

Abstract
Background
Growth hormone‐releasing hormone (GHRH) and its receptors have been implicated in the progression of various tumors. In this study, we analyzed the carcinogenetic potential of exposure to GHRH of a nontumor human prostate epithelial cell line (RWPE‐1) as well as its transforming effect in a xenograft model.
Methods
We performed cell viability, cell proliferation, adhesion and migration assays. In addition, metalloprotease (MMP)−2 activity by means gelatin zymography, GHRH‐R subcellular location using confocal immunofluorescence microscopy and vascular endothelial growth factor (VEGF) levels by enzyme‐linked immunoassay were assessed. Besides, we developed an in vivo model in order vivo model to determine the role of GHRH on tumorigenic transformation of RWPE‐1 cells.
Results
In cell cultures, we observed development of a migratory phenotype consistent with the gelatinolytic activity of MMP‐2, expression of VEGF, as well as E‐cadherin‐mediated cell‐cell adhesion and increased cell motility. Treatment with 0.1 µM GHRH for 24 h significantly increased cell viability and cell proliferation. Similar effects of GHRH were seen in RWPE‐1 tumors developed by subcutaneous injection of GHRH‐treated cells in athymic nude mice, 49 days after inoculation.
Conclusions
Thus, GHRH appears to act as a cytokine in the transformation of RWPE‐1 cells by mechanisms that likely involve epithelial‐mesenchymal transition, thus reinforcing the role of GHRH in tumorigenesis of prostate.
Keywords: epithelial‐mesenchymal transition, GHRH, prostate cancer, RWPE‐1 cells, tumorigenesis
1. INTRODUCTION
Prostate cancer (PCa) is the second leading cause of cancer‐related deaths in Western men. 1 Multiple genetic (48%), epigenetic and environmental factors are involved in the pathogenesis of such a neoplasm. 2 , 3 , 4 , 5 More than 98% of cases show an epithelial origin where external stimuli are needed to stimulate epithelial‐mesenchymal transition (EMT). A nonmotile epithelial cell changes to a mesenchymal state with invasive capabilities in a sequential and reverse program characterized by degradation of the extracellular matrix. Specific proteases of cell‐matrix adhesion act to obtain mesenchymal characteristics and thus be able to migrate away from the original tissue. 6
In vitro model systems provide the opportunity to study in a standard and reproducible way the identification of molecular events involved in neoplastic transformation. The RWPE‐1 cell line is nontumorigenic parental line originated from normal human prostate epithelium. This cell line retains several characteristics of normal prostatic luminal epithelium including expression of androgen receptor, prostate specific antigen, responsiveness to growth stimulation by epithelial growth factor (EGF) and growth inhibition by transforming growth factor‐β as prostatic epithelium. 7 Moreover, RWPE‐1 cells do not growth in agar and not form any solid tumor when implanted into nude mice. Taken together, RWPE‐1 cell line is considered for studies on growth regulation at cellar and molecular level. Previous results show different signals that induce the RWPE‐1 cell line transformation such as radiation, oncogene, 7 , 8 organic carcinogens, and chemical products such as cadmium. 9 , 10
Growth hormone‐releasing hormone (GHRH) (44 amino acids) belongs to the secretin family of peptides that also includes vasoactive intestinal peptide (VIP) and others. GHRH is expressed in a variety of extra‐pituitary tissues such as prostate. 11 , 12 The most important sources of nonhypothalamic GHRH production is found in various tumors and their cell lines. 13 GHRH promotes metastatic phenotypes in both androgen‐responsive (lymph node carcinoma of the prostate [LNCaP]) and androgen‐unresponsive (PC3) prostate adenocarcinoma cell lines 14 and stimulates neuroendocrine differentiation in LNCaP. 15 Taken together, these results support the important role of GHRH in tumor establishment and progression in prostate cells. The effects of GHRH are mediated by binding to specific membrane receptors (GHRH and its splice variants [SVs] receptors) that belong to Gs‐protein coupled heptaelical receptors. The binding of GHRH to their receptors increases the intracellular accumulation of cAMP with special role in mitogen signaling pathways, as well as the transactivation of epidermal growth factor receptor (EGFR) and human EGFR‐2. 16
Previous observations on effects of VIP in PCa have demonstrated that this peptide may induce promotion and progression of prostate carcinoma. 17 Here, we used human prostate RWPE‐1 cell cultures as well as xenografted athymic nude mice to study the tumorigenic transformations of these nonneoplastic epithelial cells after exposure to GHRH.
2. MATERIALS AND METHODS
2.1. Cell culture
Three human prostate cell lines were used. Cell lines were obtained from the American Type Culture Collection. Nonneoplastic, immortalized adult human prostatic epithelial cells, RWPE‐1 (passages 3–10, ATCC CRL‐11609, certified by STRS analysis), are androgen‐responsive and show many characteristics of nontumor cells. The two human PCa cell lines used exhibit different features of PCa progression from early stages to androgen independence. LNCaP (passages 5–16, ATCC CRL‐1740, certified by STRS analysis) is an androgen‐responsive cancer cell line and PC3 (passages 5–16, ATCC CRL‐1435, certified by STRS analysis) is an androgen‐unresponsive cell line that may be analogous to recurrent PCa that have achieved androgen independence. RWPE‐1 cells were maintained in complete keratinocyte serum‐free medium containing 50 μg/ml bovine pituitary extract and 5 ng/ml human epidermal growth factor (EGF). LNCaP and PC3 cells were grown and maintained in RPMI‐1640 medium supplemented with 10% fetal bovine serum (FBS). All culture media contained 1% penicillin/streptomycin/amphotericin B (Life Technologies). Culture was carried out in a humidified 5% CO2 environment at 37°C.
