

Abstract
Strong reducing agents (< −2.0 V vs SCE) enable a wide array of useful organic chemistry, but suffer from a variety of limitations. Stoichiometric metallic reductants such as alkali metals and SmI2 are commonly employed for these reactions, however considerations including expense, ease of use, safety, and waste generation limit the practicality of these methods. Recent approaches utilizing energy from multiple photons or electron-primed photoredox catalysis have accessed reduction potentials equivalent to Li0 and shown how this enables selective transformations of aryl chlorides via aryl radicals. However, in some cases low stability of catalytic intermediates can limit turnover numbers. Herein we report the ability of CdS nanocrystal quantum dots (QDs) to function as strong photoreductants and present evidence that a highly reducing electron is generated from two consecutive photoexcitations of CdS QDs with intermediate reductive quenching. Mechanistic experiments suggest that Auger recombination, a photophysical phenomenon known to occur in photoexcited anionic QDs, generates transient thermally excited electrons to enable the observed reductions. Using blue LEDs and sacrificial amine reductants, aryl chlorides and phosphate esters with reduction potentials up to −3.4 V vs SCE are photo-reductively cleaved to afford hydrodefunctionalized or functionalized products. In contrast to small molecule catalysts, the QDs are stable under these conditions and turnover numbers up to 47500 have been achieved. These conditions can also effect other challenging reductions, such as tosylate protecting group removal from amines, debenzylation of alcohols, and reductive ring-opening of cyclopropanecarboxylic acid derivatives.
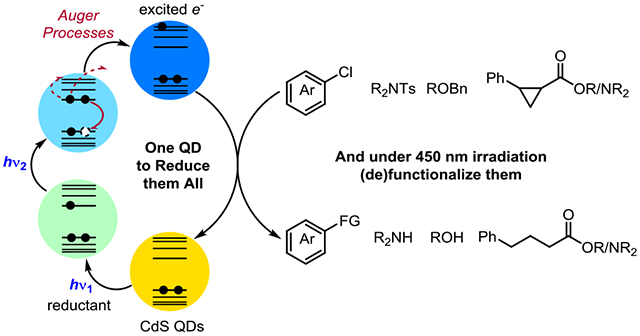
Graphical Abstract
Introduction
Photoredox catalysis has changed the way chemists think about the reactivity of common functional groups by allowing photon-gated redox events or energy transfer to catalytically convert stable substrate functionalities into reactive intermediates.1,2 Over time, structural elaboration of organic and organometallic dyes has produced a collection of effective photoredox catalysts with diverse photochemical characteristics.3,4 By expanding the electrochemical horizons of photocatalysts, increasingly inert classes of chemicals have become accessible substrates for photoredox transformations. However, the maximum energy of a visible photon (3.1 eV at 400 nm), energy losses through catalyst intersystem crossing, and other nonradiative pathways impose limits on the strength of photogenerated redox agents from single visible photon absorption (Figure 1A).5–7 Higher-energy UV irradiation can be used to generate powerful redox agents,8,9 however undesired photochemical side reactions limit the functional group tolerance of these approaches.10
Figure 1.
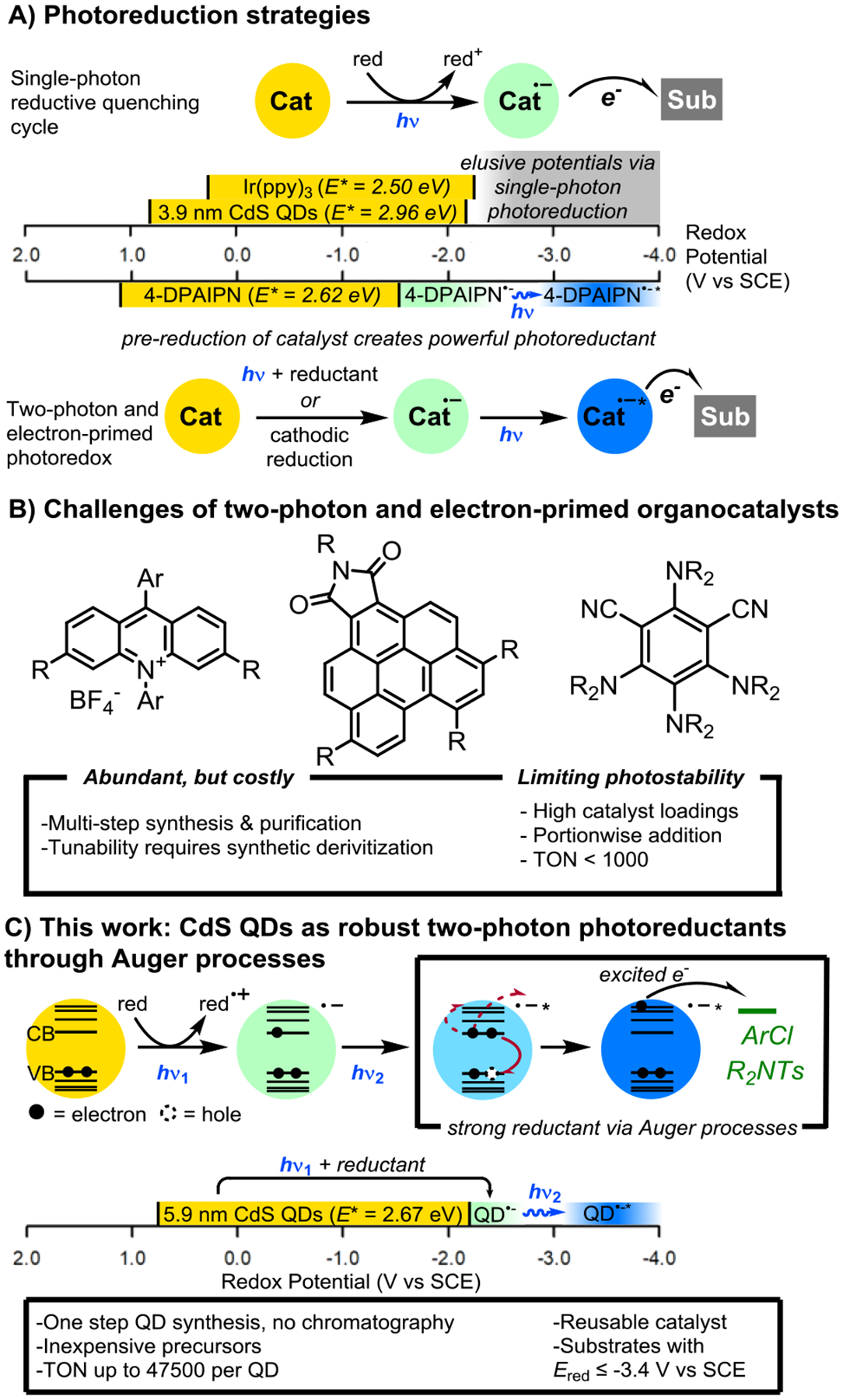
A: Contemporary strategies for photoredox-mediated reductions B: Commonly encountered challenges to existing photoreduction protocols C: This work: CdS QDs as robust highly-reducing two-photon photocatalysts for reductive organic transformations enabled by Auger recombination.
To overcome these limitations, recent innovations have provided catalytic methods allowing the input of additional energy beyond a single visible photon to generate extremely strong reducing agents (Figure 1A).11,12 One appealing approach is capturing the energy from multiple photons within a single catalyst turnover.6,12 In these “Z-scheme” reactions, initial photoexcitation and reductive quenching of the photocatalyst produces a reduced catalyst species, which can then absorb a second photon to form a powerfully reducing excited state.1,6,13–15 While this strategy has unlocked new photocatalyzed reductive transformations, such procedures present their own challenges. The photocatalyst must absorb visible photons in the ground state and after conversion to the active photoreductant via reduction, while additionally having appropriate excited state properties to drive chemistry after each successive excitation.6,15–17 In complement to this advance, others have pioneered electrochemical reduction of suitable precatalysts to form highly potent “electrochemically primed” photoredox catalysts.12,18–23 By decoupling catalyst reduction from photoexcitation, this approach has greatly expanded the pool of competent reduction-activated photoreductants to include more accessible and durable catalysts22 and enabled new selective aryl radical chemistry by spatially separating the reaction mixture from potentially problematic reductants.
Still, challenges remain for each of these approaches (Figure 1B). High catalyst loadings are currently required for most two-photon and electron-primed photoredox procedures (5–10 mol% is typical),24 owing to the tendency for the reduced catalyst intermediates to decompose,17,22,25 which also complicates mechanistic study of the operative mechanisms.17,25 Electron-primed photoredox catalysis requires a more complex apparatus, leading to challenges in vessel design and scalability. Moreover, the optimal organocatalysts for some strategies require multi-step syntheses and purification,15,26,27 and can be expensive to use in significant quantities. Therefore, while these strategies have enabled redox-initiated transformations of substrates far beyond the potentials accessible by traditional photocatalysis, these early reports underscore the need for improved photocatalysts with additional stability and access to new mechanisms of reactivity.
Semiconductor quantum dots (QDs) combine advantageous aspects of both homogeneous (high surface-volume ratio, solubility in reaction media, light penetration) and heterogeneous (durability, substrate binding) catalysts, and therefore offer new opportunities for photoredox catalysis.28,29 QDs have proven to be robust fluorophores and photocatalysts, generally exhibiting superior photostability to small-molecule dyes,30–36 however applications of QDs to organic synthesis remain underexplored. To address the need for continued development of photoredox catalysts, our group and others have been interested in new applications of QDs in organic chemistry.29,37–64 In addition to their high photostability, QDs exhibit tunable, size-dependent optical and redox properties; are made in single-step syntheses with no chromatography from abundant precursors;65 reversibly bind to many molecules at once (typically 1 – 5 ligands/nm2 of QD surface are found for closely related CdSe QDs66–68) through common organic functional groups (-CO2H, -PO3H, -SH, -NH2); can become charged with many electrons at once without decomposing;69,70 and undergo many electronic processes with no direct analogue in small-molecules.71
Inspired by reports of two-photon photoreduction mechanisms operative within commonly used photocatalyst systems,14,27,72 we envisioned that QDs could achieve a similar mode of reactivity, while also addressing the catalyst stability and availability challenges of organocatalyst-mediated photoreductions. In particular, we considered that Auger processes, a family of electronic events inaccessible to small-molecule photocatalysts which generate excited charge carriers from carrier recombination of trion or biexciton states,73–77 could be used to drive energetically demanding photoreductions of organic molecules (Figure 1C). Photoexcitation of a QD followed by reductive quenching from a suitable terminal reductant produces a “photodoped” QD.78 This is an anionic QD with the surplus electron residing in the 1Se state at the conduction band (CB) edge. The negatively charged QD can absorb a second photon at the same wavelength to produce an excited anionic “negative trion” state, with two electrons in the 1Se state and an electron-vacancy (“hole”) at the valence band (VB) edge. Negative trion states rapidly undergo Auger recombination (τ ~ 100 – 1000 ps)74,79,80 on timescales that are significantly faster than photoluminescence lifetimes. In Auger recombination, one 1Se electron acquires energy equivalent to that of the exciton at the band gap, concomitant with relaxation of the other CB electron to the valence band edge, generating a highly energetic or “hot” electron within the QD(Figure 1C). In some QDs with large bandgaps, this carrier recombination can instead result in the complete ejection of the second electron from the QD into the surrounding matrix, termed Auger ionization.77,79,81,82 While short lived,71,83 excited electrons in QDs can be transferred to nearby or adsorbed species exceptionally fast,84–86 and have been employed to improve aqueous hydrogen evolution and CO2 reduction.76,85,87,88 Auger processes are well studied phenomena in nanomaterials, however they have not been employed to generate highly reducing electrons for synthetic organic transformations requiring powerful reducing agents. We show here that CdS QDs can be employed as powerful photoreductants for organic synthesis through a two-photon mechanism involving an Auger process to generate strongly reducing electrons.
Results
We envisioned that CdS QDs could achieve this mode of reactivity by first absorbing a photon to form a neutral excited QD, then oxidizing a sacrificial organic reductant in a reductive quenching pathway to generate an anionic QD. Because one-electron reduction of QDs only results in partial bleaching of their excitonic feature, this anionic QD would be poised to absorb a second photon at the same wavelength to form a negative trion state, which undergo Auger processes to generate excited electrons. Initial studies revealed that oleate (OA) capped 5.8 – 6.0 nm CdS QDs89 were capable photocatalysts for reductive dehalogenation of electron-neutral and -rich aryl chlorides with reduction potentials up to 1300 mV more negative than the most-negative reported reduction potential of CdS QDs (EpcQD/QD– = −2.24 V vs SCE for 4.0 nm CdS QDs).90,91 To interrogate the photoreductive power of CdS QDs, optimization studies were undertaken using the reductive hydrodechlorination of 4-chloro-2,6-di-tert-butylanisole (1a) (Ered = −3.4 V vs SCE) as a challenging model reaction due to the highly negative reduction potential.18 Optimization of the reaction conditions revealed that polar aprotic solvents performed best and N,N′-dimethylpropylene urea (DMPU) provided the highest yields of dehalogenated product. Among the sacrificial reductants examined, tris(2-aminoethyl)amine (TAEA), sodium formate, and N,N-diisopropylethylamine (DIPEA) were all effective, although TAEA was the most general (Table 1, entries 1–5). Lowering the QD loading slowed the reaction rate (entry 6), while control experiments showed that QDs, reductant, and light are all required for dehalogenation activity (entries 7–9). No thermally induced reactivity was observed in the absence of light at 40 °C, an upper bound for the temperature reached by the irradiated reaction with fan cooling (entry 10). Despite their ability to coordinate to QD surfaces, the reductants NaSPh55 and ethylenediaminetetraacetic acid92 were ineffective under these reaction conditions. For additional optimization data, see the Supporting Information (Figure S1).
Table 1.
Optimization of Hydrodechlorination Reaction

| ||
|---|---|---|
| Entry | Deviation from Standard Conditions | Yield of 2aa |
| 1 | None | 95% |
| 2 | 4 equiv TAEA | 68% |
| 3 | 0.25 M | 86% |
| 4 | 4 equiv DIPEA instead of TAEA | 91% |
| 5 | 3 equiv NaCHO2 instead of TAEA | 95% |
| 6 | 0.001 mol% QDs | 74% |
| 7 | No light | 0% |
| 8 | No QDs | 0% |
| 9 | No reductant | 0% |
| 10 | No light, 40 °C | 0% |
| 11 | Ir(ppy)3 (1 mol%) instead of QDs | 2% |
| 12 | Bulk CdS (10 mol%) instead of QDs | 0% |
| 13 | 4 mmol scale, 96 hb | 87% |
| 14 | Recycled QDsc | 84% |
Corrected GC yields using 0.1 mmol of 1a. All quantities are expressed with respect to 1a. LED setups delivered 520 mW of 450 nm light to the reaction vessel.
Reaction conducted with 4 mmol 1a in Penn PhD M2 photoreactor for 96 h.
Reaction conducted using QDs collected from a previous reaction via precipitation and centrifugation. See Supporting Information for procedural details.
Substituting CdS QDs with Ir(ppy)3, among the most-reducing of commonly used molecular photocatalysts, resulted in only trace product formation (entry 11), despite exhibiting a similar reduction potential to CdS QDs (E1/2III/II = −2.19 V vs SCE for Ir(ppy)3; EpcQD/QD– = −2.15 V vs SCE for 3.9 nm CdS QDs)90 and a much longer excited state lifetime (τ = 1.3 μs for Ir(ppy)3 vs ~10 ns for Cd chalcogenide QDs).93–95 While bulk semiconductors have seen increasing utility in photoredox catalysis,96–98 in this case an equivalent mass of bulk CdS powder was completely ineffective (entry 12), showing that the catalyst morphology and quantum properties play a role in dehalogenation activity. Auger processes are vanishingly inefficient in bulk semiconductors,99,100 consistent with the inactivity of bulk CdS powder for photoreduction beyond its reduction potential. The reaction could be performed on gram-scale, although this required high intensity irradiation within a Penn PhD M2 photoreactor over 96 h to reach completion (entry 13). Finally, the QDs could be separated from the reaction mixture via standard benchtop precipitation/centrifugation procedures and reused in another reaction with minimal decrease in reaction rate (entry 14), demonstrating their durability under irradiation and strongly reducing conditions (see Supporting Information for procedural details).
