

SUMMARY
Homeostasis of neural firing properties is important in stabilizing neuronal circuitry, but how such plasticity might depend on alternative splicing is not known. Here we report that chronic inactivity homeostatically increases action potential duration by changing alternative splicing of BK channels; this requires nuclear export of the splicing factor Nova-2. Inactivity and Nova-2 relocation were connected by a novel synapto-nuclear signaling pathway that surprisingly invoked mechanisms akin to Hebbian plasticity: Ca2+-permeable AMPA receptor upregulation, L-type Ca2+ channel activation, enhanced spine Ca2+ transients, nuclear translocation of a CaM shuttle, and nuclear CaMKIV activation. These findings not only uncover commonalities between homeostatic and Hebbian plasticity but also connect homeostatic regulation of synaptic transmission and neuronal excitability. The signaling cascade provides a full-loop mechanism for a classic autoregulatory feedback loop proposed ~25 years ago. Each element of the loop has been implicated previously in neuropsychiatric disease.
In Brief
Silencing neuronal activity triggers similar molecular mechanisms as activating neurons during long-term potentiation, demonstrating Hebbian mechanisms of homeostatic spike regulation.
Graphical Abstract

INTRODUCTION
Neurons use multiple forms of plasticity to allow circuits to be flexible yet stable: Hebbian plasticity (positive feedback to strengthen already active circuit elements, as in long-term potentiation [LTP]) and homeostatic plasticity (negative feedback to boost neuronal elements deprived of activity or depress those undergoing hyperactivity). Homeostatic plasticity helps keep neuronal circuits away from extremes of silence or runaway excitation (Turrigiano and Nelson, 2004) and occurs at multiple levels of synapses, neurons, and circuits (Desai et al., 1999; Kim and Tsien, 2008; O’Leary et al., 2014; Turrigiano, 2008). Homeostatic plasticity is known to rely on changes in gene transcription (Schaukowitch et al., 2017), mRNA translation (Maghsoodi et al., 2008; Penney et al., 2012; Schanzenbächer et al., 2016), and protein stability (Ehlers, 2003). In contrast, alternative splicing (AS) of precursor mRNA has received little attention as a basis of neuronal homeostasis. This is surprising because AS is a critical intermediary between gene transcription and translation and engenders the enormous complexity of neuronal proteomes. By regulating exon inclusion, AS diversifies synaptic components, ion channels, and other key neuronal proteins (Furlanis and Scheiffele, 2018; Licatalosi and Darnell, 2010; Vuong et al., 2016). Dysregulation of AS contributes to brain disorders (Licatalosi and Darnell, 2006; Parikshak et al., 2016), possibly via altered homeostasis (Mullins et al., 2016; Ramocki and Zoghbi, 2008; Styr and Slutsky, 2018; Wondolowski and Dickman, 2013).
We asked whether AS supports homeostatic responses induced by long-term inactivity. Would this be converse to neuronal responses to hyperactivity (Ding et al., 2017; Iijima et al., 2011; Li et al., 2007; Mauger et al., 2016; Xie and Black, 2001), or would inactivity engage its own distinct mechanisms? In turn, how does AS affect neuronal activity? Answering these questions would delineate a classic homeostatic feedback loop
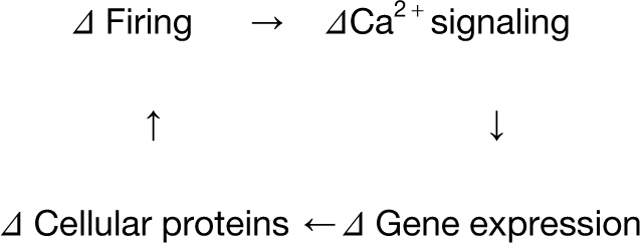
proposed more than 25 year ago at Brandeis (LeMasson et al., 1993; Marder et al., 1996; Siegel et al., 1994). This scheme embodies the feedback principles of neuronal homeostasis but has never been worked out in a full loop for any aspect of neuronal function. We focused on action potential duration (APD), fundamental in tuning neurons, synapses, and circuits. Even small changes in APD greatly affect Ca2+ influx (Borst and Sakmann, 1998; Geiger and Jonas, 2000; Llinás et al., 1982) and, thus, neurotransmitter release, gene expression, and overall neuronal function (Byrne and Kandel, 1996; Deng et al., 2013; Jackson et al., 1991; Matthews et al., 2009; Sabatini and Regehr, 1997). APD is generally prolonged by inactivity (Kim and Tsien, 2008; Trasande and Ramirez, 2007), an appropriate response to homeostatically restore Ca2+ entry, but remarkably little is known about underlying mechanisms.
Here we report that inactivity-induced homeostatic regulation of APD in excitatory neurons is controlled by AS of the large-conductance Ca2+-activated potassium channel (BK channel). We deciphered the underlying signaling mechanism: a specific change in BK AS driven by a novel cascade involving Ca2+-permeable glutamate receptors, voltage-gated Ca2+ channels, calcium/calmodulin-dependent (CaM) kinase kinase β (βCaMKK), CaM kinase IV (CaMKIV), and the splicing factor Nova-2. We find that complete silencing of an excitatory neuron triggers signaling similar to that activated during direct depolarization. Strikingly, each signaling player is encoded by a gene implicated previously in neuropsychiatric disease.
RESULTS
Homeostatic AP Broadening Driven by BK Current Attenuation
Although homeostasis of action potential (AP) frequency is well studied (Desai et al., 1999; Lee and Chung, 2014; Maffei and Turrigiano, 2008), how APD is regulated remains mysterious. This distinction is critical because spike width regulates neuronal excitability, Ca2+ influx, and neurotransmission and is governed by its own set of ion channels operating in parallel with channels controlling spike frequency (Kimm et al., 2015).
To examine homeostasis of APD, we blocked spiking of cultured cortical neurons by chronic (24- or 48-h) sodium channel blockade with tetrodotoxin (TTX) and examined APD after TTX removal. Chronically silenced neurons had a significantly longer APD than mock-treated controls (Figures 1A and S1), and AP-induced Ca2+ transients were elevated 1.5- to 2-fold in amplitude and duration (Figure 1B), as expected from APD-dependent prolongation of Ca2+ channel activation (Bischofberger et al., 2002). Thus, chronic inactivity drove compensatory increases in APD and Ca2+ entry as negative feedback autoregulation.
Figure 1. Chronic Spike Blockade-Induced BK Channel AS Is Responsible for Homeostatic Prolongation of AP Duration.

(A) Action potentials (APs) recorded from sham control- or chronic TTX-treated (48 h) neurons (left); AP duration (APD) was measured as half-width (right, n = 10). Scale bars, 20 mV, 1 ms.
(B) Single AP-elicited Ca2+ transients from control or TTX neurons transfected with GCaMP6s (left) and ΔF/F (right, n = 6).
(C) APs recorded from control (left) and TTX (right) neurons with (red) or without (black) IbTx. Scale bar, 20 mV and 1 ms.
(D) Half-widths of APs depicted in (C) (n = 8).
(E) Diagram of BK channel pore-forming α subunit and AS sites (X1–X6, arrowheads) (Pietrzykowski et al., 2008). Amino acid sequences of the sixth exon X6, E29 (purple, underlined), near the “Ca2+ bowl” (orange, underlined).
(F) RT-PCR products from control or TTX cultures with or without E29 inclusion (E29 or ΔE29, left) and percentage of products containing E29 (right; n = 8).
(G) E29 splicing in control or 60 mM K+ (60K)-treated cultures (24 h) and percentage of products containing E29 (right, n = 6).
(H) Whole-cell recordings from HEK293 cells overexpressing BK channel constructs with or without E29.
(I) Corresponding current–voltage (I-V) curves.
(J) APs from control or TTX cortical neurons transfected with plasmids encoding BK channel isoforms with or without E29. Scale bar, 20 mV and 1 ms.
(K) Pooled half-widths of APs depicted in (J) (n = 7).
For (A), (B), (D), (F), (G), (I), and (K), data are represented as mean ± SEM; *p < 0.05; **p < 0.01; N.S. represents p > 0.05.
See also Figures S1–S3.
The slowing of repolarization that drove APD prolongation was associated with blunted afterhyperpolarization (AHP) (Figure 1A). Both changes would ensue if potassium channel activity was reduced. We considered the BK channel because it regulates APD and AHP (Hu et al., 2001; Lee and Cui, 2010; Sausbier et al., 2004; Shao et al., 1999). Indeed, inhibiting BK channels with a specific blocker, iberiotoxin (IbTx), lengthened AP half-width (Figures 1C and 1D). To test whether BK channels are involved in inactivity-induced APD widening, we applied IbTx to TTX-silenced cortical cultures (48 h) just before AP recording. Strikingly, IbTx did not further widen APs (Figures 1C and 1D); the effect of inactivity largely occluded IbTx’s effect on APD. Evidently, neuronal silencing and IbTx converge on a common pathway—inhibiting BK activity in widening the AP.
Chronic Inactivity Alters BK Channel AS
Chronic inactivity might inhibit BK channels via gene expression and membrane trafficking. However, decreases in transcription, mRNA translation, or protein trafficking were ruled out by measurements of BK mRNA, total protein, and surface membrane expression (Figures S2A–S2D). BK channels also undergo AS, with profound effects on activity (Fodor and Aldrich, 2009; Shelley et al., 2013; Shipston and Tian, 2016; Xie and McCobb, 1998; Zarei et al., 2001). We assessed the expression of exons predicted to undergo splicing (X1–X6; Figure 1E) via RT-PCR (Pietrzykowski et al., 2008). In cultured cortical neurons, BK underwent AS in X3, X4, and X6 but not in X1, X2, and X5 (Figures 1F and S3A). Notably, only X6 (called E29 hereafter) splicing decreased after chronic TTX treatment (Figures 1F); AS of other exons remained unchanged (Figure S3B), narrowing our search for the basis of BK modulation.
We also tested the effect of enhanced activity by depolarization with K+-rich culture media (20, 40, and 60 mM K+; Figure S3C). Surprisingly, chronic depolarization decreased E29 exon inclusion (Figures 1G and S3D); the magnitude of shift in AS varied with strength and duration of depolarization (Figure S3D), but always in the same direction as chronic inactivity. We return later to resolve the paradox of how chronic silencing and depolarization produce similar changes in BK AS.
Depolarization- or seizure-induced changes in AS involve activation of CaM-dependent kinases (Xie and Black, 2001). In contrast, little is known about AS induced by inactivity, leading us to focus on its functional effect and underlying mechanisms.
Altered E29 Inclusion Dampens K+ Current and Broadens Spikes
E29 encodes 27 amino acids strategically located within the regulator of K+ conductance (RCK) domain, near the Ca2+ bowl, one of the Ca2+ binding sites promoting BK activation (Figure 1E). E29 inclusion affects BK responses to Ca2+ (Ha et al., 2000) and to alcohol-induced microRNAs (miRNAs) (Pietrzykowski et al., 2008). We expressed BK channels in HEK293 cells to find out whether E29 inclusion influences BK currents. BK currents were smaller with ΔE29 than with E29 (Figures 1H and 1I), whereas the membrane expression level was no different (Figures S2E–S2G). Thus, the reduction of E29 inclusion after TTX treatment likely contributes to the dampening of BK outward current.
We returned to cultured cortical neurons to test directly whether a drop in E29 inclusion leads to APD prolongation. Upon overexpression of the ΔE29 BK construct, APD was longer, in line with lower BK conductance, relative to that observed for its E29 counterpart. Furthermore, action potentials of E29-expressing neurons failed to broaden with chronic silencing. Likewise, the already widened AP of ΔE29-expressing showed no further prolongation (Figures 1J and 1K). Thus, inclusion or exclusion of E29 in overexpressed channels overrides the regulation engaged by chronic inactivity, suggesting that such regulation involves E29 splicing. Clinching this calls for understanding how the splicing is controlled.
Nova-2 Binds to the Intron Downstream of E29
AS of BK channels has been intensely studied, but how E29 inclusion is controlled remains unknown. Generally, splicing regulatory factors favor exon inclusion when binding to downstream introns but inhibit it when binding to upstream introns or the exon itself (Black, 2003; Ule et al., 2006). Our bioinformatics analysis of the intronic sequence past E29 revealed YCAY (Y=C/U) clusters (Figure 2A), consensus sequences for binding of Nova proteins, a well-studied class of neuron-specific RNA-binding proteins that regulate AS of neuronal proteins (Buckanovich and Darnell, 1997; Ule et al., 2005). Mice lacking Nova-2, the predominant isoform in the neocortex (Yang et al., 1998), are deficient in E29 inclusion (Ule et al., 2005).
Figure 2. Nova-2 Regulates E29 AS.

(A) Diagram of E29 (red box) and flanking introns (top) and biotinylated RNA probes (A1, A2, and B) created, spanning downstream intronic YCAY sequences (bottom).
(B) RNA pull-down of proteins from adult mouse brains, using the probes in (A) or empty beads with immunoblot of Nova-2 protein (n = 3).
(C) RT-PCR products from IP of Nova-2 from mouse cortical lysates. IP with IgG and an assay without reverse transcription (No RT) are controls.
(D) E29 splicing in neurons transfected with scrambled shRNA (Scr.), shRNA against the UTR of the Nova-2 gene (shRNA(UTR)), or shRNA(UTR) with exogenous shRNA(UTR)-resistant Nova-2 (Nova-2 (R)).
(E) Diagram of Nova-2 with the NLS present (WT) or absent (ΔNLS), with RNA binding domains (top: KH1, KH2, and KH3) and immunofluorescence of FLAG-tagged Nova-2 with or without the NLS. Nuclei are indicated by dashes. Scale bar, 10 μm.
(F) Diagram of the splicing reporter containing constitutive exons (black rectangles), E29 (red rectangle), and partial flanking intron sequences (horizontal lines) (top) and RT-PCR products from HEK293 cells transfected with the splicing reporter with control vector (sham), WT Nova-2 (WT), or ΔNLS-Nova-2 co-expression.
(G) E29 splicing in cultured cortical neurons infected by lentiviral Scr. or shRNAs against Nova-2 gene exposed to a sham control (Con) or 48 h TTX.
See also Figure S4.
To see whether E29 splicing is directly regulated by Nova-2, we tested whether Nova-2 binds to synthetic RNA oligonucleotides spanning the putative binding region in the downstream intron (probe A1; Figure 2A). Pull-down assays showed that probe A1 binds efficiently to endogenous Nova-2 from mouse cortical lysates. In contrast, no binding to Nova-2 was detected with a probe with mutations in putative Nova-2-binding (YCAY) sites (probe A2) or a control probe spanning a 35-bp intronic stretch upstream of A1 without YCAY motifs (probe B; Figures 2A and 2B). We asked whether Nova-2 also bound to the intron downstream of E29 in vivo, subjecting cortical lysates to RNA immunoprecipitation (IP) with Nova-2 antibodies and assaying with RT-PCR using primers flanking the YCAY sites. RT-PCR product was detected following IP with Nova-2 antibodies but not with control immunoglobulin G (IgG) (Figure 2C). No product from the Nova-2 immunoprecipitate was detected without reverse transcriptase, excluding an effect of genomic DNA contamination (Figure 2C). Thus, in vivo and in vitro experiments show that Nova-2 binds directly to the YCAY sites in the intron downstream of E29.
