

Summary
We investigated the Southern Ocean (SO) prokaryote community structure via zero‐radius operational taxonomic unit (zOTU) libraries generated from 16S rRNA gene sequencing of 223 full water column profiles. Samples reveal the prokaryote diversity trend between discrete water masses across multiple depths and latitudes in Indian (71–99°E, summer) and Pacific (170–174°W, autumn‐winter) sectors of the SO. At higher taxonomic levels (phylum‐family) we observed water masses to harbour distinct communities across both sectors, but observed sectorial variations at lower taxonomic levels (genus‐zOTU) and relative abundance shifts for key taxa such as Flavobacteria, SAR324/Marinimicrobia, Nitrosopumilus and Nitrosopelagicus at both epi‐ and bathy‐abyssopelagic water masses. Common surface bacteria were abundant in several deep‐water masses and vice‐versa suggesting connectivity between surface and deep‐water microbial assemblages. Bacteria from same‐sector Antarctic Bottom Water samples showed patchy, high beta‐diversity which did not correlate well with measured environmental parameters or geographical distance. Unconventional depth distribution patterns were observed for key archaeal groups: Crenarchaeota was found across all depths in the water column and persistent high relative abundances of common epipelagic archaeon Nitrosopelagicus was observed in deep‐water masses. Our findings reveal substantial regional variability of SO prokaryote assemblages that we argue should be considered in wide‐scale SO ecosystem microbial modelling.
Introduction
Despite being persistently cold and having little to no sunlight in the winter, considerable areas of the Southern Ocean (SO) are biologically productive regions that play central roles in nutrient recirculation, supporting primary production across global ocean scales and mediating CO2 exchange and sequestration via the biological pump (Álvarez et al., 2002; Arrigo et al., 2008; Lenton et al., 2013; Legge et al., 2015). The SO is also an important region for recirculation of climate active trace gasses such as dimethyl sulfoxide and methane (Curran and Jones, 2000; Gabric et al., 2001; Wadham et al., 2012; Thurber et al., 2020). Prokaryotes (domain Bacteria and Archaea) make up the largest proportion of microbial biomass in open SO waters and are primary drivers of biogeochemical cycling (Wilkins et al., 2013b). Even with the recent global ocean microbial surveys (Rusch et al., 2007; Duarte, 2015; Sunagawa et al., 2015; Salazar et al., 2016), the diversity of SO prokaryotes remains understudied, with previous studies performed mostly within single locations or specific oceanic regions (Selje et al., 2004; Murray and Grzymski, 2007; Alonso‐Saez et al., 2011; Grzymski et al., 2012; Wilkins et al., 2013a; Wilkins et al., 2013c; Signori et al., 2014; Yu et al., 2015; Landa et al., 2016; Milici et al., 2017; Liu et al., 2019; Liu et al., 2020). Because of the fundamental ecosystem services provided by SO prokaryotes, the development of an accurate baseline understanding of the key players and their biogeographical controls at broad, multi‐sector scales is essential for better predicting the consequences of global climate change on the SO ecosystem function and biogeochemistry. Furthermore, a general lack of knowledge of the SO prokaryotic diversity in the aphotic zone, which is estimated to globally contain up to 75% of prokaryotic biomass and production respectively, also limits our understanding of microbial functions within this critical dark end of the biological pump (Arístegui et al., 2009; Herndl and Reinthaler, 2013; Shah Walter et al., 2018).
Due to their small size, spatial ubiquity, relatively short community turnover time, constant passive movement and recirculation, identifying the distribution patterns of marine microbes, and disentangling the potential environmental (physical and chemical) and biological factors that drive these observed patterns have been a persistent challenge in marine microbial ecology (Tittensor et al., 2010; Beaugrand and Kirby, 2018; Raes et al., 2018). The prokaryotes in the SO are no exception. Traditionally, depth, latitude, season, spatial distance and water properties were thought as key contributors to influencing the distribution of marine prokaryotes (Gilbert et al., 2012; Zinger et al., 2014; Fuhrman et al., 2015; Parada and Fuhrman, 2017), but more recently advection, historical contingency, primary productivity and trophic interactions are also thought to play a role (Wilkins et al., 2013a; Fukami, 2015; Raes et al., 2018; Gralka et al., 2020).
The water properties temperature, salinity, oxygen and nutrients that are primarily used to oceanographically define water masses (i.e. water bodies with similar water properties) (Gordon, 1967; Emery, 2003) have been thought to act as boundaries delineating microbial distribution (Richardson and Schoeman, 2004; Brown et al., 2012; Wilkins et al., 2013c; Baltar and Arístegui, 2017) and past research has shown microbial communities to be distinct between water masses (Galand et al., 2010; Agogue et al., 2011; Wilkins et al., 2013a; Salazar et al., 2016; Zoccarato et al., 2016). Deep SO water masses, incorporating mainly the circumpolar deep waters and Antarctic Bottom waters (AABW) are spatially widespread. They are oceanographically categorized as common water mass types across a wide range of latitudes and sectors (i.e. different longitudinal regions, e.g. Indian, Atlantic and Pacific sectors of the SO) since they are considered uniform environments based on the water properties they are defined by. This may initially suggest the deep ocean support lower abundances and diversity of prokaryotes, however growing number of studies show high levels of diversity in deep water masses (Hewson et al., 2006; Arístegui et al., 2009; Nagata et al., 2010). This high diversity has been attributed to reduced connectivity and microbial dispersal due to sluggish deep sea currents, the presence of deep sea ridges, variable primary productivity and flux of particles from the surface, as well as water mass age (Varela et al., 2008; Schauer et al., 2010; Zinger et al., 2011; Salazar et al., 2016; Mestre et al., 2018). Much remains unknown about the community composition and drivers of observed microbial assemblage within widespread water masses that are separated across large spatial ranges of thousands of kilometres, such as within the deep SO water masses in different oceanic sectors.
Here, we explore the SO prokaryote community composition using 16S rRNA metabarcoding data associated with 223 water samples that were processed as high‐resolution, single nucleotide varying zero‐radius operational taxonomic units (zOTUs). Samples were collected from 3 to 8 depths spanning the full water column of 58 stations, representing all major water masses across the Indian and Pacific sectors of the SO during the austral summer and winter. The sequence dataset, together with distance‐based linear modelling (DistLM) with the corresponding environmental variables and distance–decay analysis allowed us to take a deep dive into (i) whether and how much do prokaryote communities in each of the hydrographically defined epi‐, meso‐ and abyssopelagic water masses differ across different SO sectors that have high spatial separation. We explored which taxonomic groups and at which taxonomic level the community variation(s) were observed, and (ii) compared deep water mass samples across oceanic sectors at an expanded scope. This is something that is not yet prevalent within the SO and provides scientific evidence of the varying community composition in oceanographically similar deep water masses in addition to exploring vertical connectivity of microbial distribution across large spatial distances. Finally, we explored (iii) which factors (such as depth, hydrographic variables and/or geographic distance) were the dominant drivers of the observed prokaryote community composition patterns.
Results and discussion
This study compared data from the Pacific and Indian sectors of the SO. Cruises were conducted during late autumn‐early winter for the Pacific sector and summer for the Indian sector. Sampling encompassed the surface to deep (bathypelagic) water spectrum and covered eight water masses within the Pacific and 10 within the Indian sector (Fig. 1), where the water masses were defined by temperature‐salinity and oxygen distribution plots (Figs. S1 and S2). Information (oceanographic voyage, sector, water mass) and number of samples collected per location are specified in the Experimental procedures section. Detailed water property profiles, definitions of SO water masses and fronts within this study are provided in supporting information. Notably, surface water salinity (32.7–33.9; PSS‐78) at 60–65°S within the Indian sector was observed to be lower than usual due to sea ice meltwater originating from the Antarctic Shelf Current (Bestley et al., 2020). Deep waters within the Pacific sector also had lower dissolved oxygen (DO) levels (252–299 μmol L−1) compared to the Indian sector (320–354 μmol L−1) (Table S1).
Fig. 1.
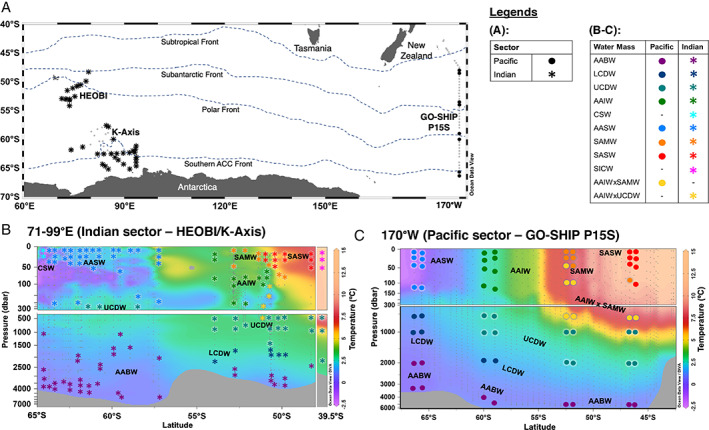
A. Map of microbial sampling stations within the HEOBI, K‐Axis and GO‐SHIP P15S transects. Stations where full‐depth microbial profile analysis were conducted are indicated based on the longitudinal sector they belong to by a closed circle (Pacific sector samples) or bold star (Indian sector samples). Frontal locations indicated are approximate latitudes only. Cross‐section showing vertical distribution of temperature at B. 71–99°E (HEOBI and K‐Axis transects) and C. 170°W (P15S transect), as well as microbial sampling locations and their distribution (indicated by coloured symbols based on the water mass they are categorized to) across the major water masses. Smaller grey dots indicate sampling points for the temperature profile. AABW – Antarctic Bottom Water; LCDW – Lower Circumpolar Deep Water; UCDW – Upper Circumpolar Deep Water; AAIW – Antarctic Intermediate Water; CSW – Continental Shelf Water; AASW – Antarctic Surface Water; SAMW – Subantarctic Mode Water; Subantarctic Surface Water; SICW – South Indian Central Water. Several samples were identified to harbour temperature‐salinity characteristics of two different water masses (regions where water mass mixing occurred): AAIWxSAWM – water with mixed characteristics of AAIW and SAMW; AAIWxUCDW – water with mixed characteristics of AAIW and UCDW.
After QC, our dataset yielded 17.5 and 14.6 million bacterial and archaeal sequencing reads respectively, and following subsampling, resulted in a total of 30 209 bacterial and 10 048 archaeal zOTUs. The alpha‐diversity indices (Table S2) and rarefaction curves (Fig. S3; generated per sample for both bacteria and archaea) indicated microbial richness to be comprehensively represented, with Good's coverage of >0.92 demonstrating that at least 92% of the total species per sample were being considered in the study (Fig. S3; Table S2). Species richness was highest in mixed water masses (i.e. AAIWxSAMW – see Fig. 1 legend and Experimental procedures for all water mass definitions) and lowest in colder AASW and CSW surface water masses for both bacteria and archaea, corroborated by Chao1 true species richness approximations (Table S2; Fig. S4). The increased species diversity observed at the mixed water masses is most likely associated with increased resource availability and higher primary productivity known to occur at frontal zones and areas of water mass transitions where there is elevated mixing and turbulence [Scales et al., 2014 and references therein].
