

Abstract
Major depressive disorder (MDD) is a highly prevalent and disabling disorder. Despite the many hypotheses proposed to understand the molecular pathophysiology of depression, it is still unclear. Current treatments for depression are inadequate for many individuals, because of limited effectiveness, delayed efficacy (usually two weeks), and side effects. Consequently, novel drugs with increased speed of action and effectiveness are required. Ketamine has shown to have rapid, reliable, and long‐lasting antidepressant effects in treatment‐resistant MDD patients and represent a breakthrough therapy for patients with MDD; however, concerns regarding its efficacy, potential misuse, and side effects remain. In this review, we aimed to summarize molecular mechanisms and pharmacological treatments for depression. We focused on the fast antidepressant treatment and clarified the safety, tolerability, and efficacy of ketamine and its metabolites for the MDD treatment, along with a review of the potential pharmacological mechanisms, research challenges, and future clinical prospects.
Keywords: (R)‐ketamine, (S)‐ketamine, ketamine, major depressive disorder (MDD)
This review aimed to clarify the safety, tolerability, and efficacy of ketamine and its metabolites for the treatment of major depressive disorder (MDD), along with a review of potential pharmacological mechanisms, research challenges, and future clinical prospects. Many novel hubba proteins and MDD‐risk proteins were found, indicating that the current pharmacological mechanisms were just the tip of the iceberg.
1. INTRODUCTION
Major depressive disorder (MDD) is a highly prevalent mental disorder and affects approximately 264 million patients worldwide, which makes it the second largest contributor to global morbidity 1 , 2 . The World Health Organization (WHO) reports approximately 800,000 suicide cases per year 3 , which suggests that MDD is a significant public health challenge.
Mounting experimental and clinical studies have indicated that patients with depression have altered neuronal serotonergic and noradrenergic functions in the central nervous system (CNS). 4 , 5 Brain‐derived neurotropic factor (BDNF) may also play an important role in depression. 6 Hypothalamic–pituitary–adrenal (HPA) axis hyperactivity is a common discovery in psychoneuroendocrinology studies of major depression. 7 Furthermore, inflammatory cytokines and endogenous metabolites are involved in the mechanisms of depression, 8 and additionally, the gut microbiome plays an critical role in depression by affecting the gut–brain axis. 9
Presently, monoamine reuptake inhibitors are the most frequently prescribed class of antidepressants; 10 however, there is a major issue with these due to the considerable lag time between the initial pharmacological effect on monoamine neurotransmitter function (up to several days) and the reduction in severity of clinical symptoms (usually a minimum of 3–8 weeks). 11 Typically, after antidepressant drug therapy, only 50% of patients experience a major reduction in symptoms. 12 Moreover, approximately 33% of all MDD cases involve treatment‐resistant depression (TRD), which is diagnosed when at least two courses of antidepressants have proven ineffective. 12 Consequently, there is a need to identify and develop new, fast‐acting, and more effective antidepressant therapeutic agents.
Currently, many antidepressants are available, but their side effects make them less than ideal. This paper presents an overview of the molecular mechanisms that lead to depression, pharmacological treatment for depression, and the current status of ketamine as a treatment for depression. It can serve as a reference for studies on depression, as well as for the research for ideal rapid‐acting antidepressants.
2. PATHOLOGICAL MECHANISMS OF DEPRESSION
2.1. Hereditary
According to previous twin and adoption studies, major depression is likely to have a heritability rate of approximately 31–42% 13 and may result from both genetic factors and the environment. 14 The gene–environment interactions are believed to be crucial in explaining the etiology of major depression. 15 However, there have been no strong and consistent genetic risk factors identified in genetic association studies due to the clinically heterogeneous nature of the disease and its complex genetic architecture. 16 In light of this, identifying the individual genes responsible for depression has proven challenging. Nevertheless, several MDD risk loci have been identified. 17 , 18 , 19 Studying a large cohort of MDD patients, Hyde et al. founded 15 genetic loci related to MDD risk in 2016. 20 Recently, Wray et al. identified 44 risk loci via the largest genome‐wide association study (GWAS) meta‐analysis on MDD so far. 21 Subsequently, Li et al. found three novel genetic loci related to the risk of MDD. 22 In addition, a recent GWAS reported 102 independent variants linked with depression, 23 and in another large cohort of individuals, these were linked to synaptic structures and neural transmission. 23 This evidence suggests that MDD is influenced by genetic factors.
2.2. Neurotransmitter systems
Neurotransmitters are thought go play a critical role in depression etiology. 4 , 24 Serotonin (5‐HT) is widely distributed throughout the nervous systems and its deficiency can lead to depression, phobias, anxiety, and other mental health disorder in vertebrates. 25 Over the past few decades, the 5‐HT hypothesis has driven research on the underlying cause of depression; with reports that depressed patients may have low brain 5‐HT levels and altered 5‐HT receptors, such as upregulated 5‐HT2 and downregulated 5‐HT1A receptors. 26 There are three possible mechanisms responsible for impaired 5‐HT1A function in depression: social isolation reducing 5‐HT1 neurotransmission, 5‐HT2 receptors inhibiting 5‐HT1 neurotransmission, and hypercortisolaemia inhibiting 5‐HT1 neurotransmission. 27 Endogenous proteins, such as BDNF and neurotrophin‐3, are related to the growth and function of 5‐HT neurons in the brains of adults. 28
In the brain, dopamine (DA) is a dominant transmitter that regulates behavior and is a precursor to epinephrine and norepinephrine (NE). 29 Numerous human and animal studies have shown that depression and DA transmission are closely related in the CNS. 30 , 31 Additionally, patients with depression have an increased level of DA transport, 32 which makes presynaptic neurons more effective at reuptake DA.
Glutamate is the primary excitatory neurotransmitter and contributes to synaptic plasticity, cognitive activities, and motivational and emotional behavior in the brain. 33 Multiple evidence suggests that depression is associated with the glutamate system. 34 , 35 Researchers have found elevated levels of glutamate in the blood, cerebrospinal fluid (CSF), and brains of patients with depression, 36 , 37 as well as N‐methyl‐D‐aspartate receptor (NMDAR) subunit disturbances in the brain. 38 , 39 The inhibition of NMDAR function has antidepressant effects and protects the hippocampal neurons from stress‐induced morphological changes. 40 Furthermore, ketamine, an NMDAR antagonists, has been found to have rapid antidepressant effects. 41 Alternatively, ketamine can enhance the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPAR) pathway by upregulating the AMPA glutamate receptor 1 subunit in hippocampal neurons. 42 Additionally, antidepressants may also affect the AMPAR pathway. 43
In contrast to glutamate, γ‐aminobutyric acid (GABA) is a primary inhibitory neurotransmitter. GABA neurons make up only a small percentage compared with glutamate neurons, but inhibitory neurotransmission is an important aspect of brain function because it balances excitatory transmission. 44 GABA neurons are widely distributed in the brain and participate in many functions, including in the regulation of anxiety, motivation, and the reward system, 45 , 46 , 47 and play an important part in alleviating the symptoms of MDD. 48 Numerous studies have demonstrated that MDD patients have defects in GABA neurotransmission function. 49 , 50 , 51 In a meta‐analysis of magnetic resonance spectroscopy studies, brain GABA levels in MDD patients were lower than those in healthy controls, but there was no difference seen between patients with depression in remission and healthy controls. 52 A study by Mann et al. found that the level of GABA in the CSF of patients with MDD was lower than that in healthy controls. 53 Several postmortem studies have demonstrated that low levels of GABA synthase and glutamic acid decarboxylase are present in the prefrontal cortex (PFC) of patients with depression. 54 , 55 There is evidence that depression is caused by an imbalance in the GABA and glutamate systems, and that GABA system activation produces antidepressant activity through the involvement of GABAA receptor mediators a2/a3. 56 , 57 A GABAA receptor mutant mouse model has shown that depression‐like behavior can be induced by altering the levels of potential GABA candidates in the brain. 58 , 59
2.3. Hypothalamic–pituitary–adrenal axis
Stress and acute challenges are factors contributing to MDD onset. 60 It has long been recognized that the HPA axis plays an key role in mammals stress response. Therefore, changes in the HPA axis during depressive illness may reflect the influence of stress and determine the manifestations of depressive symptoms. Stress triggers the release of corticotropin‐releasing hormone (CRH) from the hypothalamus followed by stimulating adrenocorticotrophic hormone (ACTH) production in the pituitary, which subsequently increases glucocorticoids secretion from the adrenal cortex. 61 The glucocorticoids interact with their receptors in multiple target tissues, such as the HPA axis, where they act as feedback inhibitors of both ACTH production in the pituitary corticotropes and CRH production in the hypothalamus.
In patients with depression, the HPA axis is overactive under stressful conditions, which results in problems such as hypercortisolemia, decreased rhythmicity, and elevated cortisol levels. 62 , 63 Disturbances of the HPA axis induced by stress have been shown to be associated with depression as a result of increased production of cortisol and insufficient inhibition of glucocorticoid receptor regulatory feedback. 64 , 65 Additionally, high cortisol levels have been linked to depression severity, particularly in cases of melancholic depression. 66 , 67 Moreover, patients with depression who could not normalize their HPA axis after treatment had a poorer clinical outcome and prognosis. 68 , 69 However, previous studies have shown that treatments that HPA axis‐regulating treatment like glucocorticoid receptor antagonists fail to alleviate the symptoms of depression. 70 , 71
2.4. Neurotrophins and neurogenesis
The findings of volumetric reductions in the hippocampus and other forebrain regions in depressed patients support the popular hypothesis that decrements in neurotrophic factors that regulate plasticity within the adult brain contribute to depression. The focus of these studies has largely been on BDNF, which plays important roles in different aspects of the nervous system, including synaptic plasticity, differentiation, maintenance, neuronal outgrowth, and repair. 72 The neurotrophin hypothesis of depression is primarily based on the theory that reduced hippocampal BDNF levels are associated with stress‐induced depression and are elevated by treatment with antidepressants. 73 , 74 Agents targeting the BDNF system have been found to produce antidepressant‐like effects. 75 , 76 Moreover, mounting research shows that BDNF levels are reduced in the postmortem peripheral blood of patients with depression, 77 , 78 , 79 , 80 , 81 and some reports have indicated that antidepressant treatment can normalize this. 82 , 83 Additionally, there is evidence that the interaction between BDNF and its receptor is related to TRD. 84 .It appears that BDNF depletion impairs neurogenesis and contributes to the onset of MDD, and the antidepressant can mitigate MDD symptoms by increasing BDNF levels in the brain.
2.5. Neuroinflammation
Some psychiatric studies over the past two decades have hypothesized that inflammation is linked to the pathogenesis and pathophysiology of major depression. Numerous early studies have found depression to be more common in patients who had autoimmune or infectious diseases than in the general population. 85 Moreover, even individuals who do not suffer from depression may exhibit depressive symptoms when exposure to cytokines, while antidepressants ease this discomfort. 86
MDD patients have been demonstrated to have increased levels of inflammatory molecules, 87 , 88 , 89 and display hallmarks of immune‐inflammatory response through evidence of elevated proinflammatory cytokines and their receptors, chemokines, and soluble adhesion molecules in their peripheral blood and CSF. 90 , 91 , 92 Peripheral inflammatory markers not only affect the state of immune activation in the CNS, which, in turn, impacts explicit behavior, but can also serve as evaluation or biological indices for antidepressant therapy. 93 , 94 Li et al. showed that tumor necrosis factor alpha (TNF‐α) levels in MDD patients were higher before treatment than those in healthy controls. When treated with venlafaxine, TNF‐α levels decreased significantly. Furthermore, there was a greater decrease in TNF‐α levels in the group of patients for whom the treatment was effective. 95 Antidepressants significantly reduced the level of peripheral interleukin‐6 (IL‐6), TNF‐α, IL‐10, and the C‐C motif ligand 2 chemokine, suggesting that antidepressants may reduce the markers of peripheral inflammation. 96 Moreover, Syed et al. found that untreated depression patients inflammatory markers were higher, and when they were treated with antidepressants, the levels of anti‐inflammatory cytokines increased; whereas in nonresponders, there was an increase in proinflammatory cytokines. 94 Various studies have also suggested that cytokine inhibitors like monoclonal antibodies may exert an antidepressant effect by blocking cytokines. 97 An imbalance between proinflammatory and anti‐inflammatory cytokines may contribute to the pathophysiology of depression.
Microglia are known to contribute to neuronal plasticity and play a role in MDD development. 98 , 99 A study by Weng et al. found a higher number of microglia in the PFC of mice intraperitoneally injected with lipopolysaccharide (LPS), whereas mouse depressive behavior also increased. 100 These researchers also observed the upregulation of IL‐1, IL‐6, and TNF‐α gene expression in the mouse PFC, which was suppressed by selective 5‐HT reuptake inhibitor (SSRI). Additionally, astrocytes have been implicated in the pathogenesis of stress‐ and LPS‐induced inflammation caused by depressive symptoms. 101 As activated microglia cause inflammation through excessive levels of proinflammatory factors and cytotoxins, depression‐like behavior may gradually develop. 98 , 102
2.6. Metabolic disorders
Patients with MDD often suffer from metabolic disorders, and those with metabolic disorders are inclined to experience depression. 103 , 104 , 105 The development of effective analytical technologies and methods for the analysis of fluids and tissues from a diseased organism allows us to gain a greater understanding of the basis for diseases. 106 Experimental findings in animal models and clinical practice indicate that metabolomics can be used to investigate the pathophysiology of depression and potential biomarkers.
Metabolomics has been shown to be an effective tool for selecting appropriated animal models to study depression. 107 Zheng et al. found 23 differentially expressed metabolites that distinguished MDD subjects from healthy control subjects, and identified five metabolites as potential biomarkers that can be used to differentiate MDD subjects accurately. 108 The key metabolites included amino acids and lipid/protein complexes, and some molecules related to lipid metabolism and energy metabolism that contributed to the discrimination between depressed patients and healthy controls were also identified. 109
To identify depression‐related biomarkers, gas chromatograph‐mass spectrometry (GC‐MS) was applied to metabolomic analysis of plasma samples collected from chronic unpredictable mild stress (CUMS‐induced) rats. Li et al. reported that 12 metabolites concentrations in the CUMS group were significantly different from those in the control group. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database revealed that CUMS treatment affected amino acid metabolism, energy metabolism, and glucometabolism. 110 Using GC‐MS‐based metabolomics, CUMS rats had lower levels of isoleucine and glycerol, whereas N‐acetylaspartate and β‐alanine levels were higher than those in control rats. 111 Furthermore, Gao et al. found that six potential biomarkers (glycine, glutamate, fructose, citric acid, glucose, and hexadecenoic acid) are closely associated with depression. 112
2.7. Microbiome–gut–brain axis
Recent attention has been drawn to the microbiota–gut–brain axis owing to its potential to regulate brain activity. Several studies have revealed that the microbiota–gut–brain axis is important for regulating mood, behavior, and neuronal transmission in the brain, 113 , 114 and is associated with MDD. 115 , 116 , 117 Several studies have suggested that depression and gastrointestinal disorders are comorbid. 118 , 119 Some antidepressants can alleviate the symptoms of people suffering from irritable bowel syndrome and other related disorders. 120 In patients with MDD, alterations in the gut microbiome have been reported, 121 , 122 and related to depression‐like behaviors 123 , 124 and brain function. 125 Studies on animals have demonstrated that stress can alter the composition and diversity of the intestinal microflora, and that this is accompanied by depressive behavior. 126 , 127 It is interesting to note that rodents display depressive behavior following fecal transplantation from human patients with depression. 124 In contrast, some probiotics have been found to ameliorate depression‐like behavior in preclinical studies 128 and to have antidepressant effects in several double‐blind, placebo‐controlled clinical trials involving patients with depression. 129 , 130
Gut microbiota can influence the brain in several ways, such as the HPA axis and the neuroendocrine‐, autonomic‐, and neuroimmune systems. 131 Recent studies have demonstrated that the gut microbiota can influence the levels of certain neurotransmitters, including 5‐HT, DA, noradrenalin, glutamate, and GABA in the gut and brain. 132 Additionally, recent studies have indicated that changes in the gut microbiota can damage the gut barrier and increase peripheral inflammatory cytokines. 133 , 134 Furthermore, short‐chain fatty acids, such as butyrate, are known to increase BDNF levels, whereas gut dysbiosis decreases BDNF levels; which could have an impact on neuronal development and synaptic plasticity. 115 There has been significant progress in research in this area, but more clinical trials are required to determine whether probiotics are effective in treating depression. In addition, the underlying mechanisms need to be elucidated.
2.8. Other systems and pathways
It is clear that a number of additional systems or pathways are also thought to play a role in the pathophysiology of depression, such as oxidant‐antioxidant imbalance, 135 mitochondrial dysfunction, 136 , 137 and circadian rhythm‐related genes; 138 especially their critical interactions (e.g., interactions between the HPA and mitochondrial metabolism 139 , 140 ), and the reciprocal interaction between oxidative stress and inflammation. 135
We still do not fully understand the causes of depressive disorders in spite of the abundance of research on the disease and numerous hypotheses nowadays. Different researchers have performed a variety of tasks related to modality from linked and complementary perspectives, which is helpful to further our understanding of depression. A comprehensive understanding of depression pathogenesis should consider interactions between various systems and pathways. Figure 1 shows the various pathological mechanisms of depression.
FIGURE 1.
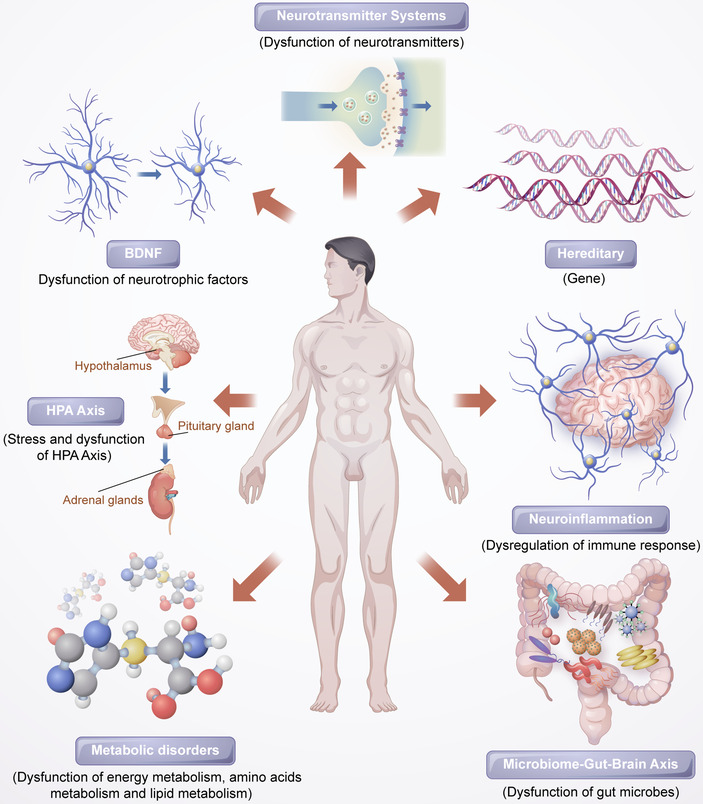
The potential underlying mechanisms of depression
3. PHARMACOLOGICAL TREATMENT OF DEPRESSION
Antidepressant medications are commonly prescribed for the treatment of MDD, including tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), serotonin noradrenaline reuptake inhibitors (SNRIs), serotonin‐2 antagonists and reuptake inhibitors (SARIs), NE reuptake inhibitors (NDRIs), specific serotonergic antidepressants (NaSSAs), and multimodal antidepressants (MMAs). Most antidepressant drugs primarily affect the brain. Monoamine neurotransmitters, 5‐HT, NE, and DA, although antidepressant drugs differ in their selectivity.