Nova-2 Is Required for E29 Splicing
Testing the necessity of Nova-2 for E29 inclusion, we knocked down Nova-2 in cultured cortical neurons using short hairpin RNAs (shRNAs) that targeted its coding sequence (CDS), or its untranslated region (UTR) (Figure S4A); the UTR-directed shRNA spared an exogenous Nova-2 construct lacking the UTR (Nova-2(R); Figure S4B). E29 inclusion was sharply reduced by lentiviral delivery of UTR-targeting shRNA but not of scrambled shRNA and not of exogenous Nova-2(R) (Figure 2D). This match to the pattern of Nova-2 knockdown and rescue (Figure S4B) suggested that Nova-2 is necessary for the AS event.
Changes in Nuclear Localization of Nova-2 Can Regulate E29 Splicing
Because Nova-2 acts outside of the nucleus (Racca et al., 2010), not just within it, we assessed the effect of cellular locale by comparing the effects of Nova-2 lacking its nuclear localization signal (ΔNLS) and with the NLS intact (wild type [WT]). Although WT Nova-2 was concentrated in the nucleus, ΔNLS-Nova-2 was mainly cytoplasmic (Figure 2E). We verified that Nova-2 directly regulates E29 inclusion using a splice reporter (Stoilov et al., 2008) containing E29 and partial flanking intron sequences (Figure 2F) in HEK293 cells (largely lacking endogenous Nova-2). In controls, E29 inclusion was knocked down by Nova-2 shRNA and rescued by Nova2(R) (Figure S4C). Critically, ΔNLS-Nova-2 largely failed to induce E29 inclusion (Figures 2F and S4C), supporting the importance of nuclear localization.
Nova-2 Is Necessary and Sufficient for TTX-Induced E29 Exclusion
To confirm in neurons that Nova-2 is critical for chronic inactivity-induced E29 reduction, we assayed the effects of chronic TTX after knockdown of Nova-2 (Figure 2G). Although scrambled lentiviral shRNA spared the reduction of E29 inclusion following 48-h TTX treatment, knockdown of Nova-2 mimicked and occluded the effect of chronic inactivity on E29 inclusion (Figure 2G). These experiments showed that Nova-2 binds directly to the intron downstream of E29 and is necessary and sufficient for regulation of E29 inclusion by inactivity.
Chronic Inactivity Reduces Nova-2 Nuclear Localization In Vitro
This sets up the question of how Nova-2 effectiveness is linked to chronic inactivity. Because Nova-2 must be in the nucleus to control splicing, its cellular relocalization is a potential control point. We assessed Nova-2 localization in cultured cortical neurons after 48-h treatment with TTX. In TTX-treated neurons, Nova-2 nuclear intensity was lower than control cells, whereas cytosolic Nova-2 intensity increased (Figure 3A), yielding a relative 2-fold drop in the nuclear/cytoplasmic ratio (Figure 3B, left). We verified these immunocytochemical findings by nuclear fractionation and immunoblot analysis (Figure 3C). After TTX exposure, nuclear Nova-2 levels fell, cytoplasmic Nova-2 rose, and total Nova-2 expression remained unchanged (Figures 3C and 3D), indicating that Nova-2 translocates from the nucleus to the cytosol upon silencing of cultured neurons. A similar shift in the nucleus/cytoplasmic ratio was seen upon chronic depolarization (Figure 3B, right); thus, the paradoxically similar regulation of E29 inclusion arises at or before Nova-2 relocation.
Figure 3. Chronic Spike Blockade Reduces Nova-2 Nuclear Localization and E29 Inclusion.

(A) Immunofluorescence of Nova-2 from neurons treated with the sham Con or 48 h TTX. Nuclei are indicated by dashes. Scale bar, 10 μm.
(B) Quantification of the nuc/cyt ratio of Nova-2 immunofluorescence intensity from neurons sham- or 48 h TTX-treated (left) and sham- or 24 h 60 mM K+ solution-treated (right) (n = 30 cells).
(C) Western blots from nuclear (left), cytosolic (center), or whole-cell fractions (right), with Nova-2, Lamin B, and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) levels.
(D) Quantification of (C) (for each condition, n = 4).
(E) Diagram of eliminated thalamic inputs to the cortex.
(F) Nova (red) localization in cortical neurons from Olig3 Cre or Olig3 Cre; TeToxf/+ mice. Scale bar, 20 μm.
(G) Quantified nuc/cyt ratio of Nova immunofluorescence intensity from the groups in (F) (n = 20 cells).
(H) Diagram of visual pathways involved in monocular deprivation (MD), with the monocular region of the primary visual cortex (V1mono) indicated (boxes).
(I) Nova-2 (red) localization in the V1mono region, contralateral (Contra) or ipsilateral (Ipsi) to visual deprived eye (MD). Scale bar, 10 μm.
(J) Quantified nuc/cyt ratio of Nova-2 immunofluorescence intensity from the groups in (I) (n = 20 cells).
(K) E29 splicing in V1mono Contra or Ipsi to the visually deprived eye (MD).
(L) Quantification of E29 splicing in (K) (n = 4).
(M) AP waveforms recorded from the V1mono region, Contra or Ipsi to the visually deprived eye (MD).
(N) Half-width of APs in (M) (n = 12–14).
For (B), (D), (G), (J), (L), and (N), data are represented as mean ± SEM; **p < 0.01; N.S. represents p > 0.05.
Chronic Inactivity Reduces Nova-2 Nuclear Localization In Vivo
We next asked whether silencing neocortical neurons in vivo caused similar changes as in dissociated cultures. Tonic inputs from the thalamus to the neocortex, which excite ongoing activity of cortical neurons, were chronically eliminated in mice by expressing tetanus toxin light chain (TeTox) in Olig3-positive thalamic neurons (Figure 3E). Nova localization was assayed in cortical neurons innervated by thalamic inputs. Nova was largely nuclear in Olig3 Cre (control) mice but excluded from the nucleus in Olig3 Cre; TeToxf/+mice (Figures 3F and 3G). Chronic removal of thalamic input led to Nova translocation from the nucleus to the cytosol of cortical neurons.
Inactivity-induced Nova-2 translocation was also observed with a monocular deprivation (MD) paradigm (Wiesel and Hubel, 1963). A lid suture was applied in juvenile mice for 5 days during the critical period of visual development (post-natal days 26–31) (Figure 3H), and subcellular localization of Nova-2 in monocular V1 was assayed by immunostaining. The nuclear intensity of Nova-2 in the deprived hemisphere (contralateral to the closed eye) was less than in the non-deprived hemisphere (ipsilateral); opposite differences were seen in cytoplasmic intensity (Figures 3I and 3J). Thus, MD-induced inactivity also induced in vivo Nova-2 translocation from the nucleus to the cytosol. Strikingly, MD also reduced E29 inclusion and increased APD in the vision-deprived hemisphere compared with its non-deprived counterpart (Figures 3K–3N). Thus, chronic inactivity in vivo leads to nucleus-to-cytosol Nova-2 redistribution, reduction of E29 inclusion, and increased APD.
CaMKIV Is Activated by Chronic Inactivity
While cellular control mechanisms of Nova-2 localization have not been reported, we considered scenarios including regulation by phosphorylation. CaMKIV was a likely candidate as a robust protein kinase, highly concentrated within the nucleus (Bito et al., 1996; Nakamura et al., 1995); CaMKIV regulates other splicing factors, such as heterogeneous nuclear ribonucleoprotein L (hnRNP L) (Liu et al., 2012), and supports homeostatic adjustments (Ibata et al., 2008). To test for involvement of CaMKIV, we looked at its status in the nucleus after chronic inactivity, monitoring phosphorylation on Thr196, its dominant activation site (Selbert et al., 1995; Tokumitsu and Soderling, 1996). Thr196 phosphorylation was higher in the nucleus of TTX-treated neurons relative to the control (Figure 4A), resulting in a 1.5-fold higher nuclear/cytoplasmic ratio (Figure 4B). Elevated activation of nuclear CaMKIV was also seen in vivo upon immunoblot analysis of samples from cortical V1, comparing lysates from monocularly deprived and non-deprived hemispheres (Figure 4C).
Figure 4. Inactivity-Induced CaMKIV Activation Leads to Nuclear Nova-2 Export.

(A) Immunofluorescence of phosphorylated CaMKIV (pCaMKIV) from neurons treated with sham Con or 48 h TTX. The nucleus is indicated (dashed white). Scale bar, 10 μm.
(B) Quantified nuc/cyt ratio of pCaMKIV immunofluorescence intensity from groups in (A) (n = 20).
(C) Western blots indicating pCaMKIV and CaMKIV levels in V1mono Contra or Ipsi to the visually deprived eye (MD).
(D) Immunofluorescence of neurons transfected with Con vectors, FLAG-tagged CA-CaMKIV (top two rows, sham Con treatment), or FLAG-tagged nuclear CaMBP4 (bottom two rows, 48 h TTX). Scale bar, 10 μm.
(E) Quantified nuc/cyt ratio of Nova-2 immunofluorescent intensity in (D) (n = 20).
For (B) and (E), data are represented as mean ± SEM; **p < 0.01.
CaMKIV Signaling Drives Nuclear Nova-2 Export
Is mimicking CaMKIV activation sufficient to induce Nova-2 translocation? To test this, we expressed constitutively activated CaMKIV (CA-CaMKIV) (Cruzalegui and Means, 1993) and assayed Nova-2 localization. In vector control-transfected neurons, Nova-2 was predominantly located in the nucleus (Figure 4D, top row). In contrast, in CA-CaMKIV-transfected neurons, Nova-2 nuclear intensity was lower, and cytoplasmic intensity was higher (Figure 4D, second row), causing a significant drop in nuclear/cytoplasmic (nuc/cyt) ratio (Figure 4E). Evidently, activation of CaMKIV is sufficient to drive Nova-2 translocation. Continuous activation of CaMKIV requires its binding to Ca2+/CaM. Accordingly, we overexpressed a nucleus-localized Ca2+/CaM trap, CaMBP4(nuc) (Cohen et al., 2016; Wang et al., 1995), before examining Nova-2 location. This intervention abolished the chronic inactivity-induced nuclear export of Nova-2 (Figures 4D, fourth row, and 4E), supporting the idea that Nova-2 translocation is regulated by activated CaMKIV. These experiments indicate that CaMKIV activation is sufficient and necessary to induce Nova-2 translocation from the nucleus to the cytosol.
CaMKIV Interaction with Nova-2
Both Nova-2 and CaMKIV are mostly located in the nucleus (Figure 5A), but whether they directly interact is unknown. To explore this, we co-expressed constructs encoding FLAG-tagged Nova-2 and GFP-tagged CaMKIV in HEK293 cells. CoIP and western blotting with anti-FLAG or anti-GFP antibodies were performed on protein lysates to detect protein-protein interaction. FLAG-tagged Nova-2 was found with IP using anti-GFP antibody; GFP-tagged CaMKIV was seen upon IP with anti-FLAG antibody (Figure 5B). No reciprocal interaction was detected upon IP of lysates of mock-transfected cells or when they were expressed individually (Figure 5B).
Figure 5. CaMKIV Phosphorylates Nova-2 and Regulates Its Nuclear Localization.

(A) Immunofluorescence of CaMKIV and Nova-2. Scale bar, 10 μm.
(B) Immunoblots of coIP lysate from HEK293 cells transfected with Nova-2-FLAG, CaMKIV-GFP, or both. Sham-transfected cells were used as Con.
(C) Immunoblots of CaMKIV and Nova-2 coIP lysate from cortical neurons. IgG was used as Con.
(D) Location of S25, T27, and S194 (red); amino acid sequences of the NLS (blue, underlined) and the partial KH2 domain are indicated.
(E–G) Quantification of phosphorylation levels of sites S25 (E), T27 (F), and S194 (G) of Nova-2 induced by CaMKIV, normalized to cells co-expressing Nova-2 and GFP (red dotted lines) (n = 3).
(H) Micrographs of FLAG-tagged wild-type (WT) and mutant Nova-2. Nuclei are indicated by white dotted circles. Scale bar, 10 μm.
(I) Quantification of the nuc/cyt ratio of the fluorescence intensity of WT or mutant Nova-2 from (H) (n = 20–62). **p < 0.01.
(J) RT-PCR products from HEK293 cells expressing the E29 splicing reporter and WT Nova-2, ΔNLS Nova-2, or S25E/T27E Nova-2.
For (E)–(G) and (I), data are represented as mean ± SEM. See also Figure S5.
The interaction was also evident even at endogenous levels of protein expression in neurons. Nova-2 was immunoprecipitated with anti-CaMKIV antibody; CaMKIV was brought down with anti-Nova-2 antibody, but no binding interactions were seen with IgG controls (Figure 5C). These results indicate that Nova-2 and CaMKIV engage in protein-protein interaction, studied as exogenous or endogenous proteins.
CaMKIV Phosphorylates Nova-2 and Regulates Its Nuclear Localization
To find out whether CaMKIV phosphorylates Nova-2, we co-expressed CA-CaMKIV (or GFP as a control) with FLAG-tagged Nova-2 in HEK293 cells and probed Nova-2 phosphorylation with mass spectrometry (liquid chromatography-tandem mass spectrometry [LC-MS/MS]) (Figures S5A–S5D and 5D5G). Compared with the control, CA-CaMKIV drove Nova-2 phosphorylation at three sites: serine 25 (site 1), threonine 27 (site 2), and serine 194 (site 3). Sites 1 and 2 lie within or next to the NLS in Nova-2, predictive of influence on nuclear localization (Harreman et al., 2004); site 3 resides in the KH2 domain, one of the RNA binding domains (Yang et al., 1998). To test whether phosphorylation affects Nova-2 location in cultured neurons, we probed the distribution of FLAG-tagged Nova-2 constructs with the sites mutated to glutamic acid (E) to mimic the negative charge phosphorylation confers or to alanine (A) to prevent phosphorylation. In contrast to the mostly nuclear positioning of WT Nova-2, Nova-2 with phosphomimetic mutations at sites 1 and 2 (1E2E) was mostly cytoplasmic (Figures 5H and 5I). Single mutations (1E or 2E) reduced Nova-2 nuclear localization, even when the other site was mutated to alanine (1E2A or 1A2E) (Figures 5H and 5I). Conversely, Nova-2 with single or double mutations to alanine (2A and 1A2A) was predominantly nuclear, like the WT (Figures 5H and 5I). In contrast, glutamate mutations at site 3 had no effect on Nova-2 localization (Figures 5H and 5I) or on Nova-2 binding to RNA (Figure S5E). These results showed that active CaMKIV reduces Nova-2 nuclear localization by phosphorylating sites 1 and 2 (S25, T27).
To test whether CaMKIV phosphorylation of Nova-2 controlled its ability to regulate E29 splicing, we co-expressed the 1E2E double mutant Nova-2 with the E29 splicing reporter in HEK293 cells. Unlike WT Nova-2, the 1E2E variant was unable to induce E29 inclusion, like Nova-2 lacking its NLS (ΔNLS; Figure 5J). Evidently, CaMKIV binds to Nova-2 in the nucleus, primed to phosphorylate serine 25 and threonine 27 sites near the NLS of Nova-2; the resulting Nova-2 nuclear exit prevents its action in splicing, reducing exon 29 inclusion.
Spontaneous Spine Depolarizations Detected with a Membrane Voltage Probe
How does chronic elimination of AP firing lead to activation of CaMKIV and nuclear exit of Nova-2? Activity blockade with TTX abolishes evoked vesicle release but spares spontaneous, AP-independent vesicle release. We asked whether spontaneous synaptic transmission might be sufficient to activate signaling to CaMKIV activation and Nova-2 translocation.