We then sought to compare the durability and potency of CdS QDs as photoreductants relative to a selection of visible light photocatalysts for two-photon and electron-primed photoreduction via the hydrodechlorination of 1a.13,14,22,101,102 While these comparisons are not exhaustive and no comparison of different catalysts optimized under different conditions is without limitations, we did our best to account for the differences in literature conditions by testing three different reductants (TAEA, DIPEA, and NaCHO2) and taking time points at both 24 and 48 h (Table 2 and Supporting Information Figure S2). In addition, to account for the different catalyst loadings and catalyst molecular weights, we calculated total turnover number (TON) and product/catalyst w/w comparisons. Consistent with its high stability,22 4-DPAIPN generally performed best of the tested molecular dyes under these conditions, affording an equal amount of 2a as the QDs when TAEA was used as the reductant (Table 2, entries 1 and 3). However, 0.002 mol% of CdS QDs outperformed all of the tested photoredox catalysts in terms of TON, mass of product formed per mass of catalyst, and generally overall yield across the different organic reductants tested.103
Table 2.
Comparison of CdS QDs with alternate two-photon catalysts

| ||||
|---|---|---|---|---|
| Entry | Photocatalyst | Yield of 2aa | TON (per cat.) | mg pdt. / mg cat. |
| 1 | 5.9 nm CdS QDs (0.002 mol%) | 95% | 47500 | 33 |
| 2 | 4-CzIPN (10 mol%) | 24% | 2.4 | 0.66 |
| 3 | 4-DPAIPN (10 mol%) | 96% | 9.6 | 2.7 |
| 4 | [Ir(dFCF3ppy)2(dtbbpy)]PF6 (2 mol%) | 54% | 27 | 5.3 |
| 5 | PDI (10 mol%) | 16% | 1.6 | 0.59 |
Corrected GC-FID yields vs n-dodecane as internal standard. For experimental details, see Supporting Information Figure S2.
Intrigued by the superiority of TAEA to other amine reductants, we conducted experiments to determine whether TAEA was interacting with the QD surface. As expected for primary amines,104–106 NMR experiments demonstrate that TAEA can displace the native oleate ligands from the QD surface (Figure 2A and Supporting Information Figure S3).67,68,107 Additionally, we observe strong negative NOE correlation between the resonances of TAEA in the presence of QDs, showing unambiguously that TAEA molecules bind dynamically to the QD surface (Figure 2B and Supporting Information Figure S4).108 Based on these results and studies of other polydentate and primary amines, TAEA presumably chelates surface-bound Cd(oleate)2 complexes present on as-synthesized QDs, removing them from the QD and subsequently binding as an L-type ligand to newly exposed Cd sites on the CdS core, yielding TAEA-capped QDs.109,110 Reducing the steric profile of QD ligands has been shown to improve the rate of redox events between QDs and redox partners due to enhanced permeability of the ligand shell,76,111 so one role of TAEA may be to increase surface accessibility of the QDs relative to the larger native oleate ligands. Swapping TAEA for non-coordinating DIPEA generally allowed for similar yields after 24–48 h but resulted in an induction period (>6 h) and less reproducible yields between QD batches (Table 1, entry 4 and Supporting Information Figure S5).
Figure 2.
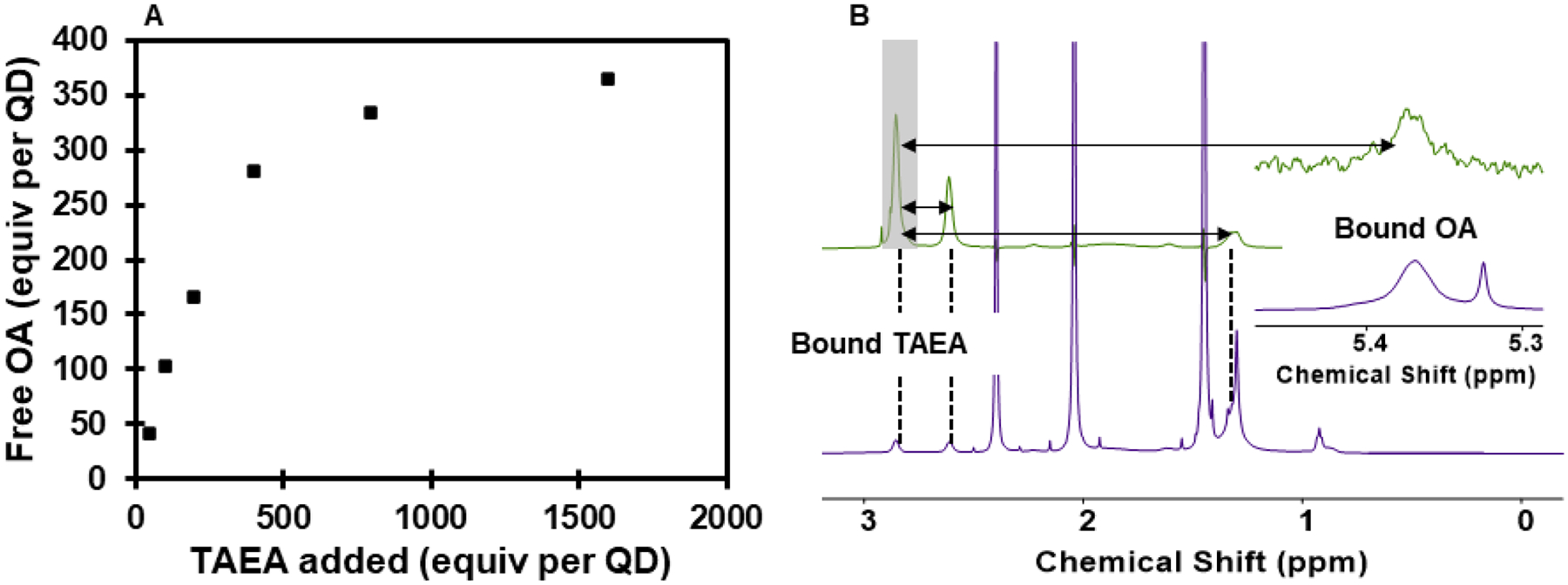
A. Displacement of OA from the surface of CdS QDs (6 × 10−6 mmol) after treatment with TAEA in toluene-d8. See Supporting Information Figure S3 for experimental details. B. Bottom spectrum (purple): 1H NMR spectrum of 6.0 nm CdS QDs (6 × 10−6 mmol) in CDCl3 after treatment with TAEA (6 × 10−2 mmol) and subsequent purification. Additional peaks are residual solvent from the sample preparation procedure. Dashed lines are guides to the eye. Top spectrum (green): Selective 1D NOESY with the 180° pulse applied to the TAEA CH2 resonance at 2.86 ppm (shaded box). Black arrows indicate negative (same-phase) NOE between the pulsed resonance and other signals. Negative NOE is observed with other TAEA resonances and residual surface-bound OA resonances which remain after treatment with TAEA, confirming the presence of TAEA on the QD surface. See Supporting Information Figure S4 for experimental details.
We hypothesize that without TAEA or other added ligands in solution, native oleate ligands slowly desorb from the QD surface under the reaction conditions, a process that occurs spontaneously in dilute QD solutions112 and after negative charging of the QD113 which may be faster or slower between QD batches depending upon variation in exact surface chemistry.109 When using DIPEA as reductant, ligand desorption may be required for substrate or reductant access to the surface,114 accounting for the observed induction period when using DIPEA. Interestingly, lowering the quantity of TAEA from 4 equivalents to 1.5 equivalents substantially increased the rate of product formation allowing for higher yields (Table 1, entries 1 and 2), while the opposite effect was observed for DIPEA (Supporting Information Figure S1), indicating that larger excesses of TAEA may reduce substrate access to the QD surface via competitive surface association.39,51,115 Attempts to observe NOE correlation between protons of DMPU or 1a in the presence of TAEA-capped QDs in CDCl3 did not yield any evidence of substrate or solvent binding to the QD surface (Supporting Information Figure S4). However, the conditions required to sufficiently solubilize TAEA-capped QDs for NMR study of the ligand shell were substantially different than the catalytic reaction conditions, under which solvent quantities of DMPU are likely to interact transiently with the QD surface. The implications of these studies with respect to mechanistic proposals are discussed later.
Mechanistic studies.
We conducted a series of studies to shed light on the mechanism by which CdS QDs catalyzed aryl chloride photoreductions significantly beyond their reduction potentials. We considered five main mechanisms for the reduction of 1a (Figure 3): (A) chloride abstraction by aminoalkyl radicals generated in situ from oxidation of TAEA; (B) reduction of 1a by a photoexcited neutral QD; (C) reduction of 1a by a ground-state anionic QD; (D) reduction of 1a by a hot electron or ionized electron generated by Auger processes; and (E) reduction of 1a by a hot electron generated by direct photoexcitation of a 1Se or a surface-trapped electron to a higher excited state. In the case of mechanisms B-E involving electron transfer to 1a, the product is formed following rapid fragmentation of the nascent radical anion to afford an aryl radical which forms product after hydrogen atom transfer from solvent or reductant.
Figure 3.
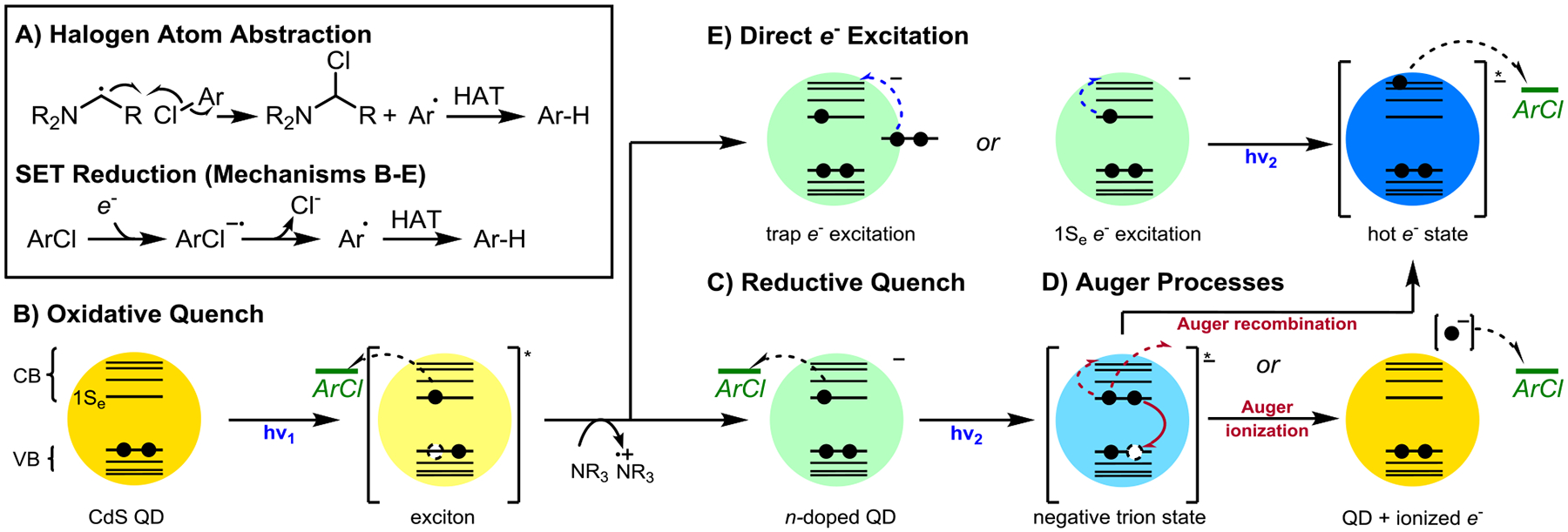
Mechanistic possibilities for aryl chloride reduction. A: Halogen atom abstraction by organic radicals derived from the reductant. B: Reduction of substrate by a neutral excited QD. C: Reduction of substrate by a ground-state anionic QD after reductive quenching. D: Reduction of substrate by a hot or ionized electron generated via Auger processes of a negative trion state. E: Reduction of substrate by a hot electron generated via direct excitation of electrons in the 1Se state or surface trap states.
Mechanism A. Halogen atom transfer.
Our findings are inconsistent with a halogen atom transfer mechanism: we observed that aminoalkyl radicals, generated using the procedure of Leonori from TAEA and sodium persulfate, did not convert 1a to 2a (Supporting Information Figure S6).116,117 This is consistent with Leonori’s findings that aryl chlorides are recalcitrant to halogen abstraction by this mechanism. Furthermore, we found that sodium formate could replace TAEA as a terminal reductant affording the product in nearly identical yield (Table 1, entries 2 and 5). The product of formate oxidation (HCOO•) is known to undergo rapid reaction with excess formate to produce CO2•–, which has been used as an SET reductant for aryl chlorides.14,118 While CO2•– is a strong reducing agent (Ered = −2.2 V vs SCE),119 it is incapable of reducing substrates with more negative reduction potentials than −2.1 vs SCE.14,118 We also note that aminoalkyl radicals, a decomposition intermediate of oxidized amine reductants, are not strong enough reductants to reduce these substrates directly via SET (Eox ~ −1.1 V vs SCE).120 This is consistent with a QD-mediated SET reduction mechanism, regardless of reductant choice.121–123
Mechanism B. Oxidative quenching mechanism.
Our findings are inconsistent with an oxidative quenching mechanism. That mechanism would require the excited-state QD to directly donate an electron to 1a (Figure 3B), but Stern-Volmer quenching studies indicate that aryl chloride 1a did not quench the PL (i.e., 1a is not reduced by the excited state of the neutral QD). Instead, TAEA was found to quench the photoluminescence (PL) of neutral QDs (Supporting Information Figure S7). QD surface modification by TAEA is unlikely to be responsible for the observed PL quenching, because Z-type displacement of Cd(oleate)2 from the QD surface by primary amines is accompanied by amine coordination to exposed Cd sites, which enhances QD PL. Therefore, the observed PL quenching by TAEA indicates a reductive quenching mechanism. This rules out oxidative quenching by 1a as in mechanism B, and suggests the intermediacy of anionic QDs (Mechanisms C-E in Figure 3).
Mechanisms C-E. Intermediacy of anionic QDs.
Mechanisms C-E require the generation of charged QDs that are stable long enough to react with 1a (Mechanism C) or to absorb a second photon (Mechanisms D and E). We were able to generate stable populations of anionic photodoped QDs by irradiation in the presence of the catalytically competent amine reductants DIPEA (Figure 4A) or LiBHEt3 (Supporting Information Figure S8)73,124 under air-free conditions, demonstrating the feasibility of anionic QDs as a catalytic intermediate in these mechanisms. These charged QDs display bleaching of the two lowest energy excitonic features centered at 464 and 448 nm due to occupation of the 1Se electron state at the conduction band (CB) edge.125 Spectral changes at higher-energy transitions were also visible, as consistent with literature reports of doped CdS QDs.126 These photodoped QDs displayed similar spectral changes to those prepared using the established chemical reduction with Na/biphenyl (Supporting Information Figure S9).127 The anionic QDs were stable under nitrogen and were quenched by introduction of oxygen (air). We also observed reversible photodoping of QDs in the absence of reductant, consistent with recent findings that CdS QDs can become photodoped through oxidation of their capping ligands or surface-bound water molecules without added reductants.126,128 While this phenomenon can be used to generate populations of reduced QDs for spectroscopic study, added chemical reductants are required for catalytic transformations. No reduction of 1a to 2a occurs in the absence of terminal reductant (amine or formate) because these reductant-free photodoping pathways cannot provide enough electrons to produce a measurable amount of product in the catalytic reaction (Table 1, entry 9). Additionally, we could observe the 1Se-1Pe transition of the doped electron within the CB of the anionic QDs centered at 1500 cm−1, consistent with other reports of CdS QDs doped with electrons (Figure 5).124,127
Figure 4.