3.1. TCAs
TCAs were developed to improve the mood of depressed patients in the 1950s. 141 TCAs inhibit the neuronal uptake of NE and 5‐HT. 142 , 143 The treatment response to TCAs results in a higher availability of 5‐HT or NE at postsynaptic receptor sites. Even though TCAs have increasingly been replaced by SSRIs and other new safer antidepressants, they remain an excellent option for some patients whose depression does not improve after treatment with less potent antidepressants. Among TCAs, the antagonistic effects of adrenergic, muscarinic, and histaminergic receptors are responsible mainly for the side effects of dizziness, memory impairments, and drowsiness. 142 , 143 , 144
3.2. MAOIS
MAOIs were among the first antidepressants to be licensed for managing depression. MAOIs inhibit the monoamine oxidase enzyme, thus demonstrating that depression is a neurochemical disorder that can be managed with medication that correct underlying neurotransmitter imbalances. 145 MAOIs are rarely prescribed today, or they are the last antidepressants used because of hypertensive crisis caused by severe and potentially fatal food and drug interactions. 146
3.3. SSRIs
SSRIs, known as fluoxetine, sertraline, citalopram, and paroxetine, are the most commonly type of antidepressants around the world to treat depression and are prescribed as the first‐line treatment. SSRIs work by decreasing the reuptake of 5‐HT, so that more of it remains at the receptor sites to alleviate mood. 147 Nowadays, people suffering adverse reactions to one SSRI may find it helpful to switch to another drug from this group. SSRIs are safer than MAOIs and TCAs, have fewer side effects, and are less likely to cause death from overdose owing to the lack of affinity for acetylcholine receptors and amines receptors. 146 Nevertheless, many side effects, including nausea, insomnia, and sexual dysfunction, are caused by SSRIs. 148
3.4. SNRIs
SNRIs, such as venlafaxine and duloxetine, work in the same way as TCAs as they inhibit the 5‐HT and NE reuptake at the respective transporters. Nevertheless, SNRIs have little or no pharmacological action at adrenergic (α1, α2, and β), histamine (H1), muscarinic, DA, or postsynaptic 5‐HT receptors, differently from TCAs. 149 , 150 , 151 , 152 , 153 According to some reports, SNRIs may be more effective at treating MDD than SSRIs. Comparatively, these differences are modest. 154 , 155 SNRIs have comparable clinical tolerability and a similar prevalence of sexual dysfunction compared with other antidepressant drug treatments. 155 , 156
3.5. SARIs
SARIs such as trazodone and its analog nefazodone are antidepressants that have the ability to inhibit the reuptake of 5‐HT and NE, interact with α1‐adrenoceptors, and have no effect on histaminergic or cholinergic receptors. 157 SARIs have comparative efficacy and lower rate of induced sexual dysfunction compared to other classes of antidepressant for treatment of MDDs. 158
3.6. NDRIs
The NDRI bupropion is the only antidepressant that has a dual action on NE and DA neurotransmitter systems, which is very different from other antidepressant drugs (i.e., TCAs, SSRI, and SNRI). Bupropion has the strongest binding affinity for DA transporters compared to that of NE transporters and minimal or no binding affinity for 5‐HT transporters or other pre‐ and postsynaptic receptors. 152 , 159 , 160 , 161 , 162 Clinical research has demonstrated that bupropion is as effective as other antidepressant drugs for treating MDD. Bupropion is well tolerated, and its most frequent side effects are dry mouth, nausea, and insomnia. 163 , 164 , 165 Additionally, bupropion has the lowest incidence of sexual dysfunction compared to that of TCAs, MAOIs, SSRIs, and SNRIs. 156 , 166
3.7. NaSSA
Mirtazapine (Remeron, Zispin) is the NaSSA class approved in many counties for its use in the treatment of major depression. 167 Mirtazapine increases central noradrenergic and serotonergic activity by blocking α2 adrenoceptors and selectively inhibiting 5‐HT2 and 5‐HT3 receptors. 167 , 168 Mirtazapine appears to exert its effect faster than antidepressant reuptake inhibitors during the acute treatment phase of major depression. 169 , 170 The main side effect of mirtazapine is weight gain, which seems to occur earlier in the course of the treatment, and becomes less of an issue as the treatment continues. 171
3.8. MMAs
Vortioxetine as well as vilazodone belong to the chemical class of the piperazines and is a new class of antidepressant drugs called MMA agents because vortioxetine exhibits high binding affinity for multiple 5‐HT receptors (such as 5‐HT1A, 5‐HT1B, 5‐HT3A, 5‐HT7, and 5‐HTtransporters). 172 , 173 , 174 Thus, vortioxetine may affect the activity of various neurotransmitter systems, including 5‐HT, NE, DA, acetylcholine, histamine, glutamate, and GABA. 175 Vortioxetine is comparable to other antidepressants in terms of its clinical efficacy and tolerability, with nausea and headaches being the most common side effects. Vortioxetine appears to be associated with low risk of sexual dysfunction and weight gain. 176 , 177 , 178 , 179 Vortioxetine has been shown to enhance cognitive functioning through its action at 5‐HT3 and 5‐HT1A receptors in clinical and preclinical studies. 174 , 180 , 181 , 182
Although there are several classes of antidepressant drug, the benefits of current available treatments for depression are limited due to the low response rates, delayed therapeutic effects, and multiple side effects. Their long therapeutic delays (up to 3 weeks) and low rates of remission (approximately 30%) have prompted the search for more effective therapies. 183 Therefore, A more effective, faster‐acting, and nonmonoaminergic‐based antidepressant medication is urgently needed. The noncompetitive NMDA receptor antagonist ketamine, which has consistently been proven to produce rapid and sustained antidepressant effects and alleviated suicidal ideation in MDD patients in multiple clinical studies, 184 , 185 has shown to be the most promising novel glutamatergic‐based treatment for MDD.
4. DISCOVERY OF THE FAST‐ACTING ANTIDEPRESSANT ACTIONS OF KETAMINE
4.1. The discovery of ketamine as an antidepressant
Treatment failures and delays in clinical improvement with traditional antidepressants, developed based on the monoamine hypothesis of depression, prompted the discovery and development of antidepressants using multiple target discovery strategies. As shown in Table 1, we list antidepressants approved between 2000 and 2021. (R,S)‐ketamine (hereafter referred to as ketamine) is a phenylcyclohexylamine derivative (mol.wt. = 237.73) initially characterized by Lodge et al. 186 as an NMDARs antagonist (Ki = 0.53 μM for NMDARs), which supported the glutamate hypothesis of depression and its implications for antidepressant treatments. Evidence that glutamatergic agents might have antidepressant efficacy dates back as far as 60 years ago. 187 Recently, the revolutionary discovery and approval of the fast‐acting antidepressant ketamine has marked a landmark in the field of psychiatry in the past half century. However, the development of ketamine's rapid and sustained antidepressant effects for the treatment of MDD has experienced a tortuous process that has led to new insights into novel antidepressants (as shown in Figure 2A). The first publication on the administration of ketamine in humans was reported in 1965; 188 subsequently, ketamine became commercially available for human consumption in 1970, 189 and was widely utilized as an intravenous anesthetic drug. However, ketamine was subsequently taken off the market in 1978 because of its psychotomimetic/psychodysleptic side effects. 190
TABLE 1.
Antidepressants approved from 2000 to 2021
| Effective constituent | Mechanisms of action | Adaptation disease | Listing country and time |
|---|---|---|---|
| Aripiprazole |
Partial agonist of D2 receptor Partial agonist of 5‐H1A receptor Partial agonist of 5‐HT2A receptor |
Adjuvant treatment of MDD |
American (2002) European (2004) Japan (2006) China (2006) |
| Escitalopram | SSRI | MDD |
American (2002) European (2001) Japan (2011) China (2005) |
| Duloxetine | SNRI | MDD |
American (2004) European (2004) Japan (2010) China (2006) |
| Quetiapine |
Partial agonist of D2 receptor Partial agonist of 5‐HT2A receptor |
Adjuvant treatment of MDD |
American (2007) European (2010) Japan (2012) China (2008) |
| Agomelatine | Melatonin | Depression |
European (2009) China (2011) |
| Desmethylvenlafaxine | SNRI | Depression | American (2008) |
| Vilazodone |
SNRI Partial agonist of 5‐H1A receptor |
MDD | American (2011) |
| Levomilnacipran | SNRI | MDD | American (2013) |
| Vortioxetine |
SNRI Antagonist of 5‐HT3,5‐HT7,5‐HT1A receptor |
MDD |
American (2013) European (2013) China (2017) |
| Brexpiprazole |
Partial agonist of D2 receptor Partial agonist of 5‐HT1A receptor Antagonist of 5‐HT2A receptor |
Adjuvant treatment of MDD |
American (2015) Japan (2018) European (2018) |
| Esketamine | Antagonist of NMDAR | TRD |
American (2019) European (2019) |
| Brexanolone | GABAA receptor modulator | Postpartum depression | American (2019) |
GABA, γ‐aminobutyric acid; 5‐HT1A, Serotonin 1A; 5‐HT2A, Serotonin 1A; 5‐HT3, Serotonin 3; 5‐HT7, Serotonin 7; 5‐HT2A, Serotonin 2A; MDD, major depressive disorder; NMDAR ,N‐methyl‐d‐aspartate receptor; SNRI, serotonin and norepinephrine reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TRD, treatment‐resistant depression.
FIGURE 2.
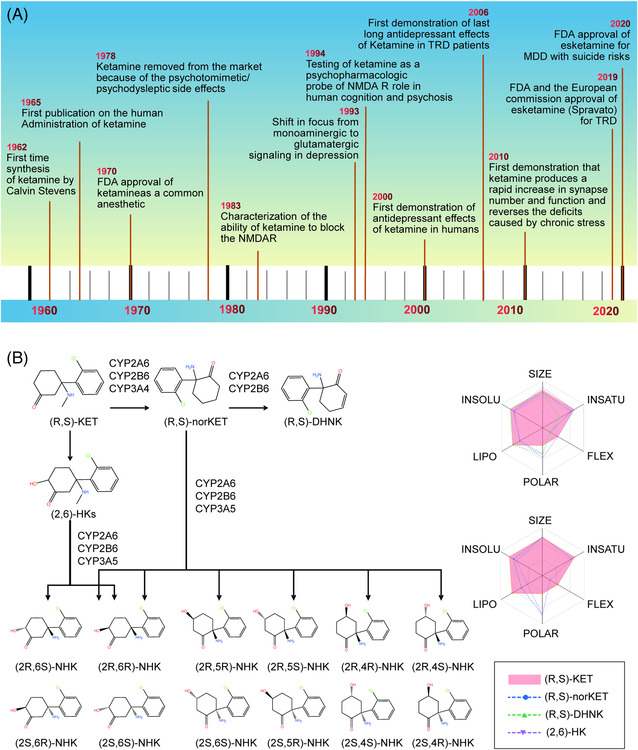
Timeline of the key events history of ketamine and its metabolic formation of hydroxynorketamines from ketamine. (A) The key events of the ketamine findings and development for MDD. (B) The ADME parameters, pharmacokinetic properties, drug‐like nature, and medicinal chemistry friendliness of ketamine metabolites predicted by SwissADME. The radar plot reflects the physicochemical properties in regard to six aspects: SIZE (molecular weight), INSOLU (solubility), LIPO (lipophilicity), POLAR (polarity), FLEX (flexibility), and INSATU (insaturation). The structure formula of Figure 2B was drawn by ChemDraw20.0.
Drug repurposing of ketamine found that its antidepressant actions had been identified in a 1975 preclinical study, 191 and a clinical study was conducted in 1973 by Khorramzadeh and Lotfy. 192 Notably, the first placebo‐controlled study in humans showing ketamine's fast antidepressant actions was reported in 2000 193 and its long‐lasting antidepressant effects were reported in 2006. 194 Ketamine was once again the subject of interest in antidepressant due to reports that subanesthetic levels of the drug (0.5 mg/kg), administered intravenously (i.v.), generated a rapid and sustained antidepressant function in patients with MDD. 193 Subsequently, Zarate and colleagues corroborated this finding with their own double‐blind, placebo‐controlled, crossover investigation of unipolar/bipolar depression cases. 194 , 195 Following this seminal study, multiple studies have validated the rapid action of ketamine in the treatment of MDD/TRD/unipolar/bipolar depression. 193 , 194 , 196 , 197 Emerging evidence demonstrates that the antidepressant effect of ketamine occurs within hours of its administration and typically lasts for days to weeks depending on individual cases. 185 , 194 , 198 Most notably, this rapid action is remarkably effective against suicidal thoughts. Randomized controlled trials (RCTs) have confirmed a reliable ketamine‐driven antidepressant function in TRD cases. 194 , 196 , 199 , 200 , 201 , 202 , 203 , 204 , 205 Notwithstanding, the antidepressant functions provided by ketamine are transient, with a typical time frame being 7 days following one infusion dose, 194 , 196 , 199 , 200 and approximately 18–19 days after multiple infusion doses. 197 , 206 Retrospective data from real‐world studies find that true‐life medical information exhibited 44% responsive rates following six i.v. ketamine doses in cases of TRD with multiple comorbidities. 207 Moreover, ketamine has exhibited antisuicide functions and anti‐anhedonic effects. 184 , 208 , 209 , 210 , 211 Moreover, its pharmacology is different from that of well‐established antidepressant drugs and consequently deemed to be a major advancement in mood‐disorder therapeutics over the preceding 60 years. 212 Importantly, an enantiomer of racemic ketamine, (S)‐ketamine, was shown to be effective in an antidepressant clinical trial, 202 , 203 , 204 , 205 and to decrease suicidal ideation in patients suffering from depression. 213 . Although four of the five esketamine ((S)‐isomer) phase 3 clinical trials found no difference with placebo, 214 , 215 the United States Food and Drug Administration (FDA) decided to grant approval based on the positive results of one trial and promising data from one of the other trials. Esketamine intranasal spray (Spravato) was approved by the United States FDA in March 2019 as an adjunctive treatment for TRD cases of MDD in a supervised setting, 216 , 217 and in 2020 for patients with MDD and suicidal risks(https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐new‐nasa‐spray‐medication‐treatment‐resistant‐depression‐available‐only‐certified).
4.2. The composition and metabolic form of ketamine
Ketamine is a racemic mixture of equal amounts of (S)‐ketamine and (R)‐ketamine. Following demethylation of ketamine to norketamine, norketamine is further metabolized to hydroxynorketamines (HNKs) and dehydronorketamine (DHNK). As shown in Figure 2B, the metabolic formation of HNKs from ketamine and the absorption, distribution, metabolism, and excretion (ADME) parameters, pharmacokinetic properties, drug‐like nature, and medicinal chemistry friendliness of ketamine metabolites were predicted by SwissADME. (S)‐Norketamine is a metabolite of (S)‐ketamine formed by the cytochrome P450(CYP) enzymatic complex. A preclinical study showed that, similar to (S)‐ketamine, (S)‐norketamine produced antidepressant actions in different rodent depression models, 218 , 219 and in contrast to ketamine and (S) ketamine did not produce detrimental side effects such as the risk of misuse/abuse, prepulse inhibition (PPI) issues, exacerbated baseline γ‐oscillations, or a decrease in parvalbumin (PV) immunoreactivity within the medial PFC (mPFC) in mice. 218 , 220 , 221 In such in vivo models, (S)‐norketamine was found to be equivalent in potency to (S)‐ketamine in terms of antidepressant‐type activities, although it had reduced potency in comparison to (R)‐ketamine. Therefore, (S)‐norketamine appears to have a higher safety profile than the parent compound (S)‐ketamine for human antidepressant function 218 , 219 , 220 and is thus a safer alternative. However, recent findings on the superiority of (R)‐ketamine versus (S)‐ketamine in animal models of depression are in agreement with previous studies by Hashimoto et al., which revealed that (R)‐ketamine has shown to have greater potency and long‐term antidepressant actions than (S)‐ketamine in several rodent models of depression. 222 , 223 (2R,6R)‐HNK, the main metabolite of (R)‐ketamine, plays a critical role in the rapid‐acting effects of ketamine. 224 , 225 , 226 Importantly, it has attracted especially high interest as a candidate antidepressant in its own right. 226 (2R,6R)‐HNK was found to induce antidepressant‐type functions with no adverse events in rodents. 227 Nevertheless, recent preclinical studies found that (2R,6R)‐HNK had no rapid or sustained antidepressant effects in animal studies. 228 , 229 , 230 Currently, there is still an considerable debate regarding the antidepressant effects of (2R,6R)‐HNK. Even so, a phase I clinical trial for (R)‐ketamine and (2R, 6R)‐HNK was initiated in early 2019. 231 It would be interesting to directly compare (R)‐ketamine, (S)‐ketamine, and (2R, 6R)‐HNK in patients with depression.
Ketamine has been clinically used as an anesthetic since the 1970s. However, indications for its use as an antidepressant and the molecular mechanisms of its metabolites are still emerging. Several molecular and cellular targets have been identified, yet the pharmacokinetics, pharmacodynamics, candidate targets, and side effects should be investigated further in order to understand the detail neurobiological mechanisms underlying the effects of ketamine and its metabolites. As shown in the Table 2, we summary the published clinical studies of intravenous ketamine and intranasal esketamine in depressive patients.
TABLE 2.
