

Abstract
Parkinson’s disease (PD) is a neurological disorder in which oxidative stress and reactive oxygen species productions are proposed to be involved in its pathogenesis. Despite considerable advancement in Selenium’s (Se) molecular biology and metabolism, we do not know much about the cell type-specific pattern of Se distribution in the brain of PD humans and experimental animals. Although, there is plenty of evidence around the role of Se deficiency in PD’s pathogenesis impacting lipid peroxidation and reducing glutathione (GSH) and glutathione peroxidase (GPX). It has been suggested that Se has an inducible role in selenium-dependent GPX activity in PD animals and humans. However, calcium as a second messenger regulates the neuron cells’ essential activities, but its overloading leads to cellular oxidative stress and apoptosis. Therefore, Se’s antioxidant role can affect calcium signaling and alleviate its complications. There are signs of Se and Selenoproteins incorporation in protecting stress oxidative in various pathways. In conclusion, there is convincing proof for the crucial role of Se and Calcium in PD pathogenesis.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12291-022-01031-1.
Keywords: Parkinson's disease, Calcium signaling, Selenium, Oxidative stress, Glutathione peroxidase (GPX)
Introduction
Although the brain occupies less than 2% of the body’s mass, it has the most metabolical function among all body organs [1]. However, according to its great metabolic activity, it is significantly defenseless to peroxidation [2]; also, lack of antioxidant enzyme activities in the brain increases oxidative stress within specific neurons [3]. Antioxidant enzymes have an essential role in the pathogenesis of neurodegenerative disorders characterized by neuronal inflammation and neuronal death among the aging population, including Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal lobar dementia, and Multiple Sclerosis (MS) [4–6]. PD, however, is a progressive neurodegenerative disorder affecting patients’ motor functions while causing non-motor symptoms ranging from cognitive impairment to gastrointestinal problems [7] [8–10]. The molecular basis underlying neurodegenerative diseases’ pathogenesis is increasingly being exposed, such as aggregation of unfolded or misfolded proteins [11], dysfunction of the ubiquitin-proteasome system [12, 13], and changing metal homeostasis (especially Calcium Signaling) [14].
Calcium (Ca2+), as a second messenger, regulates the neuron cells’ essential activities and participates in depolarization signals and synaptic activities. During neurodegenerative disorders, neurons’ ability to maintain an adequate energy level might compromise Ca2+ homeostasis. Numerous theories have been proposed to describe the role of Ca signaling in PD. Still, it has been shown that the autonomous activities of neurons, which are sustained by their specific Cav1.3 L-type channel subunits, are responsible for the basal metabolic stress [15].
On the other hand, precise trace element balance is vital for a healthy nervous system and neuronal susceptibility to excitability. Numerous articles proposed that Selenium (Se) may play a crucial role in developing PD [16, 17]. Se is an ingredient of several antioxidant enzymes such as thioredoxin reductase (TrxR) and glutathione peroxidase (GPX) [18], and its biological role has been mainly attributed to its presence as the 21st amino acid, selenocysteine (Sec) [19–21]. Se also plays a crucial role in the stress oxidative defense system through its inherent antioxidant activity, though this element’s function is not clear yet.
Even in Se deficiency, the brain has the most significant superiority to intake Se [22]. Besides, the antioxidant activity of particular selenoproteins is of distinct interest in neurodegeneration disorders [23]. This article reviews the current data and essential theories regarding Selenium’s effects on oxidative Stress-induced molecular pathways through calcium signaling in PD.
Oxidative stress
The reactive species production is one of the uncontrolled damaging biomolecules’ principles, such as DNA, lipids, proteins, and carbohydrates or altered metal homeostasis [24]. Numerous of these processes are series reactions beginning by a radical species delivered to full target biomolecules [25–28]. Two main types of free radical species are reactive oxygen species (ROS) and reactive nitrogen species (NO) [29]. ROS includes hydrogen peroxide (H2O2), hydroxyl radical (•OH), and Superoxide (O2•−) [30].
Besides, NO is generated endogenously through the reduction of l-arginine to l‐citrulline via three nitric oxide synthase (NOS) isoforms: endothelial NOS (eNOS), neuronal NOS (nNOS), and inducible NOS (iNOS) [31]. As a neurotransmitter and neuromodulator, NO protects against Microglia penetration to the brain and alerts for threatening neuronal cells [32, 33]. Microglia enhances NO generation via iNOS/NO signaling and increases calcium entrance throughout transient-receptor-potential-vanilloid type-II (TRPV II) channels via PKG/PI3K dependent pathway [34]. Besides, microglia generates H2O2 and cytokines, including interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α). These cytokines induce more ROSand iNOS production and excess NO radicals in microglia [35]. The cytokines also activate nNOS, phospholipase A2, and calpains (a member of the Ca2+-stimulated proteinases family), turning xanthine dehydrogenase into xanthine oxidase and producing O2 and H2O2 [36–38]. Furthermore, the intracellular Ca2+ elevation prevents mitochondrial functions with the production of mitochondrial O2. Thus, excess superoxide radicals react with NO radicals to produce peroxynitrite, contributing to neuronal damage in neurodegenerative disorders [39–41].
Oxidative stress and PD
Experimental evidence has shown that oxidative stress leads to PD, but the process related to neuronal degeneration in substantia nigra pars compacta (SNc) remained unknown [42].
Degeneration of SNc is advanced in PD patients, and only late-stage components of pathogenesis are detectable. The Glutathione (GSH) level in advanced PD is lower than age‐matched control tissues [43], which probably indicates that impairments of the antioxidant system in PD may be related to the vulnerability of the SNc to oxidative mechanisms [44]. However, the cause of losing nigral GSH is not understood clearly yet. Nevertheless, a hypothesis shows that the loss of nigral GSH is not due to an impairment of GSH synthesis and defects of enzyme systems in the GSH oxidation-reduction cycle; but is due to leakage of impaired GSH, which still preserved its function. Furthermore, the formation of the glutamyl and cysteinyl peptides of GSH with dopamine occurs and can be detected in PD patients’ brains. Noticeably, converting these peptides to molecular species prevents mitochondrial activity, which might be toxic to dopaminergic cells. Whatever is the reason for the losing GSH, it makes cells more vulnerable to toxin actions and potentiates the toxic impacts of glial cell activation toward dopaminergic neurons [45, 46].
Except for GSH levels, there is no evidence demonstrating that oxidative stress can initiate cell death in PD. However, oxidative stress is present in measurable quantities when the neuronal loss is marked [47], and it may be linked to other processes related to cell death, including mitochondrial impairment, inflammation, and the toxic effects of NO [48, 49]. Through inhibition of oxidative stress, prevention of apoptotic cell death is possible, whether as a direct or indirect mediated alteration; therefore, dopaminergic cells are supplied by various protective mechanisms [50]. Still, dopaminergic cells may be overcome by an extra oxidative load, and protective mechanisms’ failure may allow endogenous oxidative processes to damaged cells [51–53].
In conclusion, the evidence for the role of oxidative stress in PD is overwhelming, and it can lead to rising oxidative damage in the SNc. Oxidative stress, however, is not separated from other components of dopaminergic cell degeneration. The basic nature of free radicals production and the protective mechanisms interfere with the nigral cell degeneration process [54].
Role of Calcium in PD
Calcium (Ca2+) is one of the second messengers that regulate cells’ essential activities [55]. Developing various pathways to couple the Ca2 + signal to their biochemical functions is vital to neurons [56].
