

Abstract
Mobile elements and repetitive genomic regions are sources of lineage-specific genomic innovation and uniquely fingerprint individual genomes. Comprehensive analyses of such repeat elements, including those found in more complex regions of the genome, require a complete, linear genome assembly. We present a de novo repeat discovery and annotation of the T2T-CHM13 human reference genome. We identified previously unknown satellite arrays, expanded the catalog of variants and families for repeats and mobile elements, characterized classes of complex composite repeats, and located retroelement transduction events. We detected nascent transcription and delineated CpG methylation profiles to define the structure of transcriptionally active retroelements in humans, including those in centromeres. These data expand our insight into the diversity, distribution, and evolution of repetitive regions that have shaped the human genome.
Graphical Abstract
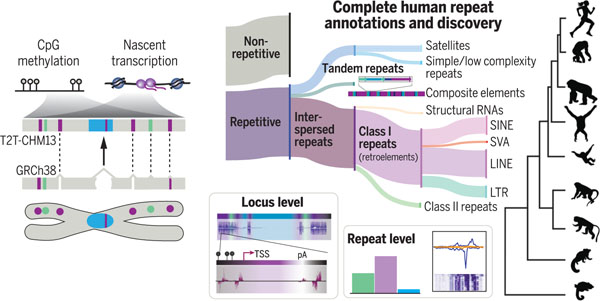
Telomere-to-telomere assembly of CHM13 supports repeat annotations and discoveries. The human reference T2T-CHM13 filled gaps and corrected collapsed regions (triangles) in GRCh38. Combining long read–based methylation calls, PRO-seq, and multilevel computational methods, we provide a compendium of human repeats, define retroelement expression and methylation profiles, and delineate locus-specific sites of nascent transcription genome-wide, including previously inaccessible centromeres. SINE, short interspersed element; SVA, SINE–variable number tandem repeat–Alu; LINE, long interspersed element; LTR, long terminal repeat; TSS, transcription start site; pA, polyadenylation signal.
RESEARCH ARTICLE SUMMARY
INTRODUCTION:
Transposable elements (TEs), repeat expansions, and repeat-mediated structural rearrangements play key roles in chromosome structure and species evolution, contribute to human genetic variation, and substantially influence human health through copy number variants, structural variants, insertions, deletions, and alterations to gene transcription and splicing. Despite their formative role in genome stability, repetitive regions have been relegated to gaps and collapsed regions in human genome reference GRCh38 owing to the technological limitations during its development. The lack of linear sequence in these regions, particularly in centromeres, resulted in the inability to fully explore the repeat content of the human genome in the context of both local and regional chromosomal environments.
RATIONALE:
Long-read sequencing supported the complete, telomere-to-telomere (T2T) assembly of the pseudo-haploid human cell line CHM13. This resource affords a genome-scale assessment of all human repetitive sequences, including TEs and previously unknown repeats and satellites, both within and outside of gaps and collapsed regions. Additionally, a complete genome enables the opportunity to explore the epigenetic and transcriptional profiles of these elements that are fundamental to our understanding of chromosome structure, function, and evolution. Comparative analyses reveal modes of repeat divergence, evolution, and expansion or contraction with locus-level resolution.
RESULTS:
We implemented a comprehensive repeat annotation workflow using previously known human repeats and de novo repeat modeling followed by manual curation, including assessing overlaps with gene annotations, segmental duplications, tandem repeats, and annotated repeats. Using this method, we developed an updated catalog of human repetitive sequences and refined previous repeat annotations. We discovered 43 previously unknown repeats and repeat variants and characterized 19 complex, composite repetitive structures, which often carry genes, across T2T-CHM13. Using precision nuclear run-on sequencing (PRO-seq) and CpG methylated sites generated from Oxford Nanopore Technologies long-read sequencing data, we assessed RNA polymerase engagement across retroelements genome-wide, revealing correlations between nascent transcription, sequence divergence, CpG density, and methylation. These analyses were extended to evaluate RNA polymerase occupancy for all repeats, including high-density satellite repeats that reside in previously inaccessible centromeric regions of all human chromosomes. Moreover, using both mapping-dependent and mapping-independent approaches across early developmental stages and a complete cell cycle time series, we found that engaged RNA polymerase across satellites is low; in contrast, TE transcription is abundant and serves as a boundary for changes in CpG methylation and centromere substructure. Together, these data reveal the dynamic relationship between transcriptionally active retroelement subclasses and DNA methylation, as well as potential mechanisms for the derivation and evolution of new repeat families and composite elements. Focusing on the emerging T2T-level assembly of the HG002 X chromosome, we reveal that a high level of repeat variation likely exists across the human population, including composite element copy numbers that affect gene copy number. Additionally, we highlight the impact of repeats on the structural diversity of the genome, revealing repeat expansions with extreme copy number differences between humans and primates while also providing high-confidence annotations of retroelement transduction events.
CONCLUSION:
The comprehensive repeat annotations and updated repeat models described herein serve as a resource for expanding the compendium of human genome sequences and reveal the impact of specific repeats on the human genome. In developing this resource, we provide a methodological framework for assessing repeat variation within and between human genomes. The exhaustive assessment of the transcriptional landscape of repeats, at both the genome scale and locally, such as within centromeres, sets the stage for functional studies to disentangle the role transcription plays in the mechanisms essential for genome stability and chromosome segregation. Finally, our work demonstrates the need to increase efforts toward achieving T2T-level assemblies for nonhuman primates and other species to fully understand the complexity and impact of repeat-derived genomic innovations that define primate lineages, including humans.
Studies of mobile elements and repeat arrays have long shown that eukaryotic genomes are in constant flux (1). Transposable element (TE) insertions and repeat-mediated structural rearrangements can influence gene regulation, create new coding structure, and affect chromosome stability. Transposition, expansion, and contraction of repeats generate species-specific genomic innovations (1, 2), major evolutionary transitions (3), and human- and primate-specific adaptations (4). Together, TEs and other forms of repetitive DNA, constituting more than half of the human genome, are the largest contributor to human genetic variation and affect human health (5) owing to their roles in deleterious copy number variants (CNVs), structural variants (SVs), insertions, deletions, and alterations to gene transcription and splicing.
A major challenge in tracking and understanding repeat structure, function, and variation is that large complex repeats, sequences in tandem arrays, and recent insertions by TEs have been largely impenetrable to available sequencing and assembly technologies. Despite this challenge, a species-agnostic repeat database (the Dfam database) (6), manual curation (7), and the development of improved algorithms for repeat discovery (8, 9) have laid the groundwork underlying efforts to create and finish a complete map and catalog of the repertoire of human repeats.
Previous assemblies of a reference human genome contained gaps and collapsed repeats (10). Capitalizing on recent advances in ultralong sequencing and assembly methods, the Telomere-to-Telomere (T2T) Consortium generated a complete human reference genome on the basis of the pseudo-haploid genome of an androgenetic hydatidiform mole (CHM13hTERT cell line, hereafter CHM13) (11). This assembly, T2T-CHM13v1.1, resulted in the addition of more than 200 mega–base pairs (Mbp) of DNA and resolution of collapsed and unassembled regions in previous reference genomes. The gap-filled and decompressed regions, representing 8% of the human genome, are dominated by tandemly arrayed repeats [such as in the alpha satellite arrays that are found in higher-order repeat arrays (HORs) within centromeres (12)] and complex repeats in pericentromeres, subtelomeres, and some chromosome arms (i.e., acrocentrics). T2T-CHM13 supported additional annotations for human repetitive sequences residing in previously unassembled regions of the human reference GRCh38 and added repeat annotations for low copy repeats genome-wide. In total, we identify 53.9% of the T2T-CHM13 assembly as repetitive. Here we highlight key advances from this resource, while illustrating the power of combining multiple approaches and tools to enhance genomic discoveries.
Eukaryotic repeats are classified into two main types on the basis of their genomic organization: tandem repeats and interspersed repeats (Fig. 1A) (6, 13). Tandem repeats are further subdivided into satellites and simple repeats; satellites are often further defined by their regional chromosomal distribution (centromeric, for example). With the exception of pseudogenes retroposed from structural RNAs (tRNAs, ribosomal RNAs, etc.), interspersed repeats largely refer to TEs, which are classified on the basis of their mechanism of propagation (6, 14–16). Class I elements are spread within genomes via retrotransposition and are further subdivided into two broad subclasses. One subclass consists of long interspersed elements (LINEs) and long terminal repeat (LTR) elements, which typically encode their own catalyzing enzymes. The other consists of short interspersed elements (SINEs) and the composite retroelement SINE-VNTRAlus (SVAs), both of which are nonautonomous, relying on LINE-encoded proteins for retrotransposition. Class II elements are those that are mobilized through transposase, helicase, or recombinase and include TEs such as Tc1-Mariner and hAT. These varied repeat types constitute a major portion, and in some cases the majority [for example, 85% of wheat genomes (17)], of eukaryotic genome sequences.
Fig. 1. T2T-CHM13 assembly supports identification of previously unknown repeat families and complex epigenetic signatures.
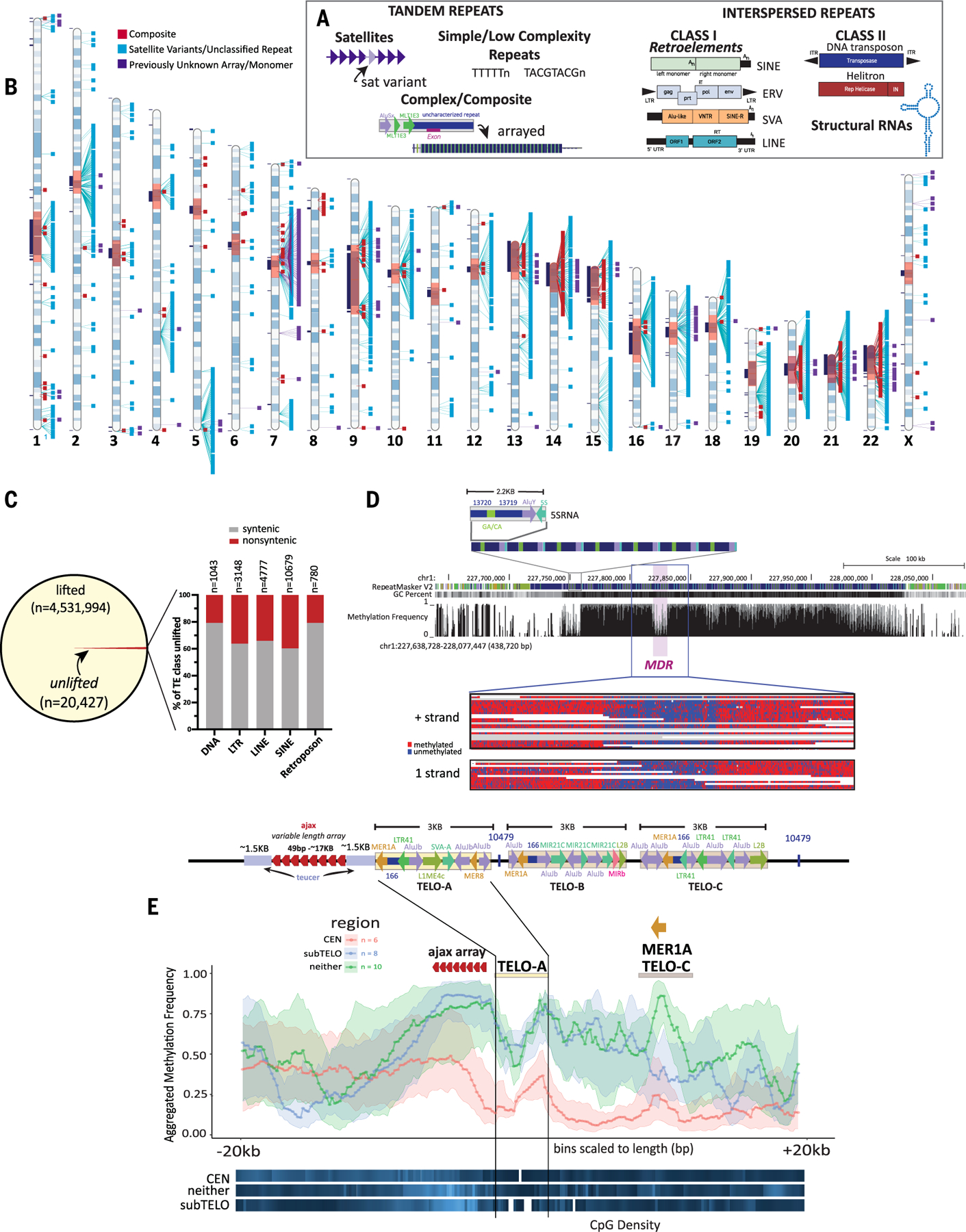
(A) Schematic illustrating examples of tandem repeats, including satellites, simple and low complexity repeats and composites, and interspersed repeats, including class I and class II TEs, and structural RNAs. (B) Ideogram of CHM13 indicating the locations of annotated composite elements (red), satellite variants and unclassified repeats (aqua), and arrays or monomers of sequences found within those arrays (purple). Gaps in GRCh38 with no synteny to T2T-CHM13 (11) are shown in black boxes to the left of each chromosome, centromere blocks [including centromere transition regions (12)] are indicated in orange. (C) (Left) The number of TEs lifted and unlifted from T2T-CHM13 to GRCh38. (Right) Bar plot showing percentage of TEs by class (DNA, LTR, LINE, SINE, and retroposon) that were unlifted from T2T-CHM13 gap-filled regions (nonsyntenic, red) and syntenic regions (gray); the n values show the number of elements within each class affected. (D) (Top) T2T-CHM13 genome browser showing the 5SRNA_Comp subunit structure and array. RepeatMaskerV2 track, CG percentage, and methylation frequency tracks are shown. The MDR is indicated. (Bottom) A zoomed image of individual nanopore reads showing consistent hypomethylation in the MDR (chr1:227,818,289–227,830,789) and hypermethylation in the flanking regions (chr1:227,804,021–227,845,689). Both positive (top) and negative (bottom) strand aligning reads show the same methylation pattern. (E) (Top) Each T2T-CHM13 TELO-composite element consists of a duplication of a teucer repeat (blue) separated by a variable 49-bp (ajax) repeat array (red arrowheads) and three different composite subunits (TELO-A, -B, and -C). Repeat and TE annotations are shown. Some copies of TELO-composite contain the previously unknown repeat “10479” between the TELO-A and TELO-C subunits and/or after the TELO-C subunit. (Bottom) Metaplot of aggregated methylation frequency (average methylation of each bin across the region, 100 bins total) centered on the TELO-A subunit, ±20 kbp, grouped by chromosomal location (orange, centromeric; blue, subtelomeric; green, interstitial). CpG density for each group is indicated at the bottom (white, no CpG; dark blue, low CpG; bright blue, high CpG). The location of the ajax repeat array and the MER1A element within the TELO-C subunit are indicated.
