

Abstract
Ticks are the second most important vector capable of transmitting diseases affecting the health of both humans and animals. Amblyomma testudinarium Koch 1844 (Acari: Ixodidae), is a hard tick species having a wide geographic distribution in Asia. In this study, we analyzed the composition of A. testudinarium whole mitogenomes from various geographical regions in Japan and investigated the population structure, demographic patterns, and phylogeographic relationship with other ixodid species. In addition, we characterized a potentially novel tick species closely related to A. testudinarium from Myanmar. Phylogeographic inference and evolutionary dynamics based on the 15 mitochondrial coding genes supported that A. testudinarium population in Japan is resolved into a star‐like haplogroup and suggested a distinct population structure of A. testudinarium from Amami island in Kyushu region. Correlation analysis using Mantel test statistics showed that no significant correlation was observed between the genetic and geographic distances calculated between the A. testudinarium population from different localities in Japan. Finally, demographic analyses, including mismatch analysis and Tajima’s D test, suggested a possibility of recent population expansion occurred within Japanese haplogroup after a bottleneck event. Although A. testudinarium has been considered widespread and common in East and Southeast Asia, the current study suggested that potentially several cryptic Amblyomma spp. closely related to A. testudinarium are present in Asia.
Keywords: Amblyomma, cryptic species, mitogenome, phylogeography, population expansion, ticks
1. INTRODUCTION
Ticks are obligatory ectoparasites that feed on the blood of various vertebrate species including mammals, birds, and reptiles (Brites‐Neto et al., 2015). There are three major tick families described, namely, Ixodidae, Argasidae, and Nuttalliellidae (Horak et al., 2002). Some species of Ixodidae (hard ticks) and Argasidae (soft ticks) are implicated in the transmission of several bacterial, viral, and protozoan pathogens to humans and animals. Ticks are widely distributed around the world, especially in warm humid regions to remain hydrated and subsequently undergo metamorphosis (Vail & Smith, 1998).
Amblyomma is the third biggest genus of Ixodidae ticks with more than 130 species (Guglielmone et al., 2014), some of which act as disease vectors of many pathogens, for instance Rickettsia tamurae and Rickettsia raoultii causing spotted fever (Fournier et al., 2006; Piotrowski & Rymaszewska, 2020), Ehrlichia chaffeensis causing ehrlichiosis (Cao et al., 2000), and severe fever with thrombocytopenia syndrome (SFTS) virus causing SFTS in humans (Suh et al., 2016). The geographical distribution of Amblyomma species is expanding due to anthropogenic activities, climate change, and increased geographical ranges of animal hosts (Childs & Paddock, 2003). In the United States, Amblyomma americanum populations have been constantly enlarging that contributed to change in the epidemiology of spotted fever group rickettsiosis in the country (Dahlgren et al., 2016; Sagurova et al., 2019). In Africa, Amblyomma species transmit Ehrlichia ruminantium and Rickettsia africae that cause heartwater disease and African tick‐bite fever in animals and humans, respectively (Kelly et al., 1996; Yunker, 1996). Furthermore, although very little is known about the diseases caused by pathogens transmitted by this group of ticks (Jabin et al., 2019), there are 14 Amblyomma species in Asia (Voltzit, 2002), and at least three species, including Amblyomma geoemydae, Amblyomma helvolum, and A. testudinarium are known to carry pathogens (Fournier et al., 2006; Sumrandee et al., 2014; Takano et al., 2012).
Amblyomma testudinarium Koch, 1844 (Acari: Ixodidae), is widely distributed in the Asian countries including Malaysia, India, Thailand, Laos, Vietnam, Indonesia, Borneo, Sarawak, the Philippines, Japan, Korea, Mainland China, and Taiwan (Kang et al., 1981; Lim et al., 2018). In Japan, A. testudinarium distribution was limited to the warm areas of Kanto region and the southwestern part of Japan, including Chubu, Kinki, Chugoku, Shikoku, Kyushu, and Okinawa regions (Takada, 1990). In recent years, A. testudinarium was infesting on grazing cattle and brown bears in the northern parts of Japan such as Aomori and Hokkaido prefectures, which were suspected to be carried by the migratory birds (Nakao et al., 2021; Terada et al., 2019). Amblyomma testudinarium is known as a vector for R. tamurae, a causative agent of spotted fever rickettsiosis in humans (Fournier et al., 2006; Imaoka et al., 2011), and SFTS virus in Japan. Recently, a novel thogotovirus, named as Oz virus, was isolated from A. testudinarium collected in Ehime prefecture, Japan (Ejiri et al., 2018). Cases of human bites have been reported elsewhere (Isohisa et al., 2011; Nakao et al., 2017). Moreover, the cases of tick‐associated rash illness (TARI) showing manifestation of erythema migrans (EM) without association of Lyme disease have been reported in the patients who were bitten by this tick species (Natsuaki et al., 2014).
High‐throughput sequencing techniques are very functional in species identification and population genetics of ticks (Burger et al., 2012, 2013, 2014). Comparative whole mitochondrial genome (mitogenome) sequence analyses have been used to identify key genetic features among tick species (Kelava et al., 2021; Regilme et al., 2021; Wang et al., 2019). Genetic characterization of complete mitogenome sequences is a core study nowadays due to its highly conserved structure, the high mutation rate, low recombination rate, and maternal inheritance. Recently, two studies reported the complete whole mitogenome of A. testudinarium, which enables the comparative analysis in a higher resolution between tick species (Chang et al., 2020; Nakao et al., 2021) than previously implemented intraspecies phylogenetic analyses targeting partial sequences of 16S ribosomal RNA gene (rDNA), 12S rDNA, and cytochrome c oxidase I gene (COI) (Chao et al., 2017; Yamauchi et al., 2012).
In the present study, we describe the population structure of A. testudinarium based on whole mitogenome sequences obtained from Japan. We investigated the relationships between genetic distances of A. testudinarium samples and the corresponding geographical locations from which they were collected. No geographic clustering was detected among A. testudinarium sequences from Japan, except the samples from Amami island in Kyushu region. Additionally, we characterized a potentially novel Amblyomma species from Myanmar and investigated its genetic relationships with A. testudinarium based on the parsimony informative sites of their mitogenomes.
2. MATERIAL AND METHODS
2.1. Specimen collection and DNA extraction
A total of 39 tick specimens were included in this study. Thirty ticks were collected from 22 collection sites in four geographical blocks (Kinki, Chugoku, Shikoku, and Kyushu) in Japan and nine specimens from Putao town in Myanmar (Table 1, Figure 1). All tick samples from Japan were collected from the vegetations (n = 30), while those from Myanmar were collected from water buffaloes (Bubalus bubalis) (n = 5) and cattle (Bos taurus) (n = 4) (Table 1). The collected ticks were kept at 4°C until transferred to the laboratory for further examinations. Species were identified morphologically using standard taxonomic keys (Yamaguti et al., 1971) and a related article (Chamuah et al., 2016) under a stereomicroscope. Then, each tick was cut in half with a sterile stainless‐steel blade. One half was crushed with stainless‐steel beads using a Micro Smash MS‐100R (TOMY, Tokyo, Japan) at 2500 rpm, and the DNA was extracted using a blackPREP Tick DNA/RNA Kit (Analytikjena, Germany) as described previously (Thu et al., 2019).
TABLE 1.
Geographic origin, collection source, and developmental stage/sex of Amblyomma ticks
| Geographic block | Prefecture | Collection site | Collection source | Number | Data source | Accession number(s) | Stage/sex |
|---|---|---|---|---|---|---|---|
| Hokkaido | Hokkaido | Site 1 | Brown bear | 1 | Nakao et al. (2021) | LC553841 | Female |
| Kinki | Mie | Site 1 | Vegetation | 2 | This study | LC554771, LC554772 | Nymph |
| Site 2 | 2 | LC554773, LC554774 | Nymph | ||||
| Site 3 | 2 | LC554775, LC554776 | Nymph | ||||
| Nara | Site 1 | 1 | Kelava et al. (2021) | MT371798 | Nymph | ||
| Wakayama | Site 1 | 1 | This study | LC554769 | Female | ||
| Site 2 | 1 | LC554770 | Female | ||||
| Chugoku | Hiroshima | Site 1 | Vegetation | 1 | This study | LC554788 | Male |
| Site 2 | 1 | LC554789 | Male | ||||
| 1 | LC554787 | Female | |||||
| Shimane | Site 1 | 1 | LC554777 | Male | |||
| Site 2 | 1 | LC554778 | Female | ||||
| Shikoku | Ehime | Site 1 | Vegetation | 1 | This study | LC554780 | Male |
| Site 2 | 2 | LC554781, LC554782 | Nymph | ||||
| Site 3 | 1 | LC554783 | Female | ||||
| Kochi | Site 1 | 1 | LC554779 | Male | |||
| Kyushu | Nagasaki | Site 1 | Vegetation | 1 | This study | LC554790 | Male |
| Kumamoto | Site 1 | 1 | LC554766 | Nymph | |||
| Miyazaki | Site 1 | 2 | LC554761, LC554762 | Nymph | |||
| Site 2 | 2 | LC554763, LC554764 | Nymph | ||||
| Kagoshima | Site 1 | 1 | LC554767 | Nymph | |||
| Site 2 | 1 | LC554765 | Female | ||||
| Site 2 | 1 | LC554768 | Nymph | ||||
| Amami (Kagoshima) | Site 1 | 1 | LC554786 | Nymph | |||
| Site 2 | 2 | LC554784, LC554785 | Nymph | ||||
| Myanmar | Putao | Site 1 | Cattle | 2 | This study | LC633546 | Male |
| LC633547 | Female | ||||||
| Water buffalo | 2 | LC633548, LC633549 | Male | ||||
| Site 2 | Cattle | 1 | LC633550 | Male | |||
| 1 | LC633554 | Female | |||||
| Water buffalo | 3 | LC633551–LC633553 | Male | ||||
| Total | 41 | ||||||
FIGURE 1.
