities, but also allows a more detailed characterization of SHM in the CDR3s. TRUST4 assembles longer receptor sequences, which improves antibody isotype identification and association with downstream immune signals. Together, these improved features of TRUST4 motivated us to re-examine the immune receptor repertoires from TCGA tumors.
Immune checkpoint blockade (ICB) therapy has revolutionized tumor treatment by reactivating T cells and the immune system to eliminate cancer cells [16]. Studies also showed that B cells could promote immunotherapy response by forming tertiary lymphoid structures in tumors [17–19]. Therefore, we also extracted the immune receptor repertoires in 915 RNA-seq samples from seven ICB studies across four cancer types and investigated the repertoire difference between immunotherapy responders and non-responders.
To facilitate the characterization of immune repertoires under various oncology and immuno-oncology settings, we also developed an open-access web server VisualizIRR (Visualized Immune Repertoire Report). Existing immune repertoire databases, such as TCRdb [20] and VDJdb [21], focus on the receptor sequences without the ability to associate them with clinical information. In VisualizIRR, we processed immune repertoire data from various resources including TCGA and immunoACCESS, integrating metadata from immunotherapy trials. We curated de-identified clinical information for each sample from their original manuscripts, which allows the users to interactively associate immune repertoires with different clinical metadata.
Materials and Methods
Sequencing data
The 10,970 RNA-seq samples from TCGA were downloaded through Genomic Data Commons, along with the gene expression file, of which 10,096 were from tumor tissue, 724 were from adjacent tumor sites and the 150 acute myeloid leukemia (LAML) samples were from blood (more details in Table S1). We collected seven immunotherapy studies from Riaz [22], Gide [23], McDermott [24], Hugo [25], Mariathasan [26], Van Allen [27] and Kim [28]. In the Riaz study, the authors split the patients into naive (treatment naive) and progressive (progressed after Ipilimumab treatment) groups, so we indicated them as Riaz_naive and Riaz_prog in this work. The 915 RNA-seq samples from these seven immunotherapy studies were all from tumor tissues (Table S1). For the immune repertoire reconstruction, TRUST4 was applied on the alignment BAM files, which were available in TCGA and were generated by STAR [29] on the human hg38 reference genome for the seven immunotherapy cohorts.
Immune receptor repertoire analysis
We applied TRUST4 v1.0.0 to obtain TCRs and BCRs, along with their abundances, for each sample. We explored the diversity of TCR and BCR in TCGA and immunotherapy cohorts, where diversity was defined as the number of clonotypes per thousand CDR3 reads (CPK) as in our previous study [8]. The advantage of CPK is that it normalizes the effect of sequencing depths and immune cell infiltration in different samples. Lower CPK means fewer clonotypes given the same number of total CDR3 abundance and may suggest clonal expansion. In addition to CPK, we also measured Shannon entropy () and clonality () for each TCGA sample, where N is the number of distinct clonotypes. When computing the correlation of CPK and other genes’ expression, partial Spearman’s rank correlations were performed by controlling for the tumor purity of each sample. P-values were adjusted by the Benjamini-Hochberg method. Gene ontology analysis of genes highly correlated with CPK was performed by the clusterProfiler package v3.10.1 [30], with significance thresholds set at p-value < 0.01 and q-value < 0.05. TCGA RNA-seq sample immune cell deconvolution results based on MCPcounter and CIBERSORT-ABS were downloaded from TIMER2 [31].
We clustered highly similar IGH CDR3 clusters within each sample and investigated the SHM patterns based on these clusters. To establish the threshold for the cluster similarity, we computed the similarities for two CDR3s from different samples of the same cancer type as the null distribution. Due to the large number of IGH CDR3s found in each sample, we randomly selected at most one million pairs from each cancer type for the similarity analysis. Since small IGH CDR3 clusters contained too few sequence variations, we only considered the 1.3 million clusters with more than 30 CDR3s each to compute the substitute matrix. For each CDR3, we further exclude the first three residues, as they are highly conserved. Then, we followed the 5.0 version of the BLOSUM Matrices to produce the amino-acid substitution matrix. To study the co-evolution pattern in SHMs, we aligned TRUST4’s full-length assemblies to their corresponding IGHV genes and utilized AbRSA [32] to index the variations. We applied CCMpred v0.3.2, a well-established pseudo-likelihood maximization (PLM) tool, to discover the direct couplings between pairs of columns in the alignment results. The direct couplings indicated the co-evolution patterns in IGHs. We considered the 30 most confident co-evolution pairs from each IGHV gene analysis and computed the fraction for a pair across 67 IGHV genes with enough TRUST4 full-length assemblies as the consensus co-evolution pair confidence. To illustrate the residues in co-evolution, a 3D model of a heavy chain was modeled by Modeller (Version 9.0) [33] using the consensus sequence of the IGHV genes, and colored with the variation ratio at each position. The figure was plotted with VMD (version 1.9) [34].
To explore the association between immune features and immunotherapy outcomes, the fold change of immune features (including diversity, richness of IGH and TRB, expression of CTLA4, PD-1, PD-L1, etc.) were calculated between responders and non-responders for each cohort. P-value was given by the two-sided Wilcoxson signed-rank test. The p-values are combined with Fisher’s method or adjusted by the Benjamini-Hochberg method depending on the application. To increase the statistical power from all the samples, we used mixed-effect logistic regression to test the effect of each immune feature on immunotherapy response and regarded the cohort as random effects.
VisualizIRR
In order to curate a comprehensive collection of immune repertoire information for VisualizIRR, we collected data from multiple assays, including RNA-seq, TCR-seq, and BCR-seq. The RNA-seq immune repertoire cohorts were sourced from published immunotherapy studies and TCGA, and the repertoires were reconstructed with TRUST4. We included the results for the receptor chains of the human, being IGH, IGK, IGL, TRA, TRB, TRD, and TRG. The TCR-seq and BCR-seq samples were obtained from the cancer immunotherapy section of the immuneACCESS database. We included both human and mouse data. Human data includes the IGH and TRB loci and mouse data includes the TRB locus. Meta information was obtained by manually cleaning the sample tags available in immuneACCESS projects associated with the cohorts. If the cohort had no or incomplete clinical information from immuneACCESS, we extracted meta information from associated studies. The meta sheets we utilized when constructing the VisualizIRR cohort collection were manually constructed amalgamations of these two sources.
We then developed an R script that combs through each cohort and performs statistical analysis on the locus level. The R script largely automated the analysis of each cohort via the inclusion of a cohort-specific configuration file. For RNA–seq-derived data, partial CDR3s were not included in analysis. This script produces sample-level and cohort-level analysis of CDR3 nucleotide sequence length distribution. Out-of-frame nucleotide sequences were also removed and compressed on the CDR3 amino acid sequence level to produce amino acid sequence length distribution analysis. Top amino acid sequence clonotypes were collected along with their associated frequencies within the repertoire. Clonotypes with out-of-frame nucleotide sequences were filtered out from the remainder of the analysis. Top V-gene and J-gene frequencies were collected, as well as combinational top V-gene and J-gene frequencies. Top C-gene frequencies were collected for the IGH locus and top D-gene frequencies were collected for the IGH and TRB loci.
To associate immune repertoire diversity and clinical conditions, we calculated commonly used diversity-associated statistics on the sample locus level, with the majority of calculations utilizing the TCR and BCR repertoire analysis R package immunarch v0.6.5 [35]. Calculated statistics include Shannon entropy measure, effective number of types, Gini coefficient, Gini-Simpson index, inverse Simpson index, Chao1 index, clonal proportion, cumulative proportion clonotypes, CPK, and clonality. We also included average CDR3 length and unique CDR3 count. For IGH analysis which includes C-gene information, compartmentalization by isotype is also available. Pooled IGK and IGL analysis is also performed. The analysis for each cohort is exported to its own directory with a data structure that allows VisualizIRR to parse it appropriately.
We utilized existing TCR annotation databases VDJdb and McPAS-TCR to perform CDR3 annotation analysis. We merged the entries of the two databases by CDR3 amino acid sequence and associated species or pathology, with the names of these categories being unified between the databases. We then collected CDR3 overlap between samples and the merged database, with each overlapping CDR3 being weighted by its frequency in the sample repertoire. These overlap values were summed by associated species or pathology in a table. A special column was generated for the sum of all cancer-associated CDR3s. Top non-cancer and top cancer columns were kept intact, while the rest were summed in another special miscellaneous column.
