

Abstract
The pursuit of highly sensitive and specific cancer diagnostics based on cell-free (cf) nucleic acids isolated from minimally invasive liquid biopsies has been an area of intense research and commercial effort for at least two decades. Most of these tests detect cancer-specific mutations or epigenetic modifications on circulating DNA derived from tumor cells (ctDNA). Although recent FDA approvals of both single and multi-analyte liquid biopsy companion diagnostic assays are proof of the tremendous progress made in this domain, using ctDNA for the diagnosis of early-stage (stage I/II) cancers remains challenging due to several factors, such as low mutational allele frequency in circulation, overlapping profiles in genomic alterations among diverse cancers, and clonal hematopoiesis. This review discusses these analytical challenges, interim solutions, and the opportunity to complement ctDNA diagnostics with microbiome-aware analyses that may mitigate several existing ctDNA assay limitations.
Keywords: circulating tumor DNA, early detection, cell-free DNA, cancer screening, cancer detection, microbiome
Graphical Abstract
Genomic and metagenomic features for minimally invasive, blood-based detection of cancers. Here we discuss established and emerging methods of analyzing and diagnosing cancers through blood-based non-invasive liquid biopsy assays with attention to the biological parameters that may set a physical limit on the limit-of-detection for ctDNA-based liquid biopsy assays.
1. Introduction – The Need for Early Detection of Cancer
Cancer is the second leading cause of death globally, responsible for an estimated 9.6 million deaths in 2018 and untold suffering for patients and their families.1 Longitudinal survival statistics have overwhelmingly demonstrated that early detection of cancer, when the malignant neoplasm is confined to the tissue of origin, is associated with improvement of patient outcomes and reduced patient costs.2,3 For instance, early-stage detection of colorectal cancer yields a 90% 5-year survival rate, compared to only 14% survival with late-stage detection.4–6 Similarly, nearly all patients with breast and prostate cancers diagnosed at early stages can expect to live at least 5-years post-diagnosis.4 Highly aggressive cancer types may benefit even more from early detection. Pancreatic ductal adenocarcinoma carries a long-term survival rate of <8.5%, but documented cases of incidental pancreatic cancer diagnoses at early stages have shown 5-year survival rates over 80%, nearly a 10-fold improvement.7 These survival and cost benefits of early stage detection have motivated public cancer screening programs.8 For example, in the United States, invasive squamous cervical cancer incidence and deaths have declined 70% since 1950 due to early detection of precancerous and cancerous lesions through Pap smear testing.9 However, not all cancer types are easily accessible for screening, leading to later stage detection with higher morbidity and mortality. Lung cancer, the most prevalent cancer worldwide and one of the most aggressive cancers to treat, is plagued by late diagnosis by poor screening methods, and has a corresponding 5-year survival rate of less than 15%.10,11 These data demonstrate that early detection of cancer can tremendously improve patients’ clinical outcomes and accelerate the course of therapeutic intervention to address malignant processes before genomic lesions accumulate and tumor metastases occur.
2. The Promise of Liquid Biopsy for Cancer Diagnostics
Conventional cancer diagnostics and management rely on obtaining images and biopsies of primary tumors or their metastases to characterize their molecular subtype and genomic aberrations.12 Large-scale sequencing efforts, such as The Cancer Genome Atlas (TCGA) or Pan-Cancer Atlas of Whole Genomes (PCAWG), have catalogued the multi-omic changes underlying tissue-specific malignant transformation and revealed shared and distinct features between cancer types. When compared to new samples, these cancer atlases can inform prognoses and, at times, suggest tailored therapeutic options for patients based on their molecular profiles, offering the opportunity to provide personalized cancer medicine.
Many challenges exist, however, with obtaining routine tissue biopsies in certain anatomical regions due to local vasculature, delicate tissue structures, and other surgical complexities. Genetic heterogeneity between multiple malignant lesions may also limit prognostic conclusions or therapy choices when sampling genomic aberrations from only one site. Moreover, collection of all lesions in circumstances of metastasis is usually unfeasible due to impacts on patient morbidity and quality of life. Collecting longitudinal biopsies of solid tumors to characterize response to treatment further risks compounded tissue damage, particularly since subsequent biopsies may reveal non-malignant tissue.
“Liquid biopsies” attempt to address several of these tissue biopsy limitations and are based on the premise that cell-free nucleic acids (cfNAs) enter circulation through cell turnover or cell death. Circulating nucleic acids can be obtained from a routine and easily accessible blood sample, and because multiple malignant lesions are connected by systemic blood flow, sampling of cfNAs potentially captures all genetic alterations from multiple tumors without performing tissue biopsies on each. Paired with TCGA and PCAWG maps of cancer type-specific alterations and matched patient survival, cfNAs could be used to prognose patients and tailor their treatments.13–15 Liquid biopsies are not restricted to blood, and could include urine, saliva or other biofluids, depending on the targeted cancer.16–19 The minimally invasive nature of liquid biopsies is highly advantageous and permits successive, serial sampling while maintaining strong concordance with the genomic and epigenomic lesions identified in matched solid tumor biopsies.20–22
Current development efforts to create liquid biopsy diagnostics for cancer encompass a wide array of analytes, including circulating tumor-derived DNA (ctDNA), RNA (ctRNA), microRNAs, circular DNAs and RNAs, exosomes, and intact circulating tumor cells (CTCs). However, the main research and commercial focus of liquid biopsy-based assay development has focused on ctDNA due to its stability and interpretability when compared against tumor mutation profiles.17,23–25 There are numerous sources of ctDNA, ranging from tumor cell apoptosis and necrosis to secretion of exosomes or other vesicular structures.26 Because each genome equivalent of ctDNA should bear the genetic mutations of the tumor cell where it was derived, deep sequencing of bloodborne ctDNA can provide a minimally invasive means of detecting and characterizing early-stage malignant processes.27,28
The presence of cell-free DNA (cfDNA) fragments in blood was first reported by Mandel and Métais in 1948, but it was not until 46 years later in 1994 that two seminal reports by Sorenson et al. and Vasioukhin et al. set the stage for modern uses of blood-based liquid biopsies for oncological indications.29–31 An increase in serum cfDNA concentration from patients with established cancers prompted Sorenson and colleagues to test for mutated KRAS sequences in plasma cfDNA of patients with pancreatic ductal adenocarcinoma.30,32–34 Using allele-specific primers, Sorenson et al. detected mutated KRAS sequences in plasma cfDNA and showed that the KRAS mutation was identical to that found in the patients’ tumors, thereby demonstrating that the mutant KRAS fragment in plasma was of tumor origin.30 Similarly, Vasioukhin et al. demonstrated PCR-based identification of NRAS point mutations in plasma of patients with myeloid disorders. Noting that the presence of RAS mutations in patients with myeloid disorders was a poor prognostic factor, the authors suggested the opportunity of plasma sequencing to diagnose and prognose a patient without an invasive bone marrow biopsy. They also predicted that “since the presence or absence of mutations might reflect the success of clinical remission by chemotherapy, the easy accessibility of plasma for a detection test would be very useful in monitoring the disease,” prescient in light of subsequent development of the field.31 Parallel advances in cfDNA analysis for noninvasive prenatal testing further demonstrated the promise of liquid biopsy for early stage cancer detection when incidental identification of cancers in presymptomatic pregnant women was reported by two groups.35,36 In these studies, fetal aneuploidy analysis via high-throughput sequencing of maternal plasma cfDNA identified copy number variations that could not be attributed to the fetal genome. Further examination of these women via orthogonal technologies, such as magnetic resonance imagining, revealed the presence of various cancers, unintended yet serendipitous demonstrations that cfDNA analysis could detect cancers in the absence of overt clinical symptoms.
Nearly thirty years later, blood-based liquid biopsies in oncology are now becoming routine for management of established disease in clinical research settings.37 Detection of mutant ctDNA alleles, either alone or in combination with other circulating biomarkers, has revolutionized cancer diagnostics; however, significant biological and technical hurdles remain to attain the requisite sensitivity for reliable detection of early-stage cancers. In this review, we discuss current applications of liquid biopsy in oncology and the analytical challenges germane to using liquid biopsies for early-stage cancer detection.