The varied modes of propagation of such repeats, from simple insertion events to promoting nonallelic recombination, facilitate genomic diversity, often in bursts of activity followed by periods of neutral evolution. Furthermore, organismal defense mechanisms that have evolved to counter the deleterious effects of mobilization, such as DNA methylation, can influence the sequence evolution of targeted elements. Repeats represent the nexus of evolutionary forces, the selfishness of mobile elements, and the cellular mechanisms marshaled to silence them. The genomic turbulence engendered by repeats makes them the most challenging genomic regions to study. However, insights from studies of these regions have revealed regulatory and coding domains critical to organismal life histories and human health. A full accounting of repeat domains permitted by a gapless telomere-to-telomere DNA sequence is therefore essential to a full understanding of the origins and function of the human genome.
Results
Comprehensive repeat annotations for a complete human genome
We developed a computational pipeline to discover previously unknown repeat annotations and tandem arrays while reducing false positives from pseudogenes, segmental duplications, and Dfam overlaps (18) (fig. S1). At each step, computational analysis was supplemented by manual curation and polishing. In total, 49 previously unidentified repeat types from Repeat Modeler were curated, including 27 repeats (Fig. 1B and fig. S2) as well as 22 potentially older TE repeats whose alignment scores precluded classification and were thus set aside (table S1). Among the 27 identified repeats were one previously unknown centromeric satellite [86.6% of base pairs found within centromere regions defined in (11, 12)] and 10 repeats classified into five variants of known satellites [three centromere transition satellites (GSATII, HSAT5v1, and HSAT5v2) and two interstitial satellites (SATR1 and SST1)] and five previously unknown repetitive sequences. Manual curation identified an additional 13 interstitial satellite arrays (and monomers of the satellites); three repetitive sequences, all of which were previously unknown and unclassified; and 19 composite elements (including 16 curated composite subunits), defined as a repeating unit consisting of three or more repeated sequences, including TEs, simple repeats, composite subunits, and satellites (fig. S2). In total, 62 repeat entries were classified and submitted to Dfam as previously unannotated human repeats, with 19 elements added as a “composite” track for the T2T-CHM13v1.1 genome browser (Table 1 and table S2).
Table 1. Complete genome assembly supported discovery and refinement of human repeat annotations.
Repeats identified through RepeatModeler and manual curation (RMv2) shown in counts and base pairs, by category, for T2T-CHM13v1.1 and GRCh38 (excluding the Y chromosome) (Fig. 1B and table S2).
| Repeat category | CHM13v1.1 |
GRCh38 (excluding Y) |
||
|---|---|---|---|---|
| RMv2 |
RMv2 |
|||
| Count | Bp | Count | Bp | |
| Composite subunits | 4,446 | 2,805,296 | 1,162 | 536,979 |
| Unclassified repeats | 1,234 | 1,025,084 | 783 | 570,985 |
| Satellite variants | 730 | 568,841 | 671 | 591,294 |
| Monomers of arrayed satellites | 11,900 | 1,127,758 | 2,651 | 415,508 |
| Total | 18,310 | 5,526,979 | 5,267 | 2,114,766 |
This updated repeat library yielded annotations of human repeats within regions previously unresolved in GRCh38 and provided copy number support to identify additional, previously unnoticed repeat elements genome-wide (Fig. 1B and fig. S2). Using this T2T-CHM13–based repeat library, the T2T-CHM13v1.1 assembly was fully annotated for all repeat classes, resulting in 1.65 giga–base pairs (Gbp) of repeat annotations (53.94% of the genome), of which 168.3 Mbp are found within the 182.1 Mbp of gap-filled T2T-CHM13 genomic sequence (92.4%), representing added annotations, and 5.5 Mbp of which are previously unknown human repeats that we identified genome-wide (Table 1; tables S2 and S3; and fig. S3). Reannotation of GRCh38 (without the Y chromosome) using the T2T-CHM13 repeat database resulted in annotation of 2,114,766 bp of previously uncataloged repeats (Table 2 and table S3), demonstrating the utility of a T2T-level assembly in supporting more comprehensive repeat annotations. Additionally, reannotation of the GRCh38 Y chromosome revealed previously unidentified annotations consisting of six composite elements, eight satellite arrays, 156 satellite variants, and six unclassified repeats, totaling 161,055 bp in repeat annotations discovered through this study (fig. S4 and table S4).
Table 2. Repeat annotations are more refined for CHM13v1.1.
Kilo–base pairs of repeat annotations, by repeat class and family, for different human genome assemblies with T2T-CHM13v1.1 RMv2, GRCh38 RMv2, and GRCh38 Dfam3.3 only. Note that AluJb is included in the Alu repeat family category.
| Repeat class | Repeat family | CHM13v1.1 |
GRCh38 (excluding Y) |
||||
|---|---|---|---|---|---|---|---|
| RMv2 |
RMv2 |
Dfam3.3 |
|||||
| Kbp | % of assembly | Kbp | % of assembly | Kbp | % of assembly | ||
| SINE | 5S-Deu-L2 | 221.0 | 0.0072 | 218.2 | 0.0075 | 210.9 | 0.0072 |
| Alu | 308,309.7 | 10.0926 | 304,734.4 | 10.4283 | 305,457.4 | 10.4531 | |
| MIR | 80,937.9 | 2.6495 | 80,726.2 | 2.7625 | 79,989.0 | 2.7373 | |
| tRNA-Deu | 48.4 | 0.0016 | 48.1 | 0.0016 | 46.1 | 0.0016 | |
| tRNA-RTE | 549.0 | 0.0180 | 546.9 | 0.0187 | 525.9 | 0.0180 | |
| tRNA | 1.6 | 0.0066 | 1.6 | 0.0069 | 1.5 | 0.0067 | |
| Retroposon | SVA | 4,654.7 | 0.1524 | 4,507.8 | 0.1543 | 4,520.7 | 0.1547 |
| LINE | CR1 | 10,817.9 | 0.3541 | 10,805.0 | 0.3698 | 10,571.1 | 0.3618 |
| Dong-R4 | 120.1 | 0.0039 | 121.0 | 0.0041 | 115.2 | 0.0039 | |
| I-Jockey | 15.7 | 0.0005 | 15.6 | 0.0005 | 14.8 | 0.0005 | |
| L1 | 512,421.5 | 16.7742 | 507385.6 | 17.3633 | 507,866.7 | 17.3797 | |
| L1-Tx1 | 49.6 | 0.0016 | 50.3 | 0.0017 | 49.1 | 0.0017 | |
| L2 | 104,083.4 | 3.4072 | 103,819.6 | 3.5528 | 102,055.0 | 3.4924 | |
| RTE-BovB | 872.7 | 0.0286 | 875.3 | 0.0300 | 843.2 | 0.0289 | |
| RTE-X | 3,195.6 | 0.1046 | 3,190.0 | 0.1092 | 3,097.8 | 0.1060 | |
| Penelope | 68.0 | 0.0022 | 68.4 | 0.0023 | 63.1 | 0.0022 | |
| LTR | ERV1 | 83,480.5 | 2.7327 | 82,370.2 | 2.8188 | 82,641.0 | 2.8281 |
| ERVK | 8,611.5 | 0.2819 | 8,370.9 | 0.2865 | 8,468.0 | 0.2898 | |
| ERVL | 59,049.8 | 1.9330 | 58,682.4 | 2.0082 | 58,646.0 | 2.0069 | |
| ERVL-MaLR | 110,751.8 | 3.6255 | 110,098.6 | 3.7677 | 109,957.5 | 3.7629 | |
| Gypsy | 4,843.6 | 0.1586 | 4,826.3 | 0.1652 | 4,629.3 | 0.1584 | |
| Undefined | 3,172.7 | 0.1039 | 3,176.1 | 0.1087 | 3,081.7 | 0.1055 | |
| DNA | Crypton | 44.4 | 0.0015 | 45.1 | 0.0015 | 44.5 | 0.0015 |
| Crypton-A | 21.7 | 0.0007 | 22.0 | 0.0008 | 20.6 | 0.0007 | |
| Kolobok | 65.7 | 0.0021 | 65.7 | 0.0022 | 63.9 | 0.0022 | |
| MULE-MuDR | 1,008.0 | 0.0330 | 985.5 | 0.0337 | 986.8 | 0.0338 | |
| Merlin | 40.3 | 0.0013 | 40.6 | 0.0014 | 40.0 | 0.0014 | |
| PIF-Harbinger | 68.5 | 0.0022 | 70.0 | 0.0024 | 67.6 | 0.0023 | |
| PiggyBac | 540.6 | 0.0177 | 541.4 | 0.0185 | 539.5 | 0.0185 | |
| TcMar | 44.9 | 0.0015 | 45.5 | 0.0016 | 45.2 | 0.0015 | |
| TcMar-Mariner | 2,961.4 | 0.0969 | 2,888.2 | 0.0988 | 2,872.1 | 0.0983 | |
| TcMar-Pogo | 4.5 | 0.0001 | 4.2 | 0.0001 | 4.0 | 0.0001 | |
| TcMar-Tc1 | 135.8 | 0.0044 | 135.8 | 0.0046 | 134.1 | 0.0046 | |
| TcMar-Tc2 | 1,678.1 | 0.0549 | 1,666.6 | 0.0570 | 1,665.6 | 0.0570 | |
| TcMar-Tigger | 37,999.9 | 1.2439 | 37,725.0 | 1.2910 | 37,557.8 | 1.2853 | |
| hAT | 561.8 | 0.0184 | 561.6 | 0.0192 | 543.1 | 0.0186 | |
| hAT-Ac | 653.4 | 0.0214 | 634.7 | 0.0217 | 610.5 | 0.0209 | |
| hAT-Blackjack | 2,608.8 | 0.0854 | 2,603.9 | 0.0891 | 2,589.8 | 0.0886 | |
| hAT-Charlie | 46,980.9 | 1.5379 | 46,779.6 | 1.6008 | 46,468.2 | 1.5902 | |
| hAT-Tag1 | 476.1 | 0.0156 | 476.5 | 0.0163 | 463.2 | 0.0159 | |
| hAT-Tip100 | 12,250.5 | 0.4010 | 12,034.5 | 0.4118 | 11,915.0 | 0.4077 | |
| hAT-hAT19 | 1.8 | 0.0001 | 1.7 | 0.0001 | 1.6 | 0.0001 | |
| Undefined | 1,201.8 | 0.0393 | 1,202.9 | 0.0412 | 1163.1 | 0.0398 | |
| Kilo–base pairs of TEs | 1,405,826.3 | 1,393,370.3 | 1,390,841.6 | ||||
| Kilo–base pairs of non-TEs | 241,986.3 | 122,998.7 | 118,576.2 | ||||
| Assembled kilo–base pairs | 3,054,815.5 | 2,922,175.7 | 2,922,175.7 | ||||
| TEs masked | 46.02 | 47.68 | 47.60 | ||||
| Non-TEs masked | 7.92 | 4.21 | 4.06 | ||||
| % repeatmasked | 53.941 | 51.892 | 51.654. | ||||
The reannotated GRCh38 and annotated T2T-CHM13 were compared with reverse liftOver coordinates (CHM13 to GRCh38) to identify TE insertions specific to CHM13 (18). TEs found in CHM13 but not in GRCh38were further grouped into those that are in gap-filled regions (nonsyntenic overlap) or those that are potentially polymorphic between these two genomes or were collapsed in the GRCh38 assembly (syntenic overlap but missing in GRCh38) (Fig. 1C).
Across 4,531,994 TEs with lifted coordinates (i.e., shared between T2T-CHM13 and GRCh38), 118,787 lifted TE pair annotations were discordant between the two genomes (fig. S5A); 82.3% of these (97,719 discordant liftOver pairs) were typically short loci with low scores and therefore of questionable discordance (19), and/or subtle subfamily reclassifications (fig. S5B and table S5). Among the 20,427 unlifted TEs specific to T2T-CHM13, all TE classes are represented (Fig. 1C), with 35.2% of TE sites specific to gap-filled regions in T2T-CHM13 (7194 total TEs) (tables S5 and S6). Unlifted TE sites are found genome-wide, with a higher density on the acrocentric chromosomes 13, 14, 15, 21, and 22 (fig. S5C and table S7).
Composite elements shape the human genome and local methylation
Composite structural elements contribute to human diversity and disease through structural variation and copy number variation, particularly when exonic regions are “captured” in a core unit (20). We annotated 19 composite repeat elements (table S2 and figs. S6 to S11) in T2T-CHM13, each composed of three or more repeated sequences, including TEs, simple repeats, composite subunits, and satellites (18). Most composites are found in a tandem array only on a single chromosome (figs. S6, A to F, and S7, B to G), and in eight cases, each core unit contains protein-coding annotations (fig. S7), indicating that unequal crossing-over events and concerted evolution among composite units contribute to the expansion or contraction of gene families within humans (table S2).
One composite, 5SRNA_Comp, consists of a portion of the 5S RNA, an AluY, and two subunit repeats as an array of 128 repeating units with high sequence similarity (most share 98 to 100% identity) on chromosome 1 (fig. S9, A and B). Using methylation profiles developed for T2T-CHM13 and long read–based methylation clusters (21), we find that the methylation pattern of the 5SRNA_Comp is not consistent across the array; rather we find a drop in methylation, which we called a methylation dip region (MDR), internal to the array, similar to the centromere dip region (CDR) identified in higher-order arrays of alpha satellites in T2T-CHM13 (21) (Fig. 1D). The location of the MDR is not linked to DNA sequence, as neither the GC content nor sequence identity is variable across repeat units in this array (Fig. 1D and fig. S9B), suggesting that other epigenetic factors may facilitate the drop in methylation.
We annotated a highly complex composite, TELO_Comp, that consists of multiple satellite arrays and other composites (Fig. 1E), with instances found on 10 chromosomes (figs. S12 and S13) at interstitial, pericentromeric, and subtelomeric loci. The canonical TELO_Comp consists of three 3-kbp (kilo–base pair) composites (TELO-A, -B, and -C subunits), each containing multiple TEs, downstream of a variable-length array of a 49-bp satellite repeat unit, ajax, bounded by a duplicated sequence, teucer (Fig. 1E). In-depth analysis of the overall structure of the subunits across all loci and phylogenetic analyses of the TELO-A subunit (18) (fig. S12 and table S8) indicate that subtelomeric units are a monophyletic group of recent origin, likely by segmental duplication events (fig. S13A and tables S8 and S9), whereas interstitial and pericentromeric units are polyphyletic. Moreover, each subtelomeric unit contains the ajax array proximal to the telomere, indicating that inverted orientations are favored at subtelomeric loci. Location-specific repeat diversification in subunit content and structure as well as ajax and teucer repeat copy numbers, which each retain high sequence identity (figs. S13, B and C, S14, and S15, and tables S10 and S11), reveal differential evolutionary forces acting on TELO_Comp loci on the basis of chromosome location.