Additional processing occurred for TCGA analysis. Cancer type and subtype-level CDR3 distribution and segment usage was calculated, as well as survival analysis based on samples containing above- and below-median statistical values. Overview analysis, containing tumor vs normal sample statistical values from different cancer types and subtypes, is precalculated.
We developed an HTML web portal that utilizes widely used web development libraries, including jQuery, Plotly.js, and dataTables. The portal contains repertoire analysis modules which dynamically display the currently user-selected cohort and clinical conditions. Figures are displayed using Plotly.js so that they can be dynamically adjusted and exported. VisualizIRR is freely available at http://cistrome.org/visualizirr without the need for registration or login. It has been tested in Firefox, Google Chrome, and Apple Safari browsers as well as MacOS, Windows, and Linux operating systems.
Data availability
TCGA data were downloaded through Genomic Data Commons. The de-identified immune repertoire from TCGA is available at Zenodo: https://doi.org/10.5281/zenodo.6326136.
All immunotherapy data are available at http://cistrome.org/visualizirr.
The TRUST4 code is available at https://github.com/liulab-dfci/TRUST4. The code for the analysis is available at https://github.com/liulab-dfci/TRUST4_analysis. VisualizIRR web server is at http://cistrome.org/visualizirr and the code for VisualizIRR is available at https://github.com/liulab-dfci/visualizirr.
Results
Characterization of TCGA immune repertoires with TRUST4
We previously published the redesigned TRUST algorithm (version 4) with substantial improvements in efficiency and sensitivity [11]. When applied to TCGA, TRUST4 recovered over 35 million complete CDR3 sequences across all TCR and BCR chains from the 10,970 TCGA samples (Table S2). Over 90% of high-quality IGH CDR3s found by TRUST3 were also recovered by TRUST4. This ratio was much higher than the 20% overlap ratio between the IGH CDR3s from the paired tumor and adjacent normal samples. In addition, TRUST4 consistently produced about seven times more complete CDR3s than previously published TRUST versions across TCGA samples (Figure 1a), which could lead to more accurate downstream analysis.
Figure 1. General results from TRUST4 on TCGA RNA-seq data.
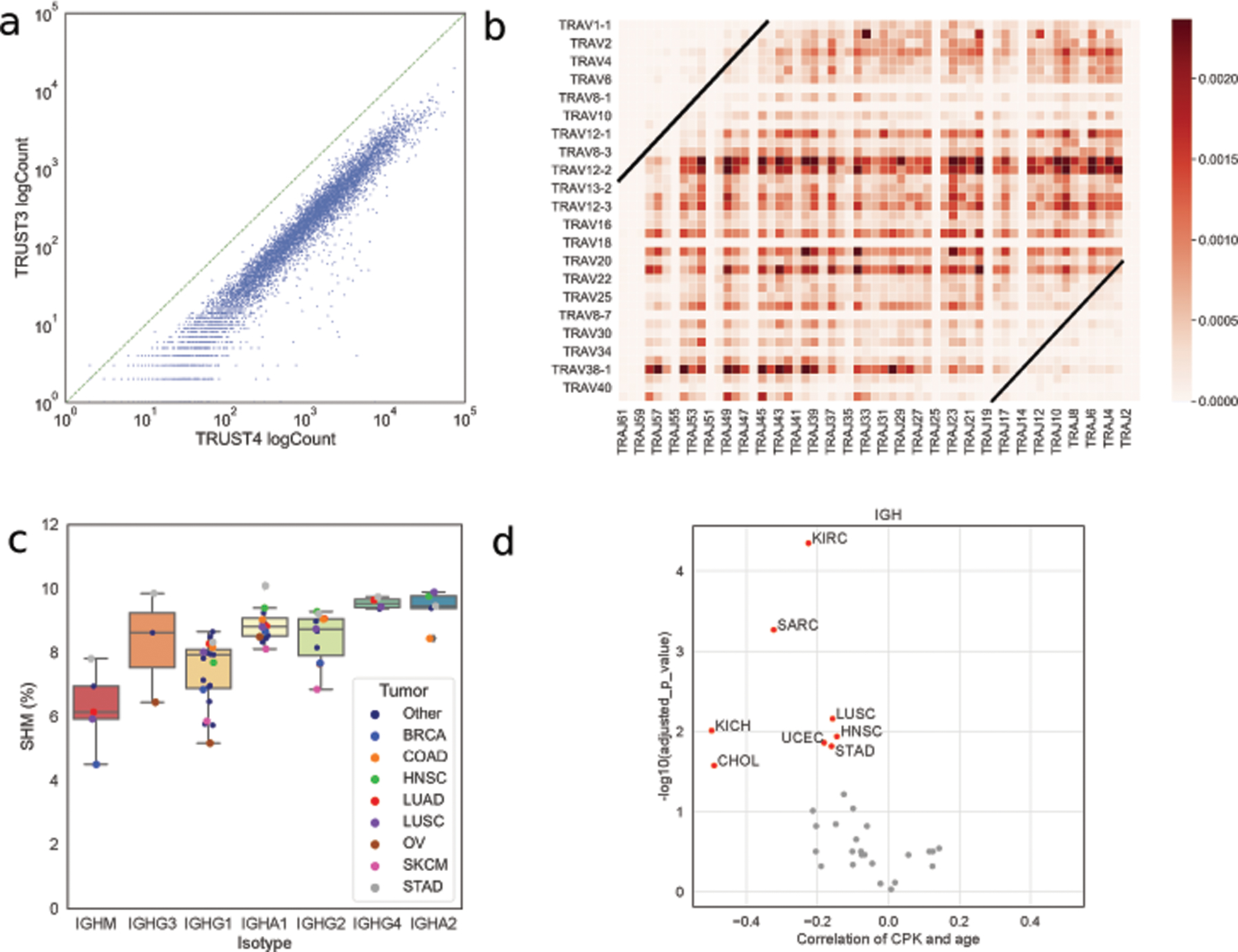
(a) Number of complete IGH CDR3s from TRUST3 and TRUST4 for each sample. (b) The combination frequency for TRAV and TRAJ genes, where the genes are sorted by the genome position. (c) SHM and isotype usage in different cancer types. Tumors with fewer than 200 samples are shown by smaller black dots(“other”). (d) Spearman correlation between age and IGH CPK corrected by tumor purity.
We analyzed the V gene and J gene pairing pattern on different chains and noticed that V-J pairing in T-cell receptor α (TRA) showed similar distances to the center (Figure 1b). In other words, the V-J recombination begins on the closest pair of V-J genes, and, if earlier attempts are unproductive, synchronously progresses to V-J genes further away from the center until successful recombination. This joining pattern was observed in mice [36] and used in a previous study to model α/β chain generation [37]. Due to the limited number of J genes on other chains (6, 5, 7, 14 for IGH, IGK, IGL, and TRB, respectively, as compared to 60 for TRA) and the involvement of D genes in TRB and IGH, we could not observe the joining pattern on other chains (Figure S1).
B cells improve BCR binding affinity against antigens by evolving their receptor sequences through SHM. TRUST4 reported full-length assemblies, whose sequences contain all the CDRs (CDR1/2/3) and framework regions. For BCR, the alignment similarity between the assembled V gene and the reference V gene could reflect the accumulated SHMs. We observed a higher accumulated SHM rate in the downstream isotypes, in agreement with the chronological splicing of constant genes in isotype switch [38] (Figure 1c). SHM rates across different isotypes seem to be higher in stomach adenocarcinoma (STAD), head and neck squamous cell carcinoma (HNSC), and colon adenocarcinoma (COAD) (Figure 1c). This may arise from the exposure to diverse food antigens in the gastrointestinal system or the gut microbiome, both of which could stimulate more activated and diverse B cells.
We explored the correlation between patient age and immune repertoire diversity (represented as clonotypes per thousand T-cell or B-cell CDR3 reads or CPK) in tumors as we previously proposed [8]. Immunosenescence, a process of immune dysfunction which occurs with age, was reported in the lymphoid tissues [39] and in peripheral blood mononuclear cells [40,41]. We observed a negative Spearman correlation between BCR IGH diversity and age in 26 out of the 33 cancer types in TCGA, of which eight had statistical significance after multiple hypothesis correction (Figure 1d). There was a similar negative association between T-cell receptor β (TRB) diversity and age in TCGA (Figure S2), although less statistically significant due to the lower number of TCRs extracted from the tumors. Together, our observations suggest that immunosenescence also occurs in non-lymphoid tissues.
