

Abstract
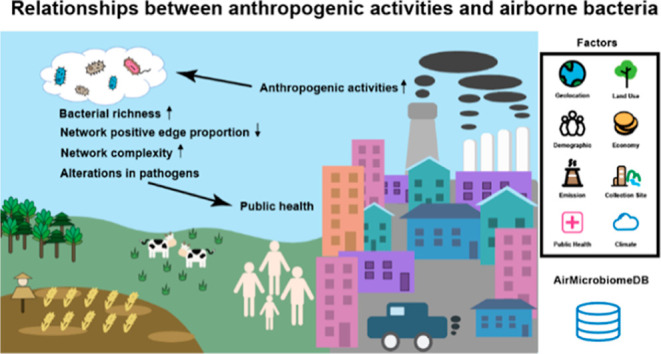
Airborne microbiome alterations, an emerging global health concern, have been linked to anthropogenic activities in numerous studies. However, these studies have not reached a consensus. To reveal general trends, we conducted a meta-analysis using 3226 air samples from 42 studies, including 29 samples of our own. We found that samples in anthropogenic activity-related categories showed increased microbial diversity, increased relative abundance of pathogens, increased co-occurrence network complexity, and decreased positive edge proportions in the network compared with the natural environment category. Most of the above conclusions were confirmed using the samples we collected in a particular period with restricted anthropogenic activities. Additionally, unlike most previous studies, we used 15 human-production process factors to quantitatively describe anthropogenic activities. We found that microbial richness was positively correlated with fine particulate matter concentration, NH3 emissions, and agricultural land proportion and negatively correlated with the gross domestic product per capita. Airborne pathogens showed preferences for different factors, indicating potential health implications. SourceTracker analysis showed that the human body surface was a more likely source of airborne pathogens than other environments. Our results advance the understanding of relationships between anthropogenic activities and airborne bacteria and highlight the role of airborne pathogens in public health.
Keywords: anthropogenic activities, airborne bacteria, pathogens, PM2.5 concentration, NH3 emissions
Short abstract
With large-scale air samples, we used 15 quantitative human production process factors to uncover novel relationships between anthropogenic activities and airborne bacteria from multiple aspects and highlighted the role of airborne pathogens in public health.
Introduction
Bacteria, among the most important components of bioaerosol particles, are ubiquitous in the air with a density of 104 to 108 cells per cubic meter.1,2 In bioaerosols, bacteria can contribute to the formation of cloud condensation nuclei and influence the air quality, weather, and global climate. Additionally, extensive studies have shown that airborne bacteria have profound implications for public health by colonizing the skin, mucous membranes, digestive system, and respiratory tracts.3,4 They can subsequently act as pathogens or triggers to induce various diseases such as cardiovascular diseases,5 respiratory diseases,6 and even lung cancers.7 As a result of their free mobility, airborne bacteria are important in microbial exchange among the biosphere and other environments, such as soil and marine environments,8 thereby facilitating the transfer of antibiotic resistance genes (ARGs), virulence factors, and functional genes.9
Considering the potential importance of airborne bacteria in public health, numerous studies have devoted considerable efforts to investigate factors influencing airborne bacterial composition and biodiversity, including meteorological parameters10,11 and anthropogenic activities.12 However, knowledge of airborne bacterial communities, especially their associations with anthropogenic activities, remains limited due to the inconsistent results yielded by previous studies. For example, several studies13,14 found a positive correlation between airborne bacterial concentration and air pollutant levels, whereas opposite results were reported by others.15,16 After reviewing 11 studies that involved urban–rural comparisons, Flies et al.12 found that seven studies showed higher airborne bacterial abundance in rural areas, while two studies reported the opposite conclusion, and the other two observed no significant difference. Additionally, a few studies have investigated the relationship between airborne microorganisms and other anthropogenic activities, such as land use type,17,18 urban greenness,19 and human occupants.20 However, most studies used qualitative classification to describe anthropogenic activities. Using quantitative factors to characterize anthropogenic activities and assess their associations with airborne bacterial communities remains elusive.
These inconsistent results may be due to limited sample sizes and the wide heterogeneity residing in the airborne microbiome at different geophysical locations and sampling times, which calls for large-scale analysis within a unified process framework. Massive accumulation of air samples using 16S rRNA high-throughput sequencing provides an excellent opportunity to examine the airborne bacterial community globally. To reveal general trends, we collected 3226 air samples from 42 studies, including 29 samples of our own, covering 15 countries from 2006 to 2018. With this data set, we aimed to explore the relationships between airborne microbial communities and anthropogenic activities from three aspects: First, we analyzed the characteristics of airborne microbiomes in environments with different anthropogenic activity intensities. Second, different from qualitative factors used in most previous studies, we described anthropogenic activities with 15 quantitative human-production process factors and explored their associations with airborne bacteria, especially pathogens. Third, we investigated whether pathogens, bacterial toxins, and ARGs in airborne microbial communities are related to anthropogenic sources. Overall, this study advances our understanding of relationships between anthropogenic activities and airborne bacteria and helps elucidate the critical role of airborne pathogens in public health.
Methods
Multi-Study Integration of Air Samples
Studies that met the following criteria were included: (1) focused on airborne microbial communities, (2) used high-throughput amplicon sequencing to target 16S rRNA genes in bacteria, and (3) data were sequenced on Illumina platforms. We excluded several studies due to (1) unavailable raw data, (2) poor data quality, short read length, low read count, or low sequence assignment compared to other studies, or (3) fewer than 10 samples (Figure S1a). Finally, 3197 air samples from 41 qualified studies were collected from the public database, including the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra), the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home), and Figshare (https://figshare.com/).
Sample Collection, DNA Extraction, Polymerase Chain Reaction Amplification, and Sequencing
A total of 29 air samples were collected from the Zhongguancun District (39°59′20.16″N, 116°18′34.47″E, approximately 1.5 m above the ground) next to a traffic road and the Peking University Conservation Biology Building (39°59′50.07″N, 116°18′21.04″E, approximately 4.5 m above the ground). Seventeen samples were collected continuously during and after the 2014 Beijing Asia-Pacific Economic Cooperation (APEC) summit. Approximately, the equal number of samples was collected from both sites. The other 12 samples were collected in 2015. Sampling was conducted using two high-volume air samplers (Laoying Co., Ltd., Qingdao, China) equipped with fine particulate matter (PM2.5) fractionating inlets. Ambient air was drawn in at an average flow rate of 1.05 m3/min for 23 h (21:00 to 20:00 the next day) per sampling day. PM2.5 was collected on 20 × 25 cm2 quartz filters (Whatman Inc., Maidstone, UK). All filters were sterilized in a muffle furnace at 450 °C for 6 h, prior to sampling. Sterilization operations and microbial DNA extraction were conducted according to the protocol reported by Cao et al.21 The V3–V4 region of 16S rRNA genes was amplified by polymerase chain reaction (PCR) (95 °C for 3 min, followed by 27 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, and a final extension at 72 °C for 10 min) using primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′).22,23 Purified amplicons were paired-end-sequenced (2 × 250 bp) on an Illumina MiSeq platform according to standard protocols. Bacterial 16S rRNA gene sequences have been deposited in the NCBI SRA database under accession number PRJNA759379.
Sequencing Data Preprocessing and Identification of Potential Pathogens
Any sequence with an average Phred score <20, containing ambiguous bases after quality trimming, and having ≤75 bp was excluded using QIIME24 version 1.9.1 (Figure S1a). Chimeras were identified using Vsearch25 against the Gold database (http://drive5.com/uchime/gold.fa) and eliminated. Closed reference operational taxonomic units (OTUs) were determined at a 97% similarity level with UCLUST.26 The taxonomy of each 16S rRNA gene sequence was analyzed using the Ribosomal Database Project27 classifier version 2.12 against the Greengenes database (gg_13_5).
We identified potential pathogens in each sample according to the protocol reported by Chen et al.28 High-quality bacterial 16S rRNA gene sequences were blasted against the reference pathogenic sequence database28 with an E-value (<1 × 10–10) and a percentage identity (>99%).
