

Abstract
Heart failure is a devastating clinical syndrome, but current therapies are unable to abolish the disease burden. New strategies to treat or prevent heart failure are urgently needed. Over the past decades, a clear relationship has been established between poor cardiac performance and metabolic perturbations, including deficits in substrate uptake and utilization, reduction in mitochondrial oxidative phosphorylation and excessive reactive oxygen species production. Together, these perturbations result in progressive depletion of cardiac adenosine triphosphate (ATP) and cardiac energy deprivation. Increasing the delivery of energy substrates (e.g., fatty acids, glucose, ketones) to the mitochondria will be worthless if the mitochondria are unable to turn these energy substrates into fuel. Micronutrients (including coenzyme Q10, zinc, copper, selenium and iron) are required to efficiently convert macronutrients to ATP. However, up to 50% of patients with heart failure are deficient in one or more micronutrients in cross‐sectional studies. Micronutrient deficiency has a high impact on mitochondrial energy production and should be considered an additional factor in the heart failure equation, moving our view of the failing myocardium away from an “an engine out of fuel” to “a defective engine on a path to self‐destruction.” This summary of evidence suggests that supplementation with micronutrients—preferably as a package rather than singly—might be a potential therapeutic strategy in the treatment of heart failure patients.
Keywords: deficiency, heart failure, micronutrients, mitochindrial dysfunction

Introduction
Low plasma concentrations of several micronutrients have been associated with reduced quality of life and adverse outcomes in heart failure (HF) [1, 2, 3, 4]. Most patients with HF consume less than the recommended daily amount of several micronutrients [5, 6], with intake of vitamin D (97% of patients), selenium (95%), zinc (65%) and iron (46%) being most often inadequate [7]. Up to 50% of patients are deficient in one or more micronutrients in cross‐sectional studies [5, 7, 8]. Moreover, patients with HF may have reduced intestinal absorption, increased urinary excretion due to diuretics and defective renal glomerular or tubular function due to oxidative and pro‐inflammatory stress, which exacerbates micronutrient deficiency [5, 9]. Available clinical evidence supports the usefulness of supplementation with some micronutrients to improve HF management in addition to evidence‐based pharmacological therapy [10]. The consensus statement from the Heart Failure Society of America (HFSA) Scientific Statements Committee has already outlined the potential benefit for HF patients when optimizing nutritional status [11]. However, European Society of Cardiology (ESC) HF guidelines do not recommend micronutrient supplementation, other than correcting iron deficiency with intravenous iron [12].
Reduction in bioenergetic capacity plays a major role in the development and worsening of HF [13, 14]. Macronutrients such as fatty acids, lactic acid and carbohydrates are the main energy sources for cardiomyocytes and are consumed in large quantities. Micronutrients—including vitamins, minerals and essential amino acids—are also necessary to convert macronutrients to adenosine triphosphate (ATP), but are required in very small amounts, which a healthy diet normally provides. The mitochondrial electron transport chain (mtETC) requires coenzyme Q10, zinc, copper, selenium and iron for efficient ATP production [15]. Micronutrient deficiency in HF may contribute to defective mitochondrial function and reduced synthetic capacity for ATP.
Despite the identification of a high prevalence of micronutrient deficiencies in HF and their association with an adverse prognosis, with the exception of iron, very few randomized trials of micronutrient interventions have been conducted [16]. Therefore, a little information is provided on micronutrients in HF management guidelines [17, 18]. In this review, we focus on five key micronutrients involved in mitochondrial ATP production (Table 1).
Table 1.
Five key micronutrients involved in mitochondrial adenosine triphosphate (ATP) production and deficiency in heart failure
| Micronutrient | Physiological role | Role in the ETC | Prevalence of deficiency in HF | Associations with deficiency in HF | Clinical response to supplements in HF |
|---|---|---|---|---|---|
| Iron |
|
|
|||
| Selenium |
|
|
|||
| Zinc |
|
|
|
|
|
|
|
||||
|
|
||||
| Copper |
|
|
|
||
| CoQ10 |
|
|
|||
|
Abbreviations: ACE, angiotensin‐converting enzyme; ATP, adenosine triphosphate; CV, cardiovascular; ETC, electron transport chain; HF, heart failure; LVEDV, left ventricular end‐diastolic volume; LVEF, left ventricular ejection fraction; NR, not reported; NYHA, New York Heart Association; RCT, randomized controlled trial; SOD, superoxide dismutase.
Mitochondrial dysfunction as common denominator
Micronutrients serve as cofactors or part of essential amino acids in enzymatic reactions because they facilitate the transition between oxidized and reduced states. The systemic and cellular levels of some micronutrients—iron and copper in particular—must be balanced, as deficiency leads to loss of activity of crucial enzymes, but their accumulation may be toxic. Micronutrients are particularly important for normal mitochondrial function, especially in mitochondria‐rich tissues such as the myocardium. Oxidative phosphorylation is the process by which ATP is formed by the transfer of electrons from 1,4‐dihydronicotinamide adenine dinucleotide (NADH) or dihydroflavine‐adenine dinucleotide (FADH2) to oxygen (O2) by a series of electron carriers [64]. Mitochondrial oxidative phosphorylation is responsible for approximately 90% of the ATP produced in cardiomyocytes. An abundance of iron (complexes I, II, III and IV), selenium (redox and antioxidant), zinc (mitochondrial antioxidant), copper (complex IV) and coenzyme Q10 (CoQ10) (complexes I, II and III) is essential for the normal function of the mtETC (Fig. 1).
Fig. 1.
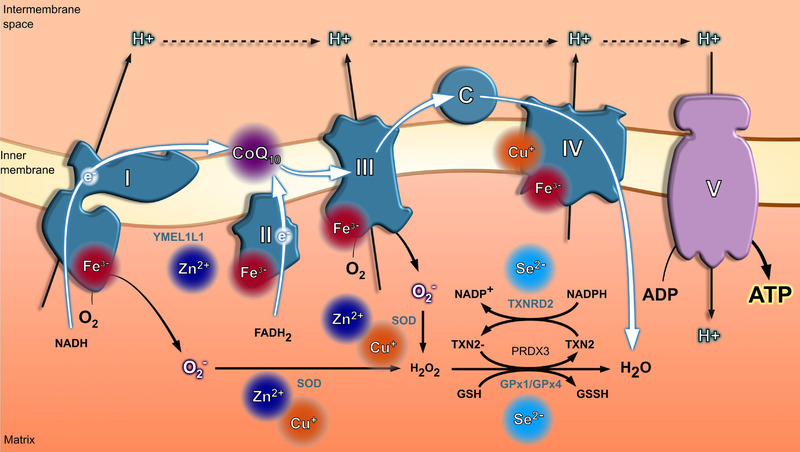
Micronutrients in the mitochondrial electron transport change (mtETC). The electron transport chain (ETC) starts with a proton transfer (H+) mediated by complexes I and II, which promotes an electrochemical gradient across the mitochondrial membrane. Complex III (ubiquinol‐cytochrome c oxidoreductase or CIII) forms the central part of the mitochondrial respiratory chain, oxidizing CoQ10 and reducing cytochrome c while pumping protons from the matrix to the intermembrane space through the so‐called Q‐cycle mechanism. Finally, four cytochrome C molecules deliver an electron to complex IV (cytochrome c oxidase or CIV), being carried by the complex and transfer them to one dioxygen molecule, converting the molecular oxygen to two molecules of water. The electrochemical gradient is used by complex V (adenosine triphosphate [ATP] synthesis) to promote the generation of ATP from the available adenosine diphosphate (ADP). Although the ETC is a quite efficient mechanism to promote energy formation, the proton gradient generation results in an elevated reactive oxygen species (ROS) production due the O2 oxidation into O2 − (superoxide anion radical), H2O2 and OH (hydroxyl radical), which are the toxic products of respiration. Micronutrients present a key role in the proton gradient generation (CoQ10) and electron carrier transfer among the different complexes (Fe3+ and Cu+). Furthermore, Cu+, Zn2– and Se2− participate in the oxidant scavenger system, decreasing toxic mitochondrial ROS. Abbreviations: FADH2, flavin adenine dinucleotide; GPXs, glutathione peroxidases; GSH glutathione; reduced NADH, nicotinamide adenine dinucleotide; PRDX3, peroxiredoxin 3; SOD, superoxide dismutase; TXN2, thioredoxin 2; TXNRDs, thioredoxin reductases.
Iron
Iron is essential for oxygen transport and ATP production, but an excess of nonferritin‐bound iron in cells is potentially extremely toxic. Iron accepts and donates electrons, switching between its bivalent (Fe2+) and trivalent (Fe3+) form [65], leading to formation of reactive oxygen species (ROS). Cells require protection from free iron, provided by ferritin intracellularly and transferrin in the plasma [25, 66]. Most of the body's iron is bound to hemoglobin or myoglobin, or it is stored intracellularly bound to ferritin or—especially in macrophages and the liver—bound to hemosiderin. A much smaller amount of iron is also found bound to carrier proteins such as transferrin or to nonheme enzymes involved in oxidation–reduction reactions and the transfer of electrons (cytochromes and catalase) in mitochondria [65]. The ability of iron to engage in redox reactions makes it a widely utilized cofactor in many key biochemical processes including oxygen delivery and storage, mitochondrial oxidative phosphorylation, DNA replication and repair, lipid metabolism and chromatin modification [19]. Specialized transporters, mitoferrin 1 and 2, move iron from the cytoplasm into the mitochondria [67]. Mitochondria play a key role in iron metabolism in which they synthesize heme, assemble iron–sulfur (Fe/S) proteins and participate in cellular iron regulation [68]. Iron is directly involved in several complexes of the mtETC, containing Fe/S clusters (complexes I, II, III and IV) and heme (complexes II, III and IV), enabling oxidative phosphorylation by ATP synthase within the mitochondria [69], but iron is also a critical component of many other heme‐containing enzymes involved in energy production [70]. Activity of mitochondrial complexes I–III, which predominantly contain Fe/S clusters, was shown to be adversely affected by deferoxamine (DFO) treatment, while the exclusively heme‐based complexes IV and V showed unaltered activity (Hoes et al. [20]). These findings were confirmed by Khechaduri et al. [71], who concluded that despite global mitochondrial dysfunction, heme levels were maintained above baseline in human failing hearts, and Melenovsky et al. [72], who also showed a decreased activity of mitochondrial complexes I and III in HF patients. However, the underlying mechanism regulating heme conservation remains unclear.