Meta-analysis of aggregated methylation frequency across the TELO_Comp units (±20 kbp) (Fig. 1E) shows that the ajax satellite array is hypermethylated across all elements, with a discernible drop in methylation across TELO-A subunits and peak of methylation in theMER1A unit in elements containing TELO-C. Subtelomeric and interstitial TELO_Comp elements share similar methylation profiles, with higher methylation levels across the entire element, whereas pericentromeric TELO_Comp units have lower overall methylation levels. This indicates that local epigenetic states affect overall methylation levels but do not change relative levels within the ajax array and TELO subunits. Comparison of aggregated methylation frequency across TELO_Comp units at the same loci in the human diploid assembly for HG002 (fig. S16) (21) show that overall methylation levels are higher across TELO_Comp elements, including those found in centromeres, as expected from global differences in methylation level between T2T-CHM13 and HG002. However, the overall methylation pattern for the TELO_Comp elements (Fig. 1E and fig. S16) is retained, indicating it is an epigenetic signature of this repeat in humans.
Transcriptional, epigenetic, and structural differences define TEs across the human genome
Precision nuclear run-on sequencing (PRO-seq) (22) detects nascent transcription from RNA polymerases with nucleotide resolution at genome scale. The resulting read density profiles quantitatively reflect the occupancy of active polymerases across the genome. Sites of accumulating RNA polymerase activity (22, 23), such as promoter-proximal pause sites, 3′ cleavage and polyA regions, splice junctions, and enhancers, indicate points of transcription regulation (22, 24). In addition, because PRO-seq captures RNA synthesis before mechanisms that affect RNA stability take place, unprocessed and unstable RNAs can be detected with high sensitivity. Capitalizing on the single-base resolution of PRO-seq and CpG methylation profiles (21), we define profiles of RNA polymerase activity that distinguish different families of retroelements (Fig. 1A). We assessed PRO-seq signal, CpG methylation density, CpG site density, and sequence divergence from the consensus for each element within each subfamily, further classified as full-length or truncated and grouped by relative age (fig. S17 and tables S12 and S13) (18). For each element type, density profiles were correlated with known features of specific repeats.
Across all full-length retroelements in T2T-CHM13, PRO-seq density profiles show signals of RNA polymerase accumulation (Fig. 2, A to E, and fig. S18). AluY elements show two signal peaks; the first corresponds to the known RNA pol III promoter site within the first monomer, while the second, broader peak within the second monomer indicates the site of a second, ancient 7SL RNA promoter (25), whose presence might promote polymerase pausing (Fig. 2A). The peak distribution closely mimics the relative size of the left and right Alu monomers and thus reflects the dimerization of Alu. Although active transcription continues in truncated AluY elements, there is no longer a visible signal of promoter exclusivity, and RNA polymerase signal spreads across the element. Full-length AluY elements retain a similar methylation profile and show low divergence levels corresponding with low, single-copy k-mer density. Truncated and older elements (AluJ and AluS) (table S13 and figs. S18 to S22) show broad methylation profiles with low CpG content and higher divergence (Fig. 2A). Transcriptionally active retroelement families wherein the majority of full-length elements show high PRO-seq signal (AluY, SVA, and L1Hs; Fig. 2, purple lines in parallel plots) do have some full-length members that exhibit the full diversity of transcriptional activity, likely influenced by local chromatin or epigenetic features of surrounding insertion sites.
Fig. 2. Transcriptional profiles of TEs are highly correlated with sequence divergence and epigenetic features.
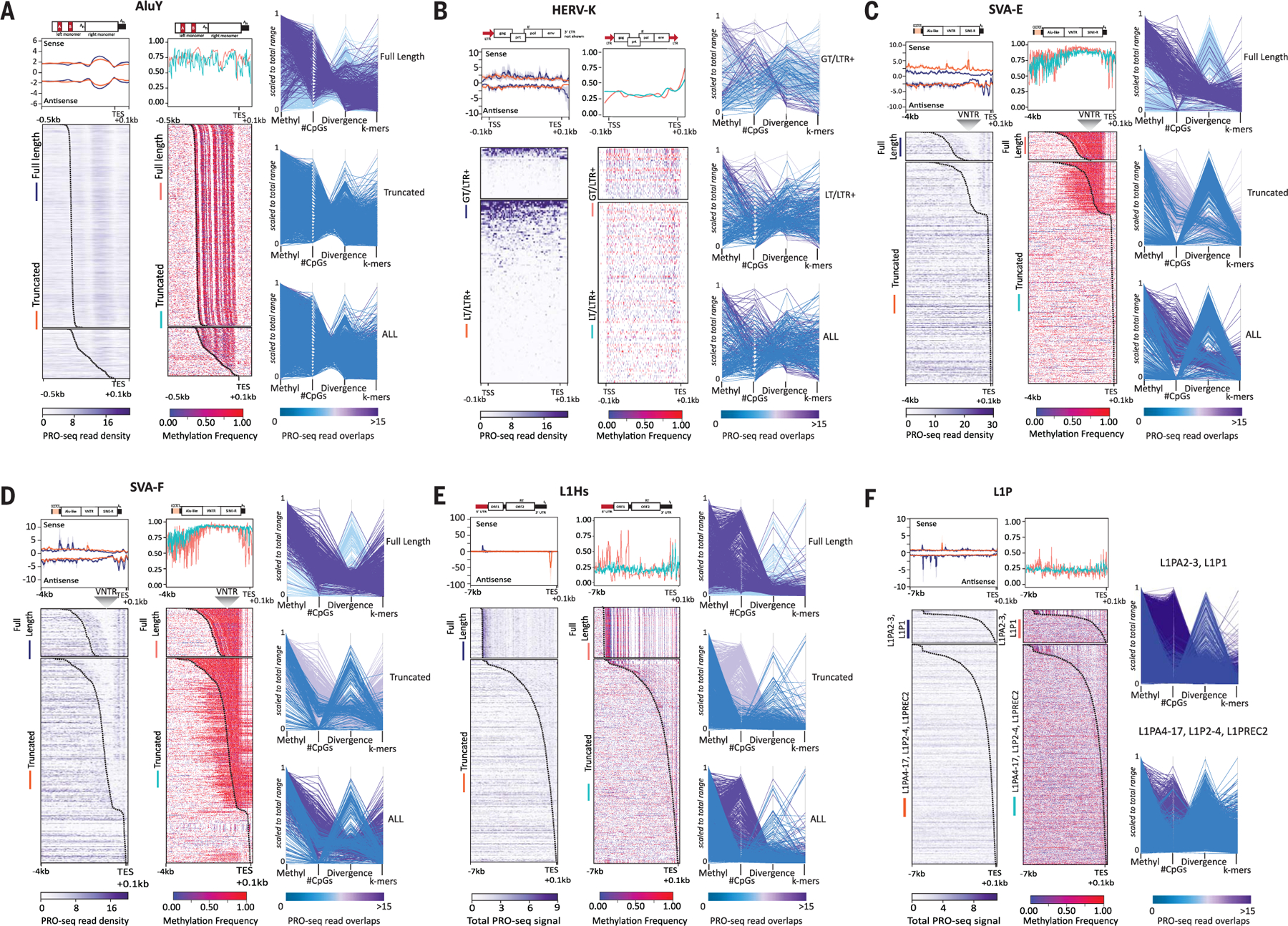
(A to F) RNA polymerase occupancy, methylation levels, CpGs, and divergence for (A) AluY, (B) HERV-K, (C) SVA-E, (D) SVA-F, (E) L1Hs, and (F) L1P elements from CHM13. Heatmaps of (left panel) T2T-CHM13 PRO-seq density (Bowtie2 default “best match,” purple scale) and average profiles showing sense and antisense strands (upper panels, standard error shown in gray) and (right panel) methylated CpGs (red–purple scale, aggregated frequency per site) for TEs grouped by their length [(A) to (E)] [fulllength (FL) and truncated (TR)] or L1PA subfamily [(F), all truncated)]. HERV-K groups are delineated as follows: >7500 bp elements (GT) and <7500 bp elements (LT) with both 5′ and 3′ long-terminal repeats (LTR+). (HERV-K elements with only one or no LTR are shown in fig. S18C). Both GT and LT/LTR+ HERV-K elements are scaled. All other TEs are anchored to the 3′ end, with a specified distance from the anchor (bottom left). Standard error for composite (gray), TSS (transcription start site), TES (transcription end site), location of the VNTR (variable number tandem repeat) within SVA are indicated. A dotted line is included on the heatmap denoting the static −0.1 kbp from the end of the annotated element. Representative schematic of elements and respective subcomponents are shown above the composite profile, scaled to the TES; red blocks indicate previously known promoter regions. (Right side of each panel) Parallel plots for each TE are shown, highlighting each group of TEs (FL/TR, or L1P subfamily; HERV-K plots represent LTRs only). Vertical axes represent scaled values for average methylation, number of CpG sites, and divergence from RepeatMasker consensus sequences for each instance of the element. Coloration by the number of overlapping PRO-Seq reads where purple represents the highest read overlap and blue the lowest, on the scale matching each plot.
Whereas PRO-seq signal is detected in truncated HERV-Ks (human endogenous retrovirus type K) that retain LTRs [LT (less than 7500 bp in length)/LTR+] (18), signal is reduced and completely lost in truncated elements without LTRs, as expected (26). Full-length HERV-Ks [GT (greater than 7500 bp in length)/LTR+] (18) generally have low methylation levels despite higher CpG content than the LT HERV-Ks, albeit with nonsignificant P values (Fig. 2B and figs. S18 to S20). Given the low number of HERV-K elements and high identity among 5′ and 3′ LTRs of HERV-K elements (GT range 0.21 to 23%, average 12.05%; LT range 1.98 to 28.96%, average 11.58%), discerning a clear 5′ promoter signal was not possible. Furthermore, SVA_E and SVA_F elements, the only SVA elements in the human genome that retain mobility (27, 28), both show similar PRO-seq peaks (Fig. 2, C and D), which distinguishes them from their truncated counterparts SVA_A, SVA_B, SVA_C, and SVA_D (figs. S18 to S22).
We find evidence for RNA polymerase promoter proximal pausing at the 5′ end of the SVA element at predicted transcription start sites (TSSs) (29). Notably, we find PRO-seq peak signal at the 3′ end within the HERV-K/LTR5a–derived portion of the element, overlapping with the Kruppel-associated box (KRAB)–containing zinc finger proteins (KZFPs) controlled enhancer activity (TEEnhancer) identified in this region (Fig. 2, C and D, gray arrowheads) that contributes to human-specific early embryonic transcription (30). While some truncated SVA_F elements retain the 5′ promoter signal, most SVA elements retain the 3′ signal (Fig. 2, C and D, and figs. S18, S20, and S21) and thus may also retain the ability to modulate gene expression.
L1Hs elements, a major contributor to human structural variation (31), show a strong promoter-proximal pause signal at the 5′ end (32) (Fig. 2E). This site also contains a methylation peak followed by a hypomethylated TSS, delineating full-length L1Hs elements from their truncated counterparts (Fig. 2E and figs. S18 to S22). As elements become inactivated through 5′ truncation (33, 34) and increased divergence, CpG content and transcriptional signal drops considerably (Fig. 2, E and F, and figs. S18 to S22), indicating that CpGs are likely targeted for methylation and subsequent deamination from cytosine to thymine.
To extend our analyses and demonstrate the applicability of this approach in studying other complex repeats in the human genome, we focused on the TE-derived macrosatellite SST1 [also called MER22 (35) and NBL2 (36, 37)]. SST1 has demonstrated meiotic instability (38), and its methylation status is of clinical relevance to multiple cancer types (39–42). SST1 arrays are variable in the human population (38), and our annotations identify about a twofold increase over the 342 loci (315,515 bases) (table S14) identified in GRCh38 (excluding the Y chromosome, which carries an additional 587 loci) (fig. S4). Randomized Axelerated Maximum Likelihood (RAxML) phylogenetic analysis with representative loci subsampled from the 16 autosomes on which SST1 resides (18) (Fig. 3, A and B, and table S15) showed that the array situated on the long (q) arm of chromosome 19 represents the ancestral SST1 in the human genome and carries a propensity for centromere seeding and array size expansions or contractions across primate lineages (35, 43).
Fig. 3. Transcriptional, epigenetic, and structural differences define SST1 elements across the human genome.