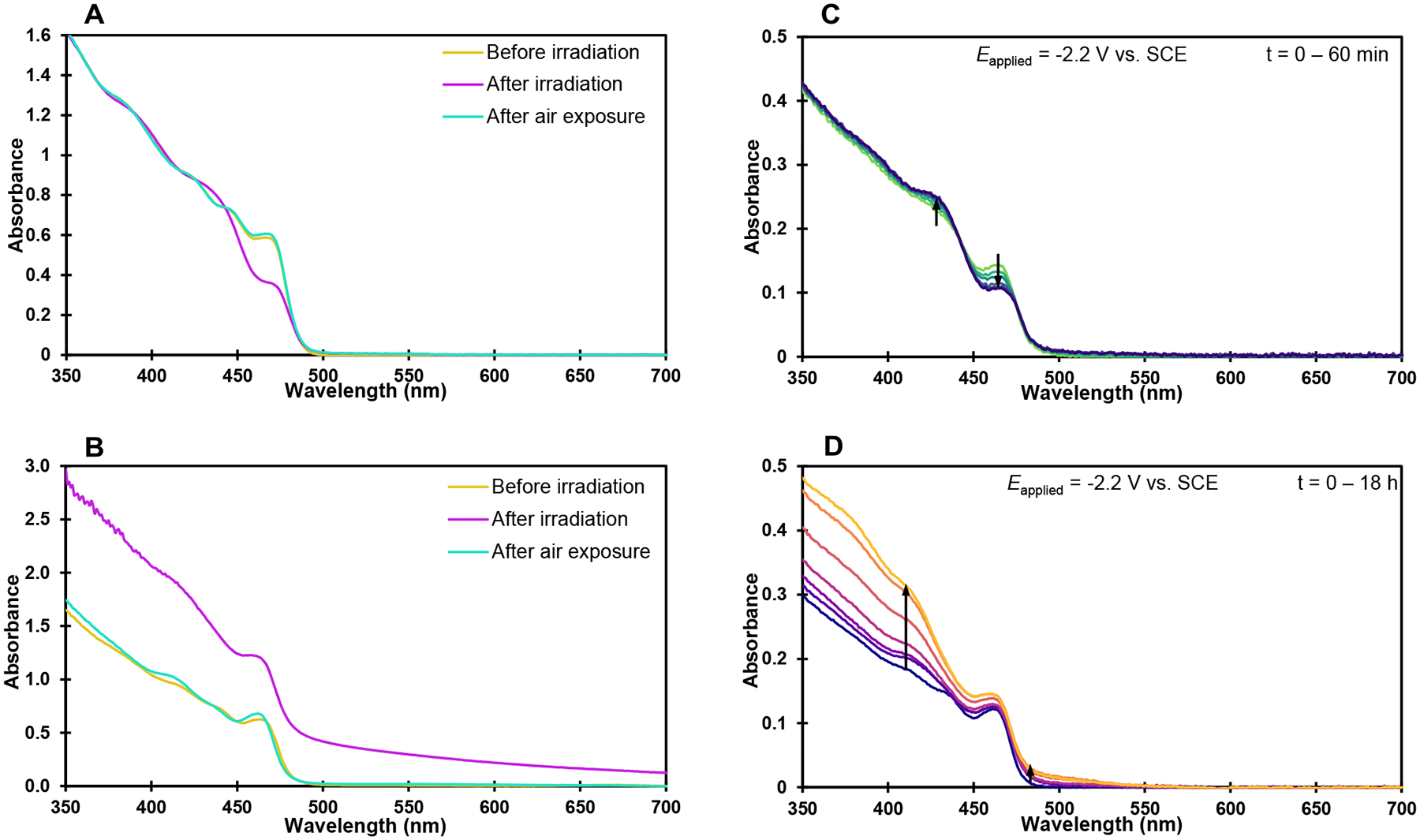
A: Air-reversible photodoping of CdS QDs in toluene by DIPEA (1000 equiv per QD) B: UV-vis spectra of catalytic reaction mixture after 1 h of irradiation employing DIPEA as reductant C: Absorbance changes during electrochemical reduction of CdS QDs; t = 0 min (light green) to 60 min (dark blue). D: Absorbance changes during electrochemical reduction of CdS QDs; t = 0 h (dark purple) to 18 h (light orange). For experimental details, see Supporting Information.
Figure 5.
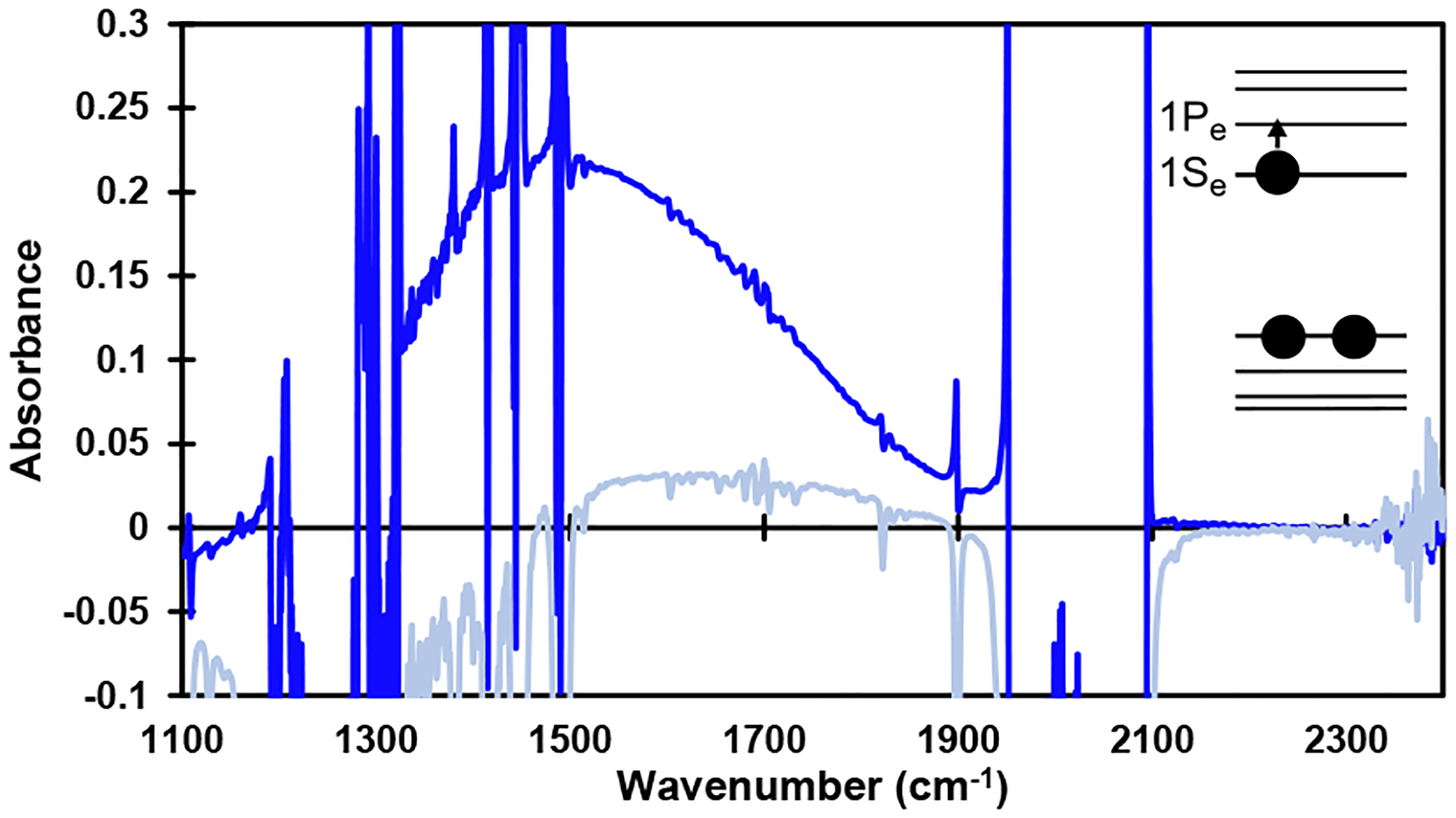
Infrared feature centered at 1500 cm−1 formed upon photodoping of 6 nm CdS QDs, assigned to the 1Se-1Pe transition of the doped electron within the conduction band. Dark blue trace: QDs after photodoping. Light blue trace: QDs after air exposure. See Supporting Information Figure S10 for experimental details.
Slight modification of the catalytic reaction conditions to allow complete solubility of the QDs enabled UV-vis measurement of the reaction mixture to gather information about QD speciation (Figure 4B and Supporting Information Figure S11). Under irradiation with excess reductant, QDs in the catalytic reaction exhibit bleaching of the lowest energy excitonic features, consistent with photogeneration of anionic QDs. Simultaneously, the reaction mixture exhibits a broad absorbance enhancement across the entire visible spectrum (Figure 4B, pink trace), similar to spectral changes reported in core/shell ZnSe/CdS QDs after prolonged irradiation in the presence of excess DIPEA.41 Absorbance tails in the visible spectrum have also been observed following chemical doping of CdSe QDs by Na/biphenyl, ascribed to the broadening and red shifting of the excitonic features by the doped electrons and excitations of electrons in surface trap states.126,129 Light scattering due to QD aggregation may contribute to the broad feature, however both observed spectral changes were fully reversible after exposure to oxygen (Figure 4B, green trace), strongly suggesting that they arise from the presence of injected electrons residing in the CB (the exciton bleaching) as well as newly formed surface states (the additional broad features).113,129
To confirm the origin of these spectral changes within the catalytic reaction, we undertook spectroelectrochemistry studies, as previously employed to study CdSe QDs70,130,131 and deeply reducing photocatalytic systems.14,22 Consistent with reports of CdS band positions, we found that cathodic reduction of 5.9 nm CdS QDs at −2.2 V vs SCE was sufficient to electrochemically dope the QDs with ~0.5 electrons per QD within 1 h (based on the magnitude of absorbance bleaching of the 1Se feature at 464 nm),73 mirroring exactly the spectral changes observed in photodoping experiments (Figure 4C, green to dark blue traces and Supporting Information Figure S12). The excitonic bleaching was accompanied by shifting of the higher energy absorbance features as observed in photodoped QDs (compare to Figure 4A).
When the experiment was repeated at a longer time scale, the excitonic bleaching was accompanied by a sub-band gap absorbance tail as well as further shifting and broadening of QD features between 350 and 450 nm throughout the electrolysis (Figure 4D, purple to orange traces), increasing in intensity over time, resembling the spectral changes previously observed in the catalytic reaction (Figure 4B). Sub-bandgap absorbance tails in the visible in good agreement with those observed in our experiments (Figure 4D) have been documented in spectroscopic studies of other reduced Cd-based QDs.113,124,127,129,132–134 They are commonly attributed to the spontaneous reduction of surface sites (namely Cd2+ ions in Cd-rich QDs) by electrons in the 1Se state, or directly by the reducing agent.113,135 Reduction of the QD surface atoms leads to the introduction of surface-localized trap states filled with electrons with energy levels inside the band gap.113,136 The presence of electrons in surface states introduces localized dipoles that interact with the polarizable QD exciton via the Stark effect, causing broadening of excitonic absorbance and PL features137,138 when QDs are reduced by various means, potentially contributing to the observed absorbance tail. Doped electrons within the 1Se state or surface states may also exhibit transitions to higher energy states within the CB which could contribute to the observed features,113,129,134 however the infrared feature corresponding to the 1Se-1Pe transition (Figure 5) occupies most of the oscillator strength of the doped 1Se electrons.129
These studies strongly suggest that sequential photodoping cycles139 during the catalytic reaction generate QDs with populations of electrons in the 1Se state and nascent mid-gap surface states which may form due to in-situ QD surface modification.135,140 We observed that the QD solutions remained optically clear throughout the spectroelectrochemistry experiments and note that the electrolysis-induced shifts in the QD absorbance features do not resemble wavelength-dependent Rayleigh scattering (scattering cross section ∝ λ−4), suggesting that light scattering did not majorly convolute the absorbance spectra (see Supporting Information Figure S12 for images of QDs during the experiments).
Mechanism C. Direct reduction of 1a by anionic QDs.
Our findings are inconsistent with reduction of 1a by anionic QDs without additional photon energy (Mechanism C). Electron-primed photoredox catalysis was used as a mechanistic tool to decouple QD doping from photoexcitation.18,19,22 We found that reactions utilizing a sacrificial anode (Mg(+)/RVC(−), −2.2 V vs SCE), instead of organic reductants, formed product 2a only when both QDs and light were present (Table 3, entries 1–4). Because of the notably short lifetimes of photoexcited organic radical ions, it has been proposed that thermally induced electron transfer from a ground-state reduced catalyst species (requiring only a single photon and reductant to generate the reduced catalyst) may be the operative mechanism of reduction for some substrates in reports of two-photon chemistry.16 The necessity of light in our electron-primed photoredox studies (Table 3, entries 3–4) cleanly demonstrates that electrochemically generated anionic QDs do not reduce 1a via thermally activated electron transfer at room temperature or 40 °C, an upper bound for the temperature reached by the reaction vessel under irradiation with fan cooling. We also observed further evidence that TAEA is serving as both a reductant and QD-stabilizing ligand:141 QD decomposition could be observed over the course of the experiments, resulting in diminished yields relative to the ordinary photocatalytic reaction (Table 1, entry 1 vs Table 3, entry 1). These results demonstrate that anionic QDs must be photoexcited to reduce 1a at the employed temperatures, ruling out mechanism C and implicating the generation of hot or ionized electrons higher in energy than the CB 1Se state.
Table 3.
Electron-Primed Photoredox Studies.
| ||
|---|---|---|
| Entry | Conditions | Yield of 2aa |
| 1 | 456 nm LED lamp | 65% |
| 2 | 456 nm LED lamp, no QDs | 0% |
| 3 | no light | 0% |
| 4 | no light, 40 °C | 0% |
| 5 | 525 nm LED lampb | 9% |
| 6 | 650 nm LED lamp | 0% |
Corrected GC yield for reactions with 0.2 mmol 1a. Average power consumption of LED lamps was 50 W. See Supporting Information for experimental details.
500 nm long-pass filter employed to prevent overlap with neutral exciton.
Mechanisms D and E. Two-photon mechanisms.
The necessity for irradiation of electrochemically generated anionic QDs to form product confirms the need for multiple photons per turnover in the purely photochemical reaction (Mechanisms D and E). Our photodoping and spectroelectrochemistry studies established that modification of the QD surface in tandem with continuous electron injection into QDs results in a population of electrons in the 1Se state as well as surface-localized trap states, resulting in excitonic bleaching and an absorbance tail. To determine whether selective excitation of the absorbance tail could induce product formation, we employed electron-primed photocatalysis using a 525 nm light source with a 500 nm long-pass filter to selectively irradiate the absorbance tail not present in the neutral QDs, leading to small amounts of product formation (Table 3, entry 5), consistent with the low absorbance of the doped QDs at >500 nm wavelengths. Finally, employing a 650 nm light source led to zero product formation (Table 3, entry 6), due to the negligible absorbance of QDs at red wavelengths under an applied potential of −2.2 V vs SCE. The photoreduction of 1a by anionic QDs is most effective when the QDs are irradiated at the bandgap transition, illustrating the importance of bandgap excitation of anionic QDs in order to access the active reducing species.
Mechanisms D. Evidence for Auger processes from PL measurements.
The main difference between mechanisms D (involving Auger processes) and mechanism E (direct excitation of electrons at non-bandgap transitions) concerns the nature and fate of the excited state formed after excitation of anionic QDs. We conducted steady state and time-resolved photoluminescence measurements which demonstrate that Auger recombination or ionization (Mechanisms D) occurs following irradiation of photodoped anionic CdS QDs, which form under our catalytic conditions. It is well known that bandgap excitation of anionic QDs initially produces negative trion states, which exhibit characteristic differences from the excited states of neutral QDs.73,77,142 Neutral excitons and negative trion states both undergo radiative decay, however the availability of fast nonradiative Auger pathways to negative trion states results in their diminished photoluminescence relative to the neutral exciton. We observe drastic steady-state PL quenching of QD samples following photodoping by amine reductants (Figure 6A and Supporting Information Figure S13), which was partially reversible after air exposure, a finding previously assigned to efficient Auger recombination of negative trions formed by excitation of similar photodoped anionic QDs.124 Analysis of time-resolved PL decay of QDs before and after photodoping (Figure 6B) shows multiexpoential decay in all QD samples, consistent with other reports of CdS QDs (Supporting Information Figure S14).126 Before photodoping, the QD sample exhibited decay components of τ ~ 16 ns, assigned to radiative decay lifetimes measured in similar neutral QDs, as well as faster decay components of τ ~ 1 ns. After photodoping, the τ ~ 16 ns component is nearly eliminated, resulting in an approximate threefold decrease in the average PL lifetime of the QDs (see Supporting Information Figure S14 for fitting data). This is indicative of Auger events: anionic QDs exhibit shorter fluorescent lifetimes than neutral QDs because nonradiative Auger recombination or ionization of negative trion excited states occurs more rapidly than fluorescent decay, effectively outcompeting fluorescence in photodoped QDs. After oxygen exposure, partial recovery of the neutral PL decay kinetics is observed, due to partial scavenging of electrons from doped CdS states, while surface-modification of the QDs may prevent full recovery of the initial PL dynamics. Our PL data are fully consistent with reported indications of Auger processes in photodoped QDs,74,124,126 demonstrating the occurrence of this mechanism under our conditions.