Depth, water mass and sector define unique prokaryote communities
As found in previous studies (Field et al., 1997; López‐García et al., 2001; DeLong et al., 2006; Brown et al., 2009; Fu et al., 2013), the bacterial and archaeal communities were first, strongly structured by depth; large composition differences between communities above and below the mixed layer depth (MLD) were observed through their beta‐diversity visualized in the nMDS (Fig. 2). This observation was supported by significant (P bacteria and P archaeea ≤ 0.0001) permutational multivariate ANOVA (PERMANOVA) tests (Table S3A) where the heterogeneity was not likely to be an effect of multivariate dispersion (Tests of homogeneity of dispersions [PERMDISP] – bacteria: F 1,221 = 9.0934, p = 0.0053; archaea: F 1,220 = 4.2361, p = 0.0548) (Table S3A). Relative abundance plots of the bacterial and archaeal community composition (Fig. 3) further detailed this community composition difference; the communities below the MLD displayed higher relative abundances of several clades, namely, Deltaproteobacteria, SAR406/Marinimicrobia, Chloroflexi, Planctomycetes, Actinobacteria for the bacterial community (Fig. 3A) and Nitrosopelagicus, Thermoplasmatota MGIIb‐O1 and MGIIb‐O1 for the archaea (Fig. 3B). Conversely, higher relative abundances of Alphaproteobacteria (SAR11 Clade Ia, Rhodobacteraceae), Bacteroidetes (Flavobacteria), Nitrosopumilus and Thermoplasmatota MGIIb‐O2 were observed in communities above the MLD (Figs. 3 and 4B and C). We also found DO (a co‐variable of depth) to correlate most strongly with bacterial and archaeal alpha diversity (Fig. S5), which further reinforces the depth‐related structuring of the prokaryote community in this study along with the DistLM analyses (Table S5) and dbRDA ordinations (Fig. 5A and D) which were used to assess the environmental and geographical predictors that best explained community distributions. The first axis of dBRDA ordinations corresponded with the strong separation of communities above and below the MLD observed in the nMDS (Fig. 2) and reflected large disparities between their oxygen levels (dbRDA1; bacteria r oxygen = 0.71, archaeal r oxygen = 0.57; Fig. S1, S2) These analyses demonstrated oxygen and depth significantly (p = 0.001) explain the largest and second‐largest proportion of variation in both the bacterial (DistLM: oxygen – 22%; depth – 13%) and archaeal (DistLM: oxygen – 19%; depth – 17%) communities (Table S5).
Fig. 2.
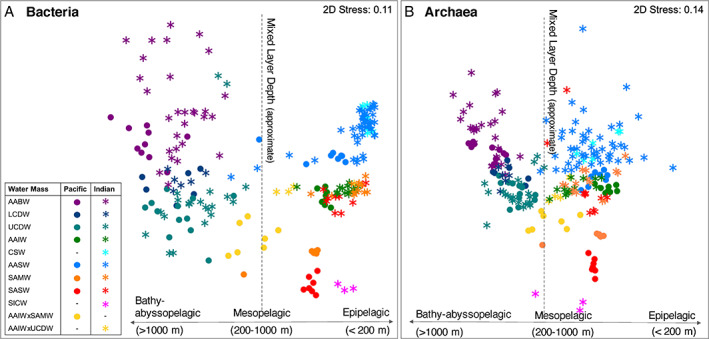
nMDS plots of (A) bacterial and (B) archaeal 16S rRNA gene communities distribution patterns generated based on zOTU abundance tables. Water mass name acronyms are as indicated in Fig. 1.
Fig. 3.
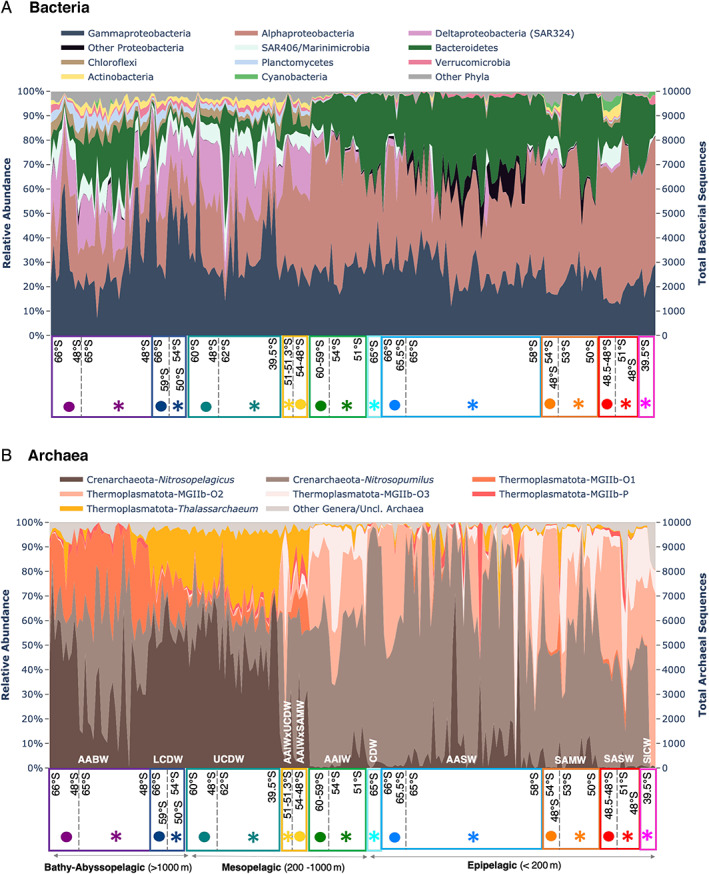
Relative abundance of major (A) bacteria and (B) archaea detected from the different water masses at phylum/class level. ‘Other phyla’ or ‘Other genera’ are bacterial phylum or archaeal genera with overall mean relative abundances of <0.5% across all water masses. ‘Unclassified archaea’ include archaeal zOTUs that were classified with bootstrap confidence values <50%. For each water mass type, samples are arranged by decreasing latitude from left to right, first for the Pacific and followed by Indian sector samples. Water column depth categories: Bathy‐Abyssopelagic (>1000 m), Mesopelagic (200–1000 m), and Epipelagic (<200 m) provide a rough depth indication of the different water masses. Full charts showing each of the 223 samples on the x‐axis are in Fig. S6. Water mass name acronyms are as indicated in Figs. 1 and 2 legends.
Fig. 4.
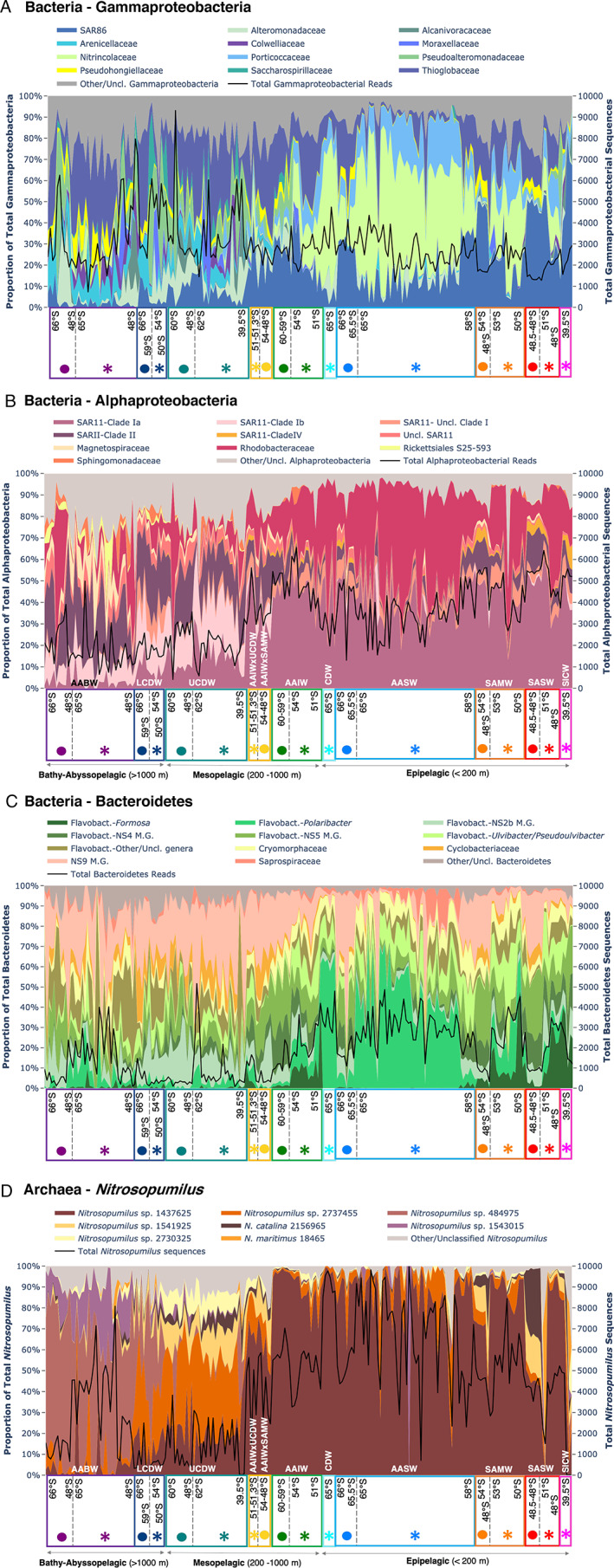
Detailed charts of the main families within (A) Gammaproteobacteria, (B) Alphaproteobacteria and (C) Bacteroidetes. Relative abundances of main species/zOTUs within the archaeal Nitrosopumilus genus are shown in (D). Samples are arranged from high to low latitude in each water mass for the Pacific and Indian sector as indicated by their respective sector and water mass symbols (see Fig. 1/Fig. 2 legends). M.G. – Marine Group.
Fig. 5.