2.2. Confocal immunofluorescence microscopy
Cells were cultured on glass coverslips. The cells were fixed and permeabilized with methanol at −20°C for 15 min and incubated in 1.5% goat serum in phosphate buffered saline (PBS) at room temperature for 1 h. Afterwards, the cells were incubated for 1 h at room temperature with pGHRH and SVs receptors (working dilution 1:2000) antibodies. After washing with PBS, cells were incubated with goat anti‐rabbit and anti‐mouse‐FITC secondary antibodies (1:2000) (Alexa, Invitrogen) for 1 h in darkness. Cells were washed again with PBS and the coverslips were mounted with FluorSave™ Reagent (Calbiochem). Color detection was performed with a LEICA TCS‐SL confocal laser scan microscope.
2.3. Cell viability studies
RWPE‐1 cells were grown to 70%–80% confluence, harvested with trypsin/ethylenediaminetetraacetic acid (EDTA) solution and seeded at low concentration (50,000 cells per well) in 24‐well plates for 24 h. The culture medium was then removed and replaced with RPMI‐1640 medium containing 1% antibiotic/antimycotic (penicillin/streptomycin/amphotericin B) and 0% FBS for 24 h. Cells were treated for 24 h with GHRH (10−9–10−5 M). Cell viability was determined by tetrazolium assay, which measures the reduction of substrate MTT [3‐(4,5‐dimethylthiazol‐2‐yl)2,5‐diphenyltetrazolium bromide] to a dark blue formazan product by mitochondrial dehydrogenases in living cells. MTT (5 mg/ml) (Sigma–Aldrich) was added to each well and the mixture incubated for 3 h at 37°C in darkness. The medium was replaced, and the dark blue formazan precipitate was dissolved with 0.2 N HCl in isopropanol. The absorbance was read at 570 nm in a plate reader (ELX 800, Bio‐Tek Instruments). Results were expressed as the relative percentage of absorbance compared with control cells.
2.4. Cell proliferation assay
RWPE‐1 cells were placed in 24‐well plates (104 cells per well) and maintained in serum‐free K‐SFM medium for 24 h. Then, they were stimulated with 0.1 μM GHRH (1–44) (NeoMPS) for 24 h. After those cells were labeled with 10 μM bromodeoxyuridine (BrdU) for 30 min. Thereafter, cells were washed with PBS, fixed with ice‐cold absolute ethanol, and stored at −20°C for 30 min or at 4°C for a maximum of 1 week. The fixative was removed by centrifugation and pellets were washed with PBS. DNA was partially denatured by incubation with 1 M HCl for 30 min at room temperature and then cells were washed three times with PBS containing 0.05% Tween‐20 (pH 7.4) and 0.1% bovine serum albumin (BSA). Afterwards, cells were incubated with 20 ml of anti‐BrdU monoclonal antibody with FITC (BD Bioscience) for a 30‐min period in the dark. For flow cytometry analysis, cells were stained with propidium iodide (PI) staining solution (50 mg/ml PI and 10 mg/ml RNase). The number of BrdU‐positive cells was counted using a FACSCalibur cytometer (BD Bioscience). The results obtained were analyzed using the Cyflogic v 1.2.1 program.
2.5. Cell adhesion assay
Concentrated stock of type I collagen solution was diluted with 10 mM glacial acetic acid and coated onto 96‐well plates for 1 h at 37°C. Plates were washed twice with PBS (pH 7.4). Cells were harvested with 0.25% trypsin/0.2% EDTA and collected by centrifugation. Afterwards, cells were resuspended in K‐SFM medium/0.1% (wt/vol) BSA (pH 7.4) at a concentration of 2.5 × 105 cells/ml. Then, cells were plated and those cells that were not attached were removed at indicated times by aspiration. Cell adhesion was quantified by adding 1 mg/ml solution of MTT for 4 h incubation. Dimethyl sulfoxide (100 µl) was added to each well to dissolve the formazan precipitate and absorbance at 540 nm with a reference wavelength at 630 nm was reported.
2.6. Protein isolation and Western blotting
Cells (3 × 106 cells in 100 mm cell culture dishes) were incubated with 0.1 μM GHRH for different time periods. Cells were washed twice with ice‐cold PBS and then harvested, scraped into ice‐cold PBS, and pelleted by centrifugation at 500g for 5 min at 4°C. The cells were kept on ice for 30 min in a solution containing 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 0.5 M NaCl, 0.5% Nonidet NP‐40, 2 mM phenyl methylsulphonylfluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 5 μg/ml pepstatin. Thereafter, the cells were pelleted by centrifugation at 4000g for 5 min at 4°C. Then, they were resolved on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), and blotted onto a nitrocellulose membrane (BioTrace/NT, Pall) overnight in 50 mM Tris‐HCl, 380 mM glycine, 0.1% SDS, and 20% methanol. Antibodies against GHRH‐R (batch number: JH‐2321/5) and SVs (batch number: JH2317/5) were raised in the laboratory of one of us (AVS). Rabbit anti‐pituitary GHRH receptor (pGHRHR) (1:4000) and anti‐SVs (1:4000), mouse anti‐E‐cadherin (at# 610181, RRID: AB_397580; BD Biosciences C), anti‐PCNA (Cat# 18‐0110, RRID: AB_86659; Innovative Research) antibodies were added followed by incubation for 1 h at room temperature. After treatment for 1 h at room temperature with the corresponding HRP‐labelled secondary antiserum (1:4000) (BD Biosciences), the signals were detected with enhanced chemiluminescence reagent (Pierce) using β‐actin antibody (BD Biosciences) as a loading control.