Figure 6.
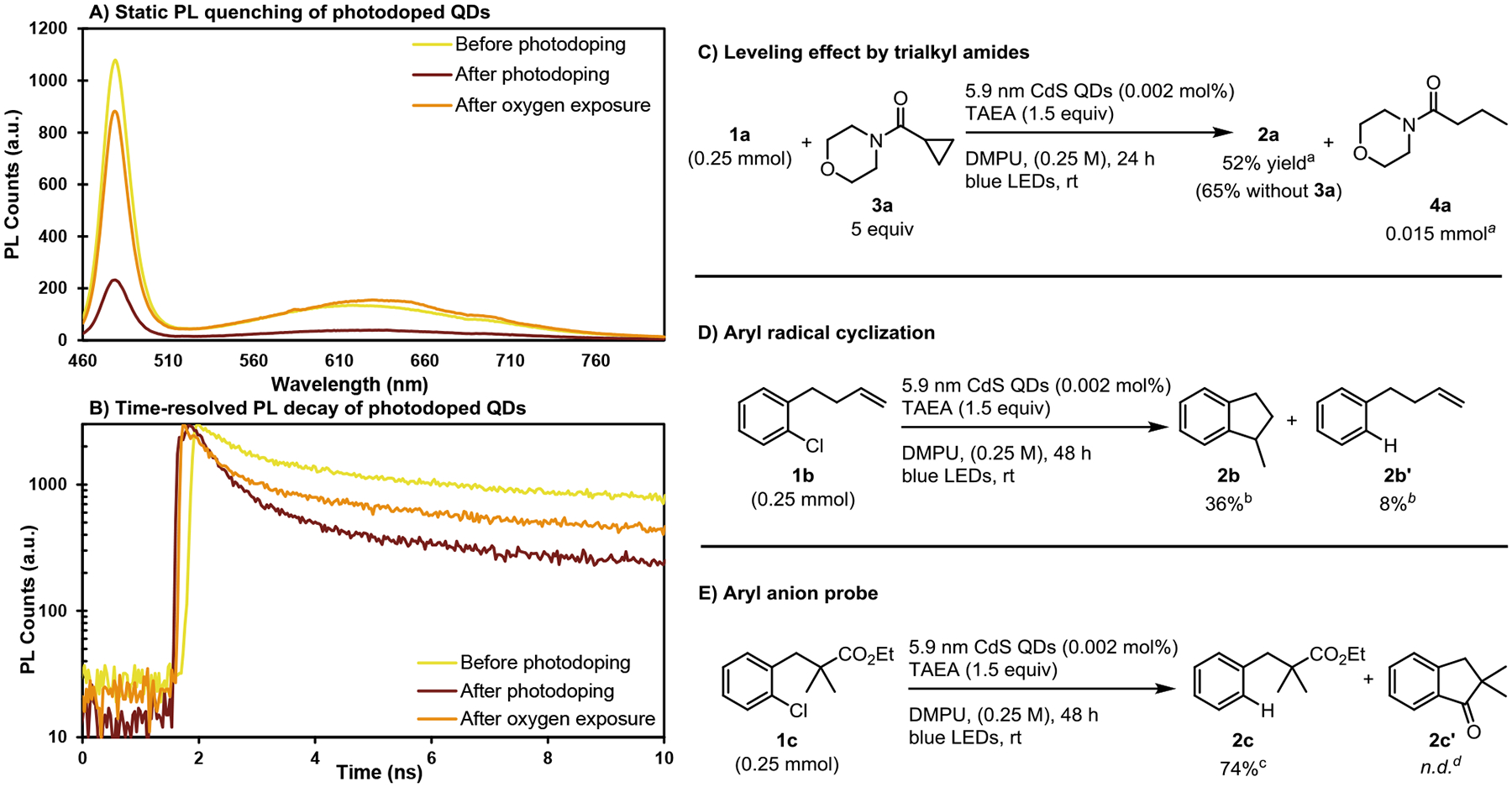
A) PL quenching in photodoped CdS QDs. B) Time-resolved PL decay of photodoped CdS QDs. C) Inhibition of reduction by amides. D) Cyclization of radical clock E) Probe for over-reduction of aryl radicals to anions. a Corrected GC-FID yields vs n-dodecane. b 1H NMR yields vs CH2Br2. c Isolated yield. d No indanone cyclization product was detected via SFC-MS. See Supporting Information for experimental details.
Potential role of DMPU solvent.
The inferior performance of amide solvents and other aprotic solvents relative to DMPU led us to consider that a solvent reduction mechanism could be operative, potentially assisting the extraction of highly reducing electrons from the QD before their relaxation (e.g., formation of DMPU•– or DMA•–, species that have been observed when alkali metals are dissolved in these solvents and which could act as redox mediators to shuttle electrons to the substrates).143,144 This could impose a solvent-dependent leveling effect on the potentials accessible by the QDs/TAEA system. The different performance would arise from differences in the reducing power of the respective radical anions because ureas like DMPU are harder to reduce to the corresponding radical anion than other carbonyl derivatives.145–147 To explore this possibility, we tested the photoreduction of 1a using DMPU as solvent in the presence of amide 3a, which we reasoned could act as a probe for the reduction of amides to amidyl radical anions. We found that the addition of 3a reduced the yield of dehalogenation product 2a, while 0.015 mmol (6 mol% relative to 1a) of ring-opened amide 4a was formed. (Figure 6C, Supporting Information Figure S15). While we cannot state with certainty that DMPU radical anions play a role in productive chemistry, these results demonstrate that tertiary amides can be reduced to the corresponding radical anion and inhibit productive catalysis.
Aryl radical and aryl anion probe substrates.
While the dehalogenation reactions were assumed to arise from aryl radicals and hydrogen atom transfer, an alternative mechanism would be sequential reduction to form an aryl anion followed by proton transfer. Aryl radicals are easier to reduce than aryl halides (Ered = +0.05 V vs SCE for phenyl radical), so we considered that multiply charged QDs could potentially reduce aryl radicals to anions, as has been observed under electrochemical reduction conditions.148 To differentiate these two mechanisms, we examined the products formed from reactions with radical clock substrate 1b in the QDs/TAEA system (Figure 6D, Supporting Information Figure S16). The cyclized product 2b was observed in a 4.5:1 ratio to the uncyclized olefin 2b’, consistent with the formation and facile cyclization of aryl radicals from 1b (kcyc = 5 × 108 s−1 at 25 °C). Recognizing that the uncyclized product could arise either by reduction of intermediate aryl radicals to the corresponding anion followed by protonation, or by HAT from solvent or reductant molecules prior to cyclization, we employed 1c as an aryl anion probe (Figure 6E).149 After reduction by QDs/TAEA, 2c was obtained as the exclusive product with no formation of the indanone product 2c’ via anionic cyclization. These results together provide strong evidence that aryl anions are not formed under the reaction conditions, and that intermediate aryl radicals are rapidly quenched via HAT from solvent or reductant molecules (pseudo-first order rate constant kHAT ~ 1 × 108 s−1).
Kinetic Dependence on Light Intensity.
Kinetic studies of the reaction under differing light intensity indicate an approximate rate order of 1.4 for photons, as measured by the initial rates of reaction over the first 5 h. A ~30 min induction period is also observed, which appears to be longer with lower light intensity (Supporting Information Figure S17). Z-type surface modification of Cd chalcogenide QDs by chelating L-type multidentate ligands like TAEA proceeds to completion within a few minutes,109 so the induction period is likely not due to surface modification by TAEA. Combined with the prior observation that anionic QDs accumulate in the reaction mixture through photodoping cycles, these results are consistent with buildup of the active catalyst,6 multiply anionic QDs, through photodoping at the beginning of the reaction, followed by a regime of rate-limiting hot electron transfer from photoexcited anionic QDs to substrate or solvent molecules, as would be expected given the rapid relaxation rate of hot electrons to the band edge before reduction occurs.
Proposed Mechanism.
Based on these studies, we propose the following reduction mechanism (Figure 7): neutral QDs (I) become negatively charged after excitation and reductive quenching by TAEA to generate anionic QDs (II). II can then absorb a second photon to generate a negative trion state (III), which can undergo reductive quenching by TAEA again to return to a ground state anionic QD with an additional negative charge (II), or undergo Auger processes to generate a hot-electron state or an ionized electron (IV), which may then relax nonradiatively back to (II) (via back electron transfer in the case of an ionized electron), or reduce a substrate or solvent molecule to return to a neutral or anionic ground state (I or II). Reduced substrates then undergo subsequent fragmentation and HAT to furnish the dehalogenated product. We also expect that back electron-transfer from reduced substrate or solvent molecules to QDs may occur competitively with productive chemistry.
Figure 7.
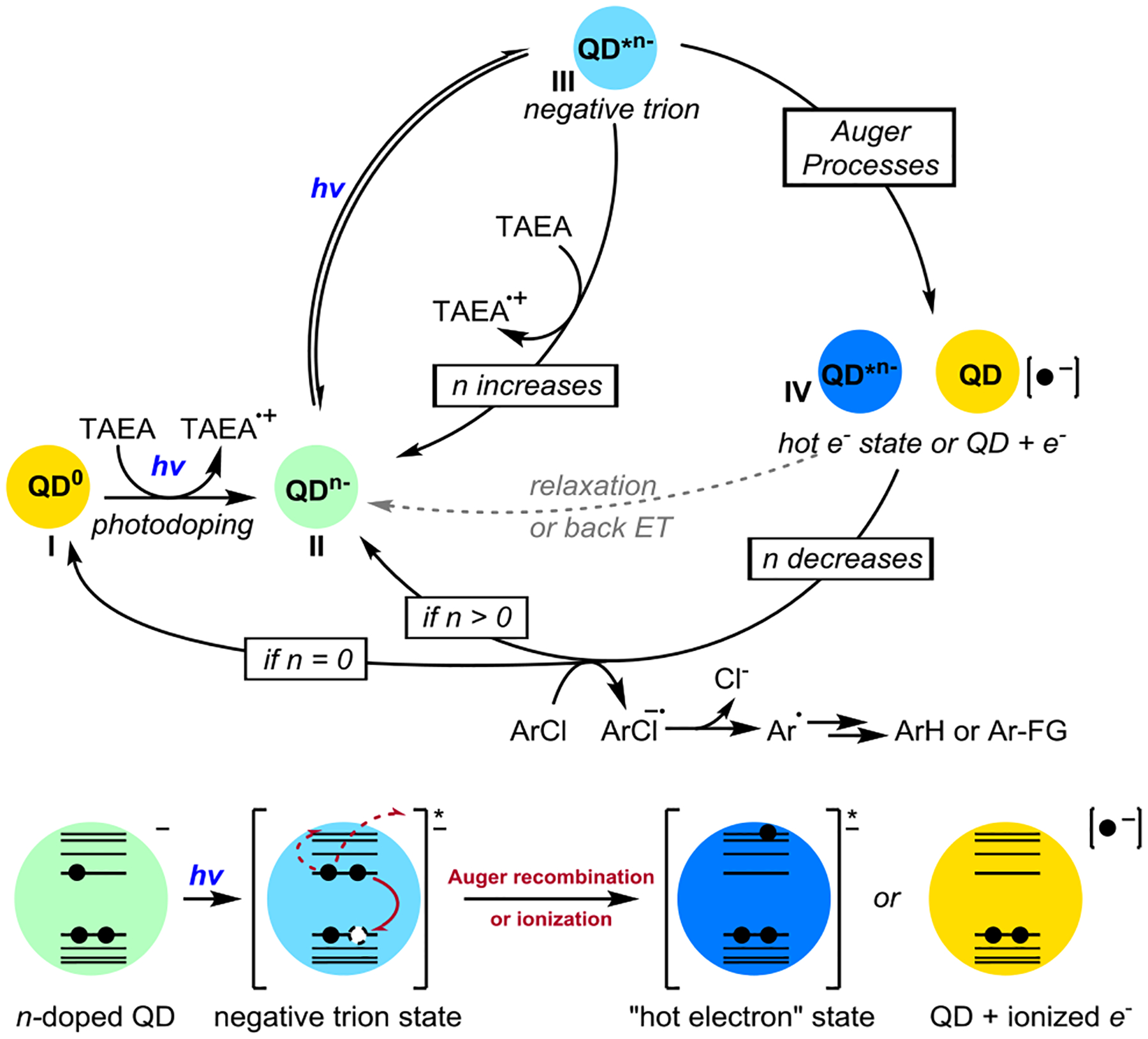
Proposed mechanism of photoreduction.
Application to organic reactions.
To explore the synthetic utility of the CdS/TAEA system, we briefly explored the generation of aryl radicals from aryl chlorides and aryl phosphate esters (Scheme 1). For the hydrodechlorination reaction (2a, 2d-2h), we found that a set of electron-rich and electron-neutral aryl chlorides could be hydrodehalogenated in high yields, while electron-rich aryl phosphate esters could be reductively cleaved to afford the arene in similar yields. Semiconductor QDs have been previously employed for reductive dehalogenation of aryl halides,37,41,150 however these protocols have been limited to substrates with reduction potentials less negative than the reduction potential of the QDs. In our system, reductions of aryl electrophiles with reduction potentials significantly more negative than CdS QDs (−2.2 V vs SCE)90 proceeded in high yields. Considering that we were using organic terminal reductants including amines (TAEA, DIPEA) and formate, we found it promising that a substrate bearing an oxidizable secondary amine (phosphate ester 5h) could be defunctionalized in 47% yield. Because a variety of aliphatic amines can serve as terminal reductants (Supporting Information Figure S1), we think this suggests that reductant pre-binding to the QD may outcompete more hindered amines.
Scheme 1.

Reaction scope for aryl radical generation.
aStandard conditions used: 5.9 nm CdS QDs (0.002 mol%), TAEA (1.5 equiv), DMPU (1 mL), blue LEDs, rt. Isolated yields unless otherwise specified. b4 mmol scale, conducted in a Penn PhD M2 photoreactor (See SI for details). cDIPEA (4 equiv) used instead of TAEA. dNaCHO2 (3 equiv) used instead of TAEA. eNMR yield vs CH2Br2.
To explore whether the aryl radicals formed could be used for C-C and C-X bond formation, we examined trapping reagents to furnish products 6, 7, and 8 in moderate yields, comparable to those reported using other aryl radical trapping conditions.19,27,41 No reduction of the ester groups was observed, with hydrodehalogenation and starting material accounting for the remaining mass balance. Consistent with the propensity of aryl radicals to undergo HAT with weak C-H bonds present on amine reductants and DMPU, we observed that employing DMSO as solvent and sodium formate as the terminal reductant for aryl radical trapping generally improved the selectivity for the desired products over hydrodehalogenation.14 We anticipate that further improvements could be made with additional optimization: the use of DMSO instead of DMPU was required for selective aryl radical functionalization but led to lower conversion, especially for electron-rich aryl electrophiles.
In addition to aryl radical formation from aryl chlorides, we also explored several other reductive transformations that require strong reductants (Scheme 2). The detosylation of p-toluenesulfonamides to amines is a common, but challenging, transformation that typically requires superstoichiometric strong reductant (e.g., SmI2,151,152 Li/Naphthalene153, or Mg/MeOH154) or anhydrous strong acid (e.g., TfOH, HBr in AcOH).155 Photocatalytic reductive cleavage was only recently reported using an acridinium catalyst (10 mol%) with UVA light (390 nm).101 This was an exciting advance because many methods rely upon tosyl-protected nitrogen, but are of limited utility due to the harsh deprotection conditions.101 In our initial examination of this reaction, several p-toluenesulfonamides were reductively cleaved to afford the free amines in 68–88% yield. Arylamine 9a and melatonin-derived 9b were completely deprotected within 24 hours, while alkylamines 9c and 9d were slower and required 72 h, presumably due to their more-negative reduction potentials (Ered ~ −2.4 V vs SCE for N-tosyl alkylamines),156 or higher propensity for back-electron transfer before fragmentation. Notably, sulfonyl-protected phenols have been reductively deprotected using CuInS2/ZnS QDs as a photocatalyst,53 however only electron-poor sulfonyl groups with reduction potentials less-negative than the employed QDs were cleaved under these conditions, in contrast to this study with CdS QDs.