Several in vitro studies have shown detrimental effects of iron deficiency on mitochondrial function and morphology in human cardiomyocytes and myoblasts [20, 72, 73]. Iron deficiency provokes a hypoxic response, resulting in mitochondrial dysfunction, reduced ATP production and impaired contractility and relaxation of cardiac myocytes [20]. Iron‐deficient mitochondria can also lead to the production of oxygen radicals (O2 −) by complexes I and III [74], causing injury to mitochondria and other organelles. Adding transferrin‐bound iron to these cells can correct these abnormalities [20]. It needs to be recognized that the circulatory, cellular and organellar systems covering iron deficiency might be subjected to separate and different regulatory mechanisms. For example, differentiating between cytosolic and mitochondrial iron content showed that mitochondrial iron was actually increased, while cytosolic iron was reduced [71].
Human mitochondrial function in patients with HF and iron deficiency has been assessed in skeletal muscle using phosphorus‐31 magnetic resonance spectroscopy (31P MRS). This technique allows for noninvasive, real‐time detection of phosphocreatine (PCr), inorganic phosphate and ATP. PCr comprises a pool of chemical energy, rapidly available during exercise, which gets replenished during periods of rest. Impaired skeletal muscle energetics—often accompanied by lower pre‐exercise PCr levels, a lower pH and a slower recovery rate of PCr after exercise—are observed both in patients with Heart Failure with reduced Ejection Fraction (HFrEF) and Heart Failure with preserved Ejection Fraction (HFpEF) [75, 76, 77]. Among patients with iron deficiency, these abnormalities are more pronounced [77]. Administration of a single dose of iron isomaltoside in a randomized, double‐blind, placebo‐controlled trial including 40 patients with HFrEF improved PCr recovery within a few weeks [78]. Future studies into organ‐, cell‐ and organelle‐specific iron regulation and pathophysiology will hopefully provide further insights [79].
Iron deficiency is the most common nutritional deficiency, estimated to affect more than 2 billion people worldwide [80]. Gastrointestinal absorption of iron is inefficient: Only 5%–10% of iron is actually taken up, and may be reduced by many factors including diet, inflammation and edema. Most patients with HF have or will develop iron deficiency [2, 21–23]. Patients with HF and iron deficiency have worse symptoms, quality of life, functional status and clinical outcomes—including mortality—irrespective of left ventricular ejection fraction (LVEF) [21, 22, 23, 24, 25, 26].
Several substantial, randomized, double‐blind, placebo‐controlled multicenter trials investigating intravenous (IV) iron—mainly with ferric carboxymaltose—in patients with HF and a reduced LVEF (HFrEF) have shown beneficial effects on symptoms, quality of life and exercise capacity [29, 30, 31]. Similar evidence does not exist for oral iron, possibly because of ineffective iron uptake and/or lack of sufficient robust trials [81, 82]. The most recent ESC guidelines for the diagnosis and treatment of acute and chronic HF recommend that all patients with HF should be tested for iron deficiency, and treatment with IV iron should be considered to improve symptoms, exercise capacity and quality of life [83]. The 2017 American Heart Association/American College of Cardiology (AHA/ACC) guidelines made similar recommendations [84]. Recently, results were published from the AFFIRM‐AHF trial (n = 1038), which investigated the effects, compared with placebo, of IV ferric carboxymaltose administered prior to discharge to patients who had been admitted with acute HF, an LVEF less than 50% and iron deficiency using the FAIR‐HF trial criteria. Patients assigned to IV iron had fewer rehospitalizations for HF, but no effect on mortality was observed [28]. Iron deficiency is also common in patients with HF and an LVEF greater than 40% (HFpEF), especially for those with more severe diastolic dysfunction, and is associated with poorer exercise capacity and quality of life [85]. An updated meta‐analysis of randomized trials including patients with iron deficiency and HFrEF suggested that IV iron reduced hospitalization for HF by about 30% (odds ratio [OR]: 0.67 [0.54–0.85]; p = 0.0007) but could not confirm an effect on cardiovascular mortality (OR: 0.89 [0.66–1.21]; p = 0.47) [27]. This meta‐analysis does not exclude the possibility of a substantial reduction in mortality with IV iron, although neither does it rule out a modest increase. Several more studies are to be completed in the next 2 years. Currently, data are lacking on the effects of iron supplementation in patients with HFpEF.
Selenium
Selenium is a component of selenocysteine [86], an amino acid that is required for the formation of selenoproteins [87, 88], including glutathione peroxidases (GPXs), thioredoxin reductases (TXNRDs) and iodothyronine deiodinases (DIOs). The function of these selenoproteins in the heart is not understood completely [1, 89–91]. Proteins from the GPX and TXNRD families are crucial antioxidant (redox) enzymes involved in preventing the harmful accumulation of intracellular hydrogen peroxide, including in the mitochondria [35, 36]. DIOs regulate the local bioactivity of thyroid hormone, which stimulates mitochondrial biogenesis, increasing myocardial mitochondrial mass, mitochondrial respiration, oxidative phosphorylation, enzyme activities, mitochondrial protein synthesis, cytochrome, phospholipid and mitochondrial DNA (mtDNA) content [32, 92]. The human selenoproteome also includes methionine sulfoxide reductase B1 (MSRB1), which promotes anti‐inflammatory cytokine expression and controls the immune response [39]. Two other selenoproteins are crucially involved in selenoprotein synthesis (selenophosphate synthetase 2 [SEPHS2]) and transport (selenoprotein P [SELENOP]) [34, 93]. SELENOP is of special interest, since it was shown to correlate very well with selenium levels [94]. SELENOP is the main protein responsible for carrying selenium in the circulatory system [91]. In the context of acute HF, it has been reported that patients with low SELENOP levels had greater risk for 30‐day rehospitalization (hazard ratio [HR]: 4.29; 95% confidence interval [CI]: 1.59–11.6), 1‐year mortality (HR: 4.13; 95% confidence interval [CI]: 1.64–10.4) and a composite endpoint of death or rehospitalization within 30 days (HR: 4.80; 95% CI: 1.80–12.8) compared with patients with higher levels [94]. Similarly, in a large Swedish prospective cohort study in the general population, patients with high SELENOP levels have significantly lower risk for all‐cause mortality (0.57, 0.48–0.69), cardiovascular mortality (0.52, 0.37–0.72) and first cardiovascular event (0.56, 0.44–0.71) [93]. The importance of other selenoproteins is uncertain [86]. Selenium deficiency impairs the ability to synthesize selenoproteins, increases oxidative stress, reduces the response to thyroxine and impairs the immune responses [95, 96]. Two well‐described and long‐known hierarchical principles can be observed in selenium availability and usage [97]. Not all tissues and selenoproteins are equally well supplied with the trace element when limited. First, selenium concentrations in the different tissues were found to vary, and selenium preferentially accumulated, and was retained efficiently, in specific organs such as the testes, adrenals and the brain. Second, comparisons of selenoprotein dependence on selenium status showed that specific selenoprotein transcripts compete for the limited amount of selenium and decrease gradually in response to deficiency [97]. Nevertheless, knowledge on the hierarchical position of the heart among other organs, and the cardiac‐specific selenoprotein hierarchy, is limited.
Selenium deprivation in human pluripotent stem cell (PSC)‐derived cardiomyocytes impairs mitochondrial respiration, biogenesis and oxidative stress [37], which may reflect the production of inactive GPX enzymes and subsequent impaired usage of glutathione [98, 99, 100]. ROS are increased in selenium‐depleted cardiomyocytes under normal culture conditions [37], which can be corrected by restoring selenium [37]. Cell apoptosis caused by reperfusion injury, which is mediated by oxidative stress, may also be exacerbated by selenium deficiency [101].
Severe selenium deficiency in humans may be associated with a rare but fatal form of dilated cardiomyopathy (DCM)—Keshan disease—that was reported from a specific geographic region where there was a very low amount of selenium in the soil and therefore in food [38]. Keshan disease is reversible with selenium supplementation [1]. Selenium soil content is very variable. For example, intakes are high in Venezuela, Canada, the United States and Japan (>100 μg/day), and much lower in some parts of Europe (∼40 μg/day) [95]. Observational studies in the general population [95, 102] suggest that selenium deficiency might be common, but data for patients with HF are scarce [47, 103, 104]. Recently, selenium deficiency (serum selenium <70 μg/L) was found to be associated with reduced exercise capacity and a substantially higher mortality in a prospective cohort study [37]. Furthermore, serum selenium concentrations of 70–100 μg/L were associated with similar adverse findings [37], suggesting that values below 100 μg/L might be considered to indicate deficiency [105, 106]. Selenium (<100 μg/L) and iron deficiency may often coincide [107], although, unlike iron, selenium is readily absorbed orally.
No substantial randomized trial of selenium supplements has yet been done in patients with HF. One placebo‐controlled, randomized trial (n = 443) investigated the effects on cardiovascular mortality of supplements of selenium together with CoQ10 given for 4 years to older Swedish citizens, some of whom had HF (KiSel‐10 study; n = 443; age 70–88 years) [108]. Despite discontinuing supplements after 4 years, cardiovascular mortality was lower at 10 years in those who had been assigned to supplementation (HR: 0.51; 95% CI: 0.36–0.74; p = 0.0003). Although most patients had cardiovascular problems, only a minority had HF. Additionally, one very small randomized controlled trial (RCT) has been performed in patients with HF from Iran. Garakyaraghi et al. [109] supplemented 32 patients with congestive heart failure (CHF) with a combination of 90 mg CoQ10 and 200 μg selenium per day for 3 months. This led to favorable effects on New York Heart Association (NYHA) classification, LVEF and myocardial performance index compared to the placebo group [109]. Besides the low number of included subjects, it cannot be concluded whether these effects are a result of selenium supplementation or CoQ10 use as they were given coincidentally, especially as no serum levels of either were measured. A meta‐analysis of nutrients with potential antioxidant properties suggested that only supplements that included selenium reduced cardiovascular disease (CVD) mortality (relative risk [RR]: 0.77; 95% CI: 0.62–0.97; p = 0.02) [110], even though the KiSel‐10 trial was not included. Supplements of other antioxidants—including vitamins A, C, and E, β‐carotene and retinol—were not associated with a lower mortality. Nonetheless, there is uncertainty and a paucity of robust data [111, 112, 113]. Only patients with selenium deficiency might benefit from supplements [98, 110]. Molecular evidence suggests that a serum selenium >100 μg/L is required for optimal GPX activity [34, 114–117]. A meta‐analysis that stratified studies into regions with higher (North‐ and South‐America) or lower (Europe and Asia) soil selenium content suggested a greater reduction in mortality with selenium supplements when soil selenium content was low (relative risk [RR]: 0.88; 95% CI: 0.78–0.98; p = 0.02), but a possible increase in mortality when selenium soil content was high (RR: 1.06; 95% CI: 1.01–1.12; p = 0.03) [118]. The hypothesis that only patients who have evidence of selenium deficiency benefit from supplements is plausible but should be evaluated in a well‐designed clinical trial.