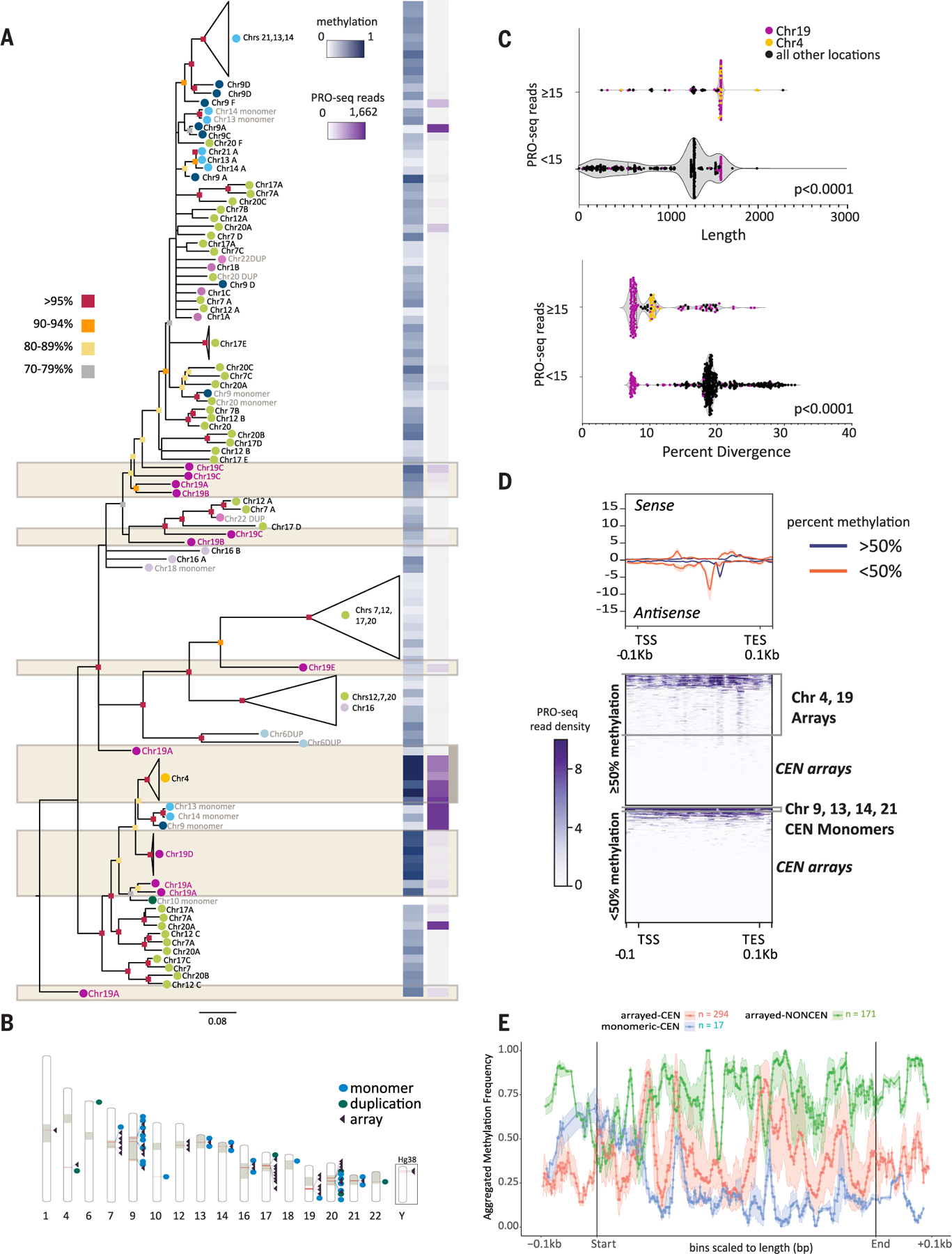
(A) RAxML phylogenetic analysis of SST1 elements [subsampled to represent each chromosomal location and aligned using MAFFT (107)] (tables S14 to S17). Bootstrap values are indicated by color (as per key to the left) at the base of each node. Branch lengths indicate distances and unresolved nodes were collapsed. “Chr#” followed by letters A to F indicates the array designation by T2T-CHM13 chromosome unless SST1 is present as a monomer or as duplicons (DUP) (indicated in gray text). Colored circles by chromosome labels indicate phylogenetic clusters (e.g., chromosomes 7, 12, 17, and 20 in green and chromosomes 13, 14, and 21 in aqua). (Right) For each SST1 sequence or group of collapsed sequences on the tree, average methylation frequency (0, hypomethylated; 1, hypermethylated) is indicated in blue, and PRO-seq read coverage is indicated in purple as per key inset. Tan boxes denote noncentromeric arrays. (B) The location of SST1 elements across T2T-CHM13 is indicated by red bars within the chromosome schematic (table S14). Tan blocks indicate centromeres and centromere transition regions as per (12). SST1 arrangement as a single monomer (blue dot), duplication (green dot), or array (purple triangle) is indicated. Locations of SST1 arrays on the Y chromosome are shown for GRCh38 (CHM13 is 46,XX). (C) Violin plot of SST1 elements shows statistically significant differences between expression levels (repeat overlap of PRO-seq reads, Bowtie2 default “best match”) and length of the element (t test, P < 0.0001) as well as percent divergence (t test, P < 0.0001). Dot colors indicate interstitial arrays on chromosome 19 (purple) and chromosome 4 (yellow) with a read overlap higher than 15. All other locations with a read overlap lower than 15 are indicated in black. Fifteen read overlap cutoffs determined by analyzing the range of read overlap among all SST1s (fig. S23). (D) T2T-CHM13 PRO-seq profiles (Bowtie2 default “best match,” upper panel) of SST1 grouped by average methylation levels (<50% and > 50%). Each element is scaled to a fixed size with standard error shading (gray), TSS, TES, and ±0.1 kbp are shown (bottom). Heatmaps (lower panels) of PRO-seq density (purple scale, normalized reads per million aggregate for sense and antisense) grouped by average methylation levels (>50%, top; <50%, bottom). Clusters of specific SST1 loci are indicated to the right. (E) Metaplot of aggregated methylation frequency (100 bins total) of SST1 elements (500 bp to 2 kbp), ±0.1 kbp, grouped by chromosomal location and arrayed versus monomeric or duplicated [orange, centromeric (CEN) array; blue, centromeric monomer; green, noncentromeric array]. Truncated noncentromeric/CEN monomers and duplications not shown; length filtering resulted in n = 1.
The number of overlapping PRO-seq reads, average methylation, and percent divergence for each SST1 element in CHM13 were compared to delineate correlations among transcriptional, epigenetic, and structural features of SST1 across genomic loci. PRO-seq revealed that the SST1 arrays on chromosome 4 and centromeric monomers on chromosomes 9, 13, and 14 are highly transcribed in comparison to other SST1 loci and are grouped in a single phylogenetic cluster (Fig. 3, A, C, and D; fig. S23; and table S16), indicating that centromeric SST1 repeat arrays are transcriptionally inactive in CHM13.
Statistical analyses of SST1 repeats showed that the highly transcribed repeats are both longer and less diverged from the consensus sequence (t test, P < 0.0001) (Fig. 3C, fig. S24, and table S17) despite their basal location in the phylogenetic tree (Fig. 3A). CpG methylation levels are high (>50%) for SST1 within chromosome 4 and 19 arrays, low (<50%) for centromeric monomers, and variable (low and high) for centromeric arrays (Fig. 3, A and D; figs. S24 and S25; and tables S16 and S17). Metaplots of aggregated methylation frequency across SST1 repeat units support this observation and indicate that while interstitial arrays and monomeric SST1s carry the same methylation frequency at their 5′ end, monomeric SST1s lose most methylation across the body of the element (Fig. 3E and figs. S25 and S26). Irrespective of this methylation pattern, heatmaps of PRO-seq density show that all highly transcribed SST1s have two internal peaks of high RNA polymerase occupancy that are closely spaced and in opposite orientations (Fig. 3D and fig. S25B), characteristic of RNA pol II promoters and enhancers.
Together, these data suggest selective pressure to retain the genomic integrity of older, less diverged SST1 arrays and monomers that are actively transcribed, whereas silenced repeats found in centromeric arrays are more susceptible to sequence variation. Contrary to expectations that CpG methylation renders repeats transcriptionally silent (44, 45), we find that high levels of average methylation across interstitial, arrayed SST1s define these transcribed repeats on chromosome 4 (Fig. 3, A, D, and E) and bear a resemblance to methylation patterns observed over gene bodies (46, 47). Chromosomal instability and cancerous phenotypes associated with demethylation and/or transcription of SST1 repeats have been reported (48, 49), indicating a need to delineate patient-specific and locus-specific annotations for SST1 (39, 50).
The transcriptional landscape of human centromeres
Centromere transcription is integral to proper centromere function, affecting the loading of newly synthesized centromere protein A (CENP-A) histones (51–57). Although evidence suggests that RNA is a critical component of the epigenetic cascade leading to faithful CENP-A assembly, an assessment of nascent transcription across human centromeres has been lacking. The availability of high confidence centromere annotations for T2T-CHM13 (12, 58, 59) provides an opportunity to assess transcription and active RNA polymerase activity across previously unresolved regions of a human genome reference: the centromere and the pericentromere. To capitalize on the T2T-level assembly and the resolution of PRO-seq at single nucleotides, we developed genome-dependent and genome-independent approaches to define the landscape of centromere transcription (18) (figs. S27 and S28).
We observed low levels of satellite transcription (figs. S29 to S33 and table S18), indicating that RNA polymerase occupancy at centromeric satellites in CHM13 is lower than that observed for all other repeat types. The low levels of satellite transcription are not explained by differences in genomic abundance between satellite repeats and other repeats. Indeed, after normalizing the observed PRO-seq levels with shuffled reads, satellite transcription is the lowest among all other repeat types (fig. S33), indicating genome-wide repression of centromere satellite transcription, including the CENP-A–containing HORs (12).
Given that centromere transcription and CENP-A deposition are dynamic processes (60), we tested whether repeat transcription varied across the cell cycle. After synchronization and release into mitosis, we find that repeat transcription across the genome drops in mitosis (Fig. 4A and fig. S34). SINEs, LINEs, and LTRs increase transcription rates at the 1-hour time point and reach a steady state by 1.5 hours, coincident with the transition to G1 after CENP-A loading. Notably, satellite transcripts are detected, but at low levels across the cell cycle (Fig. 4A and figs. S29 to S34). We used available datasets to determine whether the low level of satellite transcription was specific to CHM13 or its early developmental stage. Across cell types and developmental stages, retroelements show dynamic PRO-seq profiles, yet satellite transcription remains low (Fig. 4B and figs. S35 and S36). Across all cell types and time points, alpha satellites within the CENP-A–containing HORs (12) show generally higher PRO-seq signal than do degenerate HOR alpha satellite arrays (dHORs) and monomers or interstitial alpha satellites (MONs) (fig. S37). Thus, although nascent transcription is low, transcription from alpha satellites is detectable within the HOR domain that demarcates the active centromere (Fig. 4C). The low level of detectable transcripts within the active HOR domains contrasts with the transcriptional level of pericentromeric satellite arrays where satellite transcripts promote the recruitment of chromatin modifiers to maintain the heterochromatic status of these domains (61).
Fig. 4. Centromere landscape is characterized by the transcription of TEs rather than satellites.
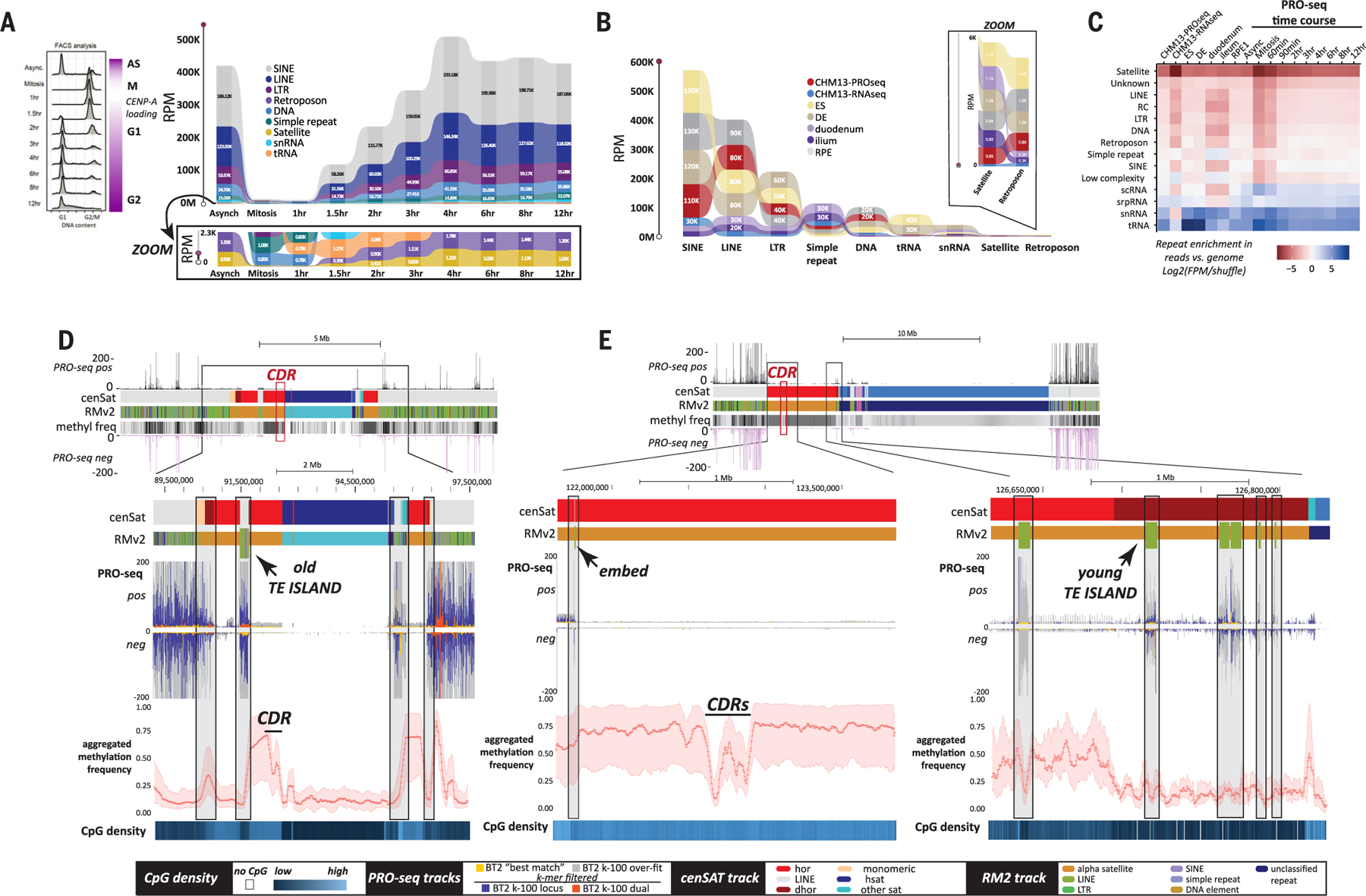
(A) (Left) Cell sorting data showing the stages of the cell cycle after synchronization and release. (Right) Ribbon plots of repeat abundance in PRO-seq data [shown as reads per million (RPM)] assessed by CASK method in asynchronous and synchronized HeLa cells collected at time points across the cell cycle (key in inset). A zoomed image shows the reads for the lower range of expressed repeats, including all satellites classified in T2T-CHM13 (tan). (B) Ribbon plot of repeat abundance in PRO/ChRO-seq data, shown as RPM, assessed by CASK method across different developmental stages and samples. Datasets include T2T-CHM13 PRO-seq and native RNA-seq, PRO-seq for RPE-1 (differentiated retinal pigment epithelial cells), and ChRO-seq for H9 ES (embryonic stem cells), DE (differentiated endoderm cells), duodenum tissue, and ileum tissue. A zoomed image shows the reads for the lowest of categories of repeats across all samples, including the satellites classified in T2T-CHM13. (C) Repeat enrichment across PRO-seq and RNA-seq datasets (all times points and tissues) ranked from least (red) to most enriched (blue) on the basis of k-mers normalized to genomic frequency in T2T-CHM13. (D and E) Recently active retroelements (green ticks in RM2 track) found embedded within alpha satellite HOR arrays (red) in (D) an “old” TE island derived from segmental duplications on chromosome 3 and (E) solo embedded TEs and “young” TE islands on chromosome 1. Stranded PRO-seq profiles (Bowtie2 default “best match”) across chromosome 3 and 1 regions encompassing the centromere are shown (top). TEs are transcriptionally active (PRO-seq Bowtie2 “best match” mapping (yellow), k-100 overfit mapping (gray), and single (blue) and dual filtered (red) k-100 mapping data are indicated for both strands) and located (black boxes) at transitions in CpG methylation (metaplot at bottom; 200 bins total) and CpG density (blue, below) within the array. Key of elements in cenSAT and RM2 tracks indicated at bottom.
