

Abstract
There is an unmet need for the development and validation of biomarkers and surrogate endpoints for clinical trials in propionic acidemia (PA) and methylmalonic acidemia (MMA). This review examines the pathophysiology and clinical consequences of PA and MMA that could form the basis for potential biomarkers and surrogate endpoints. Changes in primary metabolites such as methylcitric acid (MCA), MCA:citric acid ratio, oxidation of 13C‐propionate (exhaled 13CO2), and propionylcarnitine (C3) have demonstrated clinical relevance in patients with PA or MMA. Methylmalonic acid, another primary metabolite, is a potential biomarker, but only in patients with MMA. Other potential biomarkers in patients with either PA and MMA include secondary metabolites, such as ammonium, or the mitochondrial disease marker, fibroblast growth factor 21. Additional research is needed to validate these biomarkers as surrogate endpoints, and to determine whether other metabolites or markers of organ damage could also be useful biomarkers for clinical trials of investigational drug treatments in patients with PA or MMA. This review examines the evidence supporting a variety of possible biomarkers for drug development in propionic and methylmalonic acidemias.
Keywords: biomarker, clinical trials., methylmalonic acidemia, pathophysiology, propionic acidemia, surrogate endpoint

1. INTRODUCTION
Organic acidemias include a heterogenous group of inborn errors of metabolism resulting in elevated organic acids in body fluids. 1 , 2 Propionic acidemia (PA) and methylmalonic acidemia (MMA) are the “classic” organic acidemias and will be discussed here. There are numerous challenges to conducting clinical trials in patients with inherited metabolic diseases such as organic acidemias. Due to the rarity of these conditions, a limited number of patients—at a limited number of centers—are available to enroll in clinical trials. These conditions are heterogeneous, with substantial overlap within the organic acidemias, and there can be intrafamilial variability. Patients can be diagnosed by neonatal screening within days of birth or might not be diagnosed until adulthood. Mortality rates are high, and the clinical phenotype can evolve over time within a single patient. After patients are identified and enrolled in clinical trials, it is difficult to predict the rate of disease progression, further complicating efforts to assign patients evenly to different treatment arms and identify the contribution of an experimental treatment to observed outcomes.
PA affects approximately 1:240 000 live births in United States. 3 Approximately 70% to 90% of cases of PA are neonatal. 4 , 5 , 6 , 7 Common manifestations in addition to the acute crisis—with vomiting and lethargy progressing to coma—include hypotonia, poor feeding, and delays in development. Neurological complications caused by metabolic strokes can result in movement disorders or seizures, with delays in development resulting from the chronic exposure to toxic organic acids or their downstream effects on cell functioning. Worsening metabolic acidosis, hyperammonemia, hypoglycemia, hyperketonemia, hyperglycinemia, neutropenia, and thrombocytopenia are common biochemical/laboratory findings. Without appropriate treatment, coma and death often occur. Intellectual disability, cardiac conduction abnormalities, cardiomyopathy, recurrent pancreatitis, diabetes, optic atrophy, and deafness can occur in older individuals (Figure 1). 4 , 5 , 6 , 7 , 8
FIGURE 1.
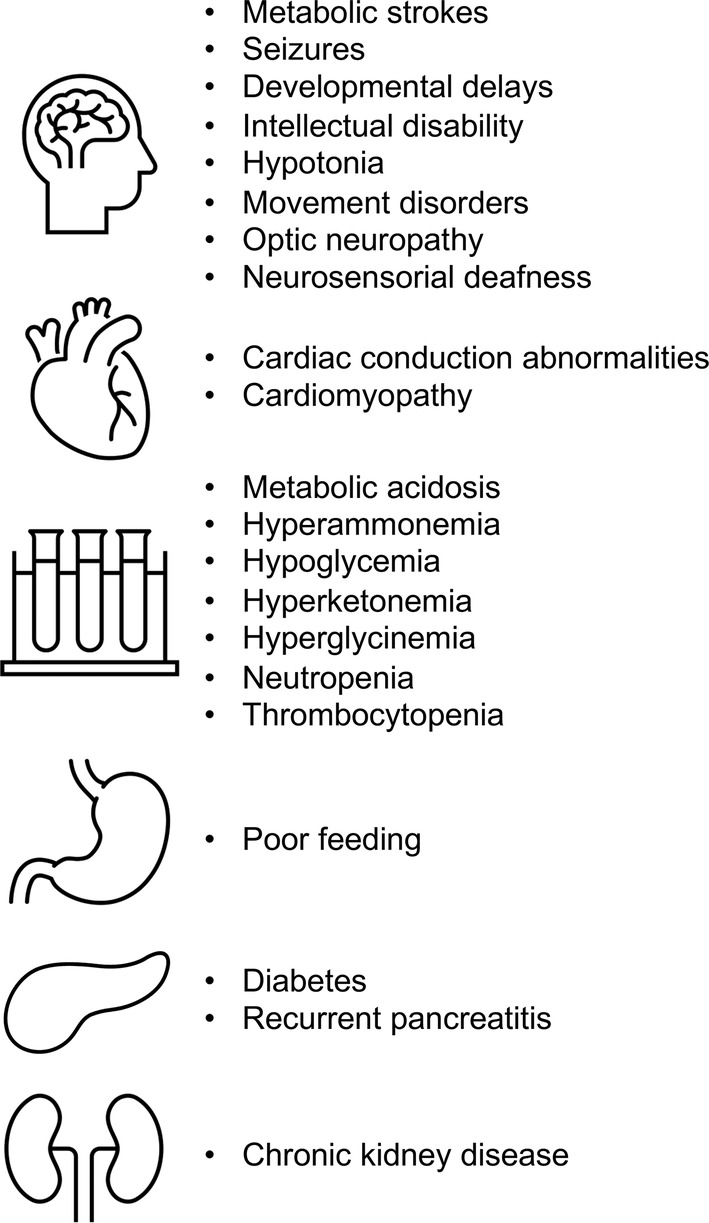
Clinical manifestations of propionic acidemia or methylmalonic acidemia
MMA occurs in 1:110 000 live births. 3 It is usually discovered through neonatal screening, but later onset variants have been described. 9 The clinical presentation of MMA is generally similar to that of PA, but end‐stage kidney disease is much more common in MMA. 10 Intellectual disability, chronic kidney disease, pancreatitis, diabetes mellitus, feeding problems, metabolic stroke, and optic neuropathy can occur.