3. Use of Liquid Biopsy in Cancer Management
Disease Prognosis
Leon and colleagues first showed in 1977 that total cfDNA concentrations (i.e., ctDNA from cancer cells + cfDNA from non-cancer cells) were higher in cancer patients than healthy controls using a competitive radioimmunoassay. They analyzed the serum of 173 patients with various cancer types and 55 healthy individuals, reporting that the average DNA concentration in cancer patients was 180 ± 38 ng/mL (50,279 haploid genome equivalents/mL, assuming 3.58 pg/haploid genome) and 13 ± 3 ng/mL (3,631 genome equivalents) in healthy controls.32 This finding has been reproduced in plasma cfDNA across numerous cancers and has served as a simple, easy to measure, diagnostic indicator. Other studies have also shown prognostic utility of cfDNA amounts. For example, Sirera et al. measured human telomerase reverse transcriptase (hTERT) in cfDNA of stage IIIB and IV non-small cell lung cancer (NSCLC) patients via real-time PCR, and found that high pretreatment hTERT level was a poor prognostic indicator for time to progression and overall survival.38
In addition to using total or per-gene cfDNA concentrations as prognostic indicators, analysis of ctDNA-specific amounts has proved to be very powerful. In a prospective study of 230 patients with resected stage II colon cancer, Tie et al. sought to determine if ctDNA could be used to triage patients who should receive postoperative adjuvant chemotherapy from those who would not benefit from the treatment. Using next generation sequencing of plasma ctDNA, the authors found that 100% of patients who had detectable ctDNA at their first postoperative visit (4–10 weeks after surgery) relapsed within three years while only 10% of the ctDNA negative group relapsed, thereby defining a group—ctDNA-positive patients—who were at extremely high risk of radiologic recurrence when not treated with adjuvant chemotherapy.39 The prognostic value of ctDNA concentration has also been demonstrated in lung, breast, melanoma, and ovarian cancers.40–44 This information empowers clinicians to determine which patients may benefit from indication-specific therapeutic options and to more effectively tailor the amount and duration of the selected therapy.
Monitoring Treatment Efficacy and Resistance
Longitudinal genomic profiling of tumors during and after patient treatment is a significant challenge for traditional tissue biopsy due to the difficulty in obtaining enough tissue for analysis, risk of permanent tissue damage from repeated sampling, and tumor heterogeneity precluding representative sampling.45,46 These problems are exacerbated if the tumors are located in anatomically difficult-to-access or dangerous locations.22 Liquid biopsy-based assays have proved an attractive alternative to traditional biopsy by being minimally invasive and, in theory, representative of all tumors that access circulation. These characteristics enable serial monitoring of tumor fate during therapy and real-time evolution of resistance mechanisms. A landmark study by Murtaza and colleagues showcased this application of liquid biopsy, analyzing serial plasma samples by whole exome sequencing to track the genomic evolution of different metastatic cancers in response to therapy.47 Plasma-derived exome samples were collected from six patients with advanced breast, ovarian, and lung cancers before and after treatment, spanning multiple treatments over the course of 1–2 years. This led to the identification of mutant alleles associated with the emergence of therapy resistance, such as an activating mutation in PIK3CA following treatment with paclitaxel; a truncating mutation in RB1 following treatment with cisplatin; and the T790M resistance-conferring mutation in EGFR following gefitinib treatment. The ability to detect these mutations through minimally-invasive, serial liquid biopsies provide important insights into selection pressures exerted on tumors during therapy and may suggest combination therapies that preempt or more effectively combat evolved resistance. The use of this technique to assess treatment response and emerging resistance has been applied extensively in non-small cell lung cancer and has shown similar value in colorectal cancer, metastatic melanoma, and gastric cancers.48–52
A recent study further suggested that liquid biopsies can provide molecular insights not observable from a single lesion tissue biopsy. Parikh and colleagues conducted a direct comparison of liquid versus tissue biopsy for detecting acquired resistance and tumor genomic heterogeneity in gastrointestinal cancers.53 In this study, plasma cfDNA from a cohort of 42 patients with molecularly characterized gastrointestinal cancers with progressive disease despite targeted therapy were analyzed. Sequence analysis identified at least one validated resistance mutation in 76% of patients, with 53% of patients exhibiting multiple resistance mutations. In total, 78 different resistance mutations were identified. Whole exome sequencing analysis of matched post-progression single lesion tissue biopsies (possible for 23 patients) identified resistance mutations in 11 out of 23 (48%) patients, whereas cfDNA analysis identified at least one resistance mutation in 20 out of 23 (87%) patients. In five cases where multiple biopsies or rapid autopsies could be performed, multiple resistance mutations were identified within distinct metastatic lesions. These results indicate that acquired resistance to targeted therapy in gastrointestinal cancer is highly heterogeneous and demonstrate that liquid biopsy can identify multiple resistance mechanisms across distinct metastatic loci within the same patient—a very advantageous feature when we consider more than 60% of solid tumor-related deaths are attributed to metastatic disease rather than advanced local disease.54
Treatment Selection – Companion Diagnostics
Blood-based liquid biopsies for cancer first became medically reimbursable with the 2016 FDA approval of the cobas EGFR Mutation Test v2, which analyzes plasma cfDNA from patients with metastatic non-small cell lung carcinoma (NSCLC) not eligible for tissue biopsy. The cobas assay is a real-time PCR test that qualitatively detects 42 defined mutations of the EGFR gene in exons 18–21, exon 19 deletions, and the T790M resistance mutation, all of which can affect efficacy of the receptor tyrosine kinase inhibitor erlotinib.55 In Phase III studies utilizing the cobas assay, patients whose plasma ctDNA were positive for exon 19 deletion and/or L858R substitution mutations had improved progression-free survival with erlotinib treatment compared to those treated with a non-targeted combination of gemcitabine plus cisplatin. The test has received further approval as a companion diagnostic with Iressa (gefitinib)—another EGFR inhibitor—for the first-line treatment of patients with NSCLC harboring exon 19 deletions or the exon 21 L858R substitution mutation, as well as with Tagrisso (osimertinib), a third-generation EGFR tyrosine kinase inhibitor that selectively targets T790M mutation, which often emerges as an evolutionary consequence of first-line EGFR inhibitor treatment.56
Another single gene qPCR assay—Qiagen’s therascreen PIK3CA RGQ PCR liquid biopsy assay—received regulatory approval in 2019 for the detection of 11 mutations in the phosphatidylinositol 3-kinase catalytic subunit alpha (PIK3CA) gene present in plasma samples of patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced, or metastatic breast cancer. This test aids clinicians in identifying breast cancer patients who may be eligible for treatment with the PIK3CA inhibitor, alpelisib, in combination with fulvestrant, a selective estrogen receptor degrader, based on PIK3CA mutations.57
FDA approval of the single gene cobas EGFR and therascreen PIK3CA assays marked a significant clinical advance in liquid biopsies and signaled a paradigm shift in how cancer biopsies are collected. More complex assays were approved by the FDA in 2020: the next-generation sequencing (NGS)-based Guardant360 CDx and FoundationOne Liquid CDx companion diagnostic assays for solid tumors. These assays use targeted hybridization-based capture to enrich ctDNA sequences from prepared plasma cfDNA NGS libraries to provide genomic alteration data on numerous analytes from a single blood draw. The Guardant360 assay was approved as a companion diagnostic for osimertinib in NSCLC using a limited panel of EGFR alterations but also received approval for tumor mutation profiling, wherein single nucleotide variants (SNVs) and insertions and deletions (indels) in 54 genes are detected; additionally, copy number variations (CNAs) in two genes and fusions in four genes are reported. FoundationOne Liquid CDx assay was approved as a companion diagnostic for three lung cancer therapies and a prostate cancer therapy; moreover, analogous to Guardant360, the FDA approved Foundation’s assay for tumor profiling, wherein genetic alterations in 324 genes are detected to help a physician develop personalized therapeutic options for the patient. The scale of Foundation Liquid CDx’s analyte panel makes it possible to perform comprehensive genomic profiling of the entire exonic region of cancer-relevant genes in advanced-stage cancers through a simple blood draw. This represents a significant technical advance and one that facilitates tumor analysis in cases where insufficient DNA or poor DNA quality is available by tissue biopsy.58,59
4. Commercial liquid biopsies for population screening or early detection
The success of liquid biopsy-based detection of advanced cancers and the aforementioned incidental detection of cancers in asymptomatic women undergoing non-invasive prenatal testing has spurred significant investment in clinical and commercial efforts in early-stage cancer detection (Table 1). Early detection tests, if sensitive enough, could be applied to symptomatic patients to substantially reduce the time between clinical presentation and disease diagnosis, thereby increasing the pre-metastasis window of opportunity for therapeutic intervention. If specific enough, these tests could also be employed as routine (e.g., yearly) screening tests in healthy individuals to identify instances of asymptomatic cancer.