To test this, we monitored the membrane potential in dendritic spines of cortical neurons using the genetically encoded voltage indicator ASAP1 (St-Pierre et al., 2014). ASAP1 fluorescence was seen in the plasma membrane of somata, dendritic shafts, and spines (Figure 6A). Decreases in ASAP1 fluorescence were linearly related to membrane depolarizations (St-Pierre et al., 2014) imposed by K+-rich external solution (Figure 6B, gray symbols). In cortical cultures acutely exposed to TTX, dendritic spines exhibited dips in relative fluorescence intensity (ΔF/F), reflecting spontaneous excitatory postsynaptic potentials (EPSPs) (Figure 6C, green exemplar trace, and 6B, pooled data). After chronic TTX, spontaneous spine depolarizations grew by ~10 mV (Figure 6C, red trace), exceeding depolarization attained with 20 mM K+ (Figure 6B, pooled data), sufficient to recruit L-type Ca2+ channel- and NMDA receptor (NMDAR)-mediated Ca2+ influx (Helton et al., 2005; Mayer et al., 1984; Nowak et al., 1984). For comparison, even larger ΔF/F transients were observed in dendritic spines of control neurons during backpropagating dendritic APs (bAPs) or somatic action potentials recorded without TTX (Figures 6C, blue and violet exemplar traces, and 6B, pooled data).
Figure 6. Chronic Spike Blockade Leads to Elevated Depolarization and Ca2+ Transients in Dendritic Spines.

(A) Micrograph of a neuron expressing ASAP1. Scale bar, 10 μm.
(B) Quantification of membrane potential (ordinate, left) of somata and spines induced by APs (AP in a soma, purple; bAP in a spine, blue) or by AP-independent synaptic transmission (spine depolarization in sham Con cultures, Vspine green; spine depolarization in 48 h TTX cultures, Vspine red), plotted against the corresponding change in ASAP1 fluorescence from the respective events (−ΔF/F). See also STAR Methods.
(C) Example traces of ASAP1 fluorescence intensity (ΔF/F) in the spines and somata in (B).
(D and F) GCaMP6s expression in Con (D) and 48 h TTX neurons (F). Active spines with Ca2+ transients (during 5 min of imaging) are indicated (red dots). Scale bar, 10 μm. See also STAR Methods.
(E and G) GaMP6s signal (spontaneous Ca2+ signals were recorded in the presence of TTX) from active spines (y axis, red dots in D and F) in Con (E) and 48 h TTX neurons (G).
(H–K) Comparison of synaptic Ca2+ transients between Con (black) and TTX (red) neurons. Shown are (H) number of active puncta (putative active spines) during 5 min of recording, (I) mean amplitude, (J) mean duration, and (K) normalized total fluorescence in Con and TTX (48 h) neurons (n = 14–15).
(L) Quantification of Ca2+ transient amplitudes in dendritic spines with wash on PhTx, APV, thapsigargin, or nimodipine (n = 8).
(M and N) AP waveforms (M) and pooled half-widths (N) after sham (black), 48 h TTX (red), or 48 h TTX with co-application of PhTx (green). Scale bars, 20 mV, 1 ms. For (H)–(L) and (N), data are represented as mean ± SEM; *p < 0.05; **p < 0.01; N.S. represents p > 0.05. See also Figure S6.
Chronic Inactivity Enhances Spontaneous Spine Ca2+ Transients
To study spontaneous Ca2+ transients in dendritic spines, we expressed the fluorescent Ca2+ indicator GCaMP6s (Figures 6D–6G). Although the soma of cortical neurons was totally silent in the presence of TTX (Figures S6A and S6B), dendritic spines remained active despite spike blockade (Figures 6D–6G and S6C). More spines were active in neurons chronically treated with TTX than in controls (Figure 6H). Likewise, the Ca2+ transients were taller, broader, and larger in area (Figures 6I–6K), whereas fluorescent punctum size and event frequency per punctum were no different (Figures S6D and S6E). Thus, chronic blockade of APs enhances spontaneous Ca2+ transients in spines, in line with optical voltage recordings.
Contributions of Various Ca2+ Pathways during Spontaneous Transmission
We characterized the elevated synaptic Ca2+ transients, mindful of increases in Ca2+-permeable, GluA1-containing AMPA receptors (AMPARs) following activity blockade (Kim and Ziff, 2014; Thiagarajan et al., 2005). Live-labeled surface GluA1 increased in neurons that had undergone chronic inactivity (Figures S6F and S6G). Elevated surface GluA1 contributed to the enlarged Ca2+ transient, indicated by a sharp drop in synaptic Ca2+ transients upon acute exposure to the GluA1 antagonist philanthotoxin (PhTx) (Figure 6L). Importantly, PhTx completely blocked chronic inactivity-induced AP prolongation (Figures 6M and 6N), indicating that GluA1 activation was critical for homeostatic regulation of APD.
Following chronic inactivity, elevated Ca2+ transients were also inhibited by blockade of NMDAR with ((2R)-amino-5-phosphonovaleric acid; (2R)-amino-5-phosphonopentanoate) APV and depletion of internal Ca2+ stores with thapsigargin (Figure 6L). This aligns with AMPAR activation recruiting Ca2+ delivery via NMDAR and intracellular Ca2+ stores (Emptage et al., 1999). Voltage-dependent L-type Ca2+ channels (CaV1) also contributed to spine Ca2+ transients during chronic inactivity, as judged by partial reduction with nimodipine (Figure 6L). A glutamate-gated cation current would create a voltage drop across the spine neck resistance (Harnett et al., 2012; Palmer and Stuart, 2009), giving rise to directly measured depolarizations and CaV1- and NMDA-mediated Ca2+ influx (Figure 6L).
We verified inactivity-induced engagement of Ca2+ signaling by testing for activation of CaMKII and CaMKI in dendritic spines, which, respectively, undergo autophosphorylation or CaM kinase kinase (CaMKK)-mediated phosphorylation in response to local Ca2+ signals (Wayman et al., 2008). Using site-specific anti-phospho-Thr antibodies, we showed CaMKII Thr286 autophosphorylation and CaMKI Thr177/178 phosphorylation, respectively (Figures S6H–S6K). Chronic inactivity augmented the intensity of both markers relative to the control (Figures S6H–S6K), providing independent biochemical evidence that local Ca2+ signaling in spines is enhanced by chronic inactivity.
The CaV1-CaMKK-CaMKIV Pathway Drives Reduced E29 Inclusion
Intensified minis and spine Ca2+ signaling could link inactivity to nuclear AS. To find out whether this involves a classical CaV1-CaMKK-CaMKIV cascade, like that engaged by depolarization (West et al., 2002), we monitored phosphorylation of CaMKIV as a pivotal step. Inactivity induced elevation of phosphorylated CaMKIV (pCaMKIV) but not in the presence of nimodipine (a CaV1 blocker), KN93 (a CaMK inhibitor), or STO-609 (a CaMKK blocker). This pattern was consistent in western blots of nuclear extracts (Figures 7A and 7B) and in nuc/cyto ratios obtained by immunocytochemistry (Figures S7A and S7B). Nova-2 localization showed a reciprocal pattern (Figures 7C, 7D, S7C, and S7D) which was mirrored by TTX-induced reduction on E29 inclusion (Figures 7E and 7F). These results supported a chain of signaling events emanating from spontaneously active spines whereby a CaV1-CaMKK-CaMKIV pathway drives reductions in nuclear Nova-2 and in E29 inclusion.
Figure 7. CaV1-CaMK Signaling Is Required for Chronic Spike Blockade-Induced Nova-2 Translocation and BK Channel AS.

(A) Expression level of pCaMKIV and CaMKIV in neurons treated with nimodipine, KN93, or STO-609.
(B) Quantification of CaMKIV activation from (A) (n = 3).
(C) Western blot indicating nuclear Nova-2 levels from neurons treated as indicated.
(D) Quantification of the nuclear Nova-2 level from (C) (n = 3).
(E) E29 splicing after 48 h TTX with other treatments as indicated.
(F) Quantification of E29 splicing from (E) (n = 4).
(G) E29 splicing after chronic depolarization (60K for 24 h) with or without nimodipine or KN93.
(H) Quantification of E29 splicing from (G) (n = 4).
(I) βCaMKK immunostaining (green) with MAP2 (red) and DAPI (blue). Nuclei are indicated by white dashed circles. Scale bar, 10 μm.
(J) Fold changes of αCaMKK and βCaMKK expression in the nucleus, cytosol, and nuc/cyt ratio after TTX (48 h) relative to Con neuron levels (n = 30).
(K) E29 splicing from neurons expressing viral shRNA constructs against endogenous βCaMKK with or without co-expression of shRNA-resistant βCaMKK (βCaMKK (R)). Scrambled shRNA was used as Con.
(L) Quantification of E29 splicing from (K) (n = 4).
(M) Diagram of the homeostatic signaling loop regulating APD. (1) Chronic spike blockade leads to upregulation of synaptic GluA1 (synaptic scaling). (2) Activation of GluA1 mediates excessive cation influx, induces membrane depolarization in dendritic spines, and facilitates opening of the NMDAR and CaV1 channels. (3) CaV1 opening leads to activation of CaMKs, including CaMKI, CaMKII, and βCaMKK. (4) βCaMKK translocates from the cytosol to nucleus and activates CaMKIV. (5) Activated CaMKIV phosphorylates nuclear Nova-2. (6) Phosphorylated Nova-2 translocates to the cytosol. (7) This leads to reduced E29 inclusion in BK channel pre-mRNA. (8) BK channel activity is inhibited when lacking E29, broadening APDs, the observed homeostatic response to chronic spike blockade.
(N) The autoregulatory feedback loop of neuronal excitability (LeMasson et al., 1993; Marder et al., 1996; Siegel et al., 1994) with components described in this study. Various genes implicated in neuropsychiatric diseases are highlighted.
For (B), (D), (F), (H), (J), and (L), data are represented as mean ± SEM; **p < 0.01; N.S. represents p > 0.05. See also Figure S7.
Activation of a CaV1-CaMKK-CaMKIV pathway by chronic inactivity appears surprising because it seemingly recapitulates effects of hyperactivity (Deisseroth et al., 2003; Ma et al., 2014). However, we found that directly imposed depolarization caused similar effects as chronic TTX on Nova-2 translocation and BK E29 exclusion (Figures S7E and S7F), with the latter effect completely prevented by nimodipine or KN93 (Figures 7G, 7H, and S7F). Thus, activation of CaV1 channels and CaMKs drives Nova-2 relocation and E29 splicing, irrespective of how the pathway is engaged.
βCaMKK Translocation Is Required for E29 Inclusion
A remaining question is how, without spiking, CaV1 activation at dendritic sites causes activation of CaMKIV in the nucleus. In excitation-transcription coupling (E-T coupling) following acute depolarization, signaling to nuclear CaMKIV involves translocation of Ca2+/calmodulin via different shuttle proteins: γCaMKII in cortical, hippocampal, and sympathetic neurons (Ma et al., 2014) and γCaMKI in parvalbumin-positive inhibitory neurons (Cohen et al., 2016). In seeking a translocator for excitation-AS coupling, we looked for an increase in nuclear level paired with a drop in cytoplasmic level during chronic inactivity. This pattern was not evident for αCaMKI, βCaMKI, γCaMKI, δCaMKI, γCaMKII, αCaMKK, and CaM itself (Figures 7J, S7G, and S7H). In contrast, levels of βCaMKK rose in the nucleus and fell in the cytosol; its nuc/cyto ratio increased by more than 50% (Figures 7I and 7J). βCaMKK can phosphorylate CaMKIV and is thus a plausible mediator of cytosol-to-nucleus signaling and nuclear CaMKIV activation. We probed βCaMKK’s involvement by shRNA knockdown. This completely blocked inactivity-induced reduction of E29 inclusion, whereas concomitantly expressing shRNA-resistant βCaMKK fully rescued it (Figure 7K and 7L). Thus, βCaMKK translocates to the nucleus and is necessary for chronic inactivity-induced E29 splicing.
DISCUSSION
We found an unexpected mechanism by which excitatory neurons modify their APs in responding homeostatically to chronic inactivity (Figure 7M). The adaptation arises from a well-defined change in splicing and is mediated by a novel signaling cascade that mobilizes Hebbian-type signaling, even in the absence of spikes. Remarkably, each element of this signaling pathway has been genetically implicated in neuropsychiatric disease (see below).
Regulation of BK Channel Splicing Selectively Controls APD
We found that splicing of BK channels is necessary and sufficient for inactivity-induced lengthening of APD. Under typical circumstances, regulatory changes in one repolarizing current (e.g., BK) would be offset by compensatory changes in others (e.g., KV2) (Kimm et al., 2015). Here, however, the key findings are null effects (Figures 1C, 1D, 1J, 1K, 6M, and 6N); without changes in spike waveform, altered recruitment of other voltage-gated channels is not expected. Kimm et al. (2015) have further shown that blockade of BK channels with IbTx spares the current-frequency relationship. Thus, mechanisms other than BK regulation are expected and seen for homeostatic adjustments of firing frequency (Lee and Chung, 2014; Lee et al., 2015). For APD, we cannot exclude ancillary changes enabled by E29 inclusion, such as BK channel phosphorylation or modified auxiliary subunits. However, the brain-dominant BK auxiliary subunit β4 is not significantly altered by TTX treatment (Lee et al., 2015).
CaV1-CaMKIV Signaling Is Engaged by Chronic Inactivity and Acute Depolarization
Elegant work shows how AS can be regulated by depolarization or activity elevation (Ding et al., 2017; Iijima et al., 2011; Mauger et al., 2016; Xie and Black, 2001). Here we show that AS is also controlled by chronic inactivity and dominates homeostatic regulation of AP shape. Strikingly, chronic inactivity-induced AS of BK, although homeostatic in outcome, relies on CaV1-CaMKK-CaMKIV signaling, like acute depolarization-induced splicing. Activation of Ca2+ signaling by silencing neuronal spiking is counterintuitive; the expectation is that Ca2+ entry would be dampened (Bridi et al., 2018). We resolved the paradox by optically tracking transient depolarizations in dendritic spines of “inactive” neurons (Figures 6A–6C), which were strong enough to activate the CaV1 channels present in dendritic spines (Stanika et al., 2016). This demystified the recruitment of CaV1 signaling, already indicated pharmacologically.
A potent set of synaptic events combine to drive homeostatic readjustment of APD. Chronic spike blockade drives enhanced spontaneous presynaptic vesicle release (Jakawich et al., 2010; Lindskog et al., 2010) and incorporation of postsynaptic, high-conductance, PhTx-sensitive GluA1 receptors (Kim and Ziff, 2014; Thiagarajan et al., 2005). This increases glutamate-gated cation influx, drives greater spine depolarization, and recruits L-type Ca2+ channels in spine heads (Obermair et al., 2004; Yasuda et al., 2003), Mg2+-unblocked NMDARs, and Ca2+ release from internal stores (Dittmer et al., 2017). Our scenario (Figure 7M) accounts for L-type channel participation in responses triggered by chronic TTX; L-type channel involvement in responses to chronic depolarization is well known (O’Leary et al., 2010). Ca2+ channel-triggered signaling in response to chronic inactivity and depolarization runs counter to conventional expectations of diametrically opposing effector actions. Our data suggest that L-type-dependent signaling can be mobilized to achieve the appropriate response irrespective of the direction of the initial perturbation.
