

Abstract
Background
To date, variants in the GBA gene represent the most frequent large‐effect genetic factor associated with Parkinson's disease (PD). However, the reason why individuals with the same GBA variant may or may not develop neurodegeneration and PD is still unclear.
Objectives
Therefore, we evaluated the contribution of rare variants in genes responsible for lysosomal storage disorders (LSDs) to GBA‐PD risk, comparing the burden of deleterious variants in LSD genes in PD patients versus asymptomatic subjects, all carriers of deleterious variants in GBA.
Methods
We used a custom next‐generation sequencing panel, including 50 LSD genes, to screen 305 patients and 207 controls (discovery cohort). Replication and meta‐analysis were performed in two replication cohorts of GBA‐variant carriers, of 250 patients and 287 controls, for whom exome or genome data were available.
Results
Statistical analysis in the discovery cohort revealed a significantly increased burden of deleterious variants in LSD genes in patients (P = 0.0029). Moreover, our analyses evidenced that the two strongest modifiers of GBA penetrance are a second variation in GBA (5.6% vs. 1.4%, P = 0.023) and variants in genes causing mucopolysaccharidoses (6.9% vs. 1%, P = 0.0020). These results were confirmed in the meta‐analysis, where we observed pooled odds ratios of 1.42 (95% confidence interval [CI] = 1.10–1.83, P = 0.0063), 4.36 (95% CI = 2.02–9.45, P = 0.00019), and 1.83 (95% CI = 1.04–3.22, P = 0.038) for variants in LSD genes, GBA, and mucopolysaccharidosis genes, respectively.
Conclusion
The identification of genetic lesions in lysosomal genes increasing PD risk may have important implications in terms of patient stratification for future therapeutic trials. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society.
Keywords: Parkinson's disease; GBA; lysosomal genes; mutation burden
Although for decades Parkinson's disease (PD) was considered the prototype of a nongenetic disease, it is now recognized as a complex multifactorial neurodegenerative disorder, with a relevant fraction of genetic heritability. About 15% of patients report a family history of PD, whereas the vast majority of patients have sporadic PD, which is likely related to the combined effect of predisposing genetic variants and environmental risk factors. 1 Heterozygous variations in GBA (encoding beta‐glucocerebrosidase, OMIM*606463), which cause the recessive lysosomal storage disorder (LSD) Gaucher disease (GD), are the most frequent large‐impact genetic risk factor for PD. Many studies have shown an increased frequency of GBA variants in PD compared to controls. 2 , 3 , 4 GBA variants were shown to confer a five‐ to seven‐fold increased risk of PD 4 , 5 and to modify PD manifestations, causing earlier age of onset, more severe cognitive dysfunction, and accelerated progression of the neurodegenerative process. 6 , 7 , 8
Concerning the mechanism relating GBA variants to PD, it has been suggested that either a chronic loss of enzymatic activity or a possible toxic gain of function of the mutated glucocerebrosidase results in lysosomal dysfunction and endoplasmic reticulum stress could contribute to disease pathogenesis. 9 However, the observation that carriers of GBA variants yielding smaller enzymatic residual activity have a more severe phenotype suggests that the pathogenic mechanism is more likely attributable to loss‐of‐function rather than gain‐of‐function effects. 10 , 11
Substantial evidence highlights a broader importance of lysosomal mechanisms in PD susceptibility and pathogenesis. 12 , 13 , 14 This idea is strengthened by multiple lines of evidence, suggesting a wider contribution of genetic variations in lysosomal genes to PD. Besides GBA, other genes involved in LSDs were associated with a higher risk of PD (eg, SMPD1, SCARB2, and GALC). 15 , 16 Additional lysosomal genes with an increased burden of variants in PD patients were recently reported (ATP13A2, LAMP1, TMEM175, and VPS13C). 17 , 18 Moreover, (1) typical motor symptoms of PD such as tremor, bradykinesia, and rigidity were described in patients affected by LSDs (GM1 and GM2 gangliosidosis, neuronal ceroid lipofuscinoses, and Fabry disease), 15 and (2) an excessive burden of LSD gene variants was found in PD patients by studying 54 LSD genes in exome data from 1156 patients versus 1679 controls. 19 Little is known about the mechanisms that underly the increased susceptibility of GBA mutation carriers to PD. The lifelong PD penetrance of GBA variants ranges between 10% and 30% 20 , 21 and significantly differs when comparing carriers of low‐ and high‐risk variants. 22 , 23 However, the reason why patients with the same variant (even within the same family) may or may not develop neurodegeneration and PD is obscure.
To shed light on this important topic, a large genome‐wide association study exploring the genetic modifiers of risk and age at onset in GBA‐PD patients was recently conducted. 24 The two main loci identified as possible modifiers in GBA carriers were SNCA and CTSB (ie, variants rs356219 and rs1293298, lying in close proximity of the two genes), both implicated in the lysosomal autophagy pathway. Interestingly, though a genetic risk score based on common PD genetic factors contributed only little to predicting the GBA‐associated risk for the disease, common variants in genes implicated in lysosomal function were found to exert the largest effect on GBA‐PD risk. 24
In this context, there must be other factors, including rare genetic variants, which are still largely unexplored, and/or environmental and aging‐related factors, involved in modulating GBA‐PD risk. We therefore decided to explore whether the burden of rare lysosomal gene variants contributes to the differences in GBA mutation penetrance, under the hypothesis that multiple LSD gene variants may contribute, in association with GBA dysfunction, to lysosomal impairment.
Patients and Methods
This study was approved by local ethics committees and was conducted according to the Declaration of Helsinki and the Italian legislation on sensitive personal data recording. All participants signed an informed consent.
