

Abstract
Previous studies suggest that microglial-expressed Apolipoprotein E (ApoE) is necessary to shift microglia into a neurodegenerative transcriptional state in Alzheimer’s Disease (AD) mouse models. On the other hand, elimination of microglia shifts amyloid beta(Aβ) accumulation from parenchymal plaques to cerebral amyloid angiopathy (CAA), mimicking the effects of global ApoE knock-out. Here, we specifically knock-out microglial-expressed ApoE while keeping astrocytic-expressed ApoE intact. When microglial-specific ApoE is knocked-out of a 5xFAD mouse model of AD, we found a ~35% increase in average Aβ plaque size, but no changes in plaque load, microglial number, microglial clustering around Aβ plaques, nor the formation of CAA. Immunostaining revealed ApoE protein present in plaque-associated microglia in 5xFAD mice with microglial-specific ApoE knockout, suggesting that microglia can take up ApoE from other cellular sources. Mice with Apoe knocked-out of microglia had lower synaptic protein levels than control mice, indicating that microglial-expressed ApoE may have a role in synapse maintenance. Surprisingly, microglial-specific ApoE knock-out resulted in few differentially expressed genes in both 5xFAD and control mice; however, some rescue of 5xFAD associated neuronal networks may occur with microglial-specific ApoE knock-out as shown by WGCNA. Altogether, our data indicates that microglial-expressed ApoE may not be necessary for plaque formation or for the microglial transcriptional shift into a Disease Associated Microglia state that is associated with reactivity to plaques but may be necessary for plaque homeostasis in disease and synaptic maintenance under normal conditions.
Keywords: Apolipoprotein E, microglia, Alzheimer’s Disease, amyloid, inflammation
Introduction
Alzheimer’s Disease (AD) is the most prevalent neurodegenerative disease and clinically manifests as memory and cognitive deficits in affected patients. A key hallmark of the disease is the progressive accumulation of amyloid-β (Aβ) plaques throughout the brain, culminating in synaptic and neuronal loss (Hardy, 2009; Jack & Holtzman, 2013). Although the mechanisms behind the emergence of AD are currently unknown, microglia have consistently been shown to play a key role in the disease as suggested by Genome Wide Association Studies which show that single nucleotide polymorphisms of genes highly enriched or exclusively associated with myeloid cells confer heightened risk of developing AD (Hansen, Hanson, & Sheng, 2018; Jansen et al., 2019; Kunkle et al., 2019; Lambert et al., 2013; Steinberg et al., 2015).
Normally involved in immune surveillance and debris clearance, microglia actively regulate plaque morphology (e.g. compaction) by clustering around Aβ plaques in multiple mouse models of AD (Bolmont et al., 2008; Condello, Yuan, Schain, & Grutzendler, 2015). A possible avenue by which microglia interact with Aβ plaques involves Apolipoprotein E (ApoE) - a glycoprotein normally involved in lipid transport. APOE is considered a major genetic risk determinant in late-onset AD (Corder et al., 1993; Genin et al., 2011; Lambert et al., 2013) with three isoforms of APOE (APOE*ε2, APOE*ε3, and APOE*ε4) being expressed in the human brain. APOE*ε4 carriers have an increased risk to develop AD (Corder et al., 1993; Saunders et al., 1993) while APOE*ε2 carriers have a decreased risk (Corder et al., 1994; Farrer et al., 1997; Reiman et al., 2020). Studies looking into APOE*ε4 effects on AD pathology revealed exacerbated plaque deposition (Castellano et al., 2011; Liu et al., 2017; Polvikoski et al., 1995; Schmechel et al., 1993; Tiraboschi et al., 2004) and synaptic degeneration and synaptic protein loss in postmortem AD patients (Arendt et al., 1997; Love et al., 2006) and mice (Liraz, Boehm-Cagan, & Michaelson, 2013; Yong, Lim, Low, & Wong, 2014), along with increased microglial reactivity to Aβ plaques (Rodriguez, Tai, LaDu, & Rebeck, 2014), and increased proinflammatory RNA signature (Shi et al., 2017). APOE*ε2 on the other hand is associated with reduced numbers of neuritic plaques (Serrano-Pozo, Qian, Monsell, Betensky, & Hyman, 2015).
With regards to the proposed mode of action by which ApoE may be affecting Aβ deposition, ApoE can directly interact with Aβ peptides and form an Aβ/ApoE complex (LaDu et al., 1994). These complexes are subsequently phagocytosed by microglia through interactions with Low density lipoprotein receptor-related protein 1 (Lrp1) or Triggering receptor expressed on myeloid cells 2 (Trem2) (Atagi et al., 2015; Bailey, DeVaux, & Farzan, 2015; Kanekiyo, Xu, & Bu, 2014; Yeh, Wang, Tom, Gonzalez, & Sheng, 2016). Our lab and others have shown ApoE immunoreactivity in dense core plaques as well as in microglial processes around the plaque cores (E. Spangenberg et al., 2019; Uchihara et al., 1995), perhaps indicative of the interactions described in the studies above. Additionally, microglial depletion in the plaque-laden 5xFAD mouse model of AD reduces plaque load (E. Spangenberg et al., 2019), suggesting that ApoE-microglia interaction may influence microglia-plaque interaction and plaque formation. This finding has been recently replicated with knock-out of the phagocytic mediators Axl and Mertk, resulting in reduced parenchymal plaque deposition and increased cerebral amyloid angiopathy (CAA), collectively suggesting that microglia play a critical role in plaque formation (Huang et al., 2021). Similarly, previous ApoE knock-out studies demonstrated a reduction in Aβ plaque load and synaptic proteins (Bales et al., 1999; Holtzman et al., 2000; Liraz et al., 2013; Ulrich et al., 2018; Yong et al., 2014), as well as a rescue in the microglial homeostatic signature (Krasemann et al., 2017), suggesting a critical link between microglia and ApoE in disease pathogenesis.
Although ApoE has been extensively studied in AD, the importance of microglial-expressed ApoE in AD has yet to be determined. Under normal conditions in the mouse brain, ApoE is primarily produced by astrocytes (Boyles, Pitas, Wilson, Mahley, & Taylor, 1985; Pitas, Boyles, Lee, Foss, & Mahley, 1987; Y. Zhang et al., 2014; Y. Zhang et al., 2016). However, in AD, microglial Apoe mRNA and single nuclei RNA are highly enriched (Keren-Shaul et al., 2017; Mathys et al., 2019; Ping et al., 2018; Rangaraju et al., 2018) while single nuclei RNA levels of astrocytic Apoe are downregulated (Mathys et al., 2019). Here, we examine the effects of constitutive microglial-specific ApoE knock-out on AD pathogenesis and homeostasis in 5xFAD mice. In 4-month-old mice, microglial-specific ApoE knock-out has no effect on plaque formation, Aβ production, or microglial and astrocytic number. At 12 months of age, the average plaque size in the hippocampus is ~35% larger than in 5xFAD controls, but found no differences in microglial interaction with plaques, nor microglial and astrocytic numbers. However, there was a reduction in microglial homeostatic marker P2ry12 intensity in microglial-expressed ApoE knock-out, 5xFAD, and 5xFAD microglial-expressed ApoE knock-out groups when compared to controls. Interestingly, ApoE-intact mice have significantly higher levels of pre- and post-synaptic markers when compared to mice with microglial-expressed ApoE depleted, 5xFAD mice, and 5xFAD mice with microglial-expressed ApoE depleted. Although modest changes in plaque size and synaptic proteins are observed, few RNA expression changes are observed; however, weighted gene co-expression analysis (WGCNA) indicates that neuronal networks downregulated in 5xFAD mice may be preserved with microglial-specific ApoE knock-out. While we show microglial-expressed ApoE is absent from our mouse model, ApoE protein still colocalizes with microglia and the surrounding plaques. Our findings indicate that microglial-expressed ApoE may not be necessary for the microglial response to Aβ plaques and that other sources of ApoE may be sufficient to induce microglial transcriptional changes in AD.
Methods
Animals
All animal experiments performed in this study were approved by the UC Irvine Institutional Animal Care and Use Committee (IACUC) and were compliant with ethical regulations for animal research and testing. The 5xFAD mouse expresses 5 familial AD genes (APP Swedish, Florida, and London; PSEN1 M146L +L286V) (Oakley et al., 2006) and is characterized by aggressive amyloid pathology throughout the brain and synaptic and neuronal loss in the subiculum. The following primers were used to genotype these animals: PS1 Forward 5′ - AAT AGA GAA CGG CAG GAG CA – 3′ and PS1 Reverse 5′ - GCC ATG AGG GCA CTA ATC AT – 3′. To generate Apoe knock-out specifically in myeloid cells, the following mice were obtained from Jackson Laboratory: Csf1r-cre (C57BL/6Tg(Csf1r-cre)1Mnz/J; Stock No: 029206) and Apoefl/fl (B6.129S6-Apoetm1.1Mae/MazzJ; Stock No: 028530). Progeny from Apoefl/fl, Csf1r-cre, and 5xFAD mice were used to produce four groups of interest. Each group of interest consisted of nearly equal numbers of male and female mice. For genotyping Csf1r-cre, the following primers were used: Forward 5′ - AGA TGC CAG GAC ATC AGG AAC CTG – 3′ and Reverse 5′ - ATC AGC CAC ACC AGA CAC AGA GAT – 3′. For genotyping Apoefl/fl, the following primers were used: Forward 5′ - GGC TTA GTG GGT AAA GGT GCT – 3′ and Reverse 5′ - GAC TAG GCA GGT GTG GAA TTA GA – 3′. All mice were sacrificed and perfused at either 4 or 12 months of age for this study. Half-brains were subsequently flash frozen for biochemical analyses or drop-fixed in 4% paraformaldehyde for immunohistochemical analyses. 4-month-old drop-fixed brains were sliced on a microtome and stored in a 1xPBS and .01% NaAzide solution at 4°C. 12 month drop-fixed brains were sliced using a microtome; however, they were then stored in a 1xPBS, 30% glycerol, and 30% ethylene glycol solution at −20°C. This allowed for the preservation of RNA in our 12-month tissue which we used for RNA in situ hybridization.