Scheme 2.
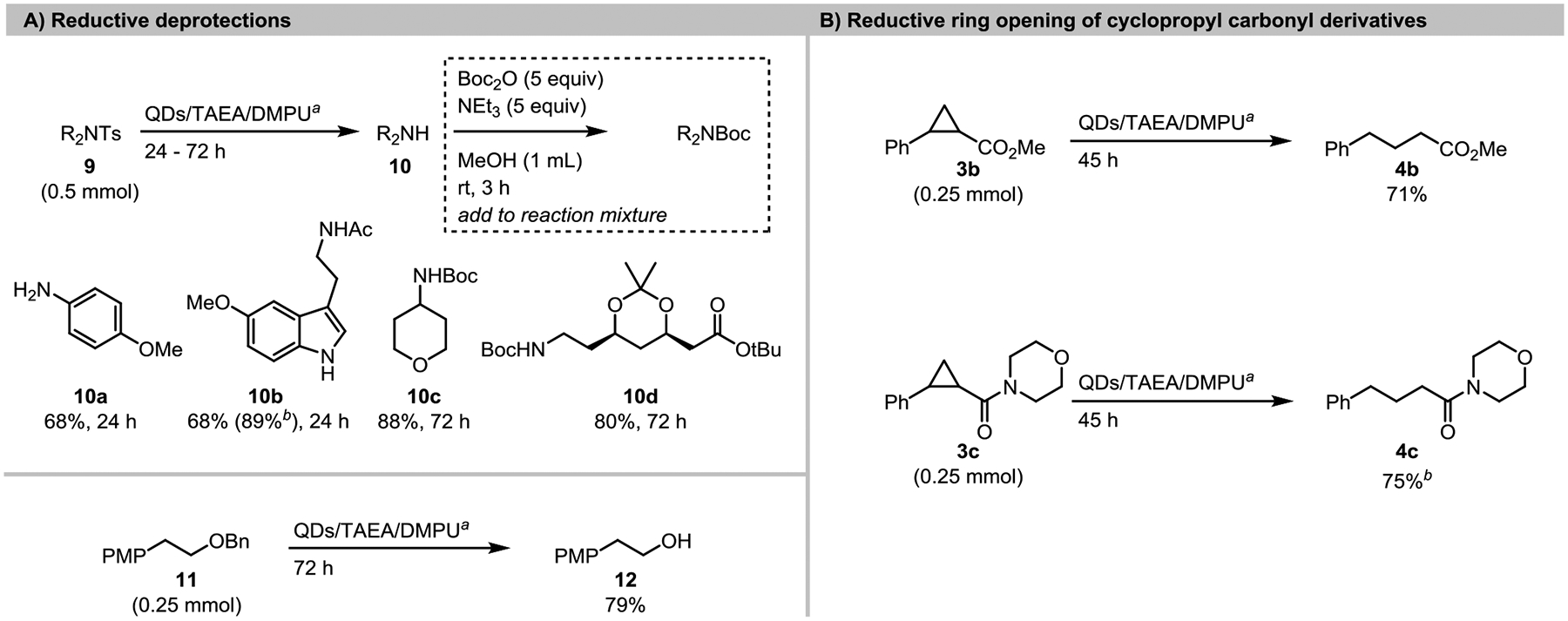
Additional reductive transformations.
aStandard conditions used: 5.9 nm CdS QDs (0.002 mol%), TAEA (1.5 equiv), DMPU (1 mL), blue LEDs, rt. Isolated yields unless otherwise specified. bNMR yield vs CH2Br2.
Besides tosylate deprotection, these conditions were also able to deprotect alkyl benzyl ether 11 to alcohol 12 in high yield. Debenzylation is commonly accomplished through Pd-catalyzed hydrogenation. However, in situations where hydrogenolysis is incompatible with other substrate functionalities, it may also be accomplished by strong stoichiometric reductants157–160, electroreduction (Ered = −3.1 V vs SCE)156, or the combination of an organic reductant and UV light.161 This approach allows clean deprotection to proceed under visible light irradiation with amine terminal reductants.
Finally, the reductive C-C bond cleavage of cyclopropyl ester 3b and amide 3c bearing beta phenyl groups could be achieved in high yield via reduction of the ester (Ered = −2.8 V vs SCE)162 or amide163 functionalities to the corresponding ketyl radical anion, followed by ring-opening to afford the distal benzylic radical anion. While the reductive ring opening of aryl cyclopropyl ketones is well-known in photoredox catalysis164–166 (Ered = −2.10 V vs SCE for phenyl cyclopropyl ketone),167 only one example exists of a photocatalyzed reductive ring opening of a more electron-rich cyclopropane carboxylic acid.15 Reductive ring openings of the analogous cyclopropanecarboxyl esters and amides have only been achieved by employing excess SmI2 activated by H2O, where care must be taken to avoid over-reduction to the alcohol.168 Reductive ring openings of cyclopropyl ketones have enabled many valuable transformations including mono169 and di-functionalizations,164,165,170–176 so we anticipate that further development of these ester and amide ring opening protocols will enable more complex transformations.
Discussion
Evidence for Dominant Auger Mechanisms.
While we have not yet directly characterized the active reducing state enabling these photoreductions, our results are most consistent with a dominant Auger mechanism (Mechanisms D). Spectral measurements of our CdS QDs after doping procedures and under various reaction conditions are consistent with the in-situ generation of anionic QDs with electron occupation of the 1Se state and surface states. After photochemical or electrochemical doping of QDs, the excitonic feature of the reduced QDs accounts for most of the absorbance of the reaction mixture at blue wavelengths. Excitation of anionic QDs produces negative trion states which are known to efficiently undergo Auger processes, generating hot or ionized electrons. This was confirmed by our ex-situ photoluminescence studies of anionic CdS QDs, which were consistent with literature reports of negative trion states undergoing Auger recombination,73,126 demonstrating the intermediacy of this process within our system. Importantly, surface modification of QDs and the introduction of electrons in trap states, which occur under our reaction conditions, do not interfere with Auger events.79,177,178 While the exact degree of negative charging of the QDs under purely photochemical conditions remains unknown, excited QDs with multiple negative charges are known to undergo Auger recombination faster than monoanionic QDs and produce longer-lived hot electrons.177 We propose that Auger processes to generate highly reducing electrons could reasonably happen from a variety of anionic QD species that may be present in the reaction. Electron-primed photoredox catalysis enabled the direct study of anionic QDs: irradiation of QDs and substrate with 456 nm light in tandem with electrochemical reduction at −2.2V vs SCE leads to effective reduction of substrate 1a, requiring QDs, light, and current to achieve any conversion. Critically, no reduction of 1a by electrochemically produced anionic QDs occurred in the absence of light at room temperature or 40 °C, ruling out a thermally activated mechanism. Employing >500 nm light to selectively excite the absorbance tail forms small amounts of product, showing that irradiation of this feature can also generate excited electrons. However, the low absorbance of this feature compared to the exciton of the doped QDs suggest that Auger processes are the dominant mechanism of electron excitation.
Electron Excitation via Auger Recombination vs Auger Ionization.
Our data are consistent with Auger events occurring from the negative trion state formed upon photoexcitation of anionic QDs, producing highly reducing electrons to drive the observed chemistry. However, we cannot currently differentiate between the prevalence of Auger processes that would lead to a hot electron at a high energy level within the conduction band or result in direct ejection of an electron from the QD into the solvent. A significant challenge of two-photon and electron-primed photoreductions is the short-lived nature of the highly reducing excited state. Rapid unimolecular relaxation or decomposition processes of excited-state radical anions typically limit the efficiency of bimolecular electron transfer to substrates.25,179 Indeed, the notably short lifetimes of excited state radical anions (typically on the order of ps) have raised questions about the active photoreductants in these systems.17,25 For example, a photoexcited catalyst species with an excited state lifetime of 10 ps will undergo productive diffusion-controlled quenching with a substrate with only 1% efficiency (1 productive event per 100 photoexcitations) at a substrate concentration of 0.25 M.6 In the case of Auger recombination, rapid relaxation of hot electrons in CB states to the band edge would present such an obstacle. The longest-lived hot electrons in semiconductor QDs exhibit lifetimes near 1 ns in core-shell QDs,177,180 however they typically relax back to the band edge within picoseconds, depending on the QD surface chemistry.85,181–183 One advantage of employing QDs in this context is their ability to bind organic molecules as ligands (up to several hundred per CdS QD). Pre-association between substrates and the QD surface could help bypass the kinetic obstacles associated with highly reducing but short-lived excited states. The ps-scale lifetimes of hot carriers in QDs, while insufficient for efficient collisional electron transfer, are sufficiently long for redox events with surface-bound or nearby species, enabling ubiquitous hot carrier chemistry in nanomaterials.76,84,85,88,184–186 Many classes of weakly coordinating molecules, including solvent, are capable of transient association with the QD surface through dispersion, electrostatic, or other noncovalent interactions.51,115,141 In principle, this could allow the observed reductions to take place through ultrafast charge transfer to adsorbed substrate or solvent molecules competitive with hot electron cooling. The observation that increasing the amount of TAEA in the catalytic reaction slows the reduction of 1a suggests that surface accessibility is important, which would be consistent with a surface-mediated reduction. While we have not yet directly observed interaction between the QDs and substrate or sub-solvent quantities of DMPU via NOE spectroscopy (Supporting Information Figure S4), significant deviation from the reaction conditions required for QD solubility in these NMR experiments makes it difficult to conclude that such interactions do not occur under the catalytic reaction conditions. The solvent quantities of DMPU present in the reaction mixture and the low steric profile of TAEA as a capping ligand render it likely that some DMPU molecules are associated with the QD surface at any given time to accept sufficiently reducing hot electrons, however we cannot currently observe this interaction. The alternative mechanistic possibility of Auger ionization, in which a CB electron is excited directly to a substrate or solvent molecule outside of the QD rather than a high-energy CB state, has been observed in many QDs in the solid state and in solution.82,187 This could be viewed as a more kinetically viable pathway due to the longer lifetime of solvated electrons (ns and longer) than hot electrons within QD states,188,189 and is also consistent with the observed solvent dependence. Differentiation between these closely related mechanistic possibilities is an area of ongoing study.
Potential of QDs for Reductive Chemistry.
These studies demonstrate that semiconductor quantum dots hold promise as versatile, robust visible light photoredox catalysts for strong reductions. The potentials accessible are already strong enough to be competitive with the best available catalysts (Table 2), and the total turnover numbers for the catalysts are an order of magnitude better than previously reported for visible light photoreductions of electron-rich aryl chlorides. The generality of this catalyst is also beneficial; one quantum dot (5.8–6.0 nm oleate-capped CdS QDs) worked for a variety of reactions (hydrodefunctionalization, heteroarylation, borylation, and stannylation of aryl chlorides; deprotection of tosyl-protected amines and benzyl-protected alcohols, and ring-opening of cyclopropanecarboxylate derivatives). While the MW of these CdS QDs is high (approximately 330 kDa of CdS and 140 kDa of oleic acid ligands for 6.0 nm CdS QDs), their productivity is high even on a mass product/mass QD scale (Table 2 and Supporting Information Figure S2). In contrast to most other small molecule dyes, CdS QDs are made in a single step process that does not require chromatography from materials that are very low cost (approximately $13 USD per gram of isolated QDs based on material costs for the described synthesis; ~2 mg of QDs are used per 0.25 mmol-scale reaction). At the end of the reaction, QDs can be easily removed from reaction media via precipitation and centrifugation to enable reuse, or easily disposed of via filtration through silica or dissolution in acid. Although cadmium is tightly regulated in drug products, we found previously that precipitation of CdSe QDs, with or without additional purification, leads to amounts of Cd in the product that are within allowed limits.37 Together, these results demonstrate how simple colloidal nanoparticles can offer advantages over small molecule molecular dyes. Looking towards the future, tailored nanomaterials optimized for catalytic performance are likely to perform even better, new materials with lower toxicity could be explored for organic chemistry,190 and surface association could be further exploited for selectivity.28,40,115
Conclusions
We have demonstrated the potential of CdS QDs with mild organic reductants and visible light (450 nm) to function as strong photoreductants for a variety of organic transformations which require strong reducing agents. Mechanistic studies implicate a similar process to reported two-photon mechanisms, wherein neutral QDs become negatively charged through excitation and reductive quenching by the terminal organic reductant. Hot or ionized electrons are then generated via excitation of the anionic QDs and subsequent Auger processes proceeding from the negative trion state. Advantages of this approach include the ease of catalyst synthesis and the high stability of CdS QDs under strongly reducing conditions, with turnover numbers of up to 47500 (per QD) achieved in a single reaction under photochemical conditions or 13000 for electron-primed photoredox catalysis. Further explorations of reductive transformations catalyzed by CdS QDs will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (R21GM141622 to DJW and TDK), the NSF (CHE-1904847 to TDK), the University of Wisconsin-Madison (DJW), and the donors to the Wayland E. Noland Chair (DJW). The instrumentation in the PBCIC was supported as follows: Thermo Q Extractive Plus was supported by the NIH (No. 1S10 OD020022); Shimadzu GCMS-QP2010S was supported by the Department of Chemistry; Bruker Avance III 500 was supported by a generous gift from Paul J. and Margaret M. Bender. We thank the entire Wickens and Yoon groups (UW-Madison) for access to their chemical inventories, Colleen Chernowsky (UW-Madison) for assistance with electron-primed photoredox catalysis, and the Stahl group (UW-Madison) for access to UV-vis instrumentation. We thank Dr. Alina Schimpf (University of California San Diego) for helpful discussions about FTIR measurements.