Study Participants
Discovery cohort: we selected 305 patients diagnosed with typical PD 25 and 207 asymptomatic (as assessed after medical examination) controls who carried a GBA variant causative for GD and/or associated with PD (Supplementary Table 1 and Supplementary Figure 1 and 2). All subjects were Italians of southern European origin. A total of 285 patients were enrolled by the Parkinson Institute Biobank of Milan, Italy (https://www.parkinson.it/biobanca), and an additional 20 patients were recruited at the IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy).
As for genotyping, 80% of PD patients and 46% of controls were identified, thanks to a recent study in which they were analyzed for the four most common PD‐related GBA variants: c.1093G > A, p.E365K; c.1223C > T, p.T408M; c.1226A > G, p.N409S; and c.1448T > C, p.L483P; and the legacy names were p.E326K, p.T369M, p.N370S, and p.L444P, respectively. These missense substitutions altogether account for up to 80% of GBA deleterious variants in PD; p.N370S and p.L444P represent recognized GD pathogenic mutations, whereas p.E326K and p.T369M are PD‐associated non‐GD variants. 26 All four genetic defects were genotyped using a multiplex allele‐specific polymerase chain reaction (PCR) assay. 23 , 27 The remaining 54% of controls were from the MIGen cohort 28 and were previously analyzed by exome sequencing; the remaining 20% of patients underwent a diagnostic next‐generation sequencing (NGS) gene panel. This approach allowed the identification of additional GBA rare variant carriers. All the identified GBA variants were confirmed by Sanger sequencing.
Replication cohorts: for replication studies, we had access to two replication cohorts. The first one (replication cohort 1) was composed of 140 GBA‐PD patients and 156 controls of European origin, collected by the Parkinson's Progression Markers Initiative (PPMI), with whole genome sequencing performed at the Laboratory of Neurogenetics, National Institutes of Health, (Bethesda, MD), and available at https://www.ppmi-info.org/ and https://amp-pd.org/. Data were processed as previously described by Iwaki et al. 29 The second cohort (replication cohort 2) comprised 110 GBA‐PD patients and 131 controls (dbGaP accession numbers: phs000668 and phs000398) of Caucasian origin bearing a PD‐related GBA variant, recruited through the Parkinson Study Group as part of the PROGENI study, using an affected sibling recruitment design (only one sibling for each family was included in our analysis). 30
Targeted NGS
DNA was extracted from peripheral blood using a Chemagic Star workstation (Hamilton, ON, Canada). Targeted NGS was performed on 401 DNAs (those not previously analyzed by whole‐exome sequencing). The target regions were captured using a custom‐designed HaloPlex Target Enrichment kit (Agilent, Santa Clara, CA), according to the manufacturer's protocol. The full coding regions and the intron–exon boundaries (25 nucleotides upstream and downstream each exon) of 50 LSD genes and 13 genes associated with PD were included for ~190 kb (Supplementary Table 2). According to Blauwendraat and colleagues, 31 only PD genes classified as “highly or very highly confident” were included. To reduce time and costs, we used a pooling strategy mixing five DNA samples for each library. The obtained libraries were sequenced (paired‐end, 2x150bp) on a NextSeq500 (Illumina, San Diego, CA). The mean depth coverage across all libraries was ~1300×. NGS data were analyzed through an in‐house pipeline: first, good‐quality reads (ie, having a read mapping quality >20) were aligned to the reference genome (release hg19, February 2009) using the Burrows‐Wheeler Aligner (http://bio-bwa.sourceforge.net/), variant calling was performed using Comprehensive Read Analysis for Identification of Single Nucleotide Polymorphisms from Pooled sequencing (https://bansal-lab.github.io/software/crisp.html), and finally, variants were annotated with Annovar (https://annovar.openbioinformatics.org/en/latest/).
Variant Selection
Independent of the sequencing procedure (exome or targeted NGS), variants covered by at least 20 reads and with a frequency <1% in the non‐Finnish European population (GnomAD v2.1.1 https://gnomad.broadinstitute.org/) were selected and classified in one of the following functional categories: (1) loss‐of‐function (LoF) variants (frameshift, nonsense, and splicing variants, ie, affecting intronic positions ±1 and ±2); (2) missense variants already reported as pathogenic in LSDs/PD; and (3) all other missense variants without a clear pathogenic role. Regarding the last group, the deleteriousness of these variants was estimated using the CADD (Combined Annotation Dependent Depletion) score (https://cadd.gs.washington.edu/), and the variants with a score >25 were retained for further analyses (Supplementary Figure 2).
Variant Validation
All candidate variants identified in the discovery cohort were confirmed by Sanger sequencing; in the case of samples multiplexed for the targeted NGS, this step also allowed the identification of the individual of the pool carrying the variation. Briefly, amplicons spanning the exons of interest were designed, and direct sequencing of the obtained PCR products was carried out using the BigDye Terminator Cycle Sequencing Ready Reaction Kit v1.1 (Thermo Fisher Scientific, Carlsbad, CA) and analyzed on an ABI‐3500 Genetic Analyzer (Thermo Fisher Scientific).
When possible, the phase of GBA variants was defined in those patients who were carriers of at least two variants in the gene. Amplicons containing the two variations were amplified and cloned in the pGEM‐T Easy vector (Promega, Madison, WI) according to the manufacturer's instructions. The obtained plasmids were used to transform JM109 competent cells (Promega); colony PCR and subsequent Sanger sequencing allowed the characterization of each allele.