Immunohistochemistry
Primary antibodies and dilutions are used as follows: anti-ionized calcium-binding adapter molecule 1 (IBA1; 1:1000; 019–19741; Wako, Osaka, Japan), anti-Aβ1–16 (6E10; 1:1000; 803001; BioLegend), PU.1 (1:500; 2266S, Cell Signaling), CD68 (1:200; MCA1957T; BioRad), anti-lysosomal associated membrane protein 1 (LAMP1; 1:200; sc-20011), anti-S100β (1:200; ab52642; Abcam), anti-glial fibrillary protein (GFAP; 1:1000; ab4674; Abcam), anti-P2ry12 (1:200; HPA014518; Sigma), anti-Apolipoprotein E (ApoE; 1:200; ab19606; Abcam) Synaptophysin (1:1000; S5768; Millipore), PSD95 (1:500; ab18258; Abcam). Amylo-Glo (TR-300-AG; Biosensis, Thebarton, South Australia, AU) was used according to manufacturer’s instructions to visualize fibrillar Aβ plaques. For Amylo-Glo staining, tissue sections were washed in 70% ethanol 1X5 minutes, followed by a 1×2 minute wash in distilled water. Sections were then 1% Amylo-Glo solution for 1×10 minutes then washed with .9% saline for 1×5 minutes and distilled water for 1×15 seconds. Sections were then briefly rinsed in 1XPBS and immersed in normal serum blocking solution (5% normal serum with 0.2% Triton-X100 in 1XPBS) for 60 minutes. Tissue was then incubated overnight in primary antibody at the dilutions described above in normal serum blocking solution at 4 degrees Celsius. The next day tissue sections were washed in 1XPBS 3×10 minutes before being placed in appropriate secondary antibody in normal serum blocking solution (1:200 for all species and wavelengths; Invitrogen) for 60 minutes. Tissue sections were then washed for 3×10 minutes in 1XPBS before tissue was mounted and coverslipped. High resolution fluorescent images were obtained using a Leica TCS SPE-II confocal microscope and LAS-X software. To capture whole brain stitches, automated slide scanning was performed using a Zeiss AxioScan.Z1 equipped with a Colibri camera and Zen AxioScan 2.3 software.
In Situ Hybridization
RNAscope In Situ Hybridization was performed following manufacturer’s instructions. Mounted tissue sections were warmed on a 40°C hot plate for 15 minutes and then dehydrated with 50%, 70%, and 100% ethyl alcohol for 5 minutes at each gradient, followed by hydrogen peroxide (Cat No.322335 ACDBio) treatment for 10 minutes. After washing the tissue with Deionized (DI) water, tissue sections were placed in boiling Target Retrieval Reagent (Cat No.322380 ACDBio) for 15 minutes and then immediately transferred to DI water. Slides were then blocked in Protease III (Cat No.322337 ACDBio) for 30 minutes at 40 °C. Apoe probe (Cat No. 313271-C2) was then added for 2 hours at 40 °C within a humidity control chamber. Signal amplification and detection reagents (Cat No.322310 ACDBio) were applied sequentially and incubated in AMP 1, AMP 2, and AMP 3 for 30 minutes each. Before adding each AMP reagent, samples were washed twice with washing buffer (Cat NO.310091 ACDBio). Respective HRPs were placed on slides for 15 minutes at 40 °C followed by 30 minutes of respective Opal dye (FP1487001KT Akoya Biosciences) for 30 minutes at 40 °C and HRP blocker for 15 minutes at 40 °C.
Aβ Elisa
To isolate protein for the Elisa, flash-frozen brain hemispheres were microdissected into cortical, hippocampal, and thalamic regions and grounded to a powder. Tissue was then homogenized in Tissue Protein Extraction Reagent (TPER (Life Technologies, Grand Island, NY)) with protease and phosphatase inhibitors present. Samples were then centrifuged at 100,000 g for 1 hour at 4 °C to generate TPER-soluble fractions. To generate formic acid fractions, protein pellets from the TPER-soluble fraction were then homogenized in 70% formic acid and centrifuged at 100,000 g for 1 hour at 4 °C, the formic acid fraction is then neutralized. Isolated protein samples were transferred to a blocked MSD Mouse (4G8) Aβ triplex ELISA plate (Aβ1–38, Aβ1–40, Aβ1–42) and incubated for two hours at room temperature with an orbital shaker. The plate was then washed, and measurements were obtained using a SECTOR Imager 2400, per the manufacturer’s instructions (Meso Scale Discovery, Gaithersburg, MD).
RNA Sequencing
Whole transcriptome RNA sequencing (RNA-Seq) libraries were produced from hippocampal tissue of Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice sacrificed at 4 and 12 months of age. RNA was isolated with an RNA Plus Universal Mini Kit (Qiagen, Valencia, USA) according to the manufacturer’s instructions. Library preparation, RNA-seq, and read mapping analysis were performed by Novagene Co. Gene expression was analyzed using Limma, edgeR, and org.Mm.eg.db packages (Robinson, McCarthy, & Smyth, 2010) with expression values normalized into FPKM (fragments per kilobase of transcript per million mapped reads). Differentially-expressed genes were selected by using false discovery rate (FDR) <0.05. Heatmaps were created using Morpheus (Morpheus, https://software.broadinstitute.org/morpheus) and volcano plots were created using VolcaNoseR (Goedhart & Luijsterburg, 2020).
Weighted correlation network analysis:
Network analysis was performed using weighted gene co-expression analysis (WGCNA) package in R (B. Zhang & Horvath, 2005). First, bi-weighted mid-correlations were calculated for all gene pairs, and then used to generate an eigengene network matrix, which reflects the similarity between genes according to their expression profiles. This matrix was then raised to power β (β=16). Modules were defined using specific module cutting parameters (minimum module size = 100 genes, deepSplit = 4 and threshold of correlation = 0.2). Modules with a correlation greater than 0.8 were merged. We used first principal component of the module, called signed bicor network, to correlate AD genotype, sex, and Apoe genotype. Hub genes were defined using intra-modular connectivity (kME) parameter of the WGCNA package. Gene enrichment analysis: Gene-set enrichment analysis was done using enrichR (Kuleshov et al., 2016).
Data Analysis and Statistics
Both male and female mice were used in all statistical analyses. Plaque, microglial, S100β astrocyte, PSD95, and Synaptophysin counts were measured via the spots function and plaque volume, Lamp1 area, and 6E10 volume and P2ry12 intensity were measured via the surfaces function on Imaris version 9.6. GFAP and Cd68 % area were measured via Image J. 1 FOV was used per animal per brain region for each analysis listed above. Microglial and astrocytic colocalization to Apoe RNA, and microglial and plaque colocalization to ApoE protein was measured using the colocalization function followed by the surfaces function in Bitplane Imaris 9.6. The colocalization function created a new channel which showed colocalized signal and the subsequent surface function then measured the volume of the total signal in the channel. The total volume was then normalized to the total volume of IBA1, GFAP or Amylo-Glo for microglia, astrocytes, and plaques, accordingly. Statistical analysis was accomplished using Prism GraphPad (v9.0.0). To compare two groups, the unpaired Student’s t-test was used. To compare all four groups at a single timepoint or in a single brain region, one-way ANOVA with Tukey’s multiple comparison correction was performed. To compare all four groups at either different timepoints or brain regions, two-way ANOVA with Tukey’s multiple comparison correction was used. For all analyses, statistical significance was accepted at p < 0.05. and significance expressed as follows: *p < 0.05, **p< 0.01, ***p < 0.001.
Results
Microglial-specific Apoe knock-out does not significantly alter gene expression in mice
To determine the role of microglial-expressed ApoE in AD pathogenesis we undertook a strategy to specifically knock-out Apoe from microglia in 5xFAD mice. To that end, we utilized a Cre mouse driver line targeting colony stimulating factor 1 receptor (Csf1r) expressing cells (Csf1r-cre); Csf1r is exclusively expressed by microglia in the adult brain, and produces >95% recombination when combined with reporter lines (E. E. Spangenberg et al., 2016). Csf1r-cre mice were bred with the 5xFAD mouse model of AD and Apoefl/fl mice (Fig. 1a, a’). Four groups were created for all subsequent analyses: Apoefl/fl (Control), Csf1r-cre/Apoefl/fl (Csf1r-Apoe-KO), 5xFAD+/−/Apoefl/fl (5xFAD), and 5xFAD+/−/Csf1r-cre/Apoefl/fl (5xFAD/Csf1r-Apoe-KO). Mice were sacrificed at 4 and 12 months of age to observe changes in early and late stage 5xFAD pathology. To confirm successful knock-out of microglial-expressed ApoE, we performed in situ hybridization via RNAscope for Apoe mRNA on 12-month-old mice (Fig. 1b). Notably, microglial-expressed ApoE was highly upregulated in 5xFAD mice but was nearly absent in 5xFAD/Csf1r-Apoe-KO mice (Fig. 1C). Apoe colocalized with GFAP+ astrocytes in the hippocampus, however; Control and Csf1r-Apoe-KO mice had significantly higher levels of GFAP-Apoe colocalization than 5xFAD and 5xFAD/Csf1r-Apoe-KO mice (Fig. 1d). These results confirm that microglia express ApoE in 5xFAD mice, and that ApoE expression in microglia is successfully knocked-out with the addition of the Csf1r-cre driver. We performed bulk-tissue RNA sequencing of microdissected hippocampi from Control and Csf1r-Apoe-KO mice to determine if deletion of Apoe from microglia would result in unexpected broad effects on hippocampal gene expression at 4 and 12 months of age (Fig. 1e, f). We found only 7 differentially expressed genes (Gm9825, Hmga1b, Atp6v0c-ps2, Rps2-ps10, Gm14292, Gm11361) between Control and Csf1r-Apoe-KO mice at 12 months of age, indicating the loss of microglial-expressed ApoE does not broadly alter gene expression in the brain. Of these genes, there are 5 pseudogenes, 1 predicted gene (Gm11361), and 1 gene, Hmga1b, that encodes a non-histone chromatin protein involved in gene transcription and can interact with p53 to inhibit apoptotic activity (Pierantoni et al., 2006).
Figure 1: Csf1r-cre Apoefl/fl mouse model specifically knocks-out microglial-expressed ApoE with few changes in hippocampal gene expression.