TE annotations for T2T-CHM13 show that members of retroelement subfamilies known to contain full-length and, in some cases, transpositionally active members are found within centromeric HOR satellite arrays and retain their PRO-seq signal (fig. S38 and table S19). We find evidence for multiple types of TE–alpha satellite associations across T2T-CHM13 (table S19); all chromosomes have TE insertions within alpha satellites, but several lack TEs within HORs (e.g., all acrocentric chromosomes). We also find “older” TE islands within HORs, derived from segmental duplications (Fig. 4D and fig. S39), recent insertions of TEs within HORs, and aggregates of TEs that appear to form emerging TE islands (Fig. 4E, right, and fig. S40). Single insertions of TEs found within HORs, dHORs, and monomeric regions (table S19) remain transcriptionally active (Fig. 4, D and E, and fig. S38) yet show limited evidence of transcription of adjacent alpha satellites (Fig. 4E and figs. S39 and S40), indicating that read-through transcription from embedded TEs may affect alpha satellites, but not in the arrays underlying the CDR, the region defined by CENP-A enrichment (Fig. 4E, left) (12).
Given the higher proportion of L1Hs insertions in HORs and work showing a link between L1 transcription and neocentromere formation (57, 62), we compared embedded L1Hs within HORs to those found in dHORs, monomers, and chromosome arms to determine whether L1Hs embeds retained their TE signatures or were “overwritten” by their local chromatin environment. We find no statistical evidence that L1Hs within HORs and dHORs deviate in length, divergence, or average methylation from those found outside of these regions (figs. S41 and S42 and table S20). However, L1Hs within monomeric segments of alpha satellites are both more diverged and less methylated than L1Hs that are in HORs (P < 0.05), dHORs (P < 0.01), or not embedded at all (P ≤ 0.001) and show less transcription than their counterparts elsewhere in the genome, including those in the HOR and dHOR (figs. S38 and S42).
Although we find no clear link between alpha satellite transcription and the CENP-A domain overlapping the CDR (12, 21), transcription detected from embedded TEs marks shifts in methylation frequencies across satellite domains, establishing putative TE boundaries. Whether and how TEs facilitate these shifts is unknown. In previous work, the activity and copy number of TEs has been linked to alterations in methylation levels within centromeres in interspecific hybrids, resulting in chromosome instability (63), indicating that a balance of methylation is required for centromere stabilization. With the technological advances presented in the assembly and annotation of the T2T-CHM13 human reference, comparative studies across other species will aide in revealing how the structure of the satellite-dense centromeres of human differs from that of TE-enriched centromeres in other species (64) and how these differences affect centromere function and chromosome evolution.
Putative TE-driven genomic DNA transductions and their evolutionary consequences
The complete sequence provided by T2T-CHM13 revealed previously unknown patterns of repeat expansions across the short (p) arms of acrocentric chromosomes. In T2T-CHM13 (11), we discovered previously unannotated repeat arrays of a 64-nucleotide sequence (Fig. 1B) present in high copy numbers on the p arms of acrocentric chromosomes 14, 15, 21, and 22 (11) and in single or low copy number (<5) on eight other chromosomes (Fig. 5A and tables S2 and S21). A solo monomer resides on chromosome 10, with all other occurrences adjacent to an AluSx3 element (thus, with Alu satellite, or WaluSat). The lack of identity among 5′ and 3′ sequences of the chromosome 10 locus and the AluSx-WaluSat loci on all other chromosomes (fig. S43), coupled with phylogenetic analyses across primates (figs. S43 to S46), indicates that an ancestral duplication of the chromosome 10 locus was followed by a mobile element insertion to form the AluSx-WaluSat unit in the last shared ancestor with Catarrhini.
Fig. 5. TE activity affects genomic repeat diversity in CHM13.
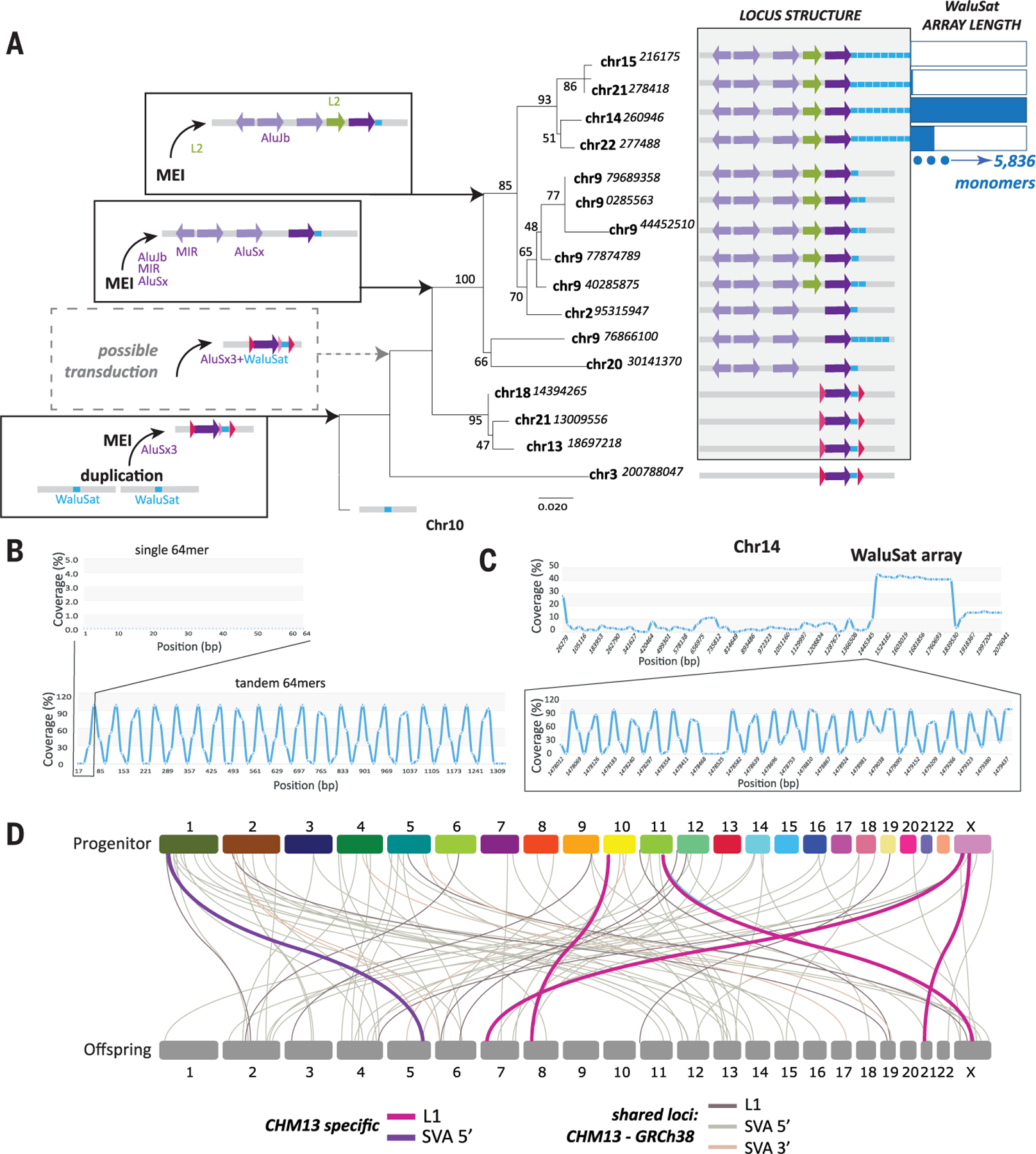
(A) Maximum likelihood (ML) phylogenetic analyses of the AluSx3-WaluSat locus across T2T-CHM13. Chromosome location is indicated (starting nucleotide position shown) at each branch. Bootstrap values shown at each node, distance indicated by length of branch. Left shows the sequential order of events, initiating with a duplication of the chromosome 10 WaluSat locus followed by mobile element insertion (MEI) of an AluSx3. The identification of putative TSDs (pink, fig. S43) and a lack of identity among sequences adjacent to WaluSat on chromosome 3 and all other loci (fig. S43) may indicate that a transduction event preceded the spread of AluSx3-WaluSat across the human genome (dotted box). MEI events upstream of the AluSx3-WaluSat are concordant with phylogenetic relationships among loci and indicate that the derivation of AluSx3-WaluSat loci across other chromosomes were the result of segmental duplication events (gray shaded box). Once the AluSx3-WaluSat was duplicated to the acrocentric chromosomes 14, 15, 21, and 22, a massive expansion of the WaluSat sequence (blue boxes) occurred. The number of WaluSat monomers within each acrocentric array is indicated on the right with monomer number relative to maximum monomer count 5836 on chromosome 14. (B) G-quadruplex (G4) analysis of a single 64-mer monomer of the WaluSat sequence showed no predicted G4 structures (top), while an in silico construct of a tandem array of the WaluSat shows high G4 coverage at the junction between individual WaluSat monomers across the array. (C) G4 analysis of the p arm of chromosome 14 shows a peak in G4 predictions coincident with the WaluSat array. Bottom is a zoom inset of a subset of the array showing that the junctions between most monomers carry predicted G4 structures. (D) Transduction events predicted for CHM13 (L1, pink; SVA 5′, purple) and shared between T2T-CHM13 and GRCh38 (gray shades) are shown. Chromosome connections link progenitor and offspring locations (fig. S49).
The WaluSat sequence exists as a single monomer at eight loci, as a duplication at three loci, and in one case as a pentamer. However, once segmental duplication events placed the AluSx-WaluSat on the p arms of chromosomes 14, 15, 21, and 22, WaluSat amplified into longer arrays, ranging from 26 copies (chromosome 15) to 5836 copies (chromosome 14) (Fig. 5A). We hypothesize that the high degree of sequence similarity and copy number variation among p arm WaluSat arrays is due to frequent nonallelic or ectopic recombination events on acrocentric chromosomes (11, 65), which may be exacerbated by replication challenges associated with the predicted periodic G-quadruplex structures (66) identified at junctions of WaluSat sequences within arrays (18) (Fig. 5, B and C).
The low identity among the sequences adjacent to the chromosome 3 AluSx-WaluSat and other AluSx-WaluSat loci, along with the identification of putative target site duplications (TSDs),may indicate that a transduction event followed the Alu insertion and preceded the spread across the human genome via duplications. TE-mediated transduction (i.e., a TE transduction event), a process by which retroelements co-mobilize DNA flanking the element to new genomic loci (67–70), has been observed for L1 and SVA elements in humans (67–73). TE transduction events mediated by Alu elements are seemingly rare (74), likely because of efficient termination of RNA polymerase III on sequences with long poly-T tract lengths and nearby RNA secondary structures (75). Given the age of the initial insertion of the AluSx element, it is unknown if such an event was mediated by an RNA polymerase III or cryptic upstream RNA polymerase II promoter, or if other rearrangements specific to chromosome 3 degraded signal of shared identity with other segmental duplications (Fig. 5A, dashed box).
Beyond potentially seeding new repeat sequences across the genome, TE transductions can affect the genome through exon shuffling (67, 71, 76) and are a possible source of somatic mutations (74, 75). Here, we applied a set of computational approaches (18) (fig. S47) to annotate putative TE transduction events in T2T-CHM13. In total, we analyzed 971,993 L1s and 7068 SVAs (figs. S48 to S51). After stringent filtering for potential artifacts, such as segmental duplications and putative duplications of truncated elements, we find 65 L1, five 3′ SVA, and 11 5′ SVA transduction events (tables S21 and S22 and figs. S50 and S51).
Of these 81 annotated transduction events, 78 are shared with GRCh38 (Fig. 5D and table S23), and three appear specific to T2T-CHM13. One T2T-CHM13 TE transduction is in a region of no synteny with GRCh38 and is caused by an L1PA4, representing an older event according to Kimura-2 distances (fig. S49). Of the remaining two T2T-CHM13 TE transductions, both events are derived from the youngest, human-specific TEs, L1Hs and SVA-F, and may represent polymorphic TE transductions. However, we find the offspring TE in both GRCh38 and T2T-CHM13, yet the transduced sequence is missing in GRCh38, owing to a collapse in the sequence, highlighting the utility of a T2T-level assembly in identifying putative TE transduction events.
CHM13 serves as a reference for comparative repeat analyses across humans and other primate genomes
Studies of the link between TE activity and chromatin states can extend beyond local influences, as exemplified by LINE and SINE transcriptional activity and the chromosome-wide silencing of the X chromosome during X inactivation (77–79). Two noncoding RNAs on the X chromosome are central to the inactivation of one X in females, Xist and Tsix (80). These two loci overlap one another in a sense and antisense orientation but are in distinct topologically associating domains (TADs); Tsix is the antisense repressor of Xist, whose up-regulation leads to X inactivation (81). The bipartite structure of the locus in two TADs facilitates partitioning of the X inactivation center (XIC) and supports appropriate timing of X inactivation through Xist transcription in early development (82). Moreover, an early step in the formation of heterochromatin across the inactive X is the silencing of LINEs and SINEs within the Xist RNA compartment (77).
The scarcity of SNPs (21) in T2T-CHM13, coupled with the short reads of PRO-seq data, made it impossible to discern transcripts originating from one X allele versus the other within CHM13. However, we were able to phase reads into their individual alleles, supporting the assessment of methylation differences of TEs between the two X chromosomes in the XIC. PRO-seq signal was found across the Xist locus, whereas no signal was detected from the Tsix locus, indicating that X inactivation has proceeded, resulting in differential methylation profiles across alleles (Fig. 6A). Low methylation (Fig. 6A, blue block in cluster 2) marks the initiation of Xist transcription, followed by high methylation levels across the Xist/Tsix locus on this allele, inclusive of the interspersed repeats found across the locus (Fig. 6A and table S25). A distinct pause (indicated by a pileup of PRO-seq signal, boxed in Fig. 6A) after the termination signal of the Xist transcript unit was found that coincides with the TAD junction and delineates the Xist and Tsix domains. These data are inconsistent with a report that androgenetic hydatidiform moles lack X inactivation (83).
Fig. 6. Repetitive elements define differences between human genomes and nonhuman primates.