Prediction Analysis of ARGs and Biological Toxins
We implemented PICRUSt29 using QIIME to predict the bacterial functional profiles. PICRUSt takes BIOM-formatted OTU files as input and outputs a table of functional gene counts known as KOs [Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologs]. The nearest sequenced taxon index (NSTI)29 used to evaluate the accuracy of PICRUSt results was 0.11 ± 0.058 (average ± standard deviation) for 3226 air samples, comparable to that in other studies,30,31 suggesting that our PICRUSt results were reliable. We downloaded the lists of ARGs (https://www.genome.jp/kegg/annotation/br01600.html) and biological toxins (https://www.kegg.jp/kegg-bin/show_brite?ko02042.keg) from the KEGG database. After intersecting these lists with the KO table generated using PICRUSt, we obtained 21 ARGs (nine aminoglycoside resistance genes, one beta-lactam resistance gene, five macrolide–lincosamide–streptogramin resistance genes, two tetracycline resistance genes, and four other resistance genes) and 89 biological toxins (three type I toxins: toxins that act from the cell surface, 28 type II toxins: membrane-damaging toxins, 41 type III toxins: intracellular toxins, 4 toxins that damage the extracellular matrix, and 13 not specified).
Identifying Sources of Airborne Microbiomes
To identify the source of 3226 airborne bacterial communities, we constructed an additional source microbiome data set consisting of 16S rRNA sequences from various potential source environments, according to previous studies32,33 and the EMP Ontology of microbial environments.34 Only studies that had a sample size greater than 50 and that met the same criteria as air sample collection were included in the data set. As a result, we obtained a source microbiome data set containing 18 166 samples from 24 different studies, including the human body surface (including the skin surfaces, oral cavities, external auditory canals, inside the nostrils, and hair) (n = 1254), human gut (feces) (n = 3168), animals (n = 823), plants (n = 2726), soil (n = 3751), non-saline water (n = 4316), saline water (n = 942), and WWTPs (n = 1186). These data were analyzed with the same preprocessing workflow in QIIME as air samples.
We then used this source data set as a reference pool for SourceTracker,35 a Bayesian method for predicting the percentage of a “sink” sample from a given set of potential “source” environments. SourceTracker analysis was performed with the default settings. The 18 166 samples from different environments were defined as “source” communities, and the 3226 air samples were used as “sinks”.
Statistical Analyses
Alpha diversity indices were calculated using the “vegan” R package.36 The Wilcoxon rank-sum test was used for two group comparisons. The Benjamini–Hochberg (BH) method was applied to correct for multiple comparisons. All statistical tests were defined as significant at p ≤ 0.05. The correlation matrices between bacteria and factors were constructed by calculating Spearman’s rank correlation coefficients, and significant correlations were considered as follows: |r| ≥ 0.3 and p ≤ 0.05 (BH correction). Principal coordinate analysis (PCoA) was performed to compare the differences in the bacterial community structures, and analysis of similarities (ANOSIM) was performed to test the significance. Box plots and heatmaps were created using R (4.0.3).
Co-occurrence Network Construction
The co-occurrence networks based on the relative abundance of genera were calculated using SparCC37 and visualized with Cytoscape (3.8.0).38 Significant associations were considered as follows: |r| ≥ 0.4 and p ≤ 0.05 (BH correction). The hub genera were determined using the Cytohubba plugin with Maximal Clique Centrality algorithms.39
Data Availability
We summarized the genus profiles, pathogen profiles, KO profiles and corresponding factors of 3226 air samples, the genus profiles and corresponding factors of 18 166 source samples, and the final lists of ARGs and biological toxins in the AirMicrobiomeDB database (http://cqb.pku.edu.cn/ZhuLab/AirMicrobiomeDB/index.html).
Results
Meta-Data Set of the Integrated Airborne Bacterial Communities with Various Factors
After quality control, we acquired 237 878 906 high-quality 16S rRNA gene sequences of 3226 air samples from 42 studies, including 29 samples of our own. Importantly, all sequences were uniformly reprocessed to reduce major nonbiological variances among studies.40 To account for significant differences in sequencing depth among studies, we rarefied the OTU table to 5000 sequences per sample as a previous work41 has encouraged this threshold to be higher than 1000 to ensure the data quality and lower than 10 000 to encompass most samples. These sequences were clustered into 71 phyla, 944 genera, and 40 408 OTUs. We performed this meta-analysis mainly at the genus level, which sacrificed sensitivity to detect fine-scale differences in exchange for less data heterogeneity, thereby alleviating the influence of batch effects.40,42 Airborne bacteria from diverse environments constituted a large “pan-microbiome”, consisting of 944 genera (Table S1). The core-microbiome (defined here as genera shared among more than 90% of the studies) represented only a minority of the microbial composition (77/944, 8.16%, Table S1), indicating the extensive community heterogeneity for airborne bacteria in both space and time.
We further collected many factors for these air samples (supplementary methods and Table S2), including basic information (biosamples, bioprojects, target regions, PCR primers, particle size, particle size groups, and sequencing platforms), geolocation and time information (category, north/south, continent, country, city, longitude, latitude, indoor/outdoor, altitude above the ground level, altitude above the sea level, date, and season), demographic factors (population density, total population, and mortality rate from respiratory diseases), economic factors [gross domestic product (GDP) and GDP per capita], air pollutants (annual PM2.5 concentration and annual anthropogenic emission data of NH3, CO, OC, BC, NMVOC, NOx, CO2, and SO2), land use type (agriculture, forests, grasslands, and settlements), and natural conditions [climate, temperature, specific humidity, and the zonal (U-wind) and meridional (V-wind) components of wind vectors]. The characteristics of 42 studies are provided in Table S3, and the geographical locations are shown in Figure S1b. Large-scale air samples and corresponding factors make it possible to analyze the influence of anthropogenic activities on airborne microbes and the relationship between air microbes and human health from the aspects of demography, economy, air pollutants, and land use types.
Alterations of Air Microbial Communities in Anthropogenic Activity-Related Categories
We classified 3226 air samples into four categories according to their collection sites, including natural environments, human settlements, livestock farms, and wastewater treatment plants (WWTPs), with 390, 1855, 610, and 371 samples, respectively. The natural environment category included samples collected from various environments, such as forests, grasslands, wetlands, deserts, polar regions, oceans, and the free troposphere. The human settlement category included samples from various collection sites in urban, suburban, and rural areas, such as bedrooms, rooftops, and metro stations. Samples in the livestock farm category were collected from diverse animal farms worldwide. The WWTP samples were collected from municipal and pharmaceutical WWTPs. According to their characteristics, the last three categories are regarded as anthropogenic activity-related categories in this meta-analysis. The dominant phyla were similar among these categories, whereas the dominant genera differed (Figure S2a,b). The PCoA and ANOSIM (Figure S2c, R = 0.268 and p < 0.001) results also showed that the clusters of airborne bacterial communities at the genus level in the four categories were significantly different.
We calculated the Chao1 and Shannon indices to estimate the alpha diversity of microbial communities in different categories. The Chao1 index reflects the microbial richness, while the Shannon index measures microbial diversity from both richness and evenness. Compared to anthropogenic activity-related categories, significantly lower Chao1 and Shannon indices were observed in the natural environment category (Figure 1a,b), indicating significantly lower richness and diversity, partly due to the different bioaerosol sources.43 Notably, three studies included samples from both the human settlement and natural environment categories. Two of them (study ID: PRJDB7404 and INHALE) displayed similar trends with significant results (Figure S2d,e, p ≤ 0.05), while the other (study ID: PRJEB17915) showed no significant difference, which might be caused by the experimental setting (fully described in Figure S2 caption).
Figure 1.

Airborne bacterial communities in the four categories. The Chao1 index (a) and Shannon index (b) of the four categories. Asterisks denote Wilcoxon rank-sum test results (ns: p > 0.05, *: p ≤ 0.05, **: p ≤ 0.01, and ****: p ≤ 0.0001). The Wilcoxon rank-sum test works well with unequal sample sizes. (c) Relative abundance of potential pathogens in the four categories. The co-occurrence network in the natural environment (d), human settlement (e), livestock farm (f), and WWTP (g) categories. Natural: The natural environment category; settlement: The human settlement category; livestock: The livestock farms category; WWTP: The wastewater treatment plant category. Red line: positive correlation and blue line: negative correlation.