Zinc
Zinc is vital for many physiological functions, including growth, reproduction, antioxidant defenses and the immune system. It is a critical component of the catalytic site of more than 300 enzymes and required for the synthesis and degradation of carbohydrates, lipids, proteins and nucleic acids [41]. Zinc also influences neuronal function and hormone release [119]. Angiotensin‐converting enzyme (ACE)—a membrane metallopeptidase that requires zinc for its catalytic activity [120]—plays a key role in the production of angiotensin II and degradation of bradykinin and many other vasoactive peptides [46]. Additionally, other Zn‐dependent proteases such as aminopeptidases are also known to limit and modulate angiotensin function, showing the importance of zinc sufficiency in the proper regulation of the ACE‐associated pathways [121]. ACE inhibitors, angiotensin II receptor blockers and thiazide diuretics may all reduce serum zinc concentrations [122].
The World Health Organization (WHO) suggests that zinc deficiency is common in many regions of the world [123]. Zinc deficiency can be caused by a diet high in phytate‐containing whole grains—as phytate is a potent ligand for zinc that prevents absorption [124]—foods grown in zinc‐deficient soil, or processed foods containing little or no zinc [125]. Plasma or serum zinc concentrations may be a poor indicator of moderate, subclinical zinc deficiency [124]. In humans, zinc deficiency usually occurs in the context of deficiencies of other nutrients, such as iron.
Mitochondrial peroxiredoxin and metalloenzymes, such as superoxide dismutase (SOD), rely on zinc for antioxidant reactions [126]. When mitochondrial production of ROS exceeds the rate of restoration of antioxidant defenses by zinc‐dependent superoxide dismutase (Cu/Zn‐SOD), mitochondrial permeability is increased, leading to swelling and degeneration and, ultimately, cell death [42, 43]. Next to the activity of SOD, zinc is also involved in the dynamic control of mitochondrial activity through so‐called mitochondrial proteases. The ATP‐dependent zinc metalloprotease YME1L1 is probably the best‐described mitoprotease and was shown to be involved in mitochondrial protein quality control, mitochondrial biogenesis, mitochondrial stress responses, mitochondrial dynamics, mitophagy and apoptosis [127]. Nevertheless, the direct functional link between this protease and zinc deficiency in the heart is not yet established. When chronic inflammation is present, zinc deficiency may contribute to apoptosis and myocardial necrosis [128]. Exogenous zinc regulates the activities of several key intracellular signaling elements such as mammalian target of rapamycin (mTOR), extracellular signal‐regulated kinase (ERK) and glycogen synthase kinase‐3β (GSK‐3β). Zinc‐induced inactivation of GSK‐3β inhibits mitochondrial permeability transition pore (mPTP) opening, preventing reperfusion injury [129].
Animal studies suggest that zinc supplementation may protect against loss of systolic function and enhance diastolic function during and after ischemia‐reperfusion injury [130]. Zinc might reduce oxidative stress partly by enhancing the activity of the zinc‐binding protein, metallothionein [41, 131], leading to increased zinc uptake and release in cardiac tissue [130, 132, 133]. Furthermore, zinc deficiency might also play a role in the development of a cardiomyopathy that can be reversed by zinc supplements [41, 45, 134, 135].
In HF, zinc deficiency may be due to low dietary intake, reduced gastrointestinal absorption, and/or increased excretion as the result of neurohormonal activation [7, 135–137]. Metabolic stress may increase cellular uptake of zinc to regulate antioxidant defenses. ACE inhibitors and angiotensin II receptor blockers (ARBs) increase urinary and fecal zinc excretion [137]. Although the prevalence of zinc deficiency in patients with HF is uncertain, serum zinc concentrations have been reported to be lower than those reported as normal values for healthy volunteers (75–140 μg/dl) [44, 45, 135, 138]. A prospective observational study of 1079 patients with decompensated HF suggested that serum zinc concentrations were less than 75 μg/dl in 66% of patients and that such values were associated with a higher cardiovascular and all‐cause mortality [3]. Amongst patients with HF, serum zinc concentrations decline with worsening NYHA class, older age, and use of ACE inhibitors and ARBs [3, 44]. Low serum concentrations are also associated with hyponatremia, iron deficiency, increases in C‐reactive protein (CRP) and troponin I—suggesting inflammation and myocardial damage, respectively—and with impaired exercise capacity [3].
There is a paucity of evidence to support zinc supplements. In a small, non‐randomized, prospective study, 10 patients with malabsorption‐associated cardiomyopathy received zinc and selenium supplements (Addamel N, 10 ml given intravenously; corresponding to 300 μg selenium and 13.6 mg zinc every day for 1 week, and subsequently every week for 6 months) [45]. Eight patients with malabsorption‐associated cardiomyopathy served as controls. Monthly injections increased serum and myocardial zinc content and LVEF (from 28% to 42%; p < 0.001), which were associated with clinical improvement. In myocardial biopsies, the content of selenium and zinc and GPX activity increased, with striking improvements in cardiomyocyte degenerative changes including a reduction of cell autophagy and myofibrillolysis [45]. A recent observational study evaluated the role for zinc supplements in patients with nonischemic heart disease. Patients (n = 25) were given zinc acetate—50 mg three times a day for 10 months—and were compared with 10 healthy subjects on similar treatment. However, the results of these studies have not yet been published [139]. Recently, a case report was presented of a 24‐year‐old female who was diagnosed with anorexia nervosa and new‐onset HF [140]. Supplementing her with oral zinc (220 mg/day), together with guideline‐recommended HF therapy and anorexia‐nervosa management, resulted in improved left ventricular systolic function. This case was pointed out as the first report of low plasma zinc levels as the probable cause of cardiomyopathy, which improved after zinc supplementation. Again, well‐designed clinical trials of sufficient size enrolling patients with HF and zinc deficiency are required to understand the potential therapeutic role of zinc supplements.
Copper
There are many copper‐dependent proteins (known as “cuproenzymes”), including transcriptional regulators, chaperones, oxidoreductases, free radical scavengers and immune function modulators [49, 50–52, 141]. The adult body contains 50–120 mg of copper, mostly in muscle and the liver. Copper is absorbed in the intestine and released by the liver into bile to prevent copper toxicity [142]. Copper is extremely toxic in excess as it can destabilize Fe/S clusters [143] and increase the generation of free radicals [144]. Mutations in ATP7A or ATP7B disrupt the homeostatic copper balance, resulting in inherited autosomal recessive disorders causing copper deficiency (Menkes disease) or copper overload (Wilson's disease), respectively [145, 146, 147]. Copper deficiency can lead to problems with connective tissues, muscle weakness, anemia, low white blood cell count, immune dysfunction and neurological problems [53]. According to an analysis of data from the 2009–2012 National Health and Nutrition Survey (NHANES), 6%–15% of adults have copper intake lower than recommended [148]. However, copper deficiency is uncommon in humans except in special cases, such as individuals with celiac disease or older people with cachexia [149].
Intracellular copper deficiency increases lipoprotein peroxidation, impairs nitric oxide (NO)‐mediated endothelial dilatation and increases the risk of cardiomyocyte oxidative damage [15]. In the mitochondria, copper is an essential component of complex IV, also known as Cytochrome C oxidase (CCO) [51]. Although copper is crucial for cytochrome oxidase function, the transporters and regulators of mitochondrial copper are unknown. In rats, a copper‐deficient diet caused a 74% decrease in CCO and increased manganese superoxide dismutase (MnSOD) and GPX in isolated heart mitochondria [150, 151]. This is an indirect indication that mitochondrial ROS production was increased [151]. Moreover, specifically cardiac mitochondrial ETC function was compromised in copper‐deficient rats [55]. Additionally, a study in mice by Elsherif et al. [152] showed that early life copper deficiency in mice leads to systolic and diastolic dysfunction in association with histopathological changes in the murine heart. The changes observed were suggested to show the transition from copper deficiency–induced hypertrophy [153] to CHF [152].
Higher blood concentrations of copper have been found in patients with HF compared to healthy controls [154]. Málek et al. [155] reported a possible association between copper status and prognosis in 64 patients with HF. Patients who died or were hospitalized for HF in the following 12 months had, on average, a higher serum copper concentration than those who did not (121 vs. 104 μg/dl [19.0 vs. 16.3 μmol/L]; p < 0.001). A meta‐analysis comprising 13 studies including 1504 subjects identified an association between high serum copper and HF [156]. High serum copper concentration in HF may reflect an increase in serum ceruloplasmin, which binds up to 95% of serum copper [157]. Ceruloplasmin plays multiple roles in copper transportation, coagulation, angiogenesis, oxidative stress defense and iron homeostasis [158], and it has also been associated with cardiovascular disease in clinical studies [159]. Ceruloplasmin oxidizes Fe(II) to Fe(III) (ferroxidase) in order for it to be incorporated into transferrin, exerts antioxidant GPX activity, scavenges ROS [44] and may play a fundamental role in protection from iron‐mediated free radical injury [160]. It has also been suggested that the increase in serum copper in HF is due to copper efflux from the myocardium, related to homocysteine dynamics, creating local/tissue deficiency [161]. Of note, ceruloplasmin constitutes an acute phase reactant [162]. Therefore, an increase in serum copper and ceruloplasmin concentrations could also be the consequence of (sub‐)clinical inflammation and stress, but not necessarily the cause. However, the increase of serum copper, and secondarily of ceruloplasmin caused by myocardial infarction, seems not to be completely associated with an inflammatory response [163].
To our knowledge, there is no evidence to support copper supplements for patients with HF. Currently, patients with HFrEF (n = 200) are being recruited for a randomized trial to evaluate the effects of a copper‐binding agent called INL1 in patients with HF (TRACER‐HF) [54]. It is hypothesized that INL1 serves as a copper redistribution shuttle to transport copper from the high‐concentration gradient, for example, the circulation, to copper‐depleted tissues such as ischemic myocardial tissue.
Coenzyme Q10
CoQ10, also known as ubiquinone, is found predominantly in meat, fish and nuts. Although CoQ10 is a common component of most cell membranes, it plays a key role in the mtETC to facilitate ATP production [57]. Mitochondria, and therefore CoQ10, are present in high concentrations in the myocardium, liver and kidneys. In addition, CoQ10 protects the cell membrane against oxidation and inhibits the peroxidation of lipids and lipoproteins [56]. The oxidative stress associated with HF increases the stress on antioxidant systems [164], which may deplete CoQ10 [165, 166, 167].