Statistical Analysis
The Sequence Kernel Association Test‐Optimal (https://cran.r-project.org/web/packages/SKAT/SKAT.pdf) was implemented in R using SKAT v1.3.2.1 to determine the difference in the aggregate burden of rare LSD and PD gene variants between PD patients and controls. Concerning LSD gene variants, we performed the same analysis (adjusted for sex, age, and the 10 main principal components, when available) for each of the following categories: LoF variants, missense variants already associated with LSDs or PD, and missense variants with CADD >25. Burden analysis for each LSD gene was also performed. For chromosome‐X genes, we considered uniform and complete X‐inactivation in women and a similar effect size between men and women. Therefore, women were considered to have 0, 1, or 2 copies of an allele, whereas men were considered to have 0 or 2 copies of the same allele (ie, male hemizygotes were considered equivalent to female homozygotes).
The threshold for significance was set to P < 0.05; we considered the multiple testing issue by correcting each P according to the number of evaluated variant categories (see table legends).
Replication Studies and Meta‐Analysis
For replication studies, we extracted LSD variants from all GBA carriers of the two cohorts following the same filtering criteria used for the Italian set and performed burden analyses as described earlier.
In the meta‐analysis, we focused on all genes/variant categories/disease classes that showed a significant association in the discovery cohort. P‐values, pooled odds ratios (ORs), and confidence intervals (CIs) were calculated using the Mantel–Haenszel model. 32 A Breslow–Day test for heterogeneity (with Tarone's correction) was performed for testing differences in OR between sample populations. 33 , 34
Results
Overall Screening Results
A cohort of 512 subjects (305 patients and 207 controls, Supplementary Table 1), all carriers of at least one GBA variant, were analyzed using exome sequencing (111 individuals) or a custom NGS panel (401 individuals) comprising 50 lysosomal genes causing LSDs and 13 known PD genes (Supplementary Table 2). After the filtering step (described earlier), we found 184 different variants in 201 individuals (Supplementary Table 3), all validated by Sanger sequencing: the majority of them were missense (88%), and 12% were LoF variants (8% nonsense/frameshift and 4% splicing variants). Among missense variants, those with a CADD score comprised between 25 and 30 were the most frequent (40%), followed by missense variants already associated with LSD or parkinsonism (31%) and by those with a CADD >30 (17%).
Increased Burden of LSD Variants in GBA‐PD
Mutation burden analysis was performed separately for lysosomal and known PD genes. GBA was included in the list of LSD genes to search for additional variants other than the one used for patient enrollment. Only the second GBA variant (variant 2) was incorporated in the analysis, considering that all the analyzed subjects are carriers of at least one GBA PD‐associated variant (variant 1) according to our a priori enrollment criteria. In individuals carrying two GBA variants, variants 1 and 2 were assigned according to their severity, as specified in Supplementary Table 4.
Considering LSD genes, a significantly higher proportion of individuals carrying a deleterious variant were observed in PD patients compared to controls (30.8% and 21.7%, respectively, P = 0.0029) (Fig. 1). This result is remarkably strong considering that the power calculation taking into account the sample size of our discovery cohort and the overall number of variants identified in LDS genes indicated 63% of probability to find an association at P < 0.05. This enrichment is significant even after excluding the second GBA variant (27.5% in patients vs. 20.3% in controls, P = 0.019) (Table 1). Conversely, we found no significant difference in the number of deleterious variants in known PD genes between patients and controls (17.4% vs. 18.8%, respectively, P = 0.28) (Fig. 1).
FIG 1.
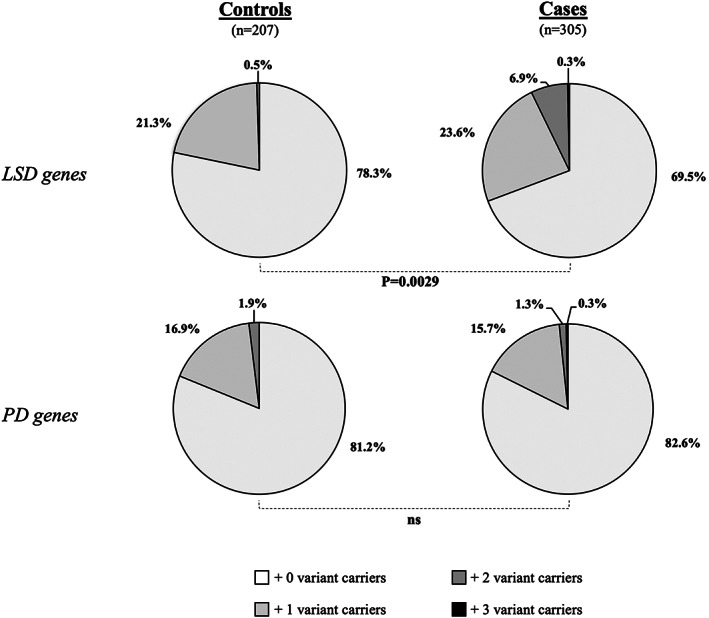
Distribution of selected variants in LSD‐ and PD‐related genes among patients and controls. SKAT‐O (Sequence Kernel Association Test‐Optimal) analyses were performed on variants identified in our cohort of GBA‐mutated patients and controls after sequencing 50 LSD and 13 PD genes.
TABLE 1.
Burden analyses of LSD variants
| Frequencies | |||
|---|---|---|---|
| Controls (%) n = 207 | PD (%) n = 305 | P* | |
| All LSD variants | 21.7 | 30.8 | 0.0029 |
| All LSD variants excluding GBA | 20.3 | 27.5 | 0.019 |
| Variant categories | |||
| LoF | 3.4 | 3.3 | 0.85 |
| Known mutations | 8.7 | 15.7 | 0.022 |
| CADD >25 | 9.7 | 16.4 | 0.041 |
| Diseases a | |||
| Sphingolipidoses | 8.2 | 12.1 | 0.23 |
| Mucopolysaccharidoses | 1.0 | 6.9 | 0.0020 |
| Neuronal lipofuscinoses | 2.9 | 4.3 | 0.57 |
| Glycoproteinoses | 1.9 | 4.6 | 0.18 |
| Integral membrane protein disorders | 5.8 | 4.9 | 0.12 |
| Posttranslational modification defects | 1.4 | 3.3 | 0.35 |
Variants in LSD genes were classified according to the diseases for which they are responsible. 35
Abbreviations: PD, Parkinson's disease; LoF, loss of function; CADD, Combined Annotation Dependent Depletion.