2.7. Wound‐healing assay
RWPE‐1 cells were incubated in 24‐well plates and a small wound area was made in the confluent monolayer with a scraper. Afterwards, cells were incubated in the absence or presence of GHRH (0.1 μM). Three representative fields of each wound were captured using a Nikon Diaphot 300 inverted microscopy at different times (0–24 h). Wound areas of untreated samples were averaged and assigned a value of 100%.
2.8. Gelatin zymography
RWPE‐1 cells were incubated in conditioned media, stimulated with 0.1 μM GHRH for different time periods and then the supernatant was collected. The samples were analyzed by a zymographic technique using 10% SDS‐PAGE containing 0.1% (wt/vol) gelatin (Sigma) as the substrate. Each lane was loaded with a total protein concentration of 3 μg and subjected to electrophoresis at 4°C. Gels were washed twice in 50 mM Tris (pH 7.4) containing 2.5% (vol/vol) Triton X−100 for 1 h, followed by two 10‐min rinses in 50 mM Tris (pH 7.4). After removal of SDS, the gels were incubated overnight in 50 mM Tris (pH 7.5) containing 10 mM CaCl2, 0.15 M NaCl, 0.1% (vol/vol) Triton X‐100, and 0.02% sodium azide at 37°C under constant gentle shaking. After incubation, the gels were stained with 0.25% Coomasie brilliant blue R‐250 (Sigma) and destained with 7.5% acetic acid in 20% methanol. The activity of MMP‐2 and MMP‐9 was semiquantitatively determined by densitometry.
2.9. Determination of vascular endothelial growth factor (VEGF)165
VEGF levels in culture media after treatment with GHRH (15 μg) were determined by using the human VEGF DuoSet (R&D Systems) according to the manufacturer's instructions. Data were normalized by the protein concentration in each sample.
2.10. Animals and preparation of RWPE‐1 cell xenografts
“In vivo” experimental procedures were carried out according to Spanish Law 32/2007, Spanish Royal Decree 1201/2005, European Directive 609/86/CEE and European Convention of Council of Europe ETS 123. Athymic male nude mice (nu/nu), 5–6 weeks old were obtained from Harlan and maintained in microisolator units on a standard sterilizable diet. Mice were housed under humidity‐ and temperature‐controlled conditions and the light/dark cycle was set at 12 h intervals. RWPE‐1 cells were incubated in the absence or presence of 0.1 µM GHRH for 24 h. Thereafter, the cells were washed with PBS, detached with 0.25% trypsin/0.2% EDTA, centrifuged at 400g, and resuspended in fresh medium at 1 × 108 cells/ml. The cell suspension was mixed with Matrigel (BD Bioscience) synthetic basement membrane (1:1, vol/vol) and then injected under the skin in the right flank (1 × 107 cells/mouse). Seven animals were used per group. After sacrifice at 49 days after subcutaneous injection, tumors were harvested.
2.11. Data analysis
Quantification of band densities was performed using the Quantified One Program (Bio‐Rad). Data shown in the figures are representative of four different experiments. The results are expressed as the mean ± SEM and were statistically evaluated following the Bonferroni's test for multiple comparisons after one or two‐way analysis of variance. The level of significance was set at p < 0.05.
3. RESULTS
3.1. Expression and subcellular location of GHRH receptors on RWPE‐1 cells
The assessment of the expression of both types of GHRH receptors was realized by Western blotting using specific antibodies against pGHRHR and its SVs in nontumor (RWPE‐1) and tumor (LNCaP and PC3) cells. The results showed expression levels for pGHRHR and SVs in all cell lines studied (Figure 1A). Pituitary GHRHR expression levels were higher in LNCaP cells and lower in PC3 cells as compared with those in RWPE‐1 cells. SVs expression levels were lower in LNCaP cells and higher in PC3 cells as compared with those in RWPE‐1 cells. Subcellular location of GHRH receptors was evaluated by Immunohistochemistry. We determined the presence of pGHRHR and SVs in both, plasma membrane and cytoplasm in all cell lines studied (Figure 1B). The results were correlated with those observed by Western blotting.
Figure 1.