CoQ10 facilitates electron transfer from complex I (NADH coenzyme Q reductase) to complex III (cytochrome bc1 complex), and from complex II (succinate dehydrogenase) to complex III [58, 59]. It may also stabilize the mitochondrial permeability transition pore and reduce apoptotic cell loss [58]. In vitro experiments show that CoQ10 supplements inhibit low‐density lipoprotein oxidation to a greater degree than other natural antioxidants, such as β‐carotene or α‐tocopherol [168, 169].
In patients with HF, lower plasma concentrations of CoQ10 are associated with poorer NYHA functional class, lower LVEF and higher plasma concentrations of NT‐proBNP [61, 62]. Furthermore, patients with more severe HF (classes III and IV) have lower plasma and myocardial levels of CoQ10, suggesting greater deficiency as the disease worsens [165, 166, 167]. In unadjusted analyses, observational studies suggest that low plasma concentrations of CoQ10 predict a higher mortality in patients with HF [61, 166, 167], although not independently of other prognostic variables. Statins reduce plasma CoQ10 concentrations, but this does not appear to account for the lack of clinical benefit of statins in advanced HF [61, 170, 171]. Most of the CoQ10 in plasma is carried in lipoproteins, the concentration of which may fall with worsening disease severity or statin therapy [57, 172].
Over the past few decades, several randomized trials of CoQ10 supplements in HF have failed to produce conclusive evidence. One of the earliest placebo‐controlled randomized trials studied the effects of 12‐month treatment with CoQ10 in patients with chronic CHF (n = 641) [173] and reported fewer HF‐related hospitalizations in those assigned to CoQ10. More recently, between 2003 and 2010 the Q‐SYMBIO enrolled 420 patients in 17 centers internationally [63]. CoQ10 added to standard HF therapy did not improve NYHA functional class, exercise capacity or NT‐proBNP at 16 weeks, but in the longer term (median follow‐up 106 weeks) reduced the primary (composite) endpoint of hospitalization for worsening HF, cardiovascular death, mechanical assist implantation or urgent cardiac transplantation (HR: 0.50; 95% CI: 0.32−0.80; p = 0.003). All‐cause mortality was also reduced (HR: 0.51; 95% CI: 0.30−0.89; p = 0.018). The KiSel‐10 trial also suggested that a combination of CoQ10 and selenium reduced cardiovascular mortality [108]. Many other, smaller trials have evaluated the effects of CoQ10, and generally found improvements in symptoms, exercise capacity and quality of life [6, 174–180]. However, the heterogeneous populations and study outcomes, different trial designs and follow‐up duration, and different preparations of CoQ10 contribute to the uncertainty of evaluating and pooling data. Furthermore, coenzyme Q10 in its native form may never reach the mitochondria after ingestion, which brings up the question of the actual mode of action of the compound considering the performed clinical studies. Mito‐Q was developed to be able to reach the mitochondria, but has shown negative effects, since the DTPP moiety causes swelling and depolarization of mitochondria [181] and was found to be associated with a decrease in Δψ m and mtDNA copy number [182]. Therefore, more in‐depth mechanistic studies are needed to assess the actual mode of action of the compound in the heart, and in parallel, to develop safe compounds that enable shuttling of coQ10 into mitochondria. Therefore, well‐designed, adequately powered trials are necessary to assess the effect of CoQ10 supplements on the well‐being and survival of patients with HF.
Multi‐micronutrient interventions
(Mal‐)nutrition of micronutrients has great impact on the human heart and especially on its ability to recover from damage, and consequently associates with prognosis. Improper micronutrient intake is frequently observed in HF patients, affecting 30%–50% of this population [5, 7]. Deficiencies in multiple micronutrients—such as vitamin A, calcium, magnesium, selenium (Se), zinc, iron, vitamin D and iodine—have been documented, without establishing a causative association between them and the onset of HF [183]. The HFSA provided valuable recommendations on diet and nutrition in its most recent guidelines [11]. Adjustment of nutritional status and energy supplementation are recommended in patients with advanced HF. It was also suggested that daily evidence‐based multi‐micronutrient supplementation should be considered for all patients with HF, particularly those receiving diuretic therapy or restricted diets [11].
Several small, double‐blind, randomized trials have evaluated the role for micro‐ and macronutritional supplementation in patients with HFrEF and ischemic heart disease [47, 48, 184]. A first trial enrolled 30 patients and randomized them to placebo or a micronutrient cocktail composed of zinc, selenium, thiamine, CoQ10 and other micronutrients. After 1 year, mean LVEF increased from 26% to 31% (p < 0.05) in the group assigned to receive micronutrients but not in those assigned to placebo. However, there was no improvement in NYHA classification in the patients receiving micronutrient therapy [47]. A second trial enrolled 38 patients and randomized them to placebo or a micronutrient cocktail. Among those assigned to supplements, left ventricular end‐diastolic volume shrank by about 5%, but LVEF did not improve [48]. A third trial involving 74 patients with chronic stable HF that assessed the effect of multi‐micronutrient supplementation showed no significant difference in LVEF, nor in quality of life, exercise capacity or biomarkers [184]. A fourth study included 41 patients who underwent coronary artery bypass surgery with an LVEF less than 40% and were randomly assigned to a double‐blind trial of MyoVive supplement or placebo. Here, supplementation was associated with a reduction in left ventricular end‐diastolic volume [48]. Although small, these trials suggest a possible beneficial effect that requires further, more thorough investigation, especially regarding the inconsistent combinations of micronutrients in these studies.
One important factor that is hampering consistency of study results is the supplementation of subjects who are nondeficient (for one or multiple micronutrients). As an example, selenoprotein activity, and therewith supplementation effectiveness, reaches plateau levels at serum selenium concentrations of approximately 125 μg/L, under a sufficiently high selenium supply [185, 186]. This implies that (multi‐)micronutrient supplementation should be assessed in a personalized, nutrient‐specific manner, since supplementing patients sufficient of certain micronutrients has limited effect (select study). A second factor that may influence the results is the geographical location of the included studies, since dietary intake of, for example, selenium is high in Canada, the United States and Japan (>100 μg/day), and much lower in some parts of Europe (∼40 μg/day) [95]. As such, selenium‐replete patients may show less benefit. Supporting this, Kuria et al. reported in a subanalysis a significant reduction of cardiovascular mortality in studies conducted with selenium in Asia (RR: 0.59, 95% CI: 0.45–0.79) and Europe (RR:0.55, 95% CI: 0.45–0.68), but not in the United States (RR: 0.93; 95% CI: 0.82–1.05) [118]. Third, there is a lack of controlled conditions regarding participants’ diet and lifestyle, which can severely impact the study outcome. For instance, many investigations estimate participants’ diet information on memory‐based methods with low scientific rigor, or participants are not advised to follow any diet recommendation [187]. As an example, minerals and/or vitamin supplementation in combination with low‐fat diets (or fasting) drastically limits micronutrient absorption. Particularly, fat‐soluble CoQ10 is not efficiently absorbed. Therefore, micronutrient supplementation should preferably occur in combination with offering balanced meals, or individualized dietary guidance or supplementation to patients with HF [188, 189, 190].
Conclusions
The failing myocardium might be “an engine out of fuel.” However, increasing the delivery of energy substrates (e.g., fatty acids, glucose, ketones) to the mitochondria [191] cannot succeed if the mitochondria cannot turn these energy substrates into fuel (ATP) without wrecking the underperforming engine (increased ROS). Micronutrient deficiency changes the paradigm from “an engine out of fuel” to “a defective engine on a path to self‐destruction.”
Conflict of interest
The authors declare that there is no conflict of interest.
Author contributions
Nils Bomer: conceptualization (lead); methodology (lead); supervision (lead). Mario G. Pavez‐Giani: conceptualization (equal); methodology (supporting). Niels Grote Beverborg: conceptualization (supporting). Peter van der Meer: conceptualization (supporting); supervision (supporting).
Acknowledgments
Nils Bomer is supported by a Dutch Research Council (NWO) Open Competition ENW‐M grant (OCENW.KLEIN.483). John G. F. Cleland is supported by a British Heart Foundation (BHF) Centre of Research Excellence Award (RE/18/6/34217). Peter van der Meer is supported by the European Research Council (ERC) Starting Grant (StG 715732).
Bomer N, Pavez‐Giani MG, Grote Beverborg N, Cleland JGF, van Veldhuisen DJ, van der Meer P. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J Intern Med. 2022;291:713–731.
Authors Nils Bomer and Mario G. Pavez‐Giani contributed equally as first authors.