Remarkably, Ca2+ contributes to activity-dependent modulation of synaptic transmission in the brain [57]. During neurodegenerative disorders, neurons’ ability to keep enough energy can affect Ca2+ homeostasis [41]. In PD, multiple neurodegeneration symptoms result from mitochondria malfunctions due to toxins’ particular effects on the respiratory chain and genetic mutations [58, 59]. Notwithstanding these effects, a distinctive feature of PD is the selective loss of dopaminergic neurons in SNc [60]. Recently it has been demonstrated that innate autonomous dopaminergic neuron activity, which their particular L-type voltage-gated calcium channel Cav1.3 subunit is responsible for the oxidative stress under physiological conditions, compensated by mitochondrial buffering. However, oxidative stress overcomes the protective mechanisms when mitochondrial functions become compromised, and the neurodegenerative process will appear [15].
The gradient of specific ions, i.e., Na+, K+, Ca2+, and Cl−, between extracellular and intracellular, leads to alteration of membrane potential toward positive values and causes action potentials which most neurons react to these small changes [61]. The diversity depends on the differential expression of voltage-dependent channels, including voltage-dependent Ca2+, voltage-activated K+, Ca2+-activated K+ currents, etc. [62]. SNc neurons involve Cav1.3 L-type Ca2+ channels [63]. These channels generate a Ca2+ influx that has the advantage of dopamine production [64]. Also, during physiological pace-making enhanced, Ca2+ entry create oxidative stress in mitochondria by enhanced ROS production [65].
On the other hand, high alpha-synuclein (α-syn, a misfolded protein that aggregates in the brain and leads to PD) disrupts cellular/ mitochondrial Ca2+ homeostasis by enhancing membrane permeability to the ions [66]. The loss of α-syn function (attributed to overexpression, silencing, or mutations) induces endoplasmic reticulum (ER)–mitochondria interaction by reduction of their tethering; Thus, α-syn losing impairs mitochondrial Ca2+ homeostasis, which is leading to PD progression (represented in the figure) [67].
Fig. 1.
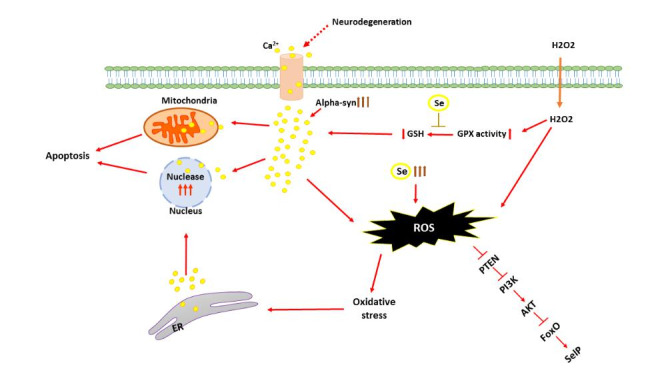
schematic view of Se effects on calcium signaling, stress oxidative and antioxidant system
SNc neurons involve Cav1.3 L-type Ca2+ channels. These channels generate Ca2+ influx that has the advantage of dopamine production. During physiological pace-making enhanced Ca2+ entry create oxidative stress in mitochondria by enhanced ROS production. On the other hand, High levels of α-syn disrupt cellular/ mitochondrial Ca2+ homeostasis. Oxidative stress causes Ca2+ influx into the cytoplasm from the extracellular environment and the ER through the cell membrane and channels. Increasing the Ca2+ level in the cytoplasm induces Ca2+ influx into mitochondria and nuclei. In mitochondria, Ca2+ impairs metabolism to induce cell death. In nuclei, Ca2+ modulates gene transcription and nucleases to control apoptosis.
The effect of oxidative stress on calcium signaling
ROS and NO species can be used as a messenger in normal cell functions [68]. Nevertheless, at oxidative stress levels, they can impair normal pathways. Such an alteration is primarily mediated by Ca2+ Signaling [69].
Oxidative stress causes Ca2+ influx into the cytoplasm from the extracellular environment and the Endoplasmic reticulum (ER) through the cell membrane and channels. Increasing the Ca2+ level in the cytoplasm induces Ca2+ influx into mitochondria and nuclei [70]. In mitochondria, Ca2+ impairs metabolism to cause cell death, but Ca2+ modulates gene transcription and nucleases in nuclei to control apoptosis [71]. Since oxidative stress is associated with PD, understanding how oxidants and antioxidants alter Ca2+ signaling help to understand the process of neurodegeneration and may lead to strategies for prevention; one of the essential antioxidants can be Se.
Selenium intake and metabolism in PD
Selenium (Se) is a micronutrient that enters into the food chain through plants, and its concentration modifies according to the available Se level of the soil [72]. Se can be in two forms of organic and inorganic. The most common inorganic types of Se are Selenite and Selenate, found in animal and plant tissues. On the other hand, the major organic forms are Selenomethionine and Se-methyl selenocysteine, found in selenium-enriched plants such as cerebral grains, grassland legumes, and wild leaks [73–75]. Selenite and selenate are converted into Sec. Besides, selenomethionine (dietary) combines in the body protein instead of methionine, then converts to Sect. [76]. Selenium compounds catabolize to the hydrogen selenide and methylate, finally secrete in the breath as dimethyl-selenide or urine as trimethyl-selenonium [77].
The total level of Se is approximately low in the human brain, but the brain can retain its Se even at prolonged times of insufficient dietary Se consumption [22, 78]. However, Se levels in various adult human brain areas differ. Gray matter areas have higher Se concentrations; the highest level belongs to putamen (1,093 ng/g dry weight) [79], but white matter regions tend to have lower Se levels (283 ng/g in Corpus callosum) [80]. Besides, Se-dependent enzymes, including Glutathione peroxidase (GPX) and thioredoxin system, are involved in all brain and nervous system parts [81]. The etiological role of Se remained unknown yet. However, evidence revealed that Se protects against ROS-induced cell damage [82]. The proposed mechanisms are principally within the function of selenoenzymes and selenoproteins [83]. Here we focus on the effects of Se in PD. Very high or deficient levels of Se might contribute to the pathogenesis of PD because this imbalance increases oxidative stress levels. Se is involved in the antioxidant system; it plays a unique role in PD’s pathogenesis [84]. Nowadays, several population-based types of research that have studied the relationship between Se and PD acclaimed that Se could be used as an independent biomarker for diagnosing PD [85–87]. Therefore, Se’s very high and low body levels may increase the oxidative stress level and contribute to PD’s pathogenesis. In this way, Gellein et al. demonstrated that the Se levels of serum samples collected after patients’ diagnosis with PD (73.0 µg/l) is lower than pre-diagnostic proportions (109.8 µg/l) [88]. Also, a research by Maass et al. on the Se level of CSF shows a higher level in PD patients (9.4 µg/l) in comparison with the control group (5.9 µg/l) [89] while, Zhao et al. acclaimed that lower range of plasma Se may reduce the risk of PD [90]. In this regard, Shahar et al. have shown in a long-term study that the level of plasma Se was not associated with PD risk but undoubtedly correlated to performance-based assessments in neurological task coordination and motor speed [91].
In addition to all this, some selenoproteins, such as SelP and GPX4, have been reported to be involved in PD’s physiopathology, which we discuss them.