Genes associated with clonal expansion
To identify genes associated with TCR and BCR diversity, we computed the correlation between immune repertoire (TCR β chain TRB and BCR heavy chain IGH) diversity and the expression of each protein-coding gene (Figure 2a, 2b). We found that most genes significantly associated with diversity are negatively correlated with diversity. Clonal expansion can decrease the immune repertoire diversity measure CPK. Therefore, we hypothesized that some of the genes negatively correlated with CPKs might be involved in or associated with T-cell and B-cell clonal expansion or activation. Indeed, pathway analysis for the top 50 correlated genes revealed these genes to be significantly enriched in T-cell and B-cell activation pathways (Figure 2c, 2d), respectively. In addition, we calculated the correlation between CPK and clonality (the latter defined as the normalized Shannon entropy), and found they were significantly negatively correlated (TRB, IGH spearman correlations ⍴=−0.82, −0.54 respectively, p values < 1e-10 both, Figure S3a). The correlation between CPK and Shannon entropy is weaker (TRB, IGH Spearman correlation =−0.05, −0.29 respectively, Figure S3b), suggesting lower CPK corresponds to clonal expansion in tissue samples.
Figure 2. Correlation between immune repertoire diversity and gene expression.
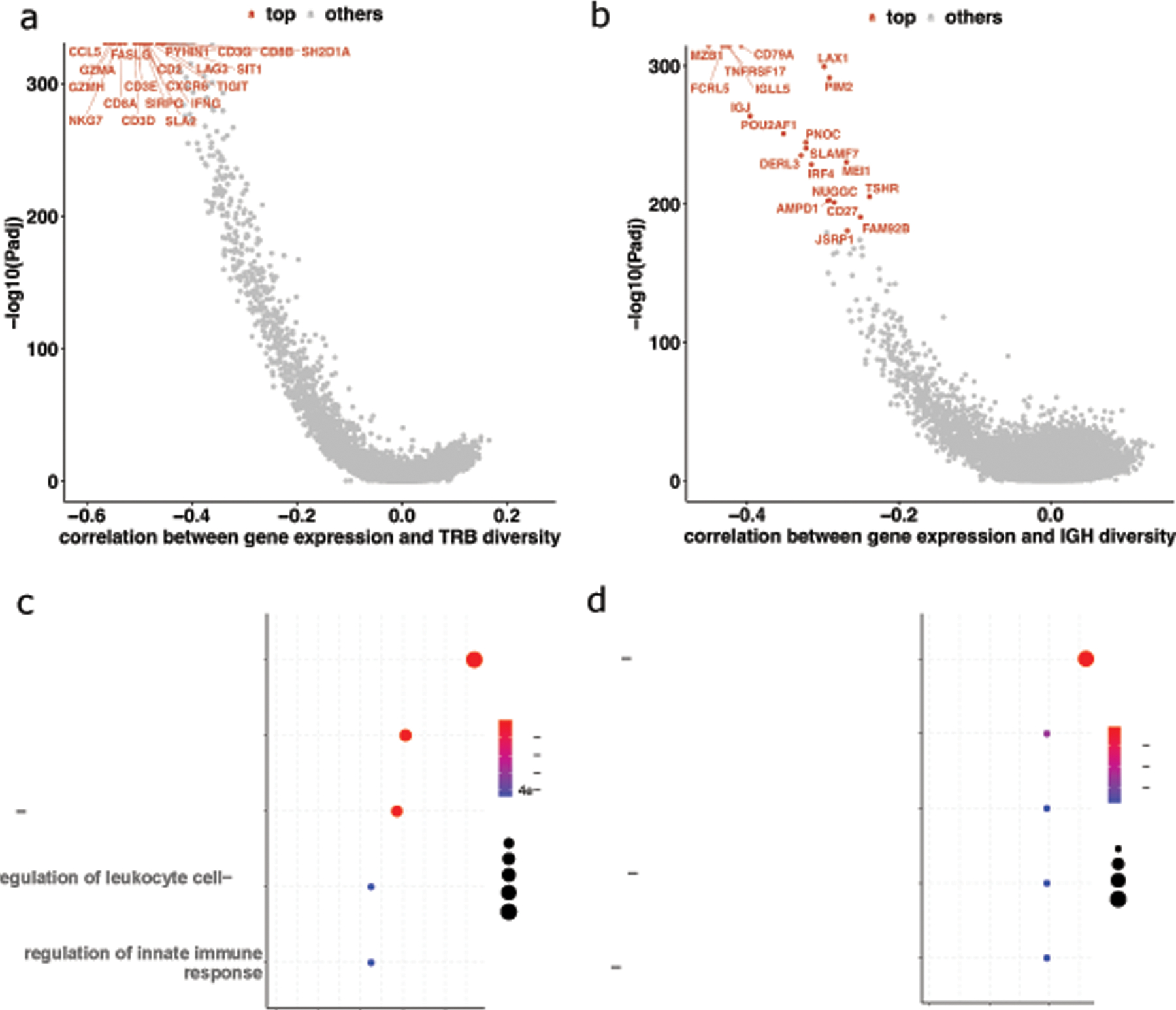
(a) Median correlations of gene expression (TPM) and TCR CPK. (b) Median correlations of gene expression (TPM) and BCR CPK. (c) Pathways from the top fifty TPM-TCR_CPK genes. (d) Pathways from the top fifty TPM-BCR_CPK genes.
Detailed examination of the top 20 genes correlated to diversity revealed their potential function in lymphocyte activation. CCL5 (median Spearman correlation ⍴=−0.565 across cancer types) was the top negatively correlated gene with TRB diversity. CCL5 encodes the chemokine guiding T cells to the tumor site and was reported to play an important role in orchestrating tumor-infiltrating lymphocytes [42]. The next two most negatively correlated genes GZMH and GZMA (median Spearman correlations ⍴=−0.556, −0.548 respectively) are granzymes expressed in cytotoxic effector cells, including CD8+ T cells. Moreover, CD8A and CD8B, the signature genes for the coreceptor of CD8+ T cells, were ranked 5 and 19 in the list (median ⍴=−0.536, −0.457 respectively). On the other hand, the CD4 gene’s highest rank was 154 in BRCA samples, and its median rank was 568 across all cancer types (median ⍴=−0.274). This suggests that CD8+ T cells might have a higher clonal expansion magnitude in the tumor environment than CD4+ T cells.
The genes negatively correlated with TCR diversity were consistent across different cancers with some notable exceptions (Figure S4a). First, thymoma (THYM) showed weak or even positive correlations between these genes and diversity, which might be explained by the role of the thymus as the T-cell maturation organ, so TRB diversity is not driven by clonal expansion. In addition, many of the top 20 diversity-associated genes also had weak to no correlations with TRB diversity in pancreatic adenocarcinoma tumors (PAAD). This might be due to tumor immune evasion through MHC-I complex protein degradation in pancreatic cancer [43], thus making antigen presentation insufficient to stimulate TCR signaling for clonal expansion. EPHB6 and HMGA2 were the two most negatively correlated genes with TRB CPK in pancreatic cancer (PAAD ⍴=−0.537, −0.450 respectively; median ⍴=−0.022,−0.026 respectively; Table S3). EPHB6 and HMGA2 have been shown to be overexpressed in T-cell acute lymphoblastic leukemia [44,45] where T cells are malignantly expanded. This may suggest abnormally high T-cell clonal expansion and dysfunction in pancreatic cancer.
A similar analysis with BCR diversity identified the genes most correlated to diversity to be related to plasma cells upon clonal expansion of memory or naive B cells. For example, MZB1 and TNFRSF17 (B-cell maturation antigen) (median ⍴=−0.451, −0.433 respectively) are known plasma cell marker genes [46]. Another top-ranked gene, FCRL5 (median ⍴=−0.427), is expressed on plasma cells [47]. It is the target of an antibody-based treatment for multiple myeloma where B cells show abnormal clonal expansion [48]. In addition to plasma cell marker genes, the top-ranked CD79A is a key gene in the MHC-II signaling pathway, which occurs during the T cell–dependent activation of B cells [49]. Although the correlation was observed across many cancer types, it is weak in diffuse large B-cell lymphoma (DLBC) where B cells exhibit malignant expansion (Figure S4b).
Amino acid residue preference in IGH somatic hypermutation
Highly similar IGH CDR3s at the nucleotide level from the same sample reflect SHMs in clonally expanded B cells of the same lineage. In order to find an appropriate threshold to cluster the highly similar IGH CDR3s, we compared the pairwise CDR3 similarities within the same patient tumor (intra-sample) and between different patient tumors (inter-sample). Similarly to the observations of our previous analysis on COAD [11], the intra-sample CDR3 similarities follow a bimodal distribution (Figure 3a). The left peak follows the inter-sample distribution which provides the null distribution of similarity among unrelated CDR3. The right peak is likely to arise from CDR3s of the same lineage, hence we used a conservative similarity threshold of 0.8 for CDR3 clustering.