Exposure to airborne pathogens can cause various diseases, including respiratory inflammation and pulmonary function impairments.44 Thus, investigating potential pathogens in air samples may help understand their threats to human health. Previous work has suggested that the relative abundance of potential pathogens is influenced by anthropogenic activities.45 We found that the proportion (3.07 ± 5.93%) of potential pathogens was significantly lower in the natural environment category than in the three anthropogenic activity-related categories (Figures 1c and S2f, p ≤ 0.05 for all comparisons). The livestock farm category harbored the highest pathogen proportion of 6.82 ± 7.82%, followed by the WWTP category (6.38 ± 11.64%) and the human settlement category (5.78 ± 6.83%). One of the three studies mentioned above (study ID: INHALE) also displayed a significantly lower relative abundance of potential pathogens in the natural environment category than in the human settlement category (Figure S2g, p ≤ 0.05), whereas the other two showed no significant difference.
Studies have shown that the co-occurrence patterns of airborne bacteria, which can reflect possible interactions among airborne bacteria, have significant disparities under different conditions.46,47 We conducted co-occurrence network analysis on airborne microbial taxa in different categories by calculating pairwise correlations. Additionally, we calculated the complexity48 and positive edge proportion (defined as the number of positive edges divided by the total number of edges in the network) to describe the topological properties of the network. The natural environment category had the lowest network complexity (724.43) with 100% positive edge proportions, indicating no negative edges (Figure 1d–g). The three anthropogenic activity-related categories, namely, human settlements, livestock farms, and WWTPs, had network complexities of 750.23, 2241.73, and 3677.00 and positive edge proportions of 98.66, 91.88, and 96.02%, indicating negative edge proportions of 1.34, 8.12, and 3.98%, respectively. As a sparse and open community, the composition and diversity of airborne bacteria are greatly affected by their sources.49,50 We speculate that airborne bacteria with positive edges tend to co-occur and are favorable to a similar origin.47 The results suggested that anthropogenic activities might enhance the complexity of the co-occurrence network and reduce positive edge proportions through introduced microbes from different origins or numerous artificial microenvironments.
People in industrialized countries spend up to 87% of their time indoors.51 The inhalable aerosol microbiomes that permeate indoors and outdoors are likely to affect residents’ health. We further focused on the human settlement category, which consisted of 1018 outdoor samples and 837 indoor samples. Significantly lower richness and diversity and higher proportions of potential pathogens were found in indoor air samples than outdoor samples (Figure S3a-d), consistent with previous studies.52−56 Co-occurrence network analysis revealed that the network complexity of indoor samples (2061.94) was higher than that of outdoor samples (464.16), whereas the positive edge proportion (90.10%) was lower than that of outdoor samples (100%) (Figure S3e,f). Considering that indoor air microbes are primarily derived from outdoors but also include occupant skin and specific indoor environments57−59 (e.g., local tap water), complex sources of indoor air microbiota may be partly responsible for the alterations in co-occurrence network characteristics.
Airborne Microbial Communities Change Dynamically with Anthropogenic Activity Restriction
During the 2014 APEC summit in Beijing, the government implemented strict air quality assurance and emission prevention measures (details described in Supporting Information S1), making it possible to investigate the alterations in airborne bacteria and pathogens when anthropogenic activities were restricted. We collected seven air samples during the APEC summit (5–12 November 2014) and 10 samples after this period (13–17 November 2014). Additionally, we examined other samples in the human settlement category and fortunately found that 12 samples could fill the gaps prior to the APEC summit (30 October to 2 November 2014) with a similar experimental setting. We found that compared to pre- and post-APEC summit samples, microbial communities during the APEC summit had lower richness and diversity and a higher relative abundance of pathogens (Figure 2a–c). The less significant but different trend of pathogens compared to the natural environment and anthropogenic activity-related categories implies a complex relationship between pathogens and anthropogenic activities.60
Figure 2.

Airborne bacterial communities in the APEC summit-related samples. The Chao1 index (a) and Shannon index (b) of the pre-APEC, during APEC, and post-APEC summit samples. The p-values of Wilcoxon rank-sum test results are shown above the boxes. (c) Relative abundance of potential pathogens in pre-APEC, during APEC, and post-APEC summit samples. The hub co-occurrence network in the pre-APEC (d), during APEC (e), and post-APEC (f) summit samples. Red line: positive correlation and gray line: negative correlation.
To investigate the alterations in relationships among microbial taxa under the restriction of anthropogenic activities, we constructed the co-occurrence networks using pre-, during, and post-APEC summit samples. The results showed that compared to samples collected pre- and post-APEC summit, the co-occurrence network of samples collected during the APEC summit had a lower complexity and higher positive edge proportion (complexity: pre-APEC summit vs APEC summit vs post-APEC summit, 16581.16 vs 13044.96 vs 22138.39 and positive edge proportion: 54.45% vs 58.79% vs 54.43%, respectively). The results confirmed our observation of comparing the natural environment and anthropogenic activity-related categories. Furthermore, we found that the hub network of samples collected during the APEC summit was quite different from that of samples collected pre- and post-APEC summit (Figure 2d–f). Before the APEC summit, the hub genera were clearly divided into two subnetworks, with positive edges within subnetworks and negative edges between subnetworks. One subnetwork consisted of eight genera. Most of them belonged to Proteobacteria and were involved in fermentation and degradation. The other subnetwork was composed of 11 genera, of which six belonged to Actinobacteria, and five belonged to Proteobacteria. Most of them are reported to be associated with low-temperature environments and/or stone surfaces, such as Blastococcus,61Janthinobacterium,62,63Kaistobacter,63Arthrobacter,63Psychrobacter,64Sphingomonas,65 and Yonghaparkia.66 During the APEC summit, the hub network was built with all positive edges, including eight hub genera. Most of them (6/8) belonged to Firmicutes. After the APEC summit, the hub network was made up of a large and a small genus subnetwork. Most (10/15) genera of the large subnetwork belonged to Firmicutes, and seven of them overlapped with the hub genera collected during the APEC summit. Half of the genera in the small subnetwork belonged to Actinobacteria.
Association of Human-Production Process Factors with Airborne Bacteria and Pathogens
From the 1855 human settlement category samples, we discarded 278 samples without detailed collection date information. For the remaining 1577 samples, we used the human-production process factors to describe anthropogenic activities, including demographic factors, economic factors, air pollutants, and land use types. Also, we did not neglect the natural conditions in this section. By calculating Spearman’s rank correlation coefficients between microbial richness (reflected by the Chao1 index) and various human-production process factors (Figure 3a), we found that microbial richness was negatively correlated with GDP per capita (r = −0.38 and p = 4.28 × 10–53), similar to the findings in previous studies,67,68 which noted a higher microbial concentration in the low socioeconomic zone than the high socioeconomic zone. On the contrary, microbial richness is positively correlated with PM2.5 concentration (r = 0.42 and p = 3.36 × 10–65), agricultural land proportion (r = 0.48 and p = 4.11 × 10–90), and NH3 emissions (r = 0.40 and p = 1.84 × 10–61). The positive correlation between PM2.5 concentration and microbial richness is supported by the previous studies,43,69,70 possibly because PM can serve as carriers,50 energy sources,47 and refuges43 for airborne bacteria, allowing them to survive. Additionally, Li et al.18 demonstrated that some land types (such as croplands) heavily affected by anthropogenic activities emitted higher airborne bacterial concentration levels than the land types (such as gardens and forests) less affected by humans. The fact that agricultural burning is one of the largest sources of PM2.571 and NH3 emissions72 also supported the above positive correlations. To alleviate the influence of statistical errors in annual data and validate the above correlations, we divided 1577 samples into four levels according to the values of different factors (Figure S4). Level 1 indicated that the values of the corresponding factors were the smallest, whereas those in level 4 were the largest. Microbial richness displayed a significant decrease when the GDP per capita level increased from 1 to 4, whereas it exhibited a noticeable increase when the NH3 and agricultural land proportion levels increased from 1 to 4, in agreement with Spearman’s correlation results. We further analyzed the PM2.5 levels and noticed an increased microbial richness as the PM2.5 levels increased from 1 to 4. The network analysis showed that the positive edge proportion decreased as the PM2.5 levels increased from 1 to 4 (Figure S5, 98.19, 94.55, 94.18, and 78.68%, respectively), while the complexity of co-occurring networks showed a significant increase, except for PM2.5 level 3 (2169.81, 3514.67, 1140.46, and 5987.41, respectively).