*SKAT‐O (Sequence Kernel Association Test‐Optimal) P‐values adjusted for age/sex. Significant P‐values are in bold (P‐values corrected for multiple testing were for variant categories, P = 0.05/3 = 0.016, and for disease categories, P = 0.05/6 = 0.0083).
To disentangle the relative contribution of LSD variants to the risk for GBA‐PD, subjects were also subclassified according to their GBA variant (only the four most common PD‐related variants were considered; RecNcil alleles were included in the p.L444P group), and for each group the same burden analysis was performed. Interestingly, a significant enrichment of variants in LSD genes was present only in PD patients who carried a non GD‐causing GBA variant (ie, the p.E326K or p.T369M variants, P = 0.0004 and P = 0.023, respectively) (Fig. 2).
FIG 2.
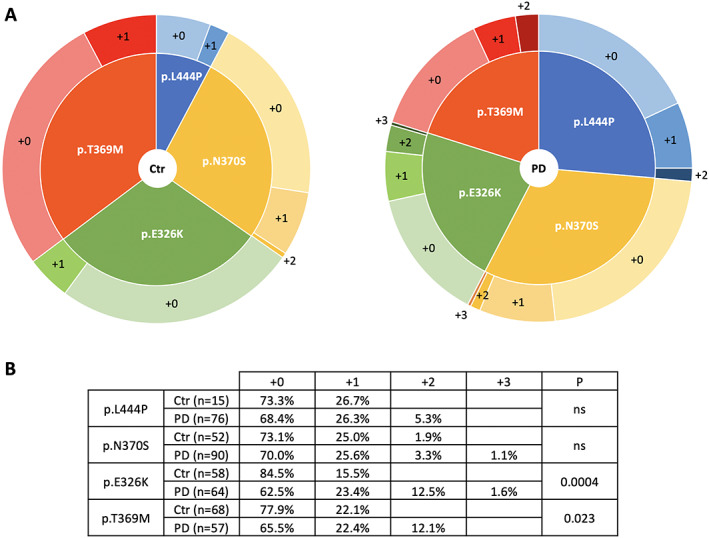
Distribution of LSD variants according to GBA genotype in PD patients and controls. (A) Graphical representation of the distribution of LSD deleterious variants among patients and controls classified depending on their GBA variant. (B) Detailed table of the obtained results. [Color figure can be viewed at wileyonlinelibrary.com]
Focusing on the different functional categories of analyzed variants, missense substitutions causing LSD were present in 15.7% of PD patients and in 8.7% of controls (P = 0.022), whereas those with CADD >25 accounted for 16.4% of PD patients compared to 9.7% of controls (P = 0.041). As for LoF variants, no difference between patients and controls was observed (3.3% vs. 3.4%, P = 0.85) (Table 1).
Burden analysis was also performed categorizing LSD genes according to the disease type for which they are responsible (Supplementary Table 2). 35 The most interesting result concerns the mucopolysaccharidosis‐related genes, in which a higher burden of deleterious variants was observed in patients compared to controls (6.9% vs. 1%, P = 0.0020) (Table 1).
Single‐Gene Analysis: A Second Variant in GBA Is the Main Risk Modulator for GBA‐PD
Analyzing the distribution of variants in different LSD genes in all subjects enrolled, we observed that the gene with the highest number of variants was GBA (n = 20), followed by GNPTAB, NPC1, and SMPD1 (all with n = 10). Other eight genes (CLN5, CTSF, GAA, GLA, GUSB, HEXB, MAN2B1, and MCOLN1) carried more than five genetic defects, whereas the vast majority of genes showed four or fewer variants (Supplementary Table 5).
Single‐gene association tests were performed only for those genes with the number of variations greater than or equal to five (Fig. 3). Variants in the GBA gene were present at a significantly higher frequency in patients (5.6%) compared to controls (1.4%, P = 0.023), suggesting that a second variant in GBA represents the strongest modulator of the disease penetrance among GBA mutation carriers. All 20 individuals carrying an additional variant in GBA were analyzed to determine the phase of the two variants. For 17 of them, a strategy combining long PCR and cloning allowed the determination of the phase: in the majority of patients (71%), an in‐trans association was experimentally confirmed (Supplementary Table 4).
FIG 3.
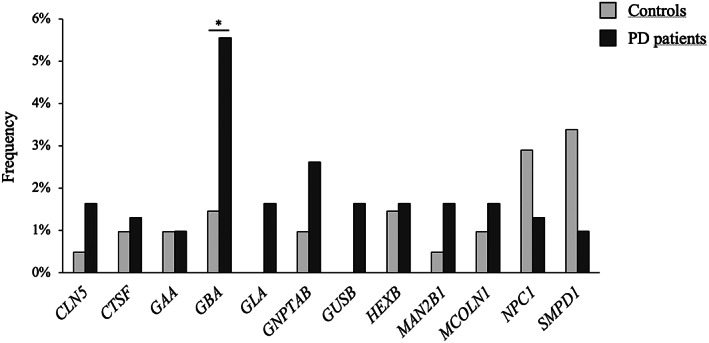
Single‐gene burden analyses. SKAT‐O (Sequence Kernel Association Test‐Optimal) single‐gene association tests were performed only on genes with a number of identified variants greater than or equal to five. * = P < 0.05 (corrected P‐value threshold = 0.05/12 = 0.0042).