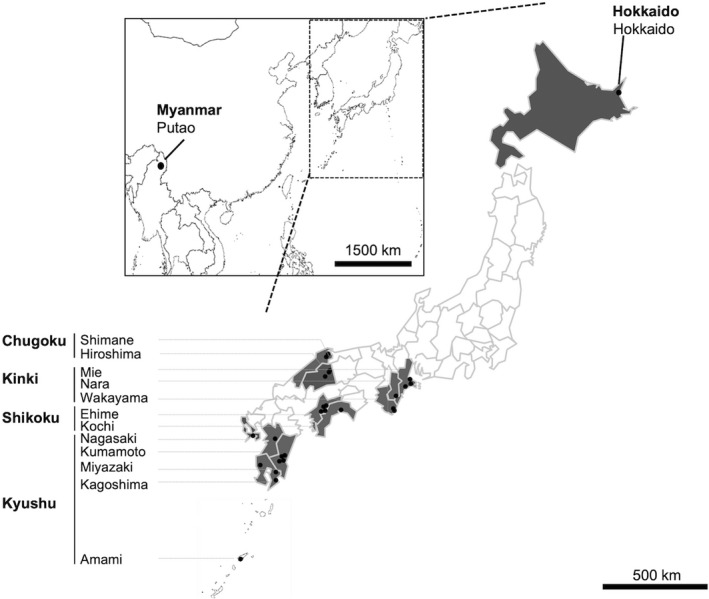
Geographic distribution of Amblyomma samples used in the present study. Sample collection sites are illustrated in dots. Samples were collected from eleven prefectures in four geographical blocks in Japan. In each prefecture, samples were collected at least from two different collection sites except for Nagasaki where only one site was explored
2.2. Next‐generation sequencing and read assembly
The entire mitogenome sequence of A. testudinarium was amplified in two PCRs (long‐ and short‐range) according to the previous study with some modifications (Kelava et al., 2021). The long‐range PCR primers (K23A: 5′‐TCCTACATGATCTGAGTTTARACCG‐3′ and ALF3: 5′‐AAATTCCTCTGAAAAGCTTAARRTACC‐3′) and the short‐range PCR primers (A_gap_AMBL_F2: 5′‐AACACTTAACATTTTCATTG‐3′ and A_gap_AMBL_R1: 5′‐ RACHAGGATTAGATACCCTWYTATT‐3′) were designed by aligning complete mitogenomes of genus Amblyomma deposited in the database. Long‐range PCR was performed in a 50‐μl reaction mixture containing 10 μl of 5 × PrimeSTAR GXL Buffer (Mg2+ Plus) (TaKaRa Bio Inc., Shiga, Japan), 4.0 μl of dNTP Mixture (2.5 mM each), 200 nM of each primer, 1.0 μl of PrimeSTAR® GXL DNA Polymerase (TaKaRa Bio Inc.), and 2.0 μl of template DNA with reaction conditions as follow: 45 cycles of 98°C for 10 s, 60°C for 15 s, and 68 °C for 10 min. Short‐range PCR was performed in a 25‐μl reaction mixture containing 12.5 μl of 2 × Gflex PCR Buffer (Mg2+, dNTP plus) (TaKaRa Bio Inc.), 0.5 μl of Tks Gflex DNA Polymerase (1.25 units/μl) (TaKaRa Bio Inc.), 200 nM of each primer, and 1.0 μl of template DNA with the reaction conditions: 94°C for 60 s, 45 cycles of 98°C for 10 s, 55°C for 15 s, 68°C for 60 s, and a final extension of 68°C for 5 min. Electrophoresis in a 1.5% agarose gel stained with Gel‐Red™ (Biotium, Hayward, CA) was used to analyze the amplified PCR products. NucleoSpin Gel and PCR Clean‐Up Kit (TaKaRa Bio Inc.) were used to purify the PCR products.
The long‐range and short‐range PCR products were mixed at equimolar concentrations in the ratio of 7:1, respectively. DNA concentration of mixed PCR products from each sample was adjusted to 0.2 ng/μl. Illumina sequencing libraries were constructed from the purified PCR amplicons with the Nextera DNA Library Prep Kit (Illumina, Hayward, CA). Illumina MiSeq platform was used for sequencing using the MiSeq reagent kit v3 for 600 cycles. CLC Genomics Workbench v20.0.4 (Qiagen, Hilden, Germany) was used to assemble the reads to obtain complete mitogenome sequence of each sample.
2.3. Phylogenetic analysis based on 16S rDNA and COI sequences
We constructed the phylogenetic trees using the previously published partial mitochondrial 16S rDNA and COI sequences of A. testudinarium samples from Taiwan and Thailand. In addition, we extracted the mitochondrial 16S rDNA and COI sequences from the complete mitogenome sequences of A. testudinarium from China, Myanmar, and Japan, six other Amblyomma spp., seven Haemaphysalis spp., and one each of Archaeocroton sphenodonti, Bothriocroton undatum, Dermacentor nitens, Dermacentor silvarum, Rhipicentor nuttalli, Rhipicephalus microplus, and Robertsicus elaphensis available in the database. Sequences of the 16S rDNA were aligned and the best fit model was selected using MEGAX software (Kumar et al., 2018). Maximum‐likelihood (ML) trees were constructed by setting the General Time Reversible (GTR) as a substitution model and gamma distribution at 5 for both the 16S rDNA and COI datasets. The phylogenetic relationship was tested by setting the bootstrap method at 1000 iterations.
Haplotypes were resolved, independently, based on the 16S rDNA sequences extracted from mitogenomes of A. testudinarium collected from Japan and the Amblyomma sp. from Myanmar using the DnaSP version 6.0 (Librado & Rozas, 2009). Median‐joining network was calculated and constructed with all parameters designated to default values in the Network version 10.2.0.0 (Bandelt et al., 1999) to understand the phylogenetic relationships among haplotypes.
2.4. Mitogenome comparison and phylogeny construction
Our complete mitogenome sequences were imported to Geneious version 10.2.6 (Biomatters Ltd., Auckland, New Zealand) and aligned with the mitogenome sequences of A. testudinarium, A. geoemydae, Amblyomma javanense, Amblyomma fimbriatum, Amblyomma triguttatum, A. americanum, Amblyomma sculptum, Haemaphysalis concinna, Haemaphysalis formosensis, Haemaphysalis flava, Haemaphysalis japonica, Haemaphysalis hystricis, Haemaphysalis longicornis, Haemaphysalis inermis, A. sphenodonti, B. undatum, D. nitens, D. silvarum, R. nuttalli, R. microplus, and R. elaphensis. A total of 13 protein coding gene (PCG) and two rDNA sequences were extracted from each mitogenome sequence and concatenated together.
Sequences of the concatenated 15 mitochondrial genes of Amblyomma samples collected from Japan and Myanmar along with those obtained from the GenBank were aligned using MAFFT software (Katoh & Standley, 2013). A substitution model was selected using PHYML 3.0 software relying on the Akaike Information Criterion (Guindon et al., 2010). A pairwise identity analysis was conducted by calculating the percentage of residues that are identical between sequences in Geneious software.
Bayesian phylogenetic tree was constructed using BEAST version 1.4. as a cross‐platform program for Bayesian analysis of molecular sequences using Markov chain Monte Carlo (MCMC). We modeled the evolution of sequences by using the GTR nucleotide substitution model with discrete gamma‐distributed rate variation and assuming a constant evolution rate all over the tree by selecting the strict clock model. We assume that the Bayesian skyline coalescent model was a demographic model in a Bayesian framework. The Bayesian skyline plot analysis was performed using a chain length of 50 million generations sampled every 50,000 MCMC steps with a pre‐burn‐in of 500,000. Maximum clade credibility (MCC) was selected by using the TreeAnnotator (Drummond & Rambaut, 2007). The MCC tree was illustrated using FigTree version 1.4.4 (http://beast.bio.ed.ac.uk/figtree), and the branch length of the tree was transformed to proportional and supported by the posterior values. To estimate the differences between A. testudinarium sequences in relation to the geographical differences, we performed a nonmetric multidimensional scaling (NMDS) analysis and visualized the results in space using the R package Bios2mds (Pele et al., 2012).
2.5. Population genetic structure analyses
The analysis of molecular variance (AMOVA) (Excoffier et al., 2007) in Arlequin software version 3.5.2.2 was used to evaluate the genetic variance among and within the populations of A. testudinarium collected from Amami island in Kyushu region vs other localities in Japan. We adjusted the number of permutations at 1000, and the difference was considered significant when tested at p < 0.05 level based on the calculated fixation indices (F‐statistics). F ST estimates the degree of differentiation within the population where the closer F ST is to 0, the greater the extent of allelic fixation or identity within populations (Holsinger & Weir, 2009). F SC estimates the differentiation among populations within the group to which they are assigned. The closer F SC is to 1, the more heterogeneity among populations exists. In case a strong population genetic structure exists at the population scale being analyzed, F SC should be high relative to F ST.
2.6. Spatial and demographic evolutionary dynamics analyses
The historical expansion dynamics of A. testudinarium populations were estimated by calculating the mismatch distribution based on 15 concatenated mitochondrial genes in Arlequin software (Rogers & Harpending, 1992). We compared the observed and expected distributions of pairwise nucleotide variations between individuals. Multimodal distribution is thought to be associated with a constant population size, whereas unimodal distribution is representing a sudden expansion. The deviation of observed mismatch from the individual expansion model was assessed by estimating the sum of square deviation (SSD); however, the Harpending’s raggedness index (RI) was applied to assess the smoothness of the haplotype frequency distribution (Harpending, 1994) where significant RI indicates a poor fit of the data to the expansion model.
Departure from the neutral model of evolution was evaluated through calculating Tajima’s D statistics (Tajima, 1989) in the Arlequin software based on the infinite‐size model by quantifying the difference between the average pairwise nucleotide differences and polymorphic sites. Significantly negative value of Tajima’s D suggests a positive selection or exhibiting a sudden population expansion after a recent bottleneck event. Significantly positive value of Tajima’s D means an increase of average pairwise genetic diversity in a population, indicating a balancing selection model or a population contraction events such as population subdivision. Nonsignificant and near zero values of Tajima’s D mean a constant population size.
Mantel test was used to test whether there is a geographic separation between A. testudinarium populations from different geographic origins and to evaluate the correlation between genetic variation and geographic distance of the tick population. R function “mantel.rtest” in ape package was used (Wynn et al., 2016). Scatter plot of the correlation was created using the R package ggplot2 (Ginestet, 2011; Villanueva & Chen, 2019).
2.7. Data accessibility
The complete mitogenome sequences of 30 A. testudinarium and 9 Amblyomma sp. were deposited in the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp) with accession numbers of LC554761‐LC554790 (samples from Japan) and LC633546‐LC633554 (samples from Myanmar).
3. RESULTS
3.1. Features of the sequenced mitochondrial genomes
The length of assembled mitogenomes from 30 Amblyomma specimens from Japan and 9 from Myanmar ranged from 14,830 to 14,839 bp. A total of 37 encoded genes were detected including 13 PCGs, 22 transfer RNA (tRNA) genes, and two rDNAs. Two control regions were identified in the mitogenome. There was no difference in gene rearrangement among the 39 newly sequenced Amblyomma mitogenomes.
3.2. Nucleotide identities based complete mitogenome sequences
Generated nucleotide sequences of 30 collected ticks from Japan and 9 from Myanmar were aligned with the reference mitogenome sequences of A. testudinarium from Japan (accession number: MT371798) along with six Amblyomma spp. and 14 other ixodid ticks including seven Haemaphysalis spp., one each of A. sphenodonti, B. undatum, D. nitens, D. silvarum, R. nuttalli, R. microplus, and R. elaphensis. Comparative molecular identity analysis of the assembled nucleotide sequences from Japan and Myanmar showed an average nucleotide identity of 74.72% (71.00–91.48) and 74.56% (71.23–79.55) with the other 20 ixodid species, respectively (Table 2). The average nucleotide identity of mitogenome sequences from Japan was 99.80% with the reference mitogenome sequence of A. testudinarium (accession number: MT371798) collected from Nara, Japan. The average nucleotide identity of the mitogenome sequences from Myanmar was 91.32% with the same reference mitogenome of A. testudinarium (accession number: MT371798) (Table 2). In addition, the average nucleotide identities of Amblyomma mitogenome sequences from Japan and Myanmar were, respectively, 79.50% and 79.28% to A. testudinarium from China (accession number: MT029329) and 78.45% and 78.37% to A. javanense (accession number: NC_043872) (Table 2).