Table 1.
Liquid biopsy technologies for early detection of cancer and monitoring of disease recurrence. Each of these tests employs cfDNA, either alone or in combination with other circulating biomarkers, to detect cancer, perform tumor mutation profiling, or detect cancer recurrence.
| Company | Cancer Application(s) | Patient Status | Product Name | Technology Description |
|---|---|---|---|---|
| Biocept | Lung and breast cancer mutation detection | Previously diagnosed | Target Selector™ | Multiplex PCR amplification via “Switch-Blocker” technology: suppression of wild-type gene sequences focuses PCR on mutant sequences of interest. Amplification is followed by amplicon sequencing.101 |
| Clinical Genomics | Colorectal cancer recurrence | Previously diagnosed and treated | Colvera® | Methylation-specific PCR-based detection of methylated BCAT1 and IKZF1 ctDNA post-bisulfite treatment.102 |
| Delfi | Multi-cancer screening | No prior diagnosis of cancer | TBD | cfDNA whole genome NGS followed by fragmentation analysis.91 |
| Epigenomics | Colorectal cancer detection | No prior diagnosis of cancer | Epi proColon® | Methylation-specific PCR-based detection of methylated Septin9 post-bisulfite treatment.60 |
| Foundation Medicine (Roche) | Tumor mutation profiling | Previously diagnosed | FoundationOne® | Hybridization probe enrichment of ctDNA mutations followed by NGS.58 |
| Freenome | Colorectal cancer detection | No prior diagnosis of cancer | PreEmpt CRC™ | Multi-analyte detection (ctDNA, cfRNA, proteins) with machine learning classification.103 |
| Grail | Multi-cancer screening | No prior diagnosis of cancer | Galleri™ | Bisulfite sequencing: bisulfite treatment of cfDNA, targeted methylation hybridization probe enrichment of ctDNA followed by NGS.66 |
| Guardant | Multi-cancer screening (with a first indication in colorectal cancer) | No prior diagnosis of cancer | LUNAR-2 | Multi-omic analysis of cfDNA incorporating methylation, mutation, and fragmentomic analyses.104 |
| Inivata | Tumor mutation profiling | Previously diagnosed | InVisionSeq™ | Tagged-amplicon deep sequencing (TAm-Seq).105 |
| Micronoma | Lung cancer diagnosis | No prior diagnosis of cancer | Oncobiota™ | Metagenomic sequencing of total cfDNA followed by machine learning-based disease classification.93 |
| Natera | Colorectal cancer molecular residual disease, recurrence monitoring | Previously diagnosed and treated | Signatera™ | Personalized mutation detection: mutations present in patient’s tumor(s) are used to design custom ctDNA sequencing panels.106 |
| Nucleix | Lung cancer diagnosis | No prior diagnosis of cancer | Lung EpiCheck® | Methylation-specific PCR-based detection of methylated markers post-bisulfite treatment.107 |
| Thrive (Exact Science) | Multi-cancer screening | No prior diagnosis of cancer | CancerSEEK | Multiplex amplicon sequencing + proteomic biomarker detection.63 |
| VolitionRX | Lung, colorectal, and prostate cancers; single cancer, ELISA-based screening | No prior diagnosis of cancer | Nu-Q™ | ELISA-based quantification of nucleosome-bound cfDNA.108 |
To date, the only blood-based liquid biopsy assay that has obtained FDA approval for cancer diagnosis is Epigenomics’s Epi proColon colorectal cancer screening assay.60 This is a PCR assay for the qualitative detection of methylated Septin9 ctDNA isolated from 3.5 milliliters of patient plasma, since methylation of certain CpG motifs in the promoter region of the SEPT9_v2 transcript has been associated with colorectal cancer but not healthy tissue. Specifically, the Epigenomics assay utilizes bisulfite-treatment of isolated cfDNA and methylation-specific primers to detect the presence of methylated Septin9.61 The test obtained FDA approval in April 2016 and is indicated to screen adults 50 years or older, defined as the average risk for colorectal cancer, who are unable or unwilling to undergo routine screening tests (e.g., flexible sigmoidoscopy, colonoscopy, or stool tests). As patient participation in conventional colorectal screening programs is suboptimal, a minimally-invasive blood test may boost patient testing and concomitant identification of pre-metastatic colorectal cancer lesions. However, despite the regulatory approval of the Epi proColon assay, the Centers for Medicare and Medical Services recently ruled that the Epi proColon assay does not meet the sensitivity (≥ 74%) and specificity (≥ 90%) thresholds established by two existing assays—fecal immunochemical tests and the multi-analyte stool DNA test Cologuard—to qualify for national reimbursement coverage, a decision that will undoubtedly reduce clinical uptake and utilization of this first blood-based ctDNA cancer diagnostic.62
In contrast to the single biomarker Epi proColon assay, an array of multi-analyte blood-based liquid biopsy cancer diagnostics are currently in development (Table 1). For example, CancerSEEK (Thrive Earlier Detection) is a blood test that combines ctDNA and protein biomarkers to screen for eight types of cancer, five of which have no existing approved screening tests (ovarian, liver, stomach, pancreatic, and esophageal cancer).63 The CancerSEEK assay utilizes multiplex PCR analysis of cfDNA derived from 7.5 milliliters of patient plasma to achieve detection of driver mutations at 2,001 genomic loci across 16 genes. Amplicon sequencing data is then combined with the protein concentrations of eight cancer-associated protein biomarkers (cancer antigen 125 (CA-125), carcinoembryonic antigen (CEA), cancer antigen 19–9 (CA19–9), prolactin (PRL), hepatocyte growth factor (HGF), osteopontin (OPN), myeloperoxidase (MPO), and tissue inhibitor of metalloproteinases 1 (TIMP-1)) to arrive at a cancer classification. In a test of 1,005 patients previously diagnosed with stage I-III breast, colorectal, gastric, liver, lung, esophageal, ovarian, or pancreatic cancer, a median of 70% of patients tested positive for cancer via CancerSEEK. Sensitivities ranged from 33% for breast cancer to 98% for ovarian cancer with excellent (>99%) specificity overall. By leveraging machine learning, Cohen et al. developed a classification system based on sequencing and proteomics data that could localize the source of the cancer to two anatomic sites in a median of 83% of CancerSEEK-positive patients, or to a single anatomic site or single organ in a median of 63% of the CancerSEEK-positive patients. Tissue of origin (TOO) determination is a critical component of any early cancer detection platform, as circulating genomic and/or proteomic signatures of tumors may be apparent well-before localized tumors can be pinpointed radiologically. However, since the CancerSEEK study population consisted largely of symptomatic cancers, it will be of great interest to see if future embodiments of this method can provide both a means of cancer detection and TOO localization when extended to asymptomatic individuals with early stage disease; alternatively, as shown in the second generation CancerSEEK DETECT-A study, TOO localization may be better addressed by orthogonal means, such as through PET-CT.64 Although the DETECT-A study used an earlier version of CancerSEEK that did not employ machine learning, Thrive, since acquired by Exact Sciences, recently completed an unpublished 3000 subject clinical trial (Detecting Cancers Earlier Through Elective Plasma-based CancerSEEK Testing (ASCEND)), consisting of 1000 subjects with known or suspected cancer and 2000 non-cancer controls. The results from the ASCEND trial should clarify how their assay incorporating machine learning works with early stage cancers.65
In contrast to Thrive’s assay, which combines driver gene mutations and cancer-associated proteomic markers to arrive at a diagnosis, GRAIL Inc. (pending Illumina acquisition) uses differential DNA methylation of genomic CpG sites to discriminate among different cancers and cancer versus non-cancer samples.66 GRAIL has set an ambitious goal to accurately screen for more than 50 unique cancer types from a single sample through targeted bisulfite sequencing analysis of ctDNA methylation patterns. DNA methylation-based biomarkers have been explored in many disease areas but may prove particularly useful in liquid biopsy-based cancer diagnostics as a means of determining which ctDNA fragments are truly tumor-derived. While most driver mutations in oncogenes (e.g., TP53, KRAS) are common among cancers regardless of their TOO, CpG methylation profiles are highly specific to tissues and tumors-derived therefrom, potentially enabling a more exact diagnosis of cancer.67,68 In addition, there are millions of CpG sites throughout the human genome whose methylation states (methylated versus unmethylated) can comprise a cancer-specific signature whereas canonical ctDNA mutations are limited in copy number/genome and therefore impose a sensitivity limitation for detection.69