Figure 3.

Relationship between airborne bacterial communities and factors. (a) Spearman’s correlation matrix of the microbial diversity (Chao1 index and Shannon index) is associated with different factors. Note: The correlations for the Shannon index are weak (correlation coefficient |r| < 0.3) but have the same trend as that of the Chao1 index. (b) Pairwise Spearman’s correlation matrix of pathogenic genera with factors. Pathogenic genera could be divided into four clusters according to their correlations with factors. Only associations with Spearman’s correlation coefficients |r| ≥ 0.3 and adjust p ≤ 0.05 were retained. Asterisks denote correlation test results (*: adjust p ≤ 0.05).
To determine the correlations between microbial richness and factors across different years and sites, we classified the human settlement category samples according to their collection years and sites. In the year groups, after excluding those with inadequate samples (exclusion criteria: the number of samples in a year did not exceed 200 or did not cover more than three studies), four groups (2014, 2015, 2016, and 2017 groups) remained. In the site groups, the human settlement category samples were primarily collected in China (mainly from Beijing and Hong Kong), the USA, Sweden, and Japan. Therefore, we selected the samples from these five sites. Most of the correlation results of the various year and site groups were consistent with those of the 1577 human settlement-category samples (Figure S6a).
Potential pathogens in aerosols can cause various respiratory diseases. It is crucial to explore the influence of human-production process factors on potential pathogens. In the human settlement category samples, 2.57% of all sequences were identified as potential pathogens and were affiliated with 35 genera (330 species). The relative abundance of potential pathogens was 5.42 ± 6.84% across the human settlement category, comparable with other studies.73 Intriguingly, pathogenic genera could be divided into four distinct clusters according to their correlations with different factors (Figure 3b). Cluster I comprised Acinetobacter, Corynebacterium, and Mycobacterium. These genera were positively related to temperature, specific humidity, and forest proportion. Epidemiological studies have reported community-acquired pneumonia due to Acinetobacter baumannii in tropical regions, especially during warm and humid months.74Corynebacterium and Mycobacteria are reported to survive better in warm and humid environments,75−77 consistent with their positive correlations with temperature and specific humidity. Bacteroides and Salmonella formed cluster II and were positively related to the PM2.5 concentration, agricultural land proportion, and NH3 emissions. The positive correlation between Bacteroides and PM2.5 concentration was supported by a previous study,78 which disclosed that the abundance of Bacteroides sequences was higher on dust event days. Cluster III was made up of Staphylococcus and Serratia. They were positively related to temperature, specific humidity, and factors related to human occupations and air pollutants, such as settlement land proportion, population density, CO2, NMVOC, and SO2 emissions. Staphylococcus was the dominant pathogenic genus (1.12 ± 2.59%) and frequently colonized the skin and mucous membranes of humans. Cluster IV consisted of Bartonella and Listeria. They were positively related to the forest proportion and negatively related to the agricultural land proportion, population density, and most air pollutants (PM2.5 concentration, NMVOC, SO2, CO2, NH3, OC, BC, and NOx emissions), in contrast to cluster III. Bartonella was transmitted by vectors. Listeria monocytogenes, the major human pathogen of the genus Listeria, can be found in soil, water, and plants. These characteristics of Bartonella and Listeria may explain their positive correlation with forests, which contain many plants, soil, and insects. To verify the correlations between pathogenic genus clusters and different factors, we focused on samples in the year and site groups (Figure S7). Although pathogenic genus clusters disappeared in some groups due to low occurrence, the results were roughly the same if the clusters existed. In detail, cluster I, which existed in the 2014, Beijing and USA groups, exhibited the same correlation results as those of 1577 samples, except for the negative correlation with forest proportion in the Beijing group. Cluster II appeared in the 2016 group, cluster III was present in the 2017 and Hong Kong groups, and cluster IV was present in the USA group, of which all agree with the results of 1577 samples.
We further investigated the relationship between the mortality rate from respiratory diseases and potential pathogens. The mortality rate from respiratory diseases was positively correlated with some pathogenic genera, that is, Acinetobacter (r = 0.31 and p = 1.73 × 10–32), Corynebacterium (r = 0.34 and p = 7.10 × 10–40), Mycobacterium (r = 0.47 and p = 3.12 × 10–77), and Staphylococcus (r = 0.37 and p = 1.64 × 10–45). However, it was negatively correlated with Salmonella (r = −0.31 and p = 3.41 × 10–33) and Enterococcus (r = −0.37 and p = 3.78 × 10–46). Acinetobacter species are becoming one of the major causes of nosocomial infections, including hospital-acquired and ventilator-associated pneumonia.79Corynebacterium species are also one of the main causative pathogens of bacterial pneumonia.80 The genus Mycobacterium can cause tuberculosis (Mycobacterium tuberculosis) and Mycobacterium avium complex pulmonary disease.81Staphylococcus aureus is the most typical bacterium that causes community-acquired pneumonia.82 However, Salmonella pneumonia is rare even in immunosuppressed individuals,83,84 and Enterococcus is a rare cause of pneumonia.85 These facts may help to understand their correlations with the mortality rate from respiratory diseases. Additionally, in all year and site groups, we found that the correlations of five groups (namely, the 2014, 2016, 2017, USA, and Hong Kong groups) were almost the same as those of the 1577 human settlement category samples (Figure S6b).
Airborne Bacterial Pathogens are Highly Related to Human Body Surface-Associated Microbes
Bacterial toxins can be biologically toxic to humans, and there is accumulating evidence that ARGs exist and spread through airborne transmission.86,87 It is necessary to investigate the correlations of ARGs, bacterial toxins, and bacterial pathogens with the source of airborne bacteria. To identify the potential source of air samples, we constructed an additional source microbiome data set consisting of 18 166 samples from 24 studies (see “Methods”, Table S4) in various possible source environments, including the human body surface (including the skin surfaces, oral cavities, external auditory canals, inside the nostrils, and hair) (n = 1254), human gut (feces) (n = 3168), animals (n = 823), plants (n = 2726), soil (n = 3751), non-saline water (n = 4316), saline water (n = 942), and WWTPs (n = 1186).
Using these 18 166 samples as the source pool, SourceTracker analysis was performed to determine the potential source of 1577 human settlement category air samples. We found that the relative abundance of potential pathogens was significantly positively correlated with the relative contribution of microbes from the human body surface (r = 0.55 and p = 4.60 × 10–123) rather than the human gut, animals, or other environments (Figure S6c), indicating that airborne pathogens were more likely to from the human body surface. The proportion of airborne ARGs was negatively correlated with the relative contribution of human gut-associated bacteria (r = −0.42 and p = 1.03 × 10–65) and animal-associated bacteria (r = −0.36 and p = 6.51 × 10–49) but showed a positive correlation with that of plant-derived bacteria (r = 0.41 and p = 1.45 × 10–64). On the contrary, the proportions of toxic genes and plant-derived bacteria were negatively correlated (r = −0.34 and p = 4.85 × 10–43). These results suggested that the contribution of plant-derived bacteria to ARGs in aerosols was higher than that of human gut-associated and animal-associated bacteria, while its contribution to toxic genes was limited. These significant correlations were present in most of the year and site groups (Figure S6d).
Discussion
In this work, we collected, reprocessed, and analyzed 3226 air samples from 42 studies. Different from qualitative factors used in most previous studies, we used 15 quantitative human-production process factors to discriminate the intensity of anthropogenic activities. By collecting large-scale air samples and various factors, we established globally generalizable relationships between anthropogenic activities and airborne microbial communities in different environments, whereas past studies generally focused on airborne microbial communities at a specific geographical location for a short period of time and sometimes yielded inconsistent results.