Footnotes
Supporting Information. Additional optimization and characterization data, experimental procedures, and characterization data for all isolated compounds (PDF) This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (2).Yoon TP; Ischay MA; Du J Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem 2010, 2, 527–532. [DOI] [PubMed] [Google Scholar]
- (3).Glaser F; Wenger OS Recent progress in the development of transition-metal based photoredox catalysts. Coord. Chem. Rev 2020, 405, 213129. [Google Scholar]
- (4).Mdluli V; Diluzio S; Lewis J; Kowalewski JF; Connell TU; Yaron D; Kowalewski T; Bernhard S High-throughput Synthesis and Screening of Iridium(III) Photocatalysts for the Fast and Chemoselective Dehalogenation of Aryl Bromides. ACS Catal 2020, 10, 6977–6987. [Google Scholar]
- (5).Qiao Y; Schelter EJ Lanthanide Photocatalysis. Acc. Chem. Res 2018, 51, 2926–2936. [DOI] [PubMed] [Google Scholar]
- (6).Glaser F; Kerzig C; Wenger OS Multi-Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods. Angew. Chem. Int. Ed 2020, 59, 10266–10284. [DOI] [PubMed] [Google Scholar]
- (7).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Speck F; Rombach D; Wagenknecht H-A N-Arylphenothiazines as strong donors for photoredox catalysis – pushing the frontiers of nucleophilic addition of alcohols to alkenes. Beilstein J. Org. Chem 2019, 15, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Discekici EH; Treat NJ; Poelma SO; Mattson KM; Hudson ZM; Luo Y; Hawker CJ; de Alaniz JR A highly reducing metal-free photoredox catalyst: design and application in radical dehalogenations. Chem. Commun 2015, 51, 11705–11708. [DOI] [PubMed] [Google Scholar]
- (10).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kim D; Teets TS Strategies for accessing photosensitizers with extreme redox potentials. Chem. Phys. Rev 2022, 3, 021302. [Google Scholar]
- (12).Liu J; Lu L; Wood D; Lin S New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ghosh I; Ghosh T; Bardagi JI; König B Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 2014, 346, 725. [DOI] [PubMed] [Google Scholar]
- (14).Chmiel AF; Williams OP; Chernowsky CP; Yeung CS; Wickens ZK Non-innocent Radical Ion Intermediates in Photoredox Catalysis: Parallel Reduction Modes Enable Coupling of Diverse Aryl Chlorides. J. Am. Chem. Soc 2021, 143, 10882–10889. [DOI] [PubMed] [Google Scholar]
- (15).Cole JP; Chen D-F; Kudisch M; Pearson RM; Lim C-H; Miyake GM Organocatalyzed Birch Reduction Driven by Visible Light. J. Am. Chem. Soc 2020, 142, 13573–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zeman CJ; Kim S; Zhang F; Schanze KS Direct Observation of the Reduction of Aryl Halides by a Photoexcited Perylene Diimide Radical Anion. J. Am. Chem. Soc 2020, 142, 2204–2207. [DOI] [PubMed] [Google Scholar]
- (17).Marchini M; Gualandi A; Mengozzi L; Franchi P; Lucarini M; Cozzi PG; Balzani V; Ceroni P Mechanistic insights into two-photon-driven photocatalysis in organic synthesis. Phys. Chem. Chem. Phys 2018, 20, 8071–8076. [DOI] [PubMed] [Google Scholar]
- (18).Cowper NGW; Chernowsky CP; Williams OP; Wickens ZK Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc 2020, 142, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kim H; Kim H; Lambert TH; Lin S Reductive Electrophotocatalysis: Merging Electricity and Light To Achieve Extreme Reduction Potentials. J. Am. Chem. Soc 2020, 142, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Caby S; Bouchet LM; Argüello JE; Rossi RA; Bardagi JI Excitation of Radical Anions of Naphthalene Diimides in Consecutive- and Electro-Photocatalysis**. ChemCatChem 2021, 13, 3001–3009. [Google Scholar]
- (21).Chen Y-J; Lei T; Hu H-L; Wu H-L; Zhou S; Li X-B; Chen B; Tung C-H; Wu L-Z Tandem photoelectrochemical and photoredox catalysis for efficient and selective aryl halides functionalization by solar energy. Matter 2021, 4, 2354–2366. [Google Scholar]
- (22).Chernowsky CP; Chmiel AF; Wickens ZK Electrochemical Activation of Diverse Conventional Photoredox Catalysts Induces Potent Photoreductant Activity**. Angew. Chem. Int. Ed 2021, 60, 21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tay NES; Lehnherr D; Rovis T Photons or Electrons? A Critical Comparison of Electrochemistry and Photoredox Catalysis for Organic Synthesis. Chem. Rev 2022, 122, 2487–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) The highest catalyst TONs for reductive conPET and electron-primed photoredox procedures proceeding via photoexcitation of in-situ generated photocatalyst radical anions are ~100 for organocatalysts and ~3000 for Ru-based dyes. See:Bell JD; Murphy JA Recent Advances in Visible Light-Activated Radical Coupling Reactions Triggered by (i) Ruthenium, (Ii) Iridium and (Iii) Organic Photoredox Agents. Chem. Soc. Rev 2021, 50, 9540–9685. [DOI] [PubMed] [Google Scholar]; (b) Glaser F; Kerzig C; Wenger OS Multi-Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods. Angew. Chem. Int. Ed 2020, 59, 10266–10284. [DOI] [PubMed] [Google Scholar]; (c) Barham JP; König B Synthetic Photoelectrochemistry. Angew. Chem. Int. Ed 2020, 59, 11732–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu J; Lu L; Wood D; Lin S New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Naumann R; Lehmann F; Goez M Micellized Tris(Bipyridine)Ruthenium Catalysts Affording Preparative Amounts of Hydrated Electrons with a Green Light-Emitting Diode. Chem. – Eur. J 2018, 24, 13259–13269. [DOI] [PubMed] [Google Scholar]
- (25).Rieth AJ; Gonzalez MI; Kudisch B; Nava M; Nocera DG How Radical Are “Radical” Photocatalysts? A Closed-Shell Meisenheimer Complex Is Identified as a Super-Reducing Photoreagent. J. Am. Chem. Soc 2021, 143, 14352–14359. [DOI] [PubMed] [Google Scholar]
- (26).White AR; Wang L; Nicewicz DA Synthesis and Characterization of Acridinium Dyes for Photoredox Catalysis. Synlett 2019, 30, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xu J; Cao J; Wu X; Wang H; Yang X; Tang X; Toh RW; Zhou R; Yeow EKL; Wu J Unveiling Extreme Photoreduction Potentials of Donor–Acceptor Cyanoarenes to Access Aryl Radicals from Aryl Chlorides. J. Am. Chem. Soc 2021, 143, 13266–13273. [DOI] [PubMed] [Google Scholar]
- (28).Kodaimati MS; McClelland KP; He C; Lian S; Jiang Y; Zhang Z; Weiss EA Viewpoint: Challenges in Colloidal Photocatalysis and Some Strategies for Addressing Them. Inorg. Chem 2018, 57, 3659–3670. [DOI] [PubMed] [Google Scholar]
- (29).Yuan Y; Jin N; Saghy P; Dube L; Zhu H; Chen O Quantum Dot Photocatalysts for Organic Transformations. J. Phys. Chem. Lett 2021, 12, 7180–7193. [DOI] [PubMed] [Google Scholar]
- (30).Bailes J Photostability of Semiconductor Quantum Dots in Response to UV Exposure. In Nanoparticles in Biology and Medicine: Methods and Protocols; Ferrari E, Soloviev M, Eds.; Springer US: New York, NY, 2020; pp 343–349. [DOI] [PubMed] [Google Scholar]
- (31).Das A; Han Z; Haghighi MG; Eisenberg R Photogeneration of hydrogen from water using CdSe nanocrystals demonstrating the importance of surface exchange. Proc. Natl. Acad. Sci 2013, 110, 16716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gould TJ; Bewersdorf J; Hess ST A Quantitative Comparison of the Photophysical Properties of Selected Quantum Dots and Organic Fluorophores. Z. Für Phys. Chem 2008, 222, 833–849. [Google Scholar]
- (33).Resch-Genger U; Grabolle M; Cavaliere-Jaricot S; Nitschke R; Nann T Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [DOI] [PubMed] [Google Scholar]
- (34).Bruchez Marcel; Moronne Mario; Gin Peter; Weiss Shimon; Alivisatos A. Paul. Semiconductor Nanocrystals as Fluorescent Biological Labels. Science 1998, 281, 2013–2016. [DOI] [PubMed] [Google Scholar]
- (35).Hahn MA; Tabb JS; Krauss TD Detection of Single Bacterial Pathogens with Semiconductor Quantum Dots. Anal. Chem 2005, 77, 4861–4869. [DOI] [PubMed] [Google Scholar]
- (36).Urban JM; Chiang W; Hammond JW; Cogan NMB; Litzburg A; Burke R; Stern HA; Gelbard HA; Nilsson BL; Krauss TD Quantum Dots for Improved Single-Molecule Localization Microscopy. J. Phys. Chem. B 2021, 125, 2566–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Caputo JA; Frenette LC; Zhao N; Sowers KL; Krauss TD; Weix DJ General and Efficient C–C Bond Forming Photoredox Catalysis with Semiconductor Quantum Dots. J. Am. Chem. Soc 2017, 139, 4250–4253. [DOI] [PubMed] [Google Scholar]
- (38).Huang C; Li X-B; Tung C-H; Wu L-Z Photocatalysis with Quantum Dots and Visible Light for Effective Organic Synthesis. Chem. – Eur. J 2018, 24, 11530–11534. [DOI] [PubMed] [Google Scholar]
- (39).Zhang Z; Edme K; Lian S; Weiss EA Enhancing the Rate of Quantum-Dot-Photocatalyzed Carbon–Carbon Coupling by Tuning the Composition of the Dot’s Ligand Shell. J. Am. Chem. Soc 2017, 139, 4246–4249. [DOI] [PubMed] [Google Scholar]
- (40).Jiang Y; Wang C; Rogers CR; Kodaimati MS; Weiss EA Regio- and diastereoselective intermolecular [2+2] cycloadditions photocatalysed by quantum dots. Nat. Chem 2019, 11, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Pal A; Ghosh I; Sapra S; Koenig B Quantum Dots in Visible-Light Photoredox Catalysis: Reductive Dehalogenations and C-H Arylation Reactions Using Aryl Bromides. Chem. Mater 2017, 29, 5225–5231. [Google Scholar]
- (42).Zhang Z; Rogers CR; Weiss EA Energy Transfer from CdS QDs to a Photogenerated Pd Complex Enhances the Rate and Selectivity of a Pd-Photocatalyzed Heck Reaction. J. Am. Chem. Soc 2020, 142, 495–501. [DOI] [PubMed] [Google Scholar]
- (43).Hu Q; Yu X; Gong S; Chen X Nanomaterial catalysts for organic photoredox catalysis-mechanistic perspective. Nanoscale 2021, 13, 18044–18053. [DOI] [PubMed] [Google Scholar]
- (44).Hu J; Pu T-J; Xu Z-W; Xu W-Y; Feng Y-S Cadmium Sulfide Quantum-Dot-Photocatalyzed Cascade Cyclization of Functionalized Difluoromethyl Chlorides with Unactivated Olefins. Adv. Synth. Catal 2019, 361, 708–713. [Google Scholar]
- (45).Wang D-Y; Yin Y-Y; Feng C-W; Rukhsana; Shen Y-M Advances in Homogeneous Photocatalytic Organic Synthesis with Colloidal Quantum Dots. Catalysts 2021, 11, 275. [Google Scholar]
- (46).Yuan Y; Zhu H; Hills-Kimball K; Cai T; Shi W; Wei Z; Yang H; Candler Y; Wang P; He J; Chen O Stereoselective C−C Oxidative Coupling Reactions Photocatalyzed by Zwitterionic Ligand Capped CsPbBr3 Perovskite Quantum Dots. Angew. Chem. Int. Ed 2020, 59, 22563–22569. [DOI] [PubMed] [Google Scholar]
- (47).Huang C; Qiao J; Ci R-N; Wang X-Z; Wang Y; Wang J-H; Chen B; Tung C-H; Wu L-Z Quantum dots enable direct alkylation and arylation of allylic C(sp3)–H bonds with hydrogen evolution by solar energy. Chem 2021, 7, 1244–1257. [Google Scholar]
- (48).Qiao J; Song Z-Q; Huang C; Ci R-N; Liu Z; Chen B; Tung C-H; Wu L-Z Direct, Site-Selective and Redox-Neutral α-C−H Bond Functionalization of Tetrahydrofurans via Quantum Dots Photocatalysis. Angew. Chem. Int. Ed 2021, 60, 27201–27205. [DOI] [PubMed] [Google Scholar]
- (49).Huang C; Ci R-N; Qiao J; Wang X-Z; Feng K; Chen B; Tung C-H; Wu L-Z Direct Allylic C(sp3)−H and Vinylic C(sp2)−H Thiolation with Hydrogen Evolution by Quantum Dots and Visible Light. Angew. Chem. Int. Ed 2021, 60, 11779–11783. [DOI] [PubMed] [Google Scholar]
- (50).Chai Z; Zeng T-T; Li Q; Lu L-Q; Xiao W-J; Xu D Efficient Visible Light-Driven Splitting of Alcohols into Hydrogen and Corresponding Carbonyl Compounds over a Ni-Modified CdS Photocatalyst. J. Am. Chem. Soc 2016, 138, 10128–10131. [DOI] [PubMed] [Google Scholar]
- (51).Jensen SC; Bettis Homan S; Weiss EA Photocatalytic Conversion of Nitrobenzene to Aniline through Sequential Proton-Coupled One-Electron Transfers from a Cadmium Sulfide Quantum Dot. J. Am. Chem. Soc 2016, 138, 1591–1600. [DOI] [PubMed] [Google Scholar]
- (52).Jiang Y; Weiss E Colloidal Quantum Dots as Photocatalysts for Triplet Excited State Reactions of Organic Molecules. J. Am. Chem. Soc 2020, 142, 15219–15229. [DOI] [PubMed] [Google Scholar]
- (53).Perez KA; Rogers CR; Weiss EA Quantum Dot-Catalyzed Photoreductive Removal of Sulfonyl-Based Protecting Groups. Angew. Chem. Int. Ed 2020, 59, 14091–14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).McClelland KP; Weiss EA Selective Photocatalytic Oxidation of Benzyl Alcohol to Benzaldehyde or C-C Coupled Products by Visible-Light-Absorbing Quantum Dots. Acs Appl. Energy Mater 2019, 2, 92–96. [Google Scholar]
- (55).Xi Z-W; Yang L; Wang D-Y; Pu C-D; Shen Y-M; Wu C-D; Peng X-G Visible-Light Photocatalytic Synthesis of Amines from Imines via Transfer Hydrogenation Using Quantum Dots as Catalysts. J. Org. Chem 2018, 83, 11886–11895. [DOI] [PubMed] [Google Scholar]
- (56).Xi Z-W; Yang L; Wang D-Y; Feng C-W; Qin Y; Shen Y-M; Pu C; Peng X Visible Light Induced Reduction and Pinacol Coupling of Aldehydes and Ketones Catalyzed by Core/Shell Quantum Dots. J. Org. Chem 2021, 86, 2474–2488. [DOI] [PubMed] [Google Scholar]
- (57).Lei T; Wei S-M; Feng K; Chen B; Tung C-H; Wu L-Z Borylation of Diazonium Salts by Highly Emissive and Crystalline Carbon Dots in Water. ChemSusChem 2020, 13, 1715–1719. [DOI] [PubMed] [Google Scholar]
- (58).Simlandy AK; Bhattacharyya B; Pandey A; Mukherjee S Picosecond Electron Transfer from Quantum Dots Enables a General and Efficient Aerobic Oxidation of Boronic Acids. ACS Catal 2018, 8, 5206–5211. [Google Scholar]
- (59).Muralirajan K; Kancherla R; Bau JA; Taksande MR; Qureshi M; Takanabe K; Rueping M Exploring the Structure and Performance of Cd–Chalcogenide Photocatalysts in Selective Trifluoromethylation. ACS Catal 2021, 11, 14772–14780. [Google Scholar]
- (60).Chakraborty IN; Roy S; Devatha G; Rao A; Pillai PP InP/ZnS Quantum Dots as Efficient Visible-Light Photocatalysts for Redox and Carbon–Carbon Coupling Reactions. Chem. Mater 2019, 31, 2258–2262. [Google Scholar]
- (61).Li J; Zhao J; Ma C; Yu Z; Zhu H; Yun L; Meng Q Visible-Light-Driven Oxidative Cleavage of Alkenes Using Water-Soluble CdSe Quantum Dots. ChemSusChem 2021, 14, 4985–4992. [DOI] [PubMed] [Google Scholar]
- (62).Chandrashekar HB; Maji A; Halder G; Banerjee S; Bhattacharyya S; Maiti D Photocatalyzed borylation using water-soluble quantum dots. Chem. Commun 2019, 55, 6201–6204. [DOI] [PubMed] [Google Scholar]
- (63).Wu X; Xie S; Liu C; Zhou C; Lin J; Kang J; Zhang Q; Wang Z; Wang Y Ligand-Controlled Photocatalysis of CdS Quantum Dots for Lignin Valorization under Visible Light. ACS Catal 2019, 9, 8443–8451. [Google Scholar]
- (64).Zhu X; Lin Y; San Martin J; Sun Y; Zhu D; Yan Y Lead halide perovskites for photocatalytic organic synthesis. Nat. Commun 2019, 10, 2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).CdS QDs are made in one step using standard benchtop synthesis techniques from CdO ($40/mol), oleic acid ($10/mol) and sulfur ($1.45/mol) in octadecene ($40/L). They are isolated by precipitation and stored as solutions in toluene. See Supporting Information for further details.