GRAIL has dedicated considerable resources towards constructing a database of DNA methylation patterns from thousands of individuals with different cancer types, healthy non-cancer controls, and non-cancer controls with other medical conditions. By applying machine learning algorithms to these data, GRAIL has developed classifiers to distinguish cancer-associated methylation patterns from non-cancer patterns and have used these classifiers on large-scale clinical studies. In a recent publication detailing the results of their clinical validation study featuring liquid biopsy analyses for over 50 cancer types, GRAIL’s classifier was able to predict the TOO in 96% of samples with an apparent cancer-signature (344/359) and, of these, 93% (321/344) were accurate classifications, a compelling demonstration of the diagnostic power of epigenetic signatures in liquid biopsies.66 Cancer diagnostic tests based on alternative ctDNA methylation status (i.e., 5-methylcytosine and 5-hydroxylmethylcytosine) have been described by others with similar degrees of success in identifying the TOO.70–73
Although GRAIL’s assay specificity (99.3%) was impressive, the reported sensitivities for early stage (I and II) cancers leave room for improvement, particularly for what is intended to serve as a population-wide cancer screening diagnostic. Stage I cancers were detected with a sensitivity of just 18% (95% confidence interval (CI), 13–25%) and stage II cancers 43% (CI, 35–51%). Such low sensitivities for stage I and II cancers are neither surprising nor unique to GRAIL’s assay, as ctDNA-based diagnostic assays have reported similar difficulties in detecting early-stage cancers that partially stem from the paucity of cancer-derived DNA in circulation at any given time. One potential exception is the pan-cancer screening assay, PanSeer, developed by Singlera Genomics and colleagues that analyzes DNA methylation patterns at 10,613 CpG sites across 477 genomic regions and can reportedly identify the presence of certain cancers (colorectal, esophageal, liver, lung, and stomach) through liquid biopsy up to four years prior to conventional diagnosis methods.73 Since reporting these results in June of last year, Singlera has raised $150 million USD in Series B financing to expand PanSeer testing with a large prospective study of healthy individuals to determine if their screening assay can reduce cancer deaths in a cost-effective manner.74
5. Technical and biological hurdles for early detection via ctDNA
Much has been written about the technical difficulties associated with early-stage ctDNA cancer diagnostics.75–79 Principal among these difficulties is that small, localized tumors, which do not exhibit ample cell turnover, do not release sufficient amounts of DNA into the circulation. As discussed above, plasma from cancer patients contains on average more cfDNA than healthy controls, but, in nearly all cases where cfDNA concentrations have been measured, the cancer cohorts consisted of patients whose tumors were already diagnosed by conventional means. Thus, it is difficult to ascertain from the existing literature how much ctDNA may be present in asymptomatic individuals harboring tumors below the limit of detection of existing imaging modalities.
Furthermore, there may be a substantial difference between how much ctDNA an early-stage diagnostic can theoretically detect versus what it needs to detect to make a substantial improvement in patient care. Diamandis and Fiala have argued that tumors ≤5 millimeters in diameter are an optimal size for early and curable cancer detection. Using breast cancer as a model, they review data showing that ~6% of tumors of 5 millimeters in diameter progress but are poorly detected by mammography in 26% of cases.28 They further suggest that liquid biopsy detection of a tumor 10 millimeters in diameter would not represent a diagnostic improvement, as more than 90% of breast cancer tumors 10 millimeters in diameter or larger are currently detected by mammographic screening. Likewise, low dose computed tomography can identify potential cancerous lung nodules with diameters as small as 4 millimeters.80 Thus, if a liquid biopsy assay can diagnose tumors in the 5–10 mm diameter range (approximately 0.06–0.5 cm3 or 6×106-1×108 cells/tumor), per Diamandis and Fiala’s assessment, it would represent a true advance in diagnostic capability.
But is it reasonable to expect liquid biopsy assays to detect tumors of such small size and cellularity? Diamandis and Fiala argue that overcoming this challenge is highly unlikely based on recent estimates comparing ctDNA amounts and tumor volumes. For instance, Abbosh et al. measured the mutant allele frequency (MAF) in patients with early stage I lung cancers and showed that 10 cm3 (27 millimeter diameter) tumors had an average MAF of just 0.1%, or 1 mutant DNA copy per 1000 non-mutant copies.52 Extrapolating this measurement, Diamandis and Fiala calculate that an early stage tumor of 5 millimeter diameter would contribute only 1 mutant DNA genome equivalent (GE) per every 160,000 GE in circulation. Assuming that an asymptomatic patient with early-stage cancer has a cfDNA concentration similar to healthy individuals, or ~6200 haploid GE per 4 milliliters of plasma from a 10 milliliter blood draw, then one would need to draw and analyze over 100 milliliters of blood to detect a tumor 5 millimeters in diameter—an unlikely scenario. These calculations are consistent with a recent mathematical model of ctDNA shedding.81 Support for these calculations are also found in Phallen et al. and Cohen et al., wherein successful instances of liquid biopsy detection of early-stage cancers were observed only in symptomatic patients with MAFs more than 0.01%, suggesting that ctDNA-based detection requires at least a 10–12.5 millimeter diameter tumor for enough ctDNA to be present in the circulation, which is not an improvement over conventional screening methods.27,63,28 Nonetheless, caveats to this mathematical argument may exist. If cfDNA concentration in early stage, asymptomatic individuals correlates more strongly with tumor metabolic activity than size, then small tumors may still shed sufficient DNA for detection. In addition, the MAF measured in the above studies could be a function of the sensitivity of the techniques employed, as technique-dependent differences in MAF for various cancers and stages have been previously observed.82,83
Another major complicating factor in ctDNA analysis that has drawn a lot of attention in recent years is a phenomenon referred to as clonal hematopoiesis (CH).84–86 In CH, white blood cells that constitute the main source of cfDNA in circulation accumulate somatic mutations in specific oncogenes (e.g., KRAS, JAK2, and TP53) and copy number alterations in genes associated with hematological malignancies as people age. While only about 1% of all patients with CH develop a hematological malignancy, the cfDNA originating from clonal leukocytes can be a confounding factor in ctDNA analysis when CH-derived mutations are falsely interpreted as originating from the tumor under investigation.87 For example, in a study of patients with advanced EGFR-mutant non-small cell lung cancer, Oxnard and colleagues found that most JAK2 mutations identified in plasma cfDNA were traced to peripheral blood cells from these same patients.88 More recently Razavi et al. carried out an extensive sequencing study to fully characterize the source of mutations found in ctDNA.89 To this end, they performed ultra-deep sequencing (60,000X depth) of 508 genes in ctDNA isolated from 124 patients with various metastatic cancers, genomic DNA isolated from patient-paired white blood cells, and, when available, matched tumor tissue. The same analyses were performed on 47 non-cancer controls (i.e., cfDNA and paired WBCs) and collectively revealed the confounding nature of CH on ctDNA analyses. Specifically, 82% of the mutations found in the cfDNA of non-cancer control subjects and 53% of the mutations found in ctDNA of cancer patients were also identified in their respectively matched WBC-derived genomic DNA, providing solid evidence that CH is a major reservoir of cfDNA mutations and tremendously diminishes assay specificity. Since aging is a risk factor for both CH and carcinogenesis, Razavi et al. strongly recommended that any ctDNA assay incorporate a paired analysis of patient-derived WBC genomic DNA to ensure that leukocyte-originating mutations are not misinterpreted as tumor-derived signals. It remains to be seen, however, how these findings may affect recent regulatory approvals for Guardant Health’s and Foundation Medicine’s ctDNA-based tumor mutation profiling. Epigenetic changes with age that could potentially confound methylome-based ctDNA diagnostics also are unknown.