In 2001, a working group of the National Institutes of Health Director's Initiative on Biomarkers and Surrogate Endpoints defined a biomarker as a laboratory result that “is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.” 11 The working group noted that biomarkers may have the greatest value in early efficacy and safety evaluations such as in vitro studies in tissue samples, in vivo studies in animal models, and early‐phase clinical trials to establish proof of concept. 11
As summarized by Gulbukan et al., there are several key parameters for reliable and informative biomarkers in the clinical setting. 12 A biomarker needs to have clinical and analytical validity. A biomarker should be measured by tests that are reliable, accurate, and reproducible and capable of distinguishing between the pathologic and healthy state. A biomarker should also be able to indicate changes in the status of the disease in a consistent manner, without being influenced by outside parameters. A clinically validated biomarker that satisfies these conditions can be used to indicate the prognosis of a disease or an individual's response to a therapeutic intervention. To have the greatest clinical utility, a clinical biomarker ideally should be measurable in biological samples obtained by less invasive methods such as urine, plasma, serum, stool, or saliva, rather than in tissue biopsies.
Potential biomarkers in patients with propionic acidemia (PA) or methylmalonic acidemia (MMA) are readily identified from the disruptions of metabolic pathways in PA/MMA (Figure 2). Metabolites that accumulate behind the primary metabolic block include organic acids (propionic acid, 3‐hydroxypropionic acid, 2‐MCA), glycine conjugates (propionylglycine, tiglylglycine) and acylcarnitines (C2, C3) (Table 1). Additional metabolic abnormalities can be induced by the primary deficiency (eg, ammonium, lactic acid, amino acids, tricarboxylic acid [TCA] cycle intermediates), thought to derive in most cases from inhibition of the urea cycle due to acetyl‐CoA depletion with consequent defective synthesis of N‐acetylglutamic acid and the enzymes of the mitochondrial tricarboxylic acid cycle by accumulated primary metabolites (Table 1). This review summarizes these potential primary and secondary metabolite abnormalities, as well as other biochemical effects of PA/MMA, and implications for diagnosis, timely adjustment of treatment regimens, prediction of patient metabolic status, and evaluation of the effects of organ transplantation in patients with PA/MMA. Potential utility of these biomarkers as surrogate endpoints for clinical trials is also discussed.
FIGURE 2.
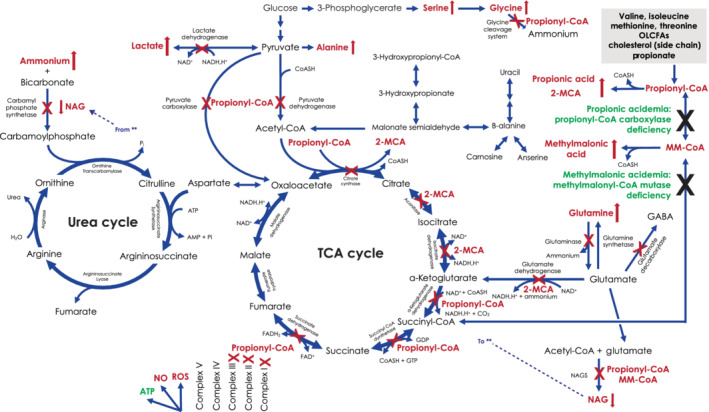
Metabolic pathways affected by propionic acidemia or methylmalonic acidemia. Genetic mutations causing propionyl‐CoA carboxylase deficiency result in propionic acidemia (accumulation of propionic acid), and mutations causing methylmalonyl‐CoA mutase deficiency result in methylmalonic acidemia (accumulation of methylmalonic acid). Both conditions result in accumulation of propionyl‐CoA, which inhibits the TCA cycle and urea cycle as indicated by “X.” Conversion of excess propionyl‐CoA to 2‐MCA leads to additional inhibitory effects on the TCA cycle, as indicated. By‐products that accumulate in patients with propionic acidemia or methylmalonic acidemia are indicated with red text and red arrows. 2‐MCA, 2‐methylcitric acid; CoA/CoASH, coenzyme A; CPS, carbamyl phosphate synthetase; GABA, gamma‐aminobutyric acid; GDP, MMA, methylmalonic acid; MM‐CoA, methylmalonyl‐coenzyme A; NAD[PH], nicotinamide adenine dinucleotide [phosphate]; NAG, N‐acetylglutamate; NO, nitrous oxide; OLCFA, odd‐numbered long‐chain fatty acid; Pi, inorganic phosphate; propionyl‐CoA, propionyl‐coenzyme A; ROS, reactive oxygen species; TCA cycle, tricarboxylic acid cycle (also known as the citric acid cycle or Krebs cycle)
TABLE 1.
Biochemical effects of propionic acidemia and methylmalonic acidemia: potential biomarkers for early drug development
| Association | Potential biomarkers | Usual change a |
|---|---|---|
| Primary metabolites |
|
↑ |
|
↑ | |
|
||
| acetylcarnitine (C2) | ↓ | |
| palmitoylcarnitine | ↓ | |
| propionylcarnitine (C3) and C3/C2 ratio | ↑ | |
| methylmalonylcarnitine (only in patients with MMA) | ↑ | |
|
↓ | |
|
↑ | |
|
↑ | |
| Secondary metabolites |
|
↓ |
|
↓ | |
|
↑ | |
|
↑ | |
|
↓ | |
|
||
| alanine:serine | ↑ | |
| alanine:lysine | ↓ | |
| alanine:(phenylalanine + tyrosine) | ↓ | |
| glycine | ↑ | |
| Other effects |
|
↑ |
|
↑ | |
|
↑ | |
|
↑ | |
|
↑ |
A uniform classification for all markers is difficult because it depends on circumstances. For example, methylmalonylcarnitine is not always elevated in patients with MMA.
2. BIOMARKERS—PRIMARY METABOLITES
2.1. Organic acids
2.1.1. Methylcitric acid
Methylcitric acid (MCA) is a tricarboxylic acid derived from the condensation of propionyl‐coenzyme A (CoA), with oxaloacetic acid, an intermediate of the Krebs cycle, by the enzyme citrate synthase. 13 MCA behaves as a potent inhibitor of glutamate oxidation by inhibiting glutamate dehydrogenase activity and as a permeability transition inducer, disturbing mitochondrial energy homeostasis. 14 MCA‐induced deleterious mitochondrial effects might contribute to the pathogenesis of brain damage in patients with PA and MMA. 14 Plasma MCA is generally considered a useful biomarker for the diagnosis and monitoring of inherited diseases of propionic acid metabolism because MCA levels are directly correlated with disease burden in PA. 15 , 16
Higher MCA levels are seen in patients with severe phenotype and significant long‐term complications, and MCA levels are generally higher in patients with PA than with MMA. 13 Disease burden shows a direct correlation with MCA and fibroblast growth factor 21 (FGF21; see Section 4.1) in both PA and MMA. 13 In MMA, MCA is higher in elderly patients and, along with FGF21 and plasma methylmalonic acid, negatively correlate with GFR. 13 In both diseases, MCA correlates with ammonium, glycine, lysine, C3, C3/C2, and C3/C16. 13 Decreases in MCA from baseline are similar after liver transplant (in patients with PA/MMA) or kidney transplant (in patients with MMA), and greater after combined liver/kidney transplant (in patients with MMA). 13 However, MCA concentrations are highly variable and affected by dietary protein intake and kidney function, 17 which might limit the use of MCA as a surrogate endpoint, particularly in patients with impaired kidney function. Additionally, plasma levels of MCA are not influenced by metabolic status. 13
2.1.2. Methylcitric acid:citric acid ratio (MCA:CA)
Both PA and MMA can lead to pathologic accumulation of propionyl‐CoA and MCA. In healthy individuals with physiologic levels of propionyl‐CoA, oxaloacetic acid in mitochondria reacts with acetyl‐CoA to produce citric acid, a normal Krebs cycle intermediate. The competitive synthesis of citric acid and MCA through the same enzymatic mechanism implies that an increase in MCA production should be accompanied by a decrease in citric acid levels. Thus, an increase in the ratio of MCA (indicative of PA or MMA) to citric acid (indicative of normal function) in plasma has been suggested as an important indicator to assess the disease course and/or severity in patients. 18 The use of MCA:CA ratio, which depends on two competitively synthesized and inversely related markers, might allow for improved ascertainment of patients with milder disease variants associated with stable MCA elevations.