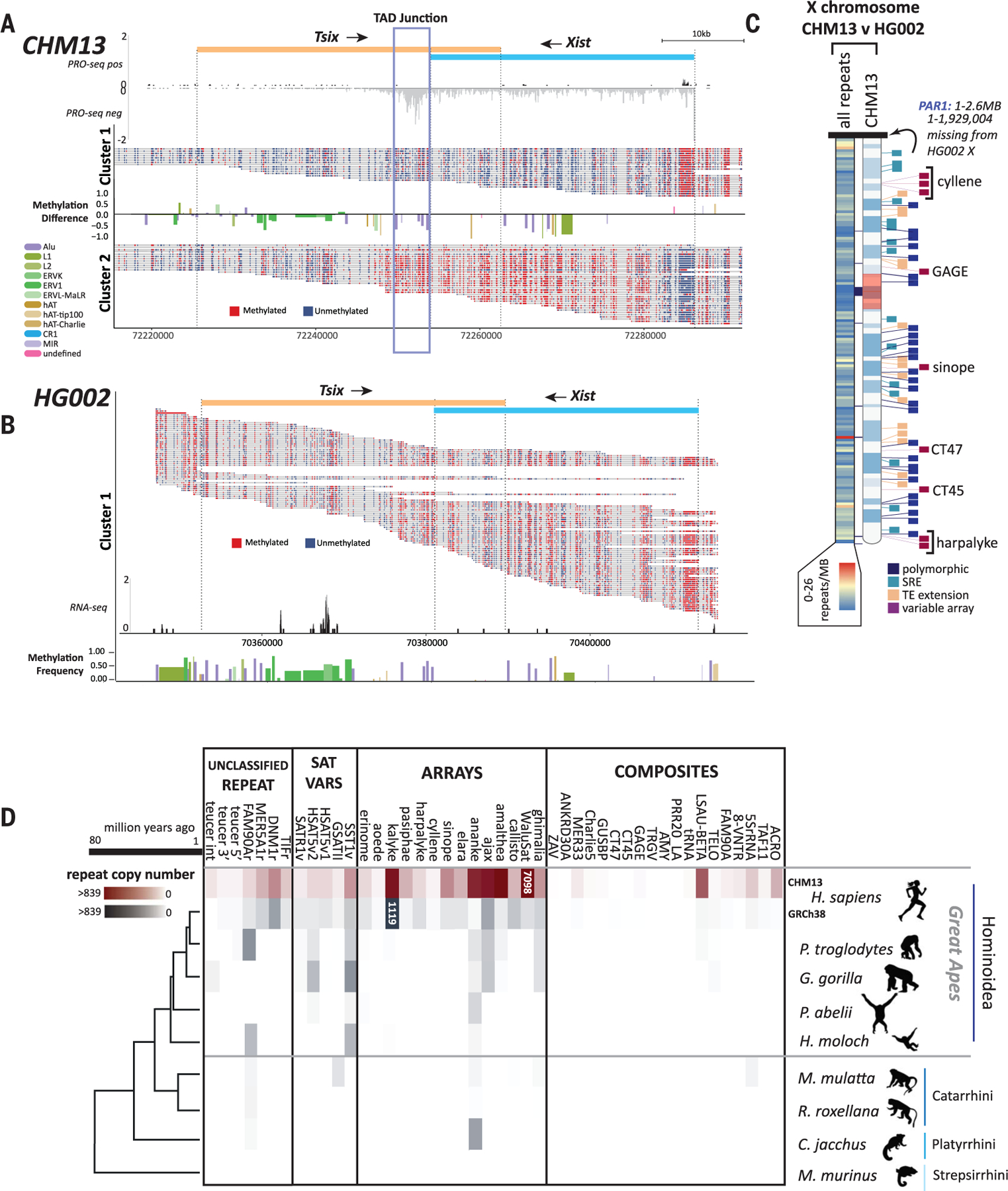
Single read methylation profiles were extracted, and reads were clustered on the basis of the methylation state of the Xist promoter from (A) T2T-CHM13 and (B) HG002. Differences in repeat methylation were calculated by taking the average methylation per repeat and subtracting cluster 2 repeats from cluster 1 repeats. Directionality of Xist/Tsix transcript units are indicated (top). Normalized PRO-seq reads show a marked pileup of RNA pol II at the predicted TAD boundary at the 3′ end of the Xist transcript [(A), blue box]. (B) Normalized RNA-seq reads across the single cluster for HG002 show no transcriptional signal for Xist. (C) Heatmap of chromosome X showing the location of all repeat differences between the Xs of HG002 and T2T-CHM13 (left) and the location of the top four categories of repeat differences: polymorphic (insertion/deletion), SRE (short repeat extension), TE extension, and variable array length (right ideogram). Gaps between T2T-CHM13 and GRCh38 are indicated with black blocks between the heatmap and ideogram. (D) Copy numbers of previously unknown human repeat annotations identified in T2T-CHM13 grouped by repeats, variants of known satellites, tandemly arrayed sequences, and composite element (inclusive of subunits) for T2T-CHM13 (maroon), GRCh38, and genomes for other primates from the Hominoidea, Catarhini, and Platyrrhini lineages (gray). Heatmap scale denotes number of repeats within the array (0 to 839). Array sizes >839 are indicated within colored blocks. Phylogenetic relationship and millions of years since divergence are indicated on the bottom. Not shown: variants of known centromeric satellites [but see (12)] and the repeat annotation for an AluJb (121) fragment, which could not reliably be delineated in copy number from other closely related full-length AluJb elements.
We also compared both the XIC and chromosome-wide repeat content of the chromosome X from T2T-CHM13 and HG002 (XY). As expected, the XIC in HG002 shows high methylation across the locus and only a single allelic cluster, with no detectable transcripts across the Tsix/Xist domain (Fig. 6B and table S25). Sequence comparison of the 269,020 repeats assessed between the haploid X of HG002 and T2T-CHM13 (Fig. 6C and tables S26 and S27), excluding the pseudoautosomal region (see fig. S52 for T2T-CHM13 PAR annotations), currently unassembled in HG002, uncovered 778 repeat differences, of which 70% were simple repeats and 21% were TEs (64 of which were length outliers) (18) (fig. S53). Collectively, these data demonstrate that the depth of repeat annotations based on the T2T-CHM13 assembly can serve as a reference for studying human variation inclusive of repeats that affect local and regional chromatin, gene expression, and gene copy numbers.
While many of the previously unidentified repeat classifications coincide with gaps filled in the T2T-CHM13 assembly, these data supported genome-wide annotation of previously undiscovered repeats and TEs (Fig. 1B). To determine whether these repeat classifications were specific to humans, we searched for or-thologous sequences in the human reference GRCh38 and available genome assemblies for primates representing the great apes (Pan troglodytes, Gorilla gorilla, Pongo abelii), Hominoidea (Hylobates moloch), Catarrhini (Macaca mulatta, Rhinopithecus roxellana), Platyrrhini (Callithrix jacchus), and Strepsirrhini (Microcebus murinus) (18).
When comparing copy numbers of repeat annotations between T2T-CHM13 and long-read, high-quality assemblies available for other great apes (chimpanzee, gorilla, and orangutan) (84), we still find an increase in copy number across most of the repeats identified herein (Fig. 6D, fig. S2, and table S28). Many repeats appear only as monomers in other primate genomes or are absent in Strepsirrhini, Platyrrhini, Catarrhini, and lesser apes; these reduced counts are largely influenced by the quality of these assemblies and potentially high rates of divergence among repeats, and they highlight the need for telomere-to-telomere assembly approaches for comparative analyses (85). Finally, eight of the repeats identified herein are human-specific, with an additional 11 found only as monomers in other species (Fig. 6D and table S28).
Conclusions
The assembly of the complete, telomere-to-telomere human genome reference facilitated development of an atlas of repeats that make up >53% of the human genome. Through this collaborative effort, we have developed a resource of human repeat annotations and methods to guide future efforts in exploring the complexities of repeat biology in human and other primate genomes. We focused on repeat sequence, CpG methylation, and transcriptional annotation; updated repeat models and implemented repeat modeling tools that supported the identification of previously unknown satellite arrays; expanded the catalog of variants for known repeats and TEs; and developed annotations for complex, composite repeat elements. Deeper exploration of such repeats revealed the complexity of genetic mechanisms that affect repeats during different phases of their life cycle and thus illustrate the myriad mechanisms by which they are major contributors to defining the structure and content of the human genome.
For example, we found that a TE insertion event captured a short sequence, WaluSat, in a primate ancestor. Subsequent segmental duplications of the region carrying this composite TE-sat repeat spread the sequence across several human chromosomes, including four of the acrocentric chromosomes. The satellite portion of the repeat expanded to almost 0.5 Mbp of sequence on the acrocentric chromosomes, resulting in the alteration of the structure of this portion of the chromosome into regions dense with G4s, which are potentially functional elements (86). This example highlights the need for future functional studies dissecting the impact of repeats on the local chromosome environment, such as replication timing, local transcription, DNA damage and repair processes, and establishing TAD boundaries. Moreover, this example lays the groundwork for exploring the impact of local environments (such as gene-poor regions as found on the acrocentric arms of human chromosomes) on sequence constraint and mutation rates for emergent repeats.
We provide a high-confidence functional annotation of repeats across the human genome. For example, we find that the tandemly arrayed TE-derived satellite SST1 carries distinctive methylation and transcriptional profiles, including an enhancer embedded in each unit, found only in specific arrays on chromosomes 19 and 4. These arrays are hypervariable in the human population, and alterations in their activity have been linked to cancer (36, 48). However, a full understanding of copy number variation, epigenetic instability, and transcription of SST1 elements has been hampered by a lack of complete annotations of copies of these elements elsewhere in the genome. Our functional annotation revealed transcriptional signatures of both promoters and enhancers within active SST1 elements that may affect local transcription and chromatin structures. Moreover, this enhancer implicates SST1 in defining cellular partitions, such as paraspeckles and phase-separated condensates (36, 87), that could have an impact on other genomic loci.
Combined with defining the linear order and content of centromeric sequences (12), we find that engaged RNA polymerase signal is low across centromeric satellites arranged in arrays, irrespective of stages of the cell cycle or development. Rather, active transcription is detected in embedded retroelements coinciding with shifts in methylation states that demarcate active centromere domains. To date, the centromere biology field has been limited by a lack of a linear assembly across human centromeres, challenging the development of models to describe genetic and epigenetic elements that define centromeric chromatin. Our data, in concert with centromere annotations (12), reveal that these high-density repeat regions are not static in sequence, epigenetic, or transcriptional activity and that there is a high degree of substructure across the centromeric regions that affect function. Comparing the landscape of the variable centromere forms across domains of life, and in human disease, will reveal the complex life cycle of centromeres (64).
Studies of human genetic variation have been relatively blind to repeat variation among individuals, particularly arrayed and complex repeats, as these types of sequences are recalcitrant to short-read sequencing technologies, mapping, and functional annotation methodologies. As a prospective of the utility of complete reference genomes in studying human genetic variation, we compared two T2T X chromosomes. We find 218 kbp of repeat differences between these two chromosomes (0.18% of the chromosome, excluding the 1.9-kbp PAR), including repeat variation in complex arrays that carry exonic material and thus affect gene dosage. Thus, comparative analyses of T2T-level assemblies reveal the potential for discovering an even wider range of repeat variation across the 46 chromosomes that constitute the human genome.
Finally, our work demonstrates the need to increase efforts toward achieving T2T-level assemblies for nonhuman primates to fully understand the complexity and impact of repeat-derived genomic innovations that define primate lineages, including humans. Although we find repeat variants that appear enriched or specific to the human lineage, in the absence of T2T-level assemblies from other primate species, we cannot truly attribute these elements to specific human phenotypes. Thus, the extent of variation described herein highlights the need to expand the effort to create human and nonhuman primate pangenome references to support exploration of repeats that define the true extent of human variation.
Materials and methods summary
Repeat model discovery
RepeatMasker4.1.2-p1 (88) with the Dfam3.3 repeat library and RepeatModeler2.0.1 (8) were used to define repeats across the genome, further refined using extensive manual curation, as described in (18). This database was used to generate a final mask of the T2T-CHM13v1.1 assembly. ULTRA (9) was used to improve the accuracy of tandemly repetitive satellite annotations. Gaps of >5 kb in T2T-CHM13v1.1 repeat annotations were identified with BEDtools (89) and manually curated. Monomer structure was confirmed using self-alignment plots. Repeat models were further refined to remove any false positives (e.g., fragments of other TEs, pieces of simple repeats), as described in (18).
Composite elements
We defined a composite element as a repeating unit consisting of three or more repeated sequences (TEs, simple repeats, subunits, and/or satellites) found as a tandem array in at least one genomic location. A composite subunit is a previously unknown repeat annotation that is found within a composite. Whereas the locations of some composite elements within a family are present as a single copy and thus are likely segmental duplications derived by nonallelic homologous recombination (90), a composite family is distinguished by the presence of composite elements in an array in at least one location.
LiftOver/reverse liftOver analyses
LiftOver chains were generated from LASTZ alignments between GRCh38 and T2T-CHM13v1.1 and X chromosomes of T2T-CHM13 and HG002 with considerations as per (18). Reverse liftOver was performed from repeat annotations in both assemblies; BEDtools (89) was used to intersect the T2T-CHM13 coordinates with regions lacking synteny to GRCh38. Results were parsed into one of five categories: full match (i.e., SINE/Alu/AluSx), class match (i.e., SINE/Alu), family match (i.e., SINE), no match, and those set aside and subject to extensive manual curation to identify correct matches.
Methylation metaplots
Nanopore CpG methylation data for T2T-CHM13 and HG002 was processed as in (21). CpG methylation frequency was calculated by fraction of methylated reads to total coverage within bins in T2T-CHM13 or HG002 with the BSgenome Bioconductor package (21, 91). Multiples of three bins were further smoothed with the “rollmean” function from the R package Zoo. Methylation clustering was performed by selecting all reads spanning a locus and using the mclust (v5.4.7) R package with the “VII” model to cluster methylation calls across the locus (92). CpG density heatmaps were calculated by counting the total number of CpG sites per position relative to the repeat start and end and dividing by the total number of repeats in each group. Methylation single-read plots were generated in the ggplot2 R package using geom_rect() to plot individual reads with methylated CpGs as red and unmethylated CpGs as blue.
Identification and classification of full-length and truncated TEs
Full-length elements of recently active TE families [AluY, L1Hs, HERV-K, and SVA_E/F (93)] were retrieved from the RepeatMasker output and cross-referenced with PRO-seq data and CpG methylation data as per (18). All retroelement classes were grouped into relative age categories based on divergence and phylogenetic distribution (6, 88, 94–99). LINEs, SINEs, and retroposons were grouped by subfamily; LTRs were grouped by family.
PRO-seq
For each of two PRO-seq replicates, cells were processed as per (18, 22). PRO-seq libraries were prepared as previously described (22) with minor modifications (100). Permeabilized cells were mixed with permeabilized Drosophila S2 nuclei in all 4-biotin-NTP run-on reactions. After amplification, libraries were polyacrylamide gel electrophoresis (PAGE)–purified to remove adapter-dimers and select molecules between 140 and 650 bp. Libraries were sequenced on an Illumina NextSeq 550 (single-end, 75 bp). Rawfastq fileswere trimmed for quality, length, and adapters using cutadapt (101) and reverse complemented using the fastx-toolkit (102). Bowtie2 (103) alignment to Dm6 was used to remove Drosophila spike-in reads; remaining reads were aligned to T2T-CHM13 using default (“best match”) parameters (and k-100 for comparison); multimapping alignment files were subjected to single-copy k-mer filtering and processed into beds with BEDtools (89) for subsequent normalization with nonmitochondrial alignments to obtain counts in reads per million mapped (RPMM) as described in (18). Complementary analyses were performed on read data (unmapped) as outlined below and in (18).