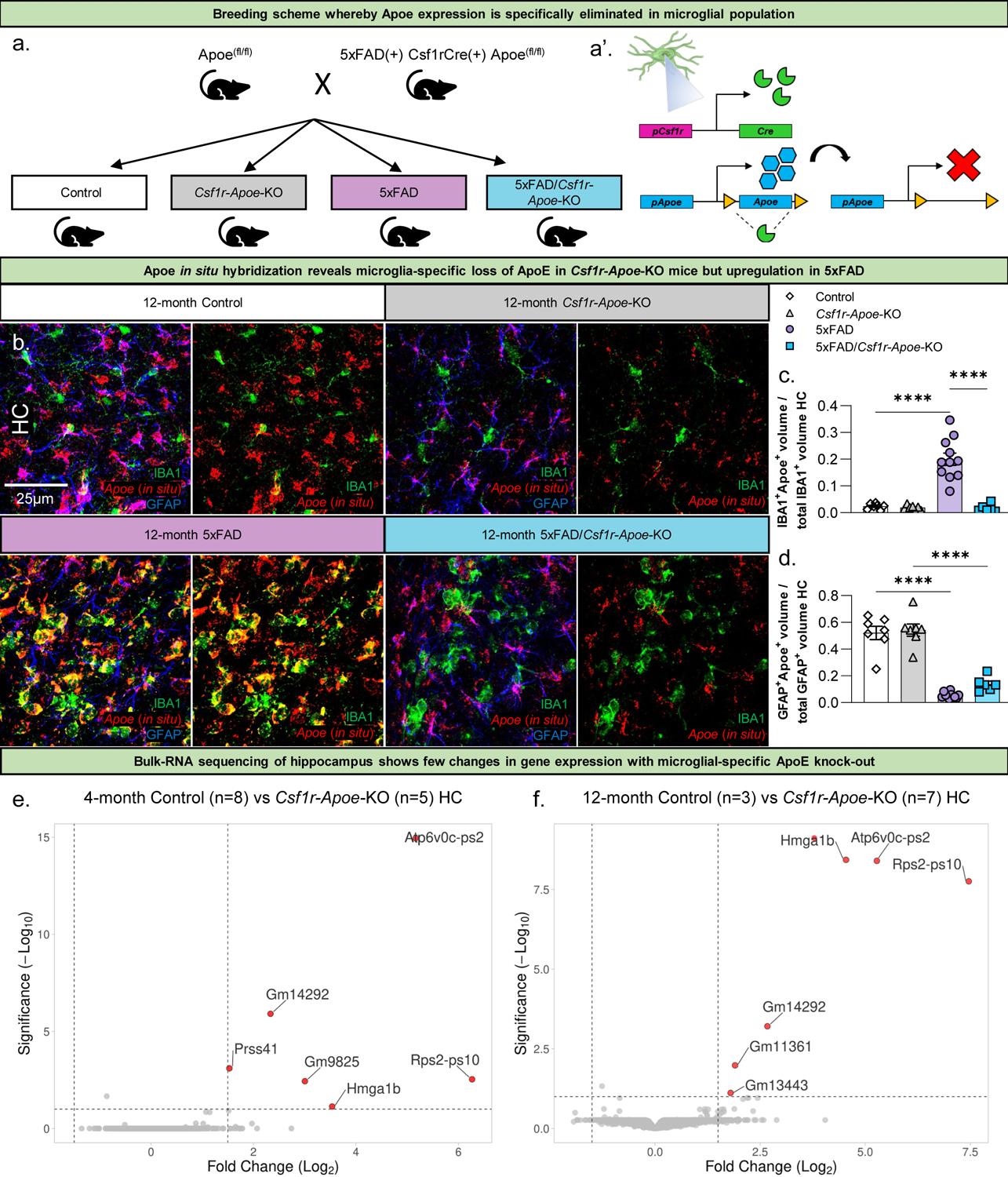
Schematic of experimental design (a). Mice homozygous for Apoefl/fl are crossed with mice that are heterozygous for 5xFAD and Csf1r-cre, and homozygous for Apoefl/fl to create four groups of interest (all groups are Apoefl/fl): Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO. Mice were sacrificed at 4 and 12 months of age for subsequent analysis. Representative 63x images of IBA1 and GFAP immunofluorescence with Apoe in situ hybridization (b). Microglial Apoe is significantly upregulated in 12-month-old 5xFAD (n=11) mice but is depleted in 5xFAD/Csf1r-Apoe-KO (n=6) mice in the hippocampus (c). GFAP+ astrocytic Apoe is present at high levels in 12-month-old Control (n=7) and Csf1r-Apoe-KO (n=7) mice, but significantly reduced in 5xFAD (n=11) and 5xFAD/Csf1r-Apoe-KO (n=6) mice in the hippocampus (d). Volcano plot comparing differentially expressed genes in the hippocampus between Control (4 months n=8; 12 months n=3) and Csf1r-Apoe-KO (4 months n=5; 12 months n=7) groups shows few genes are differentially expressed with microglial-expressed ApoE knock-out in 4- and 12-month-old mice (e, f). Statistical analysis for c, d used a one-way ANOVA with Tukey’s multiple comparisons correction. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; # 0.05 < p < 0.1.
Microglial-specific ApoE knock-out significantly increases plaque volume in aged 5xFAD mice
We next investigated the role of microglial-expressed ApoE in plaque formation and homeostasis by performing immunohistochemistry (IHC) using Amylo-Glo (amyloid-specific dye for dense core plaques), and 6E10 (APP/Aβ; Fig. 2a, b). At 4- and 12-months of age, we found no differences in dense core plaque counts between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice in the hippocampus or somatosensory cortex (Fig. 2c, d). However, 12-month-old mice lacking microglial-expressed ApoE had larger dense core plaques in the hippocampus compared to 5xFAD mice with intact microglial-expressed ApoE (P < .01) (Fig. 2e, f). No differences were seen in 6E10+ diffuse plaques between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice (Fig. 2g). Notably, no CAA was observed in 5xFAD mice, nor was CAA induced in 5xFAD/Csf1r-Apoe-KO mice (Fig. S1a). No differences in insoluble or soluble Aβ1–40 and Aβ1–42 were seen between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice at both 4 and 12 months of age, indicating that microglial-expressed ApoE may not contribute to overall Aβ levels in the mouse brain (Fig. 2h–k). Importantly, for all analyses above there were no significant sex differences. Altogether, microglial-specific ApoE knock-out modestly affects plaque size, but not number of Aβ plaques or overall Aβ load.
Figure 2: Microglial-specific ApoE knock-out increases average plaque size while plaque number and Aβ levels are unchanged in a 5xFAD mouse model.
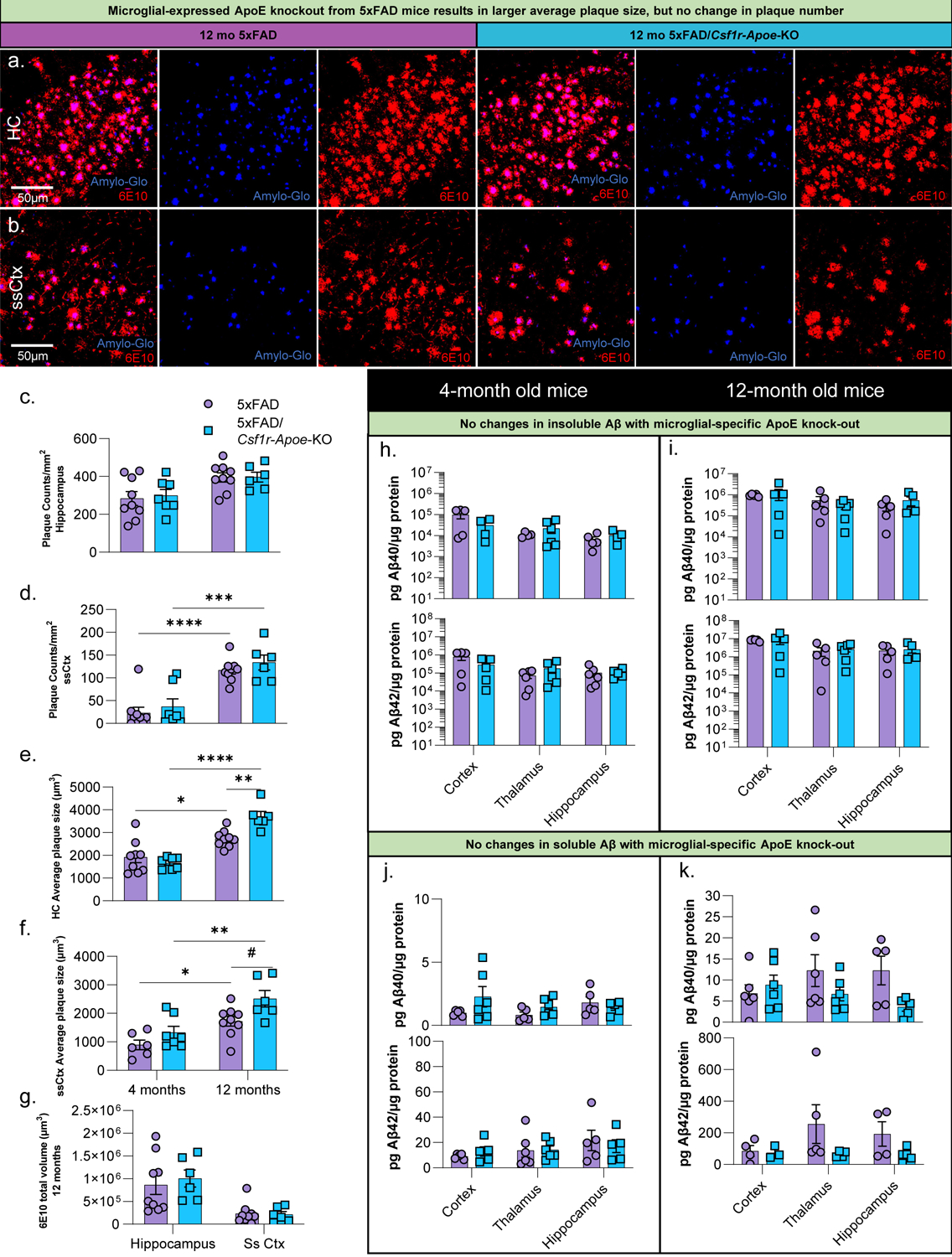
Representative 20x images of Amylo-Glo (dense-plaque core) dye staining and 6E10 (diffuse amyloid plaques) immunofluorescence in 12-month-old 5xFAD and 5xFAD/Csf1r-Apoe-KO mice in the hippocampus (a) and somatosensory cortex (b). No difference in plaque number was seen in the hippocampus between 5xFAD (4months n=9; 12 months n=9) and 5xFAD/Csf1r-Apoe-KO (4 months n=7; 12 months n=6) mice (c) or somatosensory cortex (d). At 12 months of age, plaque volume was significantly increased in 5xFAD/Csf1r-Apoe-KO mice in the hippocampus (e) but not in the somatosensory cortex (f). (g) No changes in diffuse Aβ volume (6E10) associated with microglial-expressed Apoe knock-out were observed in the hippocampus or somatosensory cortex. No differences in insoluble Aβ40 or Aβ42 were present between 5xFAD (4 and 12 months: n= 5–6/brain region) and 5xFAD/Csf1r-Apoe-KO (4 and 12 months: n= 5–6/brain region) mice in the cortex, thalamus, or hippocampus at 4- (h) or 12-months (i) of age. Additionally, no difference in soluble Aβ40 or Aβ42 were observed between 5xFAD 5xFAD (4 and 12 months: n= 5–6/brain region) and 5xFAD/Csf1r-Apoe-KO 5xFAD (4 and 12 months: n= 3–6/brain region) in all regions at 4- (j) and 12-months (k) of age. Statistical analysis used a two-way ANOVA with Tukey’s multiple comparisons correction. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; # 0.05 < p < 0.1.