The clinical study for the antidepressant effects of ketamine
| Study design | Diagnosis/patients | Sample size | Ketamine or metabolitesadministration | Other medications | Comparator | Key outcome measures/Instrument | Result/conclusions | Reference |
|---|---|---|---|---|---|---|---|---|
| Randomized, Double‐blind, placebo‐controlled | Unipolar or bipolar depression | 9 | 0 or 0.5 mg/kg over 40 min i.v. (single dose of ketamine) | Subjects were drug free | Saline | HAMD‐25 | Significant improvements in depressive symptoms within 72 h after ketamine but not placebo infusion. | 193 |
|
Randomized, Double‐blind, placebo‐controlled Crossover |
MDD | 18 | 0 or 0.5 mg/kg over 40 min i.v. (single dose of ketamine) | Subjects were drug free | Saline | HAMD‐21 | Of subjects treated with ketamine, 71% responded after 1 day, and 35% maintained a response for ≥1 week. | 194 |
| Randomized, placebo‐controlled continuation | Unipolar TRD | 26 | 0.5 mg/kg over 40 min i.v. (open‐label, single dose of ketamine) | 2 h before ketamine: Randomized, lamotrigine or placebo 72 h after ketamine: Responders randomized, riluzole or placebo | MADRS |
Responses were observed in 65% of subjects at 24 h and 54% of subjects at 72 h. Lamotrigine did not attenuate the mild, transient side effects of ketamine, and did not enhance its antidepressant effects. Riluzole did not prevent relapse in the first month after ketamine treatment. |
185 | |
| Randomized, Double‐blind, placebo‐controlled | Unipolar TRD | 42 | 0.5 mg/kg over 40 min i.v. (open‐label, single dose of ketamine) | Randomized, placebo or riluzole, starting after ketamine infusion | MADRS |
At 4–6 h after the ketamine infusion, 62% of subject had responded. The average time to relapse was approximately 17.2 days in the ketamine‐riluzole group and 9.8 days in the ketamine‐placebo group. |
201 | |
| Multiple dose, open‐label, three times weekly over 12 days | TRD | 24 | 0.5 mg/kg over 40 min i.v. of ketamine | Subjects were drug free | MADRS |
70.8% of subjects were responders; response was sustained for the duration of the study. Median time to relapse in responders was 18 days. |
197 | |
| Double‐blind, randomized, | MDD | 73 | 0.5 mg/kg 40 min infusion of ketamine | Midazolam | MADRS | The likelihood of response at 24 h was greater with ketamine than with midazolam with response rates of 64% and 28%, respectively. | 196 | |
| Randomized, double‐blind, crossover study | MDD,TRD, 21–65 years | 18 | 50 mg of racemic ketamine(once per week) | 0.9% saline solution | MADRS | 8 of 18 patients (44%) 24 h after ketamine administration compared with 1 of 18 (6%) after placebo | 232 | |
| Double‐blind, multicenter, proof‐of‐concept study | MDD,19‐64 years | 68 | 84 mg of esketamine (56 mg if intolerance) twice weekly for 4 weeks | Placebo | MADRS |
Change from baseline in MADRS total score to 4 h, 24 h, and 25 day |
213 | |
|
Randomized, multicenter, double‐blind, and active‐controlled; fixed dosing |
Adults with TRD; age group = 18–64 |
346 |
Esketamine 56 mg or 84 mg given intranasally two times per week for 4 weeks |
Subjects were treated with OAD (duloxetine, escitalopram, sertraline, or venlafaxine) | Placebo plus OAD |
MADRS; CGI‐S; SDS; PHQ‐9; GAD‐7; EQ‐5D5L; CADSS; BPRS; MOAA/S; GADR; PWC |
No statistically significant difference was seen between treatment with ESK plus OAD group compared to placebo plus OAD group |
215 |
|
Randomized, multicenter, double‐blind, and active‐controlled; flexible dosing |
Adults with TRD; age group = 18–64 |
223 |
Esketamine 56 mg or 84 mg given intranasally two times per week for 4 weeks |
Subjects were treated with OAD duloxetine, escitalopram, sertraline, or venlafaxine) | Placebo plus OAD |
MADRS; SDS; PHQ‐9; GAD‐7; EQ‐5D‐5L; CGIS; C‐SSRS; CADSS; BPRS; MOAA/S; CGADR; PWC |
Treatment with ESK plus OAD was associated with a significantly greater change in MADRS score compared to placebo plus OAD |
233 |
|
Randomized, multicenter, double‐blind, and active‐controlled; flexible dosing |
Adults with TRD; age group ≥ 65 years |
138 |
Esketamine 28 mg or 56 mg or 84 mg given intranasally two times per week for 4 weeks |
Subjects were treated with OAD (duloxetine, escitalopram, sertraline, or venlafaxine, daily for 4 weeks) | Placebo plus OAD |
MADRS; CGI‐S; PHQ‐9; SDS; CSSRS; CADSS; BPRS; CSCB; HVLT‐R; MOAA/S; CGADR; PWC |
No statistically significant difference was seen between treatment with ESK plus OAD Group compared to placebo plus OAD group |
202 |
|
Randomized withdrawal design, double‐blind, multicenter, active controlled |
Adults with TRD; age group = 18–64 | 705 |
56 mg or 84 mg intranasally twice a week of esketamine |
OAD were used |
MADRS used, and the relapse time was assessed between the two treatment groups |
Significantly delayed relapse of depressive symptoms observed in esketamine plus OAD group |
203 | |
|
Long‐term (one year) study, multicenter, Open‐label; phase 3 |
Adults with TRD; ≥18 years | 802 |
Esketamine 28 mg (for ≥65 years), 56 or 84 mg given intranasally twice weekly during the 4‐week induction phase (given along with OAD) |
OAD (duloxetine, escitalopram, sertraline, or venlafaxine) were used |
MADRS; CSCB; DET; IDN; OCL; ONB; GMLT; HVLT‐R; CSSRS; CADSS; BPRS; MOAA/S; BPIC‐SS; PWC; PHQ‐9; SDS; CGI‐S |
Improvement in depressive symptoms was found to be sustained in patients with TRD |
204 | |
| Double‐blind, phase 3 studies | MDD with acute suicidal ideation or behavior | 456 | Esketamine 84 mg or placebo nasal spray twice weekly for 4 weeks | Comprehensive standard of care, including hospitalization and newly initiated or optimized antidepressants |
MADRS scale and clinical global impression severity of suicidality‐revised were used to evaluate changes from baselines at 24 h after the first dose |
Esketamine plus comprehensive standard of care rapidly reduces depressive symptoms in patients with major depressive disorder who have acute suicidal ideation or behavior | 205 |
HDRS, Hamilton Depression Rating Scale; MADRS, Montgomery–Asberg Depression Rating Scale; TRD, treatment‐resistant depression; OAD, oral antidepressant; MDD, Major depressive disorder; IV, Intravenous; IN, Intranasal; mg, Milligrams; kg, Kilograms; SD, standard deviation; SE, Standard error; CI, confidence interval; LSMD, least square mean difference; AD, antidepressants; BPIC‐SS, bladder pain‐interstitial cystitis symptoms scale; BPRS, brief psychiatric rating scale; CADSS, clinician‐administered, dissociative states scale; CGADR, clinical global assessment of discharge readiness; CGI‐S, clinical global impression severity; CGI‐I, clinical global impression improvement; CSCB, Cog state computerized battery; C‐SSRS, Columbia suicide severity rating scale; EQ‐5D‐5L, EuroQol‐5 dimension‐5 level; DET, detection task; EWPS, Endicott work productivity scale; GAD‐7, generalized anxiety disorder 7‐item; GADR, global assessment of discharge readiness; GMLT, Groton maze learning test; HAM‐A, Hamilton anxiety rating scale; HVLT‐R, Hopkins verbal learning test‐revised; IDN, identification task; LFT, liver function tests; MOAA/S, modified observer's assessment of alertness/sedation; OCL, one card learning; ONB, one back; PHQ‐9, patient health questionnaire 9‐item; PRISE, patient‐rated inventory of side effects; PWC, physician withdrawal checklist; QIDS‐SR16, quick inventory for depressive symptomatology self‐report 16‐item; SDS, Sheehan disability scale; SF‐12, short form health survey; SHAPS, Snaith–Hamilton pleasure scale; YMRS, Young mania rating scale; 2 BACK, two back task; CADSS, the clinician administered dissociative states scale.
5. EFFICACY AND SAFETY OF KETAMINE
5.1. Efficacy of ketamine
Ketamine is essentially a noncompetitive NMDARs antagonist that blocks open channel pores at phencyclidine binding regions, thereby stopping cation (mainly calcium) flow and thwarting neuron excitation/depolarization. Multiple RCTs investigating the subanesthetic ketamine dose (40‐min infusion of 0.5 mg/kg) have been conducted in MDD/TRD cases. 193 , 196 Compared with placebo, this dose led to antidepressant action in TRD of a bipolar nature typically on mood‐stabilizer therapies with no exacerbated affective switching onto hypo/mania. 195 Ketamine was also found to rapidly alleviate suicidal thoughts. 234 , 235 Since few experimentally validated therapies with rapid response exist for suicide risk, ketamine represents a putative novel antidepressant drug with fast‐acting efficacy in this respect, particularly for emergency/acute cases of this nature. Consequently, subanesthetic infusion‐based dosage regimens for ketamine provided proof‐of‐concept effectiveness and a good safety/tolerability profile within small studies. 197 , 236 Ketamine's antidepressant functions were clinically successful; in one study, 33% of TRD cases achieved remission and approximately 50–75% of such cases exhibited alleviation of clinical symptoms following one initial dose, with even better results obtained following multiple‐dose treatment regimens. 237 Non‐i.v.‐based routes of administration were also investigated (intramuscular/subcutaneous/oral/sublingual/intranasal), and a wide range of effectiveness levels were found, usually with reduced adverse events compared to i.v. infusions. In addition, certain ketamine metabolites have also been linked to antidepressant responses. 238 Before the FDA approval of esketamine, its safety faced challenges due to ketamine's regulation of opioid receptors, 239 , 240 which raised concerns about its potential for abuse. 241 A recent report 242 revealed that acute opioid receptor antagonism by naltrexone decreased the antidepressant action of ketamine, which may indicate that long‐term use of ketamine for depression could lead to abuse problems. Interestingly, chronic naltrexone pretreatment did not diminish the ketamine's antidepressant effects and showed good tolerance in five patients with MDD recruited for the study. 243 Furthermore, chronic concurrent use of buprenorphine, methadone, or naltrexone did not inhibit antidepressant activity. 244 These divergent results can be explained by long‐term versus acute opioid blocker administration. The binding affinity of ketamine for opioid receptors and the role and precise mechanism of opiates in the antidepressant effects of ketamine should be further investigated.
5.2. Safety of ketamine
The opioid receptor blocker naltrexone may block the constitutive inhibition of cAMP by opioid receptors, and cAMP‐mediated neuronal nitric oxide synthase (nNOS) activation may be involved in the downregulation of mammalian target of rapamycin (mTOR) signaling, which plays a critical role in the rapid‐acting antidepressant effects of ketamine. 245 , 246 A series of studies have confirmed the safety and effectiveness of ketamine, and its use as an antidepressant has been globally recognized. In December 2019, the European Commission approved esketamine for patients with MDD and failed antidepressant treatment with at least two drugs, 217 , 247 despite doubts regarding its effectiveness, 248 , 249 which was ultimately determined in three acute‐phase studies and two maintenance‐phase studies. One phase 3 study of 200 cases treated with esketamine as adjunct therapy with another antidepressant demonstrated major mood‐lifting at 4 weeks in comparison to placebo. 233 However, these studies did not reach the targeted therapeutic endpoints. 215 Notably, studies of acute cases treated with esketamine involved patients with more severe depressive conditions than those indicated by FDA approval for antidepressant treatment with adjunctive medications (https://fda.gov/downloads/AdvisoryCommittees/CommitteesMettingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM630970.pdf(2019). Two maintenance investigations monitored patients treated with esketamine on a weekly/biweekly dosage regimen for 12 months, 203 and contributed positive datasets. Presently, Janssen (New Blueswick, New Jersey, USA) is conducting additional clinical trials to evaluate esketamine's safety profile for 5‐year treatment regimens. 250 Esketamine presently requires administration in tandem with risk evaluation and mitigation strategy guidelines because of its previously noted fleeting dissociative/psychotomimetic adverse events risk and possible abuse/misuse when administered at antidepressant doses. Notably, (R)‐ketamine has also been indicated for rapid antidepressant actions, with an increased tolerance profile compared to esketamine. 231 The latest research reports indicate that (R)‐ketamine generates long‐term antidepressant function with none of the adverse effects caused by (S)‐ketamine. 222 , 251 Perception pharmaceuticals have been conducting a phase I investigation on this drug since 2019, although outcome data are still pending. 231 Even though (R)‐ketamine is viewed as a potentially effective treatment for TRD, no clinical trials evaluating the efficacy and safety have been done to date, which highlights the need for further research.
6. KETAMINE ADVERSE‐EFFECT PROFILE
A single infusion dose of ketamine is typically well tolerated, although it can induce temporary adverse events during the initial hours post‐first dosing, 252 the most common being visual disturbances, dysphoria, dissociation, anxiety, and euphoria. Other side effects include nausea, vomiting, dizziness, drowsiness, hypertension, and tachycardia. 231 , 253 , 254 , 255 Due to the short half‐life of ketamine, such adverse events fade within minutes following discontinuation of transfusion dosing, resulting in total remission within 120 min. 256 Swainson et al. described a set of adverse effects linked to the intranasal administration of esketamine. 250 Ketamine/esketamine side effects that have emerged during the treatment of MDD can be classified as psychiatric (dissociation/psychotomimetic), neurologic/cognitive, hemodynamic, genitourinary, and leading to abuse risks, even from a single dose of ketamine, with cumulative effects following multiple doses, although not well investigated. 257 Severe physical adverse events included sedation, dizziness, light‐headedness, nausea, poor coordination, vomiting, and headache, which were mostly self‐limited. Adverse psychiatric events typically occur in an acute manner (irritability, agitation, anxiety, and mood elevation) and are typically short‐lived (dissociation, disorganized thought, altered perceptions/hallucinations/illusions, emotional withdrawal, and suspiciousness), and their severity is affected by the dose, dosage regimen, and administration route. Cognitive impairment traits were also noted. 257
Although this drug can be effective in MDD, it is essential for physicians to be aware that such adverse events can and will occur. 258 , 259 Furthermore, ketamine is called ‘‘Special K’’ in the narcotics‐consuming population due to its popularity as a powerful recreational drug of abuse, 260 typically leading to temporary cognitive impairment, 185 although long‐term abuse leads to neurotoxicity. 261 , 262 Prolonged ketamine abuse at excessive doses can cause urological manifestations and exacerbate the severity of adverse effects. The risk of chronic abuse of ketamine as a recreational narcotic leading to cognition/affective impairments that include depressive conditions remains a major issue of concern, and consequently raises doubts about the suitability of ketamine for treating MDD on a prolonged basis. 263 Recognition of the mechanism for ketamine‐directed antidepressant functions could aid in the development of novel fast‐operating drugs with reduced adverse effects.
Dissociation, psychoses, and cognitive adverse effects tend to be susceptible to ketamine enantiomer presence, although no proper comparative analyses have been conducted to assess this. 231 , 264 Murine receptor investigations revealed that (R)‐ketamine rapidly induced antidepressant function, presented an improved adverse event profile compared to esketamine, and enhanced phencyclidine‐driven cognitive impairment side effects within murines. 222 , 226 , 264 Such adverse effects could intensify depending on the route of administration and dosage regimen. Additional side effects associated with ketamine include neurotoxicity, bladder toxicity, and tolerance to prolonged exposure to ketamine‐based infusions. 265 The development of ketamine metabolites as antidepressants to avoid these adverse effects is an ongoing research strategy. (S)‐Ketamine has been approved in the United States and Europe because of fewer side effects; Nevertheless, some concerns remain about its efficacy and side effects. (S)‐ketamine has a greater affinity for NMDARs than (R)‐ketamine and this may contribute significantly to its clinical activity, especially when given orally. 266 Surprisingly, increasing preclinical and clinical evidence suggests that (R)‐ketamine may be more effective at treating depression with less side effects than (S)‐ketamine. 222 , 251 , 267 , 268 , 269 , 270 Pharmacological studies are needed to investigate the specific cellular and molecular mechanisms underlying the antidepressant effects and side effects of ketamine and its metabolites to discover fast‐acting antidepressants without undesirable side effects.
7. PHARMACOLOGICAL PROFILE OF KETAMINE AND ITS UNDERLYING MECHANISM
Intense focus has been placed on understanding the pharmacology behind the antidepressant actions of ketamine, mainly through in vivo investigations that unravel novel mechanisms (as shown in Figure 3). Ketamine's direct targets are NMDARs that are expressed all over the brain and crucial to brain function. Ketamine produces antidepressant actions by affecting the critical brain's reward and mood circuitry regions, specifically involving the PFC, hippocampus, nucleus accumbens (NAc), ventral tegmental area (VTA), and lateral habenula (LHb). 246 , 271 , 272 , 273 , 274 , 275 , 276 , 277 , 278 , 279 , 280 , 281 , 282 Postmortem studies have demonstrated that the PFC and hippocampal circuitry are dysregulated in depression, including alterations in structure, markers of glutamatergic and GABAergic neurotransmission, and connectivity with downstream structures, 44 as well as reduced synaptic markers and number of synapses in the PFC and hippocampus. 282 In addition, a decrease in prefrontal and hippocampal volumes has been demonstrated, which correlated with the length of illness. 283
FIGURE 3.
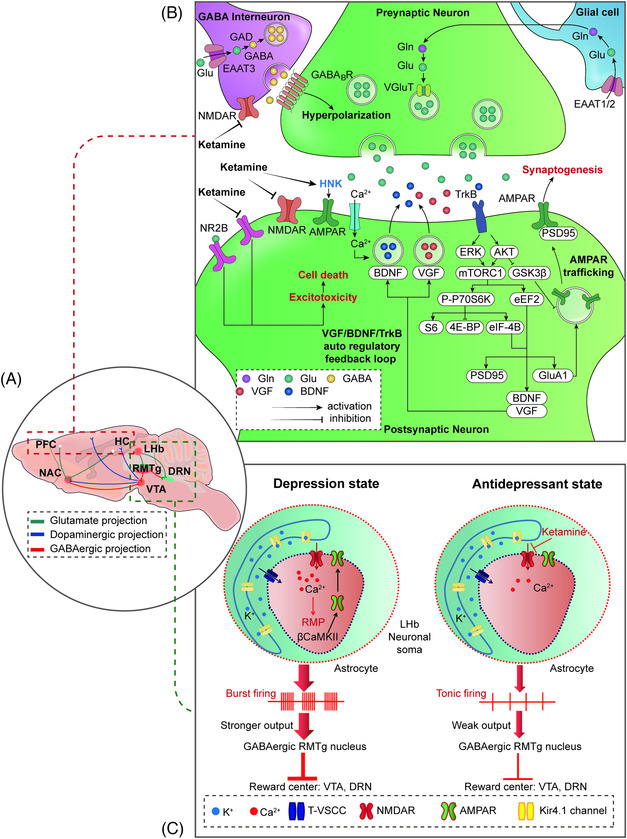
Ketamine pharmacological profile and its underlying mechanisms for rapid‐acting antidepressant action. (A) The neurocircuits implicated in the ketamine rapid antidepressant action. (B) Four potential mechanisms underlying the rapid and sustained antidepressant action of ketamine in the PFC and hippocampus: (1) Disinhibition of glutamate release from GABAergic interneurons by blocking the presynaptic NMDARs in the mPFC and hippocampus. (2) Inhibition of the extra‐synaptic NMDARs subunit (NR2B) of the pyramidal neurons in the cortex. (3) Inhibition of the spontaneous NMDARs‐mediated miniature excitatory postsynaptic current (mEPSCs) at rest in the PFC and hippocampus. (4) Direct AMPARs triggering. (C) Inhibition of NMDAR‐dependent neuron burst firing in the LHb.
Molecular biology, morphology, anatomy, electrophysiology, and pathophysiology 284 , 285 , 286 , 287 , 288 strongly implicate the hippocampus‐PFC circuit in MDD, which is a central hub regulated by the amygdala (glutamatergic projection) and dorsal raphe nucleus (DRN) (serotonergic projection). In contrast to the changes in the hippocampus and PFC in MDD, the volume, neuroplasticity, and neurotrophic factors in the NAc were increased, 273 , 289 , 290 supporting the hypothesis that stress‐induced NAc hypertrophy may be associated with the pathophysiology of MDD. Furthermore, rewarding stimuli perceived in the hippocampus and PFC are relayed by excitatory glutamate projections to the NAc; however, the regulation of the NAc is complicated. In addition, the mesolimbic DA system is related to the MDD pathophysiology, and dopaminergic projections originate in the VTA and project to the PFC and NAc. 291 Moreover, the LHb, as the “small” region, may have “big” driving effects in the psychiatry disorders phenotype. 292 , 293 , 294 , 295 More and more evidence shows that the ‘‘anti‐reward center’’ LHb is implicated in the coding of negative emotions 296 , 297 , 298 , 299 and crucial for the treatment and pathophysiology of MDD. 300 , 301 , 302 , 303 Adverse stimuli activate excitatory glutamate neurons projecting from the LHb to the VTA, resulting in a decrease in DA output from the VTA and a decrease in reward. Hu et al. demonstrated that high‐frequency burst firing of LHb neurons may drive depression‐like behaviors in rodents 276 , 277 , 278 , 279 , 280 , 281 , 282 , 283 , 284 , 285 , 286 , 287 , 288 , 289 , 290 , 291 , 292 , 293 , 294 , 295 , 296 , 297 , 298 , 299 , 300 , 301 , 302 , 303 , 304 indicating an essential role for the LHb in the pathophysiology of MDD. Recently, it was shown that the low‐voltage‐sensitive T‐type calcium channel (T‐VSCC) blocker ethosuximide did not demonstrate rapid and sustained antidepressant effects in a CSDS model. 305 A subsequent study reported that potassium channel Kir4.1 inhibitors such as quinacrine and sertraline had no rapid and sustained antidepressant effects in a CSDS model. Hence, Kir4.1 channel inhibitors are unlikely to exert robust antidepressant effects similar to ketamine although further study needs to be conducted by using selective and potent Kir4.1 channel inhibitors. 306 Based on their different roles in the regulation of MDD, the underlying mechanisms of these brain regions in mediating the rapid and sustained antidepressant effects of ketamine have been explored recently. As shown in Table 3, multiple preclinical studies demonstrated that ketamine and/or its metabolites induce behavioral effects that predict antidepressant effectiveness.
TABLE 3.