Through comparison between the natural environment and anthropogenic activity-related categories, we found that anthropogenic activities might increase airborne microbial richness and diversity, increase relative abundance of pathogens, enhance co-occurrence network complexity, and decrease positive edge proportions in the network. These discoveries were roughly confirmed by the results of indoor/outdoor comparisons, APEC summit-related samples, and samples with different PM2.5 levels. In the anthropogenic activity-related categories, massive artificial microenvironments owing to high-intensity anthropogenic activities may contribute to diversified airborne bacteria and pathogens, which could explain the higher microbial richness and diversity compared with the natural environment category. We speculate that the complexity source of airborne bacteria is also responsible for the alterations in the co-occurrence network. Airborne bacteria are sparsely distributed in aerosols with high variability, making the microbial source an important factor in community shaping.49,50 We regard the edges between microbes in the network as a reflection of their origins. Positive edges indicate a co-occurrence pattern and are favorable to a similar origin, while negative edges are not, similar to a previous study.47 Additionally, the hub analysis during the APEC summit supported our hypothesis from another aspect. Samples collected during the APEC summit, a particular period with restricted anthropogenic activities, established a totally positively connected hub network. Meanwhile, pre- and post-APEC summit groups had hub networks that could be separated into two subnetworks by negative edges.
Unlike most previous studies, we described anthropogenic activities with 15 quantitative human-production process factors and assessed their relationship with airborne bacteria. We found that microbial richness was negatively correlated with GDP per capita and positively correlated with the PM2.5 concentration, NH3 emissions, and agricultural land proportion. Nine of 35 pathogenic genera exhibited strong associations with different human-production process factors, and they could be divided into four genus clusters according to these associations. We also observed that the mortality rate of respiratory diseases was positively correlated with a few pathogenic genera, including Acinetobacter, Corynebacterium, Mycobacterium, and Staphylococcus. Additionally, the SourceTracker analysis showed that the human body surface was a more likely source of airborne pathogens than other environments. The results of different year and site groups further verified the above observations. Our findings can help people manage daily production more pertinently to prevent public health crises.
This study also has some limitations. We included only air samples based on 16S rRNA gene sequencing and applied PICRUSt to predict functional genes. Although previous studies have validated the reliability of PICRUSt results, including ARGs, using metagenomic shotgun sequences in various environments,88−90 PICRUSt cannot completely cover the full diversity of microorganisms, especially genes located on mobile elements. Due to the consideration of full diversity, metagenomic shotgun sequencing analysis shows great potential,91 but its progress in air microbiome has been relatively slow, partly owing to the limited availability of collectible atmospheric PM and the low DNA content it contains.92 Cross-comparisons between the results of PICRUSt and metagenomic shotgun sequencing are necessary in future studies to confirm the validity of PICRUSt on the air microbiome. Another potential limitation arises from technical biases among different air microbiome data sets. We performed ANOSIM tests to evaluate the effects of environmental factors and technical bias factors on the air bacterial community structure (Table S5). We found significant effects of technical biases (i.e., primer and particle size biases), which were lower than the effects of categories, climates, and countries, but higher than those of seasons, indicating that technical biases did not overshadow environmental variations. However, a lack of standardized protocols and methods might hinder our deeper understanding of the air microbiome.43,92 Further development of standardized protocols and methods, metagenomic shotgun sequencing, and novel bioinformatics tools, such as sequencing data processing methods93−95 and microbial community models,96 is critical to improve the comparability and reproducibility of airborne microbial research and is expected to explore more hidden patterns and provide additional information on the air microbiome and public health surveillance.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2021YFC2300300 and 2017YFC1200205) and the National Natural Science Foundation of China (32070667 and 31671366). Part of the analysis was performed on a High-Performance Computing Platform of Peking University. We acknowledge ESA and C3S for land cover products and NOAA/OAR/ESRL/PSL for NCEP/DOE 2 reanalysis data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c07923.
Policies during the APEC summit; methods for factor collection; multi-study integration workflow; differences among air bacteria in the four categories; airborne bacterial communities in indoor/outdoor samples; comparison of the airborne microbial richness at different levels of factors; co-occurrence networks of air samples with different levels of PM2.5; association analysis of airborne microbial communities; association analysis of pathogenic genus clusters and factors (PDF)
Pan- and core-microbiome of airborne bacteria in the 3226 air samples (XLSX)
Metadata of the 3226 air samples (XLSX)
Studies included in this meta-analysis (XLSX)
Metadata of the source samples (XLSX)
Factors associated with airborne bacterial community structure using ANOSIM (999 permutations) of Bray-Curtis dissimilarities (XLSX)
Author Contributions
# X.Q.J, C.H.W, and J.Y.G. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Bowers R. M.; Lauber C. L.; Wiedinmyer C.; Hamady M.; Hallar A. G.; Fall R.; Knight R.; Fierer N. Characterization of airborne microbial communities at a high-elevation site and their potential to act as atmospheric ice nuclei. Appl. Environ. Microbiol. 2009, 75, 5121–5130. 10.1128/aem.00447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers R. M.; McLetchie S.; Knight R.; Fierer N. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 2011, 5, 601–612. 10.1038/ismej.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert C.; Ravard Helffer K.; Lebaron P. Skin Microbiome and its interplay with the Environment. Am. J. Clin. Dermatol. 2020, 21, 4–11. 10.1007/s40257-020-00551-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man W. H.; de Steenhuijsen Piters W. A. A.; Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. 10.1038/nrmicro.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs D. W.; Yeager R. A.; Bhatnagar A. Defining the human envirome: an omics approach for assessing the environmental risk of cardiovascular Disease. Circ. Res. 2018, 122, 1259–1275. 10.1161/circresaha.117.311230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y.; Chu J.; Li Y.; Kong X. The influence of air pollution on respiratory microbiome: A link to respiratory disease. Toxicol. Lett. 2020, 334, 14–20. 10.1016/j.toxlet.2020.09.007. [DOI] [PubMed] [Google Scholar]
- Walser S. M.; Gerstner D. G.; Brenner B.; Bünger J.; Eikmann T.; Janssen B.; Kolb S.; Kolk A.; Nowak D.; Raulf M.; Sagunski H.; Sedlmaier N.; Suchenwirth R.; Wiesmüller G.; Wollin K.-M.; Tesseraux I.; Herr C. E. W. Evaluation of exposure-response relationships for health effects of microbial bioaerosols - A systematic review. Int. J. Hyg Environ. Health 2015, 218, 577–589. 10.1016/j.ijheh.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang Q.; Guo X.; Liu L.; Guo J.; Yao J.; Zhu H. Functional genomic analysis of Hawaii marine metagenomes. Sci. Bull. 2015, 60, 348–355. 10.1007/s11434-014-0658-y. [DOI] [Google Scholar]