Microglial number and plaque association are unaltered with microglial-specific ApoE knock-out
Having demonstrated that microglial-specific ApoE knock-out has modest effects on plaque size, but not plaque number, we sought to determine the effect of Apoe knock-out on microglial and astrocytic number, as well as microglial association with Aβ plaques. To assess microglial changes, we stained tissue from 4-month and 12-month-old mice with microglial markers IBA1 (Fig. 3a, b) and Pu.1 (Fig. 3a’, b’). As expected, microglial numbers increased in 5xFAD and 5xFAD/Csf1r-Apoe-KO mice compared to Control and Csf1r-Apoe-KO mice at both 4- and 12-months of age in the hippocampus (Fig. 3e). In the somatosensory cortex, we also observed an increase in microglial number in the 5xFAD groups compared to the WT groups at 12 months of age, but not at 4 months of age (Fig. 3f). In both regions and at both timepoints, there were no differences in microglial number when comparing the 5xFAD with 5xFAD/Csf1r-Apoe-KO mice and Control with Csf1r-Apoe-KO mice, indicating that microglial-specific ApoE knock-out does not alter microglia number (Fig. 3e, f). Previously, microglia have been shown to actively regulate plaque compaction (Bolmont et al., 2008; Condello et al., 2015; E. Spangenberg et al., 2019). Having demonstrated that microglial-specific ApoE knock-out in 5xFAD mice resulted in significantly larger plaques, we sought to determine if Apoe deficient microglia were reacting differently to plaques. However, we found no difference in the number of plaque-associated microglia per μm of plaque diameter between our 5xFAD and 5xFAD/Csf1r-Apoe-KO mice in the hippocampus or somatosensory cortex, indicating microglial-expressed ApoE does not impact microglial presence around Aβ plaques (Fig. 3c, d). Overall, while we do see changes in plaque size in microglial-expressed ApoE deficient mice, no changes in microglia number or plaque association were observed.
Figure 3: No microglial or astrocytic changes associated with microglial-specific ApoE knock-out.
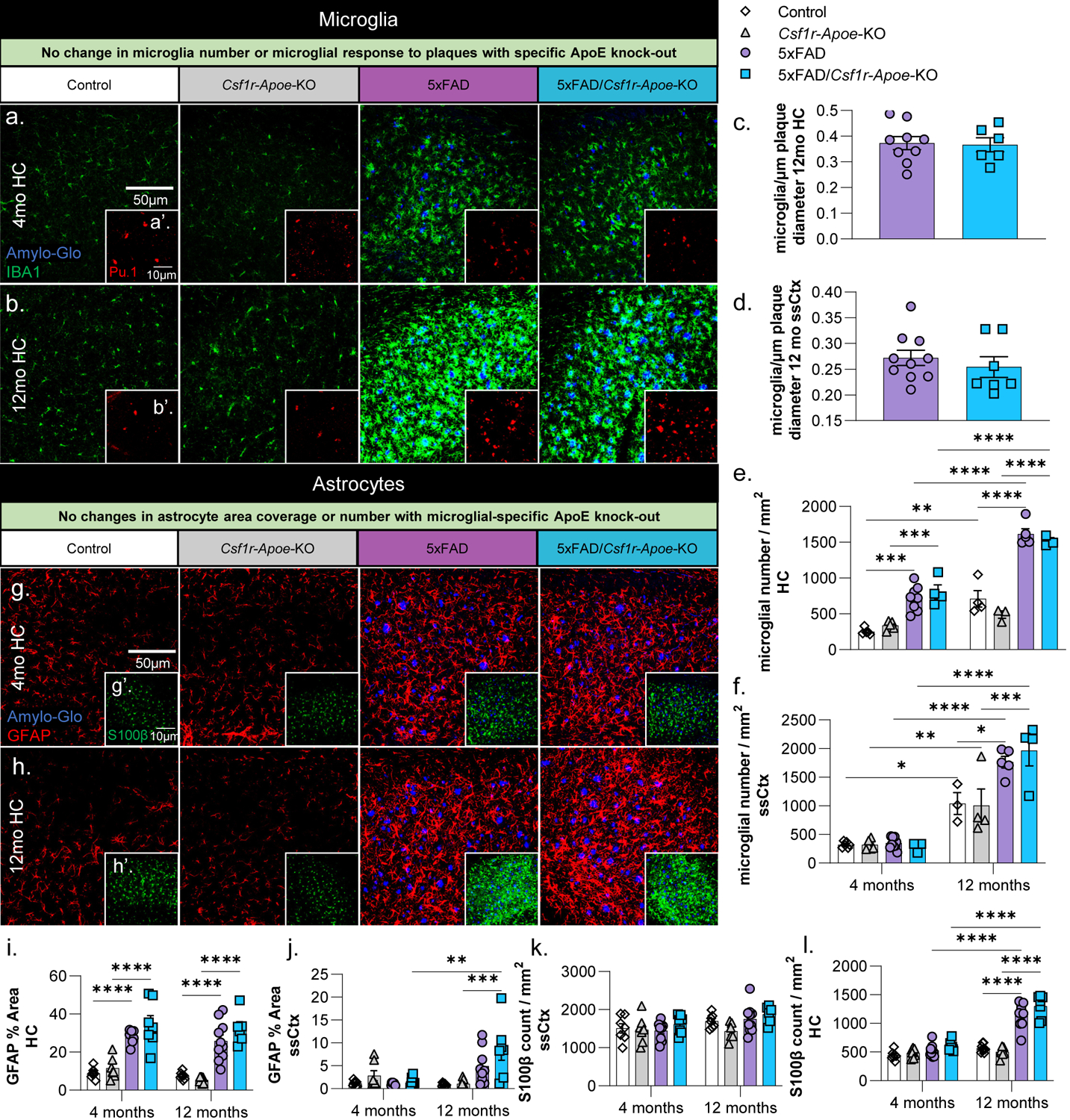
Representative 20x images of Amylo-Glo dye staining and IBA1 (microglia) immunofluorescence in Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice at 4- (a) and 12-months (b) of age in the hippocampus. Additional Amylo-Glo staining and Pu.1 (microglia) immunofluorescence images of all groups at 4- (a’) and 12-months (b’) of age in the hippocampus. No difference in the number of microglia/plaque diameter (μm) was observed between 5xFAD (n=9) and 5xFAD/Csf1r-Apoe-KO (n=7) mice in the hippocampus (c) nor somatosensory cortex (d). Microglial number between Control and 5xFAD mice is increased at 12-months of age in but no differences in microglial number were seen between Control (4 months n=5; 12 months n=4) and Csf1r-Apoe-KO (4 months n=5; 12 months n=3) or 5xFAD (4 months n=8; 12 months n=5) and 5xFAD/Csf1r-Apoe-KO (4 months n=4; 12 months n=4) mice in the hippocampus (e) nor somatosensory cortex (f). Representative 20x images of Amylo-Glo dye staining and GFAP (reactive astrocytes) immunofluorescence in all four groups at 4- (g) and 12-months (h) of age in the hippocampus. Additional Amylo-Glo staining and S100β (astrocyte cell bodies) immunofluorescence images of all groups at 4- (g’) and 12-months (h’) of age in the hippocampus. We observed significant increases in GFAP percent area quantifications in 5xFAD (4 months n=8; 12 months n=10) and 5xFAD/Csf1r-Apoe-KO (4 months n=7; 12 months n=6) groups compared to the Control (4 months n=7; 12 months n=8) and Csf1r-Apoe-KO (4 months n=7; 12 months n=7) in the hippocampus (i) at 4 and 12 months of age, and in the somatosensory cortex (j) at 12 months of age. In both the hippocampus and somatosensory cortex, no differences in astrocyte percent area were seen between Control and Csf1r-Apoe-KO groups or 5xFAD and 5xFAD/Csf1r-Apoe-KO groups. S100β astrocyte counts show no differences between Control and Csf1r-Apoe-KO groups or 5xFAD and 5xFAD/Csf1r-Apoe-KO groups in the hippocampus (k) or somatosensory cortex (l). Statistical analysis used a two-way ANOVA with Tukey’s multiple comparisons correction and a two-tailed t-test for the microglia/plaque diameter (μm) quantification. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; # 0.05 < p < 0.1.