Meta‐Analysis Confirmed the Role of Lysosomal Genes as Modulators of PD Penetrance
To confirm our results, the same burden analyses for LSD variants were performed in two independent replication cohorts of similar size. These were composed of European‐descent individuals, for 250 GBA‐PD patients and 287 GBA‐mutated asymptomatic controls, all with exome or genome data available.
In replication cohort 1, we observed suggestive evidence of an enrichment in LSD variants in PD patients (33.6% vs. 27.6% in patients and controls, respectively, P = 0.16), mainly driven by variations in genes responsible for sphingolipidoses and neuronal lipofuscinoses (Table 2A). In replication cohort 2, we replicated the significant burden of LSD variants in PD patients compared to asymptomatic GBA carriers (34.5% vs. 24.4%, P = 0.00051); this enrichment was also confirmed excluding all the second GBA variants from the analysis (Table 2A). In both replication cohorts, the single‐gene analysis confirmed the major role as a modulator of PD penetrance of a second mutation in the GBA gene (replication cohort 1: 7.9% vs. 2.5%, P = 0.032, and replication cohort 2: 8.2% vs. 0.8, P = 0.00068) (Table 2A).
TABLE 2.
Replication studies and meta‐analysis
| A. Replication studies | ||||||
|---|---|---|---|---|---|---|
| Replication cohort 1 | Replication cohort 2 | |||||
| Frequencies | Frequencies | |||||
| Controls (%) n = 156 | PD (%) n = 140 | P a | Controls (%) n = 131 | PD (%) n = 110 | P a | |
| All LSD variants | 27.6 | 33.6 | 0.16 | 24.4 | 34.5 | 0.00051 |
| All LSD variants excluding GBA | 25 | 28.6 | 0.78 | 24.4 | 27.3 | 0.0078 |
| Variant categories | ||||||
| LoF | 1.9 | 4.3 | 0.11 | 4.6 | 5.5 | 0.61 |
| Missense | 25.6 | 30.7 | 0.28 | 19.8 | 30.0 | 0.00051 |
| Diseases | ||||||
| Sphingolipidoses | 7.7 | 14.3 | 0.051 | 8.4 | 17.3 | 0.0033 |
| Mucopolysaccharidoses | 8.3 | 5.0 | 0.16 | 3.1 | 7.3 | 0.025 |
| Neuronal lipofuscinoses | 5.1 | 2.9 | 0.07 | 5.3 | 1.8 | 0.17 |
| Glycoproteinoses | 3.2 | 4.3 | 0.82 | 3.1 | 7.3 | 0.014 |
| Integral membrane protein disorders | 3.2 | 6.4 | 0.77 | 3.8 | 0.9 | 0.14 |
| Posttranslational modification defects | 1.9 | 4.3 | 0.90 | 1.5 | 1.8 | 0.69 |
| Genes b | ||||||
| GBA | 2.5 | 7.9 | 0.032 | 0.8 | 8.2 | 0.00068 |
| B. Meta‐analysis | |||||||
|---|---|---|---|---|---|---|---|
| P a (discovery) | P a (replication1) | P a (replication2) | Pooled P | Pooled OR c | 95% CI for pooled OR c | P for heterogeneity d | |
| All LSD variants | 0.0029 | 0.16 | 0.00051 | 0.0063 | 1.42 | 1.10–1.83 | 0.83 |
| All LSD variants excluding GBA | 0.019 | 0.78 | 0.0078 | 0.077 | 1.27 | 0.97–1.64 | 0.75 |
| Mucopolysaccharidoses | 0.0020 | 0.16 | 0.025 | 0.038 | 1.83 | 1.04–3.22 | 0.0052 |
| GBA | 0.023 | 0.032 | 0.00068 | 0.00019 | 4.36 | 2.02–9.45 | 0.56 |
Significant adjusted SKAT‐O (Sequence Kernel Association Test‐Optimal) P‐values are in bold (P‐values corrected for multiple testing were for variant categories, P = 0.05/3 = 0.016; for disease classes, P = 0.05/6 = 0.0083; and for single‐gene analysis, P = 0.05/2 = 0.025). P‐values <0.05 are in italics.
Genes with significant burden test in the discovery cohort.
Pooled ORs/CIs were calculated using the Mantel–Haenszel model.
P‐values for heterogeneity were calculated using Breslow–Day test with Tarone's correction.
We also performed a meta‐analysis on the variant categories that were significantly enriched in the discovery cohort (Table 2B; Supplementary Figure 3). As for LSD deleterious variants, we observed a pooled OR of 1.42 (95% CI = 1.10–1.83, P = 0.0063). The effect of variants in mucopolysaccharidosis genes seemed to be even higher, with an OR of 1.83 (95% CI = 1.04–3.22, P = 0.038). Finally, a second mutation in the GBA gene emerged as the genetic risk factor, with the highest impact on the modulation of the penetrance of GBA mutations (OR = 4.36, 95% CI = 2.02–9.45, P = 0.00019).
Discussion
A major finding of our work is that the overall burden of deleterious variants in lysosomal genes is significantly higher in GBA‐PD patients compared to asymptomatic GBA‐variant carriers. This signal is mostly due to the presence of a second deleterious variant in GBA or in one of the genes related to mucopolysaccharidoses (MPS). Interestingly, no significant increased burden of variants was found for the set of the known PD genes we analyzed. This is in line with what was reported by Blauwendraat and colleagues, 24 who found that the contribution of known PD loci in modulating the risk of GBA‐PD is not large. A partially surprising result is the finding that only missense defects (associated with LSDs or showing a CADD >25) seem to play an important role in increasing the risk of PD, whereas LoF alleles in lysosomal genes are equally distributed among patients and controls. With all the caution required considering the small number of null alleles found, this could be related to the expression of a dysfunctional lysosomal enzyme in the organelle, which could add further stress by promoting lysosomal dysfunction and/or protein aggregation.