- Rahav E.; Paytan A.; Chien C.-T.; Ovadia G.; Katz T.; Herut B. The impact of atmospheric dry deposition associated microbes on the southeastern mediterranean sea surface water following an intense dust storm. Front. Mar. Sci. 2016, 3, 127. 10.3389/fmars.2016.00127. [DOI] [Google Scholar]
- Rao Y.; Li H.; Chen M.; Huang K.; Chen J.; Xu J.; Zhuang G. Community structure and influencing factors of airborne microbial aerosols over three Chinese cities with contrasting social-economic levels. Atmosphere 2020, 11, 317. 10.3390/atmos11040317. [DOI] [Google Scholar]
- Dong L.; Qi J.; Shao C.; Zhong X.; Gao D.; Cao W.; Gao J.; Bai R.; Long G.; Chu C. Concentration and size distribution of total airborne microbes in hazy and foggy weather. Sci. Total Environ. 2016, 541, 1011–1018. 10.1016/j.scitotenv.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Flies E. J.; Clarke L. J.; Brook B. W.; Jones P. Urbanisation reduces the abundance and diversity of airborne microbes - but what does that mean for our health? A systematic review. Sci. Total Environ. 2020, 738, 140337. 10.1016/j.scitotenv.2020.140337. [DOI] [PubMed] [Google Scholar]
- Li W.; Yang J.; Zhang D.; Li B.; Wang E.; Yuan H. Concentration and community of airborne bacteria in response to cyclical haze events during the fall and midwinter in Beijing, China. Front. Microbiol. 2018, 9, 1741. 10.3389/fmicb.2018.01741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.; Li Y.; Li W.; Xie Z.; Fan C.; Liu P.; Deng S. Bacterial community structure in atmospheric particulate matters of different sizes during the haze days in Xi’an, China. Sci. Total Environ. 2018, 637–638, 244–252. 10.1016/j.scitotenv.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Gao M.; Qiu T.; Jia R.; Han M.; Song Y.; Wang X. Concentration and size distribution of viable bioaerosols during non-haze and haze days in Beijing. Environ. Sci. Pollut. Res. Int. 2015, 22, 4359–4368. 10.1007/s11356-014-3675-0. [DOI] [PubMed] [Google Scholar]
- Xu C.; Wei M.; Chen J.; Wang X.; Zhu C.; Li J.; Zheng L.; Sui G.; Li W.; Wang W.; Zhang Q.; Mellouki A. Bacterial characterization in ambient submicron particles during severe haze episodes at Ji’nan, China. Sci. Total Environ. 2017, 580, 188–196. 10.1016/j.scitotenv.2016.11.145. [DOI] [PubMed] [Google Scholar]
- Docherty K. M.; Pearce D. S.; Lemmer K. M.; Hale R. L. Distributing regionally, distinguishing locally: examining the underlying effects of local land use on airborne bacterial biodiversity. Environ. Microbiol. 2018, 20, 3529–3542. 10.1111/1462-2920.14307. [DOI] [PubMed] [Google Scholar]
- Li X.; Chen H.; Yao M. Microbial emission levels and diversities from different land use types. Environ. Int. 2020, 143, 105988. 10.1016/j.envint.2020.105988. [DOI] [PubMed] [Google Scholar]
- Li H.; Wu Z.-F.; Yang X.-R.; An X.-L.; Ren Y.; Su J.-Q. Urban greenness and plant species are key factors in shaping air microbiomes and reducing airborne pathogens. Environ. Int. 2021, 153, 106539. 10.1016/j.envint.2021.106539. [DOI] [PubMed] [Google Scholar]
- Adams R. I.; Bhangar S.; Pasut W.; Arens E. A.; Taylor J. W.; Lindow S. E.; Nazaroff W. W.; Bruns T. D. Chamber bioaerosol study: outdoor air and human occupants as sources of indoor airborne microbes. PLoS One 2015, 10, e0128022 10.1371/journal.pone.0128022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C.; Jiang W.; Wang B.; Fang J.; Lang J.; Tian G.; Jiang J.; Zhu T. F. Inhalable Microorganisms in Beijing’s PM2.5 and PM10 Pollutants during a Severe Smog Event. Environ. Sci. Technol. 2014, 48, 1499–1507. 10.1021/es4048472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N.; Tan G.; Wang H.; Gai X. Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. Eur. J. Soil Biol. 2016, 74, 1–8. 10.1016/j.ejsobi.2016.02.004. [DOI] [Google Scholar]
- Mori H.; Maruyama F.; Kato H.; Toyoda A.; Dozono A.; Ohtsubo Y.; Nagata Y.; Fujiyama A.; Tsuda M.; Kurokawa K. Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 2014, 21, 217–227. 10.1093/dnares/dst052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G.; Kuczynski J.; Stombaugh J.; Bittinger K.; Bushman F. D.; Costello E. K.; Fierer N.; Peña A. G.; Goodrich J. K.; Gordon J. I.; Huttley G. A.; Kelley S. T.; Knights D.; Koenig J. E.; Ley R. E.; Lozupone C. A.; McDonald D.; Muegge B. D.; Pirrung M.; Reeder J.; Sevinsky J. R.; Turnbaugh P. J.; Walters W. A.; Widmann J.; Yatsunenko T.; Zaneveld J.; Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T.; Flouri T.; Nichols B.; Quince C.; Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 2016, 4, e2584 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Garrity G. M.; Tiedje J. M.; Cole J. R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. 10.1128/aem.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; An X.; Li H.; Su J.; Ma Y.; Zhu Y.-G. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ. Int. 2016, 92–93, 1–10. 10.1016/j.envint.2016.03.026. [DOI] [PubMed] [Google Scholar]
- Langille M. G. I.; Zaneveld J.; Caporaso J. G.; McDonald D.; Knights D.; Reyes J. A.; Clemente J. C.; Burkepile D. E.; Vega Thurber R. L.; Knight R.; Beiko R. G.; Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoisington A.; Maestre J. P.; Kinney K. A.; Siegel J. A. Characterizing the bacterial communities in retail stores in the United States. Indoor Air 2016, 26, 857–868. 10.1111/ina.12273. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Pan X.; Xiao H.; Xiao H. Structural Characteristics and Functional Implications of PM2.5 Bacterial Communities During Fall in Beijing and Shanghai, China. Front. Microbiol. 2019, 10, 2369. 10.3389/fmicb.2019.02369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams R. I.; Bateman A. C.; Bik H. M.; Meadow J. F. Microbiota of the indoor environment: a meta-analysis. Microbiome 2015, 3, 49. 10.1186/s40168-015-0108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N.; Liang P.; Wu C.; Wang G.; Xu Q.; Xiong X.; Wang T.; Zolfo M.; Segata N.; Qin H.; Knight R.; Gilbert J. A.; Zhu T. F. Longitudinal survey of microbiome associated with particulate matter in a megacity. Genome Biol. 2020, 21, 55. 10.1186/s13059-020-01964-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. R.; Sanders J. G.; McDonald D.; Amir A.; Ladau J.; Locey K. J.; Prill R. J.; Tripathi A.; Gibbons S. M.; Ackermann G.; Navas-Molina J. A.; Janssen S.; Kopylova E.; Vázquez-Baeza Y.; González A.; Morton J. T.; Mirarab S.; Zech Xu Z.; Jiang L.; Haroon M. F.; Kanbar J.; Zhu Q.; Jin Song S.; Kosciolek T.; Bokulich N. A.; Lefler J.; Brislawn C. J.; Humphrey G.; Owens S. M.; Hampton-Marcell J.; Berg-Lyons D.; McKenzie V.; Fierer N.; Fuhrman J. A.; Clauset A.; Stevens R. L.; Shade A.; Pollard K. S.; Goodwin K. D.; Jansson J. K.; Gilbert J. A.; Knight R. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D.; Kuczynski J.; Charlson E. S.; Zaneveld J.; Mozer M. C.; Collman R. G.; Bushman F. D.; Knight R.; Kelley S. T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J.; Blanchet F. G.; Kindt R.; Legendre P.; Minchin P. R.; O’hara R. B.; Simpson G. L.; Solymos P.; Stevens M. H. H.; Wagner H.. vegan: Community ecology package. https://CRAN.R-project.org/package=vegan (accessed May 01, 2021).