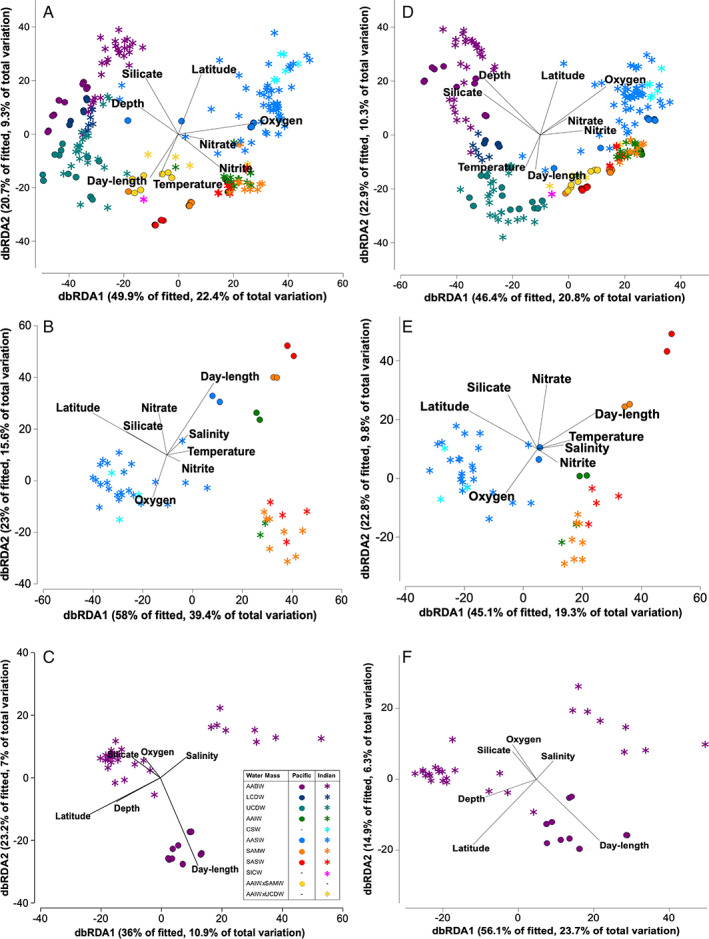
Distance‐based redundancy analysis (dbRDA) ordination of fitted values from DistLM models of the correlation between bacterial (A–C) and archaeal (D–F) community composition with environmental and geographical variables. Model ordinations of bacterial and archaeal communities from all water masses (A, D) as well as community subsets from surface water samples (B, E) and AABW (C, F) are shown. The effect of each environmental/geographical (predictor) variable is represented by vectors on two visualized axes explaining the most community variation. Relative strength of the correlation between the predictor variable with the dbRDA axes corresponds to vector length, while correlation of the predictor variable to the visualized axes corresponds to vector direction. Surface samples analyzed include all samples taken from depths <20 m from all stations. Water mass name acronyms are as indicated in Fig. 1.
Persistent oceanographic features such as water masses can act as ecological boundaries, limiting prokaryote dispersal and/or successful colonization, and thus are known to structure microbial communities (Abell and Bowman, 2005b; Giebel et al., 2009; Galand et al., 2010; Zinger et al., 2011), but the effects of sector on structuring microbial communities within the same water mass are poorly investigated. Following depth, the prokaryote community in this study was also strongly structured by water mass and oceanic sector (hereafter referred to simply as ‘sector’), as shown in the nMDS plots and PERMANOVA tests (Fig. 2; Table S3B). PERMANOVA indicated sector to structure 9 the prokaryotes more strongly above the MLD while the effects of water mass characteristics explained a greater proportion of the prokaryote community variation below the MLD (Table S3B; p ≤ 0.0001 for all tests). There was evidence for interaction between water mass and sector effects in the epipelagic community (p ≤ 0.001), though effects from the interaction of the two factors explained smaller proportions of community variation compared to either sector or water masses alone (Table S3B). The effects of water mass and water mass × sector on the archaeal community above the MLD and the effects of sector and water mass × sector on the archaeal community below the MLD may have also been influenced by heterogeneity in multivariate dispersion (Table S3C).
Dominant bacterial and archaeal clades in the different water masses and sectors
The prevailing bacterial and archaeal clades at higher taxonomic levels in the various water masses above and below the MLD are illustrated in Fig. 3 and as briefly summarized in the previous section. We further explored the dominant (high relative abundance/sequence numbers) bacteria (Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes) and archaea (Nitrosopumilus) at finer taxonomic levels (Fig. 5). In brief, some notable distribution patterns include those from the SAR11 Clade 1a (13%–61% of Alphaproteobacteria). Out of 246 474 949 SAR11 sequences from all samples, 36.3% belonged to a single Clade 1a SAR11 zOTU, which displayed 100% similarity over the analyzed 435 bp V1–V3 region with Candidatus Pelagibacter ubique isolate HTCC1062. SAR11 Clade 1a, known to be dominant only in epipelagic waters (Giovannoni, 2017), was also observed to be still relatively abundant (10%–20%) down to 2000 m (and Fig. 5B). Rhodobacteraceae (3%–74% of Alphaproteobacteria) were dominated mainly by the genus Planktomarina and Loktanella. Planktomarina, belonging to the widely distributed RCA cluster was previously isolated from the North Sea where it is abundant but have also been found in the West Antarctic Peninsula (WAP) and central Kerguelen Plateau (Giebel et al., 2011; Alcaman‐Arias et al., 2018; Liu et al., 2020).
Overall, the archaeal community comprised primarily of the phylum Crenarchaeota (formerly Thaumarchaeota) and Thermoplasmatota (formerly Marine Group II, referred to as MGII in this article) with a slightly more prevalent presence of Crenarchaeota across water masses and sectors, except for SICW where the archaeal community comprised almost completely of MGII (Fig. 3B). The effects of sector in structuring community composition were observed and discussed in the following sections.
Community composition of surface waters vary between the Indian and Pacific sectors
Sector was a key factor structuring the prokaryote community above the MLD (Table S3B), and its effects were observed through zonal geographical variations in dominant bacterial and archaeal taxa. As observed from Figs. 3 and 5, samples from the same water masses and similar depths harboured different communities at the Indian versus Pacific sectors even when observed at various taxonomic levels. Notably, Flavobacteria, a prevailing Bacteroidetes throughout this dataset and key player in phytoplankton‐derived organic matter degradation (Kirchman, 2002), was dominated by Polaribacter in the Indian AASW, but by the NS4, NS5 and NS2b groups in the Pacific AASW (Fig. 4C, Fig. S6d). With the Gammaproteobacteria, higher relative abundances of Thioglobaceae were observed within the Pacific while Porticoccaceae and Nitrincolaceae families were more abundant in the Indian sector (Fig. 4C, Fig. S6b, Fig. S7C). SAR324 and Marinimicrobia, clades known to be more common in the deeper waters with roles in carbon fixation, sulfur cycling and nitrous oxide reduction (Swan et al., 2011; Wright et al., 2014; Hawley et al., 2017), were only abundant in epipelagic SAMW‐SASW of the Pacific sector (SAR324: 1.2%–15%; Marinimicrobia: 6.1%–8.8%; Fig. 3A, Fig. S6a, Fig. S7A). For the archaea, species‐level variations of the dominant Nitrosopumilus genus were observed between sectors in the SAMW‐SASW (15%–85% of all archaea; Fig. 4D, Fig. S6f, Fig. S7F), with substantial percentages of Nitrosopumilus catalina (4%–36% of all Nitrosopumilus) and Nitrosopumilus sp. 1541925 found only in Pacific sector samples compared to Indian SAMW‐SASW which comprised almost entirely of Nitrosopumilus sp. 1437525 (Fig. 4D, Fig. S7F). The sectorial variation of these taxa was also corroborated by a substantial contribution to overall community dissimilarity between the water mass they were sampled from in the Pacific sector with the same corresponding water mass in the Indian sector (pair‐wise grouped SIMPER analysis, Table S4).
When we correlated environmental and geographical predictors to the surface prokaryote community distributions with DistLM and dBRDA plots, both types of predictors combined explained substantial proportions of prokaryote compositional variation (DistLM – bacteria: 68%, archaea: 42%; Table S9). Significant (p = 0.0001) proportions of the community variation were explained by latitude (bacteria – 32%, archaea – 16%), oxygen (bacteria – 26%, archaea – 16%) and day‐length (bacteria – 25%, archaea – 13%) (Table S5). We noted that strong correlations of the prokaryote community composition to day‐length were observed on the second dBRDA axis (Fig. 5B and E), which also showed the separation of Pacific and Indian sector samples that reflected the differences in the dominant taxa observed between sectors (i.e. Flavobacteria, N. catalina and several others as detailed in Figs. 3 and 4) within the same water masses (AASW, SAMW, SASW). While this does not conclusively disentangle the effects of sector (ocean circulation/distance) and seasonality, the strong correlation of day‐length, a strong proxy for seasonal variations (Gilbert et al., 2010) to the surface prokaryote community variations suggests seasonality as a plausible driver contributing to the observed sector specificity. Seasonality has been shown to widely influence microbial diversity patterns of some taxa in various ocean basins (Treusch et al., 2009; Gilbert et al., 2012; Giovannoni and Vergin, 2012; Cram et al., 2015; Signori et al., 2017) and the effects of seasonality on microbial diversity patterns could also be linked to seasonal energy production (Manganelli et al., 2009; Raes et al., 2020). Some taxa, such as N. catalina, is known to be less tolerant to warm temperatures (Ahlgren et al., 2017) and probably thrived better and exhibited higher relative abundance in colder winter waters when the Pacific sector was sampled.
The significant contribution of oxygen in the DistLM analysis (Table S5) indicated that the varying oxygen levels between Pacific and Indian SAMW/SASW could explain the sector specificity of taxa such as SAR324 and Marinimicrobia. A phylogenetic tree of the SAR324 sequences to those from other published studies found that the Pacific SAMW/SASW SAR324 were closely related to Clade II SAR324 (Fig. S9). Clade II SAR324 is known to be restricted to low‐latitude, deep waters with lower oxygen availability, congruent with the lower oxygen levels in Pacific SAMW/SASW (Figs. S1, S2; Table S1) that are linked to the oceanic circulation and sub‐Antarctic regions of the Pacific being a water mass formation region (Sloyan and Rintoul, 2001; Talley et al., 2016).
Sectorial variation of the deep‐water community
While PERMANOVA indicated that sector structured a smaller proportion of the prokaryote community variation below the MLD (Table S3B), several dominant bacterial and archaeal taxa were observed to exhibit sectorial variability. Sectorial variability was distinct at family level for Bacteroidetes, where there were higher relative abundances of Bacteroidetes within Indian AABW (Fig. S7A; up to 41% of all bacteria in some samples) with a substantially larger proportion of Polaribacter at higher latitudes (>60°S; up to 32% of all Bacteroidetes) compared to Pacific AABW which had a lower proportion (up to 9.6% of all bacteria) and varying composition of Bacteroidetes (Figs. 3A and 4C, Fig. S8). For the archaea, sectorial variation was also observed in the AABW where there were substantially higher proportions of Nitrosopumilus in the Indian sector (up to 81% of all archaea) compared to the Pacific (up to 19% of all archaea) where Nitrosopelagicus was more dominant (up to 76% of all archaea) (Figs. 3 and 4D, Fig. S7B; see also pair‐wise SIMPER, Table S4). While PCA indicated a strong sectorial variability of the environmental parameters (Fig. S10C) that initially suggested they could explain the observed sectorial community variation, DistLM showed that environmental and geographical predictors combined only explained 30% and 40% of the bacterial and archaeal community variation respectively (Table S5). Spatial distance alone was also not a key factor driving community variation. This was illustrated by distance–decay plots (Fig. S11C‐D) showing that both AABW bacteria and archaea had weak to no correlation with geographical distance compared to a relatively strong negative correlation observed between surface prokaryotes with geographical distance (Fig. S11A‐B; bacteria: ρ = −0.69, p < 0.001; archaea: ρ = −0.39, p < 0.001).