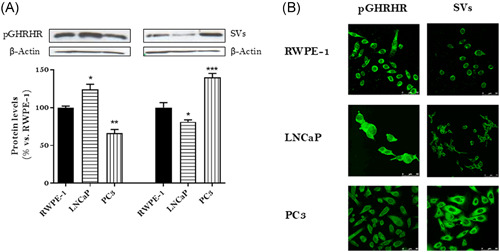
Expression and subcellular location of GHRH receptors in RWPE‐1, LNCaP and PC3 cells. (A) Expression levels of GHRH receptors were evaluated by Western blotting using specific antibodies for pGHRHR and SVs. The diagram represents the mean ± SEM of four experiments. *p < 0.05; **p < 0.01; ***p < 0.001 compared with those obtained in RWPE‐1 cells. (B) Immunofluorescence detection of pGHRHR and SVs. Cells were fixed and incubated with anti‐pGHRHR and anti‐SVs as described in Section 2.2. After an examination by confocal microscopy, the intensity of the emitted fluorescence was measured using image performing software LCS‐SL. Results are representative of four independent experiments. Cells were observed at ×200 magnification, scale bar = 20 μm. GHRH, growth hormone‐releasing hormone; SV, splice variant [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Effect of GHRH on cell viability and cell proliferation in RWPE‐1 cells
The effect of GHRH (1–44) on cell viability of RWPE‐1 cells was assessed by MTT assays for 24 h (Figure 2A). Treatment with 0.1 μM GHRH significantly increased the viability by 14% versus control. To compare the effect of GHRH on cell proliferation, BrdU incorporation assays were performed for 24 h (Figure 2B). GHRH provoked a significantly increase of proliferation (by 78% vs. control) in RWPE‐1 cells.
Figure 2.
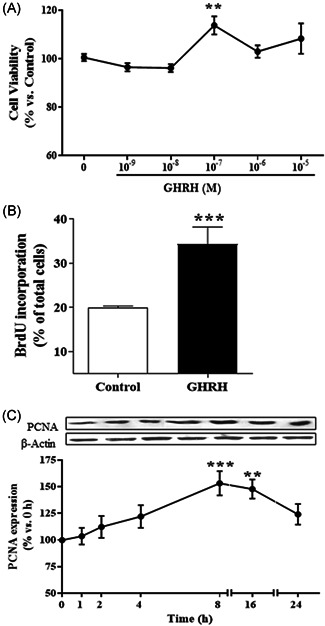
Effect of GHRH on cell viability and cell proliferation in RWPE‐1 cells. (A) RWPE‐1 cells were incubated in the absence or presence of 0.1 μM GHRH at different concentration (10−9–10−5 M) for 24 h. GHRH provoked a significant increase of cell viability at 10−7 M (n = 4). (B) RWPE‐1 cells were incubated in the absence or presence of 0.1 μM GHRH and 10 μM bromodeoxyuridine (BrdU). GHRH increased BrdU incorporation after 24 h‐treatment (n = 4). (C) Expression of PCNA and β‐actin levels were evaluated by Western blotting at indicated times (1–24 h). GHRH (1–44) (0.1 μM) augmented the cell proliferation marker at 8 and 16 h. A representative experiment is shown. The bar diagrams represent the mean ± SEM of four experiments. **p < 0.01; ***p < 0.001 compared with control. GHRH, growth hormone‐releasing hormone
Changes induced by GHRH in cell proliferation may be due to variations on the expression of specific markers such as the proliferating cell nuclear antigen (PCNA). In this context, the hormone GHRH significantly increased the expression of PCNA in a time‐dependent manner from 1 to 24 h, with the maximum value of expression at 8 h (53%, p < 0.001) in RWPE‐1 cells (Figure 2C).
3.3. Effect of GHRH on cell adhesion in RWPE‐1 cells
These studies were carried out with RWPE‐1 cells that were incubated in the absence or presence of 0.1 μM GHRH on a collagen‐coated plate. Studies were carried out for 5–80 min in serum‐free medium with 0.1% BSA to minimize any effects due to a new matrix synthesis or matrix components in serum. Human prostate RWPE‐1 cells rapidly adhered to collagen basement in a time‐dependent manner. The maximal adhesion was achieved at about 80 min. Treatment with GHRH resulted in a slight loss of cell adhesion (Figure 3A).
Figure 3.
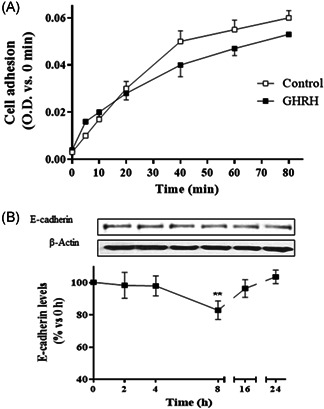
Effect of GHRH on cell adhesion in RWPE‐1 cells. (A) Cells were plated onto coated culture wells in the presence or absence of 0.1 µM GHRH; after the indicated times, MTT (1 mg/ml) was added for 4 h followed by aspiration and addition of 0.1 ml DMSO; the absorbance was measured at 530/640 nm (n = 4). (B) Expression of E‐cadherin and β‐actin levels was evaluated by Western blotting at indicated times (2–24 h). GHRH (0.1 μM) significantly decreased the cell adhesion marker at 8 h. A representative experiment is shown. The bar diagrams represent the mean ± SEM of four experiments. **p < 0.01 compared with control. DMSO, dimethyl sulfoxide; GHRH, growth hormone‐releasing hormone
We also determined whether GHRH regulated protein expression of E‐cadherin. For this purpose, RWPE‐1 cells were incubated with 0.1 μM GHRH up to 24 h. Measurement of E‐cadherin protein levels by means of Western blot analysis and further densitometry revealed that GHRH significantly decreased the expression of E‐cadherin at 8 h of incubation (Figure 3B).