The preclinical study for the antidepressant effects of ketamine and metabolites
| Species | Drug | Depression model | Administration paradigm | Route of administration | Timing | system | Effect | Reference |
|---|---|---|---|---|---|---|---|---|
| Mouse | (2R,6R)‐HNK | Chronic social defeat stress model | i.p. | 10 mg/kg | 1 h, 24 h posttreatment | Mouse hippocampus | Increased BDNF levels, decreased eEF2 phosphorylation | 226 |
| / | (2R,6R)‐HNK | / | / | 50 μM | 30 min exposure | Mouse primary neurons | Decreased eEF2 phosphorylation | 307 |
| Mouse | (2R,6R)‐HNK | / | i.p. |
30 mg/kg 10 and 50 nM |
30 min posttreatment 1 h exposure |
Mouse prefrontal cortex, rat primary neurons |
Increased BDNF release, Increased p‐mTOR, p‐ERK | 308 |
| Mouse | Ketamine | The learned helplessness (LH) model | i.p. | 10 mg/kg | 2–72 h posttreatment | mPFC | Enhanced glutamate‐evoked dendritic spinogenesis | 278 |
| Adult male mice |
Ketamine (2R,6R)‐HNK |
/ | i.p. |
10,30 mg/kg 20 mg/kg |
1, 24, and 144 h after posttreatment 1 h after exposure |
Mouse primary neurons |
induced hippocampal synaptic plasticity depends on 4E‐BPs |
225 |
| Transgenic mice and C57BL/6J mice | Ketamine | Chronic CORT exposure | i.p. | 10 mg/kg | 24 h after exposure | Mouse mPFC |
restoring lost spines and rescuing coordinated ensemble activity in PFC microcircuits |
277 |
| Mouse | (S)‐Ketamine | CSDS model | i.p. | 10 mg/kg | 7 days after injection | Mouse prefrontal cortex and hippocampus |
induced dendritic spine density and synaptogenesis |
309 |
|
C57BL/6 mice Male cLH rats |
Ketamine | CRS depression model | i.p. | 10 mg/kg | 1 h after drug delivery | LHb |
blocked bursting in the lateral habenula |
276 |
| Male C57BL/6 mice |
Ketamine (2R,6R)‐HNK |
/ | i.p. |
3 mg/kg 10 mg/kg |
1 day after injection | NAc | Impaired Long‐term potentiation (LTP) in the NAc | 310 |
| C57BL/6 mice | Ketamine | / | i.p. | 3 mg/kg | 30 min, 3 h, and 24 h after posttreatment | hippocampus |
reduced the level of eEF2 phosphorylation and strengthened synaptic responses |
311 |
| Sprague‐Dawley rats | Ketamine | / | i.p. | 10 mg/kg | 1 day after posttreatment | mPFC |
VEGF signaling mediated the rapid antidepressant actions of ketamine |
279 |
| Adult male Sprague‐Dawley rats | Ketamine | / | i.p. | 10 mg/kg | 30 min or 1 week after posttreatment |
mPFC Hippocampus |
activated of the vHipp–mPFC pathway | 280 |
| Rats | ketamine | chronic stress rats | i.p. |
100 nM 10 mg/kg |
30 min after ketamine treatment | Hippocampal |
Induced HDAC5 phosphorylation and nuclear export in Hippocampal Neurons |
281 |
| Rats | ketamine | / | i.p. | 10 mg/kg | 30 min, 1 h, 2 h, and 6 h after posttreatment | The prefrontal cortex |
Activated the mTOR pathway |
246 |
BDNF, brain derived neurotrophic factor; CSDS, chronic social defeat stress; eEF2, eukaryotic elongation factor 2; ERK, extracellular signal‐regulated kinase; HDAC5, histone deacetylase 5; LHb, lateral habenula; mTOR, mammalian target of rapamycin; mPFC, medial prefrontal cortex; NAc, nucleus accumbens.
Based on the above‐mentioned information, we summarized the molecular mechanisms behind ketamine's antidepressant effects as follows: (1) inhibition of glutamate release by blocking presynaptic NMDARs of GABAergic interneurons in the mPFC and hippocampus; 312 , 313 , 314 (2) inhibition of extra‐synaptic NMDAR subunits (NR2B) in cortical pyramidal neurons; 315 , 316 (3) inhibition of spontaneous NMDAR‐mediated miniature excitatory postsynaptic currents (mEPSCs) in the PFC and hippocampus; 311 , 317 and (4) triggering of prosynaptogenic AMPARs. 226 , 318 These four hypotheses mainly focus on the dependence or independence of NMDARs, and subsequent AMPAR activation is suggested to play a role in the rapid antidepressant actions of ketamine. Ketamine activates synaptogenic intracellular signaling, including the rapid release of neurosecretory protein VGF (nonacronymic) and BDNF and subsequent activation of tropomyosin receptor kinase B (TrkB)‐mediated AKT and/or extracellular signal‐regulated kinase (ERK)/mTORC1 signaling, thereby enhancing the VGF/BDNF/TrkB autoregulatory feedback loop in the rapid and sustained antidepressant effects of ketamine. (5) Inhibition of NMDAR‐dependent burst firing of neurons in the LHb. The finding that ketamine blocks burst firing in the LHb to rapidly relieve depression confirms that the LHb may be the trigger subregion in the rapid‐acting antidepressant‐like effects of ketamine. (6)Transforming growth factor β1(TGF‐β1) system. TGF‐β1 in microglia may be linked to the antidepressant properties of (R)‐ketamine in animal models of depression. 319
7.1. Underlying molecular signaling in the rapid and sustained antidepressant action of ketamine
Imbalance of inhibitory and excitatory neurotransmission in the PFC and hippocampus has been involved in depression and convergent evidence from clinical and preclinical studies indicates that dysfunction of glutamatergic and GABAergic systems may contribute the pathophysiology of MDD. 44 , 56 MDD is generally accompanied by low GABA levels or GABAergic interneuron numbers, possibly disinhibiting glutamate release. 320 Ketamine was reported to inhibit presynaptic NMDARs of GABAergic interneurons present in excitatory neurons within the hippocampus and/or mPFC regions, resulting in the release of tonic inhibition that subsequently leads to increased firing of pyramidal neurons, augmenting synaptic transmission, thus orchestrating quick‐acting antidepressant functions. 44 , 313 , 314 Importantly, the burst of glutamate is thought to occur via blockade of NMDARs on GABA interneurons, which are more sensitive to the open‐channel blocking actions of ketamine. 314 This mechanism reveals that downstream AMPAR activation is dependent on ketamine‐induced presynaptic‐mediated glutamate release. Enhanced AMPARs triggering, together with ketamine‐based blockade of extra‐synaptic NMDARs, begins and aids postsynaptic triggering of neuroplasticity‐linked AMPARs subunit expression and synaptic intensity, together with synaptogenesis. 310 , 321 , 322
Many rapid antidepressants, including ketamine, produce postsynaptic membrane depolarization that initiates intracellular secondary signaling transduction cascades, leading to the enhanced BDNF and VGF rapid release 273 , 323 and subsequent activation of TrkB‐mediated AKT and/or ERK/mTORC1 signaling, 309 , 324 indicating that TrkB may be the key regulator underlying ketamine‐induced rapid antidepressant actions. More recent findings confirm that TrkB is required for ketamine‐induced synaptic potentiation 325 and TrkB activation‐induced allosteric facilitation of BDNF signaling is a common mechanism for rapid antidepressant actions. 326 Additionally, TrkB‐dependent adult hippocampal progenitor differentiation mediates a sustained ketamine antidepressant response. 327 It is worth noting that the production of BDNF and VGF enhances the VGF/BDNF/TrkB autoregulatory feedback loop in ketamine's rapid‐acting and sustained antidepressant efficacy. 323 The production of VGF and BDNF may also be regulated by mTORC1; notably, downregulation of mTORC1 signaling pathway has been identified in postmortem tissue from individuals with MDD 328 and directly targeting mTORC1 produces rapid antidepressant action, 329 which supports the idea that mTORC1 is the common downstream kinase involved in the rapid‐acting antidepressant effects of ketamine and (2R,6R)‐HNK in rodents. 246 , 308 mTORC1 controls various neuronal functions, particularly through eukaryotic initiation factor 4E‐binding proteins (4E‐BPs) and eukaryotic elongation factor 2 kinase (eEF2K), which regulate protein synthesis through downstream effects. A more recent report demonstrated that brain 4E‐BPs and downstream initiation of mRNA translation are pivotal targets of ketamine and (2R,6R)‐HNK. 225 Preclinical studies have also identified eEF2K signaling as essential for the rapid antidepressant action of ketamine. 307 , 330 , 331 Ketamine deactivates eEF2K and thereby decreases the amount of phosphorylated eEF2K, eliciting desuppression of dendritic protein translation, ultimately triggering synaptic upscaling. 311 , 332 , 333
In addition to the regulation of presynaptic NMDARs, postsynaptic NMDARs also play a vital role in the antidepressant actions of ketamine. In particular, NR2B‐containing heterotetramers, the primary subunits of NMDARs, are mainly activated by ambient glutamate 334 and mediate synaptic homeostasis via suppression of protein synthesis. 335 Thus, antagonism of the NR2B subtype may be a promising target for developing novel antidepressants with more powerful effects and quicker onset compared with traditional antidepressants. 336 Ketamine also suppresses protein synthesis and produces rapid antidepressant actions through the extrasynaptic NR2B‐dependent mechanism. 315 , 316 Ketamine appears to exert its antidepressant effects by blocking NMDARs‐mediated miniature excitatory postsynaptic currents at rest, leading to deactivation of the calcium‐/calmodulin‐dependent kinase eEF2K, resulting in dephosphorylation of eEF2 and subsequent desuppression of BDNF, VGF, and AMPARs subunit GluA1 protein translation. 323 , 337 , 338 Hypofunction of the midbrain reward center has been reported in depression. 339 Recently, the reward centers (including the VTA and DRN) were found to be inhibited by the LHb dependent on the GABAergic rostromedial tegmental (RMTg) nucleus, 340 , 341 indicating that the LHb has thought to be an important brain region in the pathophysiology of depression.
LHb neurons were previously classified as silent, tonic firing, and burst firing types, 342 , 343 and an increase in burst firing neurons and spikes in burst mode in the LHb was characterized as the novel pathogenesis of MDD. Preclinical and clinical studies have revealed that the LHb is metabolically hyperactive, 344 , 345 and alleviation of burst activity in the LHb may be sufficient to prevent depressive‐like symptoms. Because LHb bursts depend on NMDARs, the rapid antidepressant pharmacological actions and unique mechanisms of ketamine in the LHb have caused wide public concern. A pair of studied from Hailan Hu's laboratory 276 , 304 demonstrated that ketamine blocks the burst firing of neurons in the LHb in an NMDARs‐dependent manner. The potential mechanisms indicate that ketamine can quickly alleviate symptoms of depression by disinhibiting reward centers through blocking LHb bursts. This striking finding might explain the mechanism of LHb bursts and provide new insights into the development of novel antidepressant targets in the LHb.
7.2. Convergent onset target: AMPARs and trafficking regulation
Ketamine is a full antagonist of NMDARs, and its structural basis on human NMDARs has been described by cryoelectron microscope. 346 However, whether the rapid‐acting antidepressant actions of ketamine and its metabolites relay on NMDARs have been questioned. Unlike ketamine, other NMDARs antagonists (lanicemine, memantine, and N2O) do not show significant antidepressant properties, 347 indicating that other additional mechanism may involve in the antidepressant regulation of ketamine. Notably, recent finding of NMDARs suppression‐independent antidepressant actions of ketamine metabolites 226 indicate a novel mechanism underlying ketamine's unique antidepressant actions; therefore, future studies should not be limited to NMDARs antagonists, because this suggests that ketamine's mechanisms for rapid‐acting antidepressant‐like effects is complicated. Additional targets within the glutamatergic system include inotropic and metabotropic receptors and glutamate transporters. The affinity of (S)‐ketamine for NMDARs is approximately fourfold greater than (R)‐ketamine, 348 and may explain the greater potential anesthetic effects and greater undesirable psychotomimetic side effects than (R)‐ketamine. However, (R)‐ketamine showed greater potency and longer term antidepressant actions than (S)‐ketamine, 223 indicating that the anesthetic and psychotomimetic actions of ketamine are mediated primarily by the blockade of NMDARs. Importantly, except to blocking NMDARs, the rapid and long‐lasting antidepressant actions of ketamine and/or (2S,6S;2R,6R)‐HNK are also dependent on the activation of AMPARs. 226 In addition, synaptic plasticity changes involving AMPARs are thought to underlie the long‐term antidepressant actions of ketamine. 72 , 349 The AMPARs‐based long‐term antidepressant function of (2R,6R)‐ HNK through the exclusion of intense bonding attractiveness for NMDARs 226 has been found, which implicate underlying AMPARs‐mediated maintenance of synaptic potentiation in the sustained antidepressant effects. The expression, distribution, and trafficking of AMPARs play a critical role in mediating the majority of fast excitatory synaptic transmissions and neuropsychiatry disorders. The metabolite of ketamine seems to be linked to intense immediate expansion in excitatory neurotransmission through AMPARs triggering, continued through sustained maintenance by upregulation of GluA1 and GluA2 AMPARs synaptic subunits. 226 , 350 Convergent evidence supports the hypothesis that AMPARs‐triggering/mTOR/BDNF/VGF signaling orchestrates ketamine‐driven synaptogenesis and antidepressant function. 42 , 351 , 352
By increasing BDNF and VGF release, ketamine upregulates the surface AMPARs subunits expression 353 required for the increase in synaptic efficacy and the antidepressant effects. 354 , 355 In addition to AMPARs‐mediated BDNF and VGF release, AMPARs‐mediated 5‐HT release may also be involved in the rapid antidepressant‐like actions of ketamine. Ketamine can increase the levels of extracellular 5‐HT in the mPFC via AMPAR activation, 356 , 357 and the antidepressant‐like actions of ketamine are blocked by pretreatment with a 5‐HT‐depleting agent 358 , 359 and 5‐HT1A receptor antagonist. 360 , 361 In contrast, selective stimulation of 5‐HT1A receptors in the mPFC exerts rapid and sustained antidepressant‐like effects via activation of AMPAR/BDNF/mTOR signaling in mice, which provides evidence for the targeting of 5‐HT1A receptor in the treatment of MDD. 362 However, pretreatment with the AMPARs inhibitor NBQX does not block the antidepressant actions of monoamine‐based antidepressants. 352 , 363 AMPAR is the specific target of current findings of rapid antidepressants (e.g., ketamine, GluN2B‐NMDARs antagonists, 4‐chlorokynurenic acid, GLYX‐13, scopolamine, mGluR2/3 antagonists, GABAAR‐NAMs, and (2R,6R)‐HNK). 360 , 364 , 365 In support of this hypothesis, AMPARs‐positive allosteric modulators have been found to induce antidepressant‐like responses in rodents, 366 , 367 , 368 which further makes AMPARs a promising target for the development of new antidepressant drug.
Previous studies have found that the AMPARs trafficking regulated by glycogen synthase kinase‐3 (GSK3) 369 , 370 and numerous associates between GSK3 and depression have been reviewed, 371 which suggest that abnormally active GSK3 contributes to susceptibility to depression and inhibition of GSK3 may as one potential downstream target of ketamine in the antidepressant process. Ketamine treatment in rodents has been reported to inhibit cerebral glycogen synthase kinase‐3β(GSK‐3β), a pharmacological pathway shared by lithium. 43 , 369 , 372 GSK‐3β also phosphorylates postsynaptic density‐95 (PSD‐95) protein, which regulates AMPARs trafficking. 373 These interactions raise the possibility and confirmed that ketamine increases membrane AMPARs subunits by its inhibitory effect on GSK‐3β dependent on the phosphorylation of PSD‐95. 42 Except for the direct targeting of AMPARs in rapid antidepressant actions, AMPARs trafficking is believed to underlie higher brain functions and has been involved in a large number of psychiatric disorders, including MDD. 42 , 367 , 374 Because relatively little is known about these mechanisms of action, it is of significant importance to elucidate the underlying molecular mechanisms that regulate AMPARs trafficking in antidepressants.
8. SUGGESTED ROUTES OF ADMINISTRATION AND DOSAGE REGIMENS OF KETAMINE
A variety of administration routes are available for ketamine. 375 , 376 Intranasal administration is considered a more attractive option because it is less invasive, causes rapid systemic absorption in to the body, and is not affected by hepatic metabolism compared with intravenous administration. One RCT focusing on intranasal ketamine administration supported the feasibility of this route of administration; 232 however, a similar study was cancelled due to decreased tolerability. 377 Alternative clinical‐based studies have described that despite the experimental findings, maintenance doses of intranasal ketamine can be of clinical utility in cases with no other therapeutic possibilities. 378 , 379 Intranasal esketamine resolves several adverse effect challenges, allowing it to be approved by multiple regulatory bodies on a global scale. However, the issue of reduced effectiveness remains. 380 Investigations on sublingual ketamine were described in a newly published review article, 381 although such investigations had large dose‐range variations and did not consider the decreased bioavailability present in oral drug formats, thus underestimating effectiveness. 382
Intramuscular/subcutaneous routes could be feasible for ketamine delivery, 383 although few investigations have been conducted. Although the main route of administration of ketamine remains i.v., it can also be delivered through subcutaneous, intramuscular, transdermal, intranasal, intrarectal, or oral routes. Bioavailability of drugs differs depending on their administration route. The bioavailability profiles are as follows: i.v. route (100%), intranasal (45%), sublingual (30%), oral (20%), intramuscular (93%), and rectal (30%). Ketamine is highly metabolically processed, with a plasma redistribution half‐life of 4 min and plasma terminal half‐life lasting 2.5 h. 256 Risk‐benefit evaluations have increased the focus on ketamine for patients suffering from extreme depressive conditions. However, due to the lack of current RCTs/proper placebos, reduced datasets regarding long‐term outcomes, and possible risks, ketamine treatments remain limited to the hospital scenario.
Scarce information exists regarding the optimized dosage regimen, ideal route of administration, and safety profile for repeated/long‐term use of ketamine. 384 , 385 Reduced dosing (2 mg/kg) and other routes of administration (intramuscular/intranasal) can contribute to antidepressant functions that are equivalent to standardized 0.5 mg/kg IV doses. 232 , 386 Ketamine‐induced antidepressant function is extended via multiple dosing regimens. 197 , 252 Datasets from pilot studies indicate that a limit of 6 i.v. infusions (0.5 mg/Kg) given three times weekly across a 14‐day timespan have good tolerance profiles and extend ketamine‐induced antidepressant functions accordingly. 197
9. CHALLENGES FOR USE OF KETAMINE IN MDD
Even though several psychiatrists/anesthesiologists presently administer ketamine in outpatient scenarios, major hurdles still persist regarding widespread use. Best practice standardizations need to be implemented for optimized routes of drug administration, particularly with regard to doses and dosage regimens. Multiple investigations have focused on other routes of administration, such as intranasal esketamine, although comparative analyses for various models have not been performed. Regarding dose optimization, excluding a small (n = 4) placebo‐regulated crossover investigation, all RCTs on TRDs and bipolar depressive disorders employed a dosage of 0.5 mg/kg. However, the dose‐response curve for ketamine‐driven antidepressant effect is currently being evaluated within a multicenter, psychoactive, placebo‐regulated, parallel‐design study using midazolam (0.045 mg/kg) and different ketamine infusion‐based doses. It must be emphasized that since ketamine's antidepressant function is fleeting, plans for maintaining responses/circumventing relapses are essential in clinical settings. One plan is the introduction of multiple doses (boosters), similar to maintenance treatments in electroconvulsive therapy. However, few investigations have focused on multiple‐infusion ketamine therapies for MDD or only described less than 10 infusions across 12–21‐day time frames. The risks of abuse/possible sustained adverse effects (such as cognitive issues or urinary cystitis) can increase with multiple dosing regimens. This requires intense monitoring and appropriate consultation.
Ketamine's antidepressant effect can be inflated through minimal responses to i.v. saline placebo. However, ketamine variations were not considerable, stemming from standardized placebo responses during MDD trials. Although midazolam is a more suitable placebo than saline, it has its own shortcomings, namely, reduced acute dissociative adverse effects, compromising trial blindness within savvy participating patients. Subsequent research hurdles include the development of improved controls compared to midazolam and formal evaluation of randomization expectations.
Another challenge is the recognition of subgroups with increased antidepressant responses to ketamine. Multiple nonredundant clinical predictive factors for ketamine‐driven antidepressant activities are known, including Body Mass Index, family history of alcohol abuse within close relatives, and dimensional anxious depression states. Apart from such variables, multiple genomic, innate neurobiological, and fringe factors have also been demonstrated to be associated with the antidepressant effectiveness of ketamine. However, very few investigations have merged such factors/datasets to enhance prediction potential and recognize minute influences. Stemming from MDD heterogeneity, such combination routes should be conducted through multicenter ketamine MDD consortiums to maximize population cohort sizes of aggregated subgroups regarding possible mechanism‐based investigations.
Additional concerns include confirmation of the sensitive/specific targets of ketamine and its metabolites. Such models could enhance the knowledge of ketamine antidepressant pharmacology, together with its use in glutamate‐dependent drug screens. In addition, the construction and analysis of the compound‐target‐pathway network and the protein–protein interaction (PPI) network of ketamine metabolites are shown in Figures 4 and 5. Many novel hubba proteins and MDD‐risk proteins were found, indicating that the current pharmacological mechanisms were just the tip of the iceberg.
FIGURE 4.