Linked to: N. Girerd J Intern Med 2022; https://doi.org/10.1111/joim.13455
References
- 1. Loscalzo J. Keshan disease, selenium deficiency, and the selenoproteome. N Engl J Med. 2014;370(18):1756–60. 10.1056/NEJMcibr1402199 [DOI] [PubMed] [Google Scholar]
- 2. Klip IT, Comin‐Colet J, Voors AA, Ponikowski P, Enjuanes C, Banasiak W, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J. 2013;165(4):575‐82.e3. [DOI] [PubMed] [Google Scholar]
- 3. Yoshihisa A, Abe S, Kiko T, Kimishima Y, Sato Y, Watanabe S, et al. Association of serum zinc level with prognosis in patients with heart failure. J Card Fail. 2018;24(6):375–83. [DOI] [PubMed] [Google Scholar]
- 4. Roberts CGP, Ladenson PW. Hypothyroidism. Lancet 2004;363(9411):793–803. [DOI] [PubMed] [Google Scholar]
- 5. McKeag NA, McKinley MC, Harbinson MT, McGinty A, Neville CE, Woodside JV, et al. Dietary micronutrient intake and micronutrient status in patients with chronic stable heart failure. J Cardiovasc Nurs. 2017;32(2):148–55. [DOI] [PubMed] [Google Scholar]
- 6. Sciatti E, Lombardi C, Ravera A, Vizzardi E, Bonadei I, Carubelli V, et al. Nutritional deficiency in patients with heart failure. Nutrients 2016;8:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hughes CM, Woodside JV, McGartland C, Roberts MJ, Nicholls DP, McKeown PP. Nutritional intake and oxidative stress in chronic heart failure. Nutr Metab Cardiovasc Dis. 2012;22(4):376–82. [DOI] [PubMed] [Google Scholar]
- 8. McKeag NA, McKinley MC, Woodside J V, Harbinson MT, McKeown PP. The role of micronutrients in heart failure. J Acad Nutr Diet. 2012;112(6):870–86. [DOI] [PubMed] [Google Scholar]
- 9. van der Wal HH, Grote Beverborg N, Dickstein K, Anker SD, Lang CC, Ng LL, et al. Iron deficiency in worsening heart failure is associated with reduced estimated protein intake, fluid retention, inflammation, and antiplatelet use. Eur Heart J. 2019;40(44):3616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cicero AFG, Colletti A, von Haehling S, Vinereanu D, Bielecka‐Dabrowa A, Sahebkar A, et al. Nutraceutical support in heart failure: a position paper of the International Lipid Expert Panel (ILEP). Nutr Res Rev. 2020;33(1):155–79. [DOI] [PubMed] [Google Scholar]
- 11. Vest AR, Chan M, Deswal A, Givertz MM, Lekavich C, Lennie T, et al. Nutrition, obesity, and cachexia in patients with heart failure: a consensus statement from the Heart Failure Society of America Scientific Statements Committee. J Card Fail. 2019;25(5):380–400. [DOI] [PubMed] [Google Scholar]
- 12. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution. Eur J Heart Fail. 2016;18(8):891–975. 10.1002/ejhf.592 [DOI] [PubMed] [Google Scholar]
- 13. Wong A‐P, Niedzwiecki A, Rath M. Myocardial energetics and the role of micronutrients in heart failure: a critical review. Am J Cardiovasc Dis. 2016;6(3):81–92. [PMC free article] [PubMed] [Google Scholar]
- 14. Katz AM. Cellular mechanisms in congestive heart failure. Am J Cardiol. 1988;62(2):3A–8A. [DOI] [PubMed] [Google Scholar]
- 15. Witte KK, Clark AL. Micronutrients and their supplementation in chronic cardiac failure. An update beyond theoretical perspectives. Heart Fail Rev. 2006;11(1):65–74. 10.1007/s10741-006-9194-4 [DOI] [PubMed] [Google Scholar]
- 16. Soukoulis V, Dihu JB, Sole M, Anker SD, Cleland J, Fonarow GC, et al. Micronutrient deficiencies an unmet need in heart failure. J Am Coll Cardiol. 2009;54(18):1660–73. [DOI] [PubMed] [Google Scholar]
- 17. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner H, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013;128(16):e240–327. 10.1161/CIR.0b013e31829e8776 [DOI] [PubMed] [Google Scholar]
- 18. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37(27):2129–200. [DOI] [PubMed] [Google Scholar]
- 19. Puig S, Vergara SV, Thiele DJ. Cooperation of two mRNA‐binding proteins drives metabolic adaptation to iron deficiency. Cell Metab. 2008;7(6):555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoes MF, Grote Beverborg N, Kijlstra JD, Kuipers J, Swinkels DW, Giepmans BNG, et al. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur J Heart Fail. 2018;20(5):910–9. http://doi.wiley.com/10.1002/ejhf.1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, et al. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J. 2010;31(15):1872–80. 10.1093/eurheartj/ehq158 [DOI] [PubMed] [Google Scholar]
- 22. von Haehling S, Gremmler U, Krumm M, Mibach F, Schön N, Taggeselle J, et al. Prevalence and clinical impact of iron deficiency and anaemia among outpatients with chronic heart failure: the PrEP Registry. Clin Res Cardiol. 2017;106(6):436–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cleland JGF, Zhang J, Pellicori P, Dicken B, Dierckx R, Shoaib A, et al. Prevalence and outcomes of anemia and hematinic deficiencies in patients with chronic heart failure. JAMA Cardiol. 2016;1(5):539–47. [DOI] [PubMed] [Google Scholar]
- 24. Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J. 2012;34(11):816–29. 10.1093/eurheartj/ehs224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. von Haehling S, Ebner N, Evertz R, Ponikowski P, Anker SD. Iron deficiency in heart failure: an overview. JACC Hear Fail. 2019;7(1):36–46. [DOI] [PubMed] [Google Scholar]
- 26. Anker SD, Kirwan B‐A, van Veldhuisen DJ, Filippatos G, Comin‐Colet J, Ruschitzka F, et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron‐deficient heart failure patients: an individual patient data meta‐analysis. Eur J Heart Fail. 2018;20(1):125–33. [DOI] [PubMed] [Google Scholar]
- 27. Graham FJ, Pellicori P, Ford I, Petrie MC, Kalra PR, Cleland JGF. Intravenous iron for heart failure with evidence of iron deficiency: a meta‐analysis of randomised trials. Clin Res Cardiol. 2021;110:1299–307. 10.1007/s00392-021-01837-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ponikowski P, Kirwan B‐A, Anker SD, McDonagh T, Dorobantu M, Drozdz J, et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: a multicentre, double‐blind, randomised, controlled trial. Lancet 2020;396(10266):1895–904. 10.1016/S0140-6736(20)32339-4 [DOI] [PubMed] [Google Scholar]
- 29. Anker SD, Comin Colet J, Filippatos G, Willenheimer R, Dickstein K, Drexler H, et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med. 2009;361(25):2436–48. 10.1056/NEJMoa0908355 [DOI] [PubMed] [Google Scholar]
- 30. van Veldhuisen DJ, Ponikowski P, van der Meer P, Metra M, Böhm M, Doletsky A, et al. Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation 2017;136(15):1374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ponikowski P, van Veldhuisen DJ, Comin‐Colet J, Ertl G, Komajda M, Mareev V, et al. Beneficial effects of long‐term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J. 2014;36(11):657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marín‐García J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascul Pharmacol. 2010;52(3–4):120–30. [DOI] [PubMed] [Google Scholar]
- 33. Schomburg L, Köhrle J. On the importance of selenium and iodine metabolism for thyroid hormone biosynthesis and human health. Mol Nutr Food Res. 2008;52(11):1235–46. 10.1002/mnfr.200700465 [DOI] [PubMed] [Google Scholar]
- 34. Schomburg L. The other view: the trace element selenium as a micronutrient in thyroid disease, diabetes, and beyond. Hormones. 2020;19:15–24. 10.1007/s42000-019-00150-4 [DOI] [PubMed] [Google Scholar]
- 35. Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase‐1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15(7):1957–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16(11):1323–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bomer N, Grote Beverborg N, Hoes MF, Streng KW, Vermeer M, Dokter MM, et al. Selenium and outcome in heart failure. Eur J Heart Fail. 2020;22(8):1415–23. 10.1002/ejhf.1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang GQ, Ge KY, Chen JS, Chen XS. Selenium‐related endemic diseases and the daily selenium requirement of humans. World Rev Nutr Diet. 1988;55:98–152. [DOI] [PubMed] [Google Scholar]
- 39. Lee BC, Lee S‐G, Choo M‐K, Kim JH, Lee HM, Kim S, et al. Selenoprotein MsrB1 promotes anti‐inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci Rep. 2017;7(1):5119. 10.1038/s41598-017-05230-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alehagen U, Johansson P, Björnstedt M, Rosén A, Dahlström U. Cardiovascular mortality and N‐terminal‐proBNP reduced after combined selenium and coenzyme Q10 supplementation: a 5‐year prospective randomized double‐blind placebo‐controlled trial among elderly Swedish citizens. Int J Cardiol. 2013;167(5):1860–6. [DOI] [PubMed] [Google Scholar]
- 41. Choi S, Liu X, Pan Z. Zinc deficiency and cellular oxidative stress: prognostic implications in cardiovascular diseases. Acta Pharmacol Sin. 2018;39(7):1120–32. 10.1038/aps.2018.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamalov G, Ahokas RA, Zhao W, Zhao T, Shahbaz AU, Johnson PL, et al. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol. 2010;55(3):248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sensi SL, Ton‐That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci. 2003;100(10):6157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexanian I, Parissis J, Farmakis D, Athanaselis S, Pappas L, Gavrielatos G, et al. Clinical and echocardiographic correlates of serum copper and zinc in acute and chronic heart failure. Clin Res Cardiol. 2014;103(11):938–49. 10.1007/s00392-014-0735-x [DOI] [PubMed] [Google Scholar]
- 45. Frustaci A, Sabbioni E, Fortaner S, Farina M, del Torchio R, Tafani M, et al. Selenium‐ and zinc‐deficient cardiomyopathy in human intestinal malabsorption: preliminary results of selenium/zinc infusion. Eur J Heart Fail. 2012;14(2):202–10. 10.1093/eurjhf/hfr167 [DOI] [PubMed] [Google Scholar]
- 46. Hooper NM, Lambert DW, Turner AJ. Discovery and characterization of ACE2—a 20‐year journey of surprises from vasopeptidase to COVID‐19. Clin Sci. 2020;134(18):2489–501. 10.1042/CS20200476 [DOI] [PubMed] [Google Scholar]
- 47. Witte KKA, Nikitin NP, Parker AC, von Haehling S, Volk H‐D, Anker SD, et al. The effect of micronutrient supplementation on quality‐of‐life and left ventricular function in elderly patients with chronic heart failure. Eur Heart J. 2005;26(21):2238–44. [DOI] [PubMed] [Google Scholar]