Figure 3. Somatic hypermutations in IGH maturation.
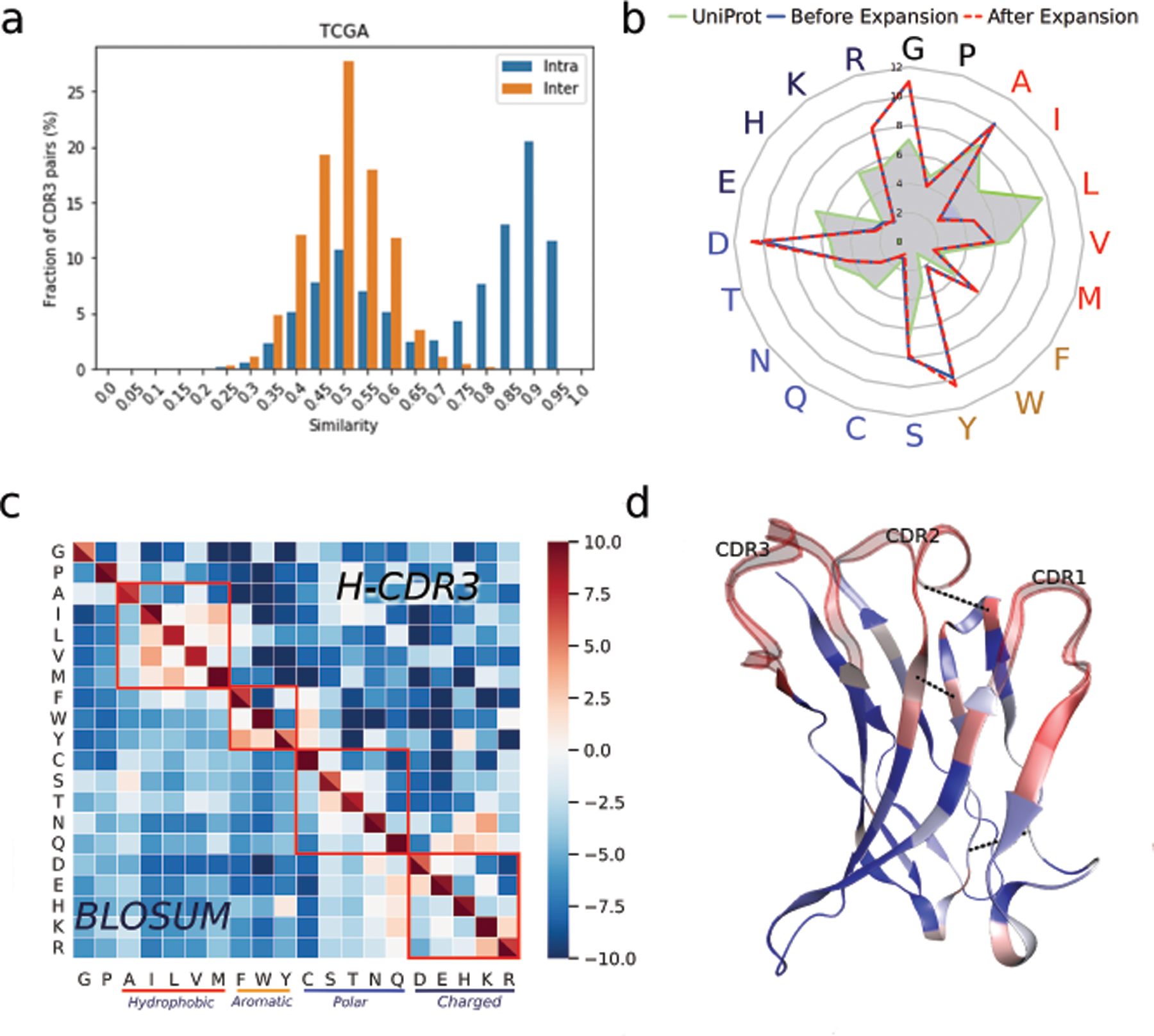
(a) Pairwise IGH CDR3 similarities within the same sample (intra-sample) and cross samples (inter-sample). (b) Amino acid usage in clonally expanded IGH CDR3, naive IGH CDR3 and proteins from the UniProt database. (c) BLOSUM matrix computed from IGH CDR3 clusters and the public BLOSUM100. (d) Co-evolution in SHM. SHM rates are shown by color (red high, blue low). Top three co-evolved residues are connected by blue dashed lines. Protein structure is based on IGHV1–69-2, which has the most full-length assemblies available.
Next, we sought to investigate amino acid residue composition in naive and expanded IGH CDR3s in the tumors. We found that residue usage in the CDR3 to be different from that of proteins in the UniProt database [50] (Figure 3b, Table S4), potentially supporting the unique function of IGH CDR3s in antigen recognition. From each CDR3 cluster, the relatively naive (since we might miss the original naive B-cell clone) and clonally expanded CDR3s can be readily distinguished. For example, in each cluster, the most abundant CDR3 can be regarded as the expanded CDR3, and the CDR3 with the earliest isotype and the fewest mutations from the germline can be annotated as the naive CDR3. We observed that Tyrosine (Tyr, Y) not only was among the most frequently used residues in CDR3s, but also was the favored residue (6% more frequent) in expanded CDR3s compared to naive CDR3s (Figure 3b, Table S4). Our observation is consistent with a theoretical proposal of Tyr as a favorable residue for antibody-antigen binding [51]. This is because Tyr is a hydrogen bond donor and can contribute a comparatively large hydrophobic surface to protein-protein interaction.
The CDR3 clustering also allowed us to evaluate residue substitution distributions in the CDR3s. We compared the BLOSUM matrix [52] derived from our CDR3 clusters with the public BLOSUM100 matrix which contained the general probabilities of residue substitution for evolutionarily divergent proteins. We found that CDR3 BLOSUM and BLOSUM100 to be highly consistent with each other (Figure 3c), where residues with similar physicochemical properties tended to mutate to each other. For example, a hydrophobic residue has a substantially higher probability to mutate into another hydrophobic residue than to hydrophilic ones. The pattern implied that although SHMs occur randomly, there is a strong negative selection pressure in the tumors to eliminate the CDR3s deviating from the baseline physicochemical properties of the naive antibodies with initial binding.
The above analysis revealed the patterns of single residue mutations in CDR3s, and we further investigated SHMs constrained by protein structure. Whereas CDR3 is a single loop on the IGH chain, the V gene constitutes the major part of the IGH protein structure by providing two additional binding loops and seven beta sheets, thus it was the focus of our structural analysis. To this end, we employed the pseudo-likelihood maximization (PLM) method CCMPred [53] to compute the co-evolution of residue substitution in SHM based on the BCR sequences with shared V genes. The most significant co-evolutions between the residues in the framework regions are in proximity according to the 3D protein structure (Figure 3d, Table S5). For example, positions 82 and 67 of an IGHV gene are 24 residues apart but the estimated spatial distance is only 6.93Å, comparable with the average amino acid contour length of 4Å [54]. This observation suggests a structural restraint of SHMs of the global BCR sequences to maintain the original folding and interactions. The co-evolution information in BCR sequences can be particularly helpful to advance antibody design and engineering.
Immune mechanisms involving IgG antibodies
Since the antibody constant region is important for downstream immune signaling, we next investigated the association between different Ig isotypes and tumor immune signals in TCGA data. Our previous work [10] showed that IgG1 and IgG3 (denoted as IgG1+IgG3) antibody fractions are positively correlated with CD16a expression level, a surrogate of natural killer (NK) cell activation. This observation suggested that IgG1+IgG3 antibodies could promote antibody-dependent cellular cytotoxicity (ADCC) to control tumor growth. Besides ADCC, complement-dependent cytotoxicity (CDC) is another essential immune mechanism to eliminate malignant cells [55]. The classical CDC pathway is initiated by the binding of the C1Q molecule to IgM or IgG antibodies which are bound to surface tumor-associated antigens (TAAs) on the target cells. A cascade of events follows, which results in the formation of a membrane attack complex (MAC) and target cell lysis (Figure 4a). We observed a positive correlation between IgM, IgG3 and IgG1 fractions and the expression of C1QA, C1QB, and C1QC across TCGA cancer types (Figure 4b, Figure S5a), consistent with these three isotypes being the most effective in activating the CDC pathway [55]. The correlations between C2~C9’ expression and isotype fractions were weaker and sometimes opposite from that between C1Q and isotypes (Figure S5b). This may be due to the fact that soluble C2~C9 proteins are mostly expressed in the liver [56].