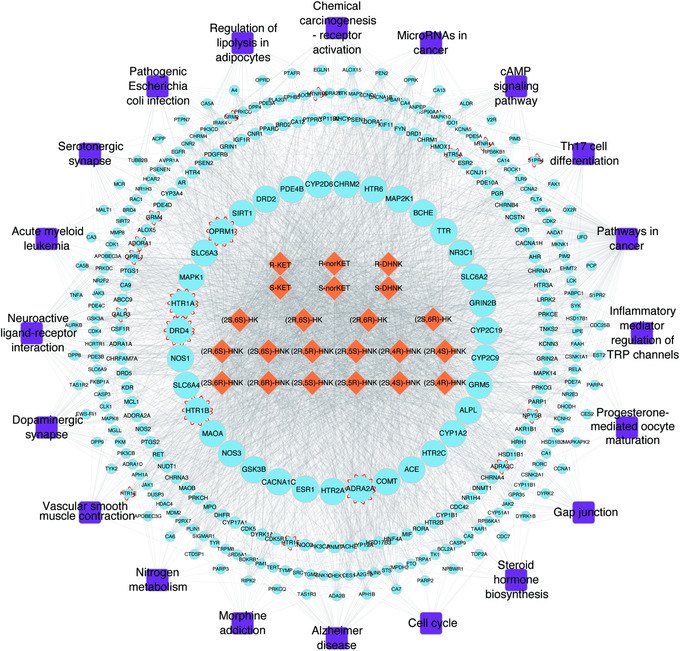
The construction and analysis of the Compound‐Target‐Pathway. The candidate ketamine metabolites (yellow diamond mesh node) are divided into the following groups based on the metabolic processes: (R,S)‐KET, (R,S)‐norKET, (R,S)‐DHNK, (2,6)‐HKs, and (2,6; 2,5; 2,4)‐HNKs. The figure was drawn by Cytoscape.
FIGURE 5.
The protein–protein interaction (PPI) networks of ketamine metabolites. The figure was drawn by Cytoscape.
Long‐term use of intravenous ketamine and intranasal esketamine was associated with an increased potential risk of addiction and suicidal incidences occurred after termination of therapy. Further studies should be conducted to explore the clinical side effects biomarkers of long‐term use of esketamine based on the technology of pharmacogenomics and clinical pharmacology. Moreover, the joint application of ketamine combined with nondrug treatment (manual‐based psychotherapies) 387 provides a comprehensive intervention for MDD. Meanwhile, such drugs must be administered solely within institutions with the necessary infrastructure and staffing resources required for safe and reliable treatment and monitoring of the patient during their therapeutic stay.
10. CONCLUSION
The recently approved esketamine formulation for TRD reflects a significant advance in psychiatric therapeutics, whereby no novel mechanism‐based agents have been generated in nearly 50 years. The identification of ketamine and its derivatives has great potential for quick and effective depression‐condition therapies. However, multiple issues must be addressed. First, ketamine is a narcotic with severe adverse effects; thus, novel drugs with lower adverse event profiles are desired. Second, even though ketamine provides fast antidepressant functions, such functions only last for 7 days, leading to patient relapses. Novel drugs that can be utilized on a daily basis are highly desired. Third, investigations are needed to clarify how novel ketamine‐driven synapses wither within 7 days and whether specific mechanisms and/or drugs exist that could possibly maintain such synaptic presence, together with similar clinical functions. Fourth, more investigations are required on the cell‐based mechanism for ketamine functions, and other similar quick‐acting drugs, to determine essential pathophysiological dysfunctions leading to depressive conditions. Fifth, prolonged effect and safety profile for ketamine therapy (over 14 days), especially regarding multiple doses, requires further research.
Future research regarding ketamine should focus on crystallizing ketamine pharmacology, clarifying the administration profile that will induce prolonged therapeutic benefits, analyzing ketamine safety profiles in multiple‐/low‐dose formulations, and recognizing novel biomarkers for differentiating ketamine response within patient cohorts.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
ETHICS APPROVAL
Not applicable.
AUTHOR CONTRIBUTION
Chuang Wang and Jia Xu conceived the manuscript; Haihua Tian, Zhenyu Hu, and Jia Xu wrote and edited the manuscript. All authors read and approved the final manuscript. Haihua Tian and Zhenyu Hu contributed equally to this work.
ACKNOWLEDGMENTS
The work was supported by the grant of the Natural Science Funds for Distinguished Young Scholar of Zhejiang (No. LR20H090001), National Natural Science Foundation of China (82171527), Ningbo Health Branding Subject Fund (PPXK2018‐08), Medical Science and Technology Project of Zhejiang Province (No: 2019KY628), Natural Science Foundation of Zhejiang Province (No: LY21H090003), Natural Science Foundation of Ningbo (No: 2019A61029), and Medical Science and Technology Project in Ningbo (2020Y20). This project is also sponsored by KC Magna funded in Ningbo University.
Tian H, Hu Z, Xu J, Wang C. The molecular pathophysiology of depression and the new therapeutics. MedComm. 2022;3:e156. 10.1002/mco2.156
Contributor Information
Jia Xu, Email: xujia@nbu.edu.cn.
Chuang Wang, Email: wangchuang@nbu.edu.cn.
DATA AVAILABILITY STATEMENT
The datasets in this study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdallah CG, Adams TG, Kelmendi B, Esterlis I, Sanacora G, Krystal JH. Ketamine's mechanism of action: a path to rapid‐acting antidepressants. Depress Anxiety. 2016;33(8):689‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruderfer DM, Walsh CG, Aguirre MW, et al. Significant shared heritability underlies suicide attempt and clinically predicted probability of attempting suicide. Mol Psychiatry. 2020;25(10):2422‐2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nemeroff CB. Recent advances in the neurobiology of depression. Psychopharmacol Bull. 2002;36(Suppl 2):6‐23. [PubMed] [Google Scholar]
- 5. Wang J, Hodes GE, Zhang H, et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat Commun. 2018;9(1):477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S. Effect of treatment on serum brain‐derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci. 2005;255(6):381‐386. [DOI] [PubMed] [Google Scholar]
- 7. Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33(6):693‐710. [DOI] [PubMed] [Google Scholar]
- 8. Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013;246:199‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Hao Y, Fan F, Zhang B. The role of microbiome in insomnia, circadian disturbance and depression. Front Psychiatry. 2018;9:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miyazaki K, Miyazaki KW, Yamanaka A, Tokuda T, Tanaka KF, Doya K. Reward probability and timing uncertainty alter the effect of dorsal raphe serotonin neurons on patience. Nat Commun. 2018;9(1):2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu B, Liu J, Wang M, Zhang Y, Li L. From serotonin to neuroplasticity: evolvement of theories for major depressive disorder. Front Cell Neurosci. 2017;11:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aleksandrova LR, Phillips AG, Wang YT. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J Psychiatry Neurosci. 2017;42(4):222‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81(3):484‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455(7215):894‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta‐analysis. Am J Psychiatry. 2000;157(10):1552‐1562. [DOI] [PubMed] [Google Scholar]
- 16. Lopez‐Leon S, Janssens AC, Gonzalez‐Zuloeta Ladd AM, et al. Meta‐analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13(8):772‐785. [DOI] [PubMed] [Google Scholar]
- 17.CONVERGE consortium. Sparse whole‐genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523(7562):588‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeng Y, Navarro P, Shirali M, et al. Genome‐wide regional heritability mapping identifies a locus within the TOX2 gene associated with major depressive disorder. Biol Psychiatry. 2017;82(5):312‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng Y, Navarro P, Fernandez‐Pujals AM, et al. A combined pathway and regional heritability analysis indicates NETRIN1 pathway is associated with major depressive disorder. Biol Psychiatry. 2017;81(4):336‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hyde CL, Nagle MW, Tian C, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48(9):1031‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wray NR, Ripke S, Mattheisen M, et al. Genome‐wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50(5):668‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li X, Luo Z, Gu C, et al. Common variants on 6q16.2, 12q24.31 and 16p13.3 are associated with major depressive disorder. Neuropsychopharmacology. 2018;43(10):2146‐2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Howard DM, Adams MJ, Clarke TK, et al. Genome‐wide meta‐analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22(3):343‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Z, Ruan M, Chen J, Fang Y. Major depressive disorder: advances in neuroscience research and translational applications. Neurosci Bull. 2021;37(6):863‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De‐Miguel FF, Trueta C. Synaptic and extrasynaptic secretion of serotonin. Cell Mol Neurobiol. 2005;25(2):297‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maes M, Yirmyia R, Noraberg J, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009;24(1):27‐53. [DOI] [PubMed] [Google Scholar]
- 27. Deakin JF, Graeff FG. 5‐HT and mechanisms of defence. J Psychopharmacol. 1991;5(4):305‐315. [DOI] [PubMed] [Google Scholar]
- 28. Xue Y, Liang H, Yang R, Deng K, Tang M, Zhang M. The role of pro‐ and mature neurotrophins in the depression. Behav Brain Res. 2021;404:113162. [DOI] [PubMed] [Google Scholar]
- 29. Babaev O, Cruces Solis H, Arban R. Dopamine modulating agents alter individual subdomains of motivation‐related behavior assessed by touchscreen procedures. Neuropharmacology. 2022:109056. [DOI] [PubMed] [Google Scholar]
- 30. Salamone JD, Ecevitoglu A, Carratala‐Ros C, et al. Complexities and paradoxes in understanding the role of dopamine in incentive motivation and instrumental action: Exertion of effort vs. anhedonia. Brain Res Bull. 2022;182:57‐66. [DOI] [PubMed] [Google Scholar]
- 31. Mercuri NB, Federici M, Rizzo FR, et al. Long‐term depression of striatal DA release induced by mGluRs via sustained hyperactivity of local cholinergic interneurons. Front Cell Neurosci. 2021;15:798464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duval F, Mokrani MC, Erb A, Gonzalez Lopera F, Danila V, Tomsa M. Neuroendocrine assessment of dopaminergic function during antidepressant treatment in major depressed patients. Brain Sci. 2021;11(4):425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaghmi A, Perez‐Mato M, Dopico‐Lopez A, Candamo‐Lourido M, Campos F, Gauthier MA. New perspectives for developing therapeutic bioconjugates of metabolite‐depleting enzymes: lessons learned combating glutamate excitotoxicity. Biomacromolecules. 2022;23:1864‐1872. [DOI] [PubMed] [Google Scholar]
- 34. Li MX, Li Q, Sun XJ, et al. Increased Homer1‐mGluR5 mediates chronic stress‐induced depressive‐like behaviors and glutamatergic dysregulation via activation of PERK‐eIF2alpha. Prog Neuropsychopharmacol Biol Psychiatry. 2019;95:109682. [DOI] [PubMed] [Google Scholar]
- 35. Chen Y, Shen M, Liu X, Xu J, Wang C. The regulation of glutamate transporter 1 in the rapid antidepressant‐like effect of ketamine in mice. Front Behav Neurosci. 2022;16:789524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tomasetti C, Montemitro C, Fiengo ALC, et al. Novel pathways in the treatment of major depression: focus on the glutamatergic system. Curr Pharm Des. 2019;25(4):381‐387. [DOI] [PubMed] [Google Scholar]
- 37. Hashimoto K. The role of glutamate on the action of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1558‐1568. [DOI] [PubMed] [Google Scholar]
- 38. Gray AL, Hyde TM, Deep‐Soboslay A, Kleinman JE, Sodhi MS. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol Psychiatry. 2015;20(9):1057‐1068. [DOI] [PubMed] [Google Scholar]
- 39. Chandley MJ, Szebeni A, Szebeni K, et al. Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. Int J Neuropsychopharmacol. 2014;17(10):1569‐1578. [DOI] [PubMed] [Google Scholar]
- 40. Musazzi L, Treccani G, Mallei A, Popoli M. The action of antidepressants on the glutamate system: regulation of glutamate release and glutamate receptors. Biol Psychiatry. 2013;73(12):1180‐1188. [DOI] [PubMed] [Google Scholar]
- 41. Kadriu B, Musazzi L, Henter ID, Graves M, Popoli M, Zarate CA, Jr . Glutamatergic neurotransmission: pathway to developing novel rapid‐acting antidepressant treatments. Int J Neuropsychopharmacol. 2019;22(2):119‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beurel E, Grieco SF, Amadei C, Downey K, Jope RS. Ketamine‐induced inhibition of glycogen synthase kinase‐3 contributes to the augmentation of alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid (AMPA) receptor signaling. Bipolar Disord. 2016;18(6):473‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gould TD, O'Donnell KC, Dow ER, Du J, Chen G, Manji HK. Involvement of AMPA receptors in the antidepressant‐like effects of lithium in the mouse tail suspension test and forced swim test. Neuropharmacology. 2008;54(3):577‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102(1):75‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang X, Liu Y, Hong X, et al. NG2 glia‐derived GABA release tunes inhibitory synapses and contributes to stress‐induced anxiety. Nat Commun. 2021;12(1):5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lowes DC, Chamberlin LA, Kretsge LN, et al. Ventral tegmental area GABA neurons mediate stress‐induced blunted reward‐seeking in mice. Nat Commun. 2021;12(1):3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farrell MR, Esteban JSD, Faget L, Floresco SB, Hnasko TS, Mahler SV. Ventral pallidum GABA neurons mediate motivation underlying risky choice. J Neurosci. 2021;41(20):4500‐4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: does GABA play a role in depression? Biol Psychiatry. 1995;38(9):578‐591. [DOI] [PubMed] [Google Scholar]
- 49. Ghosal S, Hare B, Duman RS. Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr Opin Behav Sci. 2017;14:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fee C, Banasr M, Sibille E. Somatostatin‐positive gamma‐aminobutyric acid interneuron deficits in depression: cortical microcircuit and therapeutic perspectives. Biol Psychiatry. 2017;82(8):549‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel‐Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32(2):471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schur RR, Draisma LW, Wijnen JP, et al. Brain GABA levels across psychiatric disorders: a systematic literature review and meta‐analysis of (1) H‐MRS studies. Hum Brain Mapp. 2016;37(9):3337‐3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mann JJ, Oquendo MA, Watson KT, et al. Anxiety in major depression and cerebrospinal fluid free gamma‐aminobutyric acid. Depress Anxiety. 2014;31(10):814‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guilloux JP, Douillard‐Guilloux G, Kota R, et al. Molecular evidence for BDNF‐ and GABA‐related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17(11):1130‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karolewicz B, Maciag D, O'Dwyer G, Stockmeier CA, Feyissa AM, Rajkowska G. Reduced level of glutamic acid decarboxylase‐67 kDa in the prefrontal cortex in major depression. Int J Neuropsychopharmacol. 2010;13(4):411‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lener MS, Niciu MJ, Ballard ED, et al. Glutamate and gamma‐aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2017;81(10):886‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen X, van Gerven J, Cohen A, Jacobs G. Human pharmacology of positive GABA‐A subtype‐selective receptor modulators for the treatment of anxiety. Acta Pharmacol Sin. 2019;40(5):571‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ren Z, Pribiag H, Jefferson SJ, et al. Bidirectional homeostatic regulation of a depression‐related brain state by gamma‐aminobutyric acidergic deficits and ketamine treatment. Biol Psychiatry. 2016;80(6):457‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kolata SM, Nakao K, Jeevakumar V, et al. Neuropsychiatric phenotypes produced by GABA reduction in mouse cortex and hippocampus. Neuropsychopharmacology. 2018;43(6):1445‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tan X, Zhang L, Wang D, et al. Influence of early life stress on depression: from the perspective of neuroendocrine to the participation of gut microbiota. Aging (Albany NY). 2021;13(23):25588‐25601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sukhareva EV. The role of the corticotropin‐releasing hormone and its receptors in the regulation of stress response. Vavilovskii Zhurnal Genet Selektsii. 2021;25(2):216‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol. 1991;38(5):537‐359. [DOI] [PubMed] [Google Scholar]
- 63. Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31(9):464‐468. [DOI] [PubMed] [Google Scholar]
- 64. Keller J, Gomez R, Williams G, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry. 2017;22(4):527‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gomez RG, Fleming SH, Keller J, et al. The neuropsychological profile of psychotic major depression and its relation to cortisol. Biol Psychiatry. 2006;60(5):472‐478. [DOI] [PubMed] [Google Scholar]
- 66. Lamers F, Vogelzangs N, Merikangas KR, de Jonge P, Beekman AT, Penninx BW. Evidence for a differential role of HPA‐axis function, inflammation and metabolic syndrome in melancholic versus atypical depression. Mol Psychiatry. 2013;18(6):692‐699. [DOI] [PubMed] [Google Scholar]
- 67. Nandam LS, Brazel M, Zhou M, Jhaveri DJ. Cortisol and Major depressive disorder‐translating findings from humans to animal models and back. Front Psychiatry. 2019;10:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Owashi T, Otsubo T, Oshima A, Nakagome K, Higuchi T, Kamijima K. Longitudinal neuroendocrine changes assessed by dexamethasone/CRH and growth hormone releasing hormone tests in psychotic depression. Psychoneuroendocrinology. 2008;33(2):152‐161. [DOI] [PubMed] [Google Scholar]
- 69. Mickey BJ, Ginsburg Y, Sitzmann AF, et al. Cortisol trajectory, melancholia, and response to electroconvulsive therapy. J Psychiatr Res. 2018;103:46‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stetler C, Miller GE. Depression and hypothalamic‐pituitary‐adrenal activation: a quantitative summary of four decades of research. Psychosom Med. 2011;73(2):114‐126. [DOI] [PubMed] [Google Scholar]
- 71. Aubry JM. CRF system and mood disorders. J Chem Neuroanat. 2013;54:20‐24. [DOI] [PubMed] [Google Scholar]
- 72. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid‐acting antidepressants. Nat Med. 2016;22(3):238‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10(9):1089‐1093. [DOI] [PubMed] [Google Scholar]
- 74. Li K, Shen S, Ji YT, Li XY, Zhang LS, Wang XD. Melatonin augments the effects of fluoxetine on depression‐like behavior and hippocampal BDNF‐TrkB signaling. Neurosci Bull. 2018;34(2):303‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang JJ, Gao TT, Wang Y, et al. Andrographolide exerts significant antidepressant‐like effects involving the hippocampal BDNF system in mice. Int J Neuropsychopharmacol. 2019;22(9):585‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang JQ, Mao L. The ERK pathway: molecular mechanisms and treatment of depression. Mol Neurobiol. 2019;56(9):6197‐6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM. Decreased serum brain‐derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002;109(2):143‐148. [DOI] [PubMed] [Google Scholar]
- 78. Satomura E, Baba H, Nakano Y, Maeshima H, Suzuki T, Arai H. Correlations between brain‐derived neurotrophic factor and clinical symptoms in medicated patients with major depression. J Affect Disord. 2011;135(1‐3):332‐335. [DOI] [PubMed] [Google Scholar]
- 79. Bocchio‐Chiavetto L, Bagnardi V, Zanardini R, et al. Serum and plasma BDNF levels in major depression: a replication study and meta‐analyses. World J Biol Psychiatry. 2010;11(6):763‐773. [DOI] [PubMed] [Google Scholar]
- 80. Duman RS, Monteggia LM. A neurotrophic model for stress‐related mood disorders. Biol Psychiatry. 2006;59(12):1116‐1127. [DOI] [PubMed] [Google Scholar]
- 81. Dwivedi Y. Brain‐derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat. 2009;5:433‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chiou YJ, Huang TL. Serum brain‐derived neurotrophic factors in taiwanese patients with drug‐naive first‐episode major depressive disorder: effects of antidepressants. Int J Neuropsychopharmacol. 2017;20(3):213‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Youssef MM, Underwood MD, Huang YY, et al. Association of BDNF Val66Met polymorphism and brain BDNF levels with major depression and suicide. Int J Neuropsychopharmacol. 2018;21(6):528‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li Z, Zhang Y, Wang Z, et al. The role of BDNF, NTRK2 gene and their interaction in development of treatment‐resistant depression: data from multicenter, prospective, longitudinal clinic practice. J Psychiatr Res. 2013;47(1):8‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jeon SW, Kim YK. Inflammation‐induced depression: Its pathophysiology and therapeutic implications. J Neuroimmunol. 2017;313:92‐98. [DOI] [PubMed] [Google Scholar]
- 86. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol. 2002;90(12):1279‐1283. [DOI] [PubMed] [Google Scholar]