Strikingly, the CaV1 blocker nimodipine completely abolished TTX-induced CaMK activation, Nova-2 translocation, and E29 splicing but only partially inhibited the Ca2+ transient in spines induced by chronic TTX. We suggest that other Ca2+ influx pathways contribute to Ca2+ flux (Wheeler et al., 2012), whereas only nimodipine-sensitive CaV1 channels provide critical voltage-dependent conformational (VΔC) signaling (Li et al., 2016) to help trap activated CaMKII (Wang et al., 2017).
Multiple CaM Translocators Support Different Forms of Surface-to-Nucleus Communication
Translocation of a CaMK is a recurrent theme in cytonuclear signaling in neurons, but the identity of the kinase varies (Cohen et al., 2016; Ma et al., 2014). In the present case of chronic inactivity-induced signaling, sensitivity to the selective CaMKK inhibitor STO-609 indicated that a CaMKK was involved. These findings resembled studies in C. elegans where cytonuclear signaling relies on a monomeric CaMKK (CKK-1) that translocates across the nuclear membrane (Kimura et al., 2002). Precedent exists for regulated cytonuclear distribution of βCaMKK (Cao et al., 2011; Karacosta et al., 2012; Nakamura et al., 2001), but its molecular mechanism needs elucidation. βCaMKK lacks a classic NLS, but it might rely on regulation of its nuclear export signal (NES) (Xu et al., 2012) through interaction with a resident nuclear protein.
CaMKIV was, as expected, the target of βCaMKK activation, as judged by elevation of pCaMKIV and its blockade by STO-609. Finding that CaMKIV was prebound to its substrate Nova-2 opens up the possibility of local signaling events. This would minimize the perturbation of nuclear Ca2+ regulation overall, desirable for signaling extending over hours rather than seconds to minutes. To be activated by CaMKK, CaMKIV must be bound to Ca2+/CaM. In one scenario, βCaMKK could locally transfer Ca2+/CaM to CaMKIV while retaining most of its catalytic activity, a known feature of this enzyme (Anderson et al., 1998; Edelman et al., 1996; Tokumitsu and Soderling, 1996). Consistent with such an intranuclear handoff, buffering of nuclear free Ca2+/CaM by CaMBP4nuc completely inhibited chronic inactivity-induced Nova-2 translocation (Figures 4D and 4E).
Roles of CaMKIV in Diverse Aspects of Homeostatic Plasticity
CaMKIV has been identified previously as a key player in homeostatic plasticity of excitatory neurons by Ibata et al. (2008) and Joseph and Turrigiano (2017), who found that the synaptic response to 4-h TTX treatment, a relatively brief period of inactivity, could be mimicked by CaMKIV inhibition using STO-609 or overexpression of a dominant-negative CaMKIV. Those findings might seem at odds with enhanced pCaMKIV in neurons undergoing chronic inactivity (Figures 4 and 7). However, reconciliation may be possible based on the dynamics of the homeostatic response. During TTX treatment for 24–48 h, Kim and Ziff (2014) found initial inhibition of Ca2+ signaling for at least 6 h, followed by late activation at 48 h, indicating that homeostatic modulation occurs in two phases. Perhaps early inhibition of CaMKIV, together with inhibition of calcineurin, contributes to up-scaling of AMPARs, critical for activation of synaptic NMDARs and CaV1; with further inactivity, enhanced synaptic events lead to recruitment of calcium signaling and enhanced nuclear pCaMKIV, vital for regulation of APD.
Regulation of AS by CaMKIV has been studied previously in the context of acute depolarization, acting at conserved CaMKIV-responsive RNA element (CaRRE) sequences in target precursor mRNAs (pre-mRNAs) (Iijima et al., 2011; Lee et al., 2007; Liu et al., 2012; Xie and Black, 2001). We looked for regulation of inclusion of stress-axis-regulated (STREX) exon (X4 in our study) and for CaRRE sequences flanking E29 but did not find either (Figure S3). This is not surprising in light of evidence that multiple activity-dependent splicing factors (hnRNP L, SAM68, and related STAR [signal transduction and activation of RNA] family members) have specific effects on individual pre-mRNA targets (Iijima et al., 2011). For Nova-2 and other splicing factors, puzzles remain regarding how selective control could be exerted if the splicing events are controlled by the same regulator, CaMKIV. Possible differences in the dynamics and localization of specific splicing events need further study.
Similar to Rbfox1 (Lee et al., 2009), Nova function is regulated by cytonuclear relocation (Racca et al., 2010). Nova-2 positioning is a U-shaped function of activity level, with nuclear exit favored by hyperactivity (seizure induction [Eom et al., 2013] or sustained depolarization; Figure S7E) or chronic inactivity. This echoes the U-shaped relationship of CaMKIV activation to activity. Although we focused on Nova-2 actions in the nucleus, Nova-2 has also been found in dendrites, co-localized with its target mRNAs (Racca et al., 2010). Outside of the nucleus, splicing factors might facilitate translocation of their binding partners (e.g., CaMKIV) and regulate the stability and translation of their target mRNAs (see Lee et al., 2016, for such a role of Rbfox1).
Implications for Autism, Schizophrenia, and Other Neuropsychiatric Diseases
In the specific feedback loop we circumnavigated (Figures 7M and 7N), each element is genetically implicated in autism spectrum disorder (ASD), schizophrenia, or other neuropsychiatric disorders. First, L-type Ca2+ channels have been implicated repeatedly in ASD and schizophrenia (Bhat et al., 2012; Purcell et al., 2014; Splawski et al., 2004, 2005) and affect downstream signaling of many forms (Deisseroth et al., 2003). Second, βCaMKK (gene name CAMKK2) exhibits sporadic mutations in individuals with schizophrenia with severe biochemical effects (Luo et al., 2014; O’Brien et al., 2017) and affect multiple target kinases (Marcelo et al., 2016). Third, Nova-2 mediates splicing of hundreds of pre-mRNAs, encoding proteins prominent at synaptic sites (Saito et al., 2016; Ule et al., 2005) whose patterns are altered in Fragile X, a form of ASD (Kuwano et al., 2011; Lewis et al., 2000). Finally, KCNMA1, encoding the BK channel, is causally involved in certain sporadic forms of ASD (Laumonnier et al., 2006), mental retardation, schizophrenia, and epilepsy (Du et al., 2005; Higgins et al., 2008; Zhang et al., 2006); inclusion of BK exon E29 is significantly lower in ASD samples than in matched controls (Parikshak et al., 2016). Together, these findings suggest that individual steps in the adaptive feedback pathway not only contribute to homeostatic regulation but might also go awry in neuropsychiatric disorders (Mullins et al., 2016; Figure 7N). In pinpointing players in E-AS coupling that seem to support pathogenesis, our results align with generally altered splicing in ASD (Parikshak et al., 2016), possibly arising from this kind of regulatory loop or side branches from it. Thus, correction of faulty E-AS coupling merits consideration as a therapeutic strategy (Hébert et al., 2014).
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be addressed to Lead Contact, Richard W. Tsien (richard.tsien@ nyulangone.org)
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293 cell lines (human, female) were purchased from the American Type Culture Collection (ATCC). Cells were cultured in standard Dulbeco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine. All cells used were negative for mycoplasma. The cell lines were authenticated by the source repository.
Primary cell cultures
Cortical neurons were cultured from postnatal day 0 (P0) male and female Sprague-Dawley rat pups as previously described (Li et al., 2016; Ma et al., 2014). Mixed cohorts of female and male pups were used for all experiments to minimize gender effects. The frontal cortex was isolated and washed twice in ice-cold modified HBSS (4.2 mM NaHCO3 and 1 mM HEPES, pH 7.35, 300 mOsm) containing 20% fetal bovine serum (FBS; Hyclone, Logan, UT). Samples were washed and digested for 30 min in a papain solution (2.5 mL HBSS + 145 U papain + 40 μL DNase) at 37°C with gentle shaking every 5 min. Digestion was stopped by adding 5 mL of modified HBSS containing 20% fetal bovine serum. After additional washing, the tissue was dissociated using Pasteur pipettes of decreasing diameter. The cell suspension was pelleted twice, filtered with a 70 μm nylon strainer, and plated on 10 mm coverslips coated with poly-D-lysine. The cultures were maintained in NbActiv4 (BrainBits, Springfield, IL), at 37°C in a 5% CO2 incubator. Half of the media was changed at 7 days, and once per week thereafter.
Animals
C57BL/6 male and female mice (postnatal day 26), purchased from the Charles River Laboratory, were used for monocular deprivation experiments. The Olig3Cre mouse line was a gift from Y. Nakagawa, University of Minnesota. R26floxstop-TeNT (tetoxf/f) mouse line was a gift from M. Goulding at the Salk Institute for Biological Studies. These two mouse strains were maintained on a mixed background (Swiss Webster and C57/B16). All mice were housed with a 12 hour light-dark cycle. Mixed cohorts of female and male mice were used for all experiments to minimize gender effects. Animal protocols were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee at New York University and Sun Yat-sen University.
METHOD DETAILS
Constructs
Nova-2 mutants were produced by Phusion Site-Directed Mutagenesis Kit (Thermo Scientific). BK channel without E29 was first cloned from pcDNA3-BK-GFP (Li et al., 2014) via PCR and inserted into pCKII-GFP construct with an αCaMKII promoter to restrict expression to pyramidal cells (Li et al., 2016). E29 sequence was synthesized and inserted by Phusion Site-Directed Mutagenesis Kit (Thermo Scientific). The splice reporter containing E29 and partial flanking intron sequences were cloned by PCR from genomic DNA obtained from rat brain tissue and inserted into pFlare5 vector (Stoilov et al., 2008). Other constructs, see Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit pCaMKII | Cell Signaling Technology | Cat#3361;RRID:AB_10015209 |
| Rabbit pCaMKIV | Abcam | Cat# ab59424;RRID:AB_2068253 |
| Rabbit pCaMKI | Santa Cruz | Cat#sc-28438-R;RRID:AB_667968 |
| Goat anti-βCaMKK | Santa Cruz | Cat#sc-9629;RRID:AB_2243844 |
| Rabbit anti-βCaMKK | Santa Cruz | Cat#sc-50341;RRID:AB_2068532 |
| Mouse anti-CaMKIV | Santa Cruz | Cat#sc-55501;RRID:AB_2243836 |
| Mouse anti-CaMKIV | Santa Cruz | Cat#sc-136249;RRID:AB_2275109 |
| Mouse anti-flag (DYKDDDDK Tag) | Cell Signaling Technology | Cat#8146;RRID:AB_10950495 |
| Rabbit anti-PSD95 | Synaptic Systems | Cat#124002; RRID:AB_887760 |
| Mouse anti-PSD95 | Synaptic Systems | Cat#124011; RRID:AB_10804286 |
| Goat anti-Nova-2 | Santa Cruz | Cat#sc-10546;RRID:AB_2151558 |
| Human anti-pan NOVA | anti-Nova paraneoplastic human serum | Saito et al., 2016 |
| Mouse anti-MAP2 (HM-2) | Sigma | Cat#M9942;RRID:AB_477256 |
| Mouse anti-CaM | Millipore | Cat#05–173;RRID:AB_309644 |
| Goat anti-γCaMKII | Santa Cruz | Cat#sc-1541;RRID:AB_2068234 |
| Mouse anti-CaMKI | Santa Cruz | Cat#sc-377418;RRID:AB_2069999 |
| Goat anti-βCaMKI | Santa Cruz | Cat#sc-131452;RRID:AB_2243992 |
| Rabbit anti-δCaMKI | Santa Cruz | Cat#sc-134638;RRID:AB_2070115 |
| Mouse anti-γCaMKI | Abcam | Cat#ab77046;RRID:AB_1565944 |
| Rabbit anti-γCaMKI | Thermo Scientific | Cat#PA5–19661;RRID:AB_10981875 |
| Mouse anti-αCaMKK | Santa Cruz | Cat#sc-17827;RRID:AB_2275110 |
| Rabbit anti-αCaMKK | Santa Cruz | Cat#sc-11370;RRID:AB_2068406 |
| Rabbit anti-GluR1 | Calbiochem | Cat# 04–855;RRID: AB_1977216 |
| Rabbit anti-BK channel (extracellular epitope) | Alomone labs | Cat#APC151;RRID:AB_10915895 |
| Mouse anti-PSD-95 | UC Davis/NIH NeuroMab facility | N/A |
| Guinea anti-MAP2 | Synaptic Systems | Cat# 188004;RRID:AB_2138181 |
| Rabbit anti-GFP | Abcam | Cat# ab290;RRID:AB_303395 |
| Rabbit anti-pCaMKII | PhosphoSolutions | Cat# p1005–286; RRID:AB_2492051 |
| Rabbit anti-Lamin-B1 | Cell Signaling Technology | Cat#13435;RRID:AB_2737428 |
| Rabbit anti-GAPDH | Cell Signaling Technology | Cat#5174;RRID:AB_10622025 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Tetrodotoxin (TTX) | Ascent Scientific | Cat#4368–28-9 |
| KN93 | Tocris | Cat#5215 |
| STO-609 | Tocris | Cat#1551 |
| PhTx | Tocris | Cat#2770 |
| APV | Tocris | Cat#0106 |
| nimodipine | Abcam | Cat#ab120138 |
| thapsigargin | Tocris | Cat#1138 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Phusion Site-Directed Mutagenesis Kit | Thermo Scientific | Cat# F541 |
| Dynabeads Protein G Immunoprecipitation Kit | Life Technologies | Cat#10007D |
| Magnetic RNA-Protein Pull-Down Kit | Thermo Scientific | Cat#20164 |
| Magna RIP RNA-Binding Protein Immunoprecipitation Kit | Sigma | Cat#17–700 |
| Pierce Cell Surface Protein Isolation Kit | Thermo Scientific | Cat#89881 |
| Pierce Nuclear Protein Extraction Kit | Thermo Scientific | Cat#78833 |
| CelLytic M Cell Lysis Reagent | Sigma | Cat#C3228 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse: Olig3-Cre mouse | gift from Y. Nakagawa, University of Minnesota | N/A |
| Mouse: R26floxstopTeNT | gift from M. Goulding at the Salk Institute for Biological Studies | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Probes and shRNAs, see Table S1. | This paper | N/A |
| Primers for RT-PCR and qPCR, see Table S1 | This paper | N/A |
| Primers to insert E29 and flanking introns to the splicing reporter: F: CCGGAATTCCGGC TATGTGGCAACCCTAC | This paper | N/A |
| Primers to insert E29 and flanking introns to the splicing reporter: R: CGCGGATCCGCGT CTCCTTTGACTTCCTCT | This paper | N/A |
| Primers to measure E29 splicing in the splicing reporter: F: GGAGAAGTCTGCCGTTACTGCCC TGTG (DY-782 labeled) |
This paper | N/A |
| Primers to measure E29 splicing in the splicing reporter: R: CCGTCGTCCTTGAAGAAGATGGTGC | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| mouse βCaMKK construct: Lentiviral CaMKKbeta | Green et al., 2011b | Addgene Plasmid #33322; RRID:Addgene_33322 |
| rat βCaMKK 1–460 construct: pSG5-FLAG-CaMKKbeta rat 1–460 | Green et al., 2011a | Addgene Plasmid #33324; RRID:Addgene_33324 |
| Human Nova-2 ORF | Origene | Cat# RC216200L1V, RC216200L2V |
| pcDNA3-BK-GFP | Li et al., 2014 | N/A |
| pCKII-GFP | Li et al., 2016 | N/A |
| BK channel without E29 | This paper | N/A |
| BK channel with E29 | This paper | N/A |
| splice reporter pFlare5 vector | Stoilov et al., 2008 | N/A |
| pGFP-C-shLenti βCaMKK shRNA constructs | Origene | Cat#TL711303 |
| pGFP-C-shLenti Nova2 shRNA constructs | Origene | Cat#TL508674 |
| pcDNA3.1/Puro-CAG-ASAP1 | St-Pierre et al., 2014 | Addgene Plasmid # 52519; RRID:Addgene_52519 |
| AAV-CaMKIIa-GCaMP6s-P2A-nls-tdTomato | Gift from Jonathan Ting (unpublished) | Addgene Plasmid #51086; RRID:Addgene_51086 |
| CaMKIV-GFP construct | Gift from Haruhiko Bito | N/A |
| CA-CaMKIV construct | Gifts from Tian-Ming Gao | N/A |
| CaMBP4 construct | Gifts from Tian-Ming Gao | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| pClamp 9 | Molecular Devices | https://www.moleculardevices.com/ |
| Prism | GraphPad | https://www.graphpad.com/ |
| MATLAB | Mathworks | https://www.mathworks.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
Transfection and treatment of cortical neurons
Neurons were transfected 7 to 9 days after plating using a high efficiency Ca2+-phosphate transfection method (Jiang and Chen, 2006). Experiments were performed 12–14 days after plating. For chronic spike blockade, neurons were treated with 1 μM TTX on DIV12 for 48 hours to prevent action potential. Where indicated, additional antagonists (5 μM nimodipine, 4 μM KN93 or 3 μM STO-609) were added with TTX. For chronic depolarization, neurons were treated with 20 mM K+ (20K), 40K or 60K solution (to depolarize cells; Na+ adjusted to maintain osmolarity) for 12 or 24 h. For action potential recording by patch clamp, coverslips with control or TTX-treated neurons were removed from the culture medium, washed and recorded in TTX-free artificial cerebrospinal fluid (ACSF). For imaging the action potential-independent Ca2+ transients and voltage changes in the dendrites, control or TTX-treated neurons were transferred to a TTX-containing Tyrode’s solution consisting of (in mM): 150 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, pH 7.4 with 1 μM TTX. Where indicated, antagonists were added by manual pipetting to achieve a final concentration of 10 μm PhTx, 10 μm APV, 5 μM nimodipine or 1 μM thapsigargin in TTX-containing Tyrode’s solution.