2.1.3. Methylmalonic acid
Impaired conversion of methylmalonyl‐CoA to succinyl‐CoA leads to accumulation of methylmalonic acid in the plasma of patients with MMA. Methylmalonic acid has been shown to increase DNA damage in the cerebral cortex and kidneys of rats, suggesting a possible link between methylmalonic acid and both neurotoxicity and nephrotoxicity in patients with MMA. 19 Elevated methylmalonic acid (usually 10‐ to 100‐fold elevated) leaves patients at risk for kidney disease; patients with MMA are also at risk for metabolic strokes. 20 Thus, methylmalonic acid in plasma might be useful to monitor response to treatment in patients with MMA.
In normal controls, serum MMA increases mildly with age and deteriorating kidney function. 21 , 22 In patients with MMA, levels of serum methylmalonic acid increase dramatically 23 as kidney function (measured as eGFR) decreases below 50 mL/min/1.73 m2 and are reduced by kidney or combined liver/kidney transplant. 24 , 25
2.1.4. 3‐Hydroxypropionic acid
Elevated 3‐hydroxypropionic acid in urine, combined with the presence of 2‐MCA in urine, is diagnostic of PA or MMA (if methylmalonic acid is elevated). 2 Dried blood spots from most newborns with PA or MMA also show elevation of 3‐hydroxypropionic acid. 26 During episodes of acute metabolic decompensation, increased 3‐hydroxypropionic acid in plasma is directly correlated with plasma ammonium. 16 Because it is difficult to measure clinical status by changes in 3‐hydroxypropionic acid, it is not suitable for use as a biomarker in patients with PA or MMA.
2.2. Conjugates: propionylglycine and tiglylglycine
The presence of propionylglycine and tiglylglycine in urine can be used to support a diagnosis of PA or MMA, 27 , 28 , 29 but they are not suitable for use as biomarkers. Like 3‐hydroxypropionic acid, propionylglycine levels are positively correlated with plasma ammonium, but changes in propionylglycine levels are highly variable and do not provide consistent evidence to monitor treatment outcomes in these patients. 16
2.3. Acylcarnitines
Acylcarnitines are intermediates of oxidative metabolism that consist of an acyl group esterified to a carnitine molecule. 30 They derive from acyl‐CoAs and are generated by both mitochondrial and peroxisomal enzymes, including carnitine palmitoyltransferase 1 (CPT‐1), carnitine palmitoyltransferase 2 (CPT‐2), carnitine acetyltransferase and other not fully characterized enzymes whose primary purpose is transporting long‐chain fatty acids across the mitochondrial membrane for β‐oxidation, or to bind and remove abnormal metabolites. 30 Patients with defects in branched‐chain amino acid catabolism or inherited disorders of fatty acid metabolism accumulate disease‐specific acylcarnitines in body fluids. 31
Long‐chain acylcarnitines (LCACs) are implicated in several biological processes, including cardiac function, ion balance, insulin signaling in muscle, inflammation, cellular stress, and modulation of protein kinase C. 30 However, a specific physiologic role or toxicity of C3 has not been demonstrated. Probably the most well characterized clinical application of measurement of C3 is in newborn screening, where it is elevated in MMA and PA. 32 , 33 , 34 In an affected newborn, acetylcarnitine (C2) is usually normal, and thus the C3/C2 ratio (a proxy for propionyl‐CoA/acetyl‐CoA) can be elevated up to 20‐fold. 16 A C3/C2 ratio of >0.4 suggests a diagnosis of PA/MMA, 33 but the false‐positive rate is high. 34 In general, the level of C3 reflects disease severity, but the relationship is imperfect as it can also vary according to diet and is affected by kidney disease. 17
Other acylcarnitines and ratios can also be altered in PA/MMA. 2‐Hexenoylcarnitine (C6:1) can be increased in PA. 35 Methylmalonylcarnitine (one of the compounds of C4‐DC) can be increased in MMA, 36 but not in PA, 35 and thus can be useful to differentiate the two. Methylmalonic acid, however, is not well conjugated with carnitine, and thus variable. 3‐Hydroxyhexadecenoyl‐carnitine (C16:1‐OH) is elevated in newborns with MMA. 37 Heptadecanoylcarnitine (C17) in dried blood spots is significantly higher in newborns with MMA compared with healthy newborns. 38
2.4. 13C‐propionate oxidation
Stable isotope techniques allow for determination of whole‐body protein turnover and amino acid oxidation. Potential applications in a clinical research setting include assessing the functional implications of genetic variants of unknown clinical significance, establishing guidelines for evidence‐based practice by assessing the effect of therapeutic interventions in randomized clinical trials, and monitoring therapeutic interventions in patients with disorders of intermediary amino acid catabolism. 39 Animal models of PA show markedly elevated 13C‐labeled 3‐hydroxypropionate in rat liver after infusion of 13C‐propionate. 40 13C‐propionate oxidation can also be measured in blood samples from patients with PA or MMA. 41 In primary fibroblasts, the activity of propionate incorporation in acid precipitable material has been shown to correlate negatively with PA/MMA disease severity. 42
A noninvasive “breath test” administers 13C‐propionate orally and measures the percentage of 13CO2 exhaled in 2‐hour breath samples by mass spectrometry. Exhaled 13CO2 is decreased in PA 17 and, to a lesser extent, in MMA. 43 Lower 13CO2, which indicates lower 13C‐propionate oxidation, is associated with severe PA genotypes and more adverse outcomes. 43 Patients with PA/MMA have significantly higher oxidized 13C‐propionate after liver transplant (or liver‐kidney transplant) than non‐transplanted patients. 17 , 43 In PA/MMA, 13C‐propionate breath test results correlate with plasma MCA or plasma C3. 17 , 43 Combining 13C‐propionate breath test with four clinical parameters (WBC, RBC, height z‐score, and total protein intake) has high accuracy to predict severity of PA. 17 13C‐propionate breath test also appears to be a more reliable biomarker to assess the efficacy of liver‐directed therapies compared with plasma MCA or C3 in PA, 17 or plasma methylmalonic acid in MMA. 43
3. BIOMARKERS—SECONDARY METABOLITES
3.1. Acetyl‐CoA
As discussed in Section 2.3, an increase in the C3/C2 ratio is widely used as a proxy for an increase in propionyl‐CoA relative to acetyl‐CoA in patients with PA or MMA. 16 Historically, propionyl‐CoA and acetyl‐CoA were not assessed in patients with PA or MMA due to a lack of reliable assays. In recent years, more stable assays for whole‐blood measurement of acetyl‐CoA have been developed. 44 These assays might prove to be useful to directly examine acetyl‐CoA levels as a biomarker in clinical trials of new therapies in PA or MMA; however, the potential relevance of acetyl‐CoA as a biomarker in PA or MMA has yet to be determined.