6. The potential for microbiome-based liquid biopsy diagnostics in cancer
As detailed above, extensive effort has been applied to identify, distinguish, and quantify tumor-derived DNA from non-tumor, wild-type DNA sequences to provide a sensitive and specific diagnosis of cancer. Although CH poses a significant challenge to the specificity of ctDNA-based diagnostics, alternative ctDNA analysis methods based on differential DNA methylation or DNA fragmentation patterns are being actively explored90,91. Another potential orthogonal source of cfDNA comprises non-human species, derived from bacteria, viruses, fungi, and phages that live on and within the human body. Despite many claims that tumors are sterile, recent evidence has shown overlapping immunohistochemistry, immunofluorescence, electron microscopy, and sequencing data of microbes in tumors in ~10 cancer types.92 These cancer types appear to harbor distinct microbiomes from each other, suggesting a diagnostic opportunity.93,94 Moreover, historical observations between clinical bacteremias and subsequently diagnosed colorectal cancers, despite no or minimal presenting symptoms of those cancers, have been described since the 1970s and were validated in a meta-analysis of more than 13,000 patients in Hong Kong.95,96 Recent work by Poore et al. suggests that broadening the scope of cancer-associated DNA sequences to include circulating, cell-free microbial DNA may provide an attractive means of bypassing several problems posed by CH and the low MAF inherent to early-stage tumors while also facilitating TOO classification.93
Prompted by studies detailing that some cancer types are intimately associated with microbial activity, Poore and colleagues characterized the extent and taxonomic diversity of microbial nucleic acids found across a range of human tumors in a comprehensive computational study. To accomplish this, the authors analyzed all treatment-naïve whole genome and transcriptome studies from The Cancer Genome Atlas (TCGA) for their bacterial, viral, and archaeal nucleic acid content. The TCGA dataset consisted of 18,116 samples (4,831 whole genome sequencing (WGS) and 13,285 RNA-seq) from 10,481 patients across 33 different tumor types and included non-neoplastic tumor-adjacent tissue and patient blood samples. Of the 6.40 × 1012 sequencing reads in TCGA, 7.2% were classified as non-human, of which 35.2% could be taxonomically assigned to bacteria, viruses, or archaea using a comprehensive reference microbial database of 59,974 total microbial genomes. To account for microbial nucleic acid contaminants that may have been introduced during the TCGA cohort DNA and RNA extractions, an in silico decontamination pipeline featuring statistical contaminant inference and historical known extraction kit contaminant removal was employed, discarding up to 91.3% of microbial taxa from the data in the most stringent analyses. The decontaminated datasets were then used to train stochastic gradient-boosting machine learning (ML) models (using a 70%−30% train-test split for all cancers) to discriminate between and within types and stages of cancer. The trained models could effectively discriminate one cancer versus all others (n=32 tested cancer types) and tumor versus normal adjacent tissue (n=15 types of cancer with sufficient samples) solely using microbial DNA (mbDNA) and/or microbial RNA.
Because colon cancer is epidemiologically linked to clinical bacteremia, Poore et al. explored TCGA blood-derived normal samples (cancer patient whole blood or buffy coat DNA preparations; n=1866 WGS samples across 20 cancer types) and found cancer type-specific mbDNA signatures.95,96 Additionally, the ML models trained on this blood-derived mbDNA could classify stage Ia-IIc cancers with high sensitivity and specificity.95,96 Remarkably, ML models trained solely on mbDNA features correctly classified many cancers that lacked all genomic alterations reported by the Guardant360 and FoundationOne liquid biopsy assays, demonstrating that mbDNA signatures can be truly orthogonal, cancer genome-independent biomarkers for cancer classification.
To further validate this finding and extend it to a more standardized liquid biopsy format, plasma-derived cell-free microbial DNA (cf-mbDNA) from 100 patients with stage III-IV lung (n=25), prostate (n=59), or melanoma (n=16) cancers were compared to cf-mbDNA from 69 HIV-negative, healthy patients. Isolating cf-mbDNA with a commercially available cfDNA extraction kit with dozens of experimental contamination controls, the authors applied the same TCGA microbial detection pipeline and machine learning steps as before with the exception that, due to the small sample sizes, nested leave-one-out iterative machine learning (LOOML) was performed instead of train-test splits. Again, cf-mbDNA-based signatures resulted in accurate discrimination between cancer types and cancer versus healthy, with lung and prostate cancer showing the highest and second highest performance, respectively. Specifically, 22/25 lung cancer samples were correctly classified via LOOML with 5 false positives, yielding a sensitivity and specificity of 88% and 93%, respectively. Commercial efforts are now underway to further explore cancer-associated cf-mbDNA signatures for diagnostic applications with an initial focus on lung cancer, for which effective, early-stage diagnostics are desperately needed.97 It will be critical to expand the breadth of controls to include non-healthy, non-cancer complications of the lung such as chronic obstructive pulmonary disease (COPD), sarcoidosis, interstitial fibrosis, and other conditions where microbial involvement may overlap substantially with that of lung cancer, potentially decreasing the specificity of the cf-mbDNA signature for cancer.
The development of cf-mbDNA-based diagnostics is nascent, but it is not difficult to imagine their use to complement other cancer liquid biopsies and potentially mitigate problems associated with existing ctDNA analyses (Figure 1). Because cf-mbDNA is wholly independent of the human genome, the accumulation of somatic mutations in CH that decreases ctDNA assay specificities cannot alter the specificity of cf-mbDNA signatures. Assaying cf-mbDNA may improve liquid biopsy sensitivities as well. Whereas ctDNA-based assays are limited by the frequency of targeted mutant alleles (often 1:100–1:1000 ratios of mutant:normal alleles), cf-mbDNA typically comprises 1–3.5% of the reads in a shotgun metagenomic dataset in our experience, independent of cancer stage. This is a rich data source that, when coupled with machine learning, can provide sensitive and specific cancer-specific signatures containing hundreds of features. Furthermore, because mbDNA is truly orthogonal to the human genome, mbDNA signatures can be coupled with range of cancer-associated host biomarkers—such as the driver mutations discussed above, methylomics, miRNAs, circular DNAs or RNAs, or proteomic markers—to generate new kinds of multi-analyte, multispecies assays that leverage the full breadth of circulating biomarkers in cancer.
Figure 1. Genomic and metagenomic features for minimally invasive, blood-based detection of cancers.