Effects of Selenium on Calcium Signaling
GSH is used as a substrate to synthesize the Se-dependent GPX enzyme. [92]. If the free radical generation increases equivalent to GPX enzyme activities, GSH levels decrease [93]. Researchers have shown a reduction in Ca2+ release in GSH depleted neurons, although N-acetylcysteine induces protective effects on Ca2+ release and oxidative stress in GSH depleted neurons. Besides, GSH and GPX play a crucial role in protecting cells from ROS, which is formed from the mitochondrial respiratory chain pathways [94]. Se administration in animals and humans indicates an increased GSH and GPX activity level and detoxifies reactive intermediates throughout its antioxidant role [95–97]. Hence, Se may also have protective effects on the oxidative values, Ca2+ release, and apoptosis in the neurons [98, 99]. Excessive free radicals due to oxidative stress may fundamentally stimulate voltage-gated Ca2+ channels [100]. Also, oxidative stress-induced stimulation of other ion channels leads to the calcium influx into the cytosol and induces depolarization in mitochondria that may eventually cause free radical generation. neurons display Ca2+ ion-selective channel-dependent ROS generation and Ca2+ influx following cytosolic GSH depletion[101]. Besides, evidence reveals that cytosolic GSH depletion causes lipid peroxidation, which attenuates GPX activity. A rise in cytoplasmic Ca2+ may enhance mitochondrial ROS formation during the depolarization of mitochondria. In response to a high level of cytosolic Ca2+ through activation of Ca2+ cation channels, it may provoke Ca2+-induced respiratory impairment, potentiate free radical production, inflict structural damage to mitochondria, and ultimately apoptotic cell death if antioxidants do not inhibit the Ca2+ influx [102]. In this regard, H2O2 triggers apoptotic pathways with antioxidant properties, and Se induces a protective effect on apoptotic pathways (represented in the figure) [103].
Indeed, oxidative stress and impairments in the release of Ca2+ induced by H2O2 could be improved by Se. The investigations support the neuroprotective effects of Se. It may be used in treating neurons dependent on disorders as an antioxidant element.
Selenium supplementations effects on PD.
Due to multiple studies, an Insufficient supply of Se to antioxidant enzymes in the brain may contribute to neurodegeneration pathophysiology; therefore, supplementations may potentially slow neurodegeneration by reducing oxidative stress via increasing GPX levels which is a crucial factor for the reduction of oxidative stress [104]. A piece of research has shown that administration of selenium selenite (0.1, 0.2, and 0.3 mg/kg- ip.) for a week upregulates the antioxidant status and lowers dopamine depletion, elevates GPX activity, relieves lipid peroxidation, and improves motor function of the 6-hydroxydopamine (6-OHDA) induced PD rats models. Thus, Se, as an essential micronutrient, slows neurodegeneration progress in PD [105]. Also, a considerable impairment of MPTP dopaminergic neurotransmission might reverse by Se administration (3 mg/kg) in C57BL mice [106].
In this regard, another study on experimental male Wistar rats of PQ-induced PD shows that Se (11.18 µg/L in the drinking water) maintains locomotor activity and leukocytes’ DNA integrity. Also, there is no change in DNA damage proportion in brain cells throughout the experimental groups [107]. However, this protective effect of Se on dopamine in animal models of PD was strengthened by the results of the research performed by Zafar et al., who used the 6-OHDA to induce the PD model in rats [105]. Additionally, a report of an experiment using embryonic stem cells transplantation in rats’ brains submitted to a model of 6-OHDA-induced PD explained that Se could also protect against inflammation generated in this treatment [108]. Although the outcomes of these investigations are pretty relevant, it is noticeable that these researchers have investigated the effects of the Se in experimental PD models, which differ in some aspects from PD occurring naturally. Thus, further studies are required to conclude the results to humans [109, 110].
Besides, Human studies on investigations of Se supplementation for PD are lacking. However, evidence indicates that either a deficiency or overabundance of Se might contribute to neurodegeneration, or conversely, PD pathology might impair Se mobilization in neurons. The Recommended Dietary Allowance (RDA) for Se is 70 µg /day, and the Institute of Medicine has established a Tolerable Upper Intake Level for Se at 400 mg/day [111]. Hence, meeting the RDA without excess may be cautious.
Selenoproteins in the brain
As mentioned above, Se is well maintained in the brain, even in prolonged dietary Se deficiency [22], and its functions carry out by selenoproteins [23] which are expressed in the brain; still, many questions remained about their roles in neuronal function [112]. GPX expression in glial cells will rise surrounding the damaged area in PD, consistent with its protective role against oxidative damage [113]. Besides, selenoproteins represent antioxidant activities and promote neuronal cell survival in the brain [112]. Researchers revealed the differential expression of brain selenoproteins in a mouse model of PD [114]. While 17 selenoproteins are downregulated, and none of them is upregulated in the SNc, mixed patterns of induced and suppressed expression of selenoprotein mRNAs in the cerebellum, cortex, hippocampus, and pons exist. Thus, most selenoproteins in SNc play critical roles in modulating PD [115, 116]. This idea is supported by physiological evidence generated from mouse models with innovative manipulations of selenoprotein genes.
Glutathione peroxidase (GPX)
All cells, including those of the brain, generate hydrogen peroxide, which induces oxidative stress in cells. Polyunsaturated fatty acids (PUFAs) in the brain are also subject to peroxidation, leading to cell membrane damage [117]. It is thought that the most crucial hydrogen peroxide removing enzyme in the brain is GPX, containing 20% of the total Se in the brain [118]. GPX removes hydrogen peroxide by coupling its reduction to water or alcohols with GSH oxidation [119]. The product, oxidized glutathione (GSSG), consists of two GSH linked by a disulfide bridge and can be transformed back to GSH by glutathione reductases. GPX can act on peroxides of fatty acids, changing them to alcohol [120].
GPX1 is localized in Lewy bodies [119], and its overexpression decreases the loss of tyrosine hydroxylase-positive dopaminergic neurons in SNc in mice PD models [121]. It was observed that Gpx1−/− mice intraperitoneally injected with high levels of paraquat (a quaternary nitrogen herbicide, highly toxic for humans) died within 4–6 h with severe motor symptoms and increased oxidation of proteins, lipids, NADH, and NADPH in the liver and lungs, as compared to wild-type mice that survived three days [122, 123]. Moreover, Gpx1-/- mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine face dopamine depletion and exhibit high 3-nitrotyrosine production in the striatum [124]. Hence, GPX1 can be protective at normal levels and advantageous when overexpressed to maintain dopaminergic neurons, implicating this H2O2-decomposing selenoprotein in protecting against PD in exposure to certain environmental neurotoxins within redox transition.
GPX4 has a substrate preference toward phospholipid hydroperoxides and is a crucial regulator of ferroptosis, a form of necrotic cell death characterized by iron-dependent lipid peroxidation [125]. Recent researchers acclaimed that Se renders the most critical neuroprotective role of GPX4 and its peroxidase activity [126]. Using a knock-in mouse model by replacing the selenocysteine residue of GPX4 with cysteine has shown accelerated neurodegeneration. Therefore, the oxidoreductase activity of GPX4 is essential in the protection against neurodegeneration [126]. While there is not any clear link between GPX4 and PD, such a view is supported by studies employing biochemical and cellular approaches. Evidence implicates GPX4 co-localizes with neuromelanin in SNc, and its level is increased in dystrophic axons and cortex of the PD brain [127]; GPX4 expression is raised against the separation of PARK7 (DJ-1) from its mRNA in response to oxidative stress [128]; oxidized dopamine is a covalent target mitochondrial GPX4 and diminishes its activity in dopaminergic neurons [129] leading to a progression of PD.
On the other hand, ROS couple with calcium signaling throughout the glutathione cycle [130]. Ca2+ activates NADPH oxidase to generate ROS via binding to the enzyme’s EF-hand, regulated by Ca2+ dependent phosphorylation. Besides, the Ca2+ increases NADPH level by stimulating NAD kinase and providing a substrate for NADPH oxidase [131]. In this regard, there is a hypothesis that GPX as an antioxidant might increase to prevent the excessive generation of GSH and ameliorate the oxidative stress induced by Ca2+. For proving this idea, more information and research are needed.