3.2. TCA cycle intermediates
Propionic and methylmalonic acid metabolism generates succinyl‐CoA, an important intermediate of the TCA cycle. Thus, this pathway represents an important mechanism for replenishing TCA cycle intermediates (anaplerosis). Anaplerotic deficiencies of the TCA cycle have previously been described in PA and MMA, and anaplerotic therapy has been suggested as a mechanism of treatment in these disorders. 15 , 45 These anaplerotic deficiencies are intuitive, given that the metabolic flux of propionyl‐CoA metabolism is to succinyl‐CoA (which explains decreases in ketoglutaric acid, succinic acid, and fumaric acid), leading to impaired generation of reducing equivalents for oxidative phosphorylation. Proximal defects in the TCA cycle, however, might represent defective conversion of pyruvic acid to acetyl‐CoA at the step of pyruvate dehydrogenase. 35 Thus, the TCA cycle defects in MMA cannot be solely due to limited flux of propionyl‐CoA into succinyl‐CoA. Succinic acid is usually most severely decreased, but inconsistently so and thus not particularly useful alone as an biomarker for clinical trials.
3.3. Ammonium
Ammonium is a waste product of amino acid catabolism and is also a potent neurotoxin. Hyperammonemia, which often occurs with episodes of metabolic decompensation in PA or MMA, likely results from secondary disruption of substrate balance in other biochemical pathways, including the urea and TCA cycles. 46 Accumulated metabolites (propionyl‐CoA and methylmalonyl‐CoA) likely compete with acetyl‐CoA to inhibit activity of N‐acetylglutamate synthase, thereby diminishing synthesis of carbamoylphosphate. This secondary impairment of the urea cycle can lead to elevated plasma ammonium levels, with neurotoxic effects during metabolic decompensations. 46 A chronic contributor to hyperammonemia in patients with PA or MMA is the release of ammonia from glutamine to generate glutamic acid in an attempt to replenish alpha‐ketoglutaric acid in the TCA cycle. 47
Hyperammonemia toxicity in the developing brain is due to multiple mechanisms, including disruption of amino acid and cerebral energy metabolism, and increased oxidative stress. 46 Repeated and frequent episodes of hyperammonemia (alongside metabolic decompensations) can result in impaired growth and intellectual disability, the severity of which increases with longer duration of hyperammonemia, an effect that likely is exacerbated by concurrent acidosis.
Hyperammonemia can lead to the identification and diagnosis of PA/MMA, and it might be a useful biomarker to monitor the severity of metabolic crises. 46 However, hyperammonemia is not specific for PA/MMA and it is not by itself diagnostic. It likely is also not as useful for monitoring PA/MMA at times of stability between crises.
3.4. Lactic acid
Blood lactic acid levels and urinary excretion of lactic acid increase during clinical decompensation episodes of PA. 16 , 48 However, both findings are non‐specific and are abnormal in a wide variety of chronic or acute medical conditions; thus, it has limited utility as a biomarker for PA or MMA.
3.5. Blood gas analysis
Assessment of acid‐base balance is recommended as part of regular metabolic follow‐up at clinic visits in patients with PA or MMA, and during episodes of metabolic decompensation. 2 In the chronic setting, following serum bicarbonate levels serves as a surrogate marker for chronic acidosis. Formal blood gas analysis is often necessary to monitor clinical status and response to therapy acutely, but it is not expected to have utility as a biomarker for chronic monitoring.
3.6. Amino acids
PA and MMA are associated with a variety of amino acid imbalances in end‐organs. 49 While the blood amino acid profiles can differentiate hyperammonemia in PA/MMA from that in urea cycle disorders, 46 this is better done with organic acid or acylcarnitine analysis, as discussed above. Massive elevations in plasma glycine are commonly seen in individuals with PA, while MMA patients show significantly lower glycine levels. 50 In fact, PA was originally named “non‐ketotic hyperglycinemia.” 51 It has previously been hypothesized that increased plasma glycine could be due to inhibition of the glycine cleavage system. 52 However, Anzmann et al., suggested that increased synthesis from serine may contribute to elevated plasma levels of glycine found in PA compared to MMA and unaffected individuals. 35 , 50 Elevated glycine is not specific to PA/MMA and thus is not diagnostic, though it does correlate to some extent to increases in other metabolites. 46
Alterations in biosynthesis of serine, including increases in serine de novo synthesis and serine transport, have been suggested as a novel area of cellular dysfunction in PA and MMA. 35 Abnormally low plasma serine has been reported in an individual with MMA after liver/kidney transplant. 53 , 54
Hyperlysinemia also is seen in PA, likely related to chronic hyperammonemia. 15 , 49 The precursor amino acids to propionyl‐CoA (Met, Thr, Ilu, and Val) are usually normal in patients with MMA and PA. However, iatrogenic deficiencies related to over restriction in metabolic diets can lead to their deficiency. 49 During acute metabolic decompensation, branched‐chain amino acids such as leucine increase significantly due to protein catabolism. 16 Other amino acid abnormalities have been reported including decreased glutamine in PA and MMA, 49 and alteration of alanine in MMA. 49
The ratio of alanine: serine, which is increased in patients with mitochondrial dysfunction, 54 has been shown to improve the accuracy of models to predict severity of PA. 17 Decreased ratios of alanine:lysine and alanine:(phenylalanine + tyrosine) also can be used as biomarkers of mitochondrial disease in patients with PA or MMA. 16
4. OTHER POTENTIAL BIOMARKERS
4.1. Fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15)
Long‐term mitochondrial energy dysfunction has been demonstrated in PA and MMA, 55 but histologic and biochemical assessment of a muscle biopsy to assess mitochondrial function is not practical for clinical trials or clinical practice. FGF21 is a metabolism‐regulating hormone secreted by the liver, and an elevated serum level of FGF21 has been suggested as a biomarker for mitochondrial disease. 56 It is considered stable with respect to processing and storage, has a higher specificity and sensitivity compared to other biomarkers, and its measurement is rapid and inexpensive. Plasma GDF15 is also elevated in patients in mitochondrial disorders. 57
Median plasma FGF21 concentration > 1500 pg/mL has a positive predictive value of 0.83 and a negative predictive value of 1.00 for development of long‐term complications in MMA and PA. 55 FGF21 is a highly predictive biomarker in MMA; restoration of liver MUT activity in rats decreases plasma FGF21, and this change in humans is associated with improved outcomes. 58 Plasma FGF21 in patients with MMA correlates with disease subtype, growth indices, and markers of mitochondrial dysfunction. 58 Liver and/or kidney‐transplanted patients with PA/MMA have significantly lower plasma FGF21 than non‐transplanted patients with PA/MMA. 13 A modeling study showed that plasma FGF21 and GDF15 predicted severity of PA and were lower in patients with PA after liver transplant. 17 Thus, plasma FGF21 and GDF15 might be useful surrogate endpoints for patients with PA or MMA.