Figure 4. IgG1+IgG3 antibody isotype correlation with other immune modules.
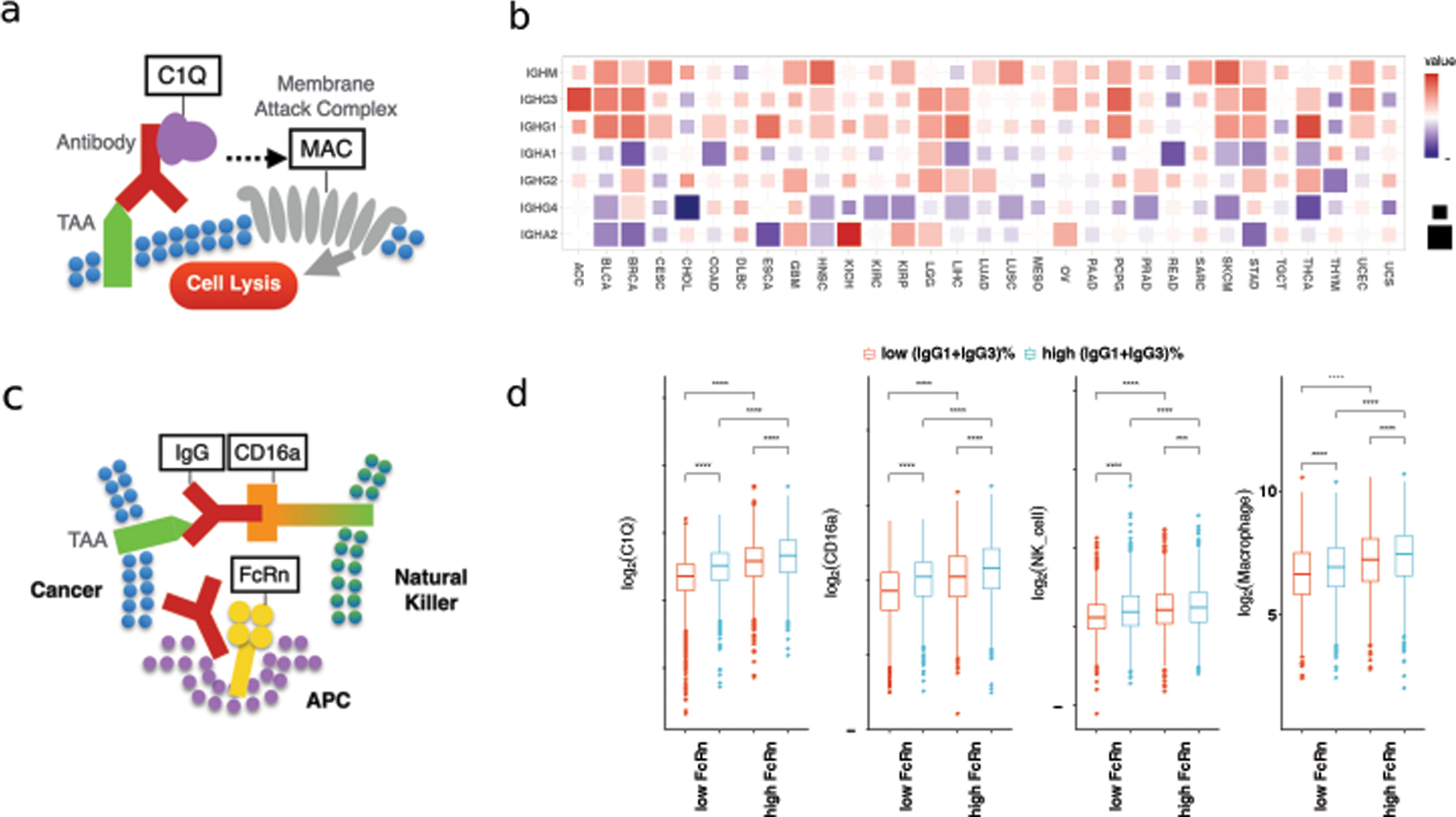
(a) Diagram of antibody and complement-dependent cytotoxicity. (b) Correlation between C1Q genes expression (sum of C1QA, C1QB and C1QC) and isotype fractions. (c) Diagram of antibody-dependent cellular cytotoxicity and FcRn. (d) C1Q genes and CD16a expression, NK cell and macrophage infiltration from MCPcounter stratified by FcRn (FCGRT) expression and IgG1+IgG3 isotype fraction. High and low groups correspond to the top 25% and lower 25% samples ranking by FcRn expression or IgG1+IgG3 fraction respectively. Statistical tests are based on two-sided Wilcoxon rank-sum test (***: p ≤ 0.001, ****: p ≤ 0.0001).
Neonatal Fc receptor (FcRn) binds to the IgG antibodies and prolongs their half-life [57] (Figure 4c). We hypothesized that higher FcRn abundance would maintain more IgG antibodies in the tumor microenvironment and result in stronger ADCC and CDC signals. Indeed, tumor samples with higher expression of the FcRn gene (FCGRT) had higher expression of CD16a and C1Q regardless of IgG fractions (Figure 4d). Furthermore, we noticed that FcRn expression had little or even negative correlation with IgG fractions across cancer types (Figure S6), suggesting that FcRn and IgG1+IgG3 antibodies could promote ADCC and CDC independently. In addition to individual gene expression, we applied MCPcounter [58] and CIBERSORT-ABS [46] to computationally deconvolve the tumor bulk RNA-seq samples. The inferred abundances of NK cells and macrophages can be regarded as the surrogates for ADCC and antibody-dependent cellular phagocytosis (ADCP). We observed the same pattern that higher IgG1+IgG3 fraction and FcRn expression related to stronger ADCC and ADCP (Figure 4d, Figure S7). Together, our findings support the antitumor role of IgG1+IgG3 antibodies and FcRn and their potential association with CDC, ADCC, and ADCP.
Immune repertoire for cancer immunotherapy studies
Previous studies using TCR-seq analyses have shown that ICB treatment can reshape T-cell receptor repertoires [6,22]. In this study, we explored the tumor-infiltrating immune repertoires extracted from RNA-seq of tumors undergoing anti–PD-1, anti–PD-L1, or anti-CTLA4 ICB treatments from seven clinical studies [22–28]. We first examined the two melanoma studies, Riaz [22] and Gide [23], both with pre-treatment and on-/post-treatment samples. We further split the Riaz study into ipiNaive (treatment-naive) and ipiProg (progressed after ipilimumab treatment) cohorts, as was done in the original study. We have established that the diversity measure CPK is significantly negatively correlated to clonality in TCGA analysis. By comparing the paired samples from the two time points, we observed trends of TRB clonal expansion and higher clonality upon ICB treatment regardless of therapy outcome in the three cohorts (Fisher’s method combined p-value 0.095 for non-responders, 0.029 for responders; Figure 5a). For BCR, the responders’ diversity tends to decrease during the treatment in the patients from Gide and Riaz_naive (Fisher’s method combined p-value 0.238 for non-responders, and 0.045 for responders), suggesting B-cell clonal expansion could be beneficial in melanoma immunotherapy. The BCR diversity remained similar in Riaz_ipiProg, which suggested pre-existing BCR clonal expansion upon the initial ipilimumab treatment.
Figure 5. Immune repertoire dynamics in immunotherapy cohorts.