- 48. Jeejeebhoy F, Keith M, Freeman M, Barr A, McCall M, Kurian R, et al. Nutritional supplementation with MyoVive repletes essential cardiac myocyte nutrients and reduces left ventricular size in patients with left ventricular dysfunction. Am Heart J. 2002;143(6):1092–100. [DOI] [PubMed] [Google Scholar]
- 49. Percival SS. Copper and immunity. Am J Clin Nutr. 1998;67(5 Suppl):1064S–8S. [DOI] [PubMed] [Google Scholar]
- 50. Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21(21):R877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim B‐E, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4(3):176–85. 10.1038/nchembio.72 [DOI] [PubMed] [Google Scholar]
- 52. Lutsenko S. Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol. 2010;14(2):211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prohaska JR. Impact of copper deficiency in humans. Ann N Y Acad Sci. 2014;1314:1–5. [DOI] [PubMed] [Google Scholar]
- 54. Innolife Co. L . Study to evaluate effects of INL1 in patients with heart failure and reduced ejection fraction (TRACER‐HF). ClinicalTrails.gov. NCT0387518; 2021. [Google Scholar]
- 55. Chen X, Jennings DB, Medeiros DM. Impaired cardiac mitochondrial membrane potential and respiration in copper‐deficient rats. J Bioenerg Biomembr. 2002;34(5):397–406. 10.1023/A:1021258204921 [DOI] [PubMed] [Google Scholar]
- 56. Sander S, Coleman CI, Patel AA, Kluger J, Michael White C. The impact of coenzyme Q10 on systolic function in patients with chronic heart failure. J Card Fail. 2006;12(6):464–72. [DOI] [PubMed] [Google Scholar]
- 57. Florkowski CM, Molyneux SL, Young JM. Coenzyme Q10 and congestive heart failure; an evolving evidence base. Kardiol Pol. 2015;73(2):73–9. [DOI] [PubMed] [Google Scholar]
- 58. Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion 2007;7:S136–45. [DOI] [PubMed] [Google Scholar]
- 59. Bentinger M, Brismar K, Dallner G. The antioxidant role of coenzyme Q. Mitochondrion 2007;7:S41–50. [DOI] [PubMed] [Google Scholar]
- 60. Molyneux SL, Florkowski CM, George PM, Pilbrow AP, Frampton CM, Lever M, et al. Coenzyme Q10: an independent predictor of mortality in chronic heart failure. J Am Coll Cardiol. 2008;52(18):1435–41. [DOI] [PubMed] [Google Scholar]
- 61. McMurray JJ V, Dunselman P, Wedel H, Cleland JGF, Lindberg M, Hjalmarson Å, et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure. J Am Coll Cardiol. 2010;56(15):1196–204. [DOI] [PubMed] [Google Scholar]
- 62. Weber C, Bysted A, Hłlmer G. The coenzyme Q10 content of the average Danish diet. Int J Vitam Nutr Res. 1997;67(2):123–9. [PubMed] [Google Scholar]
- 63. Mortensen SA, Rosenfeldt F, Kumar A, Dolliner P, Filipiak KJ, Pella D, et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q‐SYMBIO: a randomized double‐blind trial. JACC Hear Fail. 2014;2(6):641–9. [DOI] [PubMed] [Google Scholar]
- 64. Reinecke F, Smeitink JAM, van der Westhuizen FH. OXPHOS gene expression and control in mitochondrial disorders. Biochim Biophys Acta ‐ Mol Basis Dis. 2009;1792(12):1113–21. [DOI] [PubMed] [Google Scholar]
- 65. Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov. 2017;16(6):400–23. 10.1038/nrd.2016.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ebner N, von Haehling S. Iron deficiency in heart failure: a practical guide. Nutrients 2013;5(9):3730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006;440(7080):96–100. [DOI] [PubMed] [Google Scholar]
- 68. Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta ‐ Mol Cell Res. 2012;1823(9):1491–508. [DOI] [PubMed] [Google Scholar]
- 69. Asín J, Pérez‐Martos A, Fernández‐Silva P, Montoya J, Andreu AL. Iron(II) induces changes in the conformation of mammalian mitochondrial DNA resulting in a reduction of its transcriptional rate. FEBS Lett. 2000;480(2):161–4. [DOI] [PubMed] [Google Scholar]
- 70. Usselman RJ, Fielding AJ, Frerman FE, Watmough NJ, Eaton GR, Eaton SS. Impact of mutations on the midpoint potential of the [4Fe‐4S]+1,+2 cluster and on catalytic activity in electron transfer flavoprotein‐ubiquinone oxidoreductase (ETF‐QO). Biochemistry 2008;47(1):92–100. 10.1021/bi701859s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Khechaduri A, Bayeva M, Chang H‐C, Ardehali H. Heme levels are increased in human failing hearts. J Am Coll Cardiol. 2013;61(18):1884–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H, et al. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail. 2017;19(4):522–30. 10.1002/ejhf.640 [DOI] [PubMed] [Google Scholar]
- 73. Rensvold JW, Ong S‐E, Jeevananthan A, Carr SA, Mootha VK, Pagliarini DJ. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013;3(1):237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III . J Biol Chem. 2003;278(38):36027–31. [DOI] [PubMed] [Google Scholar]
- 75. van der Ent M, Jeneson JA, Remme WJ, Berger R, Ciampricotti R, Visser F. A non‐invasive selective assessment of type I fibre mitochondrial function using 31P NMR spectroscopy. Evidence for impaired oxidative phosphorylation rate in skeletal muscle in patients with chronic heart failure. Eur Heart J. 1998;19(1):124–31. [DOI] [PubMed] [Google Scholar]
- 76. Weiss K, Schär M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, et al. Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail. 2017;10(7):e004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Melenovsky V, Hlavata K, Sedivy P, Dezortova M, Borlaug BA, Petrak J, et al. Skeletal muscle abnormalities and iron deficiency in chronic heart failure (an exercise (31)P magnetic resonance spectroscopy study of calf muscle). Circ Heart Fail. 2018;11(9):e004800. [DOI] [PubMed] [Google Scholar]
- 78. Charles‐Edwards G, Amaral N, Sleigh A, Ayis S, Catibog N, McDonagh T, et al. Effect of iron isomaltoside on skeletal muscle energetics in patients with chronic heart failure and iron deficiency. Circulation 2019;139(21):2386–98. [DOI] [PubMed] [Google Scholar]
- 79. van der Meer P, van der Wal HH, Melenovsky V. Mitochondrial function, skeletal muscle metabolism, and iron deficiency in heart failure. Circulation 2019;139:2399–402. [DOI] [PubMed] [Google Scholar]
- 80. Baynes RD, Bothwell TH. Iron deficiency. Annu Rev Nutr. 1990;10(1):133–48. 10.1146/annurev.nu.10.070190.001025 [DOI] [PubMed] [Google Scholar]
- 81. Lewis GD, Malhotra R, Hernandez AF, McNulty SE, Smith A, Felker GM, et al. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA 2017;317(19):1958–66. 10.1001/jama.2017.5427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Beck‐da‐Silva L, Piardi D, Soder S, Rohde LE, Pereira‐Barretto AC, de Albuquerque D, et al. IRON‐HF study: a randomized trial to assess the effects of iron in heart failure patients with anemia. Int J Cardiol. 2013;168(4):3439–42. 10.1016/j.ijcard.2013.04.181 [DOI] [PubMed] [Google Scholar]
- 83. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail. 2016;18(8):891–975. [DOI] [PubMed] [Google Scholar]
- 84. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DEJ, Colvin MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017;136(6):e137–61. [DOI] [PubMed] [Google Scholar]
- 85. Bekfani T, Pellicori P, Morris D, Ebner N, Valentova M, Sandek A, et al. Iron deficiency in patients with heart failure with preserved ejection fraction and its association with reduced exercise capacity, muscle strength and quality of life. Clin Res Cardiol. 2019;108(2):203–11. [DOI] [PubMed] [Google Scholar]
- 86. Benstoem C, Goetzenich A, Kraemer S, Borosch S, Manzanares W, Hardy G, et al. Selenium and its supplementation in cardiovascular disease—what do we know? Nutrients 2015;7(5):3094–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lu J, Holmgren A. Selenoproteins. J Biol Chem. 2009;284(2):723–7. [DOI] [PubMed] [Google Scholar]
- 88. Rocca C, Pasqua T, Boukhzar L, Anouar Y, Angelone T. Progress in the emerging role of selenoproteins in cardiovascular disease: focus on endoplasmic reticulum‐resident selenoproteins. Cell Mol Life Sci. 2019;76(20):3969–85. 10.1007/s00018-019-03195-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. de Lorgeril M, Salen P. Selenium and antioxidant defenses as major mediators in the development of chronic heart failure. Heart Fail Rev. 2006;11(1):13–7. 10.1007/s10741-006-9188-2 [DOI] [PubMed] [Google Scholar]
- 90. Ferdous A, Wang Z V, Luo Y, Li DL, Luo X, Schiattarella GG, et al. FoxO1–Dio2 signaling axis governs cardiomyocyte thyroid hormone metabolism and hypertrophic growth. Nat Commun. 2020;11(1):2551. 10.1038/s41467-020-16345-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Al‐Mubarak AA, van der Meer P, Bomer N. Selenium, selenoproteins, and heart failure: current knowledge and future perspective. Curr Heart Fail Rep. 2021;18:122–31. 10.1007/s11897-021-00511-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bomer N, Pavez‐Giani MG, Deiman FE, Linders AN, Hoes MF, Baierl CLJ, et al. Selenoprotein DIO2 is a regulator of mitochondrial function, morphology and UPRmt in human cardiomyocytes. Int J Mol Sci. 2021;22:11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schomburg L, Orho‐Melander M, Struck J, Bergmann A, Melander O. Selenoprotein‐P deficiency predicts cardiovascular disease and death. Nutrients 2019;11:1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jujić A, Melander O, Bergmann A, Hartmann O, Nilsson PM, Bachus E, et al. Selenoprotein P deficiency and risk of mortality and rehospitalization in acute heart failure. J Am Coll Cardiol. 2019;74(7):1009–11. [DOI] [PubMed] [Google Scholar]
- 95. Rayman MP. Selenium and human health. Lancet 2012;379(9822):1256–68. [DOI] [PubMed] [Google Scholar]
- 96. Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94(3):739–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schomburg L, Schweizer U. Hierarchical regulation of selenoprotein expression and sex‐specific effects of selenium. Biochim Biophys Acta – Gen Subj. 2009;1790(11):1453–62. [DOI] [PubMed] [Google Scholar]
- 98. Alehagen U, Alexander J, Aaseth J. Supplementation with selenium and coenzyme Q10 reduces cardiovascular mortality in elderly with low selenium status. A secondary analysis of a randomised clinical trial. PLoS One 2016;11(7):e0157541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, et al. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med. 2003;349(17):1605–13. [DOI] [PubMed] [Google Scholar]