4.2. Brain injury markers
Neurological complications are common in patients with PA or MMA, but advanced neuroimaging is not available at all centers and radiographic results can lag behind the neurological changes. 59 Possible biomarkers have been identified for astroglial injury (S100B or glial fibrillary acidic protein [GFAP]) and neuronal cell body injury (neuron specific enolase [NSE] and ubiquitin C‐terminal hydrolase‐L1 [UCH‐L1]). 60 , 61 It remains to be seen if these proteins, whose presence in cerebrospinal fluid are useful to determine the presence and timing of traumatic brain injury, 60 are biomarkers for neurological complications in patients with PA or MMA. An observational cohort study (NCT04602325) is investigating systemic biomarkers of brain injury in patients with PA or MMA and other inherited disorders.
4.3. Brain natriuretic peptide
B‐type natriuretic peptide (BNP) can be produced in both cardiac atria and ventricles and is upregulated in failing ventricular myocardium. In response to increased myocardial stretch and wall stretch, ventricular myocytes secret the pro‐hormone pre‐proBNP, which is then cleaved into biologically active BNP and the inactive by product N‐terminal‐pro‐BNP (NT‐proBNP). 62 Elevated BNP can be found in many circumstances involving left ventricular dysfunction or hypertrophy, right ventricular dysfunction secondary to pulmonary disease, cardiac inflammatory or infectious diseases, endocrine diseases, and high cardiac output status without decreased LV ejection fraction. 62 However, this is an inconsistent finding in PA and likely of more limited utility in PA patients without cardiac dysfunction.
4.4. Cystatin C
Estimation of glomerular function is usually based on measurement of serum creatinine. However, patients with PA and MMA are on a protein‐restricted diet and receive lesser amounts of creatine, the precursor of creatinine, in their diet since this is mostly present in meat. For this reason, cystatin C measurements can be more sensitive to measure renal function in these patients than serum creatinine. 63 Thus, it has been recommended that laboratory measurements in patients with PA or MMA should include measurements of cystatin C, plasma uric acid, 25‐OH‐vitamin D, 1,25‐OH‐vitamin D, PTH, plasma osmolality, and 24‐hour protein excretion as part of kidney disease surveillance. 2 , 63 Plasma transthyretin (prealbumin), which correlates positively with severity of kidney disease, can also be useful as a biomarker for PA disease severity. 17
5. ADDITIONAL CONSIDERATIONS FOR DRUG DEVELOPMENT
The complexity and heterogeneity of organic acidemias provide significant challenges for clinical study design and evaluation. One key factor in the evaluation of an intervention in a controlled clinical trial is the clinical relevance of the selected study endpoints or outcome measures, together with an understanding of what comprises a minimal clinically important difference in these endpoints. 64 Correction or improvement of a biomarker does not necessarily mean that the disease course also improves. Unfortunately, many clinical trials in rare diseases have used either subclinical parameters, rather than clinical outcomes, or endpoints with unclear relevance to patient outcomes. 65 Draft guidance from FDA for drug development in rare diseases emphasizes the role of predictive biomarkers and surrogate endpoints for clinical trials of patients with rare diseases, particularly in early/mid‐phase clinical trials, or to support accelerated approval. 66 Biomarkers that are shown to be associated with—or predict—clinical outcomes may be used as surrogate endpoints for drug development. 67 , 68 , 69 However, there is no regulatory precedence for biomarkers or surrogate endpoints in the investigation of new therapies for patients with organic acidemias. The heterogeneity of the patient population may complicate the prediction of benefits of therapy, specifically in patients with milder disease. Additionally, initial trial data may show benefits in short‐term endpoints, but the real world situation may reveal different outcomes.
Several potential biomarkers have been investigated in patients with PA or MMA, including the use of biomarkers as outcomes in ongoing drug development studies (Table 2). These and other biomarkers might serve as surrogate endpoints based on evidence of their clinical relevance (Table 3), but additional research is needed to validate them.
TABLE 2.
Use of biomarkers as outcomes in drug development studies in PA or MMA
| Study (clinicaltrials.gov identifier) | Biomarkers assessed as outcomes |
|---|---|
| A Phase 2 open‐label, dose escalation study of HST5040 in subjects with propionic or methylmalonic acidemia followed by a randomized, double‐blind, placebo‐controlled, 2‐period crossover study and an open‐label, long‐term extension study (NCT04732429; ongoing) | MCA, C3, C3:C2, 3‐hydroxypropionic acid, methylmalonic acid, ammonium, anion gap |
| A first in human, dose escalation study to evaluate the safety and tolerability of BBP‐671 in healthy volunteers and patients with propionic acidemia or methylmalonic acidemia (NCT04836494; ongoing) | Whole blood acetyl‐CoA, plasma pantothenic acid, amino acids, ammonia, methylmalonic acid, MCA, brain natriuretic peptide, lactic acid, carnitines, acylcarnitines, FGF21, organic acids, TCA cycle intermediates, acylglycines |
| A global, phase 1/2, open‐label, dose optimization study to evaluate the safety, tolerability, pharmacodynamics, and pharmacokinetics of mRNA‐3705 in participants with isolated methylmalonic acidemia due to methylmalonyl‐CoA mutase deficiency (NCT04899310; ongoing) | Methylmalonic acid, MCA |
| Open‐label study of mRNA‐3927 in participants with propionic acidemia (NCT04159103; ongoing) | MCA, 3‐hydroxypropionic acid |
| A phase 1/2 open‐label clinical study of hLB‐001 gene therapy in pediatric patients with methylmalonic acidemia characterized by MMUT mutations (NCT04581785; ongoing) | Methylmalonic acid, MCA, FGF21 |
| Long‐term outcome of N‐carbamylglutamate treatment in propionic acidemia and methylmalonic acidemia (NCT01597440; terminated) | Ammonium |
Abbreviations: C2, acetylcarnitine; C3, propionylcarnitine; FGF21, fibroblast growth factor 21; MCA, methylcitric acid; TCA, tricarboxylic acid cycle (also known as the citric acid cycle or Krebs cycle).