To gauge if there were changes in microglial activation associated with microglial-specific ApoE knock-out, we stained tissue from 4-month and 12-month-old mice with Cd68, a marker for microglial lysosomes (Fig. S2a, b). At 4- and 12-months of age in the hippocampus (Fig. S2a, c) and somatosensory cortex (Fig. S2b, d) we found no significant changes in Cd68 percent area between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice. We next looked at homeostatic microglial marker P2ry12 and found a small but significant reduction in intensity when comparing the Control group with all other groups indicative of a loss of homeostatic function with disease, as expected, as well as with microglial-expressed ApoE knockout (Fig. S2e, f). Notably, this reduction was not further exacerbated with microglial-specific ApoE knock-out in tandem with AD pathology. Given the absence of Apoe mRNA from 5xFAD/Csf1r-Apoe-KO mice, we stained for ApoE protein to investigate if plaque associated microglia were taking up ApoE from other cellular sources. Colocalization of ApoE signal with either microglia or Amylo-Glo (representing dense core plaques) revealed that both contained ApoE protein in 5xFAD and 5xFAD/Csf1r-Apoe-KO mice (Supplemental Fig. S2g–i).
Under homeostatic conditions, astrocytes are the primary producer of ApoE in the mouse brain (Boyles et al., 1985; Pitas et al., 1987; Y. Zhang et al., 2014; Y. Zhang et al., 2016). We quantified astrocyte percent coverage and counts with GFAP and S100β, respectively (Fig. 3g, h). In the hippocampus, GFAP-positive astrocyte coverage was higher in 5xFAD and 5xFAD/Csf1r-Apoe-KO compared to Control and Csf1r-Apoe-KO mice at 4 and 12 months of age, as expected (Fig. 3i). The somatosensory cortex contained far less GFAP-positive astrocytes than in the hippocampus, however; GFAP-positive astrocyte coverage in 5xFAD/Csf1r-Apoe-KO mice was still significantly higher than in Csf1r-Apoe-KO mice at 12 months of age (Fig. 3j). In both regions and timepoints, there were no differences in GFAP positive astrocyte coverage when comparing the 5xFAD with 5xFAD/Csf1r-Apoe-KO mice and Control with Csf1r-Apoe-KO mice (Fig. 3i, j). S100β astrocyte counts in the hippocampus were elevated in 5xFAD and 5xFAD/Csf1r-Apoe-KO compared to Control and Csf1r-Apoe-KO in 12-month-old mice (Fig. 3k), but no differences in S100β counts were found in the somatosensory cortex (Fig. 3l). Additionally, no differences were seen between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice, and Control and Csf1r-Apoe-KO mice in either brain region (Fig. 3k, l). Importantly, for all analyses above there were no significant sex differences. Altogether, no changes in astrocyte number or coverage were observed with microglial-specific ApoE knock-out.
Microglial-specific ApoE knock-out is associated with reductions in pre- and post-synaptic markers
Recent studies indicate that Trem2-dependent microglial plaque compaction attenuates neuritic dystrophy and that knock-out of Trem2 reduces the amount of plaque-associated ApoE (Leyns et al., 2019; Parhizkar et al., 2019). We sought to determine if the lack of microglial-expressed ApoE and resultant increase in average plaque size in our 5xFAD/Csf1r-Apoe-KO would result in a change in dystrophic neurites. To that end, we observed a downward trend in Lamp1 dystrophic neurites in the hippocampus between 5xFAD and 5xFAD/Csf1r-Apoe-KO groups (Fig. 4a–c; p=.076). To assess whether synaptic changes occurred with microglial-specific ApoE knock-out, we stained all groups with the post-synaptic marker, PSD95 (Fig. 4d, e), and presynaptic marker, Synaptophysin (Fig. 4h, i). Surprisingly, 12-month-old mice had significant reductions in both PSD95 and Synaptophysin in Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice when compared to Control mice in both the hippocampus and visual cortex (Fig. 4f, g, j, k). No differences in synaptic markers were seen between 5xFAD and 5xFAD/Csf1r-Apoe-KO mice. These results suggest that microglial knock-out of ApoE drives reductions in synaptic puncta, however, synaptic puncta are not further reduced with microglial-specific ApoE knock-out in conjunction with AD pathology.
Figure 4: Microglial-specific ApoE knock-out induces pre- and post-synaptic reduction.
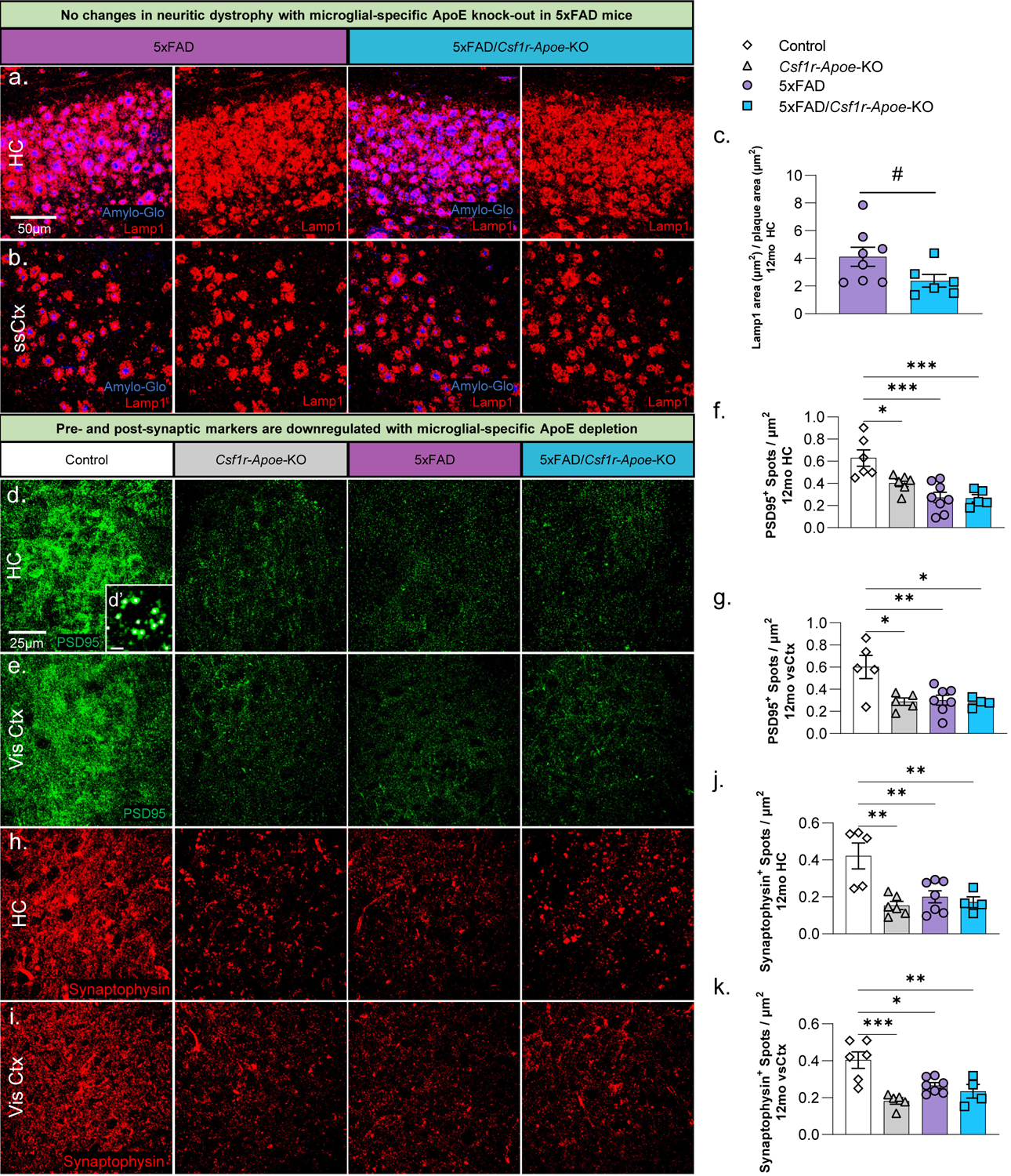
Representative 20x images of Amylo-Glo dye staining and Lamp1 (dystrophic neurites) immunofluorescence in 12-month-old 5xFAD (n=8) and 5xFAD/Csf1r-Apoe-KO (n=6) mice in the hippocampus (a) and somatosensory cortex (b). No significant change in Lamp1 area/area of plaques (μm2) was observed in the hippocampus between both groups (c). Representative 63x images of post-synaptic marker, PSD95, immunofluorescence in Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice at 12 months of age in the hippocampus (d) and visual cortex (e). Inset image (d’) shows individual post-synaptic puncta with each white dot representing one punctate. The number of PSD95 puncta was significantly reduced in the Csf1r-Apoe-KO (HC n=6; vsCtx n=5), 5xFAD (HC n=8; vsCtx n=7), and 5xFAD/Csf1r-Apoe-KO (HC n=5; vsCtx n=4) groups compared to the Control (HC n=6; vsCtx n=5) group in the hippocampus (f) and the visual cortex (g). Representative 63x images of pre-synaptic marker, Synaptophysin, immunofluorescence in all four groups in the hippocampus (h) and visual cortex (i). Like PSD95, the number of Synaptophysin puncta was significantly reduced in the Csf1r-Apoe-KO (HC n=6; vsCtx n=5), 5xFAD (HC n=7; vsCtx n=7), and 5xFAD/Csf1r-Apoe-KO (HC n=4; vsCtx n=4) groups compared to the Control (HC n=5; vsCtx n=6) group in the hippocampus (j) and the visual cortex (k). Statistical analysis used a one-way ANOVA with Tukey’s multiple comparisons correction and a two-tailed t-test for the Lamp1 quantification. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; # 0.05 < p < 0.1.