Our burden analysis in the discovery cohort also highlighted the role of MPS genes as modulators of GBA‐PD risk, which was confirmed in the meta‐analysis (OR = 1.83, 95% CI = 1.04–3.22, P = 0.038). This observation is in line with several findings relating MPS to PD: (1) the identification of GUSB (responsible for MPS type VII) as the candidate gene of a novel PD locus, 36 (2) the discovery of a common NAGLU (MPS IIIB) haplotype associated with PD (OR = 1.3), 37 and (3) the observation of α‐synuclein deposits in the brain of MPS IIIA and MPS IIIB patients. 37 , 38
Considering single‐gene analyses, we found that a second “hit” in this gene represents the most important contribution to disease risk, reaching a remarkable OR of 4.36 in the meta‐analysis (95% CI = 2.02–9.45, P = 0.00019). This was not completely unexpected, as it was previously proposed that non‐GD‐causing variants in GBA (ie, p.E326K, p.T369M) are frequently associated with a second genetic defect in the gene in PD patients and more important that age of onset is also lowered if you carry two GBA variants 24 , 39 , 40 ; it is also true that no clear‐cut results were obtained by similar investigations, 41 , 42 possibly because of the lack of statistical power in the examined cohorts. In particular, previous studies dealt with only a few dozen GBA‐PD patients and, more important, with small cohorts of healthy unrelated individuals carrying GBA mutations. We tried to overcome this problem by performing a large genotyping effort (>11,000 unrelated samples screened, all of Italian origin) to identify more than 500 individuals who are carriers of one PD‐related GBA variant. 23 In addition, we tried to assess, whenever possible, the phase of association of the two GBA variants, and in most cases we could demonstrate an in‐trans association, thus supporting the concept of a double hit in a lysosomal gene in determining a higher risk for PD.
We are aware that our work has some limitations: (1) the design of our NGS panel includes only the coding regions of the selected LSD genes, so the potential contribution of the noncoding regulatory variants in the promoter and untranslated regions, as well as in the deep intronic portions, was not assessed; in this respect, the contribution of a second variant in the GBA gene or of variants in other lysosomal genes could have been underestimated; (2) the pooled sequencing strategy might have missed some mutated alleles, even though we confirmed 100% of identified variants by Sanger sequencing; (3) patient stratification according to GBA mutations might introduce a population bias as, for example, the p.N370S variant as well as some SMPD1 variants is more frequent among individuals of Askenazi Jewish descent, whereas the p.E326K and p.T369M are more frequent among northern Europeans 43 ; and (4) replication cohorts are smaller than the discovery cohort because of the difficulty in finding patient/control samples with mutations in GBA and exome/genome data available.
In conclusion, the present study points out the contribution of deleterious variants in LSD genes as modifiers of PD penetrance in GBA carriers. Early detection of individuals with higher PD risk may be beneficial for future neuroprotective treatments.
Author Roles
Letizia Straniero played a major role in the acquisition of data; designed and conceptualized the study; analyzed and interpreted the data; drafted and revised the manuscript. Valeria Rimoldi played a major role in the acquisition of data; performed the genotyping; drafted and revised the manuscript. Edoardo Monfrini recruited patient and control samples of the discovery cohort; performed the genotyping and revised the manuscript. Salvatore Bonvegna recruited patient and control samples of the discovery cohort and revised the manuscript. Giada Melistaccio played a major role in the acquisition of data; performed the genotyping and revised the manuscript. Julie Lake played a major role in the acquisition of data; analyzed the data and revised the manuscript. Giulia Soldà played a major role in the acquisition of data and revised the manuscript Massimo Aureli played a major role in the acquisition of data and revised the manuscript Shankaracharya collected the replication cohorts and revised the manuscript. Pamela Keagle collected the replication cohorts and revised the manuscript. Tatiana Foroud collected the replication cohorts and revised the manuscript. John E. Landers collected the replication cohorts and revised the manuscript. Cornelis Blauwendraat collected the replication cohorts and revised the manuscript. Anna Zecchinelli recruited patient and control samples of the discovery cohort and revised the manuscript. Roberto Cilia recruited patient and control samples of the discovery cohort and revised the manuscript. Alessio Di Fonzo recruited patient and control samples of the discovery cohort and revised the manuscript. Gianni Pezzoli recruited patient and control samples of the discovery cohort and revised the manuscript. Stefano Duga supervised the study; designed and conceptualized the study; interpreted the data; drafted and revised the manuscript. Rosanna Asselta supervised the study; designed and conceptualized the study; analyzed and interpreted the data; drafted and revised the manuscript.
Supporting information
Supplementary Figure 1. GBA mutation distribution in the analyzed cohort.
Supplementary Figure 2. Flowchart of subject and variant selection.
Supplementary Figure 3. Forest plots of the meta‐analyses.
Supplementary Table 1. Demographic and clinical data of the discovery cohort.
Supplementary Table 2. Genes included in the sequencing panel (separate Excel file).
Supplementary Table 3. Identified variants (separate Excel file).
Supplementary Table 4. Subjects compound heterozygous for GBA mutations.
Supplementary Table 5. Distribution of variants in LSD genes.
Acknowledgments
This manuscript is dedicated in memory of Prof. Stefano Duga, whose enthusiastic support and guidance had inspired this project immensely.