TABLE 3.
Biomarkers with possible utility as surrogate endpoints for PA or MMA
| Biomarker | Disorder | Matrix | Clinical relevance | References |
|---|---|---|---|---|
| MCA | PA/MMA | Plasma | MCA predicts disease burden, long‐term complications, and impact of organ transplantation | 13 |
| MCA:citric acid | PA/MMA | Plasma | Indicator for disease course and/or severity | 18 |
| C3 | PA/MMA | Plasma | C3 (measured as C3/C2) increases by up to ~20‐fold in patients with PA/MMA | 16 |
| Methylmalonic acid | MMA | Plasma | Elevation associated with risk for kidney disease and metabolic stroke | 20 |
| Ammonium | PA/MMA | Plasma | Repeated and frequent episodes of hyperammonemia can result in impaired growth and intellectual disability | 46 |
| FGF21/GDF15 | PA/MMA | Plasma | Elevation predicts long‐term complications | 13, 55 |
| Higher in patients with severe vs mild PA; lower in patients with PA after liver transplant | 17 | |||
| Concentration correlates with disease subtype, growth indices, and markers of mitochondrial dysfunction and is not affected by kidney disease | 58 | |||
| 13C‐propionate oxidation | PA/MMA | Breath | Decreased in MMA or PA; larger decreases correlate with more severe disease | 43 |
| Significantly higher in transplanted than non‐transplanted patients with PA, MMA | 43 |
Abbreviations: C2, acetylcarnitine; C3, propionylcarnitine; FGF21, fibroblast growth factor 21; GDF15, growth differentiation factor 15; MCA, methylcitric acid; MMA, methylmalonic acidemia; PA, propionic acidemia.
The ideal surrogate endpoint for drug development would be valid both for longitudinal analysis and for continuous changes. Some biomarkers that are useful to diagnose PA/MMA or to identify acute decompensation are discrete or insensitive tests that do not provide information about the magnitude of change, limiting the utility of these biomarkers to identify clinically meaningful changes with new therapies. Additional information is needed about the use of biomarkers as surrogate endpoints based on average changes over time.
Dietary management has a central role for the treatment of many inborn errors of metabolism, such as PA and MMA. Treatment guidelines and regulatory guidance for clinical investigations of these conditions include optimizing, standardizing, and maintaining a stable diet in addition to other supportive therapy or investigational treatment. 2 , 70 A diet low in natural protein (respecting age‐appropriate total protein requirements) is recommended to improve metabolic stability. 2 However, there is no single dietary prescription for these patients, and maintenance of a low‐protein diet is particularly difficult as children age. Diet history—including a detailed description of natural protein intake, of protein provided by amino acid mixture as well as of daily caloric intake—should be recorded at each clinic visit, but inherent variability of patient recall and imprecision can complicate the collection of an accurate diet history in a clinical trial. Diet diaries can improve the accuracy and precision of diet history in drug development studies.
6. CONCLUSION AND OUTLOOK
It is highly complex to enroll and conduct a clinical trial of a new drug treatment for PA or MMA, due to the rarity, heterogeneity, mortality, and unpredictable outcomes associated with these inborn errors of metabolism. Selecting clinical endpoints for these trials is particularly challenging. Thus, there is a clear need to identify which biomarkers for PA/MMA could be surrogate endpoints to support the investigation of new therapies for these rare diseases.
CONFLICT OF INTEREST
Nicola Longo is employed by the University of Utah, which has received honoraria from CoA Therapeutics, Hemoshear, and Moderna. Jörn Oliver Sass is an official of Bonn‐Rhein‐Sieg‐University of Applied Sciences, which has received honoraria from CoA Therapeutics and Immedica Pharma AB for work performed by the author. Agnieszka Jurecka is an employee and stockholder of CoA Therapeutics. Jerry Vockley has no competing interests.
ACKNOWLEDGMENTS
Jonathan Latham of PharmaScribe, LLC received a grant from CoA Therapeutics to provide medical writing support. JV was supported in part by NIH grant R01 DK.109907.
Longo N, Sass JO, Jurecka A, Vockley J. Biomarkers for drug development in propionic and methylmalonic acidemias. J Inherit Metab Dis. 2022;45(2):132‐143. doi: 10.1002/jimd.12478
Communicating Editor: Carlos Ferreira
Funding information NIH, Grant/Award Number: R01 DK.109907; CoA Therapeutics
REFERENCES
- 1. Chapman KA. Practical management of organic acidemias. Translation Sci Rare Dis. 2019;4(3–4):121‐131. [Google Scholar]
- 2. Forny P, Horster F, Ballhausen D, et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: first revision. J Inherit Metab Dis. 2021;44(3):566‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Therrell BL Jr, Lloyd‐Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: ten‐year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113(1–2):14‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Meer SB, Poggi F, Spada M, et al. Clinical outcome and long‐term management of 17 patients with propionic acidaemia. Eur J Pediatr. 1996;155(3):205‐210. [DOI] [PubMed] [Google Scholar]
- 5. Grünert SC, Müllerleile S, de Silva L, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis. 2013;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sass JO, Hofmann M, Skladal D, Mayatepek E, Schwahn B, Sperl W. Propionic acidemia revisited: a workshop report. Clin Pediatr (Phila). 2004;43(9):837‐843. [DOI] [PubMed] [Google Scholar]
- 7. Lehnert W, Sperl W, Suormala T, Baumgartner ER. Propionic acidaemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. Eur J Pediatr. 1994;153(7 Suppl. 1):S68‐S80. [DOI] [PubMed] [Google Scholar]
- 8. Hwang WJ, Lim HH, Kim YM, et al. Pancreatic involvement in patients with inborn errors of metabolism. Orphanet J Rare Dis. 2021;16(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang L, Liu Y, Shen M, et al. A study on a cohort of 301 Chinese patients with isolated methylmalonic acidemia. J Inherit Metab Dis. 2020;43(3):409‐423. [DOI] [PubMed] [Google Scholar]
- 10. Yap S, Vara R, Morais A. Post‐transplantation outcomes in patients with PA or MMA: a review of the literature. Adv Ther. 2020;37(5):1866‐1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biomarkers Definition Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89‐95. [DOI] [PubMed] [Google Scholar]