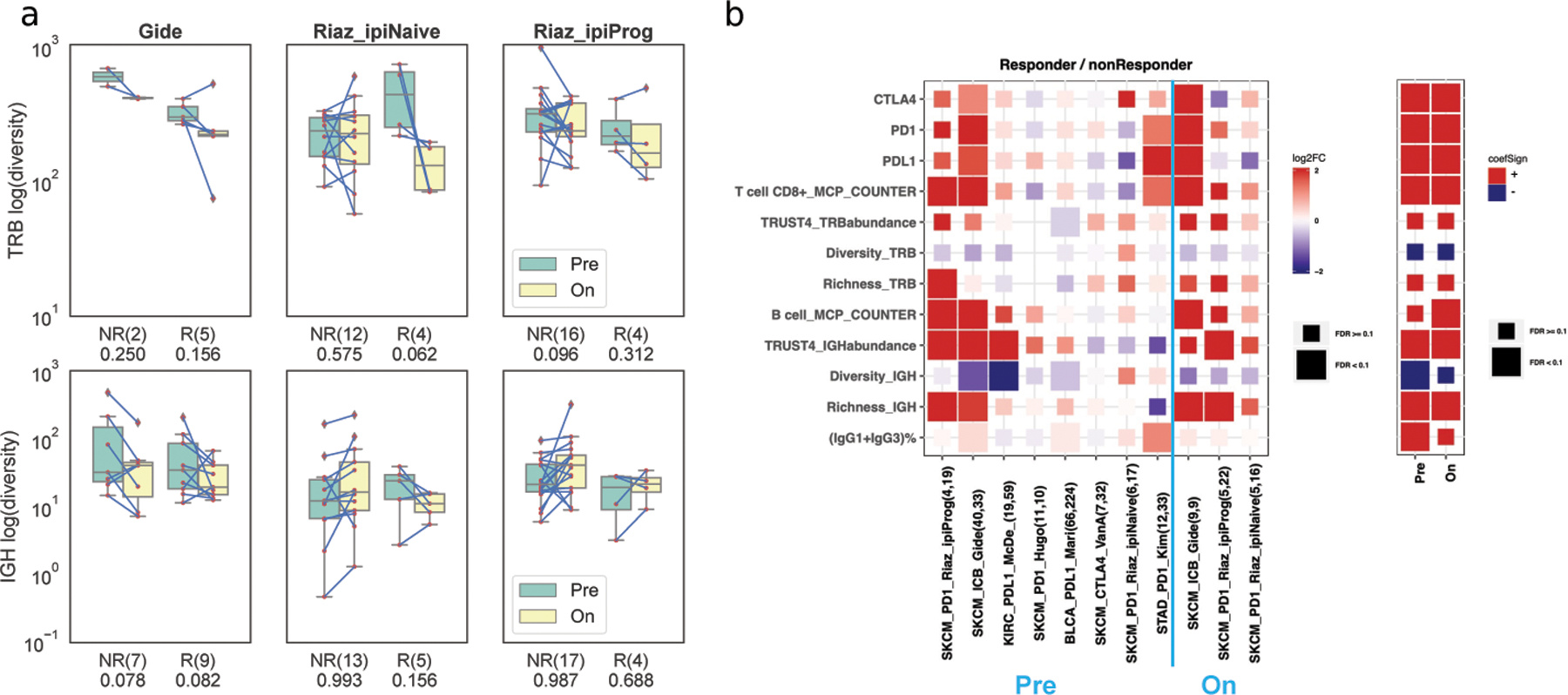
(a) Change of TRB and IGH CPK during treatment between responders (R) and non-responders (NR) in Riaz (naive, ipiProg) and Gide cohorts. Numbers under the x-axis labels are the p-values from the one-sided Wilcoxon signed-rank test. (b) Left: Fold change of gene expression and immune features between responders and non-responders in various immunotherapy cohorts, where FDR was based on the two-sided Wilcoxon rank-sum test. Right: Value and statistical significance of the gene expression and immune features from the logistic mixed-effect linear model, where the cohort was the random effect.
We extended the analysis to compare the immune repertoires between responders and non-responders across all the cohorts. We compared the fold change of various markers between responders and non-responders, before and after treatment, respectively (Figure 5b). Higher expression of immune checkpoint genes, PDCD1 (PD-1), CD274 (PD-L1) and CTLA4, were associated with better immunotherapy outcomes in Gide [23] and Kim [28]. Though responders had higher T-cell infiltration based on MCPcounter in Riaz_ipiProg, Gide, and Kim cohorts, we could not associate the TRUST4-derived TCR abundance and diversity with therapy outcome. This might be due to the limited number of samples and assembled TCRs. In BCR analysis, we observed a significant difference between responders and non-responders in B-cell infiltration, with higher infiltration in responders (Figure 5b). In addition, TRUST4-estimated IGH abundance, CDR3 richness and diversities were higher in responders in the Riaz_ipiProg, Gide, McDermott [24] and Mariathasan [26] cohorts. Responders had higher B-cell signals in these cohorts, suggesting that the B-cell immune repertoire measures could be useful for ICB treatment response prediction, as in previous studies [17–19]. Furthermore, a higher IgG1+IgG3 fraction was associated with a better ICB outcome (Figure 5b) in line with our earlier observations that IgG1+IgG3 antibodies could promote anti-tumor immunity. We repeated the analysis for the on-treatment samples, and found that T-cell infiltration, B-cell infiltration and IGH richness were also higher in responders. To combine the statistical power across different cohorts, we applied mixed-effect logistic regression using the cohort as the random effect. The analysis confirmed that the responders had significantly higher B-cell abundance, IGH CDR3 richness, clonal expansions, and IgG1+IgG3 fractions (FDR<0.1, Figure 5b right, coefficient values and statistical test results are in Table S6).
Web server for tumor immune repertoire visualization and analysis
To help researchers analyze and explore the data from this study, we organized the TRUST4 results on TCGA and immunotherapy RNA-seq samples into a website VisualizIRR (http://www.cistrome.org/visualizirr). VisualizIRR provides a user-friendly interface for users to visualize immune repertoire features under different clinical conditions on either a computer or a smartphone (Figure S8). As an example, we explored TRB diversity in TCGA and confirmed our previous observations [8] that low-grade glioma (LGG) had low TRB diversity, whereas colorectal cancers (READ, COAD), non-small-cell lung carcinomas (LUSC, LUAD), mesothelioma (MESO) and melanoma (SKCM) had relatively higher diversity (Figure 6a). With TRUST4’s highly sensitive assembly results, VisualizIRR identified THYM samples as having the highest TRB diversity, which was consistent with the role of the thymus in T-cell maturation. Furthermore, VisualizIRR shows diversity comparisons between tumor samples and adjacent normal samples for each cancer type. We found that TRBs had significantly lower diversity in COAD and LUSC tumor sites than the adjacent normal sites, reflecting the lower T-cell infiltration in these tumor samples based on MCPcounter (Figure S9). In IGH isotype analysis, VisualizIRR showed that the IgG1 fractions are positively correlated with longer patient survival in SKCM, ovarian serous cystadenocarcinoma (OV) and acute myeloid leukemia (LAML) (Figure 6b). This is consistent with the beneficial role of IgG1 antibodies in tumor control in these cancer types [41,59].
Figure 6. Characterization of immune repertoire with VisualizIRR.