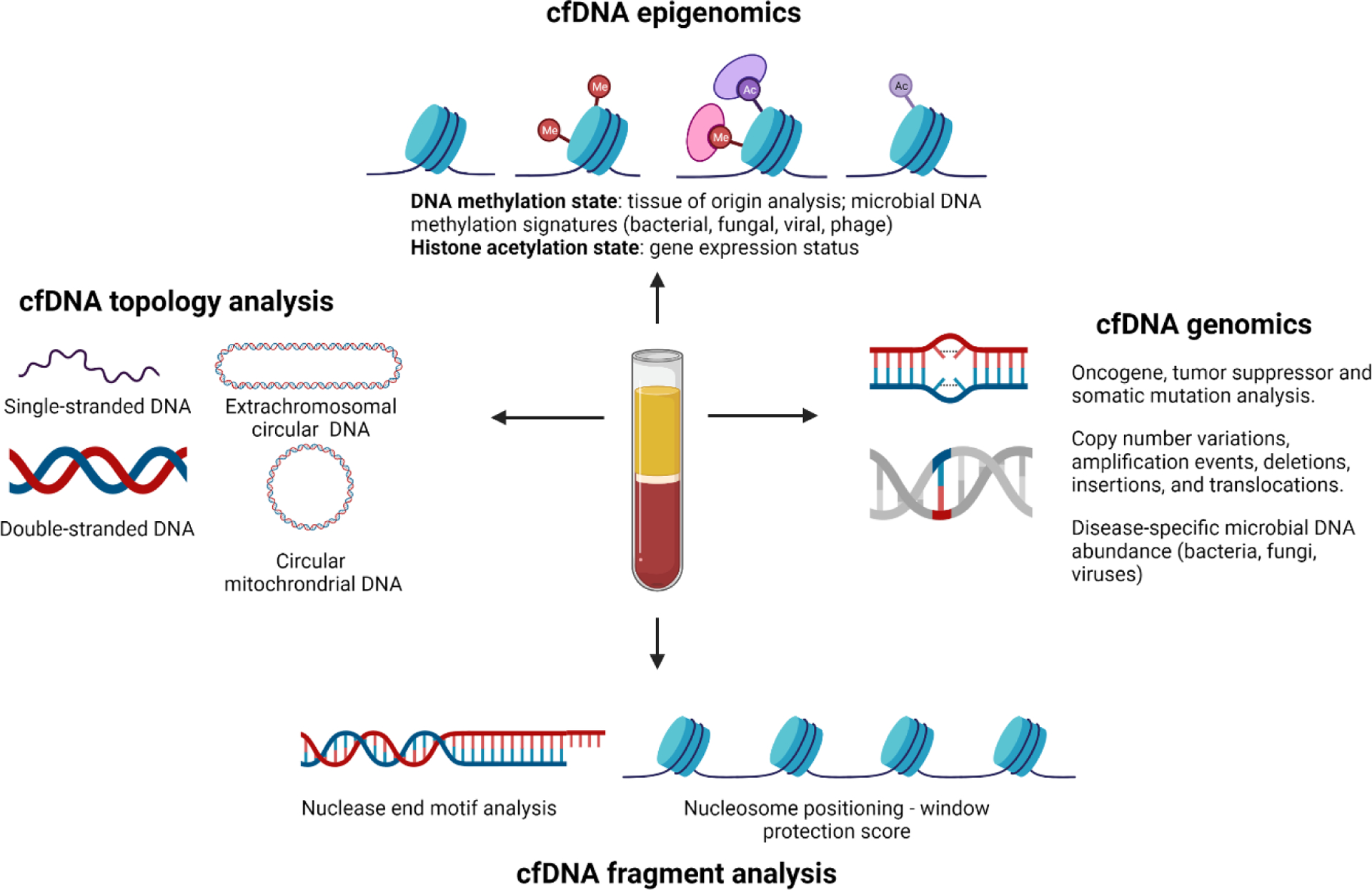
(Right) Existing commercial liquid biopsy assays focus on single gene or multi-gene analyses of ctDNA mutations present in oncogenes, tumor suppressor genes or validated biomarkers (e.g., EGFR mutations) that aid in drug selection. cfDNA shotgun metagenomic sequencing expands traditional cfDNA analyses to encompass all microbial DNA sequences present in circulation, enabling discovery of cancer-microbiome associations. (Top) Epigenetic analysis of cfDNA has traditionally focused on the methylation state of purified mammalian cfDNA, which serves as a powerful signature of ctDNA tissue of origin. Epigenetic marks on circulating microbial DNA may likewise serve to identify/enrich disease-specific microbial associations.109,110 Recent work has shown that epigenetic hallmarks of gene expression or suppression on cell-free nucleosomes (e.g., histone acetylation and methylation marks) be used as a surrogate for RNA-based transcriptional analysis.111 (Left) Single-stranded DNA library preparation methods increase sequencing reads derived from mitochondrial and microbial sources, potentially increasing coverage of cancer-associated mitochondrial DNA (mtDNA) mutations and microbial taxonomies.112,113 In addition to linear mtDNA, circular mtDNA molecules have been identified in plasma and extrachromosomal circular DNA (eccDNA) have emerged as a fascinating new class of circulating biomarkers.114–116 (Bottom) cfDNA fragment analysis aimed at elucidating tissue/disease-specific nuclease activity or nucleosome positioning throughout the genome offer new ways of extracting molecular insights that are independent from and complementary to traditional mutation-based analyses.90,117,118
7. Outlook
The last five years have demonstrated tremendous advances in the development and regulatory approval of blood-based liquid biopsy assays for minimally-invasive monitoring of cancer. Single gene assays have been quickly supplanted by comprehensive genomic profiling panels; single modality assays have substituted for multi-omic ones; and single timepoint tests have been augmented with longitudinal sampling strategies that query the time-dependent mutation status of critical cancer genes and identify recurrent disease. While consistent detection of early-stage cancers via ctDNA may always be a challenge due to the limited quantities of tumor-derived DNA in circulation, circulating microbial DNA signatures may offer a new and host genome-independent means of diagnosing cancer by itself or in combination with host-centric approaches. Combinations of these technologies may provide better cancer detection at the earliest stages of disease.
Further technical developments notwithstanding, where do liquid biopsy assays for early cancer detection have the potential to impact patient care and health care costs most immediately? We suggest that liquid biopsy assays can have an instrumental role in clinically triaging high-risk patients downstream of imaging or other clinical tests with high false positive rates for cancer. Here we consider lung cancer as a useful example. The National Lung Screening Trial (NLST) previously demonstrated that low-dose computed tomography (LDCT) reduced lung cancer deaths by 20% by improving rates of early-stage lung cancer detection in a high-risk population (age 55-80 years old with ≥ 30 pack-year smoking history and smoking cessation < 15 years).80 Based on this data, medically reimbursable lung cancer screening with annual LDCT scans was implemented in 2015 and since then the screening guidelines have been revised to include individuals aged 50-80 years old with ≥ 20 pack-year smoking history and smoking cessation <15 years.98 One consequence of LDCT screening for lung cancer, however, is the large number of unnecessary invasive procedures performed on patients with benign pulmonary masses due to LDCT’s high false positive rate for presumptive cancers: in the NLST trial, 39% of patients had a positive screening test (detection of a mass) and 96% of those positive screens were false positives.80 This false positive rate arises from the fact that it is difficult to distinguish a malignant mass (cancer) from non-malignant pulmonary nodules (such as granulomas) or benign tumors (carcinoids and hamartomas) by radiologic image analysis alone.
In turn, high false positive screening rates lead to unnecessary invasive procedures, higher systemic healthcare costs, and create morbidity and mortality risks to patients that counteract the potential benefits of screening. For example, 24% of invasive procedures performed during the NSLT revealed benign masses and 1.24% of patients with benign procedures died within 90 days of their operation.80 Considering the recently updated U.S. Preventive Services Task Force recommendation for annual LDCT screening, which extended screening eligibility up to 14.5 million individuals, the absolute number of unnecessary invasive procedures performed annually (and the burden of their associated complications) is expected to increase. This circumstance provides a window of opportunity for liquid biopsy diagnostics, wherein LDCT screening can be paired with minimally-invasive molecular assays to rule in or out the need for invasive biopsy of lung masses of unknown significance. Specifically, LDCT would enable detection of virtually all anomalous pulmonary masses ≥4 mm in size, irrespective of their clinical importance, whereas highly specific and sensitive molecular assays can differentiate between likely benign versus likely malignant masses that warrant surgical intervention.
We contrast this triage model—wherein a liquid biopsy assay is developed to address a specific clinical question—with the broader effort to utilize ctDNA detection within the context of a population-based molecular screen for multiple cancers, such as GRAIL’s Galleri test. While noble in pursuit, recent data from the Circulating Cell-free Genome Atlas study continue to demonstrate that, for most cancers tested, the paucity of ctDNA in circulation for early-stage malignancies restricts the utility of these tests for advanced Stage III and IV cancers (overall sensitivity by stage, stage I: 16.8%, stage II: 40.4%, stage III: 77.0%, stage IV: 90.1%; specificity fixed at 99%).99,100 It is important to emphasize that these validation results reflect the assay’s performance with patients already clinically diagnosed with cancer, and thus, one may anticipate the sensitivities for Stage I and Stage II cancers to decrease in population screens with asymptomatic individuals and individuals presenting with benign lesions, precancerous lesions, or non-cancer complications.