- (66).Fritzinger B; Capek RK; Lambert K; Martins JC; Hens Z Utilizing Self-Exchange To Address the Binding of Carboxylic Acid Ligands to CdSe Quantum Dots. J. Am. Chem. Soc 2010, 132, 10195–10201. [DOI] [PubMed] [Google Scholar]
- (67).Knauf RR; Lennox JC; Dempsey JL Quantifying Ligand Exchange Reactions at CdSe Nanocrystal Surfaces. Chem. Mater 2016, 28, 4762–4770. [Google Scholar]
- (68).Hens Z; Martins JC A Solution NMR Toolbox for Characterizing the Surface Chemistry of Colloidal Nanocrystals. Chem. Mater 2013, 25, 1211–1221. [Google Scholar]
- (69).Morozov Sergii; Pensa Evangelina L.; Khan Ali Hossain; Polovitsyn Anatolii; Cortés Emiliano; Maier Stefan A.; Vezzoli Stefano; Moreels Iwan; Sapienza Riccardo. Electrical control of single-photon emission in highly charged individual colloidal quantum dots. Sci. Adv 2020, 6, eabb1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ashokan A; Mulvaney P Spectroelectrochemistry of Colloidal CdSe Quantum Dots. Chem. Mater 2021, 33, 1353–1362. [Google Scholar]
- (71).Harris RD; Bettis Homan S; Kodaimati M; He C; Nepomnyashchii AB; Swenson NK; Lian S; Calzada R; Weiss EA Electronic Processes within Quantum Dot-Molecule Complexes. Chem. Rev 2016, 116, 12865–12919. [DOI] [PubMed] [Google Scholar]
- (72).Kerzig C; Guo X; Wenger OS Unexpected Hydrated Electron Source for Preparative Visible-Light Driven Photoredox Catalysis. J. Am. Chem. Soc 2019, 141, 2122–2127. [DOI] [PubMed] [Google Scholar]
- (73).Cohn AW; Rinehart JD; Schimpf AM; Weaver AL; Gamelin DR Size Dependence of Negative Trion Auger Recombination in Photodoped CdSe Nanocrystals. Nano Lett 2014, 14, 353–358. [DOI] [PubMed] [Google Scholar]
- (74).Park Y-S; Bae WK; Pietryga JM; Klimov VI Auger Recombination of Biexcitons and Negative and Positive Trions in Individual Quantum Dots. ACS Nano 2014, 8, 7288–7296. [DOI] [PubMed] [Google Scholar]
- (75).Hou X; Kang J; Qin H; Chen X; Ma J; Zhou J; Chen L; Wang L; Wang L-W; Peng X Engineering Auger recombination in colloidal quantum dots via dielectric screening. Nat. Commun 2019, 10, 1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Dong Y; Choi J; Jeong H-K; Son DH Hot Electrons Generated from Doped Quantum Dots via Upconversion of Excitons to Hot Charge Carriers for Enhanced Photocatalysis. J. Am. Chem. Soc 2015, 137, 5549–5554. [DOI] [PubMed] [Google Scholar]
- (77).Melnychuk C; Guyot-Sionnest P Multicarrier Dynamics in Quantum Dots. Chem. Rev 2021, 121, 2325–2372. [DOI] [PubMed] [Google Scholar]
- (78).Different terminology exists for describing QDs which become negatively charged by the presence of a surplus electron in the conduction band 1Se state. Here we use “photodoped” and “anionic” to describe these QDs when prepared via irradiation in the presence of reductant.
- (79).Kobayashi Y; Nishimura T; Yamaguchi H; Tamai N Effect of Surface Defects on Auger Recombination in Colloidal CdS Quantum Dots. J. Phys. Chem. Lett 2011, 2, 1051–1055. [Google Scholar]
- (80).Wu K; Lim J; Klimov VI Superposition Principle in Auger Recombination of Charged and Neutral Multicarrier States in Semiconductor Quantum Dots. ACS Nano 2017, 11, 8437–8447. [DOI] [PubMed] [Google Scholar]
- (81).Chepic DI; Efros Al. L.; Ekimov AI; Ivanov MG; Kharchenko VA; Kudriavtsev IA; Yazeva TV Auger ionization of semiconductor quantum drops in a glass matrix. J. Lumin 1990, 47, 113–127. [Google Scholar]
- (82).Padilha LA; Robel I; Lee DC; Nagpal P; Pietryga JM; Klimov VI Spectral Dependence of Nanocrystal Photoionization Probability: The Role of Hot-Carrier Transfer. ACS Nano 2011, 5, 5045–5055. [DOI] [PubMed] [Google Scholar]
- (83).Guyot-Sionnest P; Wehrenberg B; Yu D Intraband relaxation in CdSe nanocrystals and the strong influence of the surface ligands. J. Chem. Phys 2005, 123, 074709. [DOI] [PubMed] [Google Scholar]
- (84).Tisdale William A; Williams Kenrick J; Timp Brooke A; Norris David J; Aydil Eray S; Zhu X-Y Hot-Electron Transfer from Semiconductor Nanocrystals. Science 2010, 328, 1543–1547. [DOI] [PubMed] [Google Scholar]
- (85).Singhal P; Ghosh HN Hot Charge Carriers in Quantum Dots: Generation, Relaxation, Extraction, and Applications. ChemNanoMat 2019, 5, 985–999. [Google Scholar]
- (86).Apparent “anti-Kasha” chemistry proceeding through hot carriers higher in energy than band edge states, while rare in collisionally limited chemistry of organic chromophores, is extremely common in nanomaterials. For example, for colloidal semiconductor nanocrystals, hot electron transfer at the nanocrystal surface can occur in under 100 fs, which outcompetes intraband hot carrier relaxation. See: Tisdale William A.; Williams Kenrick J.; Timp Brooke A.; Norris David J.; Aydil Eray S.; Zhu X-Y Hot-Electron Transfer from Semiconductor Nanocrystals. Science 2010, 328, 1543–1547. [DOI] [PubMed] [Google Scholar]
- (87).Parobek D; Meeder JR; Puthenpurayil J; Nippe M; Son DH Breaking the short-range proximity requirement in quantum dot/molecular catalyst hybrids for CO2 reduction via long-range hot electron sensitization. J. Mater. Chem. A 2020, 8, 12984–12989. [Google Scholar]
- (88).Orrison C; Meeder JR; Zhang B; Puthenpurayil J; Hall MB; Nippe M; Son DH Efficient Redox-Neutral Photocatalytic Formate to Carbon Monoxide Conversion Enabled by Long-Range Hot Electron Transfer from Mn-Doped Quantum Dots. J. Am. Chem. Soc 2021, 143, 10292–10300. [DOI] [PubMed] [Google Scholar]
- (89).Different batches of CdS QDs yielded sizes ranging between 5.8 and 6.0 nm, however these performed equally well for the described chemistry as long as the molar loading of QDs was kept constant.
- (90).Haram SK; Quinn BM; Bard AJ Electrochemistry of CdS Nanoparticles: A Correlation between Optical and Electrochemical Band Gaps. J. Am. Chem. Soc 2001, 123, 8860–8861. [DOI] [PubMed] [Google Scholar]
- (91).Ingole PP; Markad GB; Saraf D; Tatikondewar L; Nene O; Kshirsagar A; Haram SK Band Gap Bowing at Nanoscale: Investigation of CdSxSe1–x Alloy Quantum Dots through Cyclic Voltammetry and Density Functional Theory. J. Phys. Chem. C 2013, 117, 7376–7383. [Google Scholar]
- (92).Martindale BCM; Hutton GAM; Caputo CA; Reisner E Solar Hydrogen Production Using Carbon Quantum Dots and a Molecular Nickel Catalyst. J. Am. Chem. Soc 2015, 137, 6018–6025. [DOI] [PubMed] [Google Scholar]
- (93).Gong K; Martin JE; Shea-Rohwer LE; Lu P; Kelley DF Radiative Lifetimes of Zincblende CdSe/CdS Quantum Dots. J. Phys. Chem. C 2015, 119, 2231–2238. [Google Scholar]
- (94).Gong K; Zeng Y; Kelley DF Extinction Coefficients, Oscillator Strengths, and Radiative Lifetimes of CdSe, CdTe, and CdTe/CdSe Nanocrystals. J. Phys. Chem. C 2013, 117, 20268–20279. [Google Scholar]
- (95).Holzer W; Penzkofer A; Tsuboi T Absorption and emission spectroscopic characterization of Ir(ppy)3. Chem. Phys 2005, 308, 93–102. [Google Scholar]
- (96).Gisbertz S; Pieber B Heterogeneous Photocatalysis in Organic Synthesis. ChemPhotoChem 2020, 4, 456–475. [Google Scholar]
- (97).Lin Y; Guo J; San Martin J; Han C; Martinez R; Yan Y Photoredox Organic Synthesis Employing Heterogeneous Photocatalysts with Emphasis on Halide Perovskite. Chem. – Eur. J 2020, 26, 13118–13136. [DOI] [PubMed] [Google Scholar]
- (98).Wang Y; Wei Y; Song W; Chen C; Zhao J Photocatalytic Hydrodehalogenation for the Removal of Halogenated Aromatic Contaminants. ChemCatChem 2019, 11, 258–268. [Google Scholar]
- (99).Vaxenburg R; Rodina A; Shabaev A; Lifshitz E; Efros AL Nonradiative Auger Recombination in Semiconductor Nanocrystals. Nano Lett 2015, 15, 2092–2098. [DOI] [PubMed] [Google Scholar]
- (100).Kharchenko VA; Rosen M Auger relaxation processes in semiconductor nanocrystals and quantum wells. Spectrosc. Isol. Assem. Semicond. Nanocrystals 1996, 70, 158–169. [Google Scholar]
- (101).MacKenzie IA; Wang L; Onuska NPR; Williams OF; Begam K; Moran AM; Dunietz BD; Nicewicz DA Discovery and characterization of an acridine radical photoreductant. Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).(a) Many other deeply reducing conPET catalysts have been described employing other wavelengths of light, however we elected to directly compare catalysts which function with similar wavelengths of light to the QDs (455 nm). For example, acridinium photocatalysts have been employed for two-photon photoreductions of electron-rich aryl chlorides using 390 nm light, but provided no product under our conditions. Based on this report, we calculate a turnover number of 8.2 for the dechlorination of 4-chloroanisole. See:MacKenzie IA; Wang L; Onuska NPR; Williams OF; Begam K; Moran AM; Dunietz BD; Nicewicz DA Discovery and Characterization of an Acridine Radical Photoreductant. Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).(a) While the substrate 1a has a reduction potential outside the known reducing power of some of the tested molecular photocatalysts including PDI•-*, the in-situ decomposition of PDI•- and similar molecular photocatalysts is known to produce stronger photoreductants, which may explain these results. For relevant references, see:Chernowsky CP; Chmiel AF; Wickens ZK Electrochemical Activation of Diverse Conventional Photoredox Catalysts Induces Potent Photoreductant Activity**. Angew. Chem. Int. Ed 2021, 60, 21418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rieth AJ; Gonzalez MI; Kudisch B; Nava M; Nocera DG How Radical Are “Radical” Photocatalysts? A Closed-Shell Meisenheimer Complex Is Identified as a Super-Reducing Photoreagent. J. Am. Chem. Soc 2021, 143, 14352–14359. [DOI] [PubMed] [Google Scholar]
- (104).Owen J The coordination chemistry of nanocrystal surfaces. Science 2015, 347, 615. [DOI] [PubMed] [Google Scholar]
- (105).Anderson NC; Chen Peter. E.; Buckley AK; De Roo J; Owen JS Stereoelectronic Effects on the Binding of Neutral Lewis Bases to CdSe Nanocrystals. J. Am. Chem. Soc 2018, 140, 7199–7205. [DOI] [PubMed] [Google Scholar]
- (106).De Roo J; De Keukeleere K; Hens Z; Van Driessche I From ligands to binding motifs and beyond; the enhanced versatility of nanocrystal surfaces. Dalton Trans 2016, 45, 13277–13283. [DOI] [PubMed] [Google Scholar]
- (107).Morris-Cohen AJ; Malicki M; Peterson MD; Slavin JWJ; Weiss EA Chemical, Structural, and Quantitative Analysis of the Ligand Shells of Colloidal Quantum Dots. Chem. Mater 2013, 25, 1155–1165. [Google Scholar]
- (108).Fritzinger B; Moreels I; Lommens P; Koole R; Hens Z; Martins JC In Situ Observation of Rapid Ligand Exchange in Colloidal Nanocrystal Suspensions Using Transfer NOE Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc 2009, 131, 3024–3032. [DOI] [PubMed] [Google Scholar]
- (109).Anderson NC; Hendricks MP; Choi JJ; Owen JS Ligand Exchange and the Stoichiometry of Metal Chalcogenide Nanocrystals: Spectroscopic Observation of Facile Metal-Carboxylate Displacement and Binding. J. Am. Chem. Soc 2013, 135, 18536–18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Chen PE; Anderson NC; Norman ZM; Owen JS Tight Binding of Carboxylate, Phosphonate, and Carbamate Anions to Stoichiometric CdSe Nanocrystals. J. Am. Chem. Soc 2017, 139, 3227–3236. [DOI] [PubMed] [Google Scholar]
- (111).Weinberg DJ; He C; Weiss EA Control of the Redox Activity of Quantum Dots through Introduction of Fluoroalkanethiolates into Their Ligand Shells. J. Am. Chem. Soc 2016, 138, 2319–2326. [DOI] [PubMed] [Google Scholar]
- (112).Nepomnyashchii AB; Harris RD; Weiss EA Composition and Permeability of Oleate Adlayers of CdS Quantum Dots upon Dilution to Photoluminescence-Relevant Concentrations. Anal. Chem 2016, 88, 3310–3316. [DOI] [PubMed] [Google Scholar]
- (113).Hartley CL; Dempsey JL Electron-Promoted X-Type Ligand Displacement at CdSe Quantum Dot Surfaces. Nano Lett 2019, 19, 1151–1157. [DOI] [PubMed] [Google Scholar]
- (114).Peterson MD; Jensen SC; Weinberg DJ; Weiss EA Mechanisms for Adsorption of Methyl Viologen on CdS Quantum Dots. ACS Nano 2014, 8, 2826–2837. [DOI] [PubMed] [Google Scholar]
- (115).Weiss EA Designing the Surfaces of Semiconductor Quantum Dots for Colloidal Photocatalysis. ACS Energy Lett 2017, 2, 1005–1013. [Google Scholar]
- (116).Constantin T; Zanini M; Regni A; Sheikh NS; Juliá F; Leonori D Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 2020, 367, 1021. [DOI] [PubMed] [Google Scholar]
- (117).Constantin T; Juliá F; Sheikh NS; Leonori D A case of chain propagation: α-aminoalkyl radicals as initiators for aryl radical chemistry. Chem. Sci 2020, 11, 12822–12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Hendy CM; Smith GC; Xu Z; Lian T; Jui NT Radical Chain Reduction via Carbon Dioxide Radical Anion (CO2•–). J. Am. Chem. Soc 2021, 143, 8987–8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Koppenol WH; Rush JD Reduction potential of the carbon dioxide/carbon dioxide radical anion: a comparison with other C1 radicals. J. Phys. Chem 1987, 91, 4429–4430. [Google Scholar]
- (120).Wayner DDM; McPhee DJ; Griller D Oxidation and reduction potentials of transient free radicals. J. Am. Chem. Soc 1988, 110, 132–137. [Google Scholar]
- (121).Takeda N; Poliakov PV; Cook AR; Miller JR Faster Dissociation: Measured Rates and Computed Effects on Barriers in Aryl Halide Radical Anions. J. Am. Chem. Soc 2004, 126, 4301–4309. [DOI] [PubMed] [Google Scholar]
- (122).Enemærke RJ; Christensen TB; Jensen H; Daasbjerg K Application of a new kinetic method in the investigation of cleavage reactions of haloaromatic radical anions. J. Chem. Soc. Perkin Trans 2 2001, No. 9, 1620–1630. [Google Scholar]
- (123).Costentin C; Robert M; Savéant J-M Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc 2004, 126, 16051–16057. [DOI] [PubMed] [Google Scholar]
- (124).Rinehart JD; Schimpf AM; Weaver AL; Cohn AW; Gamelin DR Photochemical Electronic Doping of Colloidal CdSe Nanocrystals. J. Am. Chem. Soc 2013, 135, 18782–18785. [DOI] [PubMed] [Google Scholar]
- (125).Schimpf AM; Knowles KE; Carroll GM; Gamelin DR Electronic Doping and Redox-Potential Tuning in Colloidal Semiconductor Nanocrystals. Acc. Chem. Res 2015, 48, 1929–1937. [DOI] [PubMed] [Google Scholar]