We thank Francesca Cancellieri for her invaluable assistance and technical support. We thank Drs Margherita Canesi, Francesca Del Sorbo, Claudio B. Mariani, Nicoletta Meucci, Giorgio Sacilotto, Silvana Tesei, and Michela Zini of the Parkinson Institute for patient referral. Finally, we thank all patients and their relatives for their contribution.
The DNA samples from patients were obtained from the Parkinson Institute Biobank (https://www.parkinson.it/biobanca), member of the Telethon Network of Genetic Biobanks (http://biobanknetwork.telethon.it/), and supported by Fondazione Grigioni per il Morbo di Parkinson, Milan, Italy. This study utilized the high‐performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (Bethesda, MD) (https://hpc.nih.gov/). Data used in the preparation of this article were obtained from the PPMI database (www.ppmi-info.org/data). As such, the investigators within PPMI contributed to the design and implementation of PPMI and/or provided data and collected samples but did not participate in the analysis or writing of this report. For up‐to‐date information on the study, visit www.ppmi-info.org. PPMI—a public–private partnership—is funded by The Michael J. Fox Foundation for Parkinson's Research and corporate sponsors, including AbbVie, AcureX Therapeutics, Allergan, Amathus Therapeutics, Avid Radiopharmaceuticals, BIAL Biotech, Biogen, Biolegend, Briston‐Myers Squibb, Calico, Celgene, Denali, 4D Pharma, GE Healthcare, Genentech, GlaxoSmithKline, Golub Capital, Handl Therapeutics, Insitro, Janssen neuroscience, Lilly, Lundbeck, Merck, Meso Scale Discovery, Neurocrine Biosciences, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, Verily, and Voyager Therapeutics. An up‐to‐date list of all PPMI Industry Partners can be found at http://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors/. Data used in the preparation of this article were obtained from the AMP PD Knowledge Platform. For up‐to‐date information on the study, visit https://www.amp-pd.org. AMP PD—a public–private partnership—is managed by the FNIH and funded by Celgene, GSK, The Michael J. Fox Foundation for Parkinson's Research, the National Institute of Neurological Disorders and Stroke, Pfizer, and Verily.
This work was supported by PRIN (Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale, grant no. 2017228L3J, program coordinator S.D.) and by the Fondazione Grigioni per il Morbo di Parkinson, Milan, Italy. L.S. was supported by a fellowship from the Fondazione Grigioni per il Morbo di Parkinson. This work was supported in part by the Intramural Research Programs of the National Institute on Aging (NIA). J.E.L. and T.F. are supported by funding from the National Institutes of Health (National Institute of Neurological Disorders and Stroke, R01NS096740).
J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics and consultant for ACI Clinical LLC sponsored by Biogen, Inc., or Ionis Pharmaceuticals, Inc. J.E.L. is also consultant for Perkins Coie LLP and may provide expert testimony. All other authors declare no conflict of interest.
Previous affiliation for Dr. Roberto Cilia: Parkinson Institute, ASST Gaetano Pini‐CTO, Milan, Italy.
Data Availability Statement
Data available on request from the authors.
References
- 1. Bandres‐Ciga S, Diez‐Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson's disease: an introspection of its journey towards precision medicine. Neurobiol Dis 2020;137:104782. 10.1016/j.nbd.2020.104782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Swan M, Saunders‐Pullman R. The association between ß‐glucocerebrosidase mutations and parkinsonism. Curr Neurol Neurosci Rep 2013;13(8):368. 10.1007/s11910-013-0368-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J, Zhang HX. Significant study of population stratification, sensitivity analysis and trim and fill analyses on GBA mutation and Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2014;165B(1):96–102. 10.1002/ajmg.b.32214 [DOI] [PubMed] [Google Scholar]
- 4. Asselta R, Rimoldi V, Siri C, et al. Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism Relat Disord 2014;20(11):1215–1220. 10.1016/j.parkreldis.2014.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361(17):1651–1661. 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winder‐Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain 2013;136(Pt2):392–399. 10.1093/brain/aws318 [DOI] [PubMed] [Google Scholar]
- 7. Brockmann K, Srulijes K, Pflederer S, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30(3):407–411. 10.1002/mds.26071 [DOI] [PubMed] [Google Scholar]
- 8. Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging 2016;37:209.e1–209.e7. 10.1016/j.neurobiolaging.2015.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Migdalska‐Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 2016;139(Suppl 1):77–90. 10.1111/jnc.13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA‐associated Parkinson's disease: the mutation matters. Ann Neurol 2016;80(5):662–673. 10.1002/ana.24777 [DOI] [PubMed] [Google Scholar]
- 11. Huh YE, Ruby Chiang MS, Locascio JJ, et al. β‐Glucocerebrosidase activity in GBA‐linked Parkinson's disease: the type of mutation matters. Neurology 2020;95(6):e685–e696. 10.1212/WNL.0000000000009989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386(9996):896–912. 10.1016/S0140-6736(14)61393-3 [DOI] [PubMed] [Google Scholar]
- 13. Moors T, Paciotti S, Chiasserini D, et al. Lysosomal dysfunction and α‐Synuclein aggregation in Parkinson's disease: diagnostic links. Mov Disord 2016;31(6):791–801. 10.1002/mds.26562 [DOI] [PubMed] [Google Scholar]