- 12. Gülbakan B, Özgül RK, Yüzbaşıoğlu A, Kohl M, Deigner H‐P, Özgüç M. Discovery of biomarkers in rare diseases: innovative approaches by predictive and personalized medicine. EPMA J. 2016;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maines E, Catesini G, Boenzi S, et al. Plasma methylcitric acid and its correlations with other disease biomarkers: the impact in the follow up of patients with propionic and methylmalonic acidemia. J Inherit Metab Dis. 2020;43(6):1173‐1185. [DOI] [PubMed] [Google Scholar]
- 14. Amaral AU, Cecatto C, Castilho RF, Wajner M. 2‐Methylcitric acid impairs glutamate metabolism and induces permeability transition in brain mitochondria. J Neurochem. 2016;137(1):62‐75. [DOI] [PubMed] [Google Scholar]
- 15. Longo N, Price LB, Gappmaier E, et al. Anaplerotic therapy in propionic acidemia. Mol Genet Metab. 2017;122(1–2):51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haijes HA, Jans JJM, van der Ham M, van Hasselt PM, Verhoeven‐Duif NM. Understanding acute metabolic decompensation in propionic and methylmalonic acidemias: a deep metabolic phenotyping approach. Orphanet J Rare Dis. 2020;15(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shchelochkov OA, Manoli I, Juneau P, et al. Severity modeling of propionic acidemia using clinical and laboratory biomarkers. Genet Med. 2021;23(8):1534‐1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Al‐Dirbashi OY, Alfadhel M, Al‐Thihli K, et al. Assessment of methylcitrate and methylcitrate to citrate ratio in dried blood spots as biomarkers for inborn errors of propionate metabolism. Sci Rep. 2019;9(1):12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrade VM, Dal Pont HS, Leffa DD, et al. Methylmalonic acid administration induces DNA damage in rat brain and kidney. Mol Cell Biochem. 2014;391(1–2):137‐145. [DOI] [PubMed] [Google Scholar]
- 20. Baker EH, Sloan JL, Hauser NS, et al. MRI characteristics of globus pallidus infarcts in isolated methylmalonic acidemia. AJNR Am J Neuroradiol. 2015;36(1):194‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mineva EM, Sternberg MR, Zhang M, et al. Age‐specific reference ranges are needed to interpret serum methylmalonic acid concentrations in the US population. Am J Clin Nutr. 2019;110(1):158‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ganji V, Kafai MR. Population reference values for serum methylmalonic acid concentrations and its relationship with age, sex, race‐ethnicity, supplement use, kidney function and serum vitamin B12 in the post‐folic acid fortification period. Nutrients. 2018;10(1):74. doi: 10.3390/nu10010074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kruszka PS, Manoli I, Sloan JL, Kopp JB, Venditti CP. Renal growth in isolated methylmalonic acidemia. Genet Med. 2013;15(12):990‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noone D, Riedl M, Atkison P, et al. Kidney disease and organ transplantation in methylmalonic acidaemia. Pediatr Transplant. 2019;23(4):e13407. [DOI] [PubMed] [Google Scholar]
- 25. Niemi AK, Kim IK, Krueger CE, et al. Treatment of methylmalonic acidemia by liver or combined liver‐kidney transplantation. J Pediatr. 2015;166(6):1455‐1461 e1451. [DOI] [PubMed] [Google Scholar]
- 26. Monostori P, Klinke G, Richter S, et al. Simultaneous determination of 3‐hydroxypropionic acid, methylmalonic acid and methylcitric acid in dried blood spots: second‐tier LC‐MS/MS assay for newborn screening of propionic acidemia, methylmalonic acidemias and combined remethylation disorders. PLoS One. 2017;12(9):e0184897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasmussen K, Ando T, Nyhan WL, et al. Excretion of tiglylglycine in propionic acidemia. J Pediatr. 1972;81(5):970‐972. [DOI] [PubMed] [Google Scholar]
- 28. Rasmussen K, Ando T, Nyhan WL, et al. Excretion of propionylglycine in propionic acidaemia. Clin Sci. 1972;42(6):665‐671. [DOI] [PubMed] [Google Scholar]
- 29. Bonafe L, Troxler H, Kuster T, et al. Evaluation of urinary acylglycines by electrospray tandem mass spectrometry in mitochondrial energy metabolism defects and organic acidurias. Mol Genet Metab. 2000;69(4):302‐311. [DOI] [PubMed] [Google Scholar]
- 30. McCoin CS, Knotts TA, Adams SH. Acylcarnitines—old actors auditioning for new roles in metabolic physiology. Nat Rev Endocrinol. 2015;11(10):617‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Sain‐van der Velden MG, Diekman EF, Jans JJ, et al. Differences between acylcarnitine profiles in plasma and bloodspots. Mol Genet Metab. 2013;110(1–2):116‐121. [DOI] [PubMed] [Google Scholar]
- 32. la Marca G, Malvagia S, Pasquini E, Innocenti M, Donati MA, Zammarchi E. Rapid 2nd‐tier test for measurement of 3‐OH‐propionic and methylmalonic acids on dried blood spots: reducing the false‐positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography‐tandem mass spectrometry. Clin Chem. 2007;53(7):1364‐1369. [DOI] [PubMed] [Google Scholar]
- 33. Cheng KH, Liu MY, Kao CH, et al. Newborn screening for methylmalonic aciduria by tandem mass spectrometry: 7 years' experience from two centers in Taiwan. J Chin Med Assoc. 2010;73(6):314‐318. [DOI] [PubMed] [Google Scholar]
- 34. Al‐Dirbashi OY, McIntosh N, Chakraborty P. Quantification of 2‐methylcitric acid in dried blood spots improves newborn screening for propionic and methylmalonic acidemias. J Med Screen. 2017;24(2):58‐61. [DOI] [PubMed] [Google Scholar]
- 35. Anzmann AF, Pinto S, Busa V, et al. Multi‐omics studies in cellular models of methylmalonic acidemia and propionic acidemia reveal dysregulation of serine metabolism. Biochim Biophys Acta Mol Basis Dis. 2019;1865(12):165538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rizzo C, Boenzi S, Inglese R, et al. Measurement of succinyl‐carnitine and methylmalonyl‐carnitine on dried blood spot by liquid chromatography‐tandem mass spectrometry. Clin Chim Acta. 2014;429:30‐33. [DOI] [PubMed] [Google Scholar]
- 37. McHugh D, Cameron CA, Abdenur JE, et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13(3):230‐254. [DOI] [PubMed] [Google Scholar]
- 38. Malvagia S, Haynes CA, Grisotto L, et al. Heptadecanoylcarnitine (C17) a novel candidate biomarker for newborn screening of propionic and methylmalonic acidemias. Clin Chim Acta. 2015;450:342‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huidekoper HH, Wijburg FA, Wanders RJA. Inborn errors of metabolism. In: Schierbeek H, ed. Mass Spectrometry and Stable Isotopes in Nutritional and Pediatric Research. Hoboken, NJ: John Wiley & Sons, Inc.; 2017. [Google Scholar]