- (126).Shulenberger KE; Keller HR; Pellows LM; Brown NL; Dukovic G Photocharging of Colloidal CdS Nanocrystals. J. Phys. Chem. C 2021, 125, 22650–22659. [Google Scholar]
- (127).Shim M; Guyot-Sionnest P N-type colloidal semiconductor nanocrystals. Nature 2000, 407, 981–983. [DOI] [PubMed] [Google Scholar]
- (128).Hu Z; Shu Y; Qin H; Hu X; Peng X Water Effects on Colloidal Semiconductor Nanocrystals: Correlation of Photophysics and Photochemistry. J. Am. Chem. Soc 2021, 143, 18721–18732. [DOI] [PubMed] [Google Scholar]
- (129).Shim M; Wang C; Guyot-Sionnest P Charge-Tunable Optical Properties in Colloidal Semiconductor Nanocrystals. J. Phys. Chem. B 2001, 105, 2369–2373. [Google Scholar]
- (130).Garoz-Ruiz J; Perales-Rondon JV; Heras A; Colina A Spectroelectrochemistry of Quantum Dots. Isr. J. Chem 2019, 59, 679–694. [Google Scholar]
- (131).Honarfar A; Mourad H; Lin W; Polukeev A; Rahaman A; Abdellah M; Chábera P; Pankratova G; Gorton L; Zheng K; Pullerits T Photoexcitation Dynamics in Electrochemically Charged CdSe Quantum Dots: From Hot Carrier Cooling to Auger Recombination of Negative Trions. ACS Appl. Energy Mater 2020, 3, 12525–12531. [Google Scholar]
- (132).Shiragami T; Ankyu H; Fukami S; Pac C; Yanaglda S; Mori H; Fujita H Semiconductor photocatalysis: visible light induced photoreduction of aromatic ketones and electron-deficient alkenes catalysed by quantised cadmium sulfide. J. Chem. Soc. Faraday Trans 1992, 88, 1055–1061. [Google Scholar]
- (133).Rajh T; Micic OI; Lawless D; Serpone N Semiconductor photophysics. 7. Photoluminescence and picosecond charge carrier dynamics in cadmium sulfide quantum dots confined in a silicate glass. J. Phys. Chem 1992, 96, 4633–4641. [Google Scholar]
- (134).Wang Congjun; Shim Moonsub; Guyot-Sionnest Philippe. Electrochromic Nanocrystal Quantum Dots. Science 2001, 291, 2390–2392. [DOI] [PubMed] [Google Scholar]
- (135).Tsui EY; Carroll GM; Miller B; Marchioro A; Gamelin DR Extremely Slow Spontaneous Electron Trapping in Photodoped n-Type CdSe Nanocrystals. Chem. Mater 2017, 29, 3754–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Bryant GW; Jaskolski W Surface Effects on Capped and Uncapped Nanocrystals. J. Phys. Chem. B 2005, 109, 19650–19656. [DOI] [PubMed] [Google Scholar]
- (137).Empedocles SA; Bawendi MG Quantum-Confined Stark Effect in Single CdSe Nanocrystallite Quantum Dots. Science 1997, 278, 2114–2117. [DOI] [PubMed] [Google Scholar]
- (138).Empedocles SA; Bawendi MG Influence of Spectral Diffusion on the Line Shapes of Single CdSe Nanocrystallite Quantum Dots. J. Phys. Chem. B 1999, 103, 1826–1830. [Google Scholar]
- (139).Wang J; Ding T; Wu K Charge Transfer from n-Doped Nanocrystals: Mimicking Intermediate Events in Multielectron Photocatalysis. J. Am. Chem. Soc 2018, 140, 7791–7794. [DOI] [PubMed] [Google Scholar]
- (140).Houtepen AJ; Hens Z; Owen JS; Infante I On the Origin of Surface Traps in Colloidal II–VI Semiconductor Nanocrystals. Chem. Mater 2017, 29, 752–761. [Google Scholar]
- (141).du Fossé I; Lal S; Hossaini AN; Infante I; Houtepen AJ Effect of Ligands and Solvents on the Stability of Electron Charged CdSe Colloidal Quantum Dots. J. Phys. Chem. C 2021, 125, 23968–23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Jha PP; Guyot-Sionnest P Trion Decay in Colloidal Quantum Dots. ACS Nano 2009, 3, 1011–1015. [DOI] [PubMed] [Google Scholar]
- (143).Langan JR; Liu KJ; Salmon GA; Edwards PP; Ellaboudy A; Holton DM The Radiation Chemistry of Organic Amides I. A Pulse Radiolysis Study of Solvated Electrons and Alkalimetal-Electron Species in Cyclic Amides. Proc. R. Soc. Lond. Ser. Math. Phys. Sci 1989, 421, 169–178. [Google Scholar]
- (144).Shkrob IA; Marin TW Electron Localization and Radiation Chemistry of Amides. J. Phys. Chem. A 2012, 116, 1746–1757. [DOI] [PubMed] [Google Scholar]
- (145).Huang H-M; McDouall JJW; Procter DJ Radical Anions from Urea-type Carbonyls: Radical Cyclizations and Cyclization Cascades. Angew. Chem. Int. Ed 2018, 57, 4995–4999. [DOI] [PubMed] [Google Scholar]
- (146).Volz N; Clayden J The Urea Renaissance. Angew. Chem. Int. Ed 2011, 50, 12148–12155. [DOI] [PubMed] [Google Scholar]
- (147).(a) Owing to difficulties in measurement of reduction potentials outside the electrochemical windows of ordinary solvents and electrolytes, there are no reports of solution-phase reduction potentials of tetraalkylureas or trialkyl amides, however gas-phase electron affinities suggest that tetraalkyl ureas should be more difficult to reduce than trialkyl amides to the corresponding radical anions.Lecomte F; Lucas B; Grégoire G; Schermann JP; Desfrançois C Urea and Methylurea Dipole-Bound Anions. Phys. Chem. Chem. Phys 2003, 5, 3120–3125. [Google Scholar]; (b) Lecomte F; Lucas B; Grégoire G; Schermann JP; Desfrançois C Structures of Amide-Water Neutral Complexes from Dipole-Bound Anion Formation. Eur. Phys. J. D 2002, 20, 449–457. [Google Scholar]
- (148).Kimura M; Miyahara H; Moritani N; Sawaki Y Electroreductive dehalogenation of chlorinated aromatic ethers. Unexpected electrogenerated base-catalyzed reactions. J. Org. Chem 1990, 55, 3897–3902. [Google Scholar]
- (149).Murphy JA; Zhou S; Thomson DW; Schoenebeck F; Mahesh M; Park SR; Tuttle T; Berlouis LEA The Generation of Aryl Anions by Double Electron Transfer to Aryl Iodides from a Neutral Ground-State Organic Super-Electron Donor. Angew. Chem. Int. Ed 2007, 46, 5178–5183. [DOI] [PubMed] [Google Scholar]
- (150).Yin H; Wada Y; Kitamura T; Sakata T; Mori H; Yanagida S Enhanced Photocatalytic Dechlorination of 1,2,3,4-Tetrachlorobenzene Using Nanosized CdS/TiO2 Hybrid Photocatalyst under Visible Light Irradiation. Chem. Lett 2001, 30, 334–335. [Google Scholar]
- (151).Alonso DA; Andersson PG Deprotection of Sulfonyl Aziridines. J. Org. Chem 1998, 63, 9455–9461. [Google Scholar]
- (152).Ankner T; Hilmersson G Instantaneous Deprotection of Tosylamides and Esters with SmI2/Amine/Water. Org. Lett 2009, 11, 503–506. [DOI] [PubMed] [Google Scholar]
- (153).Alonso E; Ramón DJ; Yus M Reductive deprotection of allyl, benzyl and sulfonyl substituted alcohols, amines and amides using a naphthalene-catalysed lithiation. Tetrahedron 1997, 53, 14355–14368. [Google Scholar]
- (154).Hyeong Lee G; Bok Choi E; Lee E; Siek Pak C An efficient desulfonylation method mediated by magnesium in ethanol. Int. J. Rapid Publ. Prelim 1993, 34, 4541–4542. [Google Scholar]
- (155).Javorskis T; Orentas E Chemoselective Deprotection of Sulfonamides Under Acidic Conditions: Scope, Sulfonyl Group Migration, and Synthetic Applications. J. Org. Chem 2017, 82, 13423–13439. [DOI] [PubMed] [Google Scholar]
- (156).Montenegro MI The electrochemical cleavage of protecting groups. Electrochimica Acta 1986, 31, 607–620. [Google Scholar]
- (157).Shi L; Xia WJ; Zhang FM; Tu YQ A Novel, Efficient, and Highly Selective O-Bn Bond Cleavage Reaction via a Rare K-Induced Electron Transfer Process. Synlett 2002, 2002, 1505–1507. [Google Scholar]
- (158).Weissman SA; Zewge D Recent advances in ether dealkylation. Tetrahedron 2005, 61, 7833–7863. [Google Scholar]
- (159).Wuts Peter G. M.; Greene Theodora W. Protection for the Hydroxyl Group, Including 1,2- and 1,3-Diols. In Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Ltd, 2006; pp 16–366. [Google Scholar]
- (160).Liu H-J; Yip J; Shia K-S Reductive cleavage of benzyl ethers with lithium naphthalenide. A convenient method for debenzylation. Tetrahedron Lett 1997, 38, 2253–2256. [Google Scholar]
- (161).Doni E; O’Sullivan S; Murphy JA Metal-Free Reductive Cleavage of Benzylic Esters and Ethers: Fragmentations Result from Single and Double Electron Transfers. Angew. Chem. Int. Ed 2013, 52, 2239–2242. [DOI] [PubMed] [Google Scholar]
- (162).Miyazaki T; Maekawa H; Yonemura K; Yamamoto Y; Yamanaka Y; Nishiguchi I Mg-promoted facile and selective intramolecular cyclization of aromatic δ-ketoesters. Tetrahedron 2011, 67, 1598–1602. [Google Scholar]
- (163).While reports of the reduction potentials of trialkyl amides are unavailable, comparison of N,N-dimethylbenzamide (Ered = −2.53 vs. SCE) with methyl benzoate (−2.29 V vs. SCE) illustrates that amides are generally harder to reduce to the radical anion than the analogous esters.
- (164).Amador AG; Sherbrook EM; Yoon TP Enantioselective Photocatalytic [3 + 2] Cycloadditions of Aryl Cyclopropyl Ketones. J Am Chem Soc 2016, 138, 4722–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Lu Z; Shen M; Yoon TP [3+2] Cycloadditions of Aryl Cyclopropyl Ketones by Visible Light Photocatalysis. J. Am. Chem. Soc 2011, 133, 1162–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Huang X; Lin J; Shen T; Harms K; Marchini M; Ceroni P; Meggers E Asymmetric [3+2] Photocycloadditions of Cyclopropanes with Alkenes or Alkynes through Visible-Light Excitation of Catalyst-Bound Substrates. Angew. Chem. Int. Ed 2018, 57, 5454–5458. [DOI] [PubMed] [Google Scholar]
- (167).Tanko JM; Li X; Chahma M; Jackson WF; Spencer JN Cyclopropyl Conjugation and Ketyl Anions: When Do Things Begin to Fall Apart? J. Am. Chem. Soc 2007, 129, 4181–4192. [DOI] [PubMed] [Google Scholar]
- (168).Szostak M; Spain M; Eberhart AJ; Procter DJ Mechanism of SmI2/Amine/H2O-Promoted Chemoselective Reductions of Carboxylic Acid Derivatives (Esters, Acids, and Amides) to Alcohols. J. Org. Chem 2014, 79, 11988–12003. [DOI] [PubMed] [Google Scholar]
- (169).Ichiyanagi T; Kuniyama S; Shimizu M; Fujisawa T Regioselective Ring-opening of Cyclopropyl Ketones with Organometallic Reagents. Chem Lett 1997, 1149–1150. [Google Scholar]
- (170).Agasti S; Beattie NA; McDouall JJW; Procter DJ SmI2-Catalyzed Intermolecular Coupling of Cyclopropyl Ketones and Alkynes: A Link between Ketone Conformation and Reactivity. J. Am. Chem. Soc 2021, 143, 3655–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Hao W; Harenberg JH; Wu X; MacMillan SN; Lin S Diastereo- and Enantioselective Formal [3 + 2] Cycloaddition of Cyclopropyl Ketones and Alkenes via Ti-Catalyzed Radical Redox Relay. J. Am. Chem. Soc 2018, 140, 3514–3517. [DOI] [PubMed] [Google Scholar]
- (172).Liu L; Montgomery J Dimerization of Cyclopropyl Ketones and Crossed Reactions of Cyclopropyl Ketones with Enones as an Entry to Five-Membered Rings. J. Am. Chem. Soc 2006, 128, 5348–5349. [DOI] [PubMed] [Google Scholar]
- (173).Tamaki T; Ohashi M; Ogoshi S [3+2] Cycloaddition Reaction of Cyclopropyl Ketones with Alkynes Catalyzed by Nickel/Dimethylaluminum Chloride. Angew. Chem. Int. Ed 2011, 50, 12067–12070. [DOI] [PubMed] [Google Scholar]
- (174).Huang H-M; McDouall JJW; Procter DJ SmI2-catalysed cyclization cascades by radical relay. Nat. Catal 2019, 2, 211–218. [Google Scholar]
- (175).Ogoshi S; Nagata M; Kurosawa H Formation of Nickeladihydropyran by Oxidative Addition of Cyclopropyl Ketone. Key Intermediate in Nickel-Catalyzed Cycloaddition. J. Am. Chem. Soc 2006, 128, 5350–5351. [DOI] [PubMed] [Google Scholar]
- (176).Tamaki T; Nagata M; Ohashi M; Ogoshi S Synthesis and Reactivity of Six-Membered Oxa-Nickelacycles: A Ring-Opening Reaction of Cyclopropyl Ketones. Chem. – Eur. J 2009, 15, 10083–10091. [DOI] [PubMed] [Google Scholar]
- (177).Wang J; Wang L; Yu S; Ding T; Xiang D; Wu K Spin blockade and phonon bottleneck for hot electron relaxation observed in n-doped colloidal quantum dots. Nat. Commun 2021, 12, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Zhu H; Song N; Lian T Charging of Quantum Dots by Sulfide Redox Electrolytes Reduces Electron Injection Efficiency in Quantum Dot Sensitized Solar Cells. J. Am. Chem. Soc 2013, 135, 11461–11464. [DOI] [PubMed] [Google Scholar]
- (179).Gosztola D; Niemczyk MP; Svec W; Lukas AS; Wasielewski MR Excited Doublet States of Electrochemically Generated Aromatic Imide and Diimide Radical Anions. J. Phys. Chem. A 2000, 104, 6545–6551. [Google Scholar]
- (180).Pandey Anshu; Guyot-Sionnest Philippe. Slow Electron Cooling in Colloidal Quantum Dots. Science 2008, 322, 929–932. [DOI] [PubMed] [Google Scholar]
- (181).Klimov VI Optical Nonlinearities and Ultrafast Carrier Dynamics in Semiconductor Nanocrystals. J. Phys. Chem. B 2000, 104, 6112–6123. [Google Scholar]
- (182).Klimov VI; McBranch DW Femtosecond 1P-to-1S Electron Relaxation in Strongly Confined Semiconductor Nanocrystals. Phys. Rev. Lett 1998, 80, 4028–4031. [Google Scholar]
- (183).Cooney RR; Sewall SL; Dias EA; Sagar DM; Anderson KEH; Kambhampati P Unified picture of electron and hole relaxation pathways in semiconductor quantum dots. Phys. Rev. B 2007, 75, 245311. [Google Scholar]
- (184).Dana J; Maity P; Ghosh HN Hot-electron transfer from the semiconductor domain to the metal domain in CdSe@CdS{Au} nano-heterostructures. Nanoscale 2017, 9, 9723–9731. [DOI] [PubMed] [Google Scholar]
- (185).Lian S; Christensen JA; Kodaimati MS; Rogers CR; Wasielewski MR; Weiss EA Oxidation of a Molecule by the Biexcitonic State of a CdS Quantum Dot. J. Phys. Chem. C 2019, 123, 5923–5930. [Google Scholar]
- (186).Shah J Hot Carriers in Semiconductor Nanostructures: Physics and Applications; Elsevier, 2012. [Google Scholar]
- (187).Kraus RM; Lagoudakis PG; Müller J; Rogach AL; Lupton JM; Feldmann J; Talapin DV; Weller H Interplay between Auger and Ionization Processes in Nanocrystal Quantum Dots. J. Phys. Chem. B 2005, 109, 18214–18217. [DOI] [PubMed] [Google Scholar]
- (188).Parobek D; Qiao T; Son DH Energetic hot electrons from exciton-to-hot electron upconversion in Mn-doped semiconductor nanocrystals. J. Chem. Phys 2019, 151, 120901. [DOI] [PubMed] [Google Scholar]
- (189).Livache C; Kim WD; Jin H; Kozlov OV; Fedin I; Klimov VI High-efficiency photoemission from magnetically doped quantum dots driven by multi-step spin-exchange Auger ionization. Nat. Photonics 2022, 16, 433–440. [Google Scholar]
- (190).Reiss P; Carrière M; Lincheneau C; Vaure L; Tamang S Synthesis of Semiconductor Nanocrystals, Focusing on Nontoxic and Earth-Abundant Materials. Chem. Rev 2016, 116, 10731–10819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.