3.4. Effect of GHRH on cell migration in RWPE‐1 cells
To evaluate whether GHRH promotes a migratory phenotype, a wound‐ healing assay was carried out. To this end, a small wound area was made in the confluent monolayer of cells. Under control conditions, the wound width was maintained for 24 h whereas administration of 0.1 μM GHRH resulted in a shorter time interval. Wound migration occurred as early as 4 h after treatment of RWPE‐1 cells with GHRH, and the complete recovery of wound area was observed at the end of the 24‐h incubation period (Figure 4A).
Figure 4.
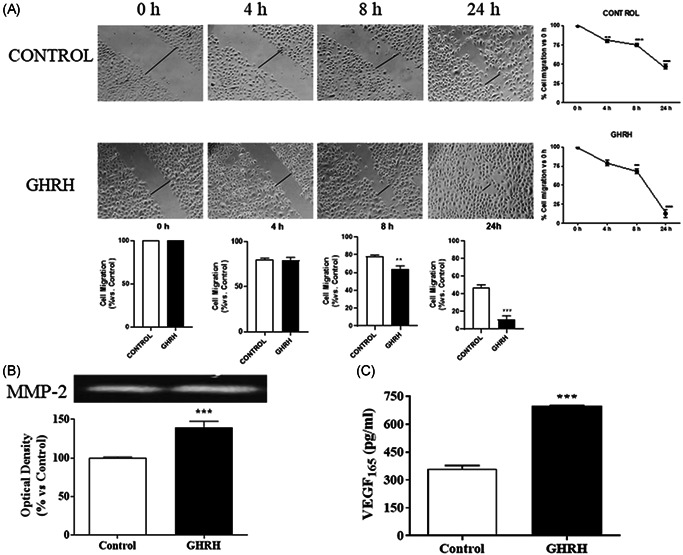
Effect of GHRH on cell migration in RWPE‐1 cells. (A) Cells were damaged by mechanical scrapping and incubated in the presence or absence of 0.1 µM GHRH for the indicated times (4–24 h). Recovery of cell monolayer wounds was followed by microscopy. Representative images from four experiments are shown. (B) Cells were incubated with the peptide for 24 h. Total protein from cell secretion was subjected to zymography to detect the gelatinases. The expression of the latent isoform of metalloproteinase (MMP)‐2 was increased by 0.1 μM GHRH. A representative experiment is shown (n = 4). (C) Secretion of the vascular endothelial growth factor (VEGF) was evaluated by ELISA after incubation with GHRH (0.1 μM). The neuropeptide augmented the secreted VEGF165 by 90% at 24 h of the treatment. The diagrams represent the mean ± SEM of four experiments. **p < 0.01; ***p < 0.001 compared with control. ELISA, enzyme‐linked immunoassay; GHRH, growth hormone‐releasing hormone
3.5. Effects of GHRH on degradation extracellular‐matrix in RWPE‐1 cells
To evaluate the effect of GHRH on MMPs signaling pathway, RWPE‐1 cells were incubated with 0.1 μM GHRH for 24 h. Gelatinolytic activity was detected by gelatin zymography in the RWPE‐1 cell line (Figure 4B). One band migrating at 66 kDa molecular mass was detected, which conceivably corresponds to the latent form of MMP‐2. This experimental approach served to estimate the effect of GHRH on the degradation of extracellular matrix. Densitometric analyses of the gelatinolytic activity of the enzyme revealed that it was significantly increased.
To study the effect of GHRH on the secretion of the VEGF (VEGF165), we analyzed the expression of such a proangiogenic factor by means of enzyme‐linked immunoassay. The results showed that GHRH significantly increased the levels of VEGF165 secreted by 90% at 24 h of GHRH treatment (Figure 4C).
3.6. Effect of GHRH on the promotion of tumorigenesis
The efficacy of GHRH on tumorigenic transformation of RWPE‐1 cells was also determined in an in vivo model. The growth of RWPE‐1 tumors implanted under the abdominal skin of athymic nude mice was followed. Two groups of seven animals each were studied. Only the GHRH‐stimulated RWPE‐1‐xenografts developed tumor masses (Figure 5).
Figure 5.