- Friedman J.; Alm E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R.; Smoot M. E.; Ono K.; Ruscheinski J.; Wang P. L.; Lotia S.; Pico A. R.; Bader G. D.; Ideker T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin C. H.; Chen S. H.; Wu H. H.; Ho C. W.; Ko M. T.; Lin C. Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J.; Kim P. J.; Ma S.; Funk C. C.; Magis A. T.; Wang Y.; Hood L.; Geman D.; Price N. D. Multi-study integration of brain cancer transcriptomes reveals organ-level molecular signatures. PLoS Comput. Biol. 2013, 9, e1003148 10.1371/journal.pcbi.1003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E. S.; Schmidt P. J.; Tremblay B. J.-M.; Emelko M. B.; Müller K. M. Enhancing diversity analysis by repeatedly rarefying next generation sequencing data describing microbial communities. Sci. Rep. 2021, 11, 22302. 10.1038/s41598-021-01636-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsó E. Microbiome in chronic obstructive pulmonary disease. Ann. Transl. Med. 2017, 5, 251. 10.21037/atm.2017.04.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Gil T.; Acuña J. J.; Fujiyoshi S.; Tanaka D.; Noda J.; Maruyama F.; Jorquera M. A. Airborne bacterial communities of outdoor environments and their associated influencing factors. Environ. Int. 2020, 145, 106156. 10.1016/j.envint.2020.106156. [DOI] [PubMed] [Google Scholar]
- Fan C.; Li Y.; Liu P.; Mu F.; Xie Z.; Lu R.; Qi Y.; Wang B.; Jin C. Characteristics of airborne opportunistic pathogenic bacteria during autumn and winter in Xi’an, China. Sci. Total Environ. 2019, 672, 834–845. 10.1016/j.scitotenv.2019.03.412. [DOI] [PubMed] [Google Scholar]
- Hosseini P. R.; Mills J. N.; Prieur-Richard A. H.; Ezenwa V. O.; Bailly X.; Rizzoli A.; Suzán G.; Vittecoq M.; García-Peña G. E.; Daszak P.; Guégan J. F.; Roche B. Does the impact of biodiversity differ between emerging and endemic pathogens? The need to separate the concepts of hazard and risk. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372, 20160129. 10.1098/rstb.2016.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.-Y.; Gao J.-F.; Pan K.-L.; Li D.-C.; Dai H.-H.; Li X. More obvious air pollution impacts on variations in bacteria than fungi and their co-occurrences with ammonia-oxidizing microorganisms in PM2.5. Environ. Pollut. 2019, 251, 668–680. 10.1016/j.envpol.2019.05.004. [DOI] [PubMed] [Google Scholar]
- Wei M.; Xu C.; Xu X.; Zhu C.; Li J.; Lv G. Characteristics of atmospheric bacterial and fungal communities in PM2.5 following biomass burning disturbance in a rural area of North China Plain. Sci. Total Environ. 2019, 651, 2727–2739. 10.1016/j.scitotenv.2018.09.399. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Jiang X.; Ni C.; Li L.; Chen L.; Wang Y.; Li M.; Wang C.; Gao L.; Zhu H.; Song J. Gut microbiota-related effects of Tanhuo decoction in acute ischemic stroke. Oxid. Med. Cell. Longev. 2021, 2021, 5596924. 10.1155/2021/5596924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smets W.; Moretti S.; Denys S.; Lebeer S. Airborne bacteria in the atmosphere: Presence, purpose, and potential. Atmos. Environ. 2016, 139, 214–221. 10.1016/j.atmosenv.2016.05.038. [DOI] [Google Scholar]
- Zhai Y.; Li X.; Wang T.; Wang B.; Li C.; Zeng G. A review on airborne microorganisms in particulate matters: Composition, characteristics and influence factors. Environ. Int. 2018, 113, 74–90. 10.1016/j.envint.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Klepeis N. E.; Nelson W. C.; Ott W. R.; Robinson J. P.; Tsang A. M.; Switzer P.; Behar J. V.; Hern S. C.; Engelmann W. H. The National Human Activity Pattern Survey (NHAPS): a resource for assessing exposure to environmental pollutants. J. Expo. Anal. Environ. Epidemiol. 2001, 11, 231–252. 10.1038/sj.jea.7500165. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Niu M.; Zheng Y.; Sun Y.; Wu Y.; Zhu T.; Shen F. Impact of outdoor air on indoor airborne microbiome under hazy air pollution: A case study in winter Beijing. J. Aerosol Sci. 2021, 156, 105798. 10.1016/j.jaerosci.2021.105798. [DOI] [Google Scholar]
- Richardson M.; Gottel N.; Gilbert J. A.; Gordon J.; Gandhi P.; Reboulet R.; Hampton-Marcell J. T. Concurrent measurement of microbiome and allergens in the air of bedrooms of allergy disease patients in the Chicago area. Microbiome 2019, 7, 82. 10.1186/s40168-019-0695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. G.; Yang J. I.; Kim E.; Geum S. W.; Park J. H.; Yeo M. K. Investigation of bacterial and fungal communities in indoor and outdoor air of elementary school classrooms by 16S rRNA gene and ITS region sequencing. Indoor Air 2021, 31, 1553–1562. 10.1111/ina.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kembel S. W.; Jones E.; Kline J.; Northcutt D.; Stenson J.; Womack A. M.; Bohannan B. J.; Brown G. Z.; Green J. L. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 2012, 6, 1469–1479. 10.1038/ismej.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams R. I.; Miletto M.; Lindow S. E.; Taylor J. W.; Bruns T. D. Airborne bacterial communities in residences: similarities and differences with fungi. PLoS One 2014, 9, e91283 10.1371/journal.pone.0091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Lai Y.; Tong X.; Leung M. H. Y.; Tong J. C. K.; Ridley I. A.; Lee P. K. H. Airborne bacteria in outdoor air and air of mechanically ventilated buildings at city scale in Hong Kong across seasons. Environ. Sci. Technol. 2020, 54, 11732–11743. 10.1021/acs.est.9b07623. [DOI] [PubMed] [Google Scholar]
- Meadow J. F.; Altrichter A. E.; Kembel S. W.; Kline J.; Mhuireach G.; Moriyama M.; Northcutt D.; O’Connor T. K.; Womack A. M.; Brown G. Z.; Green J. L.; Bohannan B. J. M. Indoor airborne bacterial communities are influenced by ventilation, occupancy, and outdoor air source. Indoor Air 2014, 24, 41–48. 10.1111/ina.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletto M.; Lindow SE Relative and contextual contribution of different sources to the composition and abundance of indoor air bacteria in residences. Microbiome 2015, 3, 61. 10.1186/s40168-015-0128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P.; Du R.; Ren W.; Lu Z.; Fu P. Seasonal variation characteristic of inhalable microbial communities in PM2.5 in Beijing city, China. Sci. Total Environ. 2018, 610–611, 308–315. 10.1016/j.scitotenv.2017.07.097. [DOI] [PubMed] [Google Scholar]
- Sghaier H.; Hezbri K.; Ghodhbane-Gtari F.; Pujic P.; Sen A.; Daffonchio D.; Boudabous A.; Tisa L. S.; Klenk H. P.; Armengaud J.; Normand P.; Gtari M. Stone-dwelling actinobacteria Blastococcus saxobsidens, Modestobacter marinus and Geodermatophilus obscurus proteogenomes. ISME J. 2016, 10, 21–29. 10.1038/ismej.2015.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z.-P.; Tian F.; Roux S.; Gazitúa M. C.; Solonenko N. E.; Li Y.-F.; Davis M. E.; Van Etten J. L.; Mosley-Thompson E.; Rich V. I.; Sullivan M. B.; Thompson L. G. Glacier ice archives nearly 15,000-year-old microbes and phages. Microbiome 2021, 9, 160. 10.1186/s40168-021-01106-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali P.; Chen F.; Hassan F.; Sosa A.; Khan S.; Badshah M.; Shah A. A. Bacterial community characterization of Batura Glacier in the Karakoram Range of Pakistan. Int. Microbiol. 2021, 24, 183–196. 10.1007/s10123-020-00153-x. [DOI] [PubMed] [Google Scholar]
- Dias LM; Folador ARC; Oliveira AM; Ramos RTJ; Silva A.; Baraúna RA Genomic Architecture of the two cold-adapted genera exiguobacterium and psychrobacter: evidence of functional reduction in the exiguobacterium antarcticum B7 Genome. Genome Biol. Evol. 2018, 10, 731–741. 10.1093/gbe/evy029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louati M.; Ennis N. J.; Ghodhbane-Gtari F.; Hezbri K.; Sevigny J. L.; Fahnestock M. F.; Cherif-Silini H.; Bryce J. G.; Tisa L. S.; Gtari M. Elucidating the ecological networks in stone-dwelling microbiomes. Environ. Microbiol. 2020, 22, 1467–1480. 10.1111/1462-2920.14700. [DOI] [PubMed] [Google Scholar]