- 40. Reijngoud DJ. Flux analysis of inborn errors of metabolism. J Inherit Metab Dis. 2018;41(3):309‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barshop BA, Yoshida I, Ajami A, et al. Metabolism of 1‐13C‐propionate in vivo in patients with disorders of propionate metabolism. Pediatr Res. 1991;30(1):15‐22. [DOI] [PubMed] [Google Scholar]
- 42. Willard HF, Ambani LM, Hart AC, Mahoney MJ, Rosenberg LE. Rapid prenatal and postnatal detection of inborn errors of propionate, methylmalonate, and cobalamin metabolism: a sensitive assay using cultured cells. Hum Genet. 1976;34(3):277‐283. [DOI] [PubMed] [Google Scholar]
- 43. Manoli I, Pass AR, Harrington EA, et al. 1‐(13)C‐propionate breath testing as a surrogate endpoint to assess efficacy of liver‐directed therapies in methylmalonic acidemia (MMA). Genet Med. 2021;23(8):1522‐1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Speziale R, Montesano C, De Leonibus ML, et al. Determination of acetyl coenzyme A in human whole blood by ultra‐performance liquid chromatography‐mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1083:57‐62. [DOI] [PubMed] [Google Scholar]
- 45. Knerr I, Weinhold N, Vockley J, Gibson KM. Advances and challenges in the treatment of branched‐chain amino/keto acid metabolic defects. J Inherit Metab Dis. 2012;35(1):29‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Häberle J, Chakrapani A, Ah Mew N, Longo N. Hyperammonaemia in classic organic acidaemias: a review of the literature and two case histories. Orphanet J Rare Dis. 2018;13(1):219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Filipowicz HR, Ernst SL, Ashurst CL, Pasquali M, Longo N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol Genet Metab. 2006;88(2):123‐130. [DOI] [PubMed] [Google Scholar]
- 48. Kuhara T, Shinka T, Matsuo M, Matsumoto I. Increased excretion of lactate, glutarate, 3‐hydroxyisovalerate and 3‐methylglutaconate during clinical episodes of propionic acidemia. Clin Chim Acta. 1982;123(1–2):101‐109. [DOI] [PubMed] [Google Scholar]
- 49. Scholl‐Bürgi S, Sass JO, Zschocke J, Karall D. Amino acid metabolism in patients with propionic acidaemia. J Inherit Metab Dis. 2012;35(1):65‐70. [DOI] [PubMed] [Google Scholar]
- 50. Nizon M, Ottolenghi C, Valayannopoulos V, et al. Long‐term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J Rare Dis. 2013;8:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Childs B, Nyhan WL, Borden M, Bard L, Cooke RE. Idiopathic hyperglycinemia and hyperglycinuria: a new disorder of amino acid metabolism. I. Pediatrics. 1961;27:522‐538. [PubMed] [Google Scholar]
- 52. Hayasaka K, Narisawa K, Satoh T, et al. Glycine cleavage system in ketotic hyperglycinemia: a reduction of H‐protein activity. Pediatr Res. 1982;16(1):5‐7. [DOI] [PubMed] [Google Scholar]
- 53. Vernon HJ, Sperati CJ, King JD, et al. A detailed analysis of methylmalonic acid kinetics during hemodialysis and after combined liver/kidney transplantation in a patient with mut (0) methylmalonic acidemia. J Inherit Metab Dis. 2014;37(6):899‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferreira CR, Goorden SMI, Soldatos A, et al. Deoxysphingolipid precursors indicate abnormal sphingolipid metabolism in individuals with primary and secondary disturbances of serine availability. Mol Genet Metab. 2018;124(3):204‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Molema F, Jacobs EH, Onkenhout W, Schoonderwoerd GC, Langendonk JG, Williams M. Fibroblast growth factor 21 as a biomarker for long‐term complications in organic acidemias. J Inherit Metab Dis. 2018;41(6):1179‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Suomalainen A, Elo JM, Pietilainen KH, et al. FGF‐21 as a biomarker for muscle‐manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10(9):806‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fujita Y, Ito M, Kojima T, Yatsuga S, Koga Y, Tanaka M. GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion. 2015;20:34‐42. [DOI] [PubMed] [Google Scholar]
- 58. Manoli I, Sysol JR, Epping MW, et al. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight. 2018;3(23):e124351. doi: 10.1172/jci.insight.124351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schreiber J, Chapman KA, Summar ML, et al. Neurologic considerations in propionic acidemia. Mol Genet Metab. 2012;105(1):10‐15. [DOI] [PubMed] [Google Scholar]
- 60. Berger RP, Pierce MC, Wisniewski SR, et al. Neuron‐specific enolase and S100B in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatrics. 2002;109(2):E31. [DOI] [PubMed] [Google Scholar]
- 61. Wang KK, Yang Z, Zhu T, et al. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev Mol Diagn. 2018;18(2):165‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tsai SH, Lin YY, Chu SJ, Hsu CW, Cheng SM. Interpretation and use of natriuretic peptides in non‐congestive heart failure settings. Yonsei Med J. 2010;51(2):151‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shchelochkov OA, Manoli I, Sloan JL, et al. Chronic kidney disease in propionic acidemia. Genet Med. 2019;21(12):2830‐2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lachmann R, Schoser B. The clinical relevance of outcomes used in late‐onset Pompe disease: can we do better? Orphanet J Rare Dis. 2013;8:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jurecka A, Tylki‐Szymańska A. Enzyme replacement therapy: lessons learned and emerging questions. Expert Opin Orphan Drugs. 2015;3(3):293‐305. [Google Scholar]
- 66. FDA . Rare Diseases: Common Issues in Drug Development Guidance for Industry. Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration; 2019. [Google Scholar]
- 67. FDA . Biomarker Qualification: Evidentiary Framework Guidance for Industry and FDA Staff. Silver Spring, MD: U.S. Department of Health and Human Services Food and Drug Administration; 2018. [Google Scholar]
- 68. FDA . E16 Biomarkers Related to Drug or Biotechnology Product Development: Context, Structure, and Format of Qualification Submissions. Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration; 2011. [PubMed] [Google Scholar]
- 69. EMA . Concept Paper on Predictive Biomarker‐Based Assay Development in the Context of Drug Development and Lifecycle. London, UK: European Medicines Agency; 2017. [Google Scholar]
- 70. FDA . Inborn Errors of Metabolism that Use Dietary Management: Considerations for Optimizing and Standardizing Diet in Clinical Trials for Drug Product Development Guidance for Industry. Silver Spring, MD: U.S. Department of Health and Human Services Food and Drug Administration; 2018. [Google Scholar]