- 88. Kop WJ, Gottdiener JS, Tangen CM, et al. Inflammation and coagulation factors in persons >65 years of age with symptoms of depression but without evidence of myocardial ischemia. Am J Cardiol. 2002;89(4):419‐424. [DOI] [PubMed] [Google Scholar]
- 89. Suarez EC, Krishnan RR, Lewis JG. The relation of severity of depressive symptoms to monocyte‐associated proinflammatory cytokines and chemokines in apparently healthy men. Psychosom Med. 2003;65(3):362‐368. [DOI] [PubMed] [Google Scholar]
- 90. Ambrosio G, Kaufmann FN, Manosso L, et al. Depression and peripheral inflammatory profile of patients with obesity. Psychoneuroendocrinology. 2018;91:132‐141. [DOI] [PubMed] [Google Scholar]
- 91. Mao R, Zhang C, Chen J, et al. Different levels of pro‐ and anti‐inflammatory cytokines in patients with unipolar and bipolar depression. J Affect Disord. 2018;237:65‐72. [DOI] [PubMed] [Google Scholar]
- 92. Enache D, Pariante CM, Mondelli V. Markers of central inflammation in major depressive disorder: a systematic review and meta‐analysis of studies examining cerebrospinal fluid, positron emission tomography and post‐mortem brain tissue. Brain Behav Immun. 2019;81:24‐40. [DOI] [PubMed] [Google Scholar]
- 93. Haroon E, Daguanno AW, Woolwine BJ, et al. Antidepressant treatment resistance is associated with increased inflammatory markers in patients with major depressive disorder. Psychoneuroendocrinology. 2018;95:43‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Syed SA, Beurel E, Loewenstein DA, et al. Defective inflammatory pathways in never‐treated depressed patients are associated with poor treatment response. Neuron. 2018;99(5):914‐924 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li Z, Qi D, Chen J, et al. Venlafaxine inhibits the upregulation of plasma tumor necrosis factor‐alpha (TNF‐alpha) in the Chinese patients with major depressive disorder: a prospective longitudinal study. Psychoneuroendocrinology. 2013;38(1):107‐114. [DOI] [PubMed] [Google Scholar]
- 96. Kohler CA, Freitas TH, Stubbs B, et al. Peripheral alterations in cytokine and chemokine levels after antidepressant drug treatment for major depressive disorder: systematic review and meta‐analysis. Mol Neurobiol. 2018;55(5):4195‐4206. [DOI] [PubMed] [Google Scholar]
- 97. Bavaresco DV, Uggioni MLR, Ferraz SD, et al. Efficacy of infliximab in treatment‐resistant depression: a systematic review and meta‐analysis. Pharmacol Biochem Behav. 2020;188:172838. [DOI] [PubMed] [Google Scholar]
- 98. Innes S, Pariante CM, Borsini A. Microglial‐driven changes in synaptic plasticity: a possible role in major depressive disorder. Psychoneuroendocrinology. 2019;102:236‐247. [DOI] [PubMed] [Google Scholar]
- 99. Li B, Yang W, Ge T, Wang Y, Cui R. Stress induced microglial activation contributes to depression. Pharmacol Res. 2022;179:106145. [DOI] [PubMed] [Google Scholar]
- 100. Weng L, Dong S, Wang S, Yi L, Geng D. Macranthol attenuates lipopolysaccharide‐induced depressive‐like behaviors by inhibiting neuroinflammation in prefrontal cortex. Physiol Behav. 2019;204:33‐40. [DOI] [PubMed] [Google Scholar]
- 101. Virmani G, D'Almeida P, Nandi A, Marathe S. Subfield‐specific effects of chronic mild unpredictable stress on hippocampal astrocytes. Eur J Neurosci. 2021;54(5):5730‐5746. [DOI] [PubMed] [Google Scholar]
- 102. Sochocka M, Diniz BS, Leszek J. Inflammatory response in the CNS: friend or foe? Mol Neurobiol. 2017;54(10):8071‐8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Silva DA, Coutinho E, Ferriani LO, Viana MC. Depression subtypes and obesity in adults: a systematic review and meta‐analysis. Obes Rev. 2020;21(3):e12966. [DOI] [PubMed] [Google Scholar]
- 104. Rao WW, Zong QQ, Zhang JW, et al. Obesity increases the risk of depression in children and adolescents: results from a systematic review and meta‐analysis. J Affect Disord. 2020;267:78‐85. [DOI] [PubMed] [Google Scholar]
- 105. Duarte‐Silva E, de Melo MG, Maes M, Filho A, Macedo D, Peixoto CA. Shared metabolic and neuroimmune mechanisms underlying type 2 diabetes mellitus and major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111:110351. [DOI] [PubMed] [Google Scholar]
- 106. Zhang F, Jia Z, Gao P, et al. Metabonomics study of urine and plasma in depression and excess fatigue rats by ultra fast liquid chromatography coupled with ion trap‐time of flight mass spectrometry. Mol Biosyst. 2010;6(5):852‐861. [DOI] [PubMed] [Google Scholar]
- 107. Shi B, Tian J, Xiang H, et al. A (1)H‐NMR plasma metabonomic study of acute and chronic stress models of depression in rats. Behav Brain Res. 2013;241:86‐91. [DOI] [PubMed] [Google Scholar]
- 108. Zheng P, Wang Y, Chen L, et al. Identification and validation of urinary metabolite biomarkers for major depressive disorder. Mol Cell Proteomics. 2013;12(1):207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zheng P, Gao HC, Li Q, et al. Plasma metabonomics as a novel diagnostic approach for major depressive disorder. J Proteome Res. 2012;11(3):1741‐1748. [DOI] [PubMed] [Google Scholar]
- 110. Li ZY, Zheng XY, Gao XX, et al. Study of plasma metabolic profiling and biomarkers of chronic unpredictable mild stress rats based on gas chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(24):3539‐3546. [DOI] [PubMed] [Google Scholar]
- 111. Chen G, Yang D, Yang Y, et al. Amino acid metabolic dysfunction revealed in the prefrontal cortex of a rat model of depression. Behav Brain Res. 2015;278:286‐292. [DOI] [PubMed] [Google Scholar]
- 112. Gao X, Guo B, Yang L, et al. Selection and dynamic metabolic response of rat biomarkers by metabonomics and multivariate statistical analysis combined with GC‐MS. Pharmacol Biochem Behav. 2014;117:85‐91. [DOI] [PubMed] [Google Scholar]
- 113. Sandhu KV, Sherwin E, Schellekens H, Stanton C, Dinan TG, Cryan JF. Feeding the microbiota‐gut‐brain axis: diet, microbiome, and neuropsychiatry. Transl Res. 2017;179:223‐244. [DOI] [PubMed] [Google Scholar]
- 114. Gonzalez‐Arancibia C, Urrutia‐Pinones J, Illanes‐Gonzalez J, et al. Do your gut microbes affect your brain dopamine? Psychopharmacology (Berl). 2019;236(5):1611‐1622. [DOI] [PubMed] [Google Scholar]
- 115. Suda K, Matsuda K. How microbes affect depression: underlying mechanisms via the gut‐brain axis and the modulating role of probiotics. Int J Mol Sci. 2022;23(3):1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Forssten SD, Ouwehand AC, Griffin SM, Patterson E. One giant leap from mouse to man: the microbiota‐gut‐brain axis in mood disorders and translational challenges moving towards human clinical trials. Nutrients. 2022;14(3):568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bhatt S, Kanoujia J, Mohanalakshmi S, et al. Role of brain‐gut‐microbiota axis in depression: emerging therapeutic avenues. CNS Neurol Disord Drug Targets. 2022;21. [DOI] [PubMed] [Google Scholar]
- 118. Kennedy PJ, Cryan JF, Dinan TG, Clarke G. Irritable bowel syndrome: a microbiome‐gut‐brain axis disorder? World J Gastroenterol. 2014;20(39):14105‐14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Whitehead WE, Palsson O, Jones KR. Systematic review of the comorbidity of irritable bowel syndrome with other disorders: what are the causes and implications? Gastroenterology. 2002;122(4):1140‐1156. [DOI] [PubMed] [Google Scholar]
- 120. Ruepert L, Quartero AO, de Wit NJ, van der Heijden GJ, Rubin G, Muris JW. Bulking agents, antispasmodics and antidepressants for the treatment of irritable bowel syndrome. Cochrane Database Syst Rev. 2011;(8):CD003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Knudsen JK, Bundgaard‐Nielsen C, Hjerrild S, Nielsen RE, Leutscher P, Sorensen S. Gut microbiota variations in patients diagnosed with major depressive disorder—a systematic review. Brain Behav. 2021;11(7):e02177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Aizawa E, Tsuji H, Asahara T, et al. Possible association of Bifidobacterium and Lactobacillus in the gut microbiota of patients with major depressive disorder. J Affect Disord. 2016;202:254‐257. [DOI] [PubMed] [Google Scholar]
- 123. Marin IA, Goertz JE, Ren T, et al. Microbiota alteration is associated with the development of stress‐induced despair behavior. Sci Rep. 2017;7:43859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zheng P, Zeng B, Zhou C, et al. Gut microbiome remodeling induces depressive‐like behaviors through a pathway mediated by the host's metabolism. Mol Psychiatry. 2016;21(6):786‐796. [DOI] [PubMed] [Google Scholar]
- 125. Curtis K, Stewart CJ, Robinson M, et al. Insular resting state functional connectivity is associated with gut microbiota diversity. Eur J Neurosci. 2019;50(3):2446‐2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. O'Mahony SM, Marchesi JR, Scully P, et al. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65(3):263‐267. [DOI] [PubMed] [Google Scholar]
- 127. Garcia‐Rodenas CL, Bergonzelli GE, Nutten S, et al. Nutritional approach to restore impaired intestinal barrier function and growth after neonatal stress in rats. J Pediatr Gastroenterol Nutr. 2006;43(1):16‐24. [DOI] [PubMed] [Google Scholar]
- 128. Hao Z, Wang W, Guo R, Liu H. Faecalibacterium prausnitzii (ATCC 27766) has preventive and therapeutic effects on chronic unpredictable mild stress‐induced depression‐like and anxiety‐like behavior in rats. Psychoneuroendocrinology. 2019;104:132‐142. [DOI] [PubMed] [Google Scholar]
- 129. Messaoudi M, Violle N, Bisson JF, Desor D, Javelot H, Rougeot C. Beneficial psychological effects of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in healthy human volunteers. Gut Microbes. 2011;2(4):256‐261. [DOI] [PubMed] [Google Scholar]
- 130. Rudzki L, Ostrowska L, Pawlak D, et al. Probiotic Lactobacillus Plantarum 299v decreases kynurenine concentration and improves cognitive functions in patients with major depression: a double‐blind, randomized, placebo controlled study. Psychoneuroendocrinology. 2019;100:213‐222. [DOI] [PubMed] [Google Scholar]
- 131. Foster JA, McVey Neufeld KA. Gut‐brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 2013;36(5):305‐312. [DOI] [PubMed] [Google Scholar]
- 132. Mittal R, Debs LH, Patel AP, et al. Neurotransmitters: the critical modulators regulating gut‐brain axis. J Cell Physiol. 2017;232(9):2359‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Diviccaro S, Giatti S, Borgo F, et al. Treatment of male rats with finasteride, an inhibitor of 5alpha‐reductase enzyme, induces long‐lasting effects on depressive‐like behavior, hippocampal neurogenesis, neuroinflammation and gut microbiota composition. Psychoneuroendocrinology. 2019;99:206‐215. [DOI] [PubMed] [Google Scholar]
- 134. Kiecolt‐Glaser JK, Wilson SJ, Bailey ML, et al. Marital distress, depression, and a leaky gut: translocation of bacterial endotoxin as a pathway to inflammation. Psychoneuroendocrinology. 2018;98:52‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Lindqvist D, Dhabhar FS, James SJ, et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology. 2017;76:197‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Czarny P, Wigner P, Galecki P, Sliwinski T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2018;80(Pt C):309‐321. [DOI] [PubMed] [Google Scholar]
- 137. Xie X, Yu C, Zhou J, et al. Nicotinamide mononucleotide ameliorates the depression‐like behaviors and is associated with attenuating the disruption of mitochondrial bioenergetics in depressed mice. J Affect Disord. 2020;263:166‐174. [DOI] [PubMed] [Google Scholar]
- 138. Wang XL, Yuan K, Zhang W, Li SX, Gao GF, Lu L. Regulation of circadian genes by the MAPK pathway: implications for rapid antidepressant action. Neurosci Bull. 2020;36(1):66‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Xie X, Shen Q, Yu C, et al. Depression‐like behaviors are accompanied by disrupted mitochondrial energy metabolism in chronic corticosterone‐induced mice. J Steroid Biochem Mol Biol. 2020;200:105607. [DOI] [PubMed] [Google Scholar]
- 140. Zhang LF, Shi L, Liu H, et al. Increased hippocampal tau phosphorylation and axonal mitochondrial transport in a mouse model of chronic stress. Int J Neuropsychopharmacol. 2012;15(3):337‐348. [DOI] [PubMed] [Google Scholar]
- 141. Wu Q, Xu Y, Bao Y, Alvarez J, Gonzales ML. Tricyclic antidepressant use and risk of fractures: a meta‐analysis of cohort studies through the use of both frequentist and Bayesian approaches. J Clin Med. 2020;9(8):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Glowinski J, Axelrod J. Inhibition of uptake of tritiated‐noradrenaline in the intact rat brain by imipramine and structurally related compounds. Nature. 1964;204:1318‐1319. [DOI] [PubMed] [Google Scholar]
- 143. Carlsson A, Corrodi H, Fuxe K, Hokfelt T. Effect of antidepressant drugs on the depletion of intraneuronal brain 5‐hydroxytryptamine stores caused by 4‐methyl‐alpha‐ethyl‐meta‐tyramine. Eur J Pharmacol. 1969;5(4):357‐366. [DOI] [PubMed] [Google Scholar]
- 144. Ogren SO, Fuxe K, Agnati LF, Gustafsson JA, Jonsson G, Holm AC. Reevaluation of the indoleamine hypothesis of depression. Evidence for a reduction of functional activity of central 5‐HT systems by antidepressant drugs. J Neural Transm. 1979;46(2):85‐103. [DOI] [PubMed] [Google Scholar]
- 145. Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp Clin Psychopharmacol. 2015;23(1):1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sabella D. Antidepressant Medications. Am J Nurs. 2018;118(9):52‐59. [DOI] [PubMed] [Google Scholar]
- 147. Kern DM, Cepeda MS, Castilla‐Puentes RC, Savitz A, Etropolski M. Characteristics of patients with major depressive disorder switching SSRI/SNRI therapy compared with those augmenting with an atypical antipsychotic in a real‐world setting. Curr Med Res Opin. 2021;37(8):1377‐1384. [DOI] [PubMed] [Google Scholar]
- 148. Papakostas GI. Tolerability of modern antidepressants. J Clin Psychiatry. 2008;69(Suppl E1):8‐13. [PubMed] [Google Scholar]
- 149. Bymaster FP, Dreshfield‐Ahmad LJ, Threlkeld PG, et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25(6):871‐880. [DOI] [PubMed] [Google Scholar]
- 150. Millan MJ, Gobert A, Lejeune F, et al. S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther. 2001;298(2):565‐580. [PubMed] [Google Scholar]
- 151. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283(3):1305‐1322. [PubMed] [Google Scholar]
- 152. Sanchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol. 1999;19(4):467‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Vaishnavi SN, Nemeroff CB, Plott SJ, Rao SG, Kranzler J, Owens MJ. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biol Psychiatry. 2004;55(3):320‐322. [DOI] [PubMed] [Google Scholar]
- 154. Papakostas GI, Thase ME, Fava M, Nelson JC, Shelton RC. Are antidepressant drugs that combine serotonergic and noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating major depressive disorder? A meta‐analysis of studies of newer agents. Biol Psychiatry. 2007;62(11):1217‐1227. [DOI] [PubMed] [Google Scholar]
- 155. Stahl SM, Grady MM, Moret C, Briley M. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10(9):732‐747. [DOI] [PubMed] [Google Scholar]
- 156. Clayton AH, Pradko JF, Croft HA, et al. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63(4):357‐366. [DOI] [PubMed] [Google Scholar]
- 157. Feighner JP. Mechanism of action of antidepressant medications. J Clin Psychiatry. 1999;60(Suppl 4):4‐11; discussion 12–3. [PubMed] [Google Scholar]
- 158. Fagiolini A, Comandini A, Catena Dell'Osso M, Kasper S. Rediscovering trazodone for the treatment of major depressive disorder. CNS Drugs. 2012;26(12):1033‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699‐711. [DOI] [PubMed] [Google Scholar]
- 160. Letchworth SR, Smith HR, Porrino LJ, et al. Characterization of a tropane radioligand, [(3)H]2beta‐propanoyl‐3beta‐(4‐tolyl) tropane ([(3)H]PTT), for dopamine transport sites in rat brain. J Pharmacol Exp Ther. 2000;293(2):686‐696. [PubMed] [Google Scholar]
- 161. Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol. 1994;45(1):125‐135. [PubMed] [Google Scholar]
- 162. Cusack B, Nelson A, Richelson E. Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology (Berl). 1994;114(4):559‐565. [DOI] [PubMed] [Google Scholar]
- 163. Feighner JP, Meredith CH, Stern WC, Hendrickson G, Miller LL. A double‐blind study of bupropion and placebo in depression. Am J Psychiatry. 1984;141(4):525‐529. [DOI] [PubMed] [Google Scholar]
- 164. Feighner J, Hendrickson G, Miller L, Stern W. Double‐blind comparison of doxepin versus bupropion in outpatients with a major depressive disorder. J Clin Psychopharmacol. 1986;6(1):27‐32. [PubMed] [Google Scholar]
- 165. Moreira R. The efficacy and tolerability of bupropion in the treatment of major depressive disorder. Clin Drug Investig. 2011;31(Suppl 1):5‐17. [DOI] [PubMed] [Google Scholar]
- 166. Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned‐Coughlin S. A review of the neuropharmacology of Bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Prim Care Companion J Clin Psychiatry. 2004;6(4):159‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Croom KF, Perry CM, Plosker GL. Mirtazapine: a review of its use in major depression and other psychiatric disorders. CNS Drugs. 2009;23(5):427‐452. [DOI] [PubMed] [Google Scholar]
- 168. Benjamin S, Doraiswamy PM. Review of the use of mirtazapine in the treatment of depression. Expert Opin Pharmacother. 2011;12(10):1623‐1632. [DOI] [PubMed] [Google Scholar]
- 169. Nagao K, Kishi T, Moriwaki M, et al. Comparative clinical profile of mirtazapine and duloxetine in practical clinical settings in Japan: a 4‐week open‐label, parallel‐group study of major depressive disorder. Neuropsychiatr Dis Treat. 2013;9:781‐786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Mathur A, Chowdhary A, Jain M. A comparative study of the efficacy and safety of mirtazapine versus amitriptyline in the treatment of major depression. Indian J Psychiatry. 2002;44(3):260‐265. [PMC free article] [PubMed] [Google Scholar]
- 171. Nutt DJ. Tolerability and safety aspects of mirtazapine. Hum Psychopharmacol. 2002;17(Suppl 1):S37‐41. [DOI] [PubMed] [Google Scholar]
- 172. Katona CL, Katona CP. New generation multi‐modal antidepressants: focus on vortioxetine for major depressive disorder. Neuropsychiatr Dis Treat. 2014;10:349‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Bang‐Andersen B, Ruhland T, Jorgensen M, et al. Discovery of 1‐[2‐(2,4‐dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem. 2011;54(9):3206‐3221. [DOI] [PubMed] [Google Scholar]
- 174. Mork A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666‐675. [DOI] [PubMed] [Google Scholar]
- 175. Zemanova N, Anzenbacher P, Anzenbacherova E. The role of cytochromes P450 in metabolism of selected antidepressants and anxiolytics under psychological stress. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2022;166:140‐149. [DOI] [PubMed] [Google Scholar]
- 176. Alam MY, Jacobsen PL, Chen Y, Serenko M, Mahableshwarkar AR. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open‐label, flexible‐dose, 52‐week extension study. Int Clin Psychopharmacol. 2014;29(1):36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Boulenger JP, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. J Psychopharmacol. 2012;26(11):1408‐1416. [DOI] [PubMed] [Google Scholar]
- 178. Gibb A, Deeks ED. Vortioxetine: first global approval. Drugs. 2014;74(1):135‐145. [DOI] [PubMed] [Google Scholar]
- 179. Pearce EF, Murphy JA. Vortioxetine for the treatment of depression. Ann Pharmacother. 2014;48(6):758‐765. [DOI] [PubMed] [Google Scholar]
- 180. Gibiino S, Marsano A, Serretti A. Specificity profile of venlafaxine and sertraline in major depression: metaregression of double‐blind, randomized clinical trials. Int J Neuropsychopharmacol. 2014;17(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 181. du Jardin KG, Jensen JB, Sanchez C, Pehrson AL. Vortioxetine dose‐dependently reverses 5‐HT depletion‐induced deficits in spatial working and object recognition memory: a potential role for 5‐HT1A receptor agonism and 5‐HT3 receptor antagonism. Eur Neuropsychopharmacol. 2014;24(1):160‐171. [DOI] [PubMed] [Google Scholar]
- 182. Jensen JB, du Jardin KG, Song D, et al. Vortioxetine, but not escitalopram or duloxetine, reverses memory impairment induced by central 5‐HT depletion in rats: evidence for direct 5‐HT receptor modulation. Eur Neuropsychopharmacol. 2014;24(1):148‐159. [DOI] [PubMed] [Google Scholar]
- 183. Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7(2):137‐151. [DOI] [PubMed] [Google Scholar]