Selenoprotein P
SelP, the major plasma selenoprotein, is produced by the liver as a glycoprotein, contains ten selenocysteine residues secreted either into plasma or interstitial fluid, supplied Se to tissues, and plays a role as a survival factor for neuronal cells [132]. SelP synthesis is regulated by a highly sophisticated system containing transcriptional, translational, and post-translational levels depending on Se availability [133, 134]. There are restricted data on the transcriptional regulation of SelP expression and transcription factors that indicate activated FoxO (forkhead box, class O) affects SelP expression. Thus, SelP expression may regulate by FoxO transcription factors and involve in the cellular response to stressful stimuli, such as oxidative stress [135–138].
The expression of FoxO genes is involved in stress resistance through regulating cell cycle progression, incorporating proteins to DNA repair, or activating antioxidant enzymes [139–141]. Serine/threonine kinase (AKT) - protein kinase B (PKB/AKT) is activated via phosphoinositide 3-kinases (PI3K) through various growth factors and hormones, including insulin; then it phosphorylates FoxO proteins [142, 143]. The protection from FoxO translocation-induced death is dependent on calcium signaling and calcium/calmodulin-dependent protein kinase IV. FoxO shuttling modulation may represent a mechanism that affects cell death processes through nuclear calcium signaling responses [144].
On the other hand, ROS can affect PKB/AKT signaling by preventing FoxO-dependent transcriptional activation, leading to Akt-dependent phosphorylation, so inactivate and translocate FoxO into the cytoplasm [145, 146]. The same effects have been revealed to result from insulin-mimetic signals obtained by the exposure of cells to stressful stimuli, such as Se [147–150]. The high level of Se increases ROS generation [151], and ROS can inhibit SelP production through disruption of PKB/AKT phosphorylation on PV interneurons in the hippocampus, inferior colliculus, medial septum, red nucleus, thalamic reticular nucleus, cerebellum, and choroid plexus in the brain [152].
Conclusions
Cav1.3 L-type Ca2+ channels produce Ca2+ influx, which is essential in releasing dopamine. Ca2+ entrance can lead to oxidative stress in mitochondria through ROS production. Also, Oxidative stress causes Ca2+ influx into the cytoplasm from the extracellular environment and the ER and increases the Ca2+ level in the cytoplasm, which in turn induces Ca2+ influx into mitochondria and nuclei and impairs their functions to cause cell death. Also, High levels of α-syn disrupt cellular/ mitochondrial Ca2+ homeostasis and again lead to cell death. In conclusion, Se induced a protective impact on oxidative stress and suppressed the neurons’ apoptosis throughout Ca2+ release regulation. Furthermore, oxidative stress and changes in intracellular Ca2+ release improved by Se. Thus, our review study supports the neuroprotective role of Se as an antioxidant agent, which might be effective in treating neurodegenerative disorders, especially PD.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative damage in Alzheimer’s disease: the metabolic dimension. Int J Dev Neurosci. 2000;18(4–5):417–21. doi: 10.1016/S0736-5748(00)00006-X. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida S, Inoh S, Asano T, Sano K, Kubota M, Shimazaki H, et al. Effect of transient ischemia on free fatty acids and phospholipids in the gerbil brain: lipid peroxidation as a possible cause of postischemic injury. J Neurosurg. 1980;53(3):323–31. doi: 10.3171/jns.1980.53.3.0323. [DOI] [PubMed] [Google Scholar]
- 3.Yuan X, Fu Z, Ji P, Guo L, Al-Ghamdy AO, Alkandiri A, et al. Selenium nanoparticles pre-treatment reverse behavioral, oxidative damage, neuronal loss and neurochemical alterations in pentylenetetrazole-induced epileptic seizures in mice. Int J Nanomed. 2020;15:6339. doi: 10.2147/IJN.S259134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(7):2–9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- 5.Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1):1003. [PMC free article] [PubMed] [Google Scholar]
- 6.Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–83. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- 7.Van Den Berge N, Ulusoy A. Animal models of brain-first and body-first Parkinson’s disease. Neurobiol Dis. 2022;163:105599. doi: 10.1016/j.nbd.2021.105599. [DOI] [PubMed] [Google Scholar]
- 8.Ekmekci H, Kaptan H. Camptocormia and deep brain stimulation: The interesting overlapping etiologies and the therapeutic role of subthalamic nucleus-deep brain stimulation in Parkinson disease with camptocormia. Surg Neurol Int. 2016;7(Suppl 4):103. doi: 10.4103/2152-7806.176130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015;31(3):140–9. doi: 10.1016/j.tig.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Zheng J, Wang M, Wei W, Keller JN, Adhikari B, King JF, et al. Dietary plant lectins appear to be transported from the gut to gain access to and alter dopaminergic neurons of Caenorhabditis elegans, a potential etiology of Parkinson’s disease. Front Nutr. 2016;3:7. doi: 10.3389/fnut.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoozemans JJ, Scheper W. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 2012;44(8):1295–8. doi: 10.1016/j.biocel.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 12.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40(2):427–46. doi: 10.1016/S0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 13.Forman MS, Lee VM, Trojanowski JQ. ‘Unfolding’pathways in neurodegenerative disease. Trends Neurosci. 2003;26(8):407–10. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- 14.De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–73. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 15.Calì T, Ottolini D, Brini M. Calcium signaling in Parkinson’s disease. Cell Tissue Res. 2014;357(2):439–54. doi: 10.1007/s00441-014-1866-0. [DOI] [PubMed] [Google Scholar]
- 16.Maass F, Michalke B, Willkommen D, Schulte C, Tönges L, Boerger M, et al. Selenium speciation analysis in the cerebrospinal fluid of patients with Parkinson’s disease. J Trace Elem Med Biol. 2020;57:126412. doi: 10.1016/j.jtemb.2019.126412. [DOI] [PubMed] [Google Scholar]
- 17.Pal A. Role of Copper and Selenium in Reproductive Biology: A Brief Update. Biochem Pharmacol (Los Angel) 2015;4(181):2167-0501.1000181. [Google Scholar]
- 18.Chafik A, Essamadi A, Çelik SY, Solak K, Mavi A. Characterization of an interesting selenium-dependent glutathione peroxidase (Se-GPx) protecting cells against environmental stress: The Camelus dromedarius erythrocytes Se-GPx. Biocatal Agric Biotechnol. 2019;18:101000. doi: 10.1016/j.bcab.2019.01.038. [DOI] [Google Scholar]
- 19.Lu J, Holmgren A, Selenoproteins J Biol Chem. 2009;284(2):723–7. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 20.Donovan J, Copeland PR. Threading the needle: getting selenocysteine into proteins. Antioxid Redox Signal. 2010;12(7):881–92. doi: 10.1089/ars.2009.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gladyshev VN, Arnér ES, Berry MJ, Brigelius-Flohé R, Bruford EA, Burk RF, et al. Selenoprotein gene nomenclature. J Biol Chem. 2016;291(46):24036–40. doi: 10.1074/jbc.M116.756155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Berry MJ. Selenium and selenoproteins in the brain and brain diseases. J Neurochem. 2003;86(1):1–12. doi: 10.1046/j.1471-4159.2003.01854.x. [DOI] [PubMed] [Google Scholar]
- 23.Solovyev ND. Importance of Selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J Inorg Biochem. 2015;153:1–12. doi: 10.1016/j.jinorgbio.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Repetto M, Semprine J, Boveris A. Lipid peroxidation: chemical mechanism, biological implications and analytical determination: chapter; 2012.