Historical contingencies (Fukami, 2015) could also contribute to the sectorial variability of the deep‐water prokaryotes. For instance, the sector‐specific higher abundance of Polaribacter (Fig. 4C, Fig. S7A), a dominant surface‐Antarctic coastal Bacteroidetes that is commonly associated with the degradation and remineralization of phytoplankton blooms (Abell and Bowman, 2005a; Straza et al., 2010; Landa et al., 2018; Kim et al., 2019) in AABW is very likely to be linked to/transmitted from the flourishing summer surface community (that is entrained to the abyss during AABW formation). It should be noted that varying proximity between Indian and Pacific AABW sampling sites from their bottom water formation regions may also influence the communities that are entrained from surface waters during the bottom water formation process.
Deep‐water prokaryote communities display high beta‐diversity within the same sector
Prokaryote communities from deep water masses within the same sector also displayed high heterogeneity, particularly for the bacterial community as clearly indicated by greater dispersion in the nMDS plot (Fig. 2A). The high dispersion amongst the deep water mass samples was corroborated by average Bray–Curtis similarity values of bacterial community in the respective deep water masses (AABW, LCDW, UCDW; with each sector considered separately) which ranged between 21% and 34% compared to those of samples within epipelagic water masses (SAMW, SASW, CSW, SICW, AAIW) ranging between 33% and 72% (Table S6). Archaeal beta‐diversity was lower compared to the bacterial community between deep water mass samples (Fig. 2B); average similarity of archaeal communities from deep water masses was 39%–52%, while epipelagic archaeal communities had similarity values ranging between 35% and 67% (Table S6).
The community heterogeneity was especially apparent in the AABW bacterial community from the Indian sector, with only 21% community similarity between sampling sites (Table S6) and showed a spatially inconsistent (patchy) distribution (Fig. 2A) that was not distance related (Fig. S11C). This high and inconsistent heterogeneity could be attributed to several mechanisms. Large proportions of the microbial community in the deep ocean are known to be sustained by sinking, microbially colonized organic C particles generated in the epipelagic (Herndl and Reinthaler, 2013), and as a result, the deep ocean microbial community structure can be influenced by variations in the surface microbial assemblage (Baltar and Arístegui, 2017), especially the particle‐attached surface community (Mestre et al., 2018). However, varying particle sinking speeds (Ploug et al., 2008) and the influence of trophic and functional structure of the surface ocean on the particles delivered results in intermittent and heterogeneously distributed particles colonizing the deep ocean community (Buesseler et al., 2009; Smith Jr. et al., 2018; Acinas et al., 2021), which may explain the patchy, high beta‐diversity observed (Hewson et al., 2006). Additionally, while advection has also been shown to influence beta‐diversity (Wilkins et al., 2013a) by increasing dispersal, deep water currents are generally slow (~43 m per day) (Haine et al., 1998) and, alone, will probably not effectively support long‐distance homogenizing dispersal of deepwater microbial communities, leading to heterogeneity observed between deep water sites.
Archaeal community composition reveals surprising depth distribution patterns
Several noteworthy depth distribution patterns were observed within the archaeal community. First was the abundant presence of the Crenarchaeota phylum (formerly Thaumarchaeota) in water masses across depths and sectors (15%–100%; Fig. 3B, excluding values in SICW), supporting a growing number of studies that Crenarchaeota do not only exhibit increasing abundances with depth but are also abundant in the euphotic zone, especially in polar oceans (Shiozaki et al., 2016; Sintes et al., 2016). Additionally, Crenarchaeota, which was previously found to be seasonally abundant only in Antarctic winter surface waters (Murray et al., 1998; Church et al., 2003; Kalanetra et al., 2009; Grzymski et al., 2012) were found to be abundant (mainly genus Nitrosopumilus) in both summer (Indian sector) and winter (Pacific sector) AASW in this study (Fig. 3B). Second was the high relative abundance (up to 76%) and persistence of several Nitrosopelagicus‐classified zOTUs across the deep (UCDW, LCDW and AABW) water masses (except AABW of the Indian sector), in contrast to the low relative abundance of this clade within epipelagic water masses (Fig. 3B). Current literature suggests Nitrosopelagicus is primarily found in shallow waters, and apart from another Nitrosopelagicus‐related Crenarchaeota sequence detected at 800 m of the South Atlantic Subtropical Gyre (Rinke et al., 2013), no other Nitrosopelagicus or affiliated sequences have been detected or linked to known deep‐water Crenarchaeota clades (Sintes et al., 2013; Santoro et al., 2017). Interestingly, each of the lower mesopelagic and bathy‐abyssopelagic water masses had their own specific taxonomic composition of Nitrosopumilus, which was consistently observed across both sectors (Fig. 4D). This composition was distinct from those in the epipelagic water masses that were dominantly inhabited by Nitrosopumilus sp. 2730325 (Fig. 4D).
Similar to the distribution patterns of the Crenarchaeota, we also found the phylum Thermoplasmatota (formerly Marine Group II, referred to as MGII in this article) to be abundant across depths and sectors (9%–84%) (Fig. 3B). Exceptions were in the Indian CSW where Thaumarchaeota were dominant, and Indian AASW where there was a mixed dominance of Thaumarchaeota in some sites and MGII in others (Fig. 3B). This was in contrast to the common perceptions of MGII as a surface ocean dweller (Zhang et al., 2015). Mixed patterns have been observed in previous work within the WAP – Church et al. (2003) found low numbers of MGII throughout the water column in both summer and winter samplings, while Signori et al. (2014) found MGII to be enriched in deeper summer waters. MGII abundances have also been reported to be invariable by depth within the Atlantic region (Teira et al., 2004; Herndl et al., 2005; Santoro et al., 2019). While MGII relative abundance was relatively invariable throughout the water column, the taxonomic composition of MGII was distinct in the surface and deep water masses, with MGIIb‐O2, MGIIbO3 and UBA59 prevalent in surface/above MLD water masses, while MGIIb‐O1 and Thalassarchaeum were the more common MGII found in deep/below MLD water masses (Fig. 3B, Fig. S7B). The dominance of different Crenarchaeota and Thermoplasmatota groups in the epipelagic versus those below the epipelagic water masses, especially at finer (genus to zOTU) taxonomic levels suggests niche specialization, metabolic or functional diversification of closely related groups, in‐line with the findings of some earlier studies, e.g. Francis et al. (2005), Hallam et al. (2006), Luo et al. (2014) and Rinke et al. (2019).
Concluding remarks
In summary, this article presented a comprehensive dataset that allowed us to explore in‐depth the prokaryote community diversity simultaneously across a large latitudinal, longitudinal and depth scale. The large spatial and depth coverage of this dataset allowed us to show that oceanographically similar water masses, whether in the epi‐, meso‐ or bathypelagic can exhibit variations in community composition of key prokaryote taxa across different sectors of the SO. The high prokaryote diversity and lesser expected sectorial variability in the meso‐ and bathypelagic water masses is likely linked to vertical connectivity with microbial and ecosystem activity occurring in the different epipelagic water masses (and its varying environmental conditions) it is linked to. Future studies can explore how this high diversity and heterogeneous distribution is linked to vertical connectivity and varying composition of organic matter due to differing activities in the surface.
Experimental procedures
Study region and sampling description
Seawater samples were collected during three voyages in 2016. The first two voyages were on the R/V Investigator: the Heard Earth‐Ocean‐Biosphere Interactions (HEOBI) voyage (8th January–27th February, 2016; austral summer) and Leg 1 of the GO‐SHIP P15S repeat hydrographic transect (http://www.go-ship.org; 26th April to 26th May, 2016; austral winter). The third voyage was on the Aurora Australis icebreaker: the Kerguelen Axis (K‐Axis) voyage (11th January–24th February 2016; austral summer; Fig. 1A). As our sampling was opportunistic, we could not control for a common sample collection season between sectors. Details on sample collection devices and sensors are in supplementary methods.
In all voyages, samples were collected at three to eight different depths from surface to bottom waters from 66°S to 39.5°S at longitude ranges of 71–99°E and 170–174°W (Fig. 1B and C). The depths were selected to target samples from the surface (SFC), chlorophyll maximum depth (CMX), MLD, below MLD, 500, 1000, 2000 m, and the bottom depth. For CMX and MLD, sampling depths were selected based on temperature, salinity, DO and fluorescence signal profiles. In total, 223 samples were considered within this study; 64 samples were from GO‐SHIP P15S (Pacific sector), 78 samples were from HEOBI (Indian sector) and 81 samples were from K‐Axis (Indian sector) (Table S1).
The water mass where samples were collected were identified following temperature and salinity criteria ranges in Emery (2003) and with reference to Pardo et al. (2012). The nine water masses identified are listed in Fig. 1, with detailed definitions listed in supplementary methods. The number of samples collected from each water mass is detailed in Table S2.
Hydrological variable measurements and nutrient analyses
Samples for the dissolved inorganic nutrients (silicate [Si], phosphate [PO4 3−], nitrate [NO3 −] and nitrite [NO2 −]) analyses were assayed by the CSIRO Hydrochemistry team on a Bran + Luebbe AA3 HR segmented flow analyzer following standard spectrophotometric methods (Hansen and Koroleff, 2009). Detailed methodology, detection limits and links to access the physical, biogeochemical, nutrient and metadata discussed here (temperature, salinity, DO, nitrate + nitrite [NOx], NO2 −, PO4 3− and Si) are available in supplementary methods.
DNA isolation, amplification, sequencing and sequence data analyses
Two litres of seawater was filtered through 0.22 μm pore size polyethersulfone Sterivex™ filter cartridges (Millipore, Darmstadt, Germany) for each sample and immediately stored at −80°C until DNA extraction. Each microbial sample was issued a unique ID (BPA ID) from the Australian National Data Service (Table S1). DNA was isolated using a modified phenol:chloroform:isoamyl based extraction protocol of the DNeasy PowerWater Sterivex Kit (MoBio‐Qiagen, Hilden, Germany) (Appleyard et al., 2013) and stored at −20°C.
DNA amplification and sequencing were performed at the Ramaciotti Centre for Genomics (Sydney, Australia). Prokaryote assemblages were surveyed using V1–V3 hypervariable regions of the 16S rRNA gene amplified using primers 27F–519R (bacteria) (Lane et al., 1985; Lane, 1991) and A2F–519R (Lane et al., 1985; DeLong, 1992). Amplicons were sequenced using Illumina's MiSeq dual index 300 bp paired‐end approach (Illumina, San Diego, CA, USA). All data including paired‐end reads (R1, R2), indexed reads (I1, I2) data in .fastq format, and latest sequence read abundance tables are available from the Australian Microbiome Initiative data portal (https://data.bioplatforms.com/organization/about/australian-microbiome).