Few RNA changes associated with microglial-specific ApoE knock-out in 5xFAD mice
To determine whether microglial-specific ApoE knock-out affects overall gene expression in 5xFAD mice, bulk-tissue RNA sequencing of the mouse hippocampus was performed. As expected, expression of inflammatory genes was higher in 5xFAD mice compared to Control mice at both 4 and 12 months of age (Fig. 5a, S3a). This includes genes known as disease associated microglia (DAM) genes (Keren-Shaul et al., 2017), such as Cst7, Clec7a, and Itgax, to name a few. When microglial-specific ApoE is knocked-out of 5xFAD mice, we found there is remarkably little change in gene expression at 4 and 12 months of age (Fig. 5b, S3b), indicating that microglial-specific ApoE knock-out in 5xFAD mice does not induce significant hippocampal gene expression changes at the bulk-tissue level. To explore changes in network gene expression, we utilized weighted gene co-expression network analysis (WGCNA) and identified 15 independent modules (Fig. S3c). Correlation of each module to either AD genotype (Fig. 5c) or Apoe KO genotype (Fig. 5d) revealed several modules of interest. Particularly, the turquoise module is highly correlated to AD genotype (5xFAD vs. non-5xFAD) and consists of 1398 genes, while the green module is highly correlated to both AD genotype and Apoe KO genotype (microglial-ApoE intact vs microglial-ApoE KO; correlation scores >.4) and consists of 392 genes, with the turquoise module displaying gene network changes associated with microglia (Fig. 5e). For the green and turquoise modules, eigengenes were counted and plotted (Fig. 5f, g). While the turquoise module is strongly associated with AD genotype, it is not associated with Apoe KO genotype, and is upregulated in both 5xFAD and 5xFAD/Csf1r-Apoe-KO mice. It consists of microglial genes such as Clec7a, Ctsl, and Ctsh while pathway analyses identified GO terms such as viral gene expression and viral transcription (Fig 5h). Notably, the green module is associated with both AD and Apoe KO genotypes, and is only downregulated in 5xFAD mice, not 5xFAD/Csf1r-Apoe-KO mice, suggesting that microglial-specific knockout of ApoE prevents this network of genes from being downregulated. This green module is strongly enriched for genes associated with axonogenesis, microtubule bundle formation, and chromatin remodeling, with hub genes such as Eif4g3, Ncoa1, and Huwe1 (Fig. 5i).
Figure 5: Few RNA changes associated with microglial-specific ApoE knock-out.
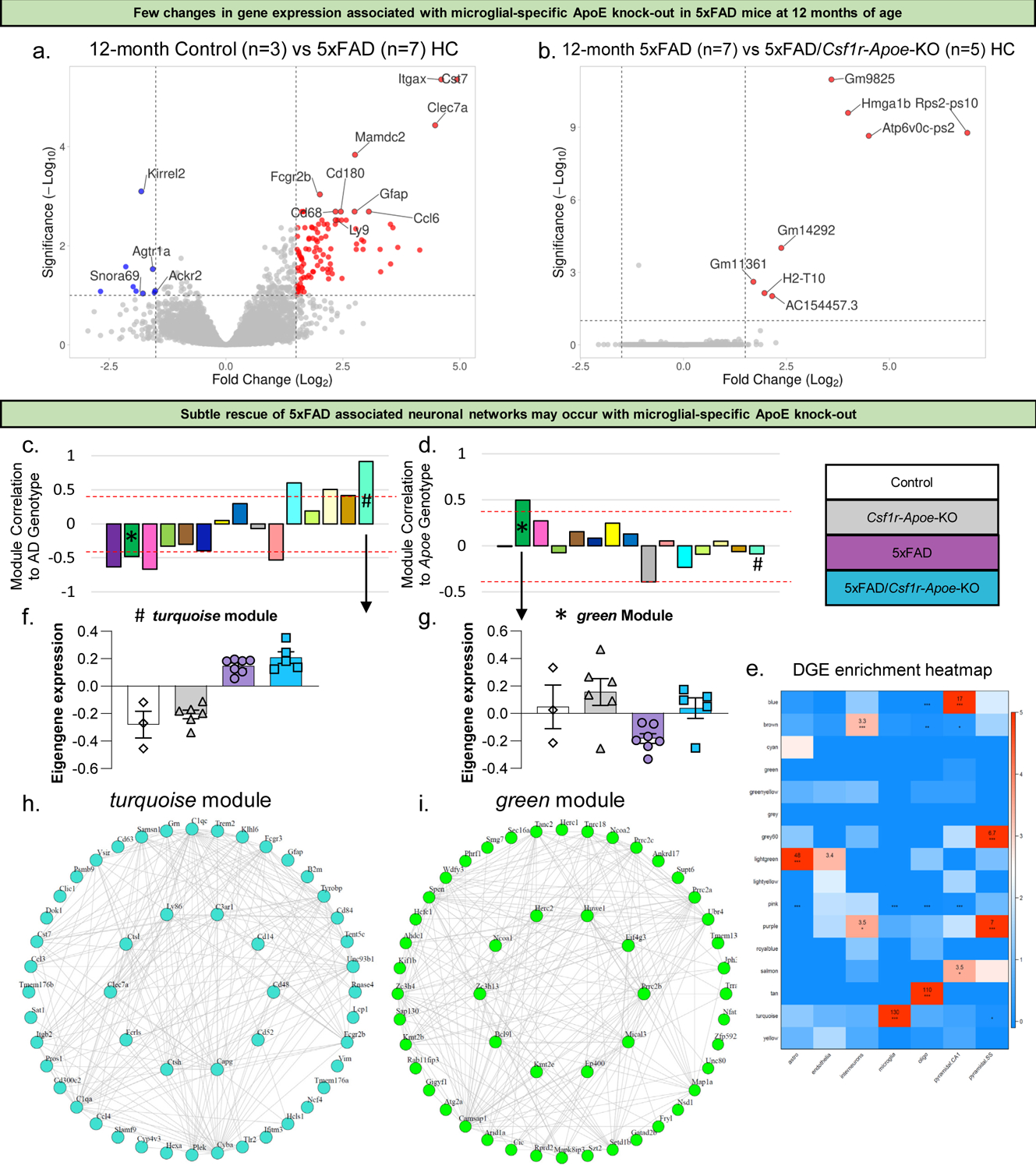
Volcano plot showing higher expression of inflammatory and other AD associated genes in the hippocampus of 5xFAD mice (n=7) compared to Control mice (n=3) at 12 months of age (a). Volcano plot showing very few changes in gene expression between 5xFAD (n=7) and 5xFAD/Csf1r-Apoe-KO (n=5) mice at 12 months of age (b). Correlation of modules generated by weighted gene correlation network analysis (WGCNA) to the AD genotype (Z-score cut-off: +/−0.4; * = green module; # = turquoise module) (c). Correlation of modules generated by WGCNA to Apoe genotype (Z-score cut-off: +/−0.4) (d). Cell-type enrichment heatmap displays genes associated with specific cell types within a given color module (e). Values provided indicate the number of genes within the network associated with that of a specific cell type. *** = 6+ genes. ** = 3+ genes. Module eigengene trajectory of gene expression value in turquoise (f) and green (g) modules. Interactive plot between hub genes extracted from the turquoise module showing a distinct AD signature (h). Interactive plot between hub genes extracted from the green module showing a distinct signature associated with Apoe knock-out (i).
Discussion
In this study, we sought to determine the role of microglial-expressed ApoE in AD pathogenesis and homeostasis. Previous studies have utilized global ApoE knock-out mouse models to elucidate the role of ApoE in AD (Bales et al., 1999; Holtzman et al., 2000; Krasemann et al., 2017; Ulrich et al., 2018); however, these studies do not delineate the role of microglial-expressed ApoE from astrocytic-expressed ApoE. Astrocytes have the capacity to associate with Aβ plaques in an ApoE-dependent fashion (Koistinaho et al., 2004) and internalize Aβ peptides (Funato et al., 1998; Matsunaga, Shirokawa, & Isobe, 2003). Therefore, there is a need to differentiate the roles of microglial- and astrocytic-Apoe in AD pathogenesis and homeostasis. To that end, we developed a mouse model that specifically eliminates microglial-Apoe expression. To accomplish this, we crossed Csf1r-cre mice with a 5xFAD mouse model and Apoefl/fl mice. The resultant progeny had Apoe expression constitutively knocked-out of microglia.
A well-studied aspect of the role of ApoE in AD is its association with Aβ. As previously noted, global ApoE knock-out has been shown to reduce the number Aβ plaques (Bales et al., 1999; Holtzman et al., 2000; Ulrich et al., 2018), a finding that we found is recapitulated by elimination of microglia (E. Spangenberg et al., 2019). Importantly, our data indicate that plaque load is not diminished, and CAA load does not change with microglial-specific loss of ApoE. Total soluble and insoluble Aβ1–40 and Aβ1–42 also remained unchanged with microglial-specific ApoE knock-out. However, we do see a significant increase in average plaque size in the hippocampus. The increase in plaque size aligns with a previous study utilizing the APP/PS1 mouse model of AD, which found that global ApoE deficiency increased Aβ plaque size, while decreasing amyloid burden (Ulrich et al., 2018). An increase in plaque size may indicate that microglial-expressed ApoE is necessary for microglial interaction with Aβ plaques, in line with data indicating that microglia are necessary for plaque compaction (Bolmont et al., 2008; Condello et al., 2015; Y. Wang et al., 2016). Interestingly, reductions in ApoE signaling prior to plaque deposition was shown to prevent Aβ accumulation in APOE*ε4 knock in mice, whereas its reduction after the emergence of plaques had no impact on plaque load, but showed an increase in average plaque size (Huynh et al., 2017), aligning directly with what we show following microglia-specific ApoE knock-out.
With microglial-specific knock-out of ApoE we found no differences in microglial number nor in the number of microglia per μm of plaque diameter, indicating that microglial clustering around plaques is unaltered in this model. Additionally, we found that astrocytic number was unchanged with microglial-specific ApoE knock-out in 5xFAD mice. Previous studies have shown that global Trem2 or Apoe knock-out decreases plaque-associated microgliosis in multiple mouse models of AD (Bales et al., 1999; Jay et al., 2017; Krasemann et al., 2017; Meilandt et al., 2020; Y. Wang et al., 2016) which is not seen with the model used in this study. Additionally, the interaction between TREM2 and ApoE has been shown to drive the dysfunctional transcriptional phenotype of microglia in AD (Krasemann et al., 2017). To explore whether this is recapitulated in our knockout model, we performed bulk-RNA sequencing on hippocampal tissue and observed very few changes in gene expression between 5xFAD mice and 5xFAD mice with microglial-specific ApoE intact. This included no changes in genes considered to be DAM associated (Cst7, Trem2, Ctss, Itgax, etc.). Additionally, there was no reduction in Apoe mRNA with microglial-specific ApoE knock-out in 5xFAD mice. Altogether, these data suggest microglial-expressed ApoE may not be necessary for the microglial transcriptional shift into a DAM state. It is important to clarify the limitations of bulk-tissue RNA sequencing as it reflects the averaged gene expression across many different cell types in the hippocampus. Worth noting, we find that microglia may utilize ApoE produced by other cells in the brain, such as astrocytes, as we find evidence for ApoE protein within plaque associated microglia in 5xFAD/Csf1r-Apoe-KO mice.