- 25.Halliwell B. Free radicals and other reactive species in disease. Els. 2001:1–9.
- 26.Weidinger A, Kozlov AV. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules. 2015;5(2):472–84. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohshima H. Genetic and epigenetic damage induced by reactive nitrogen species: implications in carcinogenesis. Toxicol Lett. 2003;140:99–104. doi: 10.1016/S0378-4274(02)00506-4. [DOI] [PubMed] [Google Scholar]
- 28.Qureshi GA, Parvez SH. Oxidative stress and neurodegenerative disorders: Elsevier; 2007.
- 29.Fubini B, Hubbard A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free Radic Biol Med. 2003;34(12):1507–16. doi: 10.1016/S0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 30.Callahan L, She Z, Nosek T. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J Appl Physiol. 2001;90(1):45–54. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- 31.Ferguson SK, Hirai DM, Copp SW, Holdsworth CT, Allen JD, Jones AM, et al. Impact of dietary nitrate supplementation via beetroot juice on exercising muscle vascular control in rats. J Physiol. 2013;591(2):547–57. doi: 10.1113/jphysiol.2012.243121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zabel MK. Microglia and Complement in Alzheimer’s Disease with Cerebral Amyloid Angiopathy. 2013.
- 33.Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229(1–2):26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 34.Maksoud MJE, Tellios V, An D, Xiang YY, Lu WY. Nitric oxide upregulates microglia phagocytosis and increases transient receptor potential vanilloid type 2 channel expression on the plasma membrane. Glia. 2019;67(12):2294–311. doi: 10.1002/glia.23685. [DOI] [PubMed] [Google Scholar]
- 35.Alam Q, Zubair Alam M, Mushtaq G, Damanhouri A, Rasool G, Amjad Kamal M, et al. Inflammatory process in Alzheimer’s and Parkinson’s diseases: central role of cytokines. Curr Pharm Design. 2016;22(5):541–8. doi: 10.2174/1381612822666151125000300. [DOI] [PubMed] [Google Scholar]
- 36.Edelstein CL, Ling H, Schrier RW. The nature of renal cell injury. Kidney Int. 1997;51(5):1341–51. doi: 10.1038/ki.1997.183. [DOI] [PubMed] [Google Scholar]
- 37.Sorg O. Oxidative stress: a theoretical model or a biological reality? CR Biol. 2004;327(7):649–62. doi: 10.1016/j.crvi.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 38.Sorg O, Horn TF, Yu N, Gruol DL, Bloom FE. Inhibition of astrocyte glutamate uptake by reactive oxygen species: role of antioxidant enzymes. Mol Med. 1997;3(7):431–40. doi: 10.1007/BF03401690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beckman JS, Chen J, Crow JP, Zu Ye Y. Reactions of nitric oxide, superoxide and peroxynitrite with superoxide dismutase in neurodegeneration. Progress in brain research. 103: Elsevier; 1994. p. 371 – 80. [DOI] [PubMed]
- 40.Li J, Su J, Li W, Liu W, Altura BT, Altura BM. Peroxynitrite induces apoptosis in canine cerebral vascular muscle cells: possible relation to neurodegenerative diseases and strokes. Neurosci Lett. 2003;350(3):173–7. doi: 10.1016/S0304-3940(03)00881-4. [DOI] [PubMed] [Google Scholar]
- 41.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–50. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 42.Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson’s disease. Front Neuroanat. 2015;9:91. doi: 10.3389/fnana.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson’s disease. Free Radic Biol Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trist BG, Hare DJ, Double KL. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell. 2019;18(6):e13031. doi: 10.1111/acel.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ikeda H, Markey CJ, Markey SP. Search for neurotoxins structurally related to 1-methyl-4-phenylpyridine (MPP+) in the pathogenesis of Parkinson’s disease. Brain Res. 1992;575(2):285–98. doi: 10.1016/0006-8993(92)90092-N. [DOI] [PubMed] [Google Scholar]
- 46.Lestienne P, Nelson J, Riederer P, Jellinger K, Reichmann H. Normal mitochondrial genome in brain from patients with Parkinson’s disease and complex I defect. J Neurochem. 1990;55(5):1810–2. doi: 10.1111/j.1471-4159.1990.tb04973.x. [DOI] [PubMed] [Google Scholar]
- 47.Franco R, Cidlowski JA. Glutathione efflux and cell death. Antioxid Redox Signal. 2012;17(12):1694–713. doi: 10.1089/ars.2012.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58(1):39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Sas K, Szabó E, Vécsei L. Mitochondria, oxidative stress and the kynurenine system, with a focus on ageing and neuroprotection. Molecules. 2018;23(1):191. doi: 10.3390/molecules23010191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–22. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 51.Caballero B, Sherman SJ, Falk T. Insights into the mechanisms involved in protective effects of VEGF-B in dopaminergic neurons. Parkinson’s Disease. 2017;2017. [DOI] [PMC free article] [PubMed]
- 52.Chinta SJ, Poksay KS, Kaundinya G, Hart M, Bredesen DE, Andersen JK, et al. Endoplasmic Reticulum Stress–Induced Cell Death in Dopaminergic Cells: Effect of Resveratrol. J Mol Neurosci. 2009;39(1–2):157–68. doi: 10.1007/s12031-008-9170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromol Med. 2008;10(4):333–42. doi: 10.1007/s12017-008-8047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jenner P. Oxidative mechanisms in nigral cell death in Parkinson’s disease. Mov disorders: official J Mov Disorder Soc. 1998;13:24. [PubMed] [Google Scholar]
- 55.Rizzuto R, Giorgi C, Romagnoli A, Pinton P. Ca2 + signaling, mitochondria and cell death. Curr Mol Med. 2008;8(2):119–30. doi: 10.2174/156652408783769571. [DOI] [PubMed] [Google Scholar]
- 56.Brini M, Calì T, Ottolini D, Carafoli E. Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci. 2014;71(15):2787–814. doi: 10.1007/s00018-013-1550-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rusakov D, Fine A. Extracellular Ca2 + depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron. 2003;37(2):287–97. doi: 10.1016/S0896-6273(03)00025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Büeler H. Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol. 2009;218(2):235–46. doi: 10.1016/j.expneurol.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 59.Schapira A, Cooper J. Mitochondrial function in neurodegeneration and ageing. Mutat Research/DNAging. 1992;275(3–6):133–43. doi: 10.1016/0921-8734(92)90018-K. [DOI] [PubMed] [Google Scholar]
- 60.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson’s disease. Neuroscience. 2011;198:221–31. doi: 10.1016/j.neuroscience.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammond C. Ionic gradients, membrane potential and ionic currents. Cell Mol Neurophysiol. 2015:39–54.