TABLE 2.
Summary of the molecular identity of Amblyomma mitogenomes
| Comparison (accession number) | Average identity (%) (min–max) |
|---|---|
| A. testudinarium from Japan vs other ixodid species a | 74.72 (71.00–91.48) |
| Amblyomma sp. from Myanmar vs other ixodid species a | 74.56 (71.23–79.55) |
| A. testudinarium from Japan vs A. testudinarium Nara strain (MT371798) | 99.80 (99.34–99.93) |
| Amblyomma sp. from Myanmar vs A. testudinarium Nara strain (MT371798) | 91.32 (91.40–91.46) |
| A. testudinarium from Japan vs A. javanense (NC_043872) | 78.45 (79.63–79.74) |
| Amblyomma sp. from Myanmar vs A. javanense (NC_043872) | 78.37 (77.16–79.63) |
| A. testudinarium from Japan vs A. testudinarium from China (MT029329) | 79.50 (79.48–79.56) |
| Amblyomma sp. from Myanmar vs A. testudinarium from China (MT029329) | 79.28 (79.26–79.32) |
| A. testudinarium from China (MT029329) vs A. javanense (NC_043872) | 83.72 (81.43–86.02) |
A total of 20 ixodid species (A. javanense, A. geoemydae, A. fimbriatum, A. triguttatum, A. americanum, A. sculptum, H. concinna, H. formosensis, H. flava, H. japonica, H. hystricis, H. longicornis, H. inermis, A. sphenodonti, B. undatum, D. nitens, D. silvarum, R. nuttalli, R. microplus, and R. elaphensis) were used to compare Amblyomma mitogenomes in the study.
3.3. Phylogenetic relationship based on mitochondrial 16S rDNA and COI
To investigate the phylogeographic relationships of Amblyomma spp., we constructed a phylogenetic tree based on the 16S rDNA sequences. The constructed ML tree showed that A. testudinarium sequences from Japan and Taiwan clustered together in one clade and were closely related to another clade containing the sequences from Amblyomma sp. collected from Myanmar and A. testudinarium from Thailand (Figure 2). Interestingly, A. testudinarium sequence from China (accession number: MT029329) did not cluster with either the A. testudinarium from Japan or Amblyomma sp. sequences from Myanmar but formed a separate cluster with A. javanense sequences from China (accession numbers: NC_043872 and MK229166). The topology of the phylogenetic trees suggested that the sequence identified as A. testudinarium in China (accession number: MT029329) is different from the other A. testudinarium sequences from Japan and Taiwan (Figure 2). In addition, the phylogenetic analysis of A. testudinarium based on the COI sequences was similar to the results of the 16S rDNA where the sequences of A. testudinarium from Japan and Taiwan clustered in a monophyletic group. Amblyomma sp. sequences from Myanmar formed a separate clade in both 16S rDNA and COI‐based phylogenetic trees. The COI sequences of A. testudinarium from China (accession number: MT029329) clustered with A. javanense but not with other A. testudinarium sequences from other Asian countries (Figure 2).
FIGURE 2.
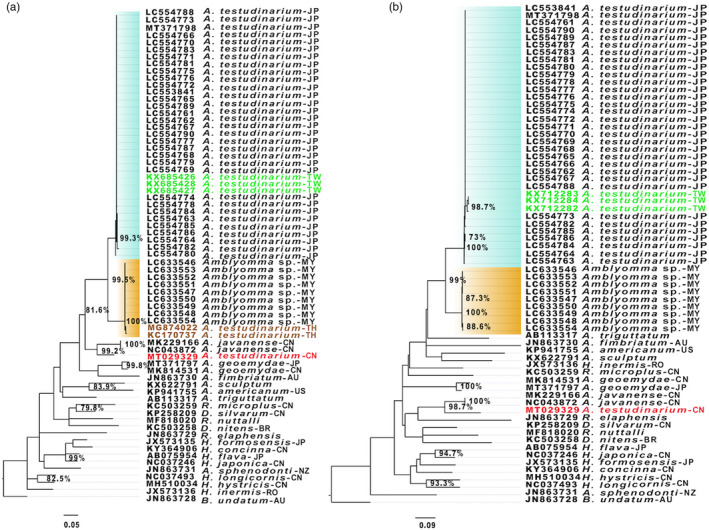
Maximum‐likelihood trees based on the mitochondrial (a) 16S rDNA and (b) COI gene. Labels in green, brown, and red indicate the sequences from Taiwan, Thailand, and China, respectively. The clade containing sequences from Japan and Taiwan is highlighted in cyan and the clade highlighted in orange represents the clade containing sequences from Myanmar. Abbreviations in the sample name refers to the country/continent of origin; AU, Australia; BR, Brazil; CN, China; JP, Japan; MY, Myanmar; NZ, New Zealand; TH, Thailand; TW, Taiwan; RO, Romania; US, USA. Bootstrap values above 70% are indicated by percentages
3.4. Phylogeographic analysis based on the concatenated 13 PCGs and two rDNAs
A phylogeographic tree was constructed with a set of 13 PCGs concatenated with two rDNAs extracted from the complete mitogenomes of 32 A. testudinarium specimens collected from eleven prefectures in Japan, nine Amblyomma sp. from Putao in Myanmar, and 19 Ixodidae species from the GenBank including six Amblyomma spp., seven Haemaphysalis spp., and one each of A. sphenodonti, B. undatum, D. nitens, D. silvarum, R. nuttalli, R. microplus, and R. elaphensis. The topology of the constructed MCC showed agreement with the constructed ML tree based on the 16S rDNA and COI in that a total of 32 A. testudinarium sequences from Japan clustered together and nine Amblyomma sp. sequences from Myanmar formed a separate cluster (Figure 3). The sequences of A. testudinarium and Amblyomma sp. were closely related to A. javanense and A. geoemydae. Most of the tree branches showed strong bootstrap support values.
FIGURE 3.
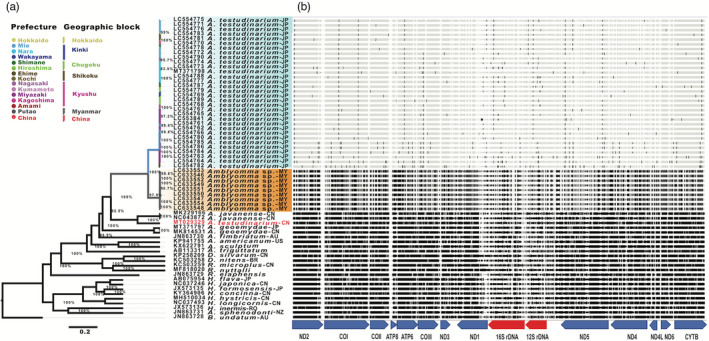
Genetic diversity and phylogenetic relationships among Amblyomma testudinarium from Japan and Amblyomma sp. from Myanmar. (a) Bayesian phylogenetic Maximum Clade Credibility (MCC) tree based on the 15 concatenated mitochondrial gene sequences. The tree was rooted to Bothriocroton undatum (accession number: JN863728). Clades of the specimen from Japan and Myanmar are heighted in cyan and orange, respectively. Branch colors represent sampling geographic origin of each sequence. (b) The nucleotide differences between 39 new mitogenome sequences of Amblyomma collected from different localities in Japan and Myanmar. Locations of single nucleotide variations are indicated as vertical lines in mitogenome sequences relative to the mitogenome sequence of A. testudinarium Nara strain (accession number: MT371798). Abbreviations in the sample names refer to the country of origin; AU, Australia; BR, Brazil; CN, China; JP, Japan; MY, Myanmar; NZ, New Zealand; RO, Romania; US, USA
3.5. Haplotyping based on the 16S rDNA of A. testudinarium from Japan
To understand the genetic population structure of A. testudinarium in Japan, we performed a haplotype analysis based on 32 16S rDNA sequences from Japan. The haplotype analysis resolved a total of 16 haplotypes, which were defined by 28 single nucleotide polymorphic sites (SNPs). Median‐joining phylogenetic network analysis for the 16 haplotypes revealed that the A. testudinarium population from Japan forms a star‐like phylogeny, with the common ancestorial haplotype (H5) in the center surrounded by the remaining haplotypes (Figure 4). The most frequent three haplotypes in the population from Japan were H2, H5, and H9 that were observed in six, six, and four samples, respectively. A total of five (H1, H2, H5, H7, and H9) were shared between different sampling sites (Figure 4).
FIGURE 4.
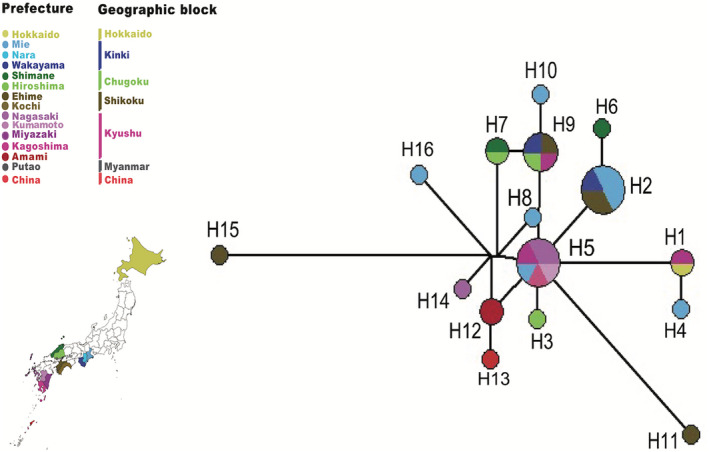
Reduced‐median‐joining phylogenetic network of the 16 haplotypes identified based on the 16S rDNA sequences. Colors in the pie represent the geographical origin of each haplotype. Node size is proportional to the number of individuals
3.6. Population structure of A. testudinarium
The phylogeographic analysis based on the 15 concatenated mitochondrial genes extracted from 32 A. testudinarium from Japan and 9 Amblyomma sp. from Myanmar, where A. javanense was used as an outgroup, showed the separation of Japan population of A. testudinarium from the Amblyomma sp. sequences from Myanmar. Interestingly, A. testudinarium sequences from Amami island and two sequences from Miyazaki prefecture were grouped and separated from the remaining Japan populations (Figure 5). In addition, one A. testudinarium sequence from Ehime prefecture was separated from the remaining sequences from Japan.
FIGURE 5.