- 100. van der Pol A, van Gilst WH, Voors AA, van der Meer P. Treating oxidative stress in heart failure: past, present and future. Eur J Heart Fail. 2019;21:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang L, Gao Y, Feng H, Zou N, Wang K, Sun D. Effects of selenium deficiency and low protein intake on the apoptosis through a mitochondria‐dependent pathway. J Trace Elem Med Biol. 2019;56:21–30. [DOI] [PubMed] [Google Scholar]
- 102. Flores‐Mateo G, Navas‐Acien A, Pastor‐Barriuso R, Guallar E. Selenium and coronary heart disease: a meta‐analysis. Am J Clin Nutr. 2006;84(4):762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. de Lorgeril M, Salen P, Accominotti M, Cadau M, Steghens JP, Boucher F, et al. Dietary and blood antioxidants in patients with chronic heart failure. Insights into the potential importance of selenium in heart failure. Eur J Heart Fail. 2001;3(6):661–9. [DOI] [PubMed] [Google Scholar]
- 104. Alexanian I, Parissis J, Farmakis D, Pantziou C, Ikonomidis I, Paraskevaidis I, et al. Selenium contributes to myocardial injury and cardiac remodeling in heart failure. Int J Cardiol. 2014;176(1):272–3. [DOI] [PubMed] [Google Scholar]
- 105. Spertus J, Peterson E, Conard MW, Heidenreich PA, Krumholz HM, Jones P, et al. Monitoring clinical changes in patients with heart failure: a comparison of methods. Am Heart J. 2005;150(4):707–15. [DOI] [PubMed] [Google Scholar]
- 106. Spertus JA, Jones PG. Development and validation of a short version of the Kansas City Cardiomyopathy Questionnaire. Circ Cardiovasc Qual Outcomes. 2015;8(5):469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Al‐Mubarak AA, Grote Beverborg N, Anker SD, Samani NJ, Dickstein K, Filippatos G, et al. A clinical tool to predict low serum selenium in patients with worsening heart failure. Nutrients 2020;12:2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Alehagen U, Aaseth J, Johansson P. Reduced cardiovascular mortality 10 years after supplementation with selenium and coenzyme Q10 for four years: follow‐Up results of a prospective randomized double‐blind placebo‐controlled trial in elderly citizens. PLoS One 2015;10(12):e0141641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Garakyaraghi M, Bahrami P, Sadeghi M, Rabiei K. Combination effects of seleniumand coenzyme Q10 on left ventricular systolic function in patients with heart failure. Iran Hear J. 2015;15(4):6–12. [Google Scholar]
- 110. Jenkins DJA, Kitts D, Giovannucci EL, Sahye‐Pudaruth S, Paquette M, Blanco Mejia S, et al. Selenium, antioxidants, cardiovascular disease, and all‐cause mortality: a systematic review and meta‐analysis of randomized controlled trials. Am J Clin Nutr. 2020; 112(6): 1642–52. 10.1093/ajcn/nqaa245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Duffield‐Lillico AJ, Reid ME, Turnbull BW, Combs GFJ, Slate EH, Fischbach LA, et al. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: a summary report of the Nutritional Prevention of Cancer Trial. Cancer Epidemiol biomarkers Prev. 2002;11(7):630–9. [PubMed] [Google Scholar]
- 112. Stranges S, Marshall JR, Trevisan M, Natarajan R, Donahue RP, Combs GF, et al. Effects of selenium supplementation on cardiovascular disease incidence and mortality: secondary analyses in a randomized clinical trial. Am J Epidemiol. 2006;163(8):694–9. [DOI] [PubMed] [Google Scholar]
- 113. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009;301(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Schomburg L, Orho‐Melander M, Struck J, Bergmann A, Melander O. Selenoprotein‐P deficiency predicts cardiovascular disease and death. Nutrients 2019;11(8):1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rayman MP. Dietary selenium: time to act. BMJ 1997;314:387–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nève J. New approaches to assess selenium status and requirement. Nutr Rev. 2000;58(12):363–9. [DOI] [PubMed] [Google Scholar]
- 117. Thomson CD, Robinson MF, Butler JA, Whanger PD. Long‐term supplementation with selenate and selenomethionine: selenium and glutathione peroxidase (EC 1.11.1.9) in blood components of New Zealand women. Br J Nutr. 1993;69(2):577–88. [DOI] [PubMed] [Google Scholar]
- 118. Kuria A, Tian H, Li M, Wang Y, Aaseth JO, Zang J, et al. Selenium status in the body and cardiovascular disease: a systematic review and meta‐analysis. Crit Rev Food Sci Nutr. 2021;61(21):3616–25. [DOI] [PubMed] [Google Scholar]
- 119. Trasobares E, Corbatón A, González‐Estecha M, Lopez‐Colón JL, Prats P, Olivan P, et al. Effects of angiotensin‐converting enzyme inhibitors (ACEi) on zinc metabolism in patients with heart failure. J Trace Elem Med Biol. 2007;21:53–5. [DOI] [PubMed] [Google Scholar]
- 120. Bünning P, Riordan JF. The functional role of zinc in angiotensin converting enzyme: implications for the enzyme mechanism. J Inorg Biochem. 1985;24(3):183–98. [DOI] [PubMed] [Google Scholar]
- 121. Alghamri MS, Morris M, Meszaros JG, Elased KM, Grobe N. Novel role of aminopeptidase‐A in angiotensin‐(1–7) metabolism post myocardial infarction. Am J Physiol Circ Physiol. 2014;306(7):H1032–40. 10.1152/ajpheart.00911.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cohen N, Golik A. Zinc balance and medications commonly used in the management of heart failure. Heart Fail Rev. 2006;11(1):19–24. 10.1007/s10741-006-9189-1 [DOI] [PubMed] [Google Scholar]
- 123. Caulfield LE, Black RE. Zinc deficiency. In: Ezzati M, Lopez AD, Rodgers A, Murray CJL, editors. Comparative quantification of health risks: global and regional burden of disease attributable to selected major risk factors, Vol. 1. Geneva: World Health Organization; 2004. p. 257–80. [Google Scholar]
- 124. Sandstead H, Freeland‐Graves J. Dietary phytate, zinc and hidden zinc deficiency. J trace Elem Med Biol organ Soc Miner Trace Elem. 2014;28(4):414–7. [DOI] [PubMed] [Google Scholar]
- 125. Solomons N. Dietary sources of zinc and factors affecting its bioavailability. Food Nutr Bull. 2001;22:138–54. [Google Scholar]
- 126. Matrix metalloproteinases: regulation and dysregulation in the failing heart Spinale FG.. Circ Res. 2002;90(5):520–30. 10.1161/01.RES.0000013290.12884.A3 [DOI] [PubMed] [Google Scholar]
- 127. Quirós PM, Langer T, López‐Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16(6):345–59. 10.1038/nrm3984 [DOI] [PubMed] [Google Scholar]
- 128. Singal PK, Kirshenbaum LA. A relative deficit in antioxidant reserve may contribute in cardiac failure. Can J Cardiol. 1990;6(2):47–9. [PubMed] [Google Scholar]
- 129. Chanoit G, Lee S, Xi J, Zhu M, McIntosh RA, Mueller RA, et al. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase‐3β. Am J Physiol Circ Physiol. 2008;295(3):H1227–33. 10.1152/ajpheart.00610.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Xu Z, Zhou J. Zinc and myocardial ischemia/reperfusion injury. BioMetals 2013;26(6):863–78. 10.1007/s10534-013-9671-x [DOI] [PubMed] [Google Scholar]
- 131. Jiang J, Han P, Zhang Q, Zhao J, Ma Y. Cardiac differentiation of human pluripotent stem cells. J Cell Mol Med. 2012;16(8):1663–8. http://doi.wiley.com/10.1111/j.1582‐4934.2012.01528.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Andrews GK. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem Pharmacol. 2000;59(1):95–104. [DOI] [PubMed] [Google Scholar]
- 133. Feng W, Benz FW, Cai J, Pierce WM, Kang YJ. Metallothionein disulfides are present in metallothionein‐overexpressing transgenic mouse heart and increase under conditions of oxidative stress. J Biol Chem. 2006;281(2):681–7. [DOI] [PubMed] [Google Scholar]
- 134. Pepersack T, Rotsaert P, Benoit F, Willems D, Fuss M, Bourdoux P, et al. Prevalence of zinc deficiency and its clinical relevance among hospitalised elderly. Arch Gerontol Geriatr. 2001;33(3):243–53. [DOI] [PubMed] [Google Scholar]
- 135. Efeovbokhan N, Bhattacharya SK, Ahokas RA, Sun Y, Guntaka RV, Gerling IC, et al. Zinc and the prooxidant heart failure phenotype. J Cardiovasc Pharmacol. 2014;64(4): 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rosenblum H, Wessler JD, Gupta A, Maurer MS, Bikdeli B. Zinc deficiency and heart failure: a systematic review of the current literature. J Card Fail. 2020;26(2):180–9. [DOI] [PubMed] [Google Scholar]
- 137. Seawell MR, Al Darazi F, Farah V, Ramanathan KB, Newman KP, Bhattacharya SK, et al. Mineralocorticoid receptor antagonism confers cardioprotection in heart failure. Curr Heart Fail Rep. 2013;10(1):36–45. 10.1007/s11897-012-0120-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ghaemian A, Salehifar E, Jalalian R, Ghasemi F, Azizi S, Masoumi S, et al. Zinc and copper levels in severe heart failure and the effects of atrial fibrillation on the zinc and copper status. Biol Trace Elem Res. 2011;143(3):1239–46. 10.1007/s12011-011-8956-6 [DOI] [PubMed] [Google Scholar]
- 139. Cowger J. The impact of zinc supplementation on left ventricular function in nonischemic cardiomyopathy. ClinicalTrials.gov. NCT 006964; 2012. [Google Scholar]
- 140. Rosenblum H, Bikdeli B, Wessler J, Gupta A, Jacoby DL. Zinc deficiency as a reversible cause of heart failure. Texas Hear Inst J. 2020;47(2):152–4. 10.14503/THIJ-17-6586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kelley DS, Daudu PA, Taylor PC, Mackey BE, Turnlund JR. Effects of low‐copper diets on human immune response. Am J Clin Nutr. 1995;62(2):412–6. 10.1093/ajcn/62.2.412 [DOI] [PubMed] [Google Scholar]
- 142. Trumbo P, Yates AA, Schlicker S, Poos M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101(3):294–301. [DOI] [PubMed] [Google Scholar]
- 143. Macomber L, Imlay JA. The iron‐sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci USA. 2009;106(20):8344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Jomova K, Valko M. Advances in metal‐induced oxidative stress and human disease. Toxicology 2011;283(2–3):65–87. [DOI] [PubMed] [Google Scholar]
- 145. van den Berghe PVE, Klomp LWJ. New developments in the regulation of intestinal copper absorption. Nutr Rev. 2009;67(11):658–72. [DOI] [PubMed] [Google Scholar]