- 62.Manzanares D, Gonzalez C, Ivonnet P, Chen R-S, Valencia-Gattas M, Conner GE, et al. Functional apical large conductance, Ca2+-activated, and voltage-dependent K + channels are required for maintenance of airway surface liquid volume. J Biol Chem. 2011;286(22):19830–9. doi: 10.1074/jbc.M110.185074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca2 + channels in heart and brain. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling. 2014;3(2):15–38. doi: 10.1002/wmts.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jefri M, Bell S, Peng H, Hettige N, Maussion G, Soubannier V, et al. Stimulation of L-type calcium channels increases tyrosine hydroxylase and dopamine in ventral midbrain cells induced from somatic cells. Stem Cells Translational Medicine. 2020;9(6):697–712. doi: 10.1002/sctm.18-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scorziello A, Borzacchiello D, Sisalli MJ, Di Martino R, Morelli M, Feliciello A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front Aging Neurosci. 2020;12:100. doi: 10.3389/fnagi.2020.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Callewaert G, D’hooge P, Ma T-Y, Del Vecchio M, Van Eyck V, Franssens V, et al. Decreased Vacuolar Ca2 + Storage and Disrupted Vesicle Trafficking Underlie Alpha-Synuclein-Induced Ca2 + Dysregulation in S. cerevisiae. Front Genet. 2020;11:266. doi: 10.3389/fgene.2020.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calì T, Ottolini D, Negro A, Brini M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J Biol Chem. 2012;287(22):17914–29. doi: 10.1074/jbc.M111.302794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiology-Cell Physiol. 2004;287(2):C246-C56. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 69.Bartosz G. Reactive oxygen species: destroyers or messengers? Biochem Pharmacol. 2009;77(8):1303–15. doi: 10.1016/j.bcp.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 70.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38(10):713–21. doi: 10.1016/S0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 71.Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Ca2 + and apoptosis. Cell Calcium. 2012;52(1):36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haug A, Graham RD, Christophersen OA, Lyons GH. How to use the world’s scarce selenium resources efficiently to increase the selenium concentration in food. Microb Ecol Health Disease. 2007;19(4):209–28. doi: 10.1080/08910600701698986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eiche E, Bardelli F, Nothstein A, Charlet L, Göttlicher J, Steininger R, et al. Selenium distribution and speciation in plant parts of wheat (Triticum aestivum) and Indian mustard (Brassica juncea) from a seleniferous area of Punjab, India. Sci Total Environ. 2015;505:952–61. doi: 10.1016/j.scitotenv.2014.10.080. [DOI] [PubMed] [Google Scholar]
- 74.Lyons M, Papazyan T, Surai P. Selenium in food chain and animal nutrition: lessons from nature-review. Asian-Australasian J Anim Sci. 2007;20(7):1135–55. doi: 10.5713/ajas.2007.1135. [DOI] [Google Scholar]
- 75.Wang C, Lovell RT. Organic selenium sources, selenomethionine and selenoyeast, have higher bioavailability than an inorganic selenium source, sodium selenite, in diets for channel catfish (Ictalurus punctatus) Aquaculture. 1997;152(1–4):223–34. doi: 10.1016/S0044-8486(96)01523-2. [DOI] [Google Scholar]
- 76.Dumont E, Vanhaecke F, Cornelis R. Selenium speciation from food source to metabolites: a critical review. Anal Bioanal Chem. 2006;385(7):1304–23. doi: 10.1007/s00216-006-0529-8. [DOI] [PubMed] [Google Scholar]
- 77.Foster SJ, Kraus RJ, Ganther HE. Formation of dimethyl selenide and trimethylselenonium from selenobetaine in the rat. Arch Biochem Biophys. 1986;247(1):12–9. doi: 10.1016/0003-9861(86)90527-8. [DOI] [PubMed] [Google Scholar]
- 78.Steinbrenner H, Sies H. Selenium homeostasis and antioxidant selenoproteins in brain: Implications for disorders in the central nervous system. Arch Biochem Biophys. 2013;536(2):152–7. doi: 10.1016/j.abb.2013.02.021. [DOI] [PubMed] [Google Scholar]
- 79.Höck A, Demmel U, Schicha H, Kasperek K, Feinendegen L. Trace element concentration in human brain. Activation analysis of cobalt, iron, rubidium, Selenium, zinc, chromium, silver, cesium, antimony and scandium. Brain. 1975;98(1):49–64. doi: 10.1093/brain/98.1.49. [DOI] [PubMed] [Google Scholar]
- 80.Ejima A, Watanabe C, Koyama H, Matsuno K, Satoh H. Determination of Selenium in the human brain by graphite furnace atomic absorption spectrometry. Biol Trace Elem Res. 1996;54(1):9–21. doi: 10.1007/BF02785316. [DOI] [PubMed] [Google Scholar]
- 81.Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97(6):1634–58. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 82.Lee M-O, Jeon H, Son M-Y, Lee SC, Cho YS. Clump-passaging-based efficient 3D culture of human pluripotent stem cells under chemically defined conditions. Biochem Biophys Res Commun. 2017;493(1):723–30. doi: 10.1016/j.bbrc.2017.08.124. [DOI] [PubMed] [Google Scholar]
- 83.Tang J-Y, He A-H, Jia G, Liu G-M, Chen X-L, Cai J-Y, et al. Protective Effect of Selenoprotein X Against Oxidative Stress-Induced Cell Apoptosis in Human Hepatocyte (LO2) Cells via the p38 Pathway. Biol Trace Elem Res. 2018;181(1):44–53. doi: 10.1007/s12011-017-1025-z. [DOI] [PubMed] [Google Scholar]
- 84.ELLWANGER JH, FRANKE SIR, BORDIN DL, HENRIQUES PRÁD. Biological functions of Selenium and its potential influence on Parkinson’s disease. Anais da Academia Brasileira de Ciências. 2016;88:1655–74. doi: 10.1590/0001-3765201620150595. [DOI] [PubMed] [Google Scholar]
- 85.Maass F, Michalke B, Willkommen D, Leha A, Schulte C, Tönges L, et al. Elemental fingerprint: Reassessment of a cerebrospinal fluid biomarker for Parkinson’s disease. Neurobiol Dis. 2020;134:104677. doi: 10.1016/j.nbd.2019.104677. [DOI] [PubMed] [Google Scholar]
- 86.Sun H. Association of soil selenium, strontium, and magnesium concentrations with Parkinson’s disease mortality rates in the USA. Environ Geochem Health. 2018;40(1):349–57. doi: 10.1007/s10653-017-9915-8. [DOI] [PubMed] [Google Scholar]
- 87.Zhang X, Liu R-P, Cheng W-H, Zhu J-H. Prioritized brain selenium retention and selenoprotein expression: Nutritional insights into Parkinson’s disease. Mech Ageing Dev. 2019;180:89–96. doi: 10.1016/j.mad.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 88.Gellein K, Syversen T, Steinnes E, Nilsen TIL, Dahl OP, Mitrovic S, et al. Trace elements in serum from patients with Parkinson’s disease — a prospective case-control study: The Nord-Trøndelag Health Study (HUNT) Brain Res. 2008;1219:111–5. doi: 10.1016/j.brainres.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Maass F, Michalke B, Leha A, Boerger M, Zerr I, Koch J-C, et al. Elemental fingerprint as a cerebrospinal fluid biomarker for the diagnosis of Parkinson’s disease. J Neurochem. 2018;145(4):342–51. doi: 10.1111/jnc.14316. [DOI] [PubMed] [Google Scholar]
- 90.Zhao H-W, Lin J, Wang X-B, Cheng X, Wang J-Y, Hu B-L, et al. Assessing plasma levels of Selenium, copper, iron and zinc in patients of Parkinson’s disease. PloS one. 2013;8(12). [DOI] [PMC free article] [PubMed]
- 91.Shahar A, Patel KV, Semba RD, Bandinelli S, Shahar DR, Ferrucci L, et al. Plasma selenium is positively related to performance in neurological tasks assessing coordination and motor speed. Mov Disord. 2010;25(12):1909–15. doi: 10.1002/mds.23218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Battin EE, Brumaghim JL. Antioxidant Activity of Sulfur and Selenium: A Review of Reactive Oxygen Species Scavenging, Glutathione Peroxidase, and Metal-Binding Antioxidant Mechanisms. Cell Biochem Biophys. 2009;55(1):1–23. doi: 10.1007/s12013-009-9054-7. [DOI] [PubMed] [Google Scholar]
- 93.Arthur JR. The glutathione peroxidases. Cell Mol Life Sci CMLS. 2001;57(13):1825–35. doi: 10.1007/PL00000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 95.Chow CK, Tappel AL. Response of Glutathione Peroxidase to Dietary Selenium in Rats. J Nutr. 1974;104(4):444–51. doi: 10.1093/jn/104.4.444. [DOI] [PubMed] [Google Scholar]
- 96.Kirana AN, Prafiantini E, Hardiany NS. Correlation Between Age, Body Mass Index, And Blood Selenium Level with Glutathione Peroxidase Activity Among Elderly in South Jakarta. 2020. 2020;4(2):5.