Sequence data analysis and accession numbers
All sequences were analyzed as part of the Australian Microbiome Initiative as previously described by Brown et al. (2018). Detailed methods used for sequence data analysis and taxonomic classification are in supplementary methods. zOTU data with single nucleotide variation between zOTUs were used to enable data analysis at the highest possible phylogenetic variation. zOTU tables were rarified to 10 000 reads per sample for subsequent statistical analysis using the ‘sub.sample’ command in MOTHUR. Genomic datasets associated with this study are accessible from NCBI Bioprojects numbers IDs PRJNA385736 and PRJNA509756. Sequence accession numbers are listed in Table S1.
Statistical analyses, diversity indices and correlation with environmental parameters
Alpha diversity matrices were calculated using the ‘scikit‐bio’ and ‘ecopy’ (Lemoine, 2015) packages on Python 3.6.1. Rarefaction curves were generated using the ‘rarecurve’ function in the R package ‘vegan’ (Oksanen et al., 2018). Good's coverage was calculated using ‘scikit‐bio’ based on the formula 1‐(F 1/N), where F 1 is the number of singletons and N is the sum of zOTU abundances. Beta‐diversity measures were used to assess spatial turnover patterns of prokaryote communities between water masses. We used PRIMER [v7.0.18; Clarke and Gorley, 2015] to generate Bray–Curtis taxonomic similarity matrices following fourth‐root transformation of zOTU abundances and visualized community differences with nMDS plots. Community variations were assessed separately for bacterial and archaeal communities below and above the mixed layer, based on either water mass or oceanographic sector (i.e. Indian or Pacific) with permutation multivariate analysis of variance (PERMANOVA) in PRIMER (Anderson et al., 2008). Two‐way crossed PERMANOVA tests were used to assess effects of factor interaction (water mass × sector) on community variation with marginal test of terms and 9999 permutations of pseudo‐F ratios. Average contribution of individual zOTUs (then grouped by phyla, family or genera) to Bray–Curtis dissimilarity between pairs of water mass × sector was calculated using the SIMPER routine with an 80% cumulative contribution cut‐off value (PRIMER). Average Bray–Curtis similarity percentages of the prokaryote community within each water mass were identified using the ANOSIM routine (PRIMER).
DistLM was used to assess the relationship between environmental [depth (pressure), oxygen, temperature, salinity, silicate, nitrite, NOx, phosphate, day‐length] and geographical (latitude and longitude) predictors that best explained the observed prokaryote beta‐diversity. DistLM models were computed using Bray–Curtis similarity matrices based on fourth root transformed bacterial and archaeal relative abundances with transformed and normalized environmental and geographical variables (see above) in PRIMER (Clarke and Gorley, 2015). Model selection procedures and criteria are detailed in supplementary methods. Models were created for all samples and subsets of surface and AABW samples. Surface samples analyzed include all samples taken from depths <20 m from all stations.
Effects of geographical distance on community variation were assessed with distance–decay plots. Methods to generate geographical distance in the distance–decay correlation plots (Fig. S11) are explained in supplementary methods.
Author Contributions
S.L.S.S., E.J.R. collected GO‐SHIP samples and executed experiments; L.J.C. collected K‐Axis samples; T.W.T. collected HEOBI samples. E.J.R., L.B. and J.v.d.K came up with a preliminary experimental design; A.B., J.v.d.K, S.S.L.S. analyzed the bioinformatics; S.S.L.S. executed statistical analysis and DistLM models, P.C.P. and B.M.S. provided physical oceanography advice. S.L.S.S. led the write‐up; all authors contributed to interpreting data, reviewing and refining the article.
Supporting information
Appendix S1: Supplementary methods
Fig. S1. A. Temperature salinity curves along the P15S transect (Pacific sector) overlayed with corresponding dissolved oxygen values, illustrating the main water masses (labelled with its corresponding acronym). Contour lines indicate potential density anomaly values. Vertical distributions of B. oxygen, C. salinity, D. nitrate + nitrite (NOx), E. nitrite, F. phosphate and G. silicate along leg 1 of the P15S transect.
Fig. S2. A. Temperature salinity curves along the HEOBI and K‐Axis voyages (Indian sector) overlayed with corresponding dissolved oxygen values, illustrating the main water masses (labelled with its corresponding acronym). Contours lines indicate potential density anomaly values. Vertical distributions of B. oxygen, C. salinity, D. nitrate + nitrite (NOx), E. nitrite, F. phosphate and G. silicate along the HEOBI and K‐Axis voyages.
Fig. S3. Rarefaction curves of observed A. bacteria and B. archaea species. Samples were subsampled to a depth of 10,000 sequences as indicated by grey dotted lines prior to subsequent analyses.
Fig. S4. Rarified mean alpha‐diversity indices for prokaryotes within different SO water masses. A. Richness and Chao1 index; B. Pielou's evenness and Shannon‐Weaver index. Water masses acronyms are as shown in Fig. 1.
Fig. S5. Plot of bacterial and archaeal community A. Richness against dissolved oxygen and B. Pielou's evenness against dissolved oxygen.
Fig. S7. Shade plots indicating overall relative abundance (at phylum/class level) of A. bacteria and B. archaea detected, within each water mass and sector they were sampled from. Detailed charts of the main families of C. Gammaproteobacteria, D. Alphaproteobacteria, E. Bacteroidetes and F. main species/zOTUs of the archaeal Nitrosopumilus genus within each water mass‐sector are also shown. ‘Other phyla’ or ‘Other genera’ are bacterial phylum or archaeal genera with overall mean relative abundances of <0.5% across all water masses. ‘Unclassified archaea’ include archaeal zOTUs that were classified with bootstrap confidence values <50%. Water mass name acronyms and water mass‐sector symbols are as indicated in Fig. 1 and Fig. 2 legends.
Fig. S8. Relative abundances of Bacteroidetes within the AABW samples. Samples are sorted by sector and latitude. Sample ID – Sector; Water Mass; Voyage.Latitude.Depth (m). P – Pacific sector; I – Indian sector; G – GO‐SHIP; KX – K‐Axis; H – HEOBI
Fig. S9. Phylogenetic tree of abundant SAR324 zOTU sequences from Pacific SAMW and SASW samples clustered with other SAR324 sequences with identified clades. Sequences with the prefix ‘AMD’ in blue are sequences from this study and are not specific to either SAMW or SASW water masses only.
Fig. S10. PCA ordination of the environmental and geographical properties of (A) all samples (B) epi‐ and mesopelagic samples and (C) bathy‐ and abyssopelagic samples considered within this study, grouped according to the water masses they were assigned to. Water mass name acronyms are as indicated in Fig. 1.
Fig. S11. Distance‐decay plots of Bray–Curtis similarity values between (A‐B) surface water and (C‐D) AABW prokaryote samples versus geographical distance between the samples.
Table S2. Alpha diversity matrices for bacterial and archaeal 16S rRNA gene sequences within the different Southern Ocean water masses.
Table S6. Average Bray–Curtis similarity percentages of the prokaryote community within the various water masses.
Table S7. Pearson correlation of environmental variables considered within this study. +Surface samples analysed include all samples taken from depths <20 m from all stations.
Fig. S6. (a) Full charts for relative abundance of major bacteria detected from the different water masses at phylum/class level, showing each of the 223 samples on the x‐axis. Samples are arranged from high to low latitude in each water mass from the Pacific (P) followed by Indian (I) sector. Detailed charts of the main families within (b) Gammaproteobacteria, (c) Alphaproteobacteria and (d) Bacteroidetes is also shown. The full chart for archaeal relative abundance is shown in (e), and a detailed chart of the main Nitrosopumilus species/zOTUs is shown in (f).
Table S1. List of samples, sampling information and contextual data for all samples collected within this study.
Table S3. A: Permutational multivariate analysis of variance using distance matrices (PERMANOVA) on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 B: Crossed two‐factor permutational multivariate analysis of variance using distance matrices (PERMANOVA) on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 C: PERMDISP on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 D: Results from PERMANOVA pairwise tests for the interaction term of Sector x Water Mass in bacterial and archaeal communities. Tests were based on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S4. SIMPER analysis of the A. bacterial and B. archaeal community, grouped by phylum/class, family and genus, indicating the contribution of each phylum/class/family/genus to community dissimilarity between the different water masses in the Pacific sector and the same corresponding water mass in the Indian sector. The analysis included higher contributing zOTUs up to a cumulative cut‐off percentage of 80%.
Table S5. Proportion of variation explained by individual predictor variations within Distance‐based linear modelling (DistLM) marginal tests. *Surface samples analysed include all samples taken from depths <20 m from all stations. ‘‐’ denotes that variable was not included in model due to high co‐linearity (Pearson's r > |0.85|) with another variable that contributed to explaining more significant proportions of community variation. ** indicate non‐significant p‐values (p > 0.05); nPerm = 9999
Acknowledgements
The authors would like to thank members of field teams, shipboard crews and logistics support personnel from the CSIRO, Australian Marine National Facility, R/V Investigator and Australian Antarctic Division involved in sample collection. This work was funded by a CSIRO Office of the Chief Executive Science Leader Fellowship (R‐04202) to L.B. as well as the Antarctic Science International Bursary 2017 to S.L.S.S. We would like to acknowledge the contribution of the Marine Microbes Project consortium (https://data.bioplatforms.com) in the generation of data used in this publication. The Marine Microbes Project is supported by funding from Bioplatforms Australia and the Integrated Marine Observing System (IMOS) through the Australian Government National Collaborative Research Infrastructure Strategy (NCRIS) and the Education Investment Fund (EIF) Super Science Initiative in partnership with the Australian research community. B.M.S. was funded by the Australian Government Department of the Environment and CSIRO through the Australian Climate Change Science Programme and by the National Environmental Science Program. Hydrographic data were collected and made publicly available by the International Global Ship‐based Hydrographic Investigations Program (GO‐SHIP; http://www.go‐ship.org/) and the national programs that contribute to it. We thank Drs. Bernadette M. Sloyan and Susan Wijffels for giving us the opportunity to sample on the GO‐SHIP P15S voyage, and Dr. Nicole Hellessey for her sampling assistance.