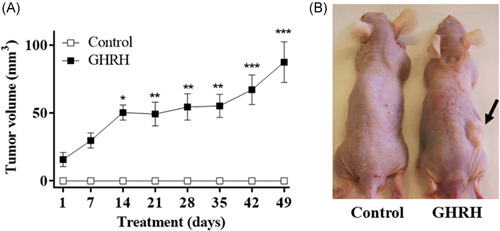
Effect of GHRH on the promotion of tumorigenesis. (A) RWPE‐1 cells were incubated in the absence or the presence of 0.1 µM GHRH for 24 h. The cell suspension was mixed with Matrigel synthetic basement membrane and then injected under the skin in the right flank (1 × 107 cells/mouse). Seven animals were used per group. Experiment lasted for 49 days. Data in each bar are the means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 versus control. (B) Image from a representative animal of each group. Tumor mass is marked with a black arrow. GHRH, growth hormone‐releasing hormone; GHRH, growth hormone‐releasing hormone [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
The physiological actions of GHRH in the prostate gland are initiated through the activation of the G‐protein coupled receptors (pGHRH and SVs receptors) which preferentially stimulate adenylate cyclase and increase intracellular AMPc levels. The presence of endogenous GHRH in human PCa cell lines has been previously described. 18 Furthermore, it should be noted that the highest expression of such a neuropeptide is found in prostate tumor specimens and in the most aggressive PCa cell lines, highlighting the importance of GHRH and its receptors in the progression of the disease. 19
Firstly, we assessed the expression and subcellular localization of the different variants of the GHRH receptors. Our results show that pGHRHR represents the majority of receptors in the non‐tumorigenic prostate cell line studied. SVs of GHRH receptors are also expressed but to a lesser extent. In this regard, it is worth noting that it has been described a greater expression of SVs of the GHRH receptor in different neoplasms including prostate tumor specimens, 20 , 21 , 22 , 23 as well as in androgen‐dependent and independent PCa cell lines. 24
Both pGHRH and SVs receptors are located mainly in the area of the plasma membrane and cell cytoplasm. This corroborates other studies carried out in human breast, ovarian and prostate carcinomas. 25
Once both the presence and the location of GHRH receptors were determined in prostate cells, we proposed to analyze the implication of GHRH in different processes that mediate the onset and progression of PCa. For this purpose, we assessed the effect of the neuropeptide GHRH on the viability of prostate cells without tumorigenic capability. The results obtained confirm that GHRH increased the viability in the studied prostate cell line. Similar results have been described in breast cells. 26 In this regard, it has been reported in the PC3 advanced PCa line that GHRH increases cell viability 27 , 28 to a greater extent than in the stimulated in non‐tumorigenic RWPE‐1 cells. The increased metabolic activity induced by GHRH may be due to mitogenic changes. Therefore, proliferation assays were carried out on the studied cell line. The augmented cell proliferation produced by GHRH could be due to the increased presence of pGHRHR in RWPE‐1 cells. The increase in the proliferation of RWPE‐1 nontumorigenic cells produced by GHRH was reflected on the augmented expression levels of the PCNA, which regulates this complex process. GHRH has a more rapid and greater effect on PCNA expression in more aggressive prostate cells than in non‐tumorigenic cells. 14 It may be due to a more active metabolism of the tumor cells and the increased presence of SVs receptors.
In normal tissue, epithelial cells are attached to each other and to the extracellular matrix. However, tumor cells “break” these junctions to become independent from other cells and “escape” from the tumor niche to other sites in the body, leading to metastasis. The first step in this process is the loss of cell adhesion, becoming one of the key processes for the study of tumor progression. In this regard, previous results demonstrate the implication of GHRH in reducing adhesion of tumor cells of advanced PCa. 14 Our results show that GHRH decreases adhesion without significantly affecting nontumor prostate cells. E‐cadherin binding of cells inhibits cell motility and maintains normal epithelial phenotype. 29 Therefore, E‐cadherin is considered an anti‐migratory protein. Furthermore, changes in E‐cadherin levels have been associated with EMT and mesenchymal‐epithelial transition processes in metastatic cancers. 30 In the same way, GHRH reduces E‐cadherin levels in nontumor cells as well as in advanced PCa cells. However, in more aggressive cells, the response to the neuropeptide occurs earlier and is of greater magnitude, which might demonstrate the start of the EMT process. 28
When tumor cells lose their adherent capacity, they can migrate, degrading the extracellular matrix that surrounds them, invading the vascular system and colonizing other tissues. In our studies, GHRH increased the migratory capacity of nontumor prostate cells at 24 h. An increase in migration after exposure to GHRH has been described in advanced PCa cells. However, in such tumor cells the response occurs earlier (8 h). 28 This is probably due to the increased presence of SVs of GHRH receptors.
The degradation of the extracellular matrix is produced by proteolytic enzymes, mainly by metalloproteases, which are highly expressed in advanced stage prostate tumor tissue. GHRH significantly altered MMP‐2 activity in nontumor prostate cells.
Tumor cells require oxygen and nutrients to proliferate. The vascularization of the tumor is a fundamental step in tumor development. It has been seen that under hypoxic conditions, as it happens in the tumor microenvironment, there is an increase in MMPs expression that positively regulates VEGF levels. 31 , 32 GHRH causes an increase in the levels of VEGF released by nontumor prostate cells.
We considered whether GHRH showed transforming capability in nontumor cells, through in vivo studies. RWPE‐1 cells are nontumorigenic cells, but after stimulation with GHRH, and subsequent inoculation in nude mice, they can generate small tumors. Previous work by our group demonstrated that neuropeptide VIP, a peptide of the GHRH family, could generate tumors in nude mice after inoculation of RWPE‐1 cells previously treated with this neuropeptide. 17 These data point to GHRH as an agent capable of transforming nontumor cells into malignant, providing more information on the oncological processes in which GHRH is involved.
The exposure of RWPE‐1 cells to GHRH results in tumorigenic transformation since these cells acquire characteristics in common with human PCa cells including hyperproliferative activity, increased gelatinolytic activity of MMP‐2, decreased cell adhesion and increased cell migration. In this regard, GHRH receptor antagonists have been reported to block the effects of GHRH. 15 , 27 , 28 All this together corroborates the important role of GHRH as a mediator in the initiation and progression of PCa.