Lentiviral transduction of cortical neurons
The production of lentivirus was performed as previously described (Ma et al., 2014), pGFP-C-shLenti constructs encoding shRNAs against Nova-2 or βCaMKK (OriGene) were transfected into 293T cells along with the packaging plasmid psPAX2 and the envelope plasmid pMD2.g. After 16 h, the medium was changed and the supernatant was collected 24 h later and cleared of cell debris by filtering through a 0.45 mm filter. The viral particles were concentrated by centrifuging the filtrate at 70,000 × g for 2 hr at 4°C using a Beckman SW28 rotor. The viral pellet was then resuspended in sterile PBS, aliquoted, and stored at −80°C. Lentivirus particles (0.5–1 μL of viral stock diluted in 20 μL of PBS per coverslip) were added to cortical cultures containing 500 μL of medium on DIV7. The experiments with overexpressed proteins or shRNAs were performed on DIV14.
Immunocytochemistry and image acquisition and analysis
Cells were fixed in ice-cold 4% paraformaldehyde in phosphate buffer with 20 mM EGTA and 4% sucrose; permeabilized with 0.1% Triton X-100; blocked with 10% normal goat serum (or donkey serum); and incubated overnight at 4°C in primary antibodies. For surface staining of GluR1 or BK channel, coverslips were fixed and blocked in 7.5% normal donkey serum for 30 minutes in the absence of Triton X-100. Surface staining with primary antibodies (Key Resources Table) was then performed for 1 hour at room temperature. Cells were then permeabilized in 0.1% Triton X-100, and stained with anti-PSD-95 and anti-MAP2 (Figure S6) overnight at 4°C. The next day, cells were washed with PBS, incubated at RT for 60 min with Alexa secondary antibodies (1:1000, Molecular Probes), washed again and mounted with ProLong Gold + DAPI (Invitrogen).
Fixed cells were imaged with a 40X or 60X oil objective on a Zeiss LSM 800 confocal microscope. Intensity quantification was performed with custom scripts in MATLAB (Mathworks) or ImageJ (NIH). For all analyses, a region of interest lacking cells was selected in each field of view as an ‘off-cell’ background, and the mean intensity was subtracted from all cellular regions of interest for each color channel. The following types of analyses were performed from at least three independent cultures in each case:
Analysis of Nova-2, pCaMKIV, βCaMKK and flag-tagged Nova-2 (e.g., Figures 3 and 4). Nuclear and cytosolic regions of interest were manually drawn while viewing only DAPI or MAP2 channels, but blinded to the Nova-2 color channel. Background-subtracted mean intensity was quantified and normalized to control conditions.
Analysis of pCaMKII, pCaMKI and surface GluR1 intensity (e.g., Figure S6) was restricted to MAP2-positive regions of interest containing a proximal dendrite (~60 μm in length, ~15 μm in width) at least 30 μm from cell soma. For GluR1, PSD-95 puncta were identified, and the average intensity of GluR1 signal intensity measured in the region of those puncta. For pCaMKII and pCaMKI, the regions of interests were manually drawn while viewing MAP2 channel but blinded to the pCaMKII color channel. The background was subtracted from pCaMKII intensity. Data represents mean ± SEM over 20 such dendrites, normalized to control conditions.
Transfection and electrophysiology of HEK293 cells
HEK293 cells were transfected via a high efficiency Ca2+- phosphate transfection method (Jiang and Chen, 2006), with constructs encoding CMV promoter-driven BK channel with (E29) or without (ΔE29) E29, or with splicing reporter constructs co-expressed with or without WT or mutant Nova-2, or with CaMKIV-GFP and flag-Nova-2 constructs, or with CA-CaMKIV and flag-Nova-2 constructs. Experiments were done 2–3 days later. Whole-cell recordings were obtained at room temperature (Axopatch 200B, Molecular Devices). Electrodes were pulled borosilicate glass capillaries (World Precision Instruments, MTW 150-F4), with 5–8 MΩ resistances, before 80% series resistance compensation.
Electrophysiological recording of action potential half-width in cultured cortical neurons
DIV 10–14 cultured cortical neurons were used to measure homeostatic changes in action potential waveforms. Cultured cortical neuron coverslips were placed into a submerged recording chamber and perfused with artificial cerebrospinal fluid (ACSF) at 4 mL/min held at room temperature (20–25°C) and bubbled with 95%/5% O2/CO2. All cells were measured in current clamp mode using borosilicate patch electrodes with tip resistance of 2–4 MΩ. Recordings were not corrected for the liquid junction potential. Data were sampled at 10 kHz and low pass filtered at 2 kHz using a Bessel filter in pClamp 9 software. Recordings were not used if the access resistance was above 25 MU or changed significantly throughout the recording (> 20%).
The ACSF contained (in mM): 3 KCl, 10 D-Glucose, 122 NaCl, 1.25 NaH2PO4, 1.3 MgCl2, 2 CaCl2, 26 NaHCO3. The internal pipette solution contained (in mM): 130 K Gluconate, 1 MgCl2, 10 HEPES, 0.3 EGTA, 10 Tris-Phosphocreatine, 4 Mg-ATP, 0.3 Na-GTP.
To minimize changes in action potential duration due to depolarizing current steps, all action potentials were evoked as a rebound spike (anodal break response) (Hodgkin and Huxley, 1952). Briefly, a 500 ms long pulse between —500 pA and —800 pA was injected into the neuron before it was allowed to rebound to resting potential. Upon return to resting potential a single action potential was usually evoked and sweeps where an action potential was successfully evoked were used for analysis. Using this method allows an action potential to be evoked from a membrane potential negative enough to maximize removal of inactivation and minimize the influence of variations in resting potential.
Action potential half width was measured using custom MATLAB scripts. Briefly, the action potential amplitude (baseline to action potential peak value) was measured and the time between the half peak amplitude on the rising and falling phases was taken to be the action potential half width.
Recording Current-Voltage relationships in HEK cells transfected with BK channels
For recordings in HEK cells a symmetrical solution (containing equal ionic concentrations in the internal pipette solution and external solution) was used containing (in mM): 10 K Gluconate, 2 KCl, 2 MgCl2, 10 HEPES, 9.93 CaCl2, 10 EGTA, 10 Glucose. A current-voltage (I-V) relationship was measured in voltage clamp mode with 250 ms long voltage steps between —100 mV and 100 mV in 10 mV increments. Custom MATLAB scripts were used offline to take current measurements when the membrane current stabilized after voltage steps.
Ca2+ imaging on dendrite and soma of cultured neurons
Neurons were transfected with GCaMP6s on DIV 7–9 and Ca2+ transients were imaged on DIV13 to 15. Images were collected with a 40X oil objective on a Zeiss 710 confocal microscope, 300 ms per frame. Intensity quantification was performed with custom scripts in MATLAB (Mathworks).
For imaging of Ca2+ transients in soma, the 488 nm fluorescence changes were quantified using regions of interest in cell soma (Figure 1B). Ca2+ transients were imaged in TTX-free Tyrode’s solution, consisting of (in mM): 150 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, pH 7.4. The amplitude of single action-induced Ca2+ transient was defined by the minimal amplitude of single Ca2+ transient that could propagate to the entire cell (soma and dendritic tree). The Ca2+ transients that arose locally in the soma but did not propagate to the dendrite was regarded as local under-threshold depolarization-induced Ca2+ transients and was eliminated from the quantification. For imaging the action potential-independent Ca2+ transients in the soma, control or TTX-treated neurons were imaged in a TTX-containing Tyrode’s solution.
For Ca2+ transients in dendrite, a ~200 μm long dendrite located at least 30 μm from cell soma was chosen. Ca2+ transients were imaged in TTX-containing Tyrode’s solution. Where indicated, antagonists were added by manual pipetting to achieve a final concentration of 10 μm PhTx, 10 μm APV, 5 μM nimodipine or 1 μM thapsigargin in TTX-containing Tyrode’s solution.
In dendritic trees, sites of spontaneous Ca2+ influx were identified as regions of interest (ROIs) which showed sparse peaks of GCaMP6s fluorescence through time, many of which spatially overlapped with spine-like protrusions in baseline GCaMP6s images highlighting dendritic trees structure (Figures 6D and 6F). ROI shape and number were obtained by automatically computing a series of operations. First, temporal stacks of fluorescence images were de-noised and the baseline GCaMP6s fluorescence was subtracted pixel-wise. Noise elimination was performed using a custom method that kept intact the non-random correlation between neighboring pixels showing coordinated activity in order to preserve localized signals from Ca2+ influx. An overly large set of putative ROIs were then identified by using the separable non-negative matrix factorization (sNMF) algorithm. sNMF works by identifying strong but temporally sparse signals and preventing the temporal redundancy between extracted signals - a good model for spontaneous Ca2+ signals in dendrites. The final ROI set was obtained by unbiasedly selecting only those with cumulated temporal fluorescence greater than the average + 3 × the standard deviation of cumulated fluorescence values drawn outside candidate ROIs.
Voltage imaging of cultured neurons
Cultured pyramidal neurons were transfected via a high efficiency Ca2+- phosphate method 7–9 days after plating (Jiang and Chen, 2006), with 1 μg pcDNA3.1/Puro-CAG-ASAP1 plasmid (St-Pierre et al., 2014). Experiments were done 5–7 days later. In voltage imaging data in Figures 6A–6C, the spontaneous AP in soma and bAP in dendritic spines were monitored by simultaneously measuring the ΔF/F of voltage sensor ASAP1 in soma and dendritic spines. We reasoned that 1) only action potential, instead of subthreshold synaptic current, could backpropagate from soma to dendritic trees and elicit simultaneous voltage changes in soma, different dendritic branches and dendritic spines; 2) due to the high on and off kinetics of ASAP1 (~2ms), high speed recording (2.87 ms / frame in our experiments) of the ΔF/F changes could reliably detect single spontaneous action potential; ΔF/F changes induced by single action potential were comparable in both amplitude and duration; ΔF/F changes induced by burst of action potentials were much larger in amplitude and duration and were discarded in our analysis. Accordingly, we named the simultaneous ΔF/F changes (voltage changes) in soma and spines as “AP in soma” and “bAP in spine,” respectively, and named the ΔF/F changes (voltage changes) that took place only in spines as “Vspine.” When measuring Vspine, TTX was acutely applied to eliminate action potentials. A narrow region of interest containing the plasma membrane of dendritic spines (30–80 μm from soma) and cell soma were chosen, and time lapse imaging with a speed of 2.87 ms per frame was performed on a Zeiss LSM 710 confocal microscope, utilizing GFP filter sets. Background-subtracted fluorescence intensity were analyzed in ImageJ (NIH). ASAP1 waveforms were plotted as −ΔF/F (to account for the negative change in ASAP1 fluorescence upon depolarization) and corrected for bleaching. The noise was filtered with LOWESS smoothing. Identical cell-selection, bleaching correction and noise-filtering parameters were used for all datasets in Figure 6. Data is plotted as mean ± SEM of 14–15 cells for each condition.
To calculate the standard fluorescence-voltage (ΔF/F-DV) curve for ASAP1, neurons were perfused high K+ Tyrode’s solutions with Na+ adjusted to maintain osmolarity. The change of fluorescence intensity (-ΔF/F) was plot against voltage according to 4 mM K+ (4K), −60 mV; 20K, −37 mV; 40K, −19 mV; 60K, −9 mV; 90K, 0 mV (Wheeler et al., 2012). The membrane potentials in different conditions were then calculated according to the peak values of −ΔF/F.
Immunoprecipitation
Immunoprecipitation was performed with Dynabeads Protein G Immunoprecipitation Kit (Life Technologies) following the kit protocol as described (Li et al., 2016). Briefly, for binding the antibody with the beads, 5 μg of antibody or control IgG was crosslinked and incubated at room temperature with 100 μL Dynabeads. The supernatant was removed with the help of a magnet to retain the bead-antibody complex, which was then washed with Ab Binding & Washing Buffer. For lysate preparation, cultured cortical neurons or HEK293 were washed with PBS and lysed on ice with IP Lysis Buffer (Pierce) containing protease and phosphatase inhibitors. The lysate was centrifuged at 16,000 g for 10 min at 4°C to pellet the cell debris. The resulting supernatant was incubated with bead-antibody complex with rotation for 30 min at room temperature. The bead-antibody-antigen complex was then washed 3 times using washing buffer. The protein complex was eluted with Elution buffer and the eluent was mixed and heated with 2 × SDS sample buffer containing β-mercaptoethanol at 90°C for 10 min.