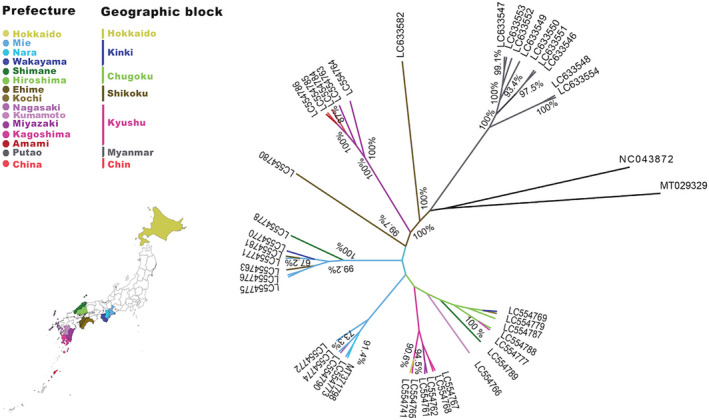
A maximum clade credibility (MCC) tree based on Bayesian approach. Tree branches are colored based on the geographic regions of sample collection sites. Tree was rooted to Amblyomma testudinarium (MT029329) and Amblyomma javanense (NC_043872) reported from China (branches are shown in bold)
Analysis of percentile identity based on the concatenated 15 mitochondrial genes of A. testudinarium used in the phylogeny revealed that three sequences of A. testudinarium from Amami clustered with two sequences from Miyazaki and showed 99.89% identity with each other, 99.66% to other samples from Miyazaki, and 99.51% to the remaining sequences from Japan. The average identity of A. testudinarium from Ehime was 99.66% (99.27–99.98) to the other sequences from Japan. The average diversity between A. testudinarium sequences from Japan and Amblyomma sp. sequences from Myanmar was 8.52%, showing 8.43% to 8.65% in comparison to the reference mitogenome A. testudinarium (accession number: MT371798) from Japan. Comparison of variation at different PCGs indicated that ATP8, ATP6, and ND2 had the highest diversity among the 15 mitochondrial genes (Table 3 and Figure 3b). In addition, the molecular diversity indices showed that A. testudinarium population in Japan is more genetically diverse than the Amblyomma sp. of Myanmar (Table S1). In addition, the nonmetric multidimension scale analysis based on the genetic distances confirmed the geographic separation and genetic distinction of A. testurinarium from Amami island and Miyazaki (two samples collected at site 2), which were clustered together in one group and separated from other samples from Japan (Figure 6). We also showed the genetic distinction and possible geographical separation of A. testurinarium from Ehime prefecture based on the NMDS analysis.
TABLE 3.
Comparison of variation at different PCGs for the Amblyomma testudinarium samples
| Gene name | Number of haplotypes | Nucleotide length (min–max) | Gene product | GC content (%) | Polymorphic sites (%) | Parsimony informative sites (%) |
|---|---|---|---|---|---|---|
| 12S rDNA | 14 | 715(711–723) | 12S ribosomal RNA | 18.1 | 19.7 (141/715) a | 8.0 (57/715) b |
| 16S rDNA | 16 | 1208 (1202–1223) | 16S ribosomal RNA | 16.8 | 20.3 (245/1208) | 8.7 (105/1208) |
| ATP6 | 10 | 663 (663‐663) | ATP synthase subunit 6 | 18.4 | 23.7 (157/663) | 10.1 (67/663) |
| ATP8 | 4 | 159 (159–162) | ATP synthase subunit 8 | 12.7 | 35.8 (57/159) | 17.0 (27/159) |
| COI | 16 | 1538 (1536–1542) | Cytochrome c oxidase subunit I | 27.1 | 18.5 (285/1538) | 7.9 (122/1538) |
| COII | 11 | 675 (675–675) | Cytochrome c oxidase subunit II | 24.0 | 22.1 (149/675) | 7.9 (53/675) |
| COIII | 14 | 778 (778–778) | Cytochrome c oxidase subunit III | 22.5 | 20.8 (162/778) | 5.8 (45/778) |
| CYTB | 19 | 1077 (1077–1077) | Cytochrome b | 21.5 | 22.7 (245/1077) | 6.5 (70/1077) |
| ND1 | 8 | 1010 (1007–1023) | NADH dehydrogenase subunit 1 | 18.8 | 22.2 (224/1010) | 7.2 (73/1010) |
| ND2 | 13 | 963 (963–963) | NADH dehydrogenase subunit 2 | 13.2 | 27.0 (260/963) | 9.9 (95/963) |
| ND3 | 6 | 342 (342–342) | NADH dehydrogenase subunit 3 | 14.0 | 23.7 (81/342) | 9.4 (32/342) |
| ND4 | 21 | 1326 (1326–1326) | NADH dehydrogenase subunit 4 | 18.7 | 26.0 (345/1326) | 9.0 (119/1326) |
| ND4L | 6 | 276 (276–276) | NADH dehydrogenase subunit 4L | 14.2 | 21.0 (58/276) | 7.2 (20/276) |
| ND5 | 23 | 1656 (1656–1656) | NADH dehydrogenase subunit 5 | 16.8 | 24.2 (400/1656) | 7.4 (122/1656) |
| ND6 | 6 | 432 (429–432) | NADH dehydrogenase subunit 6 | 12.5 | 30.1 (130/432) | 8.8 (38/432) |
Note: The three genes showing the highest diversity levels are shown in Bold.
Number of polymorphic sites/average number of base pairs.
Number of parsimony informative sites/average number of base pairs.
FIGURE 6.
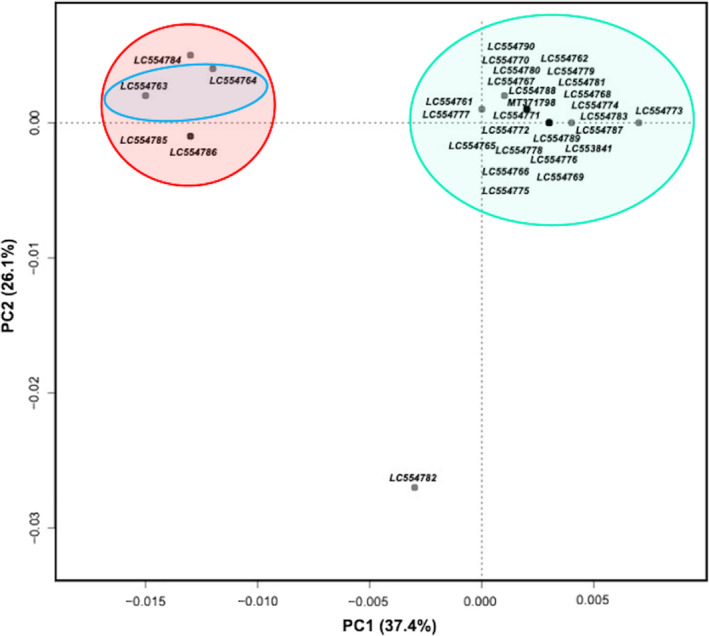
Nonmetric multidimensional scaling (NMDS) ordination plot based on the distance matrix. Red, blue, and cyan ellipsoids represent the grouping of Amblyomma testudinarium from Amami, Miyazaki (two samples collected at site 2), and the remaining Japan localities, respectively
To statistically test the population genetic structure between and within populations, we used AMOVA to determine whether the genetic structure was influenced by the geographic structure. Molecular variance results revealed that the genetic variation among‐population of A. testudinarium from Amami island (Kyushu) and the other regions in Japan indicated that the population of A. testudinarium is structured with one population present only in Amami (Kyushu), where the among‐population genetic variation (57.55%) was higher than that within‐population (42.45%). Significant global F ST values (p < 0.01) indicated significant genetic differentiation between populations from Amami and the remaining localities (p = 0.00098) (Table 4). The correlation analysis of A. testudinarium from Japan using Mantel test revealed that there is no significant correlation between the degree of genetic variation based on the pairwise genetic distances and geographic distances with r statistics = 0.11 (p‐value = 0.1779 based on 9999 replicates) (Figure 7).
TABLE 4.
Analysis of molecular variance (AMOVA) using the concatenated 15 mitochondrial genes sequences extracted from whole mitogenomes of Amblyomma testudinarium populations in Japan
| Populations | Source of variation | Degree of freedom | Sum of squares | Variance components | Percentage of variation | F ST |
|---|---|---|---|---|---|---|
| Amami island Other regions (Japan) | Among populations | 1 | 71.48 | 11.57 | 57.55 | 57.58*** |
| Within populations | 30 | 256.15 | 8.54 | 42.45 |
Note: Degree of significance: *p < 0.05, **p < 0.01, ***p < 0.0.
FIGURE 7.
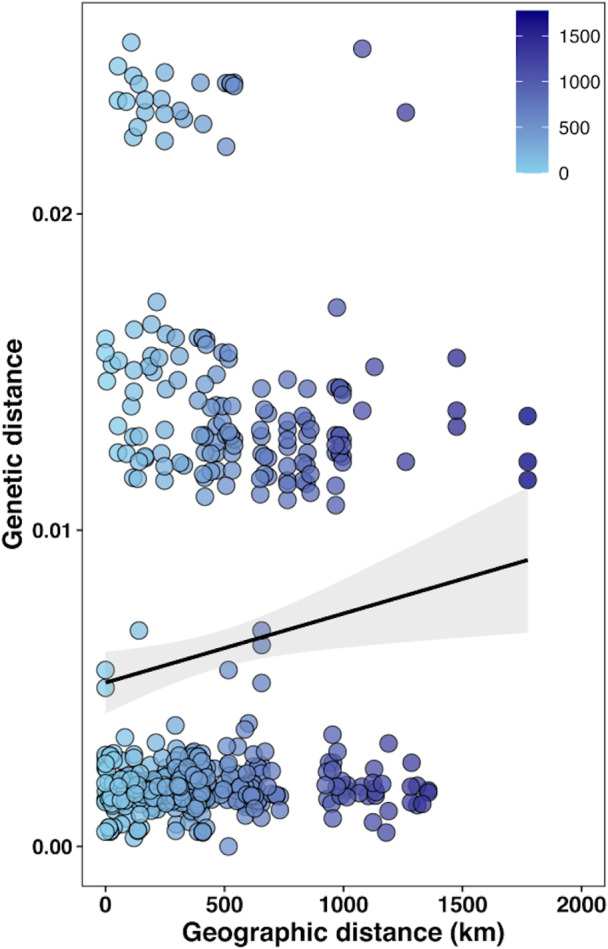
Scatter plot of genetic variation versus geographic distances in kilometer (km) between the four sampling regions in Japan. Regression line is overlayed
Results of the mismatch analysis based on pairwise nucleotide differences between 41 nucleotide sequences from Japan (n = 32) and Myanmar (n = 9) revealed that the shape of the mismatch distributions is far from unimodal distribution while analyzing either sequences of A. testudinarium from Japan or Amblyomma sp. from Myanmar, suggesting a possibility of long‐term demographic stability in each with a constant population size (Figure 8). To determine whether the observed mismatch deviated significantly from a population expansion model, we estimated the SSD and RI for the spatial and demographic expansion model. The observed mismatch pattern of A. testudinarium from Japan did not deviate significantly from that expected under population expansion scenario (SSD = 0.015; p = 0.180 and p = 0.160 for demographic and spatial expansion, respectively), and the similar result was observed for the Amblyomma sp. population from Myanmar (SSD = 0.033; p = 0.47 and p = 0.69 for demographic and spatial expansion, respectively). In addition, the RI index for A. testudinarium haplotypes in Japan was 0.008 (p = 0.49 and p = 0.63 for the demographic and spatial expansion model, respectively) and the one for Amblyomma sp. in Myanmar was 0.062 (p = 0.51 and p = 0.73 for the demographic and spatial expansion model, respectively). The results of SSD and RI indices suggested a sudden expansion of A. testudinarium in Japan and a stability of the population size of Amblyomma sp. population in Myanmar (Table 5).