- 14. Wong YC, Krainc D. Lysosomal trafficking defects link Parkinson's disease with Gaucher's disease. Mov Disord 2016;31(11):1610–1618. 10.1002/mds.26802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Do CB, Tung JY, Dorfman E, et al. Web‐based genome‐wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLoS Genet 2011;7(6):e1002141. 10.1371/journal.pgen.1002141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang D, Nalls MA, Hallgrímsdóttir IB, et al. A meta‐analysis of genome‐wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 2017;49(10):1511–1516. 10.1038/ng.3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krohn L, Öztürk TN, Vanderperre B, et al. Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol 2020;87(1):139–153. 10.1002/ana.25629 [DOI] [PubMed] [Google Scholar]
- 18. Hopfner F, Mueller SH, Szymczak S, et al. Rare variants in specific lysosomal genes are associated with Parkinson's disease. Mov Disord 2020;35(7):1245–1248. 10.1002/mds.28037 [DOI] [PubMed] [Google Scholar]
- 19. Robak LA, Jansen IE, van Rooij J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 2017;140(12):3191–3203. 10.1093/brain/awx285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012;78(6):417–420. 10.1212/WNL.0b013e318245f476 [DOI] [PubMed] [Google Scholar]
- 21. Rana HQ, Balwani M, Bier L, Alcalay RN. Age‐specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013;15(2):146–149. 10.1038/gim.2012.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gan‐Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84(9):880–887. 10.1212/WNL.0000000000001315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Straniero L, Asselta R, Bonvegna S, et al. The SPID‐GBA study: sex distribution, penetrance, incidence, and dementia in GBA‐PD. Neurol Genet 2020;6(6):e523. 10.1212/NXG.0000000000000523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blauwendraat C, Reed X, Krohn L, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain 2020;143(1):234–248. 10.1093/brain/awz350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55(3):181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malek N, Weil RS, Bresner C, et al. Features of GBA‐associated Parkinson's disease at presentation in the UKtracking Parkinson's study. J Neurol Neurosurg Psychiatry 2018;89(7):702–709. 10.1136/jnnp-2017-317348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Straniero L, Rimoldi V, Melistaccio G, et al. A rapid and low‐cost test for screening the most common Parkinson's disease‐related GBA variants. Parkinsonism Relat Disord 2020;80:138–141. 10.1016/j.parkreldis.2020.09.036 [DOI] [PubMed] [Google Scholar]
- 28. Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015;518:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwaki H, Leonard HL, Makarious MB, et al. Accelerating medicines partnership: Parkinson's disease. Genetic resource. Mov Disord 2021;36(8):1795–1804. 10.1002/mds.28549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nichols WC, Pankratz N, Marek DK, et al. Parkinson study group‐PROGENI investigators. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009;72(4):310–316. 10.1212/01.wnl.0000327823.81237.d1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol 2020;19(2):170–178. 10.1016/S1474-4422(19)30287-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719–748. 10.1093/jnci/22.4.719 [DOI] [PubMed] [Google Scholar]
- 33. Breslow NE, Day NE. Classical methods of analysis of grouped data. In: statistical methods in cancer research. Volume I ‐ the analysis of case‐control studies. IARC Sci Publ 1980;32:122–146. [PubMed] [Google Scholar]
- 34. Tarone RE. On heterogeneity tests based on efficient scores. Biometrika 1985;72:91–95. [Google Scholar]
- 35. Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers 2018;4(1):27. 10.1038/s41572-018-0025-4 Erratum in: Nat Rev Dis Primers. 2018;4(1):36. Erratum in: Nat Rev Dis Primers 2019;5(1):34 [DOI] [PubMed] [Google Scholar]
- 36. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol 2019;18(12):1091–1102. 10.1016/S1474-4422(19)30320-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winder‐Rhodes SE, Garcia‐Reitböck P, Ban M, et al. Genetic and pathological links between Parkinson's disease and the lysosomal disorder Sanfilippo syndrome. Mov Disord 2012;27(2):312–315. 10.1002/mds.24029 [DOI] [PubMed] [Google Scholar]
- 38. Hamano K, Hayashi M, Shioda K, Fukatsu R, Mizutani S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: analysis of human brain tissue. Acta Neuropathol 2008;115(5):547–559. [DOI] [PubMed] [Google Scholar]
- 39. Mallett V, Ross JP, Alcalay RN, et al. GBA p.T369M substitution in Parkinson disease: polymorphism or association? A meta‐analysis. Neurol Genet. 2016;2(5):e104. 10.1212/NXG.0000000000000104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldstein O, Gana‐Weisz M, Cohen‐Avinoam D, et al. Revisiting the non‐Gaucher‐GBA‐E326K carrier state: is it sufficient to increase Parkinson's disease risk? Mol Genet Metab 2019;128(4):470–475. 10.1016/j.ymgme.2019.10.001 [DOI] [PubMed] [Google Scholar]
- 41. Alcalay RN, Dinur T, Quinn T, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71(6):752–757. 10.1001/jamaneurol.2014.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Menozzi E, Schapira AHV. Exploring the genotype‐phenotype correlation in GBA‐Parkinson disease: clinical aspects, biomarkers, and potential modifiers. Front Neurol 2021;12:694764. 10.3389/fneur.2021.694764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y, Shu L, Sun Q, et al. Integrated genetic analysis of racial differences of common GBA variants in Parkinson's disease: a meta‐analysis. Front Mol Neurosci 2018;11:43. 10.3389/fnmol.2018.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. GBA mutation distribution in the analyzed cohort.
Supplementary Figure 2. Flowchart of subject and variant selection.
Supplementary Figure 3. Forest plots of the meta‐analyses.
Supplementary Table 1. Demographic and clinical data of the discovery cohort.
Supplementary Table 2. Genes included in the sequencing panel (separate Excel file).
Supplementary Table 3. Identified variants (separate Excel file).
Supplementary Table 4. Subjects compound heterozygous for GBA mutations.
Supplementary Table 5. Distribution of variants in LSD genes.
Data Availability Statement
Data available on request from the authors.