In conclusion, we provide strong evidence that GHRH, a neuropeptide present in the prostate gland, induces tumorigenic potential in RWPE‐1 cells, an immortalized cell line from prostatic epithelial cell origin. The use of cultured cells and tumors developed after their subcutaneous injection in athymic nude mice served to demonstrate a set of effects of GHRH at both molecular and cellular levels that likely implies EMT and reinforces the role of GHRH as a cytokine involved in prostate tumorigenesis.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We thank Dr. L. Puebla (University of Alcala) for her assistance in writing the manuscript. This work was supported by grants from Universidad de Alcalá (CCG2015/BIO‐010 to A.M.B. and CCG2014/BIO‐028 to A.M.B.).
Muñoz‐Moreno L, Carmena MJ, Prieto JC, Schally AV, Bajo AM. Tumorigenic transformation of human prostatic epithelial cell line RWPE‐1 by growth hormone‐releasing hormone (GHRH). The Prostate. 2022;82:933‐941. 10.1002/pros.24339
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941‐1953. 10.1002/ijc.31937 [DOI] [PubMed] [Google Scholar]
- 2. Nowacka‐Zawisza M, Wiśnik E. DNA methylation and histone modifications as epigenetic regulation in prostate cancer (Review). Oncol Rep. 2017;38(5):2587‐2596. 10.3892/or.2017.5972 [DOI] [PubMed] [Google Scholar]
- 3. Junejo NN, AlKhateeb SS. BRCA2 gene mutation and prostate cancer risk. Comprehensive review and update. Saudi Med J. 2020;41(1):9‐17. 10.15537/smj.2020.1.24759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramanand SG, Mani RS. Genetic, environmental, and nuclear factors governing genomic rearrangements. Adv Exp Med Biol. 2019;1210:57‐66. 10.1007/978-3-030-32656-2-3 [DOI] [PubMed] [Google Scholar]
- 5. Singh VK, Pal R, Srivastava P, Misra G, Shukla Y, Sharma PK. Exposure of androgen mimicking environmental chemicals enhances proliferation of prostate cancer (LNCaP) cells by inducing AR expression and epigenetic modifications. Environ Pollut. 2021;272:116397. 10.1016/j.envpol.2020.116397 [DOI] [PubMed] [Google Scholar]
- 6. Jonckheere S, Adams J, De Groote D, Campbell K, Berx G, Goossens S. Epithelial‐mesenchymal transition (EMT) as a therapeutic target. Cells Tissues Organs. 2021;1:2‐26. 10.1159/000512218 [DOI] [PubMed] [Google Scholar]
- 7. He M, Young CYF. Mutant epidermal growth factor receptor vIII increases cell motility and clonogenecity in a prostate cell line RWPE1. J Endocrinol Invest. 2009;32:272‐278. 10.1007/BF03346466 [DOI] [PubMed] [Google Scholar]
- 8. Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis. 1997;18(6):1215‐1223. 10.1093/carcin/18.6.1215 [DOI] [PubMed] [Google Scholar]
- 9. Achanzar WE, Brambila EM, Diwan BA, Webber MM, Waalkes MP. Inorganic arsenite‐induced malignant transformation of human prostate epithelial cells. J Natl Cancer Inst. 2002;94(24):1888‐1891. 10.1093/jnci/94.24.1888 [DOI] [PubMed] [Google Scholar]
- 10. Dasgupta P, Kulkarni P, Bhat NS, et al. Activation of the Erk/MAPK signaling pathway is a driver for cadmium induced prostate cancer. Toxicol Appl Pharmacol. 2020;401:115102. 10.1016/j.taap.2020.115102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiaris H, Chatzistamou I, Papavassiliou AG, Schally AV. Growth hormone‐releasing hormone: not only a neurohormone. Trends Endocrinol Metab. 2011;22(8):311‐317. 10.1016/j.tem.2011.03.006 [DOI] [PubMed] [Google Scholar]
- 12. Barabutis N, Schally AV. Growth hormone‐releasing hormone: extrapituitary effects in physiology and pathology. Cell Cycle. 2010;9(20):4110‐4116. 10.4161/cc.9.20.13787 [DOI] [PubMed] [Google Scholar]
- 13. Kiaris H, Koutsilieris M, Kalofoutis A, Schally AV. Growth hormone‐releasing hormone and extra‐pituitary tumorigenesis: therapeutic and diagnostic applications of growth hormone‐releasing hormone antagonists. Expert Opin Investig Drugs. 2003;12(8):1385‐1394. 10.1517/13543784.12.8.1385 [DOI] [PubMed] [Google Scholar]
- 14. Muñoz‐Moreno L, Bajo AM, Prieto JC, Carmena MJ. Growth hormone‐releasing hormone (GHRH) promotes metastatic phenotypes through EGFR/HER2 transactivation in prostate cancer cells. Mol Cell Endocrinol. 2017;446:59‐69. 10.1016/j.mce.2017.02.011 [DOI] [PubMed] [Google Scholar]
- 15. Muñoz‐Moreno L, Carmena MJ, Schally AV, Prieto JC, Bajo AM. Stimulation of neuroendocrine differentiation in prostate cancer cells by GHRH and its blockade by GHRH antagonists. Invest New Drugs. 2020;38:746‐754. 10.1007/s10637-019-00831-2 [DOI] [PubMed] [Google Scholar]