- 184. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: A midazolam‐controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Mathew SJ, Shah A, Lapidus K, et al. Ketamine for treatment‐resistant unipolar depression: current evidence. CNS Drugs. 2012;26(3):189‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Lodge D, Anis NA, Burton NR. Effects of optical isomers of ketamine on excitation of cat and rat spinal neurones by amino acids and acetylcholine. Neurosci Lett. 1982;29(3):281‐286. [DOI] [PubMed] [Google Scholar]
- 187. Crane GE. Cyloserine as an antidepressant agent. Am J Psychiatry. 1959;115(11):1025‐1026. [DOI] [PubMed] [Google Scholar]
- 188. Domino EF, Chodoff P, Corssen G. Pharmacologic effects of Ci‐581, a new dissociative anesthetic, in man. Clin Pharmacol Ther. 1965;6:279‐291. [DOI] [PubMed] [Google Scholar]
- 189. Dundee JW, Knox JW, Black GW, et al. Ketamine as an induction agent in anaesthetics. Lancet. 1970;1(7661):1370‐1371. [DOI] [PubMed] [Google Scholar]
- 190. Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Ther. 2013;19(6):370‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Sofia RD, Harakal JJ. Evaluation of ketamine HCl for anti‐depressant activity. Arch Int Pharmacodyn Ther. 1975;214(1):68‐74. [PubMed] [Google Scholar]
- 192. Khorramzadeh E, Lotfy AO. The use of ketamine in psychiatry. Psychosomatics. 1973;14(6):344‐346. [DOI] [PubMed] [Google Scholar]
- 193. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351‐354. [DOI] [PubMed] [Google Scholar]
- 194. Zarate CA, Jr. , Singh JB, Carlson PJ, et al. A randomized trial of an N‐methyl‐D‐aspartate antagonist in treatment‐resistant major depression. Arch Gen Psychiatry. 2006;63(8):856‐864. [DOI] [PubMed] [Google Scholar]
- 195. Zarate CA, Jr. , Brutsche NE, Ibrahim L, et al. Replication of ketamine's antidepressant efficacy in bipolar depression: a randomized controlled add‐on trial. Biol Psychiatry. 2012;71(11):939‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment‐resistant major depression: a two‐site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Murrough JW, Perez AM, Pillemer S, et al. Rapid and longer‐term antidepressant effects of repeated ketamine infusions in treatment‐resistant major depression. Biol Psychiatry. 2013;74(4):250‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Krystal JH, Sanacora G, Duman RS. Rapid‐acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):1133‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Coyle CM, Laws KR. The use of ketamine as an antidepressant: a systematic review and meta‐analysis. Hum Psychopharmacol. 2015;30(3):152‐163. [DOI] [PubMed] [Google Scholar]
- 200. Wilkinson ST, Ballard ED, Bloch MH, et al. The Effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta‐analysis. Am J Psychiatry. 2018;175(2):150‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Ibrahim L, Diazgranados N, Franco‐Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add‐on riluzole: results from a 4‐week, double‐blind, placebo‐controlled study. Neuropsychopharmacology. 2012;37(6):1526‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Ochs‐Ross R, Daly EJ, Zhang Y, et al. Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment‐resistant depression‐TRANSFORM‐3. Am J Geriatr Psychiatry. 2020;28(2):121‐141. [DOI] [PubMed] [Google Scholar]
- 203. Daly EJ, Trivedi MH, Janik A, et al. Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment‐resistant depression: a randomized clinical trial. JAMA Psychiatry. 2019;76(9):893‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Wajs E, Aluisio L, Holder R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment‐resistant depression: assessment of long‐term safety in a phase 3, open‐label study (SUSTAIN‐2). J Clin Psychiatry. 2020;81(3). [DOI] [PubMed] [Google Scholar]
- 205. Canuso CM, Ionescu DF, Li X, et al. Esketamine nasal spray for the rapid reduction of depressive symptoms in major depressive disorder with acute suicidal ideation or behavior. J Clin Psychopharmacol. 2021;41(5):516‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated‐dose intravenous ketamine for treatment‐resistant depression. Biol Psychiatry. 2010;67(2):139‐145. [DOI] [PubMed] [Google Scholar]
- 207. Thomas RK, Baker G, Lind J, Dursun S. Rapid effectiveness of intravenous ketamine for ultraresistant depression in a clinical setting and evidence for baseline anhedonia and bipolarity as clinical predictors of effectiveness. J Psychopharmacol. 2018;32(10):1110‐1117. [DOI] [PubMed] [Google Scholar]
- 208. Zanos P, Thompson SM, Duman RS, Zarate CA, Jr. , Gould TD. Convergent mechanisms underlying rapid antidepressant action. CNS Drugs. 2018;32(3):197‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Breen E. Ketamine for suicidal ideation: heed lessons from opiate epidemic. BMJ. 2022;376:o597. [DOI] [PubMed] [Google Scholar]
- 210. Abbar M, Demattei C, El‐Hage W, et al. Ketamine for the acute treatment of severe suicidal ideation: double blind, randomised placebo controlled trial. BMJ. 2022;376:e067194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Pulcu E, Guinea C, Cowen PJ, Murphy SE, Harmer CJ. A translational perspective on the anti‐anhedonic effect of ketamine and its neural underpinnings. Mol Psychiatry. 2022;27(1):81‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Duman RS. Ketamine and rapid‐acting antidepressants: a new era in the battle against depression and suicide. F1000Res. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Canuso CM, Singh JB, Fedgchin M, et al. Efficacy and Safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double‐blind, randomized, placebo‐controlled study. Am J Psychiatry. 2018;175(7):620‐630. [DOI] [PubMed] [Google Scholar]
- 214. Preskorn SH. Consistency of the Antidepressant effect of intranasal esketamine in phase 3 clinical trials. J Psychiatr Pract. 2021;27(2):115‐120. [DOI] [PubMed] [Google Scholar]
- 215. Fedgchin M, Trivedi M, Daly EJ, et al. Efficacy and safety of fixed‐dose esketamine nasal spray combined with a new oral antidepressant in treatment‐resistant depression: results of a randomized, double‐blind, active‐controlled study (TRANSFORM‐1). Int J Neuropsychopharmacol. 2019;22(10):616‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Cristea IA, Naudet F. US Food and Drug Administration approval of esketamine and brexanolone. Lancet Psychiatry. 2019;6(12):975‐977. [DOI] [PubMed] [Google Scholar]
- 217. Kim J, Farchione T, Potter A, Chen Q, Temple R. Esketamine for treatment‐resistant depression ‐ first FDA‐approved antidepressant in a new class. N Engl J Med. 2019;381(1):1‐4. [DOI] [PubMed] [Google Scholar]
- 218. Yang C, Kobayashi S, Nakao K, et al. AMPA receptor activation‐independent antidepressant actions of ketamine metabolite (S)‐norketamine. Biol Psychiatry. 2018;84(8):591‐600. [DOI] [PubMed] [Google Scholar]
- 219. Yokoyama R, Higuchi M, Tanabe W, et al. (S)‐norketamine and (2S,6S)‐hydroxynorketamine exert potent antidepressant‐like effects in a chronic corticosterone‐induced mouse model of depression. Pharmacol Biochem Behav. 2020;191:172876. [DOI] [PubMed] [Google Scholar]
- 220. Hashimoto K, Yang C. Is (S)‐norketamine an alternative antidepressant for esketamine? Eur Arch Psychiatry Clin Neurosci. 2019;269(7):867‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Hashimoto K. Mood, psychomotor, and cognitive function in major depressive disorder: from biomarkers to rapid‐acting antidepressants. Eur Arch Psychiatry Clin Neurosci. 2019;269(7):759‐760. [DOI] [PubMed] [Google Scholar]
- 222. Yang C, Shirayama Y, Zhang JC, et al. R‐ketamine: a rapid‐onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Zhang JC, Li SX, Hashimoto K. R (‐)‐ketamine shows greater potency and longer lasting antidepressant effects than S (+)‐ketamine. Pharmacol Biochem Behav. 2014;116:137‐141. [DOI] [PubMed] [Google Scholar]
- 224. Hess EM, Riggs LM, Michaelides M, Gould TD. Mechanisms of ketamine and its metabolites as antidepressants. Biochem Pharmacol. 2022;197:114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Aguilar‐Valles A, De Gregorio D, Matta‐Camacho E, et al. Antidepressant actions of ketamine engage cell‐specific translation via eIF4E. Nature. 2021;590(7845):315‐319. [DOI] [PubMed] [Google Scholar]
- 226. Zanos P, Moaddel R, Morris PJ, et al. NMDAR inhibition‐independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Highland JN, Zanos P, Riggs LM, et al. Hydroxynorketamines: pharmacology and potential therapeutic applications. Pharmacol Rev. 2021;73(2):763‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Yang C, Qu Y, Abe M, Nozawa D, Chaki S, Hashimoto K. (R)‐ketamine shows greater potency and longer lasting antidepressant effects than its metabolite (2R,6R)‐hydroxynorketamine. Biol Psychiatry. 2017;82(5):e43‐e44. [DOI] [PubMed] [Google Scholar]
- 229. Shirayama Y, Hashimoto K. Lack of antidepressant effects of (2R,6R)‐hydroxynorketamine in a rat learned helplessness model: comparison with (R)‐ketamine. Int J Neuropsychopharmacol. 2018;21(1):84‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Yamaguchi JI, Toki H, Qu Y, et al. (2R,6R)‐Hydroxynorketamine is not essential for the antidepressant actions of (R)‐ketamine in mice. Neuropsychopharmacology. 2018;43(9):1900‐1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Hashimoto K. Rapid‐acting antidepressant ketamine, its metabolites and other candidates: a historical overview and future perspective. Psychiatry Clin Neurosci. 2019;73(10):613‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Lapidus KA, Levitch CF, Perez AM, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76(12):970‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Popova V, Daly EJ, Trivedi M, et al. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment‐resistant depression: a randomized double‐blind active‐controlled study. Am J Psychiatry. 2019;176(6):428‐438. [DOI] [PubMed] [Google Scholar]
- 234. DiazGranados N, Ibrahim LA, Brutsche NE, et al. Rapid resolution of suicidal ideation after a single infusion of an N‐methyl‐D‐aspartate antagonist in patients with treatment‐resistant major depressive disorder. J Clin Psychiatry. 2010;71(12):1605‐1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Price RB, Iosifescu DV, Murrough JW, et al. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment‐resistant depression. Depress Anxiety. 2014;31(4):335‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Bahji A, Zarate CA, Vazquez GH. Efficacy and safety of racemic ketamine and esketamine for depression: a systematic review and meta‐analysis. Expert Opin Drug Saf. 2022:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Wilkinson ST, Toprak M, Turner MS, Levine SP, Katz RB, Sanacora G. A survey of the clinical, off‐label use of ketamine as a treatment for psychiatric disorders. Am J Psychiatry. 2017;174(7):695‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zarate CA, Jr. , Brutsche N, Laje G, et al. Relationship of ketamine's plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72(4):331‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Pacheco Dda F, Romero TR, Duarte ID. Central antinociception induced by ketamine is mediated by endogenous opioids and mu‐ and delta‐opioid receptors. Brain Res. 2014;1562:69‐75. [DOI] [PubMed] [Google Scholar]
- 240. Schatzberg AF. A word to the wise about ketamine. Am J Psychiatry. 2014;171(3):262‐264. [DOI] [PubMed] [Google Scholar]
- 241. Sanacora G, Frye MA, McDonald W, et al. A Consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry. 2017;74(4):399‐405. [DOI] [PubMed] [Google Scholar]
- 242. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175(12):1205‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Yoon G, Petrakis IL, Krystal JH. Association of combined naltrexone and ketamine with depressive symptoms in a case series of patients with depression and alcohol use disorder. JAMA Psychiatry. 2019;76(3):337‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Marton T, Barnes DE, Wallace A, Woolley JD. Concurrent use of buprenorphine, methadone, or naltrexone does not inhibit ketamine's antidepressant activity. Biol Psychiatry. 2019;85(12):e75‐e76. [DOI] [PubMed] [Google Scholar]
- 245. Wang M, Kaplin A. Explaining naltrexone's interference with ketamine's antidepressant effect. Am J Psychiatry. 2019;176(5):410‐411. [DOI] [PubMed] [Google Scholar]
- 246. Li N, Lee B, Liu RJ, et al. mTOR‐dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Mahase E. Esketamine is approved in Europe for treating resistant major depressive disorder. BMJ. 2019;367:l7069. [DOI] [PubMed] [Google Scholar]
- 248. Turner EH. Esketamine for treatment‐resistant depression: seven concerns about efficacy and FDA approval. Lancet Psychiatry. 2019;6(12):977‐979. [DOI] [PubMed] [Google Scholar]
- 249. Horowitz MA, Moncrieff J. Are we repeating mistakes of the past? A review of the evidence for esketamine. Br J Psychiatry. 2020:1‐4. [DOI] [PubMed] [Google Scholar]
- 250. Swainson J, Thomas RK, Archer S, et al. Esketamine for treatment resistant depression. Expert Rev Neurother. 2019;19(10):899‐911. [DOI] [PubMed] [Google Scholar]
- 251. Fukumoto K, Toki H, Iijima M, et al. Antidepressant potential of (R)‐ketamine in rodent models: comparison with (S)‐ketamine. J Pharmacol Exp Ther. 2017;361(1):9‐16. [DOI] [PubMed] [Google Scholar]
- 252. Blier P, Zigman D, Blier J. On the safety and benefits of repeated intravenous injections of ketamine for depression. Biol Psychiatry. 2012;72(4):e11‐12. [DOI] [PubMed] [Google Scholar]
- 253. Andrade C. Ketamine for depression, 1: clinical summary of issues related to efficacy, adverse effects, and mechanism of action. J Clin Psychiatry. 2017;78(4):e415‐e419. [DOI] [PubMed] [Google Scholar]
- 254. Xi XJ, Zeng JJ, Lu Y, et al. Extracellular vesicles enhance oxidative stress through P38/NF‐kB pathway in ketamine‐induced ulcerative cystitis. J Cell Mol Med. 2020;24(13):7609‐7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Nikayin S, Murphy E, Krystal JH, Wilkinson ST. Long‐term safety of ketamine and esketamine in treatment of depression. Expert Opin Drug Saf. 2022. (Apr 19;1‐11). [DOI] [PubMed] [Google Scholar]
- 256. Zhao X, Venkata SL, Moaddel R, et al. Simultaneous population pharmacokinetic modelling of ketamine and three major metabolites in patients with treatment‐resistant bipolar depression. Br J Clin Pharmacol. 2012;74(2):304‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Short B, Fong J, Galvez V, Shelker W, Loo CK. Side‐effects associated with ketamine use in depression: a systematic review. Lancet Psychiatry. 2018;5(1):65‐78. [DOI] [PubMed] [Google Scholar]
- 258. Schwartz J, Murrough JW, Iosifescu DV. Ketamine for treatment‐resistant depression: recent developments and clinical applications. Evid Based Ment Health. 2016;19(2):35‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Abdallah CG, Sanacora G, Duman RS, Krystal JH. Ketamine and rapid‐acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med. 2015;66:509‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Feifel D. Breaking sad: unleashing the breakthrough potential of ketamine's rapid antidepressant effects. Drug Dev Res. 2016;77(8):489‐494. [DOI] [PubMed] [Google Scholar]
- 261. Gerhard DM, Wohleb ES, Duman RS. Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today. 2016;21(3):454‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Reus GZ, Abelaira HM, Tuon T, et al. Glutamatergic NMDA receptor as therapeutic target for depression. Adv Protein Chem Struct Biol. 2016;103:169‐202. [DOI] [PubMed] [Google Scholar]
- 263. Li CR, Zhang S, Hung CC, et al. Depression in chronic ketamine users: sex differences and neural bases. Psychiatry Res Neuroimaging. 2017;269:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Muller J, Pentyala S, Dilger J, Pentyala S. Ketamine enantiomers in the rapid and sustained antidepressant effects. Ther Adv Psychopharmacol. 2016;6(3):185‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disord. 2014;155:123‐129. [DOI] [PubMed] [Google Scholar]
- 266. Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non‐competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol. 1997;333(1):99‐104. [DOI] [PubMed] [Google Scholar]
- 267. Jelen LA, Young AH, Stone JM. Ketamine: a tale of two enantiomers. J Psychopharmacol. 2021;35(2):109‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Leal GC, Bandeira ID, Correia‐Melo FS, et al. Intravenous arketamine for treatment‐resistant depression: open‐label pilot study. Eur Arch Psychiatry Clin Neurosci. 2021;271(3):577‐582. [DOI] [PubMed] [Google Scholar]
- 269. Chang L, Zhang K, Pu Y, et al. Comparison of antidepressant and side effects in mice after intranasal administration of (R,S)‐ketamine, (R)‐ketamine, and (S)‐ketamine. Pharmacol Biochem Behav. 2019;181:53‐59. [DOI] [PubMed] [Google Scholar]
- 270. Rafalo‐Ulinska A, Palucha‐Poniewiera A. The effectiveness of (R)‐ketamine and its mechanism of action differ from those of (S)‐ketamine in a chronic unpredictable mild stress model of depression in C57BL/6J mice. Behav Brain Res. 2022;418:113633. [DOI] [PubMed] [Google Scholar]
- 271. Abdallah CG, Averill LA, Collins KA, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. 2017;42(6):1210‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Fuchikami M, Thomas A, Liu R, et al. Optogenetic stimulation of infralimbic PFC reproduces ketamine's rapid and sustained antidepressant actions. Proc Natl Acad Sci U S A. 2015;112(26):8106‐8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Jiang C, Lin WJ, Sadahiro M, et al. VGF function in depression and antidepressant efficacy. Mol Psychiatry. 2018;23(7):1632‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Skiteva O, Yao N, Chergui K. Ketamine induces opposite changes in AMPA receptor calcium permeability in the ventral tegmental area and nucleus accumbens. Transl Psychiatry. 2021;11(1):530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Rincon‐Cortes M, Grace AA. Sex‐dependent effects of stress on immobility behavior and VTA dopamine neuron activity: modulation by ketamine. Int J Neuropsychopharmacol. 2017;20(10):823‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Yang Y, Cui Y, Sang K, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature. 2018;554(7692):317‐322. [DOI] [PubMed] [Google Scholar]
- 277. Moda‐Sava RN, Murdock MH, Parekh PK, et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant‐induced spine formation. Science. 2019;364(6436):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Kozorovitskiy Y. Ketamine rapidly enhances glutamate‐evoked dendritic spinogenesis in medial prefrontal cortex through dopaminergic mechanisms. Biol Psychiatry. 2021;89(11):1096‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Deyama S, Bang E, Wohleb ES, et al. Role of neuronal VEGF signaling in the prefrontal cortex in the rapid antidepressant effects of ketamine. Am J Psychiatry. 2019;176(5):388‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Carreno FR, Donegan JJ, Boley AM, et al. Activation of a ventral hippocampus‐medial prefrontal cortex pathway is both necessary and sufficient for an antidepressant response to ketamine. Mol Psychiatry. 2016;21(9):1298‐308. [DOI] [PubMed] [Google Scholar]
- 281. Choi M, Lee SH, Wang SE, et al. Ketamine produces antidepressant‐like effects through phosphorylation‐dependent nuclear export of histone deacetylase 5 (HDAC5) in rats. Proc Natl Acad Sci U S A. 2015;112(51):15755‐15760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Kang HJ, Voleti B, Hajszan T, et al. Decreased expression of synapse‐related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18(9):1413‐1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental‐degenerative divide. Neurosci Biobehav Rev. 2009;33(5):699‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Yuan N, Tang K, Da X, et al. Integrating clinical and genomic analyses of hippocampal‐prefrontal circuit disorder in depression. Front Genet. 2020;11:565749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Liu W, Ge T, Leng Y, et al. The role of neural plasticity in depression: from hippocampus to prefrontal cortex. Neural Plast. 2017;2017:6871089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Price RB, Duman R. Neuroplasticity in cognitive and psychological mechanisms of depression: an integrative model. Mol Psychiatry. 2020;25(3):530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Genzel L, Dresler M, Cornu M, et al. Medial prefrontal‐hippocampal connectivity and motor memory consolidation in depression and schizophrenia. Biol Psychiatry. 2015;77(2):177‐186. [DOI] [PubMed] [Google Scholar]