Statistical analyses and data visualization
BEDtools (89) map was used to calculate average methylation and CpG density across all repeats in RepeatMaskerV2 (RMv2) and incorporated into 3D graphs and parallel plots. Genomic datawere visualized using RIdeogram (v0.2.2) (104), Circos (v0.69–6) (105), and Circa (v1.2.2). Genome browser tracks and centromeric satellite (cenSAT) annotations for T2T-CHM13 are as described in (11, 12, 21, 65). Heatmaps for PRO-seq profiles were generated using deepTools2 (106). Normalized data were binned in 10-bp windows, and repeat elements were anchored to the 3′ end, with the exception of HERV-K, which was divided into subcategories on the basis of length and presence of dual LTRs and scaled as per (18). The maximum value per bin and composite profiles were summarized by averaging each bin across all regions in the group; standard error was estimated and is shown in gray in each composite. Methylation heatmaps for HERV-K were generated in R ggplot2 by normalizing repeat size by start and end position and using geom_tile() to plot CpG methylation frequency at each position. For all other elements, methylation heatmaps were anchored at the 3′ and using geom_tile() to plot CpG methylation frequency at each position.
SST1/L1Hs embed analyses
SST1 sequences were extracted from CHM13 annotations via BEDtools (89) and aligned with MAFFT (107). The evolutionary history was inferred by using RAxML (108) and the GTR+Gmodel (109) asmatched by jModelTest (110); 100 bootstrap replicates reported. PRO-seq density for SST1 with <15 and ≥15 reads overlapping were determined by plotting the distribution of read overlaps across all annotated SST1 elements. BEDtools (v2.29.0) (89) was used to intersect SST1/L1Hs repeats with genomic locations, methylation (21), and transcriptional data. An unpaired t test was performed to quantify differences among repeats in each group by repeat length, percent divergence, percent insertions, percent deletions, and average methylation. Violin plots were generated via GraphPad Prism (v9.1.1).
HeLa cell cycle analyses
Given the low rate of cell division and synchronization challenges in CHM13 cells, HeLa-S3 cells were used, noting the caveat that this cell line carries high levels of karyotypic instability (111). HeLa-S3 cells were arrested as per (112), mitotic cells collected and subsequently grown for the corresponding time or immediately permeabilized (mitotic sample) as described in (18). All time points were collected in replicate experiments. Before cellular permeabilization, 10% of each sample was removed, fixed in cold 75% ethanol, and stained with propidium iodide, and DNA content was analyzed using a BD FACSAria II. The flowCore package was used to read FCS files into R. PRO-seq libraries (both replicates) were prepared as previously described (22), with minor modifications as for CHM13 (18). All data were processed, mapped, and normalized as above for CHM13. Comparative and quantitative analyses are outlined below and described in (18).
H9 ChRO-seq data analyses
External chromatin run-on and sequencing (ChRO-seq) data (GSE142316) for four developmental stages in replicate (ES, DE, duodenum, and ileum) (113) of H9 cells were used for comparison comparison to CHM13. H9 ChRO-seq data was preprocessed using the PRO-seq2.0 pipeline to generate adapter-trimmed and deduplicated fastq files used for repeat composition analysis as per (18).
Preprocessing, mapping, and postprocessing of RNA-seq data (CHM13 and HG002)
Data from two replicates of CHM13 paired-end native RNA sequencing (RNA-seq) using oligoDT (12) were processed with the same workflow as the CHM13 PRO-seq data, with minor modifications as per (18). External paired-end ribodepleted RNA-seq data for HG002 (GM24385) were used for comparison, preprocessed as per CHM13 RNA-seq and mapped to a combined assembly of T2T-CHM13 autosomes, HG002 chrX, and GRCh38 chrY with Bowtie2.
Comparative analyses of transcript quantification approaches
To complement TE (herein) and centromere satellite repeat annotations (12), we implemented a three-pronged approach to define centromere transcription as described in (18): a mapping-dependent approach, in which PRO-seq (two replicates) and RNA-seq (two replicates) data were mapped and reads were intersected with single copy k-mers derived from the T2T-CHM13 assembly and whole-genome shotgun polymerase chain reaction–free reads (11, 114); a mapping-independent approach in which unmapped PRO-seq and RNA-seq reads were annotated using classification of ambivalent sequences using k-mers (CASK) and a T2T-CHM13–dependent k-mer database formed via T2T-CHM13 repeat annotations; and a genome-independent approach, in which PRO-seq and RNA-seq reads were processed through RepeatMasker using the human Dfam 3.3 library. RepeatMaskerV2 (RM2) was intersected with cenSAT annotations to identify and label repeats adjacent to alpha satellites designated HOR, dHOR, MON, or “none of the above” regions (RMv2-alpha).
To compare across these three methods, BEDtools (89) coverage was used to obtain counts of reads overlapping repeats defined in RMv2 and RMv2-alpha across all mapping methods, requiring at least 50% (~25 to 30 bp, roughly equivalent to the CASK k-mer length) of the read to overlap the repeat element [and see (18)]. The relative abundance of each repeat was similar across replicates; thus, counts from both replicates were summed. Variable bowtie mapping parameters (default, k-100, and k-100 filtered for single copy k-mers with multiple filters) on PRO-seq and RNA-seq datasets were assessed (18).
WaluSat analyses
The evolutionary history of WaluSat, AluSx, and the AluSx-WaluSat loci were inferred by using the maximum likelihood method as described (18). Dotplots were generated by comparison of 1.5-kb sequences flanking both 5′ and 3′ regions adjacent to WaluSat insertions with FlexiDot as per (18). G-quadruplex analysis was performed with G4Hunter (115).
Transduction analyses
TE transduction events were analyzed using the modified TSDfinder tool (67), filtering for artifacts such as segmental duplications and truncated elements, and refined on the basis of TE age using Kimura-2 distance parameters as described in (18).
ChrX liftOver analysis and repeat fasta comparison
Lifted T2T-CHM13 chrX to HG002 coordinates were compared (18) using a similarity score as a percentage of the max score (>90% were considered concordant, <50 bp were in-sufficient; others were considered potentially polymorphic). Sequences of interest were filtered for length differences between the liftOver coordinates. Differences were subject to manual curation depending on repeat type, and the final loci were subjected to RepeatMasker analysis.
Copy number comparison across primates
Copy number comparisons across primate genomes (18) were generated with the most recent, available primate genomes for each species: Pan troglodytes (accession: GCA_002880755.3) (84), Gorilla gorilla (accession: GCA_900006655.3) (116), Pongo abelii (accession: GCA_002880775.3) (84), Hylobates moloch (accession: GCA_009828535.2), Macaca mulatta (accession: GCA_008058575.1) (117), Rhinopithecus roxellana (accession: GCF_007565055.1) (118), Callithrix jacchus (accession: GCF_009663435) (119), and Microcebus murinus (accession: GCF_000165445.2) (120). BLAST was used to search each genome for individual instances of the corresponding repeat or composite element, requiring at least an 85% length match to the query repeat/composite monomer and a 100% match requirement across the 85% length for gap tandem arrays.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UConn Computational Biology Core for computational support. We thank the NIH Intramural Sequencing Center and the UConn Center for Genome Innovation for sequencing. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov), the UConn HPC Xanadu Cluster (https://bioinformatics.uconn.edu; https://health.uconn.edu/high-performance-computing), the Stanford Research Computing Center, the Stanford Sherlock HPC cluster, the PALMA-II HPC cluster of the University of Münster (https://confluence.uni-muenster.de/display/HPC), and the University of Montana’s Griz Shared Computing Cluster (GSCC). Special thanks to M. Diekhans for assistance with UCSC browser tracks and liftOver chain development.
Funding:
This work was supported, in part, by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (AMP). Grants from the US National Institutes of Health: R01GM123312-02 and R21CA240199 to S.J.H., G.A.H., P.G.S.G., and R.J.O.; R01HG002939 and U24HG010136 to J.M.S., J.R., A.F.A.S.; R01HG009190 to A.G. and W.T.; R01HG00990905 to C.L. and A.F.S.; R35GM128857 to L.W. and L.J.C.; R01GM132600, P20GM103546, and U24HG010136 to D.O. and T.J.W.; U24HG010263, U24HG006620, U01CA253481, and R24DK106766-01A1 to M.C.S.; R01HG002385, R01HG010169, and U01HG010971 to M.R.V. and E.E.E.; 1ZIAHG200398R01 to A.R. and A.M.P.; 1R01HG011274-01, R21HG010548-01, and U01HG010971 to K.H.M. Grants from the National Science Foundation: NSF 1613806 and NSF 1643825 to R.J.O.; NSF DBI-1627442, NSF IOS-1732253, and NSF IOS-1758800 to M.C.S. Connecticut Innovations grant 20190200 to R.J.O. Mark Foundation for Cancer Research grant 19-033-ASP to M.C.S. Stowers Institute for Medical Research grant to L.G.d.L. and J.L.G. Howard Hughes Medical Institute Hanna H. Gray Fellowship to N.A. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: K.H.M. has received travel funds to speak at symposia organized by Oxford Nanopore. K.H.M. is a scientific advisory board (SAB) member of Centaura, Inc. E.E.E. is a SAB member of Variant Bio, Inc. W.T. has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies. All other authors declare that they have no competing interests.
Data and materials availability: Sequencing data and assemblies, PRO-seq CHM13/RPE-1, RNA-seq CHM13 (NCBI BioProject PRJNA559484; www.ncbi.nlm.nih.gov/bioproject/559484); sequencing data, assemblies, and other supporting data on Amazon Web Services (11); repeat library for previously unknown repeat entries, UCSC assembly hub browser, RepeatMasterv2 Track CHM13v1.1, RepeatMaskerv2 Track GRCh38 + chrY, RepeatMaskerv2 Track HG002 chrX, RepeatMasterv2 Track Composites CHM13v1.1, RepeatMasterv2 Track previously unknown satellites and arrays CHM13v1.1, sequence alignment and nexus files for phylogenetic analyses, and all scripts and codes used herein (122); CHM13v1.1 Meryl 21-mers and 51-mers (114); PRO-seq HeLa (GSE179576); updated human repeat database (www.dfam.org). CHM13hTERT cells were obtained for research use via a material transfer agreement with U. Surti and the University of Pittsburgh.
REFERENCES AND NOTES
- 1.Chuong EB, Elde NC, Feschotte C, Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet 18, 71–86 (2017). doi: 10.1038/nrg.2016.139; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cordaux R, Udit S, Batzer MA, Feschotte C, Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proc. Natl. Acad. Sci. U.S.A 103, 8101–8106 (2006). doi: 10.1073/pnas.0601161103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koonin EV, Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. London Ser. B 371, 20150442 (2016). doi: 10.1098/rstb.2015.0442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koga A et al. , Co-opted megasatellite DNA drives evolution of secondary night vision in Azara’s owl monkey. Genome Biol. Evol 9, 1963–1970 (2017). doi: 10.1093/gbe/evx142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hancks DC, Kazazian HH Jr., Roles for retrotransposon insertions in human disease. Mob. DNA 7, 9 (2016). doi: 10.1186/s13100-016-0065-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF, The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2 (2021). doi: 10.1186/s13100-020-00230-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandes JD et al. , The UCSC repeat browser allows discovery and visualization of evolutionary conflict across repeat families. Mob. DNA 11, 13 (2020). doi: 10.1186/s13100-020-00208-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flynn JM et al. , RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A 117, 9451–9457 (2020). doi: 10.1073/pnas.1921046117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson D, Wheeler T, ULTRA: A model based tool to detect tandem repeats. ACM BCB 2018, 37–46 (2018). doi: 10.1145/3233547.3233604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaisson MJP et al. , Resolving the complexity of the human genome using single-molecule sequencing. Nature 517, 608–611 (2015). doi: 10.1038/nature13907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nurk S et al. , The complete sequence of a human genome. Science 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altemose N et al. , Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dfam, Transposable Element Classification; www.dfam.org/classification/tree.
- 14.Piégu B, Bire S, Arensburger P, Bigot Y, A survey of transposable element classification systems—A call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. Evol 86, 90–109 (2015). doi: 10.1016/j.ympev.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 15.Wicker T et al. , A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet 8, 973–982 (2007). doi: 10.1038/nrg2165 [DOI] [PubMed] [Google Scholar]
- 16.Kapitonov VV, Jurka J, A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet 9, 411–412 (2008). doi: 10.1038/nrg2165-c1 [DOI] [PubMed] [Google Scholar]
- 17.International Wheat Genome Sequencing Consortium (IWGSC), Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191 (2018). doi: 10.1126/science.aar7191 [DOI] [PubMed] [Google Scholar]
- 18. See supplementary materials and methods.