In summary, the ultimate litmus test for a liquid biopsy is whether it adds value to clinical practice by detecting cancers earlier than conventional diagnostic means while avoiding large numbers of false positives or false negatives. Existing data on the quantity of ctDNA in circulation and clinical trials of it as a diagnostic modality suggest that patient triaging opportunities are realizable but that we are not there yet with population-level screening performance. Given the complexity of cancer genome, epigenome, proteome, and microbiome landscapes, a one-size-fits-all test for population-level screening may not be realistic, just as existing conventional screens vary by cancer type. Nonetheless, we maintain an optimistic hope that novel combinations of multi-analyte, multispecies targets can address this goal in the future, providing sufficient sensitivity and specificity for early cancer detection that translates to better patient survival and quality of life.
Acknowledgments:
The figure was created with BioRender.com. We also acknowledge the patients and their families who have helped contribute toward a better understanding of this field. G.D.S.-P. is supported by a fellowship from the National Cancer Institute of the U.S. National Institutes of Health under award number F30 CA243480. R.K. is supported by the National Cancer Institute of the U.S. National Institutes of Health under award number R01 CA255206.
Abbreviations:
- cfDNA
cell free DNA
- ctDNA
circulating tumor DNA
- mbDNA
microbial DNA
Footnotes
Conflicts of interest statement: E.A., G.D.S.-P., and S.M.-M are employees of Micronoma, Inc., an in vitro diagnostics company founded to commercialize the microbiome-based diagnostic approach described in this article. G.D.S.-P. and R.K. are inventors on a U.S. patent application (PCT/US2019/059647) submitted by The Regents of the University of California and licensed by Micronoma that covers methods of diagnosing and treating cancer using microbial biomarkers in blood and cancer tissues. S.M-M, G.D.S.-P., and R.K. are founders of Micronoma and all authors report stock interest in the company. R.K. is a member of the scientific advisory board for GenCirq, holds an equity interest in GenCirq, and can receive reimbursements for expenses up to US$5,000 per year.
References
- 1.Bray F et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Crosby D et al. A roadmap for the early detection and diagnosis of cancer. The Lancet Oncology 21, 1397–1399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Etzioni R et al. The case for early detection. Nat Rev Cancer 3, 243–252 (2003). [DOI] [PubMed] [Google Scholar]
- 4.SEER Cancer Statistics Review, 1975–2017. SEER https://seer.cancer.gov/csr/1975_2017/index.html.
- 5.Zauber AG The impact of screening on colorectal cancer mortality and incidence: has it really made a difference? Dig Dis Sci 60, 681–691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel RL et al. Colorectal cancer statistics, 2020. CA: A Cancer Journal for Clinicians 70, 145–164 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Blackford AL, Canto MI, Klein AP, Hruban RH & Goggins M Recent Trends in the Incidence and Survival of Stage 1A Pancreatic Cancer: A Surveillance, Epidemiology, and End Results Analysis. JNCI: Journal of the National Cancer Institute 112, 1162–1169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy KM & Topel RH The Value of Health and Longevity. https://www.nber.org/papers/w11405 (2005) doi: 10.3386/w11405. [DOI] [Google Scholar]
- 9.Adegoke O, Kulasingam S & Virnig B Cervical Cancer Trends in the United States: A 35-Year Population-Based Analysis. J Womens Health (Larchmt) 21, 1031–1037 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allemani C et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25 676 887 patients from 279 population-based registries in 67 countries (CONCORD-2). The Lancet 385, 977–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffman RM & Sanchez R Lung Cancer Screening. Med Clin North Am 101, 769–785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVita Vincent T., Lawrence Theodore S. & Rosenberg Steven A. DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. (Wolters Kluwer, 2019). [Google Scholar]
- 13.Wang Z, Jensen MA & Zenklusen JC A Practical Guide to The Cancer Genome Atlas (TCGA). Methods Mol Biol 1418, 111–141 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Mallona I, Sierco A & Peinado MA The Pancancer DNA Methylation Trackhub: A Window to The Cancer Genome Atlas Epigenomics Data. Methods Mol Biol 1766, 123–135 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Mamatjan Y et al. Molecular Signatures for Tumor Classification: An Analysis of The Cancer Genome Atlas Data. J Mol Diagn 19, 881–891 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Cescon DW, Bratman SV, Chan SM & Siu LL Circulating tumor DNA and liquid biopsy in oncology. Nature Cancer 1, 276–290 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Baassiri A et al. Exosomal Non Coding RNA in LIQUID Biopsies as a Promising Biomarker for Colorectal Cancer. Int J Mol Sci 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Rubis G, Rajeev Krishnan S & Bebawy M Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol Sci 40, 172–186 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Heitzer E, Haque IS, Roberts CES & Speicher MR Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet 20, 71–88 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Jahangiri L & Hurst T Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers (Basel) 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartzberg LS et al. Liquid biopsy mutation panel for non-small cell lung cancer: analytical validation and clinical concordance. npj Precision Oncology 4, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overman MJ et al. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol 31, 17–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Yang T & Xiao J Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 34, 267–274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter JV et al. Blood-based microRNAs as biomarkers for the diagnosis of colorectal cancer: a systematic review and meta-analysis. Br J Cancer 116, 762–774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JL, Feizi A, Uhlén M & Nielsen J A Systematic Investigation of the Malignant Functions and Diagnostic Potential of the Cancer Secretome. Cell Rep 26, 2622–2635.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thierry AR, El Messaoudi S, Gahan PB, Anker P & Stroun M Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 35, 347–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diamandis EP & Fiala C Can circulating tumor DNA be used for direct and early stage cancer detection? F1000Res 6, 2129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandel P & Metais P Nuclear Acids In Human Blood Plasma. C R Seances Soc Biol Fil 142, 241–243 (1948). [PubMed] [Google Scholar]
- 30.Sorenson GD et al. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol Biomarkers Prev 3, 67–71 (1994). [PubMed] [Google Scholar]
- 31.Vasioukhin V et al. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol 86, 774–779 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Leon SA, Shapiro B, Sklaroff DM & Yaros MJ Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37, 646–650 (1977). [PubMed] [Google Scholar]
- 33.Shapiro B, Chakrabarty M, Cohn EM & Leon SA Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 51, 2116–2120 (1983). [DOI] [PubMed] [Google Scholar]
- 34.Maebo A [Plasma DNA level as a tumor marker in primary lung cancer]. Nihon Kyobu Shikkan Gakkai Zasshi 28, 1085–1091 (1990). [PubMed] [Google Scholar]
- 35.Amant F et al. Presymptomatic Identification of Cancers in Pregnant Women During Noninvasive Prenatal Testing. JAMA Oncol 1, 814–819 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Bianchi DW et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA 314, 162–169 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Siravegna G, Marsoni S, Siena S & Bardelli A Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14, 531–548 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Sirera R et al. Circulating DNA is a useful prognostic factor in patients with advanced non-small cell lung cancer. J Thorac Oncol 6, 286–290 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Tie J et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 8, 346ra92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Catarino R et al. Circulating DNA: Diagnostic Tool and Predictive Marker for Overall Survival of NSCLC Patients. PLoS One 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dawson S-J et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368, 1199–1209 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Lee RJ et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann Oncol 29, 490–496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JH et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann Oncol 30, 815–822 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim YM et al. Prospective study of the efficacy and utility of TP53 mutations in circulating tumor DNA as a non-invasive biomarker of treatment response monitoring in patients with high-grade serous ovarian carcinoma. J Gynecol Oncol 30, e32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coghlin CL et al. Quantitative analysis of tumor in bronchial biopsy specimens. J Thorac Oncol 5, 448–452 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Mao C et al. Blood as a Substitute for Tumor Tissue in Detecting EGFR Mutations for Guiding EGFR TKIs Treatment of Nonsmall Cell Lung Cancer: A Systematic Review and Meta-Analysis. Medicine (Baltimore) 94, e775 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murtaza M et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 497, 108–112 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Oxnard GR et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res 20, 1698–1705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spence T et al. Clinical implementation of circulating tumour DNA testing for EGFR T790M for detection of treatment resistance in non-small cell lung cancer. J Clin Pathol 74, 91–97 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diehl F et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 14, 985–990 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schreuer M et al. Quantitative assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J Transl Med 14, 95 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parikh AR et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 25, 1415–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dillekås H, Rogers MS & Straume O Are 90% of deaths from cancer caused by metastases? Cancer Med 8, 5574–5576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Research, C. for D. E. and. cobas EGFR Mutation Test v2. FDA (2018). [Google Scholar]
- 56.Health, C. for D. and R. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). FDA (2020). [Google Scholar]
- 57.Commissioner, O. of the. FDA approves first PI3K inhibitor for breast cancer. FDA https://www.fda.gov/news-events/press-announcements/fda-approves-first-pi3k-inhibitor-breast-cancer (2020). [Google Scholar]
- 58.Woodhouse R et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One 15, e0237802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aggarwal C et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol 5, 173–180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lamb YN & Dhillon S Epi proColon® 2.0 CE: A Blood-Based Screening Test for Colorectal Cancer. Mol Diagn Ther 21, 225–232 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Sun G, Meng J, Duan H, Zhang D & Tang Y Diagnostic Assessment of septin9 DNA Methylation for Colorectal Cancer Using Blood Detection: A Meta-Analysis. Pathol Oncol Res 25, 1525–1534 (2019). [DOI] [PubMed] [Google Scholar]
- 62.U.S. Centers for Medicare & Medicaid Services publish negative NCD for Epi proColon. epigenomics https://www.epigenomics.com/u-s-centers-for-medicare-medicaid-services-publish-negative-ncd-for-epi-procolon/ (2021).