- 288. Bearden CE, Thompson PM, Avedissian C, et al. Altered hippocampal morphology in unmedicated patients with major depressive illness. ASN Neuro. 2009;1(4):265‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Heshmati M, Christoffel DJ, LeClair K, et al. Depression and social defeat stress are associated with inhibitory synaptic changes in the nucleus accumbens. J Neurosci. 2020;40(32):6228‐6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wook Koo J, Labonte B, Engmann O, et al. Essential role of mesolimbic brain‐derived neurotrophic factor in chronic social stress‐induced depressive behaviors. Biol Psychiatry. 2016;80(6):469‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Nestler EJ, Carlezon WA, Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151‐1159. [DOI] [PubMed] [Google Scholar]
- 292. Hu H, Cui Y, Yang Y. Circuits and functions of the lateral habenula in health and in disease. Nat Rev Neurosci. 2020;21(5):277‐295. [DOI] [PubMed] [Google Scholar]
- 293. Flanigan ME, Aleyasin H, Li L, et al. Orexin signaling in GABAergic lateral habenula neurons modulates aggressive behavior in male mice. Nat Neurosci. 2020;23(5):638‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Hu H. Advances in molecular and circuitry mechanisms of depressive disorder—a focus on lateral habenula. Adv Exp Med Biol. 2019;1180:135‐146. [DOI] [PubMed] [Google Scholar]
- 295. Seo JS, Zhong P, Liu A, Yan Z, Greengard P. Elevation of p11 in lateral habenula mediates depression‐like behavior. Mol Psychiatry. 2018;23(5):1113‐1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Matsumoto M, Hikosaka O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature. 2007;447(7148):1111‐1115. [DOI] [PubMed] [Google Scholar]
- 297. Lammel S, Lim BK, Ran C, et al. Input‐specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491(7423):212‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R. Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron. 2012;74(3):475‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Stamatakis AM, Stuber GD. Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidance. Nat Neurosci. 2012;15(8):1105‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Li B, Piriz J, Mirrione M, et al. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470(7335):535‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Lecca S, Pelosi A, Tchenio A, et al. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression‐like phenotypes in mice. Nat Med. 2016;22(3):254‐261. [DOI] [PubMed] [Google Scholar]
- 302. Li K, Zhou T, Liao L, et al. betaCaMKII in lateral habenula mediates core symptoms of depression. Science. 2013;341(6149):1016‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Huang L, Xi Y, Peng Y, et al. A visual circuit related to habenula underlies the antidepressive effects of light therapy. Neuron. 2019;102(1):128‐142 e8. [DOI] [PubMed] [Google Scholar]
- 304. Cui Y, Yang Y, Ni Z, et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature. 2018;554(7692):323‐327. [DOI] [PubMed] [Google Scholar]
- 305. Tian Z, Dong C, Zhang K, Chang L, Hashimoto K. Lack of antidepressant effects of low‐voltage‐sensitive T‐type calcium channel blocker ethosuximide in a chronic social defeat stress model: comparison with (R)‐ketamine. Int J Neuropsychopharmacol. 2018;21(11):1031‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Xiong Z, Zhang K, Ishima T, et al. Lack of rapid antidepressant effects of Kir4.1 channel inhibitors in a chronic social defeat stress model: comparison with (R)‐ketamine. Pharmacol Biochem Behav. 2019;176:57‐62. [DOI] [PubMed] [Google Scholar]
- 307. Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. Effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546(7659):E1‐E3. [DOI] [PubMed] [Google Scholar]
- 308. Fukumoto K, Fogaca MV, Liu RJ, et al. Activity‐dependent brain‐derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)‐hydroxynorketamine. Proc Natl Acad Sci U S A. 2019;116(1):297‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Yang C, Ren Q, Qu Y, et al. Mechanistic target of rapamycin‐independent antidepressant effects of (R)‐ketamine in a social defeat stress model. Biol Psychiatry. 2018;83(1):18‐28. [DOI] [PubMed] [Google Scholar]
- 310. Yao N, Skiteva O, Zhang X, Svenningsson P, Chergui K. Ketamine and its metabolite (2R,6R)‐hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol Psychiatry. 2018;23(10):2066‐2077. [DOI] [PubMed] [Google Scholar]
- 311. Autry AE, Adachi M, Nosyreva E, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Gerhard DM, Pothula S, Liu RJ, et al. GABA interneurons are the cellular trigger for ketamine's rapid antidepressant actions. J Clin Invest. 2020;130(3):1336‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Widman AJ, McMahon LL. Disinhibition of CA1 pyramidal cells by low‐dose ketamine and other antagonists with rapid antidepressant efficacy. Proc Natl Acad Sci U S A. 2018;115(13):E3007‐E3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27(43):11496‐11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Miller OH, Yang L, Wang CC, et al. GluN2B‐containing NMDA receptors regulate depression‐like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;3:e03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Li SX, Han Y, Xu LZ, et al. Uncoupling DAPK1 from NMDA receptor GluN2B subunit exerts rapid antidepressant‐like effects. Mol Psychiatry. 2018;23(3):597‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, Kavalali ET. Acute suppression of spontaneous neurotransmission drives synaptic potentiation. J Neurosci. 2013;33(16):6990‐7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Freudenberg F, Celikel T, Reif A. The role of alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors in depression: central mediators of pathophysiology and antidepressant activity? Neurosci Biobehav Rev. 2015;52:193‐206. [DOI] [PubMed] [Google Scholar]
- 319. Zhang K, Yang C, Chang L, et al. Essential role of microglial transforming growth factor‐beta1 in antidepressant actions of (R)‐ketamine and the novel antidepressant TGF‐beta1. Transl Psychiatry. 2020;10(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Kantrowitz JT, Dong Z, Milak MS, et al. Ventromedial prefrontal cortex/anterior cingulate cortex Glx, glutamate, and GABA levels in medication‐free major depressive disorder. Transl Psychiatry. 2021;11(1):419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Zunszain PA, Horowitz MA, Cattaneo A, Lupi MM, Pariante CM. Ketamine: synaptogenesis, immunomodulation and glycogen synthase kinase‐3 as underlying mechanisms of its antidepressant properties. Mol Psychiatry. 2013;18(12):1236‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Servick K. The trespasser. Science. 2019;364(6436):118‐121. [DOI] [PubMed] [Google Scholar]
- 323. Jiang C, Lin WJ, Salton SR. Role of a VGF/BDNF/TrkB autoregulatory feedback loop in rapid‐acting antidepressant efficacy. J Mol Neurosci. 2019;68(3):504‐509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Yao W, Cao Q, Luo S, et al. Microglial ERK‐NRBP1‐CREB‐BDNF signaling in sustained antidepressant actions of (R)‐ketamine. Mol Psychiatry. 2022;27:1618‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Lin PY, Ma ZZ, Mahgoub M, Kavalali ET, Monteggia LM. A synaptic locus for TrkB signaling underlying ketamine rapid antidepressant action. Cell Rep. 2021;36(7):109513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Casarotto PC, Girych M, Fred SM, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021;184(5):1299‐1313 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Ma Z, Zang T, Birnbaum SG, et al. TrkB dependent adult hippocampal progenitor differentiation mediates sustained ketamine antidepressant response. Nat Commun. 2017;8(1):1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Jernigan CS, Goswami DB, Austin MC, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(7):1774‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Kato T, Pothula S, Liu RJ, et al. Sestrin modulator NV‐5138 produces rapid antidepressant effects via direct mTORC1 activation. J Clin Invest. 2019;129(6):2542‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Gideons ES, Kavalali ET, Monteggia LM. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc Natl Acad Sci U S A. 2014;111(23):8649‐8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Adaikkan C, Taha E, Barrera I, David O, Rosenblum K. Calcium/calmodulin‐dependent protein kinase II and eukaryotic elongation factor 2 kinase pathways mediate the antidepressant action of ketamine. Biol Psychiatry. 2018;84(1):65‐75. [DOI] [PubMed] [Google Scholar]
- 332. Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125(4):785‐799. [DOI] [PubMed] [Google Scholar]
- 333. Suzuki K, Kim JW, Nosyreva E, Kavalali ET, Monteggia LM. Convergence of distinct signaling pathways on synaptic scaling to trigger rapid antidepressant action. Cell Rep. 2021;37(5):109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246(4931):815‐818. [DOI] [PubMed] [Google Scholar]
- 335. Wang CC, Held RG, Hall BJ. SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One. 2013;8(12):e83941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N‐methyl‐D‐aspartate antagonist, CP‐101,606, in patients with treatment‐refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631‐637. [DOI] [PubMed] [Google Scholar]
- 337. Bjorkholm C, Monteggia LM. BDNF ‐ a key transducer of antidepressant effects. Neuropharmacology. 2016;102:72‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Suzuki K, Monteggia LM. The role of eEF2 kinase in the rapid antidepressant actions of ketamine. Adv Pharmacol. 2020;89:79‐99. [DOI] [PubMed] [Google Scholar]
- 339. Proulx CD, Hikosaka O, Malinow R. Reward processing by the lateral habenula in normal and depressive behaviors. Nat Neurosci. 2014;17(9):1146‐1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Ji H, Shepard PD. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor‐mediated mechanism. J Neurosci. 2007;27(26):6923‐6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron. 2009;61(5):786‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Chang SY, Kim U. Ionic mechanism of long‐lasting discharges of action potentials triggered by membrane hyperpolarization in the medial lateral habenula. J Neurosci. 2004;24(9):2172‐2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Wagner F, Weiss T, Veh RW. Electrophysiological properties of neurons and synapses in the lateral habenular complex (LHb). Pharmacol Biochem Behav. 2017;162:38‐45. [DOI] [PubMed] [Google Scholar]
- 344. Caldecott‐Hazard S, Mazziotta J, Phelps M. Cerebral correlates of depressed behavior in rats, visualized using 14C‐2‐deoxyglucose autoradiography. J Neurosci. 1988;8(6):1951‐1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Park H, Rhee J, Park K, Han JS, Malinow R, Chung C. Exposure to stressors facilitates long‐term synaptic potentiation in the lateral habenula. J Neurosci. 2017;37(25):6021‐6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Zhang Y, Ye F, Zhang T, et al. Structural basis of ketamine action on human NMDA receptors. Nature. 2021;596(7871):301‐305. [DOI] [PubMed] [Google Scholar]
- 347. Newport DJ, Carpenter LL, McDonald WM, et al. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950‐966. [DOI] [PubMed] [Google Scholar]
- 348. Domino EF. Taming the ketamine tiger. 1965. Anesthesiology. 2010;113(3):678‐684. [DOI] [PubMed] [Google Scholar]
- 349. Koike H, Chaki S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav Brain Res. 2014;271:111‐115. [DOI] [PubMed] [Google Scholar]
- 350. Shaffer CL, Dutra JK, Tseng WC, et al. Pharmacological evaluation of clinically relevant concentrations of (2R,6R)‐hydroxynorketamine. Neuropharmacology. 2019;153:73‐81. [DOI] [PubMed] [Google Scholar]
- 351. Akinfiresoye L, Tizabi Y. Antidepressant effects of AMPA and ketamine combination: role of hippocampal BDNF, synapsin, and mTOR. Psychopharmacology (Berl). 2013;230(2):291‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Maeng S, Zarate CA, Jr. , Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionic acid receptors. Biol Psychiatry. 2008;63(4):349‐352. [DOI] [PubMed] [Google Scholar]
- 353. Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ. Ketamine‐induced antidepressant effects are associated with AMPA receptors‐mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry. 2014;29(7):419‐423. [DOI] [PubMed] [Google Scholar]
- 354. Lv D, Chen Y, Shen M, et al. Mechanisms underlying the rapid‐acting antidepressant‐like effects of neuropeptide VGF (non‐acronymic) C‐terminal peptide TLQP‐62. Neuropharmacology. 2018;143:317‐326. [DOI] [PubMed] [Google Scholar]
- 355. Yu H, Li M, Zhou D, et al. Vesicular glutamate transporter 1 (VGLUT1)‐mediated glutamate release and membrane GluA1 activation is involved in the rapid antidepressant‐like effects of scopolamine in mice. Neuropharmacology. 2018;131:209‐222. [DOI] [PubMed] [Google Scholar]
- 356. Nishitani N, Nagayasu K, Asaoka N, et al. Raphe AMPA receptors and nicotinic acetylcholine receptors mediate ketamine‐induced serotonin release in the rat prefrontal cortex. Int J Neuropsychopharmacol. 2014;17(8):1321‐1326. [DOI] [PubMed] [Google Scholar]
- 357. Pham TH, Mendez‐David I, Defaix C, et al. Ketamine treatment involves medial prefrontal cortex serotonin to induce a rapid antidepressant‐like activity in BALB/cJ mice. Neuropharmacology. 2017;112(Pt A):198‐209. [DOI] [PubMed] [Google Scholar]
- 358. Fukumoto K, Iijima M, Chaki S. The antidepressant effects of an mGlu2/3 receptor antagonist and ketamine require AMPA receptor stimulation in the mPFC and subsequent activation of the 5‐HT neurons in the DRN. Neuropsychopharmacology. 2016;41(4):1046‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Gigliucci V, O'Dowd G, Casey S, Egan D, Gibney S, Harkin A. Ketamine elicits sustained antidepressant‐like activity via a serotonin‐dependent mechanism. Psychopharmacology (Berl). 2013;228(1):157‐166. [DOI] [PubMed] [Google Scholar]
- 360. Fukumoto K, Iijima M, Chaki S. Serotonin‐1A receptor stimulation mediates effects of a metabotropic glutamate 2/3 receptor antagonist, 2S‐2‐amino‐2‐(1S,2S‐2‐carboxycycloprop‐1‐yl)‐3‐(xanth‐9‐yl)propanoic acid (LY341495), and an N‐methyl‐D‐aspartate receptor antagonist, ketamine, in the novelty‐suppressed feeding test. Psychopharmacology (Berl). 2014;231(11):2291‐2298. [DOI] [PubMed] [Google Scholar]
- 361. Fukumoto K, Iijima M, Funakoshi T, Chaki S. Role of 5‐HT1A receptor stimulation in the medial prefrontal cortex in the sustained antidepressant effects of ketamine. Int J Neuropsychopharmacol. 2018;21(4):371‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Fukumoto K, Fogaca MV, Liu RJ, et al. Medial PFC AMPA receptor and BDNF signaling are required for the rapid and sustained antidepressant‐like effects of 5‐HT1A receptor stimulation. Neuropsychopharmacology. 2020;45(10):1725‐1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Wolak M, Siwek A, Szewczyk B, et al. Involvement of NMDA and AMPA receptors in the antidepressant‐like activity of antidepressant drugs in the forced swim test. Pharmacol Rep. 2013;65(4):991‐997. [DOI] [PubMed] [Google Scholar]
- 364. Burgdorf J, Zhang XL, Nicholson KL, et al. GLYX‐13, a NMDA receptor glycine‐site functional partial agonist, induces antidepressant‐like effects without ketamine‐like side effects. Neuropsychopharmacology. 2013;38(5):729‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Zanos P, Nelson ME, Highland JN, et al. A negative allosteric modulator for alpha5 subunit‐containing GABA receptors exerts a rapid and persistent antidepressant‐like action without the side effects of the NMDA receptor antagonist ketamine in mice. eNeuro. 2017;4(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Andreasen JT, Fitzpatrick CM, Larsen M, et al. Differential role of AMPA receptors in mouse tests of antidepressant and anxiolytic action. Brain Res. 2015;1601:117‐126. [DOI] [PubMed] [Google Scholar]
- 367. Shen M, Lv D, Li S, et al. Positive allosteric modulation of AMPAR by PF‐4778574 produced rapid onset antidepressant actions in mice. Cereb Cortex. 2019;29(10):4438‐4451. [DOI] [PubMed] [Google Scholar]
- 368. Gordillo‐Salas M, Pascual‐Anton R, Ren J, Greer J, Adell A. Antidepressant‐like effects of CX717, a positive allosteric modulator of AMPA receptors. Mol Neurobiol. 2020;57(8):3498‐3507. [DOI] [PubMed] [Google Scholar]
- 369. Du J, Wei Y, Liu L, et al. A kinesin signaling complex mediates the ability of GSK‐3beta to affect mood‐associated behaviors. Proc Natl Acad Sci U S A. 2010;107(25):11573‐11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Wei J, Liu W, Yan Z. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J Biol Chem. 2010;285(34):26369‐26376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Jope RS. Glycogen synthase kinase‐3 in the etiology and treatment of mood disorders. Front Mol Neurosci. 2011;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Du J, Gray NA, Falke CA, et al. Modulation of synaptic plasticity by antimanic agents: the role of AMPA glutamate receptor subunit 1 synaptic expression. J Neurosci. 2004;24(29):6578‐6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Nelson CD, Kim MJ, Hsin H, Chen Y, Sheng M. Phosphorylation of threonine‐19 of PSD‐95 by GSK‐3beta is required for PSD‐95 mobilization and long‐term depression. J Neurosci. 2013;33(29):12122‐12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Shen M, Lv D, Liu X, et al. Essential roles of neuropeptide VGF regulated TrkB/mTOR/BICC1 signaling and phosphorylation of AMPA receptor subunit GluA1 in the rapid antidepressant‐like actions of ketamine in mice. Brain Res Bull. 2018;143:58‐65. [DOI] [PubMed] [Google Scholar]
- 375. Malinovsky JM, Servin F, Cozian A, Lepage JY, Pinaud M. Ketamine and norketamine plasma concentrations after i.v., nasal and rectal administration in children. Br J Anaesth. 1996;77(2):203‐207. [DOI] [PubMed] [Google Scholar]
- 376. Andrade C. Ketamine for depression, 4: in what dose, at what rate, by what route, for how long, and at what frequency? J Clin Psychiatry. 2017;78(7):e852‐e857. [DOI] [PubMed] [Google Scholar]
- 377. Galvez V, Li A, Huggins C, et al. Repeated intranasal ketamine for treatment‐resistant depression ‐ the way to go? Results from a pilot randomised controlled trial. J Psychopharmacol. 2018;32(4):397‐407. [DOI] [PubMed] [Google Scholar]
- 378. Lee V, Archer S, Chrenek C, Swainson J. A response to: Repeated intranasal ketamine for treatment resistant depression: The way to go? Results from a pilot randomised controlled trial. J Psychopharmacol. 2019;33(2):258‐259. [DOI] [PubMed] [Google Scholar]
- 379. Andrade C. Intranasal drug delivery in neuropsychiatry: focus on intranasal ketamine for refractory depression. J Clin Psychiatry. 2015;76(5):e628‐631. [DOI] [PubMed] [Google Scholar]
- 380. Bahji A, Vazquez GH, Zarate CA, Jr. Comparative efficacy of racemic ketamine and esketamine for depression: a systematic review and meta‐analysis. J Affect Disord. 2021;278:542‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Rosenblat JD, Carvalho AF, Li M, Lee Y, Subramanieapillai M, McIntyre RS. Oral ketamine for depression: a systematic review. J Clin Psychiatry. 2019;80(3). [DOI] [PubMed] [Google Scholar]
- 382. Swainson J, Khullar A. Sublingual ketamine: an option for increasing accessibility of ketamine treatments for depression? J Clin Psychiatry. 2020;81(1). [DOI] [PubMed] [Google Scholar]
- 383. Loo CK, Galvez V, O'Keefe E, et al. Placebo‐controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatr Scand. 2016;134(1):48‐56. [DOI] [PubMed] [Google Scholar]
- 384. Li W, Zhou Y, Liu W, et al. Long‐term outcomes of repeated ketamine infusions in patients with unipolar and bipolar depression: A naturalistic follow‐up study. J Affect Disord. 2022;300:172‐178. [DOI] [PubMed] [Google Scholar]
- 385. Zheng W, Gu LM, Sun CH, et al. Comparative effectiveness of repeated ketamine infusions in treating anhedonia in bipolar and unipolar depression. J Affect Disord. 2022;300:109‐113. [DOI] [PubMed] [Google Scholar]
- 386. Chilukuri H, Reddy NP, Pathapati RM, Manu AN, Jollu S, Shaik AB. Acute antidepressant effects of intramuscular versus intravenous ketamine. Indian J Psychol Med. 2014;36(1):71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Greenway KT, Garel N, Jerome L, Feduccia AA. Integrating psychotherapy and psychopharmacology: psychedelic‐assisted psychotherapy and other combined treatments. Expert Rev Clin Pharmacol. 2020;13(6):655‐670. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets in this study are available from the corresponding author on reasonable request.