- 97.ZACHARA BA, MIKOLAJCZAK J. Effect of Various Dietary Selenium (Se) Intakes on Tissue Se Levels and Glutathione Peroxidase Activities in Lambs. J Vet Med Ser A. 1993;40(1-10):310–8. doi: 10.1111/j.1439-0442.1993.tb00632.x. [DOI] [PubMed] [Google Scholar]
- 98.Nazıroğlu M, Çiğ B, Özgül C. Neuroprotection induced by N-acetylcysteine against cytosolic glutathione depletion-induced Ca2 + influx in dorsal root ganglion neurons of mice: Role of TRPV1 channels. Neuroscience. 2013;242:151–60. doi: 10.1016/j.neuroscience.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 99.Özgül C, Nazıroğlu M. TRPM2 channel protective properties of N-acetylcysteine on cytosolic glutathione depletion dependent oxidative stress and Ca2 + influx in rat dorsal root ganglion. Physiol Behav. 2012;106(2):122–8. doi: 10.1016/j.physbeh.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 100.Pall ML. Electromagnetic fields act via activation of voltage-gated calcium channels to produce beneficial or adverse effects. J Cell Mol Med. 2013;17(8):958–65. doi: 10.1111/jcmm.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mukherjee SB, Das M, Sudhandiran G, Shaha C. Increase in Cytosolic Ca2 + Levels through the Activation of Non-selective Cation Channels Induced by Oxidative Stress Causes Mitochondrial Depolarization Leading to Apoptosis-like Death in Leishmania donovaniPromastigotes. J Biol Chem. 2002;277(27):24717–27. doi: 10.1074/jbc.M201961200. [DOI] [PubMed] [Google Scholar]
- 102.Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2 + signaling. Am J Physiology-Cell Physiol. 2006;291(5):C1082-C8. doi: 10.1152/ajpcell.00217.2006. [DOI] [PubMed] [Google Scholar]
- 103.Demelash A, Karlsson JO, Nilsson M, Björkman U. Selenium has a protective role in caspase-3-dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. Eur J Endocrinol. 2004;150(6):841–9. doi: 10.1530/eje.0.1500841. [DOI] [PubMed] [Google Scholar]
- 104.Hasani M, Djalalinia S, Khazdooz M, Asayesh H, Zarei M, Gorabi AM, et al. Effect of selenium supplementation on antioxidant markers: a systematic review and meta-analysis of randomized controlled trials. Hormones. 2019;18(4):451–62. doi: 10.1007/s42000-019-00143-3. [DOI] [PubMed] [Google Scholar]
- 105.Zafar KS, Siddiqui A, Sayeed I, Ahmad M, Salim S, Islam F. Dose-dependent protective effect of Selenium in rat model of Parkinson’s disease: neurobehavioral and neurochemical evidences. J Neurochem. 2003;84(3):438–46. doi: 10.1046/j.1471-4159.2003.01531.x. [DOI] [PubMed] [Google Scholar]
- 106.Khan HA. Selenium partially reverses the depletion of striatal dopamine and its metabolites in MPTP-treated C57BL mice. Neurochem Int. 2010;57(5):489–91. doi: 10.1016/j.neuint.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 107.Ellwanger JH, Molz P, Dallemole DR, Pereira dos Santos A, Müller TE, Cappelletti L, et al. Selenium reduces bradykinesia and DNA damage in a rat model of Parkinson’s disease. Nutrition. 2015;31(2):359–65. doi: 10.1016/j.nut.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 108.Tian P, Zhang L, Xu S, Li L, Wang W, Chen YD, et al. Selenite benefits embryonic stem cells therapy in Parkinson’s disease. Curr Mol Med. 2012;12(8):1005–14. doi: 10.2174/156652412802480880. [DOI] [PubMed] [Google Scholar]
- 109.Hassan W, Oliveira S, Noreen C, Kamdem HP, Nogueira JW, Rocha CBT. Organoselenium compounds as potential neuroprotective therapeutic agents. Curr Org Chem. 2016;20(2):218–31. doi: 10.2174/1385272819666150810222632. [DOI] [Google Scholar]
- 110.Miorelli ST, Rosa RM, Moura DJ, Rocha JC, Carneiro Lobo LA, Pêgas Henriques JA, et al. Antioxidant and anti-mutagenic effects of ebselen in yeast and in cultured mammalian V79 cells. Mutagenesis. 2008;23(2):93–9. doi: 10.1093/mutage/gem048. [DOI] [PubMed] [Google Scholar]
- 111.Zeece M. Chapter Five - Vitamins and minerals. In: Zeece M, editor. Introduction to the Chemistry of Food. Academic Press; 2020. pp. 163–212.
- 112.Burk RF, Hill KE. Selenoprotein P—Expression, functions, and roles in mammals. Biochimica et Biophysica Acta (BBA) - Gen Subj. 2009;1790(11):1441–7. doi: 10.1016/j.bbagen.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirsch EC, Hunot S, Damier P, Faucheux B. Glial cells and inflammation in parkinson’s disease: A role in neurodegeneration? Ann Neurol. 1998;44(S1):115-S20. doi: 10.1002/ana.410440717. [DOI] [PubMed] [Google Scholar]
- 114.Zhang X, Ye Y-L, Zhu H, Sun S-N, Zheng J, Fan H-H, et al. Selenotranscriptomic Analyses Identify Signature Selenoproteins in Brain Regions in a Mouse Model of Parkinson’s Disease. PLoS ONE. 2016;11(9):e0163372. doi: 10.1371/journal.pone.0163372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pitts MW, Byrns CN, Ogawa-Wong AN, Kremer P, Berry MJ. Selenoproteins in Nervous System Development and Function. Biol Trace Elem Res. 2014;161(3):231–45. doi: 10.1007/s12011-014-0060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu Z, Jing Y, Yin J, Mu J, Yao T, Gao L. Downregulation of thioredoxin reductase 1 expression in the substantia nigra pars compacta of Parkinson’s disease mice. Neural Regen Res. 2013;8(35):3275–83. doi: 10.3969/j.issn.1673-5374.2013.35.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: focus on age-related macular degeneration. Pharmacol Rep. 2013;65(2):288–304. doi: 10.1016/S1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- 118.Whanger PD. Selenium and the Brain: A Review. Nutr Neurosci. 2001;4(2):81–97. doi: 10.1080/1028415X.2001.11747353. [DOI] [PubMed] [Google Scholar]
- 119.Power JH, Blumbergs PC. Cellular glutathione peroxidase in human brain: cellular distribution, and its potential role in the degradation of Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol. 2009;117(1):63–73. doi: 10.1007/s00401-008-0438-3. [DOI] [PubMed] [Google Scholar]
- 120.Yang MS, Chan HW, Yu LC. Glutathione peroxidase and glutathione reductase activities are partially responsible for determining the susceptibility of cells to oxidative stress. Toxicology. 2006;226(2):126–30. doi: 10.1016/j.tox.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 121.Bensadoun JC, Mirochnitchenko O, Inouye M, Aebischer P, Zurn AD. Attenuation of 6-OHDA-induced neurotoxicity in glutathione peroxidase transgenic mice. Eur J Neurosci. 1998;10(10):3231–6. doi: 10.1046/j.1460-9568.1998.00345.x. [DOI] [PubMed] [Google Scholar]
- 122.Cheng W-H, Fu YX, Porres JM, Ross DA, Lei XG. Selenium-dependent cellular glutathione peroxidase protects mice against a pro-oxidant-induced oxidation of NADPH, NADH, lipids, and protein. FASEB J. 1999;13(11):1467–75. doi: 10.1096/fasebj.13.11.1467. [DOI] [PubMed] [Google Scholar]