- 16. Muñoz‐Moreno L, Arenas MI, Carmena MJ, Schally AV, Prieto JC, Bajo AM. Growth hormone‐releasing hormone antagonists abolish the transactivation of human epidermal growth factor receptors in advanced prostate cancer models. Invest New Drugs. 2014;32(5):871‐882. 10.1007/s10637-014-0131-4 [DOI] [PubMed] [Google Scholar]
- 17. Fernández‐Martínez AB, Bajo AM, Isabel Arenas M, Sánchez‐Chapado M, Prieto JC, Carmena MJ. Vasoactive intestinal peptide (VIP) induces malignant transformation of the human prostate epithelial cell line RWPE‐1. Cancer Lett. 2010;299(1):11‐21. 10.1016/j.canlet.2010.07.019 [DOI] [PubMed] [Google Scholar]
- 18. Barabutis N, Schally AV. Knocking down gene expression for growth hormone‐releasing hormone inhibits proliferation of human cancer cell lines. Br J Cancer. 2008;98(11):1790‐1796. 10.1038/sj.bjc.6604386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chopin L, Herington A. A potential autocrine pathway for growth hormone releasing hormone (GHRH) and its receptor in human prostate cancer cell lines. Prostate. 2001;49:116‐121. 10.1002/pros.1125 [DOI] [PubMed] [Google Scholar]
- 20. Busto R, Schally AV, Varga JL, et al. The expression of growth hormone‐releasing hormone (GHRH) and splice variants of its receptor in human gastroenteropancreatic carcinomas. Proc Natl Acad Sci USA. 2002;99(18):11866‐11871. 10.1073/pnas.182433099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Halmos G, Schally AV, Czompoly T, Krupa M, Varga JL, Rekasi Z. Expression of growth hormone‐releasing hormone and its receptor splice variants in human prostate cancer. J Clin Endocrinol Metab. 2002;87(10):4707‐4714. 10.1210/jc.2002-020347 [DOI] [PubMed] [Google Scholar]
- 22. Garcia‐Fernandez MO, Schally AV, Varga JL, Groot K, Busto R. The expression of growth hormone‐releasing hormone (GHRH) and its receptor splice variants in human breast cancer lines; the evaluation of signaling mechanisms in the stimulation of cell proliferation. Breast Cancer Res Treat. 2003;77(1):15‐26. 10.1023/a:1021196504944 [DOI] [PubMed] [Google Scholar]
- 23. Xiong X, Ke X, Wang L, et al. Splice variant of growth hormone‐releasing hormone receptor drives esophageal squamous cell carcinoma conferring a therapeutic target. Proc Natl Acad Sci USA. 2020;117(12):6726‐6732. 10.1073/pnas.1913433117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rick FG, Schally AV, Szalontay L, et al. Antagonists of growth hormone‐releasing hormone inhibit growth of androgen‐independent prostate cancer through inactivation of ERK and Akt kinases. Proc Natl Acad Sci USA. 2012;109(5):1655‐1660. 10.1073/pnas.1120588109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schulz S, Röcken C, Schulz S. Immunocytochemical localisation of plasma membrane GHRH receptors in human tumours using a novel anti‐peptide antibody. Eur J Cancer. 2006;42(14):2390‐2396. 10.1016/j.ejca.2006.03.027 [DOI] [PubMed] [Google Scholar]
- 26. Pozsgai E, Schally AV, Hocsak E, Zarandi M, Rick F, Bellyei S. The effect of a novel antagonist of growth hormone releasing hormone on cell proliferation and on the key cell signaling pathways in nine different breast cancer cell lines. Int J Oncol. 2011;39(4):1025‐1032. 10.3892/ijo.2011.1098 [DOI] [PubMed] [Google Scholar]
- 27. Muñoz‐Moreno L, Arenas MI, Carmena MJ, et al. Anti‐proliferative and pro‐apoptotic effects of GHRH antagonists in prostate cancer. Oncotarget. 2016;7(32):52195‐52206. 10.18632/oncotarget.10710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Muñoz‐Moreno L, Schally AV, Prieto JC, Carmena MJ, Bajo AM. Growth hormone‐releasing hormone receptor antagonists modify molecular machinery in the progression of prostate cancer. Prostate. 2018;78(12):915‐926. 10.1002/pros.23648 [DOI] [PubMed] [Google Scholar]
- 29. Arya M, Bott SR, Shergill IS, Ahmed HU, Williamson M, Patel HR. The metastatic cascade in prostate cancer. Surg Oncol. 2006;15(3):117‐128. 10.1016/j.suronc.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 30. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E‐cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645‐3654. 10.1158/0008-5472.CAN-07-2938 [DOI] [PubMed] [Google Scholar]
- 31. Gong Y, Chippada‐Venkata UD, Oh WK. Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancers (Basel). 2014;6(3):1298‐1327. 10.3390/cancers6031298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A. Positive feedback regulation between MMP‐9 and VEGF in human RPE cells. Invest Ophthalmol Vis Sci. 2007;48(9):4360‐4367. 10.1167/iovs.06-1234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.