- 146. Bandmann O, Weiss KH, Kaler SG. Wilson's disease and other neurological copper disorders. Lancet Neurol. 2015;14(1):103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. de Bie P, Muller P, Wijmenga C, Klomp LWJ. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44(11):673–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Blumberg JB, Frei B, Fulgoni VL, Weaver CM, Zeisel SH. Contribution of dietary supplements to nutritional adequacy in various adult age groups. Nutrients 2017;9(12):1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Prohaska JR. Copper. In: Erdman JW, Macdonald IA, Zeisel SH, editors. Present knowledge in nutrition. Oxford, UK: Wiley‐Blackwell; 2012. http://doi.wiley.com/10.1002/9781119946045 [Google Scholar]
- 150. Medeiros DM, Jiang Y, Klaahsen D, Lin D. Mitochondrial and sarcoplasmic protein changes in hearts from copper‐deficient rats: up‐regulation of PGC‐1α transcript and protein as a cause for mitochondrial biogenesis in copper deficiency. J Nutr Biochem. 2009;20(10):823–30. http://www.sciencedirect.com/science/article/pii/S0955286308001770 [DOI] [PubMed] [Google Scholar]
- 151. Johnson WT, Newman SMJ. Copper deficiency: a potential model for determining the role of mitochondria in cardiac aging. J Am Aging Assoc. 2003;26(1–2):19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Elsherif L, Ortines R V, Saari JT, Kang YJ. Congestive heart failure in copper‐deficient mice. Exp Biol Med. 2003;228(7):811–7. 10.1177/15353702-0322807-06 [DOI] [PubMed] [Google Scholar]
- 153. Kang YJ, Wu H, Saari JT. Alterations in hypertrophic gene expression by dietary copper restriction in mouse heart (44492). Proc Soc Exp Biol Med. 2000;223(3):282–7. https://journals.sagepub.com/doi/abs/10.1177/153537020022300310 [DOI] [PubMed] [Google Scholar]
- 154. Ford ES. Serum copper concentration and coronary heart disease among US adults. Am J Epidemiol. 2000;151(12):1182–8. 10.1093/oxfordjournals.aje.a010168 [DOI] [PubMed] [Google Scholar]
- 155. Málek F, Dvorák J, Jiresová E, Spacek R. Difference of baseline serum copper levels between groups of patients with different one year mortality and morbidity and chronic heart failure. Cent Eur J Public Health. 2003;11(4):198–201. [PubMed] [Google Scholar]
- 156. Huang L, Shen R, Huang L, Yu J, Rong H. Association between serum copper and heart failure: a meta‐analysis. Asia Pac J Clin Nutr. 2019;28(4):761–9. [DOI] [PubMed] [Google Scholar]
- 157. Hammadah M, Fan Y, Wu Y, Hazen SL, Tang WHW. Prognostic value of elevated serum ceruloplasmin levels in patients with heart failure. J Card Fail. 2014;20(12):946–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Fox PL, Mazumder B, Ehrenwald E, Mukhopadhyay CK. Ceruloplasmin and cardiovascular disease. Free Radic Biol Med. 2000;28(12):1735–44. [DOI] [PubMed] [Google Scholar]
- 159. Dadu RT, Dodge R, Nambi V, Virani SS, Hoogeveen RC, Smith NL, et al. Ceruloplasmin and heart failure in the atherosclerosis risk in communities study. Circ Heart Fail. 2013;6(5):936–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cabassi A, Binno SM, Tedeschi S, Ruzicka V, Dancelli S, Rocco R, et al. Low serum ferroxidase I activity is associated with mortality in heart failure and related to both peroxynitrite‐induced cysteine oxidation and tyrosine nitration of ceruloplasmin. Circ Res. 2014;114(11):1723–32. [DOI] [PubMed] [Google Scholar]
- 161. He W, James Kang Y. Ischemia‐induced copper loss and suppression of angiogenesis in the pathogenesis of myocardial infarction. Cardiovasc Toxicol. 2013;13(1):1–8. [DOI] [PubMed] [Google Scholar]
- 162. Grammer TB, Kleber ME, Silbernagel G, Pilz S, Scharnagl H, Lerchbaum E, et al. Copper, ceruloplasmin, and long‐term cardiovascular and total mortality (The Ludwigshafen Risk and Cardiovascular Health Study). Free Radic Res. 2014;48(6):706–15. 10.3109/10715762.2014.901510 [DOI] [PubMed] [Google Scholar]
- 163. Arenas de Larriva AP, Limia‐Pérez L, Alcalá‐Díaz JF, Alonso A, López‐Miranda J, Delgado‐Lista J. Ceruloplasmin and coronary heart disease—a systematic review. Nutrients 2020;12:3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ferrari R, Guardigli G, Mele D, Percoco G, Ceconi C, Curello S. Oxidative stress during myocardial ischaemia and heart failure. Curr Pharm Des. 2004;10(14):1699–711. [DOI] [PubMed] [Google Scholar]
- 165. Folkers K, Vadhanavikit S, Mortensen SA. Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10. Proc Natl Acad Sci USA. 1985;82(3):901–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Sharma A, Fonarow GC, Butler J, Ezekowitz JA, Felker GM. Coenzyme Q10 and heart failure: a state‐of‐the‐art review. Circ Heart Fail. 2016;9(4):e002639. [DOI] [PubMed] [Google Scholar]
- 167. Molyneux SL, Florkowski CM, George PM, Pilbrow AP, Frampton CM, Lever M, et al. Coenzyme Q10 . J Am Coll Cardiol. 2008;52(18):1435–441. [DOI] [PubMed] [Google Scholar]
- 168. Stocker R, Bowry VW, Frei B. Ubiquinol‐10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha‐tocopherol. Proc Natl Acad Sci. 19911;88(5):1646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Opie LH. The metabolic vicious cycle in heart failure. Lancet 2004;364(9447):1733–4. [DOI] [PubMed] [Google Scholar]
- 170. Cleland JGF, Squire I, Ng L. Interpretation of amino‐terminal pro‐brain natriuretic peptide levels in the HPS and the CORONA study. J Am Coll Cardiol. 2008;52:1104–5. [DOI] [PubMed] [Google Scholar]
- 171. Cleland JGF, McMurray JJ V, Kjekshus J, Cornel JH, Dunselman P, Fonseca C, et al. Plasma concentration of amino‐terminal pro‐brain natriuretic peptide in chronic heart failure: prediction of cardiovascular events and interaction with the effects of rosuvastatin: a report from CORONA (Controlled Rosuvastatin Multinational Trial in Heart Failure). J Am Coll Cardiol. 2009;54(20):1850–9. [DOI] [PubMed] [Google Scholar]
- 172. Mortensen SA, Leth A, Agner E, Rohde M. Dose‐related decrease of serum coenzyme Q10 during treatment with HMG‐CoA reductase inhibitors. Mol Aspects Med. 1997;18:137–44. [DOI] [PubMed] [Google Scholar]
- 173. Morisco C, Trimarco B, Condorelli M. Effect of coenzyme Q10 therapy in patients with congestive heart failure: a long‐term multicenter randomized study. Clin Investig. 1993;71(8 Suppl):S134–6. [DOI] [PubMed] [Google Scholar]
- 174. Fotino AD, Thompson‐Paul AM, Bazzano LA. Effect of coenzyme Q₁₀ supplementation on heart failure: a meta‐analysis. Am J Clin Nutr. 2013;97(2):268–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Langsjoen PH, Vadhanavikit S, Folkers K. Effective treatment with coenzyme Q10 of patients with chronic myocardial disease. Drugs Exp Clin Res. 1985;11(8):577–9. [PubMed] [Google Scholar]
- 176. Baggio E, Gandini R, Plancher AC, Passeri M, Carmosino G. Italian multicenter study on the safety and efficacy of coenzyme Q10 as adjunctive therapy in heart failure. Mol Aspects Med. 1994;15:s287–94. [DOI] [PubMed] [Google Scholar]
- 177. Raizner AE, Quiñones MA. Coenzyme Q(10) for patients with cardiovascular disease: JACC focus seminar. J Am Coll Cardiol. 2021;77(5):609–19. [DOI] [PubMed] [Google Scholar]
- 178. Hofman‐Bang C, Rehnqvist N, Swedberg K, Wiklund I, Åström H. Coenzyme Q10 as an adjunctive in the treatment of chronic congestive heart failure. J Card Fail. 1995;1(2):101–7. [DOI] [PubMed] [Google Scholar]
- 179. Zhao Q, Kebbati AH, Zhang Y, Tang Y, Okello E, Huang C. Effect of coenzyme Q10 on the incidence of atrial fibrillation in patients with heart failure. J Investig Med. 2015;63(5):735–9. [DOI] [PubMed] [Google Scholar]
- 180. Sobirin MA, Herry Y, Sofia SN, Uddin I, Rifqi S, Tsutsui H. Effects of coenzyme Q10 supplementation on diastolic function in patients with heart failure with preserved ejection fraction. Drug Discov Ther. 2019;13(1):38–46. [DOI] [PubMed] [Google Scholar]
- 181. Gottwald EM, Duss M, Bugarski M, Haenni D, Schuh CD, Landau EM, et al. The targeted anti‐oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol Rep. 2018;6(7):e13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Pokrzywinski KL, Biel TG, Kryndushkin D, Rao VA. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtDNA integrity. PLoS One 2016;11(12):e0168283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Cascino TM, Hummel SL. Nutrient deficiencies in heart failure: a micro problem with macro effects? J Am Heart Assoc. 2018;7(17):e010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. McKeag NA, McKinley MC, Harbinson MT, Noad RL, Dixon LH, McGinty A, et al. The effect of multiple micronutrient supplementation on left ventricular ejection fraction in patients with chronic stable heart failure: a randomized, placebo‐controlled trial. JACC Heart Fail. 2014;2(3):308–17. [DOI] [PubMed] [Google Scholar]
- 185. Hurst R, Armah CN, Dainty JR, Hart DJ, Teucher B, Goldson AJ, et al. Establishing optimal selenium status: results of a randomized, double‐blind, placebo‐controlled trial. Am J Clin Nutr. 2010;91(4):923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Fairweather‐Tait SJ, Collings R, Hurst R. Selenium bioavailability: current knowledge and future research requirements. Am J Clin Nutr. 2010;91(5):1484S‐91S. [DOI] [PubMed] [Google Scholar]
- 187. Archer E, Lavie CJ, Hill JO. The failure to measure dietary intake engendered a fictional discourse on diet‐disease relations. Front Nutr. 2018;5:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Abu‐Sawwa R, Dunbar SB, Quyyumi AA, Sattler ELP. Nutrition intervention in heart failure: should consumption of the DASH eating pattern be recommended to improve outcomes? Heart Fail Rev. 2019;24(4):565–73. 10.1007/s10741-019-09781-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ishikawa Y, Sattler ELP. Nutrition as treatment modality in heart failure. Curr Atheroscler Rep. 2021;23(4):13. 10.1007/s11883-021-00908-5 [DOI] [PubMed] [Google Scholar]
- 190. Hersberger L, Dietz A, Bürgler H, Bargetzi A, Bargetzi L, Kägi‐Braun N, et al. Individualized nutritional support for hospitalized patients with chronic heart failure. J Am Coll Cardiol. 2021;77(18):2307–19. [DOI] [PubMed] [Google Scholar]
- 191. Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356(11):1140–51. 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]