- 123.Reczek CR, Birsoy K, Kong H, Martínez-Reyes I, Wang T, Gao P, et al. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat Chem Biol. 2017;13(12):1274–9. doi: 10.1038/nchembio.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, et al. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J neuroscience: official J Soc Neurosci. 2000;20(1):1–7. doi: 10.1523/JNEUROSCI.20-01-00001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014. [DOI] [PubMed] [Google Scholar]
- 126.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell. 2018;172(3):409–22.e21. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 127.Bellinger FP, Bellinger MT, Seale LA, Takemoto AS, Raman AV, Miki T, et al. Glutathione Peroxidase 4 is associated with Neuromelanin in Substantia Nigra and Dystrophic Axons in Putamen of Parkinson’s brain. Mol Neurodegeneration. 2011;6(1):8. doi: 10.1186/1750-1326-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van der Brug MP, Blackinton J, Chandran J, Hao LY, Lal A, Mazan-Mamczarz K, et al. RNA binding activity of the recessive parkinsonism protein DJ-1 supports involvement in multiple cellular pathways. Proc Natl Acad Sci USA. 2008;105(29):10244–9. doi: 10.1073/pnas.0708518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hauser DN, Dukes AA, Mortimer AD, Hastings TG. Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic Biol Med. 2013;65:419–27. doi: 10.1016/j.freeradbiomed.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang X, Tang X, Wang M, Zhang W, Zhou B, Wang Y. ROS and calcium signaling mediated pathways involved in stress responses of the marine microalgae Dunaliella salina to enhanced UV-B radiation. J Photochem Photobiol B. 2017;173:360–7. doi: 10.1016/j.jphotobiol.2017.05.038. [DOI] [PubMed] [Google Scholar]
- 131.Harding SA, Oh SH, Roberts DM. Transgenic tobacco expressing a foreign calmodulin gene shows an enhanced production of active oxygen species. EMBO J. 1997;16(6):1137–44. doi: 10.1093/emboj/16.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burk RF, Hill KE, Motley AK. Selenoprotein metabolism and function: evidence for more than one function for selenoprotein P. J Nutr. 2003;133(5):1517S-20S. doi: 10.1093/jn/133.5.1517S. [DOI] [PubMed] [Google Scholar]
- 133.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, et al. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439–43. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 134.Papp LV, Holmgren A, Khanna KK. Selenium and selenoproteins in health and disease. Antioxid Redox Signal. 2010;12(7):793–5. doi: 10.1089/ars.2009.2973. [DOI] [PubMed] [Google Scholar]
- 135.Klotz L-O, Sánchez-Ramos C, Prieto-Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Manolopoulos K, Klotz L, Korsten P, Bornstein S, Barthel A. Linking Alzheimer’s disease to insulin resistance: the FoxO response to oxidative stress. Mol Psychiatry. 2010;15(11):1046. doi: 10.1038/mp.2010.17. [DOI] [PubMed] [Google Scholar]
- 137.Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PIH., Jr Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J. 2016;473(24):4527–50. doi: 10.1042/BCJ20160503C. [DOI] [PubMed] [Google Scholar]
- 138.Nho RS, Hergert P. FoxO3a and disease progression. World J Biol Chem. 2014;5(3):346. doi: 10.4331/wjbc.v5.i3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lombard DB, Schwer B, Alt FW, Mostoslavsky R. SIRT6 in DNA repair, metabolism and ageing. J Intern Med. 2008;263(2):128–41. doi: 10.1111/j.1365-2796.2007.01902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shalini S, Dorstyn L, Wilson C, Puccini J, Ho L, Kumar S. Impaired antioxidant defence and accumulation of oxidative stress in caspase-2-deficient mice. Cell Death Differ. 2012;19(8):1370. doi: 10.1038/cdd.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tokarz P, Kaarniranta K, Blasiak J. Inhibition of DNA methyltransferase or histone deacetylase protects retinal pigment epithelial cells from DNA damage induced by oxidative stress by the stimulation of antioxidant enzymes. Eur J Pharmacol. 2016;776:167–75. doi: 10.1016/j.ejphar.2016.02.049. [DOI] [PubMed] [Google Scholar]
- 142.Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5(12):921. doi: 10.1038/nrc1753. [DOI] [PubMed] [Google Scholar]
- 143.Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490(1–3):25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 144.Dick O, Bading H. Synaptic Activity and Nuclear Calcium Signaling Protect Hippocampal Neurons from Death Signal-associated Nuclear Translocation of FoxO3a Induced by Extrasynaptic N-Methyl-d-aspartate Receptors*. J Biol Chem. 2010;285(25):19354–61. doi: 10.1074/jbc.M110.127654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schreibelt G, Kooij G, Reijerkerk A, Van Doorn R, Gringhuis SI, Van Der Pol S, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21(13):3666–76. doi: 10.1096/fj.07-8329com. [DOI] [PubMed] [Google Scholar]
- 146.Oh H-M, Yu C-R, Dambuza I, Marrero B, Egwuagu CE. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4 + T cells. J Biol Chem. 2012;287(36):30436–43. doi: 10.1074/jbc.M112.359661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barthel A, Klotz L-O. Phosphoinositide 3-kinase signaling in the cellular response to oxidative stress. Biol Chem. 2005;386(3):207–16. doi: 10.1515/BC.2005.026. [DOI] [PubMed] [Google Scholar]
- 148.Estevez AO, Morgan KL, Szewczyk NJ, Gems D, Estevez M. The neurodegenerative effects of Selenium are inhibited by FOXO and PINK1/PTEN regulation of insulin/insulin-like growth factor signaling in Caenorhabditis elegans. Neurotoxicology. 2014;41:28–43. doi: 10.1016/j.neuro.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tarrado-Castellarnau M, Cortés R, Zanuy M, Tarragó-Celada J, Polat IH, Hill R, et al. Methylseleninic acid promotes antitumour effects via nuclear FOXO3a translocation through Akt inhibition. Pharmacol Res. 2015;102:218–34. doi: 10.1016/j.phrs.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pinto A, Speckmann B, Heisler M, Sies H, Steinbrenner H. Delaying of insulin signal transduction in skeletal muscle cells by selenium compounds. J Inorg Biochem. 2011;105(6):812–20. doi: 10.1016/j.jinorgbio.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 151.Wang X, Zhang W, Chen H, Liao N, Wang Z, Zhang X, et al. High Selenium impairs hepatic insulin sensitivity through opposite regulation of ROS. Toxicol Lett. 2014;224(1):16–23. doi: 10.1016/j.toxlet.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 152.Pitts MW, Raman AV, Hashimoto AC, Todorovic C, Nichols RA, Berry MJ. Deletion of selenoprotein P results in impaired function of parvalbumin interneurons and alterations in fear learning and sensorimotor gating. Neuroscience. 2012;208:58–68. doi: 10.1016/j.neuroscience.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.