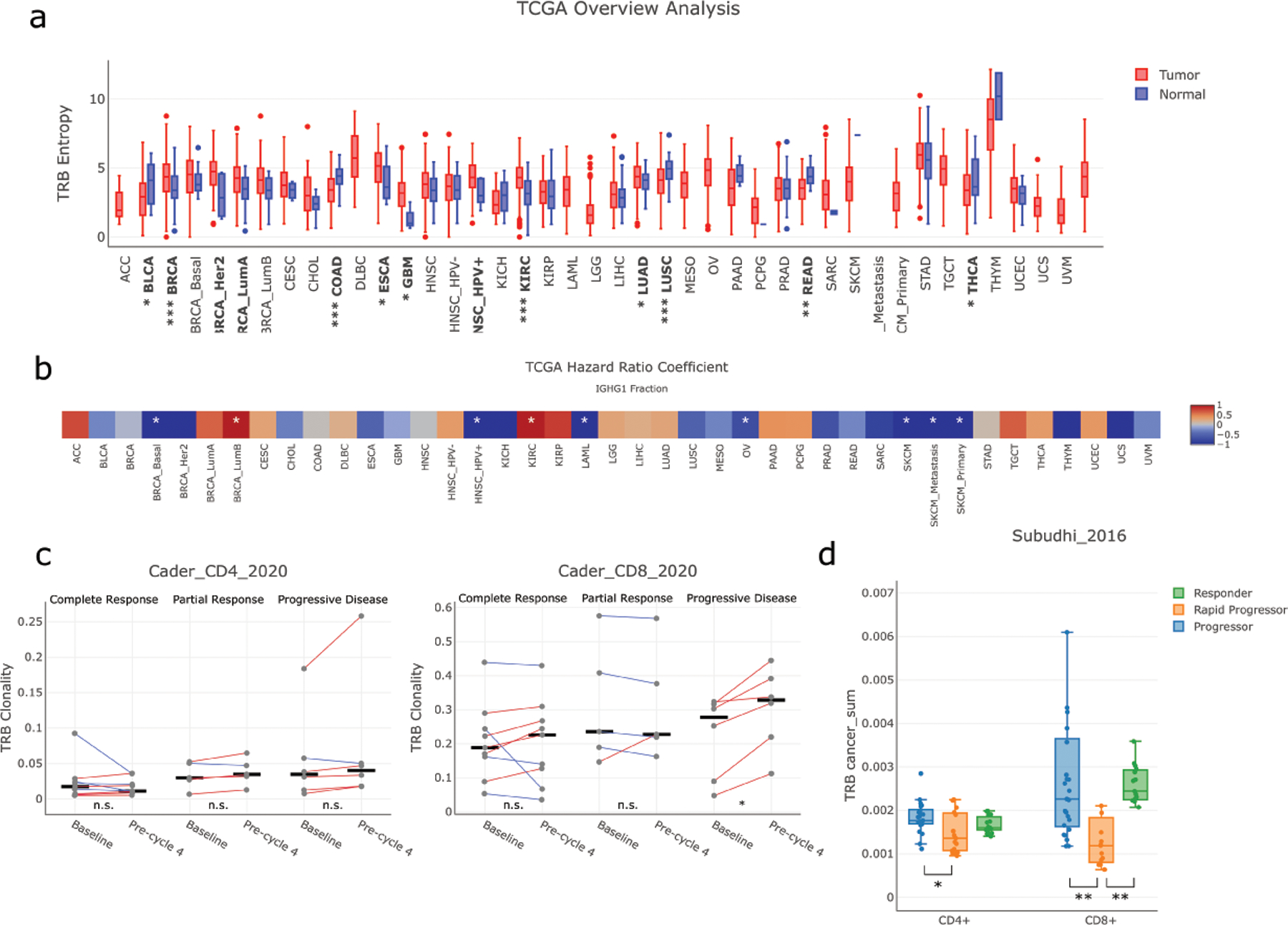
(a) TRB diversity in TCGA samples. P-values for comparing diversity between tumor samples and adjacent normal samples were computed by the Wilcoxon rank-sum test (*: p ≤ 0.05; **: p ≤ 0.005; ***: p ≤ 0.0005) (b) IgG1 fraction and patient survival in TCGA tumor samples. Color scale reflects the coefficient value from the Cox proportional hazards regression model and asterisks indicate raw p-value ≤0.05 (c) Comparisons of TRB clonality between pre-treatment and post-treatment samples in CD4+ and CD8+ sorted TCR-seq samples of a cHL immunotherapy study [6]. (d) Comparisons of the cancer-associated CDR3s between different response groups from both CD4+ and CD8+ sorted TCR-seq samples of a metastatic prostate cancer immunotherapy study. All the plots were generated by VisualizIRR.
In addition to RNA-seq immune repertoires, we collected TCR-seq and BCR-seq data from an additional 28 immunotherapy cohorts and included them in the VisualizIRR website. The website allows users to examine the immune repertoire diversity under various clinical conditions. For example, TCR-seq of peripheral blood mononuclear cell (PBMC) from healthy donors and newly diagnosed classical Hodgkin lymphoma (cHL) patients had greater TCR diversity than from relapsed/refractory patients [6](Figure S10a). VisualizIRR also has a time series feature to reproduce the result that cHL patients with complete response have increased CD4+ T-cell diversity upon PD-1 blockade treatment (Figure S10b). In contrast, CD8+ T cells were clonally expanded in cHL patients with progressive disease (Figure 6c). Since cHL tumors are known to have MHC-I deficiency, this result suggests that CD4+ instead of CD8+ T cells might be the effector cells in anti–PD-1 response in cHL.
To identify pathogens stimulating T cells in patients, we further annotated the TCRs in VisualizIRR with the antigen information from the VDJdb and McPAS-TCR [60] databases. In each sample, VisualizIRR computes the fraction for different types of pathogens, such as CMV, or tumor antigens, such as lung cancer antigen. The TCR antigen annotation provides novel insights to explore the relationship between immune repertoire and other molecular or clinical information. For example, in a PBMC TCR-seq study of metastatic prostate cancer patients undergoing anti-CTLA4 treatment [61], VisualizIRR found that responders and non-rapid progressors had significantly more TCRs targeting tumor antigens than rapid progressors (Figure 6d). This result suggests that the ability of ICB in expanding tumor-antigen-specific TCRs might be the cause of the better patient response.
Discussion
In this work, we applied TRUST4, an improved de novo TCR and BCR assembler, on TCGA and immunotherapy cohort RNA-seq data to study the immune receptor repertoire in tumors. The exploration of gene expression and immune repertoire information from RNA-seq data helped us identify the relationships of T cells and B cells with other immune pathways or processes. Our study further supports the positive correlation of IgG1+IgG3 antibodies with antitumor signals and immunotherapy outcomes and could inspire future research on establishing B cells as a biomarker for cancer patient survival or treatment predictions.
From TCGA data, we identified genes correlated with T-cell and B-cell clonal expansions such as CCL5, MZB1 and FCRL5 through correlation analysis between repertoire diversity and gene expression. Previously, there have been conflicting reports regarding the function of FCRL5 in promoting or inhibiting B-cell activation and clonal expansion [47,62]. Our analysis suggests that FCRL5 may play a more activating role in the tumor environment. We found that CD8+ T cells had more clonal expansion signals based on gene expression patterns, and this could be validated through CD4+ T-cell and CD8+ T-cell repertoire diversity analysis. CD4+ and CD8+ TCR information can be obtained after cell sorting, which is beyond the scope of the typical RNA-seq data in this study. We note that clonal expansion was measured by the CPK of TRB and IGH CDR3s, which might be affected by immune cell infiltration. For example, the lower CPK in elder patients could be due to clonal expansion from past infections or immune cell apoptosis in earlier life. Systematic evaluation of diversity measures or development of new metrics that are robust to the impact of T-cell or B-cell infiltrations are still needed.
In the SHM analysis, we found that Tyr was favored in BCR CDR3 SHMs, and the retained SHMs maintained the initial CDR3’s physicochemical properties. These discoveries could help researchers understand the binding affinity improvement during SHM and improve tumor-targeting antibody design [63]. BCR is formed by heavy and light chains, where the light chain also undergoes SHM. We observed that SHMs were constrained by the protein structure within a chain but could not analyze the impacts from the paired chain. With the development of single-cell sequencing technology, future studies can include profiling of co-evolution between heavy and light chains.
The function of B cells in cancer immunotherapies has received increasing attention in clinical studies [17–19]. The observed elevated clonal expansion of B cells and higher IgG1+IgG3 fraction in immunotherapy responders could provide additional evidence that B cells may participate in anti-tumor immune responses to ICB treatment. Further, it is likely that the B cells in the tumor are activated by cancer-associated antigens, as evidenced by isotype class switch. Characterization of these antigens may lead to novel therapeutic targets, and thus is of high clinical interest. However, due to the limited epitope annotations of BCRs, we could not computationally infer the antigens based on sequences of the clonally expanded BCRs. With the growing size of annotation databases, such as IEDB [64], or the advances of computational approaches, such as DeepCAT [65] for TCRs, we may identify the dynamics of tumor-targeted BCRs during ICB treatment in the future.
We developed VisualizIRR to consolidate various immune repertoire analyses into an interactive open-access website. We have demonstrated how an end-user could reproduce previously published immune repertoire analyses and make new observations. The strength of VisualizIRR lies in the curated profiling of numerous immunotherapy and TCGA samples, combined with a variety of commonly useful repertoire analysis techniques. Going forward, we aim to not only build upon the functionality of VisualizIRR but also expand the number of collected datasets. Some potential functionality enhancements include single-cell specific modules and cross-cohort analysis. We envision continued maintenance and development of VisualizIRR to benefit the cancer immunology and immunotherapy research communities.
Supplementary Material
Synopsis.
The immune repertoires from bulk tumor transcriptomes were computationally assembled by TRUST4 and comprehensively analyzed to improve the understanding of T cell and B cell antitumor activity and clonal dynamics in tumor infiltration and response to cancer immunotherapies.
Acknowledgement
We acknowledge the following funding sources for supporting this work: NCI grants U01CA226196 (H.L), U24CA224316 (CIDC at DFCI), 1R01CA245318 (B.L) and 1R01CA258524-01A1 (B.L).
Footnotes
Competing interests
X.S.L. conducted the work while being a faculty at DFCI, and is currently a board member and shareholder of GV20 Oncotherapy, and the full-time CEO of its subsidiary GV20 Therapeutics. X.H. conducted the work while being a postdoctoral fellow at DFCI, and is currently a full-time employee of GV20 Therapeutics.