Protein sample preparation and western blot
Nuclear and cytosolic protein fraction was performed as described (Li et al., 2014). Briefly, nuclear and cytosolic protein was extracted using the Pierce Nuclear Protein Extraction Kit (Thermo), following the manual. Neurons were collected by centrifugation at 500 g for 3 min at 4°C and were resuspended in 200 μL precooled CER I by 15 s of intense vortexing. The solution was then incubated on ice for 10 min. CER II (11 μl) was precooled and added to the solution. After 5 s of intense vortexing, the solution was incubated on ice for 1 min. After another 5 s of intense vortexing, the solution was then centrifuged at 16,000 g for 5 min at 4°C and the supernatant collected as cytosolic protein. NER (100 μl) was added to the precipitate and the sample vibrated intensively for 15 s. The solution was then incubated on ice for 10 min. The vibration and incubation were repeated four times, and then the precipitate was centrifuged at 16,000 g for 10 min at 4°C. The supernatant represented purified nuclear protein.
Plasma membrane protein extraction was performed with Pierce Cell Surface Protein Isolation Kit as described (Li et al., 2014). For surface biotinylation, neurons were chilled on ice, washed twice with ice-cold PBS, and then incubated with PBS dissolved sulfo-NHS-SS-biotin for 30 min at 4°C. Unreacted biotin was quenched by washing cells with Quenching Solution. Cultures were harvested in Lysis Buffer. Homogenates were centrifuged at 10,000 g for 2 min at 4°C. The resulting supernatant was rotated 1 hr at room temperature with NeutrAvidin Agarose. The beads were washed with Wash Buffer and analyzed by immunoblotting with each antibody.
Whole cell lysate was performed as previously described (Li et al., 2016). Cells were lysed with CelLytic M Cell Lysis Reagent (Sigma). Protein concentration was measured by the BCA Protein Assay Kit (Thermo). The lysates were combined with 2 × SDS loading buffer, then boiled for 10 min.
Protein samples were loaded onto 4%–20% gradient precast or 10% SDS-PAGE gel (Bio-Rad). Proteins were transferred to a PVDF membrane (Millipore) for 1 h at 350 mA, and membranes were incubated in Odyssey Blocking Buffer (Li-Cor) for 1 h at room temperature. The membrane was incubated overnight at 4°C with primary antibodies. The blots were washed three times in PBS containing 0.1% Tween-20 for 15 min and then incubated with IRDye-conjugated secondary antibody for 1 h in PBS, 0.1% Tween-20 at room temperature. Immunoreactivity was detected by an Odyssey imaging system (Li-Cor).
Oligo-RNA pull-down
RNA pull-down was performed with Magnetic RNA-Protein Pull-Down Kit (Thermo) following the manual. Briefly, synthesized 5′-biotinylated 2′-OMe-RNA oligonucleotides (Eurofins) (Key Resources Table) were bound to streptavidin magnetic beads (Thermo) in RNA Capture Buffer at room temperature with agitation. Cleared brain lysates from 1-month old mouse cortex (Figure 2B) or transfected HEK cells (Figure S5E) prepared in IP Lysis Buffer (Pierce) containing protease and phosphatase inhibitors mixed with Protein-RNA Binding Buffer, and then incubated with the packed beads at 4°C for 1 hr. Beads were washed three times with wash buffer and the precipitate was subjected to immunoblot analysis. Signal intensities were quantified by an Odyssey imaging system (Li-Cor).
RNA immunoprecipitation
RNA immunoprecipitation was performed with Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Sigma) following the manual. Briefly, 5 μg of anti-Nova-2 antibody or control IgG were incubated with magnetic beads in RIP wash buffer at room temperature for 30 mins. The antibody prebound beads were then washed 3 times with RIP wash buffer at room temperature. For lysate preparation, mouse cortical lysates were prepared with complete RIP Lysis Buffer. The lysates were mixed with prebound beads in RIP Immunoprecipitation Buffer containing RIP wash buffer, EDTA and RNase inhibitor, overnight with rotation at 4°C. The magnetic beads antibody-RNA binding protein complex was washed with wash buffer and then re-suspended in proteinase K buffer at 55°C for 30 minutes. Phenol:chloroform:isoamyl alcohol and chloroform were used to separate the phase and the RNA was precipitated with Salt Solution I, Salt Solution II, Precipitate Enhancer and absolute ethanol at −80°C overnight. The RNA precipitation was centrifugated and washed with 80% ethanol solution. The pellets were dried and resuspended in RNase-free water and used for further RT-PCR.
RT-PCR and Realtime qPCR
Total RNA was isolated from cortical neurons or HKE 293 cells with RNeasy Mini kit (QIAGEN) according to manufacturer’s instructions. Extracted RNA was reverse transcribed into first strand cDNA using oligo dT and Superscript III (Invitrogen). PCR was performed with AmpliTaq Gold® 360 Master Mix with DY-682 labeled primers (for X6). The PCR products were either resolved on agarose gel (X1-X5) or denatured and resolved on 6% polyacrylamide/8 M urea denaturing gels (X6). The band density was analyzed by an Odyssey imaging system (Li-Cor). qPCR was performed with SYBR-green PCR master mix (Fermentas) with primers (Table S1) using the DNA engine Opticon 2 (Bio-Rad).
Thalamic input elimination and immunohistochemistry
The Olig3Cre mouse line was a gift from Y. Nakagawa, University of Minnesota. R26floxstop-TeNT (tetoxf/f) was a gift from M. Goulding at the Salk Institute for Biological Studies. Neurotransmission is selectively blocked from the thalamic cells as expression of Cre removes a stop codon at the R26 locus (removing the flox sites) permitting the expression of the tetanus toxin light chain subunit (TeNT) only within thalamic Olig3+ neurons. Olig3Cre mice were bred with animals that were homozygous for tetoxf/f. Pups from the subsequent litters were genotyped for Cre and presence of the TeNT allele in the R26 locus. Olig3Cre-only animals were used as controls and Olig3Cre plus tetoxf/+ heterozygous animals as mutants. All mouse strains were maintained on a mixed background (Swiss Webster and C57/B16).
Mice were perfused inter cardiac with 4% PFA after being anesthetized either on ice or using Sleepaway IP administration. Brains were post-fixed for 30 minutes and cryopreserved in 30% sucrose following the perfusion and brain harvest. 16μm coronal sections were obtained using Cryostat (Leica Biosystems) and collected on super-frost coated slides, then allowed to dry overnight and stored at −20°C until use. For immunofluorescence, cryosections were thawed and allowed to dry for 5–10 min and rinsed twice in 1x PBS. They were incubated at room temperature in a blocking solution of PBST (PBS-0.1%Tx-100) and 10% normal donkey serum (NDS) for 60min, followed by incubation with primary antibodies in PBS-T and 1% NDS at 4°C overnight. Samples were then washed 3 times with PBS-T and incubated with fluorescence conjugated secondary Alexa antibodies (Life Technologies) in PBS-T with 1% NDS at room temperature for 60–90min. Slides were then incubated for 30 s with DAPI, washed 3 times with PBS-T and once with PBS. Slides were mounted with Fluoromount G (Southern Biotech) and imaged on a Zeiss LSM 510 Laser scanning microscope.
Monocular deprivation
The monocular deprivation was performed following previous study (Ma et al., 2013). Briefly, the mice (postnatal day 26) were anesthetized with the mixture of 100 mg/kg ketamine hydrochloride and 10 mg/kg xylazine hydrochloride intraperitoneally. Under microscope and illuminator, the eyelashes and the edge of eyelids were cut using the spring scissors. The eyelids of the right eye were sutured with three mattress sutures. A thin layer of xylocaine 2% Jelly and bacitracin zinc ointment was applied to the sutured eyelids. After 5 days monocular deprivation, mice brains were sliced for immunostaining or RNA isolation. Nova-2 localization or E29 splicing in the monocular region of primary cortex V1, both contralateral and ipsilateral to the visual deprived eye, was analyzed.
Protein sample preparation and mass spectrometry analysis
Protein sample preparation and mass spectrometry analysis were perform as described (Ma et al., 2014). Briefly, flag-tagged Nova-2 were co-transfected with CA-CaMKIV or GFP into HEK293 cells. 48 h after transfection, the cells were washed with ice-cold PBS and lysed with IP Lysis buffer (Thermo Fisher) containing a protease inhibitor cocktail tablet (Roche, Indianapolis, IN). The lysates were mixed with CaM kinase reaction buffer (Enzo Life Sciences) and incubated with anti-flag antibody (Cell Signaling Technology) and Protein A/G Sepharose beads. The immunoprecipitated complex was separated by SDS-PAGE. Corresponding areas showing Nova-2 bands were cut out from the Coomassie blue-stained SDS-PAGE gels. Gel pieces underwent in-gel digestion and the phosphopeptides were enriched by titanium dioxide (GL Sciences Inc., Japan) based on a previously published protocol (Rappsilber et al., 2007). Resulting peptides were subjected to Nano LC-MS/MS analysis using a Thermo Scientific EASY-nLC 1000 coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific). A self-packed 75μm × 25 cm reversed phase column (Reprosil C18, 3μm, Dr. Maisch GmbH, Germany) was used for peptide separation. Peptides were eluted by a gradient of 3%–30% acetonitrile in 0.1% formic acid over 60 min at a flow rate of 250 nL/min. The Q Exactive was operated in the data-dependent mode with survey scans acquired at a resolution of 50,000 at m/z 400. Up to the top 10 most abundant precursors from the survey scan were selected with an isolation window of 1.6 Thomsons and fragmented by higher energy collisional dissociation with normalized collision energies of 27. The maximum ion injection times for the survey scan and the MS/MS scans were 20 and 60 ms, respectively, and the ion target value for both scan modes were set to 1,000,000. Each sample was processed and analyzed in triplicate.
Phosphopeptide identification and quantitation
Phosphopeptide identification and quantitation was performed as described (Ma et al., 2014). Briefly, the raw files were processed using the MaxQuant computational proteomics platform version 1.2.7.0 for peptide identification and quantitation. The fragmentation spectra were searched against the Uniprot protein database allowing up to two missed tryptic cleavages. Carbamidomethylation of cysteine was set as a fixed modification, and phosphorylation of serine/threonine/tyrosine, oxidation of methionine and protein N-terminal acetylation were used as variable modifications for database searching. The precursor and fragment mass tolerances were set to 7 and 20 ppm, respectively. Quantitation of the phosphopeptides was performed using the ion intensity values calculated by MaxQuant for the quintuply charged peptides. Normalization of phosphopeptide intensities to global peptide intensities had no effect on calculated ratios.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using Prism (GraphPad Software). Means of two groups were compared using Student’s t test. One-way ANOVA was used to compare group means between more than two groups, followed by either Dunnett’s multiple comparisons test when all other groups were compared to the control group, or Tukey’s multiple comparisons test when means of each pair of groups were compared. All comparisons are two-sided. Statistical significance was determined as follows: *p < 0.05, **p < 0.01. Data are shown as mean ± SEM. The statistical details of all experiments, including the number of samples and p values, can be found in figures and figure legends.
DATA AND CODE AVAILABILITY
All data supporting the findings of this study and custom MATLAB code for calcium imaging are available upon request from the Lead Contact.
Supplementary Material
(A) Representative waveforms of action potential recorded after TTX treatment (24 or 48 h). (B) Quantification of half width of action potentials in the groups in (A) (n = 10. Data for control and 48 h groups is from Figure 1A).
{kind=link}
(A) Impact of chronic inactivity on alternative splicing of X1 to X5. Cultured cortical neurons were sham- or TTX-treated for 48 h. Separation of RT-PCR products obtained with specific primers flanking each alternatively spliced site (X1 to X5). Closed arrows indicate the longer RT-PCR products obtained if the alternative exon was included, open arrows indicate the shorter products if that exon was excluded. (B) Quantification of inclusion or exclusion of X1 to X6 by quantitative RT-PCR. (C) High K+ solution maintained long-term depolarization in resting membrane potential. (D) Impact of sustain depolarization on E29 inclusion. Cultured cortical neurons were sham- or high K+-treated (20mM, 40mM and 60mM, respectively) for 12 or 24 h. E29 inclusion was examined by RT-PCR.
{kind=link}
(A) Validation of the knockdown of Nova-2 expression by shRNAs. Cortical neurons were transfected with shRNAs either targeting the untranslated region (UTR) of the Nova-2 gene or targeting the Nova-2 coding sequence (CDS). Both shRNAs efficiently knocked down endogenous Nova-2 expression. (B) UTR-lacking flag-Nova-2 plasmids (Nova-2 (R)) were co-expressed with shRNAs targeting CDS (targeting three different regions) or shRNAs targeting UTR in HEK293 cells. The shRNAs targeting CDS efficiently knocked down flag-Nova-2 expression, whereas shRNAs targeting UTR did not. (C) E29 inclusion could only be induced by wild-type (WT) Nova-2, but not ΔNLS-Nova-2. WT-Nova-2-induced E29 inclusion was prevented by the shRNAs targeting the Nova-2 coding sequence (CDS), but not by shRNAs targeting the untranslated region (UTR) of the Nova-2 gene.
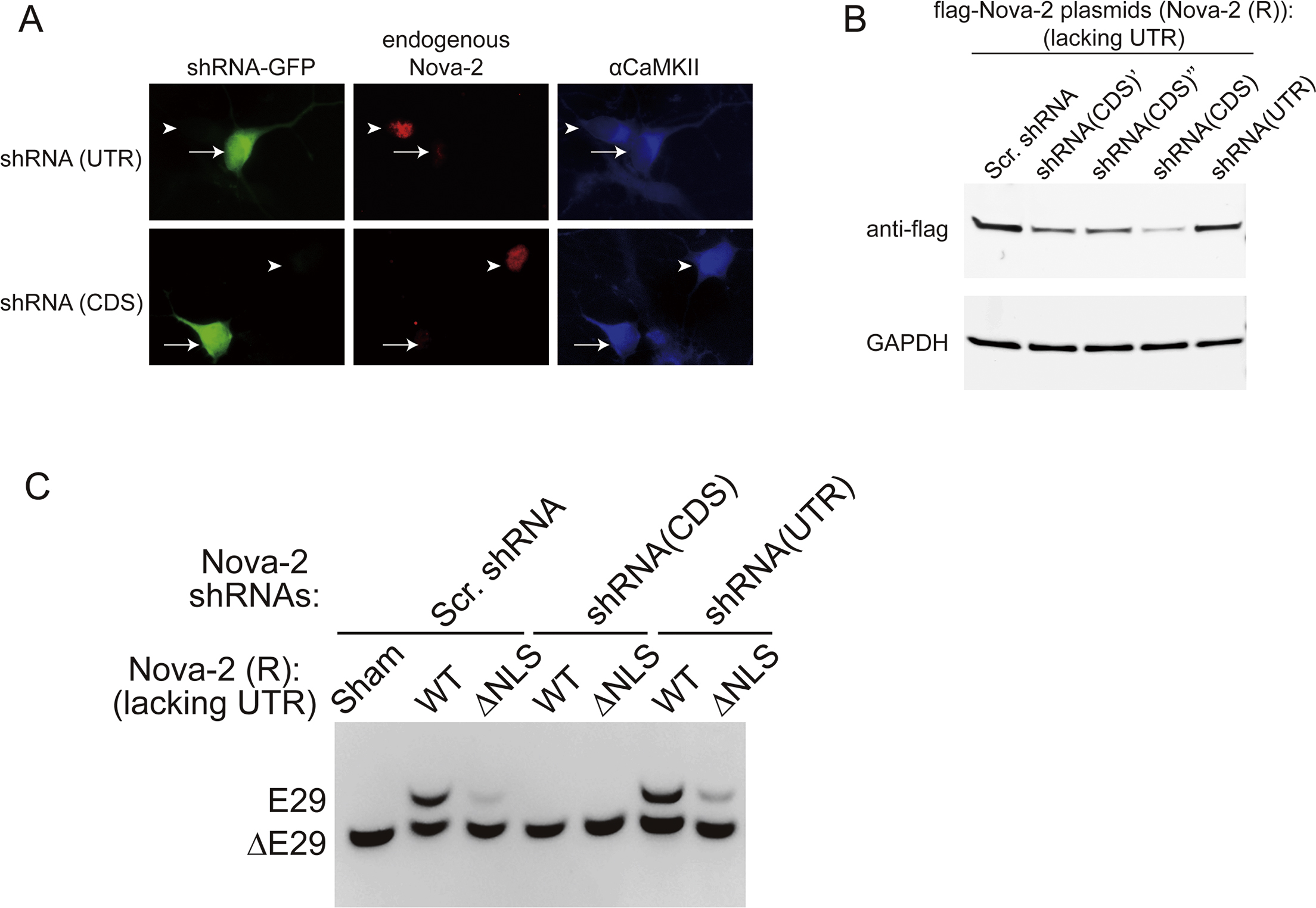{kind=link}
(A) Expression level of BK channel mRNA was assayed by RT-PCR (n = 4). (B) BK channel expression in the plasma membrane was assayed by western blotting after biotinylation-mediated membrane protein fractionation. (C) Immunofluorescence of surface BK channel. Surface BK channel was probed with antibodies targeting extracellular N terminus. (D) Quantification of intensity of surface BK channel (n = 10). (E) Overexpression of GFP-tagged BK channel isoforms in HEK293 cells. (F) Quantification of surface fluorescence intensity of GFP-tagged BK channel isoforms. (G) Expression level of GFP-tagged BK channel isoforms was assayed by western blotting after biotinylation-mediated membrane protein fractionation. Na/K ATPase, Lamin B and GAPDH are markers for plasma membrane, nucleus and cytosol.