In mouse models of AD, the absence of ApoE increases neuritic dystrophy (Ulrich et al., 2018) and synaptic loss (Lane-Donovan et al., 2016; Masliah et al., 1995). However, reductions in APOE signaling after initial plaque deposition in APOE*ε4 APP/PS1–21 mice attenuate neuritic dystrophy, suggesting a time-specific role of ApoE in AD (Huynh et al., 2017). With microglial-specific knock-out of ApoE in 5xFAD mice, we found the ratio of dystrophic neurites to plaque volume trended downward (p=.076), suggesting microglial-expressed ApoE may contribute to neuronal damage in AD.
To determine why there may be alterations in plaque size and neuritic dystrophy, we explored genetic network analysis, utilizing weighted gene co-expression analysis (WGCNA). We found 15 independent modules of which turquoise and green stood out. The turquoise module was highly correlated to AD genotype (5xFAD vs. non-5xFAD) while the green module was highly correlated to both AD and Apoe KO genotypes (microglial-ApoE intact vs microglial ApoE KO). Interestingly in the green module, eigengene expression for genes associated with this network were downregulated in 5xFAD mice but upregulated with microglial-specific ApoE knock-out. Go-term analysis revealed that these genes are involved in neuronal processes such as axonogenesis, microtubule bundle formation, and chromatin remodeling. This may indicate that microglial-specific ApoE possibly promotes these neuronal functions and could serve as a reason why we see a downward trend in neuritic dystrophy in these mice compared to 5xFAD controls.
Reductions in synaptic proteins such as PSD95 have been observed in APOE*ε4 knock-in mice and APOE*ε4 postmortem human brain tissue (Liraz et al., 2013; Love et al., 2006; Yong et al., 2014). We found both pre- and post- synaptic protein levels were reduced in microglial-specific ApoE knock-out mice and 5xFAD microglial-specific ApoE knock-out mice compared to ApoE intact controls at 12 months of age. Importantly, microglial-specific ApoE knock-out in 5xFAD mice did not further exacerbate synaptic marker loss seen in 5xFAD mice. To investigate further, we stained tissue with homeostatic microglial marker P2ry12 and found a loss in intensity associated with microglial-expressed ApoE knock-out, mirroring our synaptic protein findings (Fig. 5d–k). Recently, we have shown that dysregulation of homeostatic microglia through partial inhibition of Csf1r results in reduced P2ry12 expression and reduction in synaptic proteins (Arreola et al., 2021). In a similar manner, microglial-expressed ApoE knock-out may dysregulate microglial homeostasis potentially leading to changes in synapse maintenance and development outside of AD.
Collectively, our results indicate that microglial-expressed ApoE does not profoundly alter the course of AD pathogenesis, at least with regard to the microglial reaction to plaques and adoption of a disease-associated gene expression phenotype. However, while we explored the role of microglial-specific ApoE in AD in this study, it is worth mentioning that targeting ApoE in the mouse may have differential effects compared to studies targeting human APOE variants. Additionally, the involvement of microglial-expressed ApoE in the pathogenesis of tau pathology needs to be considered – Trem2 knock-out studies have shown differential effects on inflammation in plaque vs. tau pathology developing mouse models (Jiang et al., 2016; Leyns et al., 2017; Y. Wang et al., 2016), while recent studies have highlighted the protective effects of both global ApoE knock-out and microglial depletion on brain atrophy induced by tau (Shi et al., 2019; Shi et al., 2017). Additionally, it was shown that the removal of APOE*ε4 from astrocytes stemmed neurodegeneration and lessened tau pathology and neurodegeneration in a P301S tauopathy mouse model (C. Wang et al., 2021). These studies highlight the need to determine the role of microglial-specific knock-out of ApoE in tauopathy models.
Conclusions
Our results indicate that microglial-expressed ApoE may not be necessary to induce the microglial transcriptional shift into a Disease Associated Microglia state that is associated with reactivity to plaques, nor for plaque formation, suggesting microglia may utilize astrocytic-derived ApoE in the absence of microglial-expressed ApoE. While seemingly unnecessary for plaque formation, we found microglial-expressed ApoE may be needed for proper plaque homeostasis in AD, and knock-out may result in subtle rescue in 5xFAD associated neuronal networks. Additionally, microglial-expressed ApoE may have a role in synapse maintenance, although further experimentation is needed to determine the mechanism behind this.
Supplementary Material
Figure S1: Microglial-specific ApoE knock-out results in no changes in cerebral amyloid angiopathy (CAA). Slide-scanned images of sliced half-brains show no traces of CAA in 5xFAD and 5xFAD/Csf1r-Apoe-KO at both 4 and 12 months of age (a).
Figure S3: Few changes in gene expression associated with microglial-specific ApoE knock-out in 5xFAD mice at 4 months of age. Volcano plot showing higher expression of inflammatory and other AD associated genes in the hippocampus of 5xFAD (n=5) mice compared to Control (n=8) mice at 4 months of age (a). Volcano plot showing very few changes in gene expression between 5xFAD (n=5) and 5xFAD/Csf1r-Apoe-KO (n=4) mice at 4 months of age (b). WGCNA Dendrogram highlighting 15 independent modules (c).
Figure S2: P2ry12 intensity is reduced with microglial-specific ApoE knock-out. Representative 20x images of Amylo-Glo dye staining and IBA1 and Cd68 immunofluorescence in 12-month-old 5xFAD and 5xFAD/Csf1r-Apoe-KO mice (a, b). No significant change in Cd68 percent coverage was observed between 5xFAD (4 months n=9; 12 months n=9) and 5xFAD/Csf1r-Apoe-KO (4 months n=7; 12 months n=6) mice in the hippocampus (c) nor the somatosensory cortex (d). Representative 20x images of Amylo-Glo dye staining and P2ry12 immunofluorescence in the hippocampus in Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice at 12 months of age (e). A significant reduction in P2ry12 staining was observed in Csf1r-Apoe-KO (n=6), 5xFAD (n=10), and 5xFAD/Csf1r-Apoe-KO (n=5) mice compared to Control (n=6) mice (f). Representative 20x images of Amylo-Glo dye staining and IBA1 and ApoE immunofluorescence in 5xFAD and 5xFAD/Csf1r-Apoe-KO mice. IBA1+ApoE+ colocalized volume is unchanged between 5xFAD (n=4) and 5xFAD/Csf1r-Apoe-KO (n=3) mice (h) as is Amylo-Glo+ApoE+ colocalized volume (i). Statistical analysis used a two-way ANOVA with Tukey’s multiple comparisons correction for Cd68 % Area, a one-way ANOVA with Tukey’s multiple comparisons correction for P2ry12 intensity, and a two-tailed t-test for the ApoE quantifications. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; # 0.05 < p < 0.1.
Main Points.
Microglia-specific ApoE knockout does not alter plaque load, CAA formation, or inflammatory gene expression in 5xFAD mice.
Microglia-specific ApoE knockout leads to impaired plaque compaction in 5xFAD mice and reduced synaptic protein levels outside of disease.
Subtle rescue of 5xFAD associated neuronal networks may occur with microglial-specific ApoE knock-out
Acknowledgments:
This work was supported by the National Institutes of Health (NIH) under awards: R01NS083801 (NINDS), R01AG056768 (NIA), 1RF1AG065329 (NIA), and U54 AG054349 (NIA Model Organism Development and Evaluation for Late-onset Alzheimer’s Disease (MODEL-AD)) to K.N.G., 1F31NS111882-01A1 (NINDS) to M.A.A., F31AG059367 (NIA) and T32AG00096 (NIA) to E.E.S., 1F31AG072852-01 (NIA) to C.M.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Sources of funding did not have any role in study design, data collection or analysis, interpretation of results, or manuscript preparation or submission.
Abbreviations
- AD
Alzheimer’s Disease
- Aβ
Amyloid Beta
- APOE
Apolipoprotein E
- CAA
Cerebral Amyloid Angiopathy
- Csf1r
Colony stimulating factor 1 receptor
- DAM
Disease associated microglia
- GFAP
Glial fibrillary acidic protein
- HC
Hippocampus
- IBA1
Ionized calcium binding adaptor molecule 1
- Lrp1
Low density lipoprotein receptor-related protein 1
- PSD95
Postsynaptic density protein 95
- S100β
S100 Calcium Binding Protein B
- ssCtx
Somatosensory cortex
- Trem2
Triggering receptor expressed on myeloid cells 2
- Vis Ctx
Visual cortex
Footnotes
Ethics approval
The present study and associated protocols were approved by the UC Irvine Institutional Animal Care and Use Committee (IACUC) and were compliant with ethical regulations for animal research and testing.
Competing Interests
KNG is on the scientific advisory board of Ashvattha Therapeutic Inc. All other authors declare no competing interests.
Availability of data and materials
RNA sequencing is available on GEO servers.
