

Abstract
Multiple mitochondrial matrix enzymes playing key roles in metabolism require cofactors for their action. Due to the high impermeability of the mitochondrial inner membrane, these cofactors need to be synthesized within the mitochondria or be imported, themselves or one of their precursors, into the organelles. Transporters belonging to the protein family of mitochondrial carriers have been identified to transport the coenzymes: thiamine pyrophosphate, coenzyme A, FAD and NAD+, which are all structurally similar to nucleotides and derived from different B‐vitamins. These mitochondrial cofactors bind more or less tightly to their enzymes and, after having been involved in a specific reaction step, are regenerated, spontaneously or by other enzymes, to return to their active form, ready for the next catalysis round. Disease‐causing mutations in the mitochondrial cofactor carrier genes compromise not only the transport reaction but also the activity of all mitochondrial enzymes using that particular cofactor and the metabolic pathways in which the cofactor‐dependent enzymes are involved. The mitochondrial transport, metabolism and diseases of the cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD+ are the focus of this review.
Keywords: coenzyme, coenzyme A, flavin adenine dinucleotide, mitochondria, mitochondrial carrier family SLC25, nicotinamide adenine dinucleotide, thiamine pyrophosphate
Abbreviations
- ALS
acetolactate synthase
- BCAA
branched‐chain amino acid
- BCKDH(c)
branched‐chain α‐ketoacid dehydrogenase (complex)
- CHO
chinese hamster ovary
- CoA
coenzyme A
- COASY
coenzyme A synthase
- EPRA
expression, purification, reconstitution and transport assay
- FAD
flavin adenine dinucleotide
- FADS
flavin adenine dinucleotide synthase
- FMN
flavin mononucleotide
- MADD
multiple acyl‐CoA dehydrogenase deficiencies
- MC
mitochondrial carrier
- NAAD
nicotinic acid adenine dinucleotide
- NAD+
nicotinamide adenine dinucleotide
- NAMN
nicotinate mononucleotide
- NUDIX
nucleoside diphosphate‐linked moiety X
- OADH(c)
2‐oxoadipate dehydrogenase (complex)
- OGDH(c)
oxoglutarate dehydrogenase (complex)
- PANK
pantothenate kinase
- PAP
adenosine 3′,5′‐diphosphate
- PDC
pyruvate decarboxylase
- PDH(c)
pyruvate dehydrogenase (complex)
- RDA
recommended daily allowance
- SAM
S‐adenosylmethionine
- SM
synthetic minimal
- TCA
tricarboxylic acid
- ThMP
thiamine monophosphate
- ThPP
thiamine pyrophosphate
- ThTP
thiamine triphosphate
- THTPA
thiamine triphosphatase
- TKT
transketolase
- TPC
thiamine pyrophosphate carrier
- TPK1
thiamine pyrophosphatase 1
1. INTRODUCTION
Many enzymes use different non‐proteic cofactors, such as metal ions and organic coenzymes, that have an important role in catalysis, for example, by activating the substrates, transferring chemical groups and donating/accepting electrons or protons. Whereas many microorganisms and plants have biosynthesis capabilities for the coenzymes, animals depend on diet absorption of most of them or their precursors, in the form of vitamins or essential amino acids. Subsequently, the cofactors or their precursors and derivatives are distributed, taken up by various tissue cell types, and the precursors are modified to assume their active forms. Many of these cofactors are required in the mitochondrial matrix for enzymatic activities of vital importance in central metabolism. Some of the mitochondrial cofactors are synthesized or assembled, at least partly, in mitochondria, such as ubiquinone, lipoate, iron–sulfur clusters and heme. 1 , 2 , 3 , 4 Others have to be imported into mitochondria, such as thiamine pyrophosphate (ThPP), FAD, NAD+, CoA, pyridoxal phosphate, biotin, tetrahydrofolate and cobalamin, that are all B‐type vitamins or their derivatives: B1 (thiamine), B2 (riboflavin), B3 (niacin), B5 (pantothenic acid), B6 (pyridoxine), B7 (biotin), B9 (folic acid or folate) and B12 (cobalamins), respectively. Until now, mitochondrial coenzyme transporters have been identified for ThPP, CoA, FAD (and folate) and NAD+, and they all belong to the mitochondrial carrier protein family. 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13
Mitochondrial carriers (MCs or SLC25 in higher animals) are found in all eukaryotes in variable numbers: 35 in Saccharomyces cerevisiae, 53 in Homo sapiens and 58 in Arabidopsis thaliana. 14 , 15 , 16 , 17 They all have the N‐ and C‐termini exposed towards the cytosolic side of the mitochondrial membrane and a triple‐repeated sequence of about 100 amino acids, which contains two transmembrane segments that are linked by a signature motif sequence PX[DE]XX[KR]X[KR]X20−30[DE]GIUB2612X[WYF][KR]G (PROSITE PS50920, PFAM PF00153 and IPR00193). 18 , 19 , 20 , 21 , 22 , 23 With a few exceptions, MCs are localized to the inner membrane of mitochondria, where their about 300‐residue structure is arranged in a barrel of six‐transmembrane α‐helices (H1‐H6) around a central substrate translocation pore with gates towards the intermembrane space (cytoplasmic c‐gate) and matrix (m‐gate) sides of the membrane. 24 , 25 , 26 With this structural arrangement, MCs transport their specific substrates across the mitochondrial inner membrane with a similar mechanism (named “single binding center‐gating”, 27 “ping pong” 20 , 28 , 29 , 30 and recently “alternating access transport” mechanism 25 ) by binding the substrates in the central part of the pore and alternatively opening and closing the gates for entry and exit of the transported solute on the opposite side of the membrane. 23 , 26 , 31 , 32 , 33 , 34 , 35 Besides the above‐mentioned cofactors, different MCs have been shown to transport specific substrates, for example, phosphate, sulfate, 2‐oxoglutarate, 2‐oxoadipate, malate, citrate, aspartate, glutamate, ornithine, arginine and methylarginines, branch chain amino acids (BCAA), purine and pyrimidine nucleotides and deoxynucleotides. 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 In phylogenetic analysis, MCs are divided into three main clusters that coincide with the type of substrate they transport: nucleotides, carboxylated metabolites and amino acids. 32 , 44 It has been shown that there is a co‐variation between the type of substrate and the MC residues in the central translocation pore approximately halfway between the two gates on H4 in the so‐called contact point II (motifs G[IVLM] for nucleotides, R[QHNT] for carboxylated metabolites, and R[DE] for amino acids), which are part of the substrate‐binding site. 45 , 46 , 47 , 48 The MCs for B‐vitamin derived cofactors all belong to the nucleotide‐transporting carriers, both considering the contact II residues and their clustering in phylogenetic analysis, with the exception of the animal NAD+ transporter SLC25A51. 32 , 36 , 49 The fact that they belong to nucleotide‐transporting carriers is not surprising given that ThPP, CoA, FAD and NAD+ all enclose nucleotide or nucleotide‐like moieties (Figure 1).
FIGURE 1.