- Włodarczyk A.; Lirski M.; Fogtman A.; Koblowska M.; Bidziński G.; Matlakowska R. The oxidative metabolism of fossil hydrocarbons and sulfide minerals by the lithobiontic microbial community inhabiting deep subterrestrial kupferschiefer black shale. Front. Microbiol. 2018, 9, 972. 10.3389/fmicb.2018.00972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balyan P.; Ghosh C.; Das S.; Banerjee B. Spatio-temporal variations of indoor bioaerosols in different socio-economic zones of an urban metropolis. Pol. J. Environ. Stud. 2019, 28, 4087–4097. 10.15244/pjoes/81272. [DOI] [Google Scholar]
- Nasir Z. A.; Colbeck I.; Sultan S.; Ahmed S. Bioaerosols in residential micro-environments in low income countries: a case study from Pakistan. Environ. Pollut. 2012, 168, 15–22. 10.1016/j.envpol.2012.03.047. [DOI] [PubMed] [Google Scholar]
- Yu R.; Wang S.; Wu X.; Shen L.; Liu Y.; Li J.; Qiu G.; Zeng W. Community structure variation associated with airborne particulate matter at central south of China during hazy and nonhazy days. Atmos. Pollut. Res. 2020, 11, 162–169. 10.1016/j.apr.2019.05.016. [DOI] [Google Scholar]
- Bai W.; Li Y.; Xie W.; Ma T.; Hou J.; Zeng X. Vertical variations in the concentration and community structure of airborne microbes in PM2.5. Sci. Total Environ. 2021, 760, 143396. 10.1016/j.scitotenv.2020.143396. [DOI] [PubMed] [Google Scholar]
- Johnston H. J.; Mueller W.; Steinle S.; Vardoulakis S.; Tantrakarnapa K.; Loh M.; Cherrie J. W. How harmful is particulate matter emitted from biomass burning? A Thailand perspective. Curr. Pollut. Rep. 2019, 5, 353–377. 10.1007/s40726-019-00125-4. [DOI] [Google Scholar]
- Behera S. N.; Sharma M.; Aneja V. P.; Balasubramanian R. Ammonia in the atmosphere: a review on emission sources, atmospheric chemistry and deposition on terrestrial bodies. Environ. Sci. Pollut. Res. Int. 2013, 20, 8092–8131. 10.1007/s11356-013-2051-9. [DOI] [PubMed] [Google Scholar]
- Li H.; Zhou X.-Y.; Yang X.-R.; Zhu Y.-G.; Hong Y.-W.; Su J.-Q. Spatial and seasonal variation of the airborne microbiome in a rapidly developing city of China. Sci. Total Environ. 2019, 665, 61–68. 10.1016/j.scitotenv.2019.01.367. [DOI] [PubMed] [Google Scholar]
- Kanafani Z.; Kanj S.. Acinetobacter Infection: Treatment and Prevention; Ministry of Health, Kingdome of Saudi Arabia, 2015. [Google Scholar]
- Walsh C. M.; Gebert M. J.; Delgado-Baquerizo M.; Maestre F. T.; Fierer N. A Global Survey of Mycobacterial Diversity in Soil. Appl. Environ. Microbiol. 2019, 85, e01180-19 10.1128/AEM.01180-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevots D. R.; Marras T. K. Epidemiology of Human Pulmonary Infection with Nontuberculous Mycobacteria. Clin. Chest Med. 2015, 36, 13–34. 10.1016/j.ccm.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forouzan P.; Cohen PR Erythrasma revisited: diagnosis, differential diagnoses, and comprehensive review of treatment. Cureus 2020, 12, e10733 10.7759/cureus.10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd Aziz A.; Lee K.; Park B.; Park H.; Park K.; Choi I.-G.; Chang I. S. Comparative study of the airborne microbial communities and their functional composition in fine particulate matter (PM2.5) under non-extreme and extreme PM2.5 conditions. Atmos. Environ. 2018, 194, 82–92. 10.1016/j.atmosenv.2018.09.027. [DOI] [Google Scholar]
- Hartzell J. D.; Kim A. S.; Kortepeter M. G.; Moran K. A. Acinetobacter pneumonia: a review. MedGenMed. 2007, 9, 4. [PMC free article] [PubMed] [Google Scholar]
- Yatera K.; Mukae H. Corynebacterium species as one of the major causative pathogens of bacterial pneumonia. Respir. Invest. 2020, 58, 131–133. 10.1016/j.resinv.2020.01.008. [DOI] [PubMed] [Google Scholar]
- Kwon Y.-S.; Koh W.-J.; Daley C. L. Treatment ofMycobacterium aviumComplex Pulmonary Disease. Tuberc. Respir. Dis. 2019, 82, 15–26. 10.4046/trd.2018.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster T.; Barron S.; Teake R.; James D.; Susman M.; Kennedy C.; Singleton M.; Schuenke S.. Staphylococcus in Medical Microbiology, 4th ed. University of Texas Medical Branch at Galveston: Galveston (TX), 1996. [Google Scholar]
- Saeed N. K. Salmonella pneumonia complicated with encysted empyema in an immunocompromised youth: case report and literature review. J. Infect. Dev. Ctries. 2016, 10, 437–444. 10.3855/jidc.7069. [DOI] [PubMed] [Google Scholar]
- Azher M.; El-Kassimi F.; Chagla A. H. Pneumonia due to Salmonella typhimurium. Ann. Saudi Med. 1991, 11, 341–342. 10.5144/0256-4947.1991.341. [DOI] [PubMed] [Google Scholar]
- Li F.; Wang Y.; Sun L.; Wang X. Vancomycin-resistant Enterococcus faecium pneumonia in a uremic patient on hemodialysis: a case report and review of the literature. BMC Infect. Dis. 2020, 20, 167. 10.1186/s12879-020-4892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald T. L.Elsevier’s Integrated Review Immunology and Microbiology, Ed., 2nd ed.; Actor J. K.; Elsevier B.V, 2014; Vol. 23, p 379. [Google Scholar]
- Li N.; Chai Y.; Ying G.-G.; Jones K. C.; Deng W.-J. Airborne antibiotic resistance genes in Hong Kong kindergartens. Environ. Pollut. 2020, 260, 114009. 10.1016/j.envpol.2020.114009. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Shen L.; Ju Z.; Fu Y.; Qin S.; Cui J. The key environmental influencing factors for the change of sediment bacterial community and antibiotics resistance genes in a long-term polluted lake, China. Ecotoxicology 2021, 30, 1538–1549. 10.1007/s10646-020-02309-x. [DOI] [PubMed] [Google Scholar]
- Zaura E.; Brandt B. W.; Teixeira de Mattos M. J.; Buijs M. J.; Caspers M. P.; Rashid M.-U.; Weintraub A.; Nord C. E.; Savell A.; Hu Y.; Coates A. R.; Hubank M.; Spratt D. A.; Wilson M.; Keijser B. J.; Crielaard W. Same exposure but two radically different responses to antibiotics: resilience of the salivary microbiome versus long-term microbial shifts in feces. mBio 2015, 6, e01693 10.1128/mBio.01693-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šamanić I.; Kalinić H.; Fredotović Ž.; Dželalija M.; Bungur A.-M.; Maravić A. Bacteria tolerant to colistin in coastal marine environment: Detection, microbiome diversity and antibiotic resistance genes’ repertoire. Chemosphere 2021, 281, 130945. 10.1016/j.chemosphere.2021.130945. [DOI] [PubMed] [Google Scholar]
- Chen P.; Guo X.; Li F. Antibiotic resistance genes in bioaerosols: Emerging, non-ignorable and pernicious pollutants. J. Clean. Prod. 2022, 348, 131094. 10.1016/j.jclepro.2022.131094. [DOI] [Google Scholar]
- Behzad H.; Gojobori T.; Mineta K. Challenges and opportunities of airborne metagenomics. Genome Biol. Evol. 2015, 7, 1216–1226. 10.1093/gbe/evv064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B.; Wang F.; Wang X.; Duan L.; Zhu H. InteMAP: Integrated metagenomic assembly pipeline for NGS short reads. BMC Bioinf. 2015, 16, 244. 10.1186/s12859-015-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.-Q.; Guo J.-T.; Liu Y.-C.; Zhu H. MetaTISA: Metagenomic Translation Initiation Site Annotator for improving gene start prediction. Bioinformatics 2009, 25, 1843–1845. 10.1093/bioinformatics/btp272. [DOI] [PubMed] [Google Scholar]
- Lai B.; Ding R.; Li Y.; Duan L.; Zhu H. A de novo metagenomic assembly program for shotgun DNA reads. Bioinformatics 2012, 28, 1455–1462. 10.1093/bioinformatics/bts162. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Li X.; Yang L.; Liu C.; Wang Q.; Chi W.; Zhu H. How microbes shape their communities? A microbial community model based on functional genes. Genomics, Proteomics Bioinf. 2019, 17, 91–105. 10.1016/j.gpb.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We summarized the genus profiles, pathogen profiles, KO profiles and corresponding factors of 3226 air samples, the genus profiles and corresponding factors of 18 166 source samples, and the final lists of ARGs and biological toxins in the AirMicrobiomeDB database (http://cqb.pku.edu.cn/ZhuLab/AirMicrobiomeDB/index.html).