References
- Abell, G.C. , and Bowman, J.P. (2005a) Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol Ecol 51: 265–277. [DOI] [PubMed] [Google Scholar]
- Abell, G.C.J. , and Bowman, J.P. (2005b) Colonization and community dynamics of class Flavobacteria on diatom detritus in experimental mesocosms based on Southern Ocean seawater. FEMS Microbiol Ecol 53: 379–391. [DOI] [PubMed] [Google Scholar]
- Acinas, S.G. , Sanchez, P. , Salazar, G. , Cornejo‐Castillo, F.M. , Sebastian, M. , Logares, R. , et al. (2021) Deep ocean metagenomes provide insight into the metabolic architecture of bathypelagic microbial communities. Commun Biol 4: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agogue, H. , Lamy, D. , Neal, P.R. , Sogin, M.L. , and Herndl, G.J. (2011) Water mass‐specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol Ecol 20: 258–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren, N.A. , Chen, Y. , Needham, D.M. , Parada, A.E. , Sachdeva, R. , Trinh, V. , et al. (2017) Genome and epigenome of a novel marine Thaumarchaeota strain suggest viral infection, phosphorothioation DNA modification and multiple restriction systems. Environ Microbiol 19: 2434–2452. [DOI] [PubMed] [Google Scholar]
- Alcaman‐Arias, M.E. , Farias, L. , Verdugo, J. , Alarcon‐Schumacher, T. , and Diez, B. (2018) Microbial activity during a coastal phytoplankton bloom on the Western Antarctic peninsula in late summer. FEMS Microbiol Lett 365: fny090. [DOI] [PubMed] [Google Scholar]
- Alonso‐Saez, L. , Andersson, A. , Heinrich, F. , and Bertilsson, S. (2011) High archaeal diversity in Antarctic circumpolar deep waters. Environ Microbiol Rep 3: 689–697. [DOI] [PubMed] [Google Scholar]
- Álvarez, M. , Ríos, A.F. , and Rosón, G. (2002) Spatio‐temporal variability of air–sea fluxes of carbon dioxide and oxygen in the Bransfield and Gerlache Straits during Austral summer 1995–96. Deep‐Sea Res II Top Stud Oceanogr 49: 643–662. [Google Scholar]
- Anderson, M.J. , Gorley, R. , and Clarke, K.R. (2008) PERMANOVA+ for PRIMER: Guide to Software and Statistical Methods. Plymouth, United Kingdom: PRIMER‐E. [Google Scholar]
- Appleyard, S. , Abell, G. , and Watson, R. (2013) Tackling microbial related issues in cultured shellfish via integrated molecular and water chemistry approaches. In Seafood CRC Final Report (2011/729). Deakin, Australia: Australian Seafood Cooperative Research Centre, p. 89. [Google Scholar]
- Arístegui, J. , Gasol, J.M. , Duarte, C.M. , and Herndld, G.J. (2009) Microbial oceanography of the dark ocean's pelagic realm. Limnol Oceanogr 54: 1501–1529. [Google Scholar]
- Arrigo, K.R. , van Dijken, G.L. , and Bushinsky, S. (2008) Primary production in the Southern Ocean, 1997–2006. J Geophys Res Oceans 113: 1–27. [Google Scholar]
- Baltar, F. , and Arístegui, J. (2017) Fronts at the Surface Ocean can shape distinct regions of microbial activity and community assemblages down to the bathypelagic zone: the Azores front as a case study. Front Mar Sci 4: 252. [Google Scholar]
- Beaugrand, G. , and Kirby, R.R. (2018) How do marine pelagic species respond to climate change? Theories and observations. Annu Rev Mar Sci 10: 169–197. [DOI] [PubMed] [Google Scholar]
- Bestley, S. , van Wijk, E. , Rosenberg, M. , Eriksen, R. , Corney, S. , Tattersall, K. , and Rintoul, S. (2020) Ocean circulation and frontal structure near the southern Kerguelen plateau: the physical context for the Kerguelen Axis ecosystem study. Deep‐Sea Res II Top Stud Oceanogr 174: 104479. [Google Scholar]
- Brown, M.V. , Lauro, F.M. , DeMaere, M.Z. , Muir, L. , Wilkins, D. , Thomas, T. , et al. (2012) Global biogeography of SAR11 marine bacteria. Mol Syst Biol 8: 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M.V. , Philip, G.K. , Bunge, J.A. , Smith, M.C. , Bissett, A. , Lauro, F.M. , et al. (2009) Microbial community structure in the North Pacific Ocean. ISME J 3: 1374–1386. [DOI] [PubMed] [Google Scholar]
- Brown, M.V. , van de Kamp, J. , Ostrowski, M. , Seymour, J.R. , Ingleton, T. , Messer, L.F. , et al. (2018) Systematic, continental scale temporal monitoring of marine pelagic microbiota by the Australian marine microbial biodiversity initiative. Sci Data 5: 180130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buesseler, K.O. , Pike, S. , Maiti, K. , Lamborg, C.H. , Siegel, D.A. , and Trull, T.W. (2009) Thorium‐234 as a tracer of spatial, temporal and vertical variability in particle flux in the North Pacific. Deep‐Sea Res I Oceanogr Res Pap 56: 1143–1167. [Google Scholar]
- Church, M.J. , DeLong, E.F. , Ducklow, H.W. , Karner, M.B. , Preston, C.M. , and Karl, D.M. (2003) Abundance and distribution of planktonic archaea and bacteria in the waters west of the Antarctic peninsula. Limnol Oceanogr 48: 1893–1902. [Google Scholar]
- Clarke, K. , and Gorley, R. (2015) PRIMER Version 7: User Manual/Tutorial. Plymouth, United Kingdom: PRIMER‐E. [Google Scholar]
- Cram, J.A. , Chow, C.E. , Sachdeva, R. , Needham, D.M. , Parada, A.E. , Steele, J.A. , and Fuhrman, J.A. (2015) Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J 9: 563–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, M.A.J. , and Jones, G.B. (2000) Dimethyl sulfide in the Southern Ocean: seasonality and flux. J Geophys Res Atmos 105: 20451–20459. [Google Scholar]
- DeLong, E.F. (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89: 5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong, E.F. , Preston, C.M. , Mincer, T. , Rich, V. , Hallam, S.J. , Frigaard, N.‐U. , et al. (2006) Community genomics among stratified microbial assemblages in the Ocean's interior. Science 311: 496–503. [DOI] [PubMed] [Google Scholar]
- Duarte, C.M. (2015) Seafaring in the 21St century: the Malaspina 2010 circumnavigation expedition. Limnol Oceanogr Bull 24: 11–14. [Google Scholar]
- Emery, W.J. (2003) Water types and water masses. In Ocean Circulation. London, UK: Elsevier Science, pp. 1556–1567. [Google Scholar]
- Field, K.G. , Gordon, D. , Wright, T. , Rappé, M. , Urback, E. , Vergin, K. , and Giovannoni, S.J. (1997) Diversity and depth‐specific distribution of SAR11 cluster rRNA genes from marine planktonic bacteria. Appl Environ Microbiol 63: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis, C.A. , Roberts, K.J. , Beman, J.M. , Santoro, A.E. , and Oakley, B.B. (2005) Ubiquity and diversity of ammonia‐oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci U S A 102: 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. , Keats, K.F. , Rivkin, R.B. , and Lang, A.S. (2013) Water mass and depth determine the distribution and diversity of Rhodobacterales in an Arctic marine system. FEMS Microbiol Ecol 84: 564–576. [DOI] [PubMed] [Google Scholar]
- Fuhrman, J.A. , Cram, J.A. , and Needham, D.M. (2015) Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol 13: 133–146. [DOI] [PubMed] [Google Scholar]
- Fukami, T. (2015) Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu Rev Ecol Evol Syst 46: 1–23. [Google Scholar]
- Gabric, A.J. , Whetton, P.H. , and Cropp, R. (2001) Dimethylsulphide production in the subantarctic southern ocean under enhanced greenhouse conditions. Tellus B: Chem Phys Meteorol 53: 273–287. [Google Scholar]
- Galand, P.E. , Potvin, M. , Casamayor, E.O. , and Lovejoy, C. (2010) Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J 4: 564–576. [DOI] [PubMed] [Google Scholar]
- Giebel, H.A. , Brinkhoff, T. , Zwisler, W. , Selje, N. , and Simon, M. (2009) Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ Microbiol 11: 2164–2178. [DOI] [PubMed] [Google Scholar]
- Giebel, H.A. , Kalhoefer, D. , Lemke, A. , Thole, S. , Gahl‐Janssen, R. , Simon, M. , and Brinkhoff, T. (2011) Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J 5: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, J. , Somerfield, P. , Temperton, B. , Huse, S. , Joint, I. , and Field, D. (2010) Day‐length is central to maintaining consistent seasonal diversity in marine bacterioplankton. Nat Precedings. [Google Scholar]
- Gilbert, J.A. , Steele, J.A. , Caporaso, J.G. , Steinbruck, L. , Reeder, J. , Temperton, B. , et al. (2012) Defining seasonal marine microbial community dynamics. ISME J 6: 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni, S.J. (2017) SAR11 bacteria: the most abundant plankton in the oceans. Annu Rev Mar Sci 9: 231–255. [DOI] [PubMed] [Google Scholar]
- Giovannoni, S.J. , and Vergin, K.L. (2012) Seasonality in ocean microbial communities. Science 335: 671–676. [DOI] [PubMed] [Google Scholar]
- Gordon, A.L. (1967) Structure of Antarctic waters between 20°W and 170°W. In Antarctic Map Folio Series, Bushnell, V. (ed). New York, NY: American Geographical Society, p. 10. [Google Scholar]
- Gralka, M. , Szabo, R. , Stocker, R. , and Cordero, O.X. (2020) Trophic interactions and the drivers of microbial community assembly. Curr Biol 30: R1176–R1188. [DOI] [PubMed] [Google Scholar]
- Grzymski, J.J. , Riesenfeld, C.S. , Williams, T.J. , Dussaq, A.M. , Ducklow, H. , Erickson, M. , et al. (2012) A metagenomic assessment of winter and summer bacterioplankton from Antarctica peninsula coastal surface waters. ISME J 6: 1901–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haine, T.W.N. , Watson, A.J. , Liddicoat, M.I. , and Dickson, R.R. (1998) The flow of Antarctic bottom water to the Southwest Indian Ocean estimated using CFCs. J Geophys Res Oceans 103: 27637–27653. [Google Scholar]
- Hallam, S.J. , Mincer, T.J. , Schleper, C. , Preston, C.M. , Roberts, K. , Richardson, P.M. , and DeLong, E.F. (2006) Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol 4: e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, H.P. , and Koroleff, F. (2009) Determination of nutrients. In Methods of Seawater Analysis, Grasshoff, K. , Kremling, K. , and Ehrhardt, M. (eds). Weinheim, Germany: Wiley‐VCH, pp. 159–228. [Google Scholar]
- Hawley, A.K. , Nobu, M.K. , Wright, J.J. , Durno, W.E. , Morgan‐Lang, C. , Sage, B. , et al. (2017) Diverse Marinimicrobia bacteria may mediate coupled biogeochemical cycles along eco‐thermodynamic gradients. Nat Commun 8: 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndl, G.J. , and Reinthaler, T. (2013) Microbial control of the dark end of the biological pump. Nat Geosci 6: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndl, G.J. , Reinthaler, T. , Teira, E. , van Aken, H. , Veth, C. , Pernthaler, A. , and Pernthaler, J. (2005) Contribution of archaea to total prokaryotic production in the deep Atlantic Ocean. Appl Environ Microbiol 71: 2303–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson, I. , Steele Joshua, A. , Capone Douglas, G. , and Fuhrman Jed, A. (2006) Remarkable heterogeneity in meso‐ and bathypelagic bacterioplankton assemblage composition. Limnol Oceanogr 51: 1274–1283. [Google Scholar]
- Kalanetra, K.M. , Bano, N. , and Hollibaugh, J.T. (2009) Ammonia‐oxidizing archaea in the Arctic Ocean and Antarctic coastal waters. Environ Microbiol 11: 2434–2445. [DOI] [PubMed] [Google Scholar]
- Kim, S.J. , Kim, J.G. , Lee, S.H. , Park, S.J. , Gwak, J.H. , Jung, M.Y. , et al. (2019) Genomic and metatranscriptomic analyses of carbon remineralization in an Antarctic polynya. Microbiome 7: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman, D.L. (2002) The ecology of Cytophaga‐Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91–100. [DOI] [PubMed] [Google Scholar]
- Landa, M. , Blain, S. , Christaki, U. , Monchy, S. , and Obernosterer, I. (2016) Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. ISME J 10: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa, M. , Blain, S. , Harmand, J. , Monchy, S. , Rapaport, A. , and Obernosterer, I. (2018) Major changes in the composition of a Southern Ocean bacterial community in response to diatom‐derived dissolved organic matter. FEMS Microbiol Ecol 94: fiy034. [DOI] [PubMed] [Google Scholar]
- Lane, D.J. (1991) 16S/23S rRNA Sequencing. In Nucleic Acid Techniques in Bacterial Systematics, Stackebrandt, E. , and Goodfellow, M. (eds). New York: John Wiley & Sons, pp. 115–148. [Google Scholar]
- Lane, D.J. , Pace, B. , Olsen, G.J. , Stahl, D.A. , Sogin, M. , and Pace, N.R. (1985) Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A 82: 6955–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legge, O.J. , Bakker, D.C.E. , Johnson, M.T. , Meredith, M.P. , Venables, H.J. , Brown, P.J. , and Lee, G.A. (2015) The seasonal cycle of ocean‐atmosphere CO2 flux in Ryder Bay, West Antarctic peninsula. Geophys Res Lett 42: 2934–2942. [Google Scholar]
- Lemoine, N. (2015) EcoPy – ecological data analysis in python.