- 19.Carey KM, Patterson G, Wheeler TJ, Transposable element subfamily annotation has a reproducibility problem. Mob. DNA 12, 4 (2021). doi: 10.1186/s13100-021-00232-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pajic P et al. , Independent amylase gene copy number bursts correlate with dietary preferences in mammals. eLife 8, e44628 (2019). doi: 10.7554/eLife.44628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gershman A et al. , Epigenetic patterns in a complete human genome. Science 376, eabj5089 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahat DB et al. , Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476 (2016). doi: 10.1038/nprot.2016.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwak H, Fuda NJ, Core LJ, Lis JT, Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953 (2013). doi: 10.1126/science.1229386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wissink EM, Vihervaara A, Tippens ND, Lis JT, Nascent RNA analyses: Tracking transcription and its regulation. Nat. Rev. Genet 20, 705–723 (2019). doi: 10.1038/s41576-019-0159-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kriegs JO, Churakov G, Jurka J, Brosius J, Schmitz J, Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet 23, 158–161 (2007). doi: 10.1016/j.tig.2007.02.002 [DOI] [PubMed] [Google Scholar]
- 26.Thompson PJ, Macfarlan TS, Lorincz MC, Long terminal repeats: From parasitic elements to building blocks of the transcriptional regulatory repertoire. Mol. Cell 62, 766–776 (2016). doi: 10.1016/j.molcel.2016.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raiz J et al. , The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res 40, 1666–1683 (2012). doi: 10.1093/nar/gkr863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hancks DC, Goodier JL, Mandal PK, Cheung LE, Kazazian HH Jr., Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum. Mol. Genet 20, 3386–3400 (2011). doi: 10.1093/hmg/ddr245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancks DC, Kazazian HH Jr., SVA retrotransposons: Evolution and genetic instability. Semin. Cancer Biol 20, 234–245 (2010). doi: 10.1016/j.semcancer.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pontis J et al. , Hominoid-specific transposable elements and KZFPs facilitate human embryonic genome activation and control transcription in naive human ESCs. Cell Stem Cell 24, 724–735.e5 (2019). doi: 10.1016/j.stem.2019.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck CR, Garcia-Perez JL, Badge RM, Moran JV, LINE-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet 12, 187–215 (2011). doi: 10.1146/annurev-genom-082509-141802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swergold GD, Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol. Cell. Biol 10, 6718–6729 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokolowski M, Chynces M, deHaro D, Christian CM, Belancio VP, Truncated ORF1 proteins can suppress LINE-1 retrotransposition in trans. Nucleic Acids Res 45, 5294–5308 (2017). doi: 10.1093/nar/gkx211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brouha B et al. , Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. U.S.A 100, 5280–5285 (2003). doi: 10.1073/pnas.0831042100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fatyol K, Illies K, Szalay AA, Diamond DC, Janish C, Mer22-related sequence elements form pericentric repetitive DNA families in primates. Mol. Gen. Genet 262, 931–939 (2000). doi: 10.1007/PL00008661 [DOI] [PubMed] [Google Scholar]
- 36.Nishiyama R et al. , A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol. Ther 4, 446–454 (2005). doi: 10.4161/cbt.4.4.1622 [DOI] [PubMed] [Google Scholar]
- 37.Thoraval D et al. , Demethylation of repetitive DNA sequences in neuroblastoma. Genes Chromosomes Cancer 17, 234–244 (1996). doi: 10.1002/(SICI)1098-2264(199612)17:4<234::AID-GCC5>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- 38.Tremblay DC, Alexander G Jr., Moseley S, Chadwick BP , Expression, tandem repeat copy number variation and stability of four macrosatellite arrays in the human genome. BMC Genomics 11, 632 (2010). doi: 10.1186/1471-2164-11-632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samuelsson JK et al. , Helicase lymphoid-specific enzyme contributes to the maintenance of methylation of SST1 pericentromeric repeats that are frequently demethylated in colon cancer and associate with genomic damage. Epigenomes 1, 2 (2017). doi: 10.3390/epigenomes1010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Igarashi S et al. , A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin. Cancer Res 16, 5114–5123 (2010). doi: 10.1158/1078-0432.CCR-10-0581 [DOI] [PubMed] [Google Scholar]
- 41.Suzuki K et al. , Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 9, 199–207 (2006). doi: 10.1016/j.ccr.2006.02.016 [DOI] [PubMed] [Google Scholar]
- 42.Nagai H et al. , A novel sperm-specific hypomethylation sequence is a demethylation hotspot in human hepatocellular carcinomas. Gene 237, 15–20 (1999). doi: 10.1016/S0378-1119(99)00322-4 [DOI] [PubMed] [Google Scholar]
- 43.Hartley GA, Okhovat M, O’Neill RJ, Carbone L, Comparative analyses of gibbon centromeres reveal dynamic genus-specific shifts in repeat composition. Mol. Biol. Evol 38, 3972–3992 (2021). doi: 10.1093/molbev/msab148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bird AP, Gene number, noise reduction and biological complexity. Trends Genet 11, 94–100 (1995). doi: 10.1016/S0168-9525(00)89009-5 [DOI] [PubMed] [Google Scholar]
- 45.Yoder JA, Walsh CP, Bestor TH, Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13, 335–340 (1997). doi: 10.1016/S0168-9525(97)01181-5 [DOI] [PubMed] [Google Scholar]
- 46.Ball MP et al. , Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol 27, 361–368 (2009). doi: 10.1038/nbt.1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lister R et al. , Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). doi: 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dumbović G et al. , A novel long non-coding RNA from NBL2 pericentromeric macrosatellite forms a perinucleolar aggregate structure in colon cancer. Nucleic Acids Res 46, 5504–5524 (2018). doi: 10.1093/nar/gky263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlevaro-Fita J et al. , Ancient exapted transposable elements promote nuclear enrichment of human long noncoding RNAs. Genome Res 29, 208–222 (2019). doi: 10.1101/gr.229922.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.González B et al. , Somatic hypomethylation of pericentromeric SST1 repeats and tetraploidization in human colorectal cancer cells. Cancers 13, 5353 (2021). doi: 10.3390/cancers13215353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bobkov GOM, Gilbert N, Heun P, Centromere transcription allows CENP-A to transit from chromatin association to stable incorporation. J. Cell Biol 217, 1957–1972 (2018). doi: 10.1083/jcb.201611087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rošić S, Köhler F, Erhardt S, Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J. Cell Biol 207, 335–349 (2014). doi: 10.1083/jcb.201404097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carone DM et al. , Hypermorphic expression of centromeric retroelement-encoded small RNAs impairs CENP-A loading. Chromosome Res 21, 49–62 (2013). doi: 10.1007/s10577-013-9337-0 [DOI] [PubMed] [Google Scholar]
- 54.McNulty SM, Sullivan LL, Sullivan BA, Human centromeres produce chromosome-specific and array-specific alpha satellite transcripts that are complexed with CENP-A and CENP-C. Dev. Cell 42, 226–240.e6 (2017). doi: 10.1016/j.devcel.2017.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Catania S, Pidoux AL, Allshire RC, Sequence features and transcriptional stalling within centromere DNA promote establishment of CENP-A chromatin. PLOS Genet 11, e1004986 (2015). doi: 10.1371/journal.pgen.1004986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CC et al. , Establishment of centromeric chromatin by the CENP-A assembly factor CAL1 requires FACT-mediated transcription. Dev. Cell 34, 73–84 (2015). doi: 10.1016/j.devcel.2015.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chueh AC, Northrop EL, Brettingham-Moore KH, Choo KHA, Wong LH, LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLOS Genet 5, e1000354 (2009). doi: 10.1371/journal.pgen.1000354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Logsdon GA et al. , The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021). doi: 10.1038/s41586-021-03420-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miga KH et al. , Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). doi: 10.1038/s41586-020-2547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jansen LET, Black BE, Foltz DR, Cleveland DW, Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol 176, 795–805 (2007). doi: 10.1083/jcb.200701066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson WL et al. , RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. eLife 6, e25299 (2017). doi: 10.7554/eLife.25299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chueh AC, Wong LH, Wong N, Choo KHA, Variable and hierarchical size distribution of L1-retroelement-enriched CENP-A clusters within a functional human neocentromere. Hum. Mol. Genet 14, 85–93 (2005). doi: 10.1093/hmg/ddi008 [DOI] [PubMed] [Google Scholar]
- 63.O’Neill RJ, O’Neill MJ, Graves JA, Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393, 68–72 (1998). doi: 10.1038/29985 [DOI] [PubMed] [Google Scholar]
- 64.Klein SJ, O’Neill RJ, Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res 26, 5–23 (2018). doi: 10.1007/s10577-017-9569-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vollger MR et al. , Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rizzo A et al. , Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res 37, 5353–5364 (2009). doi: 10.1093/nar/gkp582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szak ST, Pickeral OK, Landsman D, Boeke JD, Identifying related L1 retrotransposons by analyzing 3′ transduced sequences. Genome Biol 4, R30 (2003). doi: 10.1186/gb-2003-4-5-r30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodier JL, Ostertag EM, Kazazian HH Jr., Transduction of 3′-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet 9, 653–657 (2000). doi: 10.1093/hmg/9.4.653 [DOI] [PubMed] [Google Scholar]
- 69.Pickeral OK, Makałowski W, Boguski MS, Boeke JD, Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res 10, 411–415 (2000). doi: 10.1101/gr.10.4.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Damert A et al. , 5′-Transducing SVA retrotransposon groups spread efficiently throughout the human genome. Genome Res 19, 1992–2008 (2009). doi: 10.1101/gr.093435.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xing J et al. , Emergence of primate genes by retrotransposon-mediated sequence transduction. Proc. Natl. Acad. Sci. U.S.A 103, 17608–17613 (2006). doi: 10.1073/pnas.0603224103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardner EJ et al. , The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res 27, 1916–1929 (2017). doi: 10.1101/gr.218032.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ebert P et al. , Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021). doi: 10.1126/science.abf7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tubio JMC et al. , Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345, 1251343 (2014). doi: 10.1126/science.1251343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pradhan B et al. , Detection of subclonal L1 transductions in colorectal cancer by long-distance inverse-PCR and Nanopore sequencing. Sci. Rep 7, 14521 (2017). doi: 10.1038/s41598-017-15076-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moran JV, DeBerardinis RJ, Kazazian HH Jr., Exon shuffling by L1 retrotransposition. Science 283, 1530–1534 (1999). doi: 10.1126/science.283.5407.1530 [DOI] [PubMed] [Google Scholar]
- 77.Chow JC et al. , LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141, 956–969 (2010). doi: 10.1016/j.cell.2010.04.042 [DOI] [PubMed] [Google Scholar]
- 78.Lyon MF, X-chromosome inactivation: A repeat hypothesis. Cytogenet. Cell Genet 80, 133–137 (1998). doi: 10.1159/000014969 [DOI] [PubMed] [Google Scholar]
- 79.Chaumeil J, Le Baccon P, Wutz A, Heard E, A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20, 2223–2237 (2006). doi: 10.1101/gad.380906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stavropoulos N, Lu N, Lee JT, A functional role for Tsix transcription in blocking Xist RNA accumulation but not in X-chromosome choice. Proc. Natl. Acad. Sci. U.S.A 98, 10232–10237 (2001). doi: 10.1073/pnas.171243598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nora EP et al. , Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). doi: 10.1038/nature11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Bemmel JG et al. , The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat. Genet 51, 1024–1034 (2019). doi: 10.1038/s41588-019-0412-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen X et al. , Loss of X chromosome inactivation in androgenetic complete hydatidiform moles with 46, XX karyotype. Int. J. Gynecol. Pathol 40, 333–341 (2021). doi: 10.1097/PGP.0000000000000697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kronenberg ZN et al. , High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018). doi: 10.1126/science.aar6343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewin HA et al. , Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. U.S.A 115, 4325–4333 (2018). doi: 10.1073/pnas.1720115115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guiblet WM et al. , Selection and thermostability suggest G-quadruplexes are novel functional elements of the human genome. Genome Res 31, 1136–1149 (2021). doi: 10.1101/gr.269589.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roden C, Gladfelter AS, RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol 22, 183–195 (2021). doi: 10.1038/s41580-020-0264-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smit AFA, Hubley R, Green P, RepeatMasker Open-4.0, 2013–2015; www.repeatmasker.org.
- 89.Quinlan AR, Hall IM, BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang L, Lu HHS, Chung W-Y, Yang J, Li W-H, Patterns of segmental duplication in the human genome. Mol. Biol. Evol 22, 135–141 (2005). doi: 10.1093/molbev/msh262 [DOI] [PubMed] [Google Scholar]
- 91.Pagès H, BSgenome: Software infrastructure for efficient representation of full genomes and their SNPs, R package version 1.62.0; https://bioconductor.org/packages/BSgenome.
- 92.Scrucca L, Fop M, Murphy TB, Raftery AE, Mclust 5: Clustering, classification and density estimation using Gaussian finite mixture models. R J 8, 289–317 (2016). doi: 10.32614/RJ-2016-021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xing J et al. , Mobile elements create structural variation: Analysis of a complete human genome. Genome Res 19, 1516–1526 (2009). doi: 10.1101/gr.091827.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khan H, Smit A, Boissinot S, Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res 16, 78–87 (2006). doi: 10.1101/gr.4001406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smit AF, Tóth G, Riggs AD, Jurka J, Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences. J. Mol. Biol 246, 401–417 (1995). doi: 10.1006/jmbi.1994.0095 [DOI] [PubMed] [Google Scholar]
- 96.Furano AV, Duvernell DD, Boissinot S, L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet 20, 9–14 (2004). doi: 10.1016/j.tig.2003.11.006 [DOI] [PubMed] [Google Scholar]
- 97.Konkel MK, Walker JA, Batzer MA, LINEs and SINEs of primate evolution. Evol. Anthropol 19, 236–249 (2010). doi: 10.1002/evan.20283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang H et al. , SVA elements: A hominid-specific retroposon family. J. Mol. Biol 354, 994–1007 (2005). doi: 10.1016/j.jmb.2005.09.085 [DOI] [PubMed] [Google Scholar]
- 99.Freimanis GL, thesis, University of Wolverhampton (2008). [Google Scholar]
- 100.Judd J et al. , A rapid, sensitive, scalable method for Precision Run-On sequencing (PRO-seq). bioRxiv 2020.05.18.102277 [Preprint] (2020). 10.1101/2020.05.18.102277. [DOI] [Google Scholar]
- 101.Martin M, Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12 (2011). doi: 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 102.Fastx-toolkit, FASTQ/A short-reads preprocessing tools; http://hannonlab.cshl.edu/fastx_toolkit.
- 103.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hao Z et al. , RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci 6, e251 (2020). doi: 10.7717/peerj-cs.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krzywinski M et al. , Circos: An information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009). doi: 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ramírez F et al. , deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016). doi: 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Katoh K, Misawa K, Kuma K, Miyata T, MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066 (2002). doi: 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A, RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019). doi: 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nei M, Kumar S, Molecular Evolution and Phylogenetics (Oxford Univ. Press, 2000). [Google Scholar]
- 110.Darriba D, Taboada GL, Doallo R, Posada D, jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 9, 772 (2012). doi: 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Frattini A et al. , High variability of genomic instability and gene expression profiling in different HeLa clones. Sci. Rep 5, 15377 (2015). doi: 10.1038/srep15377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Whitfield ML et al. , Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13, 1977–2000 (2002). doi: 10.1091/mbc.02-02-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hung YH et al. , Chromatin regulatory dynamics of early human small intestinal development using a directed differentiation model. Nucleic Acids Res 49, 726–744 (2021). doi: 10.1093/nar/gkaa1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cartney AMM et al. , Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. bioRxiv 2021.07.02.450803 [Preprint] (2021). 10.1101/2021.07.02.450803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brázda V et al. , G4Hunter web application: A web server for G-quadruplex prediction. Bioinformatics 35, 3493–3495 (2019). doi: 10.1093/bioinformatics/btz087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gordon D et al. , Long-read sequence assembly of the gorilla genome. Science 352, aae0344 (2016). doi: 10.1126/science.aae0344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He Y et al. , Long-read assembly of the Chinese rhesus macaque genome and identification of ape-specific structural variants. Nat. Commun 10, 4233 (2019). doi: 10.1038/s41467-019-12174-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang L et al. , A high-quality genome assembly for the endangered golden snub-nosed monkey (Rhinopithecus roxellana). Gigascience 8, giz098 (2019). doi: 10.1093/gigascience/giz098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jayakumar V et al. , An improved de novo genome assembly of the common marmoset genome yields improved contiguity and increased mapping rates of sequence data. BMC Genomics 21 (suppl. 3), 243 (2020). doi: 10.1186/s12864-020-6657-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Larsen PA et al. , Hybrid de novo genome assembly and centromere characterization of the gray mouse lemur (Microcebus murinus). BMC Biol 15, 110 (2017). doi: 10.1186/s12915-017-0439-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bandyopadhyay R, McQuillan C, Page SL, Choo KH, Shaffer LG, Identification and characterization of satellite III subfamilies to the acrocentric chromosomes. Chromosome Res 9, 223–233 (2001). doi: 10.1023/A:1016648404388 [DOI] [PubMed] [Google Scholar]
- 122.Hoyt SJ et al. , From telomere to telomere: the transcriptional and epigenetic state of human repeat elements analysis code: T2T-CHM13, Zenodo (2022); https://zenodo.org/record/5537106. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.