References
- Arendt T, Schindler C, Bruckner MK, Eschrich K, Bigl V, Zedlick D, & Marcova L (1997). Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J Neurosci, 17(2), 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola MA, Soni N, Crapser JD, Hohsfield LA, Elmore MRP, Matheos DP, … Green KN (2021). Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R(+/−) mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci Adv, 7(35). doi: 10.1126/sciadv.abg1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, … Bu G (2015). Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J Biol Chem, 290(43), 26043–26050. doi: 10.1074/jbc.M115.679043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CC, DeVaux LB, & Farzan M (2015). The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J Biol Chem, 290(43), 26033–26042. doi: 10.1074/jbc.M115.677286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, … Paul SM (1999). Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A, 96(26), 15233–15238. doi: 10.1073/pnas.96.26.15233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, … Calhoun ME (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci, 28(16), 4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, & Taylor JM (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest, 76(4), 1501–1513. doi: 10.1172/JCI112130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, … Holtzman DM (2011). Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med, 3(89), 89ra57. doi: 10.1126/scitranslmed.3002156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, & Grutzendler J (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun, 6, 6176. doi: 10.1038/ncomms7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr., … et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet, 7(2), 180–184. doi: 10.1038/ng0694-180 [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, … Pericak-Vance MA (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science, 261(5123), 921–923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, … van Duijn CM (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA, 278(16), 1349–1356. [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, … Ihara Y (1998). Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol, 152(4), 983–992. [PMC free article] [PubMed] [Google Scholar]
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, … Campion D (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry, 16(9), 903–907. doi: 10.1038/mp.2011.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, & Luijsterburg MS (2020). VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci Rep, 10(1), 20560. doi: 10.1038/s41598-020-76603-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, & Sheng M (2018). Microglia in Alzheimer’s disease. J Cell Biol, 217(2), 459–472. doi: 10.1083/jcb.201709069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem, 110(4), 1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, … Hyman BT (2000). Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol, 47(6), 739–747. [PubMed] [Google Scholar]
- Huang Y, Happonen KE, Burrola PG, O’Connor C, Hah N, Huang L, … Lemke G (2021). Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat Immunol. doi: 10.1038/s41590-021-00913-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, … Holtzman DM (2017). Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron, 96(5), 1013–1023 e1014. doi: 10.1016/j.neuron.2017.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., & Holtzman DM (2013). Biomarker modeling of Alzheimer’s disease. Neuron, 80(6), 1347–1358. doi: 10.1016/j.neuron.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, … Posthuma D (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet, 51(3), 404–413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, … Landreth GE (2017). Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci, 37(3), 637–647. doi: 10.1523/JNEUROSCI.2110-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Zhang YD, Chen Q, Gao Q, Zhu XC, Zhou JS, … Yu JT (2016). TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology, 105, 196–206. doi: 10.1016/j.neuropharm.2016.01.028 [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, & Bu G (2014). ApoE and Abeta in Alzheimer’s disease: accidental encounters or partners? Neuron, 81(4), 740–754. doi: 10.1016/j.neuron.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, … Amit I, (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169(7), 1276–1290 e1217. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, … Paul SM (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med, 10(7), 719–726. doi: 10.1038/nm1058 [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, … Butovsky O (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47(3), 566–581 e569. doi: 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, … Ma’ayan A (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res, 44(W1), W90–97. doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, … Environmental Risk for Alzheimer’s Disease, C. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet, 51(3), 414–430. doi: 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, & Frail DE (1994). Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem, 269(38), 23403–23406. [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, … Amouyel P (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet, 45(12), 1452–1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C, Wong WM, Durakoglugil MS, Wasser CR, Jiang S, Xian X, & Herz J (2016). Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J Neurosci, 36(39), 10141–10150. doi: 10.1523/JNEUROSCI.1054-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, … Holtzman DM (2019). TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci, 22(8), 1217–1222. doi: 10.1038/s41593-019-0433-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, … Holtzman DM (2017). TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A, 114(43), 11524–11529. doi: 10.1073/pnas.1710311114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liraz O, Boehm-Cagan A, & Michaelson DM (2013). ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol Neurodegener, 8, 16. doi: 10.1186/1750-1326-8-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, & Bu G (2017). ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron, 96(5), 1024–1032 e1023. doi: 10.1016/j.neuron.2017.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, & Allen SJ (2006). Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging, 27(6), 797–803. doi: 10.1016/j.neurobiolaging.2005.04.008 [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, & Roses AD (1995). Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol, 136(2), 107–122. doi: 10.1006/exnr.1995.1088 [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, … Tsai LH (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570(7761), 332–337. doi: 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga W, Shirokawa T, & Isobe K (2003). Specific uptake of Abeta1-40 in rat brain occurs in astrocyte, but not in microglia. Neurosci Lett, 342(1–2), 129–131. doi: 10.1016/s0304-3940(03)00240-4 [DOI] [PubMed] [Google Scholar]
- Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee SH, Srinivasan K, … Hansen DV (2020). Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Abeta42:Abeta40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J Neurosci, 40(9), 1956–1974. doi: 10.1523/JNEUROSCI.1871-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, … Vassar R (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci, 26(40), 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, … Haass C (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci, 22(2), 191–204. doi: 10.1038/s41593-018-0296-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierantoni GM, Rinaldo C, Esposito F, Mottolese M, Soddu S, & Fusco A (2006). High Mobility Group A1 (HMGA1) proteins interact with p53 and inhibit its apoptotic activity. Cell Death Differ, 13(9), 1554–1563. doi: 10.1038/sj.cdd.4401839 [DOI] [PubMed] [Google Scholar]
- Ping L, Duong DM, Yin L, Gearing M, Lah JJ, Levey AI, & Seyfried NT (2018). Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci Data, 5, 180036. doi: 10.1038/sdata.2018.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, & Mahley RW (1987). Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta, 917(1), 148–161. doi: 10.1016/0005-2760(87)90295-5 [DOI] [PubMed] [Google Scholar]
- Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, … Kontula K (1995). Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med, 333(19), 1242–1247. doi: 10.1056/NEJM199511093331902 [DOI] [PubMed] [Google Scholar]
- Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, … Levey AI (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener, 13(1), 24. doi: 10.1186/s13024-018-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, … Alzheimer’s Disease Genetics, C. (2020). Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun, 11(1), 667. doi: 10.1038/s41467-019-14279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, & Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GA, Tai LM, LaDu MJ, & Rebeck GW (2014). Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J Neuroinflammation, 11, 111. doi: 10.1186/1742-2094-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, … et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology, 43(8), 1467–1472. doi: 10.1212/wnl.43.8.1467 [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, … Roses AD (1993). Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A, 90(20), 9649–9653. doi: 10.1073/pnas.90.20.9649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Qian J, Monsell SE, Betensky RA, & Hyman BT (2015). APOEepsilon2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol, 77(6), 917–929. doi: 10.1002/ana.24369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, … Holtzman DM (2019). Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med, 216(11), 2546–2561. doi: 10.1084/jem.20190980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, … Holtzman DM (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673), 523–527. doi: 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, … Green KN (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun, 10(1), 3758. doi: 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, … Green KN (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain, 139(Pt 4), 1265–1281. doi: 10.1093/brain/aww016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, … Stefansson K (2015). Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet, 47(5), 445–447. doi: 10.1038/ng.3246 [DOI] [PubMed] [Google Scholar]
- Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, & Corey-Bloom J (2004). Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology, 62(11), 1977–1983. doi: 10.1212/01.wnl.0000128091.92139.0f [DOI] [PubMed] [Google Scholar]
- Uchihara T, Duyckaerts C, He Y, Kobayashi K, Seilhean D, Amouyel P, & Hauw JJ (1995). ApoE immunoreactivity and microglial cells in Alzheimer’s disease brain. Neurosci Lett, 195(1), 5–8. doi: 10.1016/0304-3940(95)11763-m [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, Song WM, … Holtzman DM (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J Exp Med, 215(4), 1047–1058. doi: 10.1084/jem.20171265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, … Holtzman DM (2021). Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. doi: 10.1016/j.neuron.2021.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, … Colonna M (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med, 213(5), 667–675. doi: 10.1084/jem.20151948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Wang Y, Tom I, Gonzalez LC, & Sheng M (2016). TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron, 91(2), 328–340. doi: 10.1016/j.neuron.2016.06.015 [DOI] [PubMed] [Google Scholar]
- Yong SM, Lim ML, Low CM, & Wong BS (2014). Reduced neuronal signaling in the ageing apolipoprotein-E4 targeted replacement female mice. Sci Rep, 4, 6580. doi: 10.1038/srep06580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, & Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol, 4, Article17. doi: 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, … Wu JQ (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 34(36), 11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, … Barres BA (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron, 89(1), 37–53. doi: 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Microglial-specific ApoE knock-out results in no changes in cerebral amyloid angiopathy (CAA). Slide-scanned images of sliced half-brains show no traces of CAA in 5xFAD and 5xFAD/Csf1r-Apoe-KO at both 4 and 12 months of age (a).
Figure S3: Few changes in gene expression associated with microglial-specific ApoE knock-out in 5xFAD mice at 4 months of age. Volcano plot showing higher expression of inflammatory and other AD associated genes in the hippocampus of 5xFAD (n=5) mice compared to Control (n=8) mice at 4 months of age (a). Volcano plot showing very few changes in gene expression between 5xFAD (n=5) and 5xFAD/Csf1r-Apoe-KO (n=4) mice at 4 months of age (b). WGCNA Dendrogram highlighting 15 independent modules (c).
Figure S2: P2ry12 intensity is reduced with microglial-specific ApoE knock-out. Representative 20x images of Amylo-Glo dye staining and IBA1 and Cd68 immunofluorescence in 12-month-old 5xFAD and 5xFAD/Csf1r-Apoe-KO mice (a, b). No significant change in Cd68 percent coverage was observed between 5xFAD (4 months n=9; 12 months n=9) and 5xFAD/Csf1r-Apoe-KO (4 months n=7; 12 months n=6) mice in the hippocampus (c) nor the somatosensory cortex (d). Representative 20x images of Amylo-Glo dye staining and P2ry12 immunofluorescence in the hippocampus in Control, Csf1r-Apoe-KO, 5xFAD, and 5xFAD/Csf1r-Apoe-KO mice at 12 months of age (e). A significant reduction in P2ry12 staining was observed in Csf1r-Apoe-KO (n=6), 5xFAD (n=10), and 5xFAD/Csf1r-Apoe-KO (n=5) mice compared to Control (n=6) mice (f). Representative 20x images of Amylo-Glo dye staining and IBA1 and ApoE immunofluorescence in 5xFAD and 5xFAD/Csf1r-Apoe-KO mice. IBA1+ApoE+ colocalized volume is unchanged between 5xFAD (n=4) and 5xFAD/Csf1r-Apoe-KO (n=3) mice (h) as is Amylo-Glo+ApoE+ colocalized volume (i). Statistical analysis used a two-way ANOVA with Tukey’s multiple comparisons correction for Cd68 % Area, a one-way ANOVA with Tukey’s multiple comparisons correction for P2ry12 intensity, and a two-tailed t-test for the ApoE quantifications. Significance indicated as * p< 0.05; ** p < 0.01; *** p < 0.001; # 0.05 < p < 0.1.
Data Availability Statement
RNA sequencing is available on GEO servers.