- Lenton, A. , Tilbrook, B. , Law, R.M. , Bakker, D. , Doney, S.C. , Gruber, N. , et al. (2013) Sea‐air CO2 fluxes in the Southern Ocean for the period 1990–2009. Biogeosciences 10: 4037–4054. [Google Scholar]
- Liu, Y. , Blain, S. , Crispi, O. , Rembauville, M. , and Obernosterer, I. (2020) Seasonal dynamics of prokaryotes and their associations with diatoms in the Southern Ocean as revealed by an autonomous sampler. Environ Microbiol 22: 3968–3984. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Debeljak, P. , Rembauville, M. , Blain, S. , and Obernosterer, I. (2019) Diatoms shape the biogeography of heterotrophic prokaryotes in early spring in the Southern Ocean. Environ Microbiol 21: 1452–1465. [DOI] [PubMed] [Google Scholar]
- López‐García, P. , López‐López, A. , Moreira, D. , and Rodríguez‐Valera, F. (2001) Diversity of free‐living prokaryotes from a deep‐sea site at the Antarctic Polar Front. FEMS Microbiol Ecol 36: 193–202. [DOI] [PubMed] [Google Scholar]
- Luo, H. , Tolar, B.B. , Swan, B.K. , Zhang, C.L. , Stepanauskas, R. , Ann Moran, M. , and Hollibaugh, J.T. (2014) Single‐cell genomics shedding light on marine Thaumarchaeota diversification. ISME J 8: 732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli, M. , Malfatti, F. , Samo, T.J. , Mitchell, B.G. , Wang, H. , and Azam, F. (2009) Major role of microbes in carbon fluxes during Austral winter in the Southern Drake Passage. PLoS One 4: e6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre, M. , Ruiz‐Gonzalez, C. , Logares, R. , Duarte, C.M. , Gasol, J.M. , and Sala, M.M. (2018) Sinking particles promote vertical connectivity in the ocean microbiome. Proc Natl Acad Sci U S A 115: E6799–E6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milici, M. , Vital, M. , Tomasch, J. , Badewien, T.H. , Giebel, H.‐A. , Plumeier, I. , et al. (2017) Diversity and community composition of particle‐associated and free‐living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait. Limnol Oceanogr 62: 1080–1095. [Google Scholar]
- Murray, A.E. , and Grzymski, J.J. (2007) Diversity and genomics of Antarctic marine micro‐organisms. Philos Trans R Soc Lond B Biol Sci 362: 2259–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A.E. , Preston, C.M. , Massana, R. , Taylor, L.T. , Blakis, A. , Wu, K. , and DeLong, E.F. (1998) Seasonal and spatial variability of bacterial and archaeal assemblages in the coastal waters near Anvers Island, Antarctica. Appl Environ Microbiol 64: 2585–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, T. , Tamburini, C. , Arístegui, J. , Baltar, F. , Bochdansky, A.B. , Fonda‐Umani, S. , et al. (2010) Emerging concepts on microbial processes in the bathypelagic ocean – ecology, biogeochemistry, and genomics. Deep‐Sea Res II Top Stud Oceanogr 57: 1519–1536. [Google Scholar]
- Oksanen, J. , Blanchet, F.G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. et al. (2018) vegan: Community Ecology R package. URL https://CRAN.R-project.org/package=vegan.
- Parada, A.E. , and Fuhrman, J.A. (2017) Marine archaeal dynamics and interactions with the microbial community over 5 years from surface to seafloor. ISME J 11: 2510–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo, P.C. , Pérez, F.F. , Velo, A. , and Gilcoto, M. (2012) Water masses distribution in the Southern Ocean: improvement of an extended OMP (eOMP) analysis. Prog Oceanogr 103: 92–105. [Google Scholar]
- Ploug, H. , Iversen, M.H. , and Fischer, G. (2008) Ballast, sinking velocity, and apparent diffusivity within marine snow and zooplankton fecal pellets: implications for substrate turnover by attached bacteria. Limnol Oceanogr 53: 1878–1886. [Google Scholar]
- Raes, E.J. , Bodrossy, L. , van de Kamp, J. , Bissett, A. , Ostrowski, M. , Brown, M.V. , et al. (2018) Oceanographic boundaries constrain microbial diversity gradients in the South Pacific Ocean. Proc Natl Acad Sci U S A 115: E8266–E8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes, E.J. , van de Kamp, J. , Bodrossy, L. , Fong, A.A. , Riekenberg, J. , Holmes, B.H. , et al. (2020) N2 fixation and new insights into nitrification from the ice‐edge to the equator in the South Pacific Ocean. Front Mar Sci 7: 389. [Google Scholar]
- Richardson, A.J. , and Schoeman, D.S. (2004) Climate impact on plankton ecosystems in the Northeast Atlantic. Science 305: 1609–1612. [DOI] [PubMed] [Google Scholar]
- Rinke, C. , Rubino, F. , Messer, L.F. , Youssef, N. , Parks, D.H. , Chuvochina, M. , et al. (2019) A phylogenomic and ecological analysis of the globally abundant marine group II archaea (Ca. Poseidoniales ord. nov.). ISME J 13: 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke, C. , Schwientek, P. , Sczyrba, A. , Ivanova, N.N. , Anderson, I.J. , Cheng, J.F. , et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- Rusch, D.B. , Halpern, A.L. , Sutton, G. , Heidelberg, K.B. , Williamson, S. , Yooseph, S. , et al. (2007) The sorcerer II Global Ocean sampling expedition: Northwest Atlantic through eastern tropical Pacific. PLoS Biol 5: e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, G. , Cornejo‐Castillo, F.M. , Benitez‐Barrios, V. , Fraile‐Nuez, E. , Alvarez‐Salgado, X.A. , Duarte, C.M. , et al. (2016) Global diversity and biogeography of deep‐sea pelagic prokaryotes. ISME J 10: 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, A.E. , Richter, R.A. , and Dupont, C.L. (2019) Planktonic marine archaea. Ann Rev Mar Sci 11: 131–158. [DOI] [PubMed] [Google Scholar]
- Santoro, A.E. , Saito, M.A. , Goepfert, T.J. , Lamborg, C.H. , Dupont, C.L. , and DiTullio, G.R. (2017) Thaumarchaeal ecotype distributions across the equatorial Pacific Ocean and their potential roles in nitrification and sinking flux attenuation. Limnol Oceanogr 62: 1984–2003. [Google Scholar]
- Scales, K.L. , Miller, P.I. , Hawkes, L.A. , Ingram, S.N. , Sims, D.W. , Votier, S.C. , and Punt, A. (2014) REVIEW: on the front line: frontal zones as priority at‐sea conservation areas for mobile marine vertebrates. J Appl Ecol 51: 1575–1583. [Google Scholar]
- Schauer, R. , Bienhold, C. , Ramette, A. , and Harder, J. (2010) Bacterial diversity and biogeography in deep‐sea surface sediments of the South Atlantic Ocean. ISME J 4: 159–170. [DOI] [PubMed] [Google Scholar]
- Selje, N. , Simon, M. , and Brinkhoff, T. (2004) A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 427: 445–448. [DOI] [PubMed] [Google Scholar]
- Shah Walter, S.R. , Jaekel, U. , Osterholz, H. , Fisher, A.T. , Huber, J.A. , Pearson, A. , et al. (2018) Microbial decomposition of marine dissolved organic matter in cool oceanic crust. Nat Geosci 11: 334–339. [Google Scholar]
- Shiozaki, T. , Ijichi, M. , Isobe, K. , Hashihama, F. , Nakamura, K. , Ehama, M. , et al. (2016) Nitrification and its influence on biogeochemical cycles from the equatorial Pacific to the Arctic Ocean. ISME J 10: 2184–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signori, C.N. , Pellizari, V.H. , Enrich‐Prast, A. , and Sievert, S.M. (2017) Spatiotemporal dynamics of marine bacterial and archaeal communities in surface waters off the northern Antarctic peninsula. Deep‐Sea Res II Top Stud Oceanogr 149: 150–160. [Google Scholar]
- Signori, C.N. , Thomas, F. , Enrich‐Prast, A. , Pollery, R.C. , and Sievert, S.M. (2014) Microbial diversity and community structure across environmental gradients in Bransfield Strait, Western Antarctic Peninsula. Front Microbiol 5: 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintes, E. , Bergauer, K. , De Corte, D. , Yokokawa, T. , and Herndl, G.J. (2013) Archaeal amoA gene diversity points to distinct biogeography of ammonia‐oxidizing Crenarchaeota in the ocean. Environ Microbiol 15: 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sintes, E. , De Corte, D. , Haberleitner, E. , and Herndl, G.J. (2016) Geographic distribution of archaeal ammonia oxidizing ecotypes in the Atlantic Ocean. Front Microbiol 7: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloyan, B.M. , and Rintoul, S.R. (2001) Circulation, renewal, and modification of Antarctic mode and intermediate water. J Phys Oceanogr 31: 1005–1030. [Google Scholar]
- Smith, K.L., Jr. , Ruhl, H.A. , Huffard, C.L. , Messie, M. , and Kahru, M. (2018) Episodic organic carbon fluxes from surface ocean to abyssal depths during long‐term monitoring in NE Pacific. Proc Natl Acad Sci U S A 115: 12235–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straza, T.R.A. , Ducklow, H.W. , Murray, A.E. , and Kirchman, D.L. (2010) Abundance and single‐cell activity of bacterial groups in Antarctic coastal waters. Limnol Oceanogr 55: 2526–2536. [Google Scholar]
- Sunagawa, S. , Coelho, L.P. , Chaffron, S. , Kultima, J.R. , Labadie, K. , Salazar, G. , et al. (2015) Structure and function of the global ocean microbiome. Science 348: 1261359. [DOI] [PubMed] [Google Scholar]
- Swan, B.K. , Martinez‐Garcia, M. , Preston, C.M. , Sczyrba, A. , Woyke, T. , Lamy, D. , et al. (2011) Potential for Chemolithoautotrophy among ubiquitous bacteria lineages in the Dark Ocean. Science 333: 1296–1300. [DOI] [PubMed] [Google Scholar]
- Talley, L.D. , Feely, R.A. , Sloyan, B.M. , Wanninkhof, R. , Baringer, M.O. , Bullister, J.L. , et al. (2016) Changes in ocean heat, carbon content, and ventilation: a review of the first decade of GO‐SHIP global repeat hydrography. Ann Rev Mar Sci 8: 185–215. [DOI] [PubMed] [Google Scholar]