- 63.Jd C et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science (New York, N.Y.) vol. 359 https://pubmed.ncbi.nlm.nih.gov/29348365/ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lennon AM et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 369, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thrive Earlier Detection Corp. Detecting Cancers Earlier Through Elective Plasma-based CancerSEEK Testing - Ascertaining Serial Cancer Patients to Enable New Diagnostic. https://clinicaltrials.gov/ct2/show/record/NCT04213326 (2021).
- 66.Liu MC et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Annals of Oncology 31, 745–759 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DNA Methylation and Complex Human Disease. (Elsevier, 2016). doi: 10.1016/C2013-0-13028-0. [DOI] [Google Scholar]
- 68.Feinberg AP, Ohlsson R & Henikoff S The epigenetic progenitor origin of human cancer. Nat Rev Genet 7, 21–33 (2006). [DOI] [PubMed] [Google Scholar]
- 69.Hanley MP et al. Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 36, 5035–5044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen SY et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Shen SY, Burgener JM, Bratman SV & De Carvalho DD Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat Protoc 14, 2749–2780 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Guler GD et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun 11, 5270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen X et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun 11, 3475 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Genomics S Singlera Genomics raises $150 million in Series B financing to promote the development and commercialization of early cancer screening products. https://www.prnewswire.com/news-releases/singlera-genomics-raises-150-million-in-series-b-financing-to-promote-the-development-and-commercialization-of-early-cancer-screening-products-301192505.html.
- 75.Babayan A & Pantel K Advances in liquid biopsy approaches for early detection and monitoring of cancer. Genome Medicine 10, 21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fiala C & Diamandis EP Circulating tumor DNA (ctDNA) is not a good proxy for liquid biopsies of tumor tissues for early detection. Clin Chem Lab Med 58, 1651–1653 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Bardelli A & Pantel K Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 31, 172–179 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Pashayan N & Pharoah PDP The challenge of early detection in cancer. Science 368, 589–590 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Mattox AK et al. Applications of liquid biopsies for cancer. Sci Transl Med 11, (2019). [DOI] [PubMed] [Google Scholar]
- 80.National Lung Screening Trial Research Team et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 365, 395–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Avanzini S et al. A mathematical model of ctDNA shedding predicts tumor detection size. Sci Adv 6, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Elazezy M & Joosse SA Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput Struct Biotechnol J 16, 370–378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Volik S, Alcaide M, Morin RD & Collins C Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol Cancer Res 14, 898–908 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Bauml J & Levy B Clonal Hematopoiesis: A New Layer in the Liquid Biopsy Story in Lung Cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 24, 4352–4354 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Chan HT et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Molecular Oncology 14, 1719–1730 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okamura R et al. High prevalence of clonal hematopoiesis-type genomic abnormalities in cell-free DNA in invasive gliomas after treatment. Int J Cancer (2021) doi: 10.1002/ijc.33481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steensma DP Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv 2, 3404–3410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu Y et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res 24, 4437–4443 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Razavi P et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 25, 1928–1937 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lo YMD, Han DSC, Jiang P & Chiu RWK Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 372, (2021). [DOI] [PubMed] [Google Scholar]
- 91.Cristiano S et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sepich-Poore GD et al. The microbiome and human cancer. Science 371, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poore GD et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Nejman D et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klein RS et al. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med 297, 800–802 (1977). [DOI] [PubMed] [Google Scholar]
- 96.Kwong TNY et al. Association Between Bacteremia From Specific Microbes and Subsequent Diagnosis of Colorectal Cancer. Gastroenterology 155, 383–390.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 97.Lung Cancer Fact Sheet. /lung-health-diseases/lung-disease-lookup/lung-cancer/resource-library/lung-cancer-fact-sheet.
- 98.US Preventive Services Task Force et al. Screening for Lung Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 325, 962–970 (2021). [DOI] [PubMed] [Google Scholar]
- 99.Klein EA et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol S0923-7534(21)02046-9 (2021) doi: 10.1016/j.annonc.2021.05.806. [DOI] [PubMed] [Google Scholar]
- 100.Chen X et al. Prognostic Significance of Blood-Based Multi-cancer Detection in Plasma Cell-Free DNA. Clin Cancer Res (2021) doi: 10.1158/1078-0432.CCR-21-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Our Technology - Biocept. https://biocept.com/technology/.
- 102.Young GP et al. A cross-sectional study comparing a blood test for methylated BCAT1 and IKZF1 tumor-derived DNA with CEA for detection of recurrent colorectal cancer. Cancer Med 5, 2763–2772 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Freenome Holdings Inc. Prevention of Colorectal Cancer Through Multiomics Blood Testing. https://clinicaltrials.gov/ct2/show/NCT04369053 (2021).
- 104.Guardant Health, Inc. Evaluation of the ctDNA LUNAR Test in an Average Patient Screening Episode. https://clinicaltrials.gov/ct2/show/NCT04136002 (2021).
- 105.Forshew T et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 4, 136ra68 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Reinert T et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol 5, 1124–1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gaga M et al. Validation of Lung EpiCheck, a novel methylation-based blood assay, for the detection of lung cancer in European and Chinese high-risk individuals. Eur Respir J 57, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rahier J-F et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin Epigenetics 9, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sánchez-Romero MA & Casadesús J The bacterial epigenome. Nat. Rev. Microbiol 18, 7–20 (2020). [DOI] [PubMed] [Google Scholar]
- 110.Adhikari S & Curtis PD DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiology Reviews 40, 575–591 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Sadeh R et al. ChIP-seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nature Biotechnology 1–13 (2021) doi: 10.1038/s41587-020-00775-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Burnham P et al. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci Rep 6, 27859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gammage PA & Frezza C Mitochondrial DNA: the overlooked oncogenome? BMC Biology 17, 53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ma M-JL et al. Topologic Analysis of Plasma Mitochondrial DNA Reveals the Coexistence of Both Linear and Circular Molecules. Clin Chem 65, 1161–1170 (2019). [DOI] [PubMed] [Google Scholar]
- 115.Zhu J et al. Molecular characterization of cell-free eccDNAs in human plasma. Sci Rep 7, 10968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kumar P et al. Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol Cancer Res 15, 1197–1205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang P et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res 30, 1144–1153 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Snyder MW, Kircher M, Hill AJ, Daza RM & Shendure J Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 164, 57–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]