FIGURE 8.
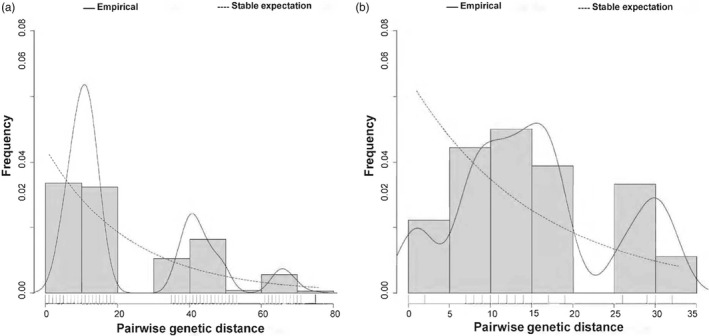
Mismatch distribution pattern for Amblyomma testudinarium from Japan and Amblyomma sp. from Myanmar based on the 15 concatenated mitochondrial gene sequences. (a) Mismatch distribution pattern for A. testudinarium sequences from Japan. (b) Mismatch distribution pattern for Amblyomma sp. sequences from Myanmar. The x‐axis shows the number of pairwise differences (genetic distance) between pairs of sequences and the y‐axis shows their frequency. Solid histograms illustrate the observed frequencies. Solid black line indicates the simulated mismatch distributions expected under demographic expansion and dotted black line indicates those expected under spatial expansion
TABLE 5.
Evolutionary dynamics of Amblyomma testudinarium from Japan and Amblyomma sp. from Myanmar identified from 15 concatenated mitochondrial gene sequences
| Statistics | Japan | Myanmar | Mean | SD |
| Spatial expansion | ||||
| SSD | 0.015 | 0.033 | 0.016 | 0.016 |
| Model (SSD) p value | 0.160 | 0.690 | 0.283 | 0.361 |
| RI | 0.008 | 0.062 | 0.023 | 0.033 |
| Model (RI) p value | 0.630 | 0.730 | 0.453 | 0.395 |
| Demographic expansion | ||||
| SSD | 0.015 | 0.033 | 0.016 | 0.016 |
| Model (SSD) p value | 0.180 | 0.470 | 0.216 | 0.237 |
| RI | 0.009 | 0.062 | 0.023 | 0.033 |
| Model (RI) p value | 0.490 | 0.510 | 0.333 | 0.288 |
Abbreviations: SSD, sum of square deviation; RI, Harpending’s raggedness index; SD, standard deviation.
Tajima’s D test assessing departure from the neutral evolutionary model based on the 15 concatenated mitochondrial genes of A. testudinarium and Amblyomma sp. showed that the Tajima’s D value was calculated to be ‐2.26 in Japan population and 0.06 for the Amblyomma sp. from Myanmar. Tajima’s D value was significant in Japan population (p‐value = 0.0), suggesting a possibility of a recent population expansion after a bottleneck; however, Tajima’s D value was not significant and near zero in the Myanmar population, indicating that the mitogenome does not reject the neutral model hypothesis (Table 6). This suggests that Amblyomma sp. in Myanmar might have experienced a weak signal of population bottleneck that reduced its diversity, or this group is characterized by a constant population size (demographic equilibrium), which could happened as the samples in Myanmar were collected from a single collection site.
TABLE 6.
Neutrality test values of Amblyomma testudinarium from Japan and Amblyomma sp. from Myanmar based on the concatenated 15 mitochondrial gene sequences
| Statistics | Japan | Myanmar | Mean | SD |
|---|---|---|---|---|
| Tajima's D test | ||||
| Sample size | 32.00 | 9.00 | 20.50 | 16.26 |
| S | 207.00 | 37.00 | 122.00 | 120.21 |
| Pi | 21.14 | 13.78 | 17.46 | 5.20 |
| Tajima's D | −2.26 | 0.06 | −1.10 | 1.64 |
| Tajima's D p value | 0.00 | 0.54 | 0.27 | 0.38 |
Abbreviations: S, singleton sites; Pi, parsimony informative sites; SD, standard deviation.
4. DISCUSSION
Phylogeographic structure of tick populations can help enrich the knowledge on the distribution of ticks and tick‐borne pathogens (Black et al., 2001). In addition, intraspecies genetic differences within geographically distinct populations of ticks has been well established (Burnard & Shao, 2019; Mechai et al., 2013; Sakamoto et al., 2014). The current work is the first to assess the genetic diversity and population structure of A. testudinarium, which is one of the most widely distributed Amblyomma in East and Southeast Asia (Voltzit, 2002) and one of the main ticks that bite humans in Japan (Natsuaki, 2021). Our study is the second to use whole mitogenomes of multiple individuals of the same tick species to assess the genetic diversity based on the regional scale after the report on Ixodes ricinus in Italy (Carpi et al., 2016). Although the earlier study detected intraspecies genetic relationships, population differentiation, and demographic history of I. ricinus, our study provides an advantage of the wide spatial scale over which our samples were collected and phylogenetic tree reconstruction using 30 new A. testudinarium mitogenome sequences as well molecular characterization of a potentially novel Amblyomma sp. closely related to A. testudinarium based on nine mitogenome sequences from Myanmar.
Only a few studies have focused on ticks in Myanmar (Petney et al., 2019) including A. testudinarium, which was reported from other countries in Southeast Asia. Taxonomy of the genus Amblyomma is currently under revision and several species were re‐assigned to a new subfamily Bothriocrotoninae (Klompen et al., 2002) or to new genera Archaeocroton and Robertsicus (Barker & Burger, 2018; Hornok et al., 2020). The species identification of Amblyomma species collected in Myanmar is hampered by the lack of reference genomic sequence data for many Amblyomma spp. including A. testudinarium as well as the lack of morphological description of A. testudinarium from Myanmar. Although A. testudinarium has been considered widespread and common in East and Southeast Asia, our study suggested that potentially several cryptic Amblyomma spp. closely related to A. testudinarium may be distributed in Myanmar, Thailand, and China.
Mitogenomes have been used as a marker in population genetics, phylogeography, and DNA barcoding (Eimanifar et al., 2018; Fourdrilis et al., 2016). Of note, mitogenomes were proven to provide a better opportunity to understand tick taxonomy than targeting only a single gene (Kelava et al., 2021). Previously, intraspecies phylogenetic relationships was investigated through targeting the partial sequences of 16S rDNA, 12S rDNA, and COI (Chao et al., 2017; Yamauchi et al., 2012). Since different genes have different evolutionary rates, the final conclusions could have been affected by the targeted gene topology of the phylogenetic tree (Liu et al., 2018). Traditionally, morphological characterization has been used for species identification and differentiation of Amblyomma ticks (Aguilar‐Dominguez et al., 2021; Namgyal et al., 2021). Now, a molecular based characterization can provide insights on the genetic variance at the base‐pair level and the genetic diversity between and within species of Amblyomma ticks (Burger et al., 2012; Lopes et al., 2016). An important advantage of targeting tick mitogenomes to study its population genetics and distribution is the significantly smaller size when compared to nuclear genomes (Nadolny et al., 2016), which enables the cost‐effective comparative genome analysis. We observed that the gene order of the mitogenome of A. testudinarium from Japan and Amblyomma sp. from Myanmar was identical to that of the other members of genus Amblyomma, which confirms that gene order and composition are highly conserved between Amblyomma species to a common ancestor (Chauve & Tannier, 2008; de Lima et al., 2017; Namgyal et al., 2021).
Although we examined tick specimens collected from six different biogeographic regions in Japan with different ecoclimatic habitats, phylogenetic analysis revealed that all sequences from Japan clustered in a monophyletic group with percentile identity of 99.36–100%. However, we demonstrated that mitogenomes derived from A. testudinarium in Japan and Amblyomma sp. ticks from Myanmar are genetically distinguished from each other and were assigned to a biphyletic group with sequence identity range of 91.35–91.57%. These results are supported by a previous report (Nakao et al., 2021), where the mitogenome of A. testudinarium from Hokkaido, the northernmost main island of Japan, was clustered with the same species from Nara in the Honshu Island in a monophyletic group, but obviously discriminated from other species of Amblyomma ticks as well other ixodid genera (Haemaphysalis, Archaeochroton, Bothriocroton, Dermacentor, Robertsicus, and Rhipicephalus) of ticks.
The results of polymorphic sites comparison showed that relying solely on one single mitochondrial gene for phylogenetic analysis of A. testudinarium might yield incorrect or incomplete conclusions (Table 3). Currently, most studies focusing on population structure and phylogenetic analysis of ticks build the final conclusions based on the results obtained by targeting more than one mitochondrial genes (Gui et al., 2021; Regilme et al., 2021). Furthermore, we showed that A. testudinarium population in Japan is genetically more diverse than the closely related Amblyomma sp. of Myanmar. In fact, the evolution of mitochondrial genes can be affected by several ecological factors such as endosymbionts and animal host diversity (Kaufman et al., 2018; Sassera et al., 2006). The difference in diversity between A. testudinarium in Japan and Amblyomma sp. in Myanmar could be attributed to the greater breadth of sampling collection sites in Japan compared to Myanmar where we collected the samples from one site. Unfortunately, we did not investigate the bacterial endosymbionts in our study, so we suggest that more studies are required to correlate the endosymbiont genetics and the mitogenome divergence in A. testudinarium.
Amblyomma testudinarium from China (accession number: MT029329) showed low sequence similarity with those from Japan but relatively high similarity with A. javanense reported from China (Table 2). This was supported by the analysis based on the 16S rDNA obtained from A. testudinarium, which included samples from Taiwan, Japan, and Thailand reported by other studies (Chao et al., 2017; Malaisri et al., 2015). We confirmed that the sequence from China (MT029329) is different from A. testudinarium from Japan, Taiwan, and Thailand (Figure 2). Collectively, these data suggest a possibility of misidentification of the tick specimen used in the previous report (Chang et al., 2020) or the presence of cryptic species related to A. javanense in China.
The 16S rDNA sequences of A. testudinarium from Japan are clustered together with those from Taiwan but not with Thailand (Figure 2). These findings suggest that natural aquatic barriers around Japan lowered the chance of A. testudinarium gene flow between East and Southeast Asia. This scenario was previously suggested for I. ricinus in continental Europe, where marine barriers around the British Isles were suggested to prevent the gene flow of I. ricinus (Al‐Khafaji et al., 2019). Our results also showed that a possible gene flow of A. testudinarium between Japan and Taiwan might have occurred presumably through the migratory birds flying through the East‐Asia Australian flyway (EAAF).
In general, geographical variations are mostly accompanied with differences in the distribution of tick hosts. The large animals are the primary hosts for A. testudinarium, while larvae and nymphs feed on small mammals, birds, reptiles, and amphibians (Kitaoka, 1974; Takada, 1990; Yamaguti et al., 1971). In Japan, adult A. testudinarium are frequently found on wild boars, which habitats are linked to those of A. testudinarium (Motoi et al., 2013). The rapid expansion of the wild boar habitats (Kodera et al., 2001) may be the reason for no clear geographic substructuring among A. testudinarium populations in Japan (Figure 6). However, future studies with larger numbers of Amblyomma samples collected from Japan and other countries are required to confirm the possible genetic distinction and pattern of geographical distribution of A. testurinarium from Amami island, Miyazaki, and Ehime prefectures.
The genetic variation within A. testudinarium populations in Japan, that was detected by the AMOVA statistics, could be attributed to selective pressures due to several ecoclimatic and environmental changes that requires adaptive genetic modifications. According to the Japan Meteorological Agency (https://www.data.jma.go.jp/gmd/cpd/longfcst/en/tourist.html), the climate in Japan ranges from subarctic in the north to subtropical in the south with Okinawa and Amami having humid and hot summers and mild winters compared to the warm summers and very cold winters in the remaining regions. It was reported previously that dynamics of arthropod vectors including ticks and the associated vector‐borne diseases are sensitive to climate change (Githeko et al., 2000). We also detected a pattern for the population differentiation of A. testudinarium in accordance with the spatial isolation where specimens collected from Amami island might have its own population in Japan. This finding could be explained by the complete geographical separation from the Japan mainland. The climate around Amami island is humid subtropical and known as Köppen climate classification (Cfa) with high precipitation throughout the year. Furthermore, the life cycle of A. testudinarium in Amami island is not clear; however, it is possible that this tick species utilizes endogenous animal species of this island as feeding hosts (Chae et al., 2017). Any of these factors may have supported the distinction of A. testudinarium tick population in Amami island.
The algorithms in our study support the expansion history of A. testudinarium in Japan. We detected a relatively high haplotype diversity in Japan, suggesting a strong possibility of sudden expansion (Amzati et al., 2018; Ray et al., 2003; Simonsen et al., 1995). By contrast, Amblyomma sp. in Myanmar was suggested to have a bottleneck historical event. Although our study did not estimate this sudden expansion and bottleneck times, relatively high diversity of A. testudinarium in Japan within population indicates that A. testudinarium colonies have been existed in Japan for long time. However, our study included samples that were collected from only a single sampling site in Myanmar, and we strongly suggest further investigations using A. testudinarium and the closely related Amblyomma spp. from all over the country to find a possible explanation of the population equilibrium observed in our study. Our finding that A. testudinarium in Japan has experienced a sudden expansion event agrees with the expected movement of migratory birds that are considered the carrying hosts of ticks in the country (Ishiguro et al., 2000; Kuo et al., 2017). This can be supported by the possible establishment of new colonies in previously unoccupied geographical locations in Japan, such as the recent detection of A. testudinarium in Hokkaido Island for the first time in history (Nakao et al., 2021).
5. CONCLUSION
Overall, we report whole mitogenome sequences of 30 A. testudinarium from Japan and 9 closely related Amblyomma species from Myanmar. Our study provides the first molecular evidence and convincing sequences to confirm the genetic identity and diversity of Amblyomma sp. in Myanmar. The lack of geographic substructuring among A. testudinarium populations in Japan suggests that geographic distance might not have an impact on the genetic variation. The data provides information about the sudden expansion event of A. testudinarium in Japan. With the advancement of high‐throughput sequencing techniques and bioinformatics, phylogeographic approaches can be used to distinguish A. testudinarium globally based on the genetic data of the whole mitochondrial genomes. The accumulation of more mitogenome sequences from different geographic regions can help in understanding the phylogeny of Amblyomma ticks present in East and Southeast Asia.
CONFLICT OF INTEREST
The authors have no competing interests to declare.
Supporting information
Table S1
ACKNOWLEDGMENTS
This study was supported by JSPS KAKENHI [17H04638, 19H03118, 19F19097, 20K21358, 20KK0151, and 22H02505], the Japan Program for Infectious Diseases Research and Infrastructure [20wm0225016j0001] from the Japan Agency for Medical Research and Development (AMED), and the JSPS Bilateral Open Partnership Joint Research Project with Myanmar.
Mohamed, W. M. A. , Moustafa, M. A. M. , Thu, M. J. , Kakisaka, K. , Chatanga, E. , Ogata, S. , Hayashi, N. , Taya, Y. , Ohari, Y. , Naguib, D. , Qiu, Y. , Matsuno, K. , Bawm, S. , Htun, L. L. , Barker, S. C. , Katakura, K. , Ito, K. , Nonaka, N. , & Nakao, R. (2022). Comparative mitogenomics elucidates the population genetic structure of Amblyomma testudinarium in Japan and a closely related Amblyomma species in Myanmar. Evolutionary Applications, 15, 1062–1078. 10.1111/eva.13426
Contributor Information
Wessam Mohamed Ahmed Mohamed, Email: wessam@czc.hokudai.ac.jp.
Mohamed Abdallah Mohamed Moustafa, Email: m.abdallah@vetmed.hokudai.ac.jp.
Ryo Nakao, Email: ryo.nakao@vetmed.hokudai.ac.jp.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp) with accession numbers of LC554761‐LC554790 (samples from Japan) and LC633546‐LC633554 (samples from Myanmar).
REFERENCES
- Aguilar‐Dominguez, M. , Romero‐Salas, D. , Sanchez‐Montes, S. , Serna‐Lagunes, R. , Rosas‐Saito, G. , Cruz‐Romero, A. , & de Leon, A. P. A. (2021). Morphometrics of Amblyomma mixtum in the State of Veracruz, Mexico. Pathogens, 10(5), 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Khafaji, A. M. , Clegg, S. R. , Pinder, A. C. , Luu, L. , Hansford, K. M. , Seelig, F. , Dinnis, R. E. , Margos, G. , Medlock, J. M. , Feil, E. J. , Darby, A. C. , McGarry, J. W. , Gilbert, L. , Plantard, O. , Sassera, D. , & Makepeace, B. L. (2019). Multi‐locus sequence typing of Ixodes ricinus and its symbiont Candidatus Midichloria mitochondrii across Europe reveals evidence of local co‐cladogenesis in Scotland. Ticks and Tick‐borne Diseases, 10(1), 52–62. [DOI] [PubMed] [Google Scholar]
- Amzati, G. S. , Pelle, R. , Muhigwa, J.‐B. B. , Kanduma, E. G. , Djikeng, A. , Madder, M. , Kirschvink, N. , & Marcotty, T. (2018). Mitochondrial phylogeography and population structure of the cattle tick Rhipicephalus appendiculatus in the African Great Lakes region. Parasites & Vectors, 11(1), 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt, H. J. , Forster, P. , & Rohl, A. (1999). Median‐joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution Molecular Biology and Evolution, 16(1), 37–48. [DOI] [PubMed] [Google Scholar]
- Barker, S. C. , & Burger, T. D. (2018). Two new genera of hard ticks, Robertsicus n. gen. and Archaeocroton n. gen., and the solution to the mystery of Hoogstraal’s and Kaufman’s “primitive” tick from the Carpathian Mountains. Zootaxa, 4500(4), 543–552. [DOI] [PubMed] [Google Scholar]
- Black, W. C. , Baer, C. F. , Antolin, M. F. , & DuTeau, N. M. (2001). Population genomics: Genome‐wide sampling of insect populations. Annual Review of Entomology, 46, 441–469. [DOI] [PubMed] [Google Scholar]
- Brites‐Neto, J. , Duarte, K. M. R. , & Martins, T. F. (2015). Tick‐borne infections in human and animal population worldwide. Veterinary World, 8(3), 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger, T. D. , Shao, R. , & Barker, S. C. (2013). Phylogenetic analysis of the mitochondrial genomes and nuclear rRNA genes of ticks reveals a deep phylogenetic structure within the genus Haemaphysalis and further elucidates the polyphyly of the genus Amblyomma with respect to Amblyomma sphenodonti and Amblyomma elaphense . Ticks and Tick‐borne Diseases, 4(4), 265–274. [DOI] [PubMed] [Google Scholar]
- Burger, T. D. , Shao, R. F. , Beati, L. , Miller, H. , & Barker, S. C. (2012). Phylogenetic analysis of ticks (Acari: Ixodida) using mitochondrial genomes and nuclear rRNA genes indicates that the genus Amblyomma is polyphyletic. Molecular Phylogenetics and Evolution, 64(1), 45–55. [DOI] [PubMed] [Google Scholar]
- Burger, T. D. , Shao, R. , Labruna, M. B. , & Barker, S. C. (2014). Molecular phylogeny of soft ticks (Ixodida: Argasidae) inferred from mitochondrial genome and nuclear rRNA sequences. Ticks and Tick‐borne Diseases, 5(2), 195–207. [DOI] [PubMed] [Google Scholar]
- Burnard, D. , & Shao, R. F. (2019). Mitochondrial genome analysis reveals intraspecific variation within Australian hard tick species. Ticks and Tick‐borne Diseases, 10(3), 677–681. [DOI] [PubMed] [Google Scholar]
- Cao, W. C. , Gao, Y. M. , Zhang, P. H. , Zhang, X. T. , Dai, Q. H. , Dumler, J. S. , Fang, L. Q. , & Yang, H. (2000). Identification of Ehrlichia chaffeensis by nested PCR in ticks from southern China. Journal of Clinical Microbiology, 38(7), 2778–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpi, G. , Kitchen, A. , Kim, H. L. , Ratan, A. , Drautz‐Moses, D. I. , McGraw, J. J. , Kazimirova, M. , Rizzoli, A. , & Schuster, S. C. (2016). Mitogenomes reveal diversity of the European Lyme borreliosis vector Ixodes ricinus in Italy. Molecular Phylogenetics and Evolution, 101, 194–202. [DOI] [PubMed] [Google Scholar]
- Chae, J. B. , Kang, J. G. , Kim, H. C. , Chong, S. T. , Lee, I. Y. , Shin, N. S. , & Chae, J. S. (2017). Identification of Tick Species Collected from Wild Boars and Habitats of Wild Boars and Domestic Pigs in the Republic of Korea. The Korean Journal of Parasitology, 55(2), 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamuah, J. K. , Bhattacharjee, K. , Sarmah, P. C. , Raina, O. K. , Mukherjee, S. , & Rajkhowa, C. (2016). Report of Amblyomma testudinarium in mithuns (Bos frontalis) from eastern Mizoram (India). Journal of Parasitic Diseases, 40(4), 1217–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Q. C. , Wu, T. T. , Cao, H. , Sun, M. Q. , Zhang, W. T. , & Xue, S. J. (2020). The complete mitochondrial genome of Amblyomma testudinarium (Ixodida: Ixodidae). Mitochondrial DNA Part B‐Resources, 5(2), 1485–1486. [Google Scholar]
- Chao, L. L. , Lu, C. W. , Lin, Y. F. , & Shih, C. M. (2017). Molecular and morphological identification of a human biting tick, Amblyomma testudinarium (Acari: Ixodidae), in Taiwan. Experimental and Applied Acarology, 71(4), 401–414. [DOI] [PubMed] [Google Scholar]
- Chauve, C. , & Tannier, E. (2008). A methodological framework for the reconstruction of contiguous regions of ancestral genomes and its application to mammalian genomes. PLoS Computational Biology, 4(11), e1000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs, J. E. , & Paddock, C. D. (2003). The ascendancy of Amblyomma americanum as a vector of pathogens affecting humans in the United States. Annual Review of Entomology, 48, 307–337. [DOI] [PubMed] [Google Scholar]
- Dahlgren, F. S. , Paddock, C. D. , Springer, Y. P. , Eisen, R. J. , & Behravesh, C. B. (2016). Expanding range of Amblyomma americanum and simultaneous changes in the epidemiology of spotted fever group rickettsiosis in the United States. American Journal of Tropical Medicine and Hygiene, 94(1), 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima, P. , Barcelos, R. M. , Klein, R. C. , Vidigal, P. , Montandon, C. E. , Fabres‐Klein, M. H. , Dergam, J. A. , & Mafra, C. (2017). Sequencing and comparative analysis of the Amblyomma sculptum mitogenome. Veterinary Parasitology, 247, 121–128. [DOI] [PubMed] [Google Scholar]
- Drummond, A. J. , & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC evolutionary biology, 7(1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimanifar, A. , Kimball, R. T. , Braun, E. L. , & Ellis, J. D. (2018). Mitochondrial genome diversity and population structure of two western honey bee subspecies in the Republic of South Africa. Scientific Reports, 8(1), 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejiri, H. , Lim, C. K. , Isawa, H. , Fujita, R. , Murota, K. , Sato, T. , Kobayashi, D. , Kan, M. , Hattori, M. , Kimura, T. , Yamaguchi, Y. , Takayama‐Ito, M. , Horiya, M. , Posadas‐Herrera, G. , Minami, S. , Kuwata, R. , Shimoda, H. , Maeda, K. , Katayama, Y. , … Sawabe, K. (2018). Characterization of a novel thogotovirus isolated from Amblyomma testudinarium ticks in Ehime, Japan: A significant phylogenetic relationship to Bourbon virus. Virus Research, 249, 57–65. [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , Laval, G. , & Schneider, S. (2007). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47–50. [PMC free article] [PubMed] [Google Scholar]
- Fourdrilis, S. , Mardulyn, P. , Hardy, O. J. , Jordaens, K. , de Frias Martins, A. M. , & Backeljau, T. (2016). Mitochondrial DNA hyperdiversity and its potential causes in the marine periwinkle Melarhaphe neritoides (Mollusca: Gastropoda). PeerJ, 4, e2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier, P. E. , Takada, N. , Fujita, H. , & Raoult, D. (2006). Rickettsia tamurae sp nov., isolated from Amblyomma testudinarium ticks. International Journal of Systematic and Evolutionary Microbiology, 56, 1673–1675. [DOI] [PubMed] [Google Scholar]
- Ginestet, C. (2011). ggplot2: Elegant graphics for data analysis. Journal of the Royal Statistical Society Series a‐Statistics in Society, 174, 245. [Google Scholar]
- Githeko, A. K. , Lindsay, S. W. , Confalonieri, U. E. , & Patz, J. A. (2000). Climate change and vector‐borne diseases: a regional analysis. Bulletin of the World Health Organ, 78(9), 1136–1147. [PMC free article] [PubMed] [Google Scholar]
- Guglielmone, A. A. , Beati, L. , Barros‐Battesti, D. M. , Labruna, M. B. , Nava, S. , Venzal, J. M. , Mangold, A. J. , Szabó, M. P. , Martins, J. R. , González‐Acuña, D. , & Estrada‐Peña, A. (2014). Ticks (Ixodidae) on humans in South America. Experimental and Applied Acarology, 40(2), 83–100. [DOI] [PubMed] [Google Scholar]
- Gui, Z. , Wu, L. , Cai, H. , Mu, L. , Yu, J. F. , Fu, S. Y. , & Si, X. Y. (2021). Genetic diversity analysis of Dermacentor nuttalli within Inner Mongolia, China. Parasites and Vectors, 14(1), 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J. F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321. [DOI] [PubMed] [Google Scholar]
- Harpending, H. C. (1994). Signature of ancient population growth in a low‐resolution mitochondrial DNA mismatch distribution. Human Biology, 66(4), 591–600. [PubMed] [Google Scholar]
- Holsinger, K. E. , & Weir, B. S. (2009). Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nature Reviews Genetics, 10(9), 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak, I. G. , Camicas, J. L. , & Keirans, J. E. (2002). The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida): a world list of valid tick names. Experimental and Applied Acarology, 28(1), 27–54. [DOI] [PubMed] [Google Scholar]
- Hornok, S. , Kontschan, J. , Takacs, N. , Chaber, A. L. , Halajian, A. , Abichu, G. , Kamani, J. , Szekeres, S. , & Plantard, O. (2020). Molecular phylogeny of Amblyomma exornatum and Amblyomma transversale, with reinstatement of the genus Africaniella (Acari: Ixodidae) for the latter. Ticks and Tick‐borne Diseases, 11(6), 101494. [DOI] [PubMed] [Google Scholar]
- Imaoka, K. , Kaneko, S. , Tabara, K. , Kusatake, K. , & Morita, E. (2011). The first human case of Rickettsia tamurae infection in Japan. Case Reports in Dermatology, 3(1), 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro, F. , Takada, N. , Masuzawa, T. , & Fukui, T. (2000). Prevalence of Lyme disease Borrelia spp. in ticks from migratory birds on the Japanese mainland. Applied and Environmental Microbiology, 66(3), 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isohisa, T. , Inakai, N. , Okuzawa, Y. , Yamada, M. , Arizono, N. , & Katoh, N. (2011). Case of tick bite with infestation of an extraordinary number of larval Amblyomma testudinarium ticks. The Journal of Dermatology, 38(11), 1110–1112. [DOI] [PubMed] [Google Scholar]
- Jabin, G. , Dewan, Y. , Khatri, H. , Singh, S. K. , Chandra, K. , & Thakur, M. (2019). Identifying the tick Amblyomma javanense (Acari: Ixodidae) from Chinese pangolin: generating species barcode, phylogenetic status and its implication in wildlife forensics. Experimental and Applied Acarology, 78(3), 461–467. [DOI] [PubMed] [Google Scholar]
- Kang, Y. B. , Suh, M. D. , & Kim, Y. H. (1981). Amblyomma testudinarium Koch, 1844: Discovery and record in Korea, and identification and redescription of male tick. Korean Journal of Veterinary Research, 21(2), 65–72. [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution, 30(4), 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, E. L. , Stone, N. E. , Scoles, G. A. , Hepp, C. M. , Busch, J. D. , & Wagner, D. M. (2018). Range‐wide genetic analysis of Dermacentor variabilis and its Francisella‐like endosymbionts demonstrates phylogeographic concordance between both taxa. Parasites & Vectors, 11(1), 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelava, S. , Mans, B. J. , Shao, R. , Moustafa, M. , Matsuno, K. , Takano, A. , Kawabata, H. , Sato, K. , Fujita, H. , Ze, C. , Plantard, O. , Hornok, S. , Gao, S. , Barker, D. , Barker, S. C. , & Nakao, R. (2021). Phylogenies from mitochondrial genomes of 120 species of ticks: Insights into the evolution of the families of ticks and of the genus Amblyomma . Ticks and Tick‐borne Diseases, 12(1), 101577. [DOI] [PubMed] [Google Scholar]
- Kelly, P. J. , Beati, L. , Mason, P. R. , Matthewman, L. A. , Roux, V. , & Raoult, D. (1996). Rickettsia africae sp. nov., the Etiological Agent of African Tick Bite Fever. International Journal of Systematic and Evolutionary Microbiology, 46(2), 611–614. [DOI] [PubMed] [Google Scholar]
- Kitaoka, S. (1974). Reports of medico‐zoological investigations in the Nansei Islands Part 2. Ticks and their seasonal prevalences in southern Amami‐oshima. Medical Entomology and Zoology, 25(1), 21–26. [Google Scholar]
- Klompen, H. , Dobson, S. J. , & Barker, S. C. (2002). A new subfamily, Bothriocrotoninae n. subfam., for the genus Bothriocroton Keirans, King & Sharrad, 1994 status amend. (Ixodida: Ixodidae), and the synonymy of Aponomma Neumann, 1899 with Amblyomma Koch, 1844. Systematic Parsitology, 53, 101–107. [DOI] [PubMed] [Google Scholar]
- Koch, C. L. (1844). Systematische übersicht über die Ordnung der Zecken. Archiv für Naturgeschichte, 10, 217–239. 10.5962/bhl.part.29560 [DOI] [Google Scholar]
- Kodera, Y. , Kanzaki, N. , Kaneko, Y. , & Tokida, K. (2001). Habitat selection of Japanese wild boar in Iwami district, Shimane Prefecture, western Japan. Wildlife Conservation Japan, 6(2), 119–129. [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C. C. , Lin, Y. F. , Yao, C. T. , Shih, H. C. , Chung, L. H. , Liao, H. C. , Hsu, Y. C. , & Wang, H. C. (2017). Tick‐borne pathogens in ticks collected from birds in Taiwan. Parasites & Vectors, 10(1), 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lim, F. S. , Loong, S. K. , Khoo, J. J. , Tan, K. K. , Zainal, N. , Abdullah, M. F. , Khor, C. S. , & Abubakar, S. (2018). Identification and characterization of Corynebacterium lactis isolated from Amblyomma testudinarium of Sus scrofa in Malaysia. Systematic and Applied Acarology, 23(9), 1838–1844. [Google Scholar]
- Liu, Z. Q. , Liu, Y. F. , Kuermanali, N. , Wang, D. F. , Chen, S. J. , Guo, H. L. , Zhao, L. , Wang, J. W. , Han, T. , Wang, Y. Z. , Wang, J. , Shen, C. F. , Zhang, Z. Z. , & Chen, C. F. (2018). Sequencing of complete mitochondrial genomes confirms synonymization of Hyalomma asiaticum asiaticum and kozlovi, and advances phylogenetic hypotheses for the Ixodidae. PLoS One, 13(5), e0197524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, M. G. , May Junior, J. , Foster, R. J. , Harmsen, B. J. , Sanchez, E. , Martins, T. F. , Quigley, H. , Marcili, A. , & Labruna, M. B. (2016). Ticks and rickettsiae from wildlife in Belize, Central America. Parasites & Vectors, 9, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisri, P. , Hirunkanokpun, S. , Baimai, V. , Trinachartvanit, W. , & Ahantarig, A. (2015). Detection of Rickettsia and Anaplasma from hard ticks in Thailand. Journal of Vector Ecology, 40(2), 262–268. [DOI] [PubMed] [Google Scholar]
- Mechai, S. , Feil, E. J. , Gariepy, T. D. , Gregory, T. R. , Lindsay, L. R. , Millien, V. , & Ogden, N. H. (2013). Investigation of the Population Structure of the Tick Vector of Lyme Disease Ixodes scapularis (Acari: Ixodidae) in Canada Using Mitochondrial Cytochrome C Oxidase Subunit I Gene Sequences. Journal of Medical Entomology, 50(3), 560–570. [DOI] [PubMed] [Google Scholar]
- Motoi, Y. , Asano, M. , Inokuma, H. , Ando, S. , Kawabata, H. , Takano, A. , & Suzuki, M. (2013). Detection of Rickettsia tamurae DNA in ticks and wild boar (Sus scrofa leucomystax) skins in Shimane Prefecture, Japan. The Journal of Veterinary Medical Science, 75(3), 263–267. [DOI] [PubMed] [Google Scholar]
- Nadolny, R. M. , Gauthier, D. T. , Gaff, H. D. , & Bermúdez, S. E. (2016). Preliminary assessment of the population genetics of Ixodes affinis (Ixodida: Ixodidae) in North and Central America. Systematic and Applied Acarology, 21(10), 1300–1308. [Google Scholar]
- Nakao, R. , Shinjo, K. , Sakiyama, T. , Ogata, S. , Kusakisako, K. , Kinoshita, G. , Naguib, D. , Chatanga, E. , Mohamed, W. , Moustafa, M. , Matsuno, K. , Ito, T. , Nonaka, N. , Sashika, M. , Tsubota, T. , & Shimozuru, M. (2021). Amblyomma testudinarium infestation on a brown bear (Ursus arctos yesoensis) captured in Hokkaido, a northern island of Japan. Parasitology International, 80, 102209. [DOI] [PubMed] [Google Scholar]
- Nakao, Y. , Tanigawa, T. , & Shibata, R. (2017). Human otoacariasis caused by Amblyomma testudinarium: Diagnosis and management: Case report. Medicine (Baltimore), 96(26), e7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgyal, J. , Lysyk, T. J. , Couloigner, I. , Checkley, S. , Gurung, R. B. , Tenzin, T. , Dorjee, S. , & Cork, S. C. (2021). Identification, Distribution, and Habitat Suitability Models of Ixodid Tick Species in Cattle in Eastern Bhutan. Tropical Medicine and Infectious Disease, 6(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuaki, M. (2021). Tick bites in Japan. The Journal of Dermatology, 48(4), 423–430. [DOI] [PubMed] [Google Scholar]
- Natsuaki, M. , Takada, N. , Kawabata, H. , Ando, S. , & Yamanishi, K. (2014). Case of tick‐associated rash illness caused by Amblyomma testudinarium . The Journal of Dermatology, 41(9), 834–836. [DOI] [PubMed] [Google Scholar]
- Pele, J. , Becu, J. , Abdi, H. , & Chabbert, M. (2012). Bios2mds: an R package for comparing orthologous protein families by metric multidimensional scaling. BMC Bioinformatics, 13, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petney, T. N. , Saijuntha, W. , Boulanger, N. , Chitimia‐Dobler, L. , Pfeffer, M. , Eamudomkarn, C. , Andrews, R. H. , Ahamad, M. , Putthasorn, N. , Muders, S. V. , Petney, D. A. , & Robbins, R. G. (2019). Ticks (Argasidae, Ixodidae) and tick‐borne diseases of continental Southeast Asia. Zootaxa, 4558(1), 1–89. [DOI] [PubMed] [Google Scholar]
- Piotrowski, M. , & Rymaszewska, A. (2020). Expansion of Tick‐Borne Rickettsioses in the World. Microorganisms, 8(12), 1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, N. , Currat, M. , & Excoffier, L. (2003). Intra‐deme molecular diversity in spatially expanding populations. Molecular Biology and Evolution, 20(1), 76–86. [DOI] [PubMed] [Google Scholar]
- Regilme, M. A. F. , Sato, M. , Tamura, T. , Arai, R. , Sato, M. O. , Ikeda, S. , Gamboa, M. , Monaghan, M. T. , & Watanabe, K. (2021). Comparative population genetic structure of two ixodid tick species (Acari:Ixodidae) (Ixodes ovatus and Haemaphysalis flava) in Niigata prefecture, Japan. Infection, Genetics and Evolution, 94, 104999. [DOI] [PubMed] [Google Scholar]
- Rogers, A. R. , & Harpending, H. (1992). Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution, 9(3), 552–569. [DOI] [PubMed] [Google Scholar]
- Sagurova, I. , Ludwig, A. , Ogden Nicholas, H. , Pelcat, Y. , Dueymes, G. , & Gachon, P. (2019). Predicted Northward Expansion of the Geographic Range of the Tick Vector Amblyomma americanum in North America under Future Climate Conditions. Environmental Health Perspectives, 127(10), 107014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, J. M. , Goddard, J. , & Rasgon, J. L. (2014). Population and demographic structure of Ixodes scapularis say in the Eastern United States. PLoS One, 9(7), e101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassera, D. , Beninati, T. , Bandi, C. , Bouman, E. A. P. , Sacchi, L. , Fabbi, M. , & Lo, N. (2006). 'Candidatus Midichloria mitochondrii', an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle. International Journal of Systematic and Evolutionary Microbiology, 56(Pt 11), 2535–2540. [DOI] [PubMed] [Google Scholar]
- Simonsen, K. L. , Churchill, G. A. , & Aquadro, C. F. (1995). Properties of statistical tests of neutrality for DNA polymorphism data. Genetics, 141(1), 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh, J. H. , Kim, H. C. , Yun, S. M. , Lim, J. W. , Kim, J. H. , Chong, S. T. , Kim, D. H. , Kim, H. T. , Kim, H. , Klein, T. A. , Johnson, J. L. , & Lee, W. J. (2016). Detection of SFTS Virus in Ixodes nipponensis and Amblyomma testudinarium (Ixodida: Ixodidae) Collected From Reptiles in the Republic of Korea. Journal of Medical Entomology, 53(3), 584–590. [DOI] [PubMed] [Google Scholar]
- Sumrandee, C. , Hirunkanokpun, S. , Doornbos, K. , Kitthawee, S. , Baimai, V. , Grubhoffer, L. , Trinachartvanit, W. , & Ahantarig, A. (2014). Molecular detection of Rickettsia species in Amblyomma ticks collected from snakes in Thailand. Ticks and Tick‐borne Diseases, 5(6), 632–640. [DOI] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123(3), 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, N. (1990). Byougen Danirui Zufu Kinpodou. Kinpodo, Inc. [Google Scholar]
- Takano, A. , Sugimori, C. , Fujita, H. , Kadosaka, T. , Taylor, K. R. , Tsubota, T. , Konnai, S. , Tajima, T. , Sato, K. , Watanabe, H. , Ohnishi, M. , & Kawabata, H. (2012). A novel relapsing fever Borrelia sp. infects the salivary glands of the molted hard tick, Amblyomma geoemydae . Ticks and Tick‐borne Diseases, 3(4), 259–261. [DOI] [PubMed] [Google Scholar]
- Terada, Y. , Takahashi, T. , Abe, T. , & Moriyama, Y. (2019). Three cases of tick infestation (Amblyomma testudinarium) on grazing cattle in Aomori Prefecture, the northern part of Honshu. Japan. Medical Entomology and Zoology, 70, 167–170. [Google Scholar]
- Thu, M. J. , Qiu, Y. , Matsuno, K. , Kajihara, M. , Mori‐Kajihara, A. , Omori, R. , Monma, N. , Chiba, K. , Seto, J. , Gokuden, M. , Andoh, M. , Oosako, H. , Katakura, K. , Takada, A. , Sugimoto, C. , Isoda, N. , & Nakao, R. (2019). Diversity of spotted fever group rickettsiae and their association with host ticks in Japan. Scientific Reports, 9(1), 1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vail, S. G. , & Smith, G. (1998). Air temperature and relative humidity effects on behavioral activity of blacklegged tick (Acari: Ixodidae) nymphs in New Jersey. Journal of Medical Entomology, 35(6), 1025–1028. [DOI] [PubMed] [Google Scholar]
- Villanueva, R. A. M. , & Chen, Z. J. (2019). ggplot2: elegant graphics for data analysis, 2nd edition. Measurement: Interdisciplinary Research and Perspectives, 17(3), 160–167. [Google Scholar]
- Voltzit, O. V. (2002). A review of Asian Amblyomma species (Acari, Ixodida, Ixodidae). Acarina, 10, 95–136. [Google Scholar]
- Wang, T. H. , Zhang, S. Q. , Pei, T. W. , Yu, Z. J. , & Liu, J. Z. (2019). Tick mitochondrial genomes: structural characteristics and phylogenetic implications. Parasites & Vectors, 12(1), 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn, M. , Kidwell, K. M. , & Merajver, S. D. (2016). The Impact of Nathan Mantel's "The Detection of Disease Clustering and a Generalized Regression Approach". Cancer Research, 76(9), 2495–2496. [DOI] [PubMed] [Google Scholar]
- Yamaguti, N. T. V. J. , Keegan, H. L. , & Toshioka, S. (1971). Ticks of Japan, Korea, and the Ryukyu Islands. Brigham Young University Science Bulletin, Biological Series, 15(1), 25–30. [Google Scholar]
- Yamauchi, T. , Takano, A. , Maruyama, M. , & Kawabata, H. (2012). Human Infestation by Amblyomma testudinarium (Acari: Ixodidae) in Malay Peninsula, Malaysia. Journal of the Acarological Society of Japan, 21(2), 143–148. [Google Scholar]
- Yunker, C. E. (1996). Heartwater in sheep and goats: a review. Onderstepoort Journal of Veterinary Research, 63(2), 159–170. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The complete mitogenome sequences of 30 A. testudinarium and 9 Amblyomma sp. were deposited in the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp) with accession numbers of LC554761‐LC554790 (samples from Japan) and LC633546‐LC633554 (samples from Myanmar).
The data that support the findings of this study are openly available in the DNA Data Bank of Japan (http://www.ddbj.nig.ac.jp) with accession numbers of LC554761‐LC554790 (samples from Japan) and LC633546‐LC633554 (samples from Myanmar).