References
- 1.Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology 2020. pp. 233–247. doi: 10.1111/imm.13176 [DOI] [PMC free article] [PubMed]
- 2.Lee J, Boutz DR, Chromikova V, Joyce MG, Vollmers C, Leung K, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med 2016;22: 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiyotani K, Mai TH, Yamaguchi R, Yew PY, Kulis M, Orgel K, et al. Characterization of the B-cell receptor repertoires in peanut allergic subjects undergoing oral immunotherapy. J Hum Genet 2018;63: 239–248. [DOI] [PubMed] [Google Scholar]
- 4.Liu S, Hou XL, Sui WG, Lu QJ, Hu YL, Dai Y. Direct measurement of B-cell receptor repertoire’s composition and variation in systemic lupus erythematosus. Genes & Immunity 2017. pp. 22–27. doi: 10.1038/gene.2016.45 [DOI] [PubMed]
- 5.Kurtz DM, Green MR, Bratman SV, Scherer F, Liu CL, Kunder CA, et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015;125: 3679–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cader FZ, Hu X, Goh WL, Wienand K, Ouyang J, Mandato E, et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat Med 2020;26: 1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cowell LG. The Diagnostic, Prognostic, and Therapeutic Potential of Adaptive Immune Receptor Repertoire Profiling in Cancer. Cancer Res 2020;80: 643–654. [DOI] [PubMed] [Google Scholar]
- 8.Li B, Li T, Pignon J-C, Wang B, Wang J, Shukla SA, et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat Genet 2016;48: 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Li T, Wang B, Dou R, Zhang J, Liu JS, et al. Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA-seq data. Nat Genet 2017;49: 482–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu X, Zhang J, Wang J, Fu J, Li T, Zheng X, et al. Landscape of B cell immunity and related immune evasion in human cancers. Nat Genet 2019;51: 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Liu XS. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat Methods 2021. doi: 10.1038/s41592-021-01142-2 [DOI] [PMC free article] [PubMed]
- 12.Mose LE, Selitsky SR, Bixby LM, Marron DL, Iglesia MD, Serody JS, et al. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V’DJer. Bioinformatics 2016;32: 3729–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol 2017;35: 908–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S-Y, Liu C-J, Zhang Q, Guo A-Y. An ultrasensitive T-cell receptor detection method for TCR-Seq and RNA-Seq data. Bioinformatics 2020. doi: 10.1093/bioinformatics/btaa432 [DOI] [PubMed]
- 15.Mandric I, Rotman J, Yang HT, Strauli N, Montoya DJ, Van Der Wey W, et al. Profiling immunoglobulin repertoires across multiple human tissues using RNA sequencing. Nat Commun 2020;11: 3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359: 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577: 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577: 561–565. [DOI] [PubMed] [Google Scholar]
- 19.Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020;577: 556–560. [DOI] [PubMed] [Google Scholar]
- 20.Chen S-Y, Yue T, Lei Q, Guo A-Y. TCRdb: a comprehensive database for T-cell receptor sequences with powerful search function. Nucleic Acids Res 2021;49: D468–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res 2018;46: D419–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171: 934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin Cancer Res 2018;24: 1260–1270. [DOI] [PubMed] [Google Scholar]
- 24.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018;24: 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016. pp. 35–44. [DOI] [PMC free article] [PubMed]
- 26.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554: 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24: 1449–1458. [DOI] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 2020;48: W509–W514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li L, Chen S, Miao Z, Liu Y, Liu X, Xiao Z-X, et al. AbRSA: A robust tool for antibody numbering. Protein Sci 2019;28: 1524–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol 2014;1137: 1–15. [DOI] [PubMed] [Google Scholar]
- 34.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14: 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
- 35.Nazarov V, Bot I, Rumynskiy E. immunomind/immunarch: 0.6.5: Basic single-cell support 2020. doi: 10.5281/zenodo.3893991 [DOI]
- 36.Aude-Garcia C, Gallagher M, Marche PN, Jouvin-Marche E. Preferential ADV-AJ association during recombination in the mouse T-cell receptor alpha/delta locus. Immunogenetics 2001;52: 224–230. [DOI] [PubMed] [Google Scholar]
- 37.Dupic T, Marcou Q, Walczak AM, Mora T. Genesis of the αβ T-cell receptor. PLoS Comput Biol 2019;15: e1006874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol 2008;26: 261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabibian-Keissar H, Hazanov L, Schiby G, Rosenthal N, Rakovsky A, Michaeli M, et al. Aging affects B-cell antigen receptor repertoire diversity in primary and secondary lymphoid tissues. Eur J Immunol 2016;46: 480–492. [DOI] [PubMed] [Google Scholar]
- 40.Britanova OV, Putintseva EV, Shugay M, Merzlyak EM, Turchaninova MA, Staroverov DB, et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 2014;192: 2689–2698. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Hu X, Wang J, Sahu AD, Cohen D, Song L, et al. Immune receptor repertoires in pediatric and adult acute myeloid leukemia. Genome Med 2019;11: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019;35: 885–900.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020;581: 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El Zawily A, McEwen E, Toosi B, Vizeacoumar FS, Freywald T, Vizeacoumar FJ, et al. The EphB6 receptor is overexpressed in pediatric T cell acute lymphoblastic leukemia and increases its sensitivity to doxorubicin treatment. Sci Rep 2017;7: 14767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Efanov A, Zanesi N, Coppola V, Nuovo G, Bolon B, Wernicle-Jameson D, et al. Human HMGA2 protein overexpressed in mice induces precursor T-cell lymphoblastic leukemia. Blood Cancer J 2014;4: e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12: 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Borrego F, Nagata S, Tolnay M. Fc Receptor-like 5 Expression Distinguishes Two Distinct Subsets of Human Circulating Tissue-like Memory B Cells. J Immunol 2016;196: 4064–4074. [DOI] [PubMed] [Google Scholar]
- 48.Elkins K, Zheng B, Go M, Slaga D, Du C, Scales SJ, et al. FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma. Mol Cancer Ther 2012;11: 2222–2232. [DOI] [PubMed] [Google Scholar]
- 49.Katikaneni DS, Jin L. B cell MHC class II signaling: A story of life and death. Hum Immunol 2019;80: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Consortium UniProt. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 2021;49: D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mian IS, Bradwell AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol 1991;217: 133–151. [DOI] [PubMed] [Google Scholar]
- 52.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992;89: 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seemayer S, Gruber M, Söding J. CCMpred--fast and precise prediction of protein residue-residue contacts from correlated mutations. Bioinformatics 2014;30: 3128–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ainavarapu SRK, Brujic J, Huang HH, Wiita AP, Lu H, Li L, et al. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys J 2007;92: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Macor P, Capolla S, Tedesco F. Complement as a Biological Tool to Control Tumor Growth. Front Immunol 2018;9: 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol 2017;188: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pyzik M, Sand KMK, Hubbard JJ, Andersen JT, Sandlie I, Blumberg RS. The Neonatal Fc Receptor (FcRn): A Misnomer? Front Immunol 2019;10: 1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol 2020. doi: 10.1038/s41577-019-0257-x [DOI] [PubMed]
- 60.Tickotsky N, Sagiv T, Prilusky J, Shifrut E, Friedman N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 2017;33: 2924–2929. [DOI] [PubMed] [Google Scholar]
- 61.Subudhi SK, Aparicio A, Gao J, Zurita AJ, Araujo JC, Logothetis CJ, et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci U S A 2016;113: 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haga CL, Ehrhardt GRA, Boohaker RJ, Davis RS, Cooper MD. Fc receptor-like 5 inhibits B cell activation via SHP-1 tyrosine phosphatase recruitment. Proc Natl Acad Sci U S A 2007;104: 9770–9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 2020;27: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 2019;47: D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beshnova D, Ye J, Onabolu O, Moon B, Zheng W, Fu Y-X, et al. De novo prediction of cancer-associated T cell receptors for noninvasive cancer detection. Sci Transl Med 2020;12. doi: 10.1126/scitranslmed.aaz3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TCGA data were downloaded through Genomic Data Commons. The de-identified immune repertoire from TCGA is available at Zenodo: https://doi.org/10.5281/zenodo.6326136.
All immunotherapy data are available at http://cistrome.org/visualizirr.
The TRUST4 code is available at https://github.com/liulab-dfci/TRUST4. The code for the analysis is available at https://github.com/liulab-dfci/TRUST4_analysis. VisualizIRR web server is at http://cistrome.org/visualizirr and the code for VisualizIRR is available at https://github.com/liulab-dfci/visualizirr.