- Teira, E. , Reinthaler, T. , Pernthaler, A. , Pernthaler, J. , and Herndl, G.J. (2004) Combining catalyzed reporter deposition‐fluorescence in situ hybridization and microautoradiography to detect substrate utilization by bacteria and archaea in the deep ocean. Appl Environ Microbiol 70: 4411–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber, A.R. , Seabrook, S. , and Welsh, R.M. (2020) Riddles in the cold: Antarctic endemism and microbial succession impact methane cycling in the Southern Ocean. Proc Biol Sci 287: 20201134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittensor, D.P. , Mora, C. , Jetz, W. , Lotze, H.K. , Ricard, D. , Berghe, E.V. , and Worm, B. (2010) Global patterns and predictors of marine biodiversity across taxa. Nature 466: 1098–1101. [DOI] [PubMed] [Google Scholar]
- Treusch, A.H. , Vergin, K.L. , Finlay, L.A. , Donatz, M.G. , Burton, R.M. , Carlson, C.A. , and Giovannoni, S.J. (2009) Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J 3: 1148–1163. [DOI] [PubMed] [Google Scholar]
- Varela, M.M. , van Aken, H.M. , Sintes, E. , and Herndl, G.J. (2008) Latitudinal trends of Crenarchaeota and bacteria in the meso‐ and bathypelagic water masses of the eastern North Atlantic. Environ Microbiol 10: 110–124. [DOI] [PubMed] [Google Scholar]
- Wadham, J.L. , Arndt, S. , Tulaczyk, S. , Stibal, M. , Tranter, M. , Telling, J. , et al. (2012) Potential methane reservoirs beneath Antarctica. Nature 488: 633–637. [DOI] [PubMed] [Google Scholar]
- Wilkins, D. , Lauro, F.M. , Williams, T.J. , Demaere, M.Z. , Brown, M.V. , Hoffman, J.M. , et al. (2013c) Biogeographic partitioning of Southern Ocean microorganisms revealed by metagenomics. Environ Microbiol 15: 1318–1333. [DOI] [PubMed] [Google Scholar]
- Wilkins, D. , van Sebille, E. , Rintoul, S.R. , Lauro, F.M. , and Cavicchioli, R. (2013a) Advection shapes Southern Ocean microbial assemblages independent of distance and environment effects. Nat Commun 4: 2457. [DOI] [PubMed] [Google Scholar]
- Wilkins, D. , Yau, S. , Williams, T.J. , Allen, M.A. , Brown, M.V. , DeMaere, M.Z. , et al. (2013b) Key microbial drivers in Antarctic aquatic environments. FEMS Microbiol Rev 37: 303–335. [DOI] [PubMed] [Google Scholar]
- Wright, J.J. , Mewis, K. , Hanson, N.W. , Konwar, K.M. , Maas, K.R. , and Hallam, S.J. (2014) Genomic properties of marine group A bacteria indicate a role in the marine sulfur cycle. ISME J 8: 455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Z. , Yang, J. , Liu, L. , Zhang, W. , and Amalfitano, S. (2015) Bacterioplankton community shifts associated with epipelagic and mesopelagic waters in the Southern Ocean. Sci Rep 5: 12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Xie, W. , Martin‐Cuadrado, A.‐B. , and Rodriguez‐valera, F. (2015) Marine group II archaea, potentially important players in the global ocean carbon cycle. Front Microbiol 6: 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinger, L. , Amaral‐Zettler, L.A. , Fuhrman, J.A. , Horner‐Devine, M.C. , Huse, S.M. , Welch, D.B. , et al. (2011) Global patterns of bacterial beta‐diversity in seafloor and seawater ecosystems. PLoS One 6: e24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinger, L. , Boetius, A. , and Ramette, A. (2014) Bacterial taxa‐area and distance‐decay relationships in marine environments. Mol Ecol 23: 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccarato, L. , Pallavicini, A. , Cerino, F. , Fonda Umani, S. , and Celussi, M. (2016) Water mass dynamics shape Ross Sea protist communities in mesopelagic and bathypelagic layers. Prog Oceanogr 149: 16–26. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary methods
Fig. S1. A. Temperature salinity curves along the P15S transect (Pacific sector) overlayed with corresponding dissolved oxygen values, illustrating the main water masses (labelled with its corresponding acronym). Contour lines indicate potential density anomaly values. Vertical distributions of B. oxygen, C. salinity, D. nitrate + nitrite (NOx), E. nitrite, F. phosphate and G. silicate along leg 1 of the P15S transect.
Fig. S2. A. Temperature salinity curves along the HEOBI and K‐Axis voyages (Indian sector) overlayed with corresponding dissolved oxygen values, illustrating the main water masses (labelled with its corresponding acronym). Contours lines indicate potential density anomaly values. Vertical distributions of B. oxygen, C. salinity, D. nitrate + nitrite (NOx), E. nitrite, F. phosphate and G. silicate along the HEOBI and K‐Axis voyages.
Fig. S3. Rarefaction curves of observed A. bacteria and B. archaea species. Samples were subsampled to a depth of 10,000 sequences as indicated by grey dotted lines prior to subsequent analyses.
Fig. S4. Rarified mean alpha‐diversity indices for prokaryotes within different SO water masses. A. Richness and Chao1 index; B. Pielou's evenness and Shannon‐Weaver index. Water masses acronyms are as shown in Fig. 1.
Fig. S5. Plot of bacterial and archaeal community A. Richness against dissolved oxygen and B. Pielou's evenness against dissolved oxygen.
Fig. S7. Shade plots indicating overall relative abundance (at phylum/class level) of A. bacteria and B. archaea detected, within each water mass and sector they were sampled from. Detailed charts of the main families of C. Gammaproteobacteria, D. Alphaproteobacteria, E. Bacteroidetes and F. main species/zOTUs of the archaeal Nitrosopumilus genus within each water mass‐sector are also shown. ‘Other phyla’ or ‘Other genera’ are bacterial phylum or archaeal genera with overall mean relative abundances of <0.5% across all water masses. ‘Unclassified archaea’ include archaeal zOTUs that were classified with bootstrap confidence values <50%. Water mass name acronyms and water mass‐sector symbols are as indicated in Fig. 1 and Fig. 2 legends.
Fig. S8. Relative abundances of Bacteroidetes within the AABW samples. Samples are sorted by sector and latitude. Sample ID – Sector; Water Mass; Voyage.Latitude.Depth (m). P – Pacific sector; I – Indian sector; G – GO‐SHIP; KX – K‐Axis; H – HEOBI
Fig. S9. Phylogenetic tree of abundant SAR324 zOTU sequences from Pacific SAMW and SASW samples clustered with other SAR324 sequences with identified clades. Sequences with the prefix ‘AMD’ in blue are sequences from this study and are not specific to either SAMW or SASW water masses only.
Fig. S10. PCA ordination of the environmental and geographical properties of (A) all samples (B) epi‐ and mesopelagic samples and (C) bathy‐ and abyssopelagic samples considered within this study, grouped according to the water masses they were assigned to. Water mass name acronyms are as indicated in Fig. 1.
Fig. S11. Distance‐decay plots of Bray–Curtis similarity values between (A‐B) surface water and (C‐D) AABW prokaryote samples versus geographical distance between the samples.
Table S2. Alpha diversity matrices for bacterial and archaeal 16S rRNA gene sequences within the different Southern Ocean water masses.
Table S6. Average Bray–Curtis similarity percentages of the prokaryote community within the various water masses.
Table S7. Pearson correlation of environmental variables considered within this study. +Surface samples analysed include all samples taken from depths <20 m from all stations.
Fig. S6. (a) Full charts for relative abundance of major bacteria detected from the different water masses at phylum/class level, showing each of the 223 samples on the x‐axis. Samples are arranged from high to low latitude in each water mass from the Pacific (P) followed by Indian (I) sector. Detailed charts of the main families within (b) Gammaproteobacteria, (c) Alphaproteobacteria and (d) Bacteroidetes is also shown. The full chart for archaeal relative abundance is shown in (e), and a detailed chart of the main Nitrosopumilus species/zOTUs is shown in (f).
Table S1. List of samples, sampling information and contextual data for all samples collected within this study.
Table S3. A: Permutational multivariate analysis of variance using distance matrices (PERMANOVA) on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 B: Crossed two‐factor permutational multivariate analysis of variance using distance matrices (PERMANOVA) on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 C: PERMDISP on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S3 D: Results from PERMANOVA pairwise tests for the interaction term of Sector x Water Mass in bacterial and archaeal communities. Tests were based on Bray‐Curtis resemblance matrices of fourth root transformed bacterial and archaeal zOTU abundance data (number of permutations = 9999). *p ≤ 0.0001, **p ≤ 0.01
Table S4. SIMPER analysis of the A. bacterial and B. archaeal community, grouped by phylum/class, family and genus, indicating the contribution of each phylum/class/family/genus to community dissimilarity between the different water masses in the Pacific sector and the same corresponding water mass in the Indian sector. The analysis included higher contributing zOTUs up to a cumulative cut‐off percentage of 80%.
Table S5. Proportion of variation explained by individual predictor variations within Distance‐based linear modelling (DistLM) marginal tests. *Surface samples analysed include all samples taken from depths <20 m from all stations. ‘‐’ denotes that variable was not included in model due to high co‐linearity (Pearson's r > |0.85|) with another variable that contributed to explaining more significant proportions of community variation. ** indicate non‐significant p‐values (p > 0.05); nPerm = 9999