{kind=link}
(A) Schematic diagram of Nova-2 protein as in Figure 7. (B to D) Mass spec results of phosphorylation level of S25, T27 and S194 sites. (E) WT and S194E Nova-2 are no different in RNA binding. Flag-tagged WT Nova-2 or S194E Nova-2 was overexpressed in HEK293 cells. RNA pull-down assay was performed from the cell lysates by probe A1 (Figure 2). The RNA binding ability of WT and S194E Nova-2 was assayed by western blotting with anti-flag antibody.
{kind=link}
(A) The activation of nuclear CaMKIV in different groups as indicated. Immunofluorescence of phosphorylated CaMKIV on Thr196 site indicating the level of CaMKIV activation by CaMKK. (B) Normalized nuclear/cytosolic ratio of pCaMKIV immunofluorescence intensity in each group as indicated. (C) The localization of Nova-2 in different groups as indicated. (D) Normalized nuclear/cytosolic ratio of Nova-2 immunofluorescence intensity in each group as indicated. (E) 12 h and 24 h high potassium media treatment (20mM, 40mM and 60mM, respectively) led to Nova-2 translocation from nucleus to cytosol. (F) Long-term depolarization reduced the inclusion of E29, which could be blocked by CaV1 blocker nimodipine and CaM kinases blocker KN-93. (G and H) Fold changes in abundance of CaM, αCaMKI, βCaMKI, γCaMKI, δCaMKI and γCaMKII in the nucleus, cytosol and nucleus/cytosol ratio. (G) Representative micrographs of different proteins in control or TTX-treated neurons. (H) Ratios of levels after TTX treatment (48 h) relative to levels in control neurons. (I) Validation of the knockdown of βCaMKK expression by shRNAs. flag-tagged βCaMKK was co-expressed with shRNAs targeting different locations of rat βCaMKK CDS. The efficiency of shRNAs was assayed by western blotting.
{kind=link}
(A) Ca2+ transients were imaged from the soma of control neurons, with no TTX present. (B) Ca2+ imaging, from the soma of control neurons (upper) or TTX-treated (48 h) neurons, was performed in the acute presence of TTX. (C) Ca2+ transients induced by action potential-independent synaptic transmission (red, imaged with TTX present) or by back-propagating action potentials (bAP) (blue, imaged without TTX). (D and E) Puncta area (indicating active spines, D) and the number of Ca2+ transients (per minute, E) in each puncta (active spine) in control and TTX-treated (48 h) neurons (n = 14 to 15). (F) The influence of chronic inactivity on surface GluA1 expression. Surface-labeling of GluA1 in the dendrites from control and TTX-treated (48 h) neurons. PSD95 as synaptic marker, MAP2 as dendrite marker. Scale bar 10 μm. (G) Quantification of surface GluA1 immunofluorescent intensity in the dendrites from control and TTX-treated (48 h) neurons. (H) The activation of CaMKII in control and TTX-treated neurons. Immunofluorescence of phosphorylated CaMKII on Thr286 site, indicating the level of autophosphorylated CaMKII. (I) Quantification of dendritic pCaMKII intensity in control and TTX groups. (J) The activation of CaMKI in control and TTX-treated neurons. Immunofluorescence of phosphorylated CaMKI on Thr177/178 site, indicating the activation of CaMKI by CaMKK. (K) Quantification of dendritic pCaMKII intensity in control and TTX groups. MAP2 as dendrite marker. Scale bar 10 μm.
{kind=link}
Highlights.
Chronic spike blockade with tetrodotoxin causes homeostatic spike broadening
Alternative splicing of BK channels by exclusion of a specific exon is responsible
Synaptic homeostasis starts CaM kinase signaling to drive nuclear exit of Nova-2
Chronic inactivity and hyperactivity can initiate similar LTP-like events
ACKNOWLEDGMENTS
We thank Dr. Robert B. Darnell for providing human anti-Nova serum and Dr. Peter Stoilov and Dr. Douglas Black for providing AS reporter constructs. We thank Xiaohan Wang and other Tsien lab members for advice and comments on the manuscript. This work was supported by research grants from the NIGMS (GM058234), NINDS (NS24067), NIMH (MH071739), NIDA (DA040484), Druckenmiller Foundation, Simons Foundation, Mathers Foundation, and Burnett Family Foundation (to R.W.T.); the National Key R&D Program of China (2018YFA0108300 to B.L.); the National Natural Science Foundation of China (81622016 and 31571034 to B.L. and 81871048 and 81741063 to L.H.); the Guangdong Natural Science Foundation (Grants for Distinguished Young Scholars 2015A030306019 to B.L. and 2018B030311034 to L.H.); and the Guangdong Provincial Key R&D Programs (Key Technologies for Treatment of Brain Disorders 2018B030332001 and Development of New Tools for Diagnosis and Treatment of Autism 2018B030335001 to L.H. and B.L.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.05.013.
REFERENCES
- Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, and Means AR (1998). Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J. Biol. Chem 273, 31880–31889. [DOI] [PubMed] [Google Scholar]
- Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, and Gould TD (2012). CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol 99, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, and Jonas P. (2002). Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J. Neurosci 22, 10593–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, and Tsien RW (1996). CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Black DL (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem 72, 291–336. [DOI] [PubMed] [Google Scholar]
- Borst JG, and Sakmann B. (1998). Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J. Physiol 506, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridi MCD, de Pasquale R, Lantz CL, Gu Y, Borrell A, Choi SY, He K, Tran T, Hong SZ, Dykman A, et al. (2018). Two distinct mechanisms for experience-dependent homeostasis. Nat. Neurosci 21, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, and Darnell RB (1997). The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol. Cell. Biol 17, 3194–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, and Kandel ER (1996). Presynaptic facilitation revisited: state and time dependence. J. Neurosci 16, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Sohail M, Liu G, Koumbadinga GA, Lobo VG, and Xie J. (2011). Differential effects of PKA-controlled CaMKK2 variants on neuronal differentiation. RNA Biol. 8, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Ma H, Kuchibhotla KV, Watson BO, Buzsáki G, Froemke RC, and Tsien RW (2016). Excitation-transcription coupling in parvalbumin-positive interneurons employs a novel CaM kinase-dependent pathway distinct from excitatory neurons. Neuron 90, 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzalegui FH, and Means AR (1993). Biochemical characterization of the multifunctional Ca2+/calmodulin-dependent protein kinase type IV expressed in insect cells. J. Biol. Chem 268, 26171–26178. [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, and Tsien RW (2003). Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol 13, 354–365. [DOI] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, and Klyachko VA (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, and Turrigiano GG (1999). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci 2, 515–520. [DOI] [PubMed] [Google Scholar]
- Ding X, Liu S, Tian M, Zhang W, Zhu T, Li D, Wu J, Deng H, Jia Y, Xie W, et al. (2017). Activity-induced histone modifications govern Neurexin-1 mRNA splicing and memory preservation. Nat. Neurosci 20, 690–699. [DOI] [PubMed] [Google Scholar]
- Dittmer PJ, Wild AR, Dell’Acqua ML, and Sather WA (2017). STIM1 Ca2+ Sensor Control of L-type Ca2+-Channel-Dependent Dendritic Spine Structural Plasticity and Nuclear Signaling. Cell Rep. 19, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Lüders HO, Shi J, Cui J, et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet 37, 733–738. [DOI] [PubMed] [Google Scholar]
- Edelman AM, Mitchelhill KI, Selbert MA, Anderson KA, Hook SS, Stapleton D, Goldstein EG, Means AR, and Kemp BE (1996). Multiple Ca(2+)-calmodulin-dependent protein kinase kinases from rat brain. Purification, regulation by Ca(2+)-calmodulin, and partial amino acid sequence. J. Biol. Chem 271, 10806–10810. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, and Fine A. (1999). Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22, 115–124. [DOI] [PubMed] [Google Scholar]
- Eom T, Zhang C, Wang H, Lay K, Fak J, Noebels JL, and Darnell RB (2013). NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. eLife 2, e00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor AA, and Aldrich RW (2009). Convergent evolution of alternative splices at domain boundaries of the BK channel. Annu. Rev. Physiol 71, 19–36. [DOI] [PubMed] [Google Scholar]
- Furlanis E, and Scheiffele P. (2018). Regulation of neuronal differentiation, function, and plasticity by alternative splicing. Annu. Rev. Cell Dev. Biol 34, 451–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, and Jonas P. (2000). Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. [DOI] [PubMed] [Google Scholar]
- Green MF, Anderson KA, and Means AR (2011a). Characterization of the CaMKKβ-AMPK signaling complex. Cell. Signal 23, 2005–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF, Scott JW, Steel R, Oakhill JS, Kemp BE, and Means AR (2011b). Ca2+/Calmodulin-dependent protein kinase kinase β is regulated by multisite phosphorylation. J. Biol. Chem 286, 28066–28079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TS, Jeong SY, Cho SW, Jeon Hk, Roh GS, Choi WS, and Park CS (2000). Functional characteristics of two BKCa channel variants differentially expressed in rat brain tissues. Eur. J. Biochem 267, 910–918. [DOI] [PubMed] [Google Scholar]
- Harnett MT, Makara JK, Spruston N, Kath WL, and Magee JC (2012). Synaptic amplification by dendritic spines enhances input cooperativity. Nature 491, 599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harreman MT, Kline TM, Milford HG, Harben MB, Hodel AE, and Corbett AH (2004). Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J. Biol. Chem 279, 20613–20621. [DOI] [PubMed] [Google Scholar]
- Hébert B, Pietropaolo S, Même S, Laudier B, Laugeray A, Doisne N, Quartier A, Lefeuvre S, Got L, Cahard D, et al. (2014). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J. Rare Dis 9, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, and Lipscombe D. (2005). Neuronal L-type calcium channels open quickly and are inhibited slowly. J. Neurosci 25, 10247–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JJ, Hao J, Kosofsky BE, and Rajadhyaksha AM (2008). Dysregulation of large-conductance Ca2+-activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p.R419X mutation. Neurogenetics 9, 219–223. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, and Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, Laake P, Pongs O, Knaus HG, Ottersen OP, and Storm JF (2001). Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J. Neurosci 21, 9585–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, and Turrigiano GG (2008). Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57, 819–826. [DOI] [PubMed] [Google Scholar]
- Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, and Scheiffele P. (2011). SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 147, 1601–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, and Augustine GJ (1991). Action potential broadening and frequency-dependent facilitation of calcium signals in pituitary nerve terminals. Proc. Natl. Acad. Sci. USA 88, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, and Sutton MA (2010). Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, and Chen G. (2006). High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat. Protoc 1, 695–700. [DOI] [PubMed] [Google Scholar]
- Joseph A, and Turrigiano GG (2017). All for one but not one for all: excitatory synaptic scaling and intrinsic excitability are coregulated by CaMKIV, whereas inhibitory synaptic scaling is under independent control. J. Neurosci 37, 6778–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, and Edelman AM (2012). A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem 287, 24832–24843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Tsien RW (2008). Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron 58, 925–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, and Ziff EB (2014). Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 12, e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T, Khaliq ZM, and Bean BP (2015). Differential regulation of action potential shape and burst-frequency firing by BK and Kv2 channels in substantia nigra dopaminergic neurons. J. Neurosci 35, 16404–16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Corcoran EE, Eto K, Gengyo-Ando K, Muramatsu MA, Kobayashi R, Freedman JH, Mitani S, Hagiwara M, Means AR, and Tokumitsu H. (2002). A CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep. 3, 962–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y, Kamio Y, Kawai T, Katsuura S, Inada N, Takaki A, and Rokutan K. (2011). Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS ONE 6, e24723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Roger S, Guérin P, Molinari F, M’rad R, Cahard D, Belhadj A, Halayem M, Persico AM, Elia M, et al. (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry 163, 1622–1629. [DOI] [PubMed] [Google Scholar]
- Lee KY, and Chung HJ (2014). NMDA receptors and L-type voltage-gated Ca2+ channels mediate the expression of bidirectional homeostatic intrinsic plasticity in cultured hippocampal neurons. Neuroscience 277, 610–623. [DOI] [PubMed] [Google Scholar]
- Lee US, and Cui J. (2010). BK channel activation: structural and functional insights. Trends Neurosci. 33, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Xing Y, Nguyen D, Xie J, Lee CJ, and Black DL (2007). Depolarization and CaM kinase IV modulate NMDA receptor splicing through two essential RNA elements. PLoS Biol. 5, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Tang ZZ, and Black DL (2009). An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 23, 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Royston SE, Vest MO, Ley DJ, Lee S, Bolton EC, and Chung HJ (2015). N-methyl-D-aspartate receptors mediate activity-dependent down-regulation of potassium channel genes during the expression of homeostatic intrinsic plasticity. Mol. Brain 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, and Martin KC (2016). Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMasson G, Marder E, and Abbott LF (1993). Activity-dependent regulation of conductances in model neurons. Science 259, 1915–1917. [DOI] [PubMed] [Google Scholar]
- Lewis HA, Musunuru K, Jensen KB, Edo C, Chen H, Darnell RB, and Burley SK (2000). Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell 100, 323–332. [DOI] [PubMed] [Google Scholar]
- Li Q, Lee JA, and Black DL (2007). Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci 8, 819–831. [DOI] [PubMed] [Google Scholar]
- Li B, Jie W, Huang L, Wei P, Li S, Luo Z, Friedman AK, Meredith AL, Han MH, Zhu XH, and Gao TM (2014). Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat. Neurosci 17, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Tadross MR, and Tsien RW (2016). Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, and Darnell RB (2006). Splicing regulation in neurologic disease. Neuron 52, 93–101. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, and Darnell RB (2010). RNA processing and its regulation: global insights into biological networks. Nat. Rev. Genet 11, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, and Tsien RW (2010). Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc. Natl. Acad. Sci. USA 107, 21806–21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Razanau A, Hai Y, Yu J, Sohail M, Lobo VG, Chu J, Kung SK, and Xie J. (2012). A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J. Biol. Chem 287, 22709–22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Sugimori M, and Simon SM (1982). Transmission by presynaptic spike-like depolarization in the squid giant synapse. Proc. Natl. Acad. Sci. USA 79, 2415–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XJ, Li M, Huang L, Steinberg S, Mattheisen M, Liang G, Donohoe G, Shi Y, Chen C, Yue W, et al. ; MooDS SCZ Consortium (2014). Convergent lines of evidence support CAMKK2 as a schizophrenia susceptibility gene. Mol. Psychiatry 19, 774–783. [DOI] [PubMed] [Google Scholar]
- Ma WP, Li YT, and Tao HW (2013). Downregulation of cortical inhibition mediates ocular dominance plasticity during the critical period. J. Neurosci 33, 11276–11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, and Tsien RW (2014). γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, and Turrigiano GG (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. J. Neurosci 28, 4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, and Chen L. (2008). Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 105, 16015–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcelo KL, Means AR, and York B. (2016). The Ca(2+)/Calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol. Metab 27, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Abbott LF, Turrigiano GG, Liu Z, and Golowasch J. (1996). Memory from the dynamics of intrinsic membrane currents. Proc. Natl. Acad. Sci. USA 93, 13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Linardakis JM, and Disterhoft JF (2009). The fast and slow afterhyperpolarizations are differentially modulated in hippocampal neurons by aging and learning. J. Neurosci 29, 4750–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger O, Lemoine F, and Scheiffele P. (2016). Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron 92, 1266–1278. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, and Guthrie PB (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263. [DOI] [PubMed] [Google Scholar]
- Mullins C, Fishell G, and Tsien RW (2016). Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89, 1131–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Okuno S, Sato F, and Fujisawa H. (1995). An immunohistochemical study of Ca2+/calmodulin-dependent protein kinase IV in the rat central nervous system: light and electron microscopic observations. Neuroscience 68, 181–194. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Okuno S, Kitani T, Otake K, Sato F, and Fujisawa H. (2001). Immunohistochemical localization of Ca(2+)/calmodulin-dependent protein kinase kinase β in the rat central nervous system. Neurosci. Res 39, 175–188. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, and Prochiantz A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–465. [DOI] [PubMed] [Google Scholar]
- O’Brien MT, Oakhill JS, Ling NX, Langendorf CG, Hoque A, Dite TA, Means AR, Kemp BE, and Scott JW (2017). Impact of genetic variation on human CaMKK2 regulation by Ca2+-calmodulin and multisite phosphorylation. Sci. Rep 7, 43264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, van Rossum MC, and Wyllie DJ (2010). Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J. Physiol 588, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary T, Williams AH, Franci A, and Marder E. (2014). Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron 82, 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, and Flucher BE (2004). Differential targeting of the L-type Ca2+ channel α 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur. J. Neurosci 19, 2109–2122. [DOI] [PubMed] [Google Scholar]
- Palmer LM, and Stuart GJ (2009). Membrane potential changes in dendritic spines during action potentials and synaptic input. J. Neurosci 29, 6897–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genomewide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney J, Tsurudome K, Liao EH, Elazzouzi F, Livingstone M, Gonzalez M, Sonenberg N, and Haghighi AP (2012). TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74, 166–178. [DOI] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, Siegelmann HT, and Treistman SN (2008). Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron 59, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racca C, Gardiol A, Eom T, Ule J, Triller A, and Darnell RB (2010). The Neuronal Splicing Factor Nova Co-Localizes with Target RNAs in the Dendrite. Front. Neural Circuits 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, and Zoghbi HY (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y. (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, and Regehr WG (1997). Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J. Neurosci 17, 3425–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Miranda-Rottmann S, Ruggiu M, Park CY, Fak JJ, Zhong R, Duncan JS, Fabella BA, Junge HJ, Chen Z, et al. (2016). NOVA2-mediated RNA regulation is required for axonal pathfinding during development. eLife 5, e14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. USA 101, 9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanzenbächer CT, Sambandan S, Langer JD, and Schuman EM (2016). Nascent Proteome Remodeling following Homeostatic Scaling at Hippocampal Synapses. Neuron 92, 358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaukowitch K, Reese AL, Kim SK, Kilaru G, Joo JY, Kavalali ET, and Kim TK (2017). An Intrinsic Transcriptional Program Underlying Synaptic Scaling during Activity Suppression. Cell Rep. 18, 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbert MA, Anderson KA, Huang QH, Goldstein EG, Means AR, and Edelman AM (1995). Phosphorylation and activation of Ca(2+)-calmodulin-dependent protein kinase IV by Ca(2+)-calmodulin-dependent protein kinase Ia kinase. Phosphorylation of threonine 196 is essential for activation. J. Biol. Chem 270, 17616–17621. [DOI] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, and Storm JF (1999). The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J. Physiol 521, 135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley C, Whitt JP, Montgomery JR, and Meredith AL (2013). Phosphorylation of a constitutive serine inhibits BK channel variants containing the alternate exon “SRKR”. J. Gen. Physiol 142, 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipston MJ, and Tian L. (2016). Posttranscriptional and posttranslational regulation of BK channels. Int. Rev. Neurobiol 128, 91–126. [DOI] [PubMed] [Google Scholar]
- Siegel M, Marder E, and Abbott LF (1994). Activity-dependent current distributions in model neurons. Proc. Natl. Acad. Sci. USA 91, 11308–11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, and Keating MT (2005). Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 102, 8089–8096, discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre F, Marshall JD, Yang Y, Gong Y, Schnitzer MJ, and Lin MZ (2014). High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat. Neurosci 17, 884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika R, Campiglio M, Pinggera A, Lee A, Striessnig J, Flucher BE, and Obermair GJ (2016). Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci. Rep 6, 34528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Lin CH, Damoiseaux R, Nikolic J, and Black DL (2008).A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl. Acad. Sci. USA 105, 11218–11223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styr B, and Slutsky I. (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci 21, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, and Tsien RW (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, and Soderling TR (1996). Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J. Biol. Chem 271, 5617–5622. [DOI] [PubMed] [Google Scholar]
- Trasande CA, and Ramirez JM (2007). Activity deprivation leads to seizures in hippocampal slice cultures: is epilepsy the consequence of homeostatic plasticity? J. Clin. Neurophysiol 24, 154–164. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, and Nelson SB (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci 5, 97–107. [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, et al. (2005). Nova regulates brain-specific splicing to shape the synapse. Nat. Genet 37, 844–852. [DOI] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, and Darnell RB (2006). An RNA map predicting Nova-dependent splicing regulation. Nature 444, 580–586. [DOI] [PubMed] [Google Scholar]
- Vuong CK, Black DL, and Zheng S. (2016). The neurogenetics of alternative splicing. Nat. Rev. Neurosci 17, 265–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Campos B, Jamieson GA Jr., Kaetzel MA, and Dedman JR (1995). Functional elimination of calmodulin within the nucleus by targeted expression of an inhibitor peptide. J. Biol. Chem 270, 30245–30248. [DOI] [PubMed] [Google Scholar]
- Wang X, Marks CR, Perfitt TL, Nakagawa T, Lee A, Jacobson DA, and Colbran RJ (2017). A novel mechanism for Ca2+/calmodulin-dependent protein kinase II targeting to L-type Ca2+ channels that initiates long-range signaling to the nucleus. J. Biol. Chem 292, 17324–17336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Lee YS, Tokumitsu H, Silva AJ, and Soderling TR (2008). Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron 59, 914–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, and Greenberg ME (2002). Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci 3, 921–931. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, and Tsien RW (2012). Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149, 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1963). Effects of visual deprivation on morphology and physiology of cells in the cats lateral geniculate body. J. Neurophysiol 26, 978–993. [DOI] [PubMed] [Google Scholar]
- Wondolowski J, and Dickman D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell. Neurosci 7, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, and Black DL (2001). A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 410, 936–939. [DOI] [PubMed] [Google Scholar]
- Xie J, and McCobb DP (1998). Control of alternative splicing of potassium channels by stress hormones. Science 280, 443–446. [DOI] [PubMed] [Google Scholar]
- Xu D, Farmer A, Collett G, Grishin NV, and Chook YM (2012). Sequence and structural analyses of nuclear export signals in the NESdb database. Mol. Biol. Cell 23, 3677–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Yin GL, and Darnell RB (1998). The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc. Natl. Acad. Sci. USA 95, 13254–13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, and Svoboda K. (2003). Plasticity of calcium channels in dendritic spines. Nat. Neurosci 6, 948–955. [DOI] [PubMed] [Google Scholar]
- Zarei MM, Zhu N, Alioua A, Eghbali M, Stefani E, and Toro L. (2001). A novel MaxiK splice variant exhibits dominant-negative properties for surface expression. J. Biol. Chem 276, 16232–16239. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li X, Zhou R, and Xing G. (2006). Possible role of potassium channel, big K in etiology of schizophrenia. Med. Hypotheses 67, 41–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative waveforms of action potential recorded after TTX treatment (24 or 48 h). (B) Quantification of half width of action potentials in the groups in (A) (n = 10. Data for control and 48 h groups is from Figure 1A).
(A) Impact of chronic inactivity on alternative splicing of X1 to X5. Cultured cortical neurons were sham- or TTX-treated for 48 h. Separation of RT-PCR products obtained with specific primers flanking each alternatively spliced site (X1 to X5). Closed arrows indicate the longer RT-PCR products obtained if the alternative exon was included, open arrows indicate the shorter products if that exon was excluded. (B) Quantification of inclusion or exclusion of X1 to X6 by quantitative RT-PCR. (C) High K+ solution maintained long-term depolarization in resting membrane potential. (D) Impact of sustain depolarization on E29 inclusion. Cultured cortical neurons were sham- or high K+-treated (20mM, 40mM and 60mM, respectively) for 12 or 24 h. E29 inclusion was examined by RT-PCR.
(A) Validation of the knockdown of Nova-2 expression by shRNAs. Cortical neurons were transfected with shRNAs either targeting the untranslated region (UTR) of the Nova-2 gene or targeting the Nova-2 coding sequence (CDS). Both shRNAs efficiently knocked down endogenous Nova-2 expression. (B) UTR-lacking flag-Nova-2 plasmids (Nova-2 (R)) were co-expressed with shRNAs targeting CDS (targeting three different regions) or shRNAs targeting UTR in HEK293 cells. The shRNAs targeting CDS efficiently knocked down flag-Nova-2 expression, whereas shRNAs targeting UTR did not. (C) E29 inclusion could only be induced by wild-type (WT) Nova-2, but not ΔNLS-Nova-2. WT-Nova-2-induced E29 inclusion was prevented by the shRNAs targeting the Nova-2 coding sequence (CDS), but not by shRNAs targeting the untranslated region (UTR) of the Nova-2 gene.
(A) Expression level of BK channel mRNA was assayed by RT-PCR (n = 4). (B) BK channel expression in the plasma membrane was assayed by western blotting after biotinylation-mediated membrane protein fractionation. (C) Immunofluorescence of surface BK channel. Surface BK channel was probed with antibodies targeting extracellular N terminus. (D) Quantification of intensity of surface BK channel (n = 10). (E) Overexpression of GFP-tagged BK channel isoforms in HEK293 cells. (F) Quantification of surface fluorescence intensity of GFP-tagged BK channel isoforms. (G) Expression level of GFP-tagged BK channel isoforms was assayed by western blotting after biotinylation-mediated membrane protein fractionation. Na/K ATPase, Lamin B and GAPDH are markers for plasma membrane, nucleus and cytosol.
(A) Schematic diagram of Nova-2 protein as in Figure 7. (B to D) Mass spec results of phosphorylation level of S25, T27 and S194 sites. (E) WT and S194E Nova-2 are no different in RNA binding. Flag-tagged WT Nova-2 or S194E Nova-2 was overexpressed in HEK293 cells. RNA pull-down assay was performed from the cell lysates by probe A1 (Figure 2). The RNA binding ability of WT and S194E Nova-2 was assayed by western blotting with anti-flag antibody.
(A) The activation of nuclear CaMKIV in different groups as indicated. Immunofluorescence of phosphorylated CaMKIV on Thr196 site indicating the level of CaMKIV activation by CaMKK. (B) Normalized nuclear/cytosolic ratio of pCaMKIV immunofluorescence intensity in each group as indicated. (C) The localization of Nova-2 in different groups as indicated. (D) Normalized nuclear/cytosolic ratio of Nova-2 immunofluorescence intensity in each group as indicated. (E) 12 h and 24 h high potassium media treatment (20mM, 40mM and 60mM, respectively) led to Nova-2 translocation from nucleus to cytosol. (F) Long-term depolarization reduced the inclusion of E29, which could be blocked by CaV1 blocker nimodipine and CaM kinases blocker KN-93. (G and H) Fold changes in abundance of CaM, αCaMKI, βCaMKI, γCaMKI, δCaMKI and γCaMKII in the nucleus, cytosol and nucleus/cytosol ratio. (G) Representative micrographs of different proteins in control or TTX-treated neurons. (H) Ratios of levels after TTX treatment (48 h) relative to levels in control neurons. (I) Validation of the knockdown of βCaMKK expression by shRNAs. flag-tagged βCaMKK was co-expressed with shRNAs targeting different locations of rat βCaMKK CDS. The efficiency of shRNAs was assayed by western blotting.
(A) Ca2+ transients were imaged from the soma of control neurons, with no TTX present. (B) Ca2+ imaging, from the soma of control neurons (upper) or TTX-treated (48 h) neurons, was performed in the acute presence of TTX. (C) Ca2+ transients induced by action potential-independent synaptic transmission (red, imaged with TTX present) or by back-propagating action potentials (bAP) (blue, imaged without TTX). (D and E) Puncta area (indicating active spines, D) and the number of Ca2+ transients (per minute, E) in each puncta (active spine) in control and TTX-treated (48 h) neurons (n = 14 to 15). (F) The influence of chronic inactivity on surface GluA1 expression. Surface-labeling of GluA1 in the dendrites from control and TTX-treated (48 h) neurons. PSD95 as synaptic marker, MAP2 as dendrite marker. Scale bar 10 μm. (G) Quantification of surface GluA1 immunofluorescent intensity in the dendrites from control and TTX-treated (48 h) neurons. (H) The activation of CaMKII in control and TTX-treated neurons. Immunofluorescence of phosphorylated CaMKII on Thr286 site, indicating the level of autophosphorylated CaMKII. (I) Quantification of dendritic pCaMKII intensity in control and TTX groups. (J) The activation of CaMKI in control and TTX-treated neurons. Immunofluorescence of phosphorylated CaMKI on Thr177/178 site, indicating the activation of CaMKI by CaMKK. (K) Quantification of dendritic pCaMKII intensity in control and TTX groups. MAP2 as dendrite marker. Scale bar 10 μm.
Data Availability Statement
All data supporting the findings of this study and custom MATLAB code for calcium imaging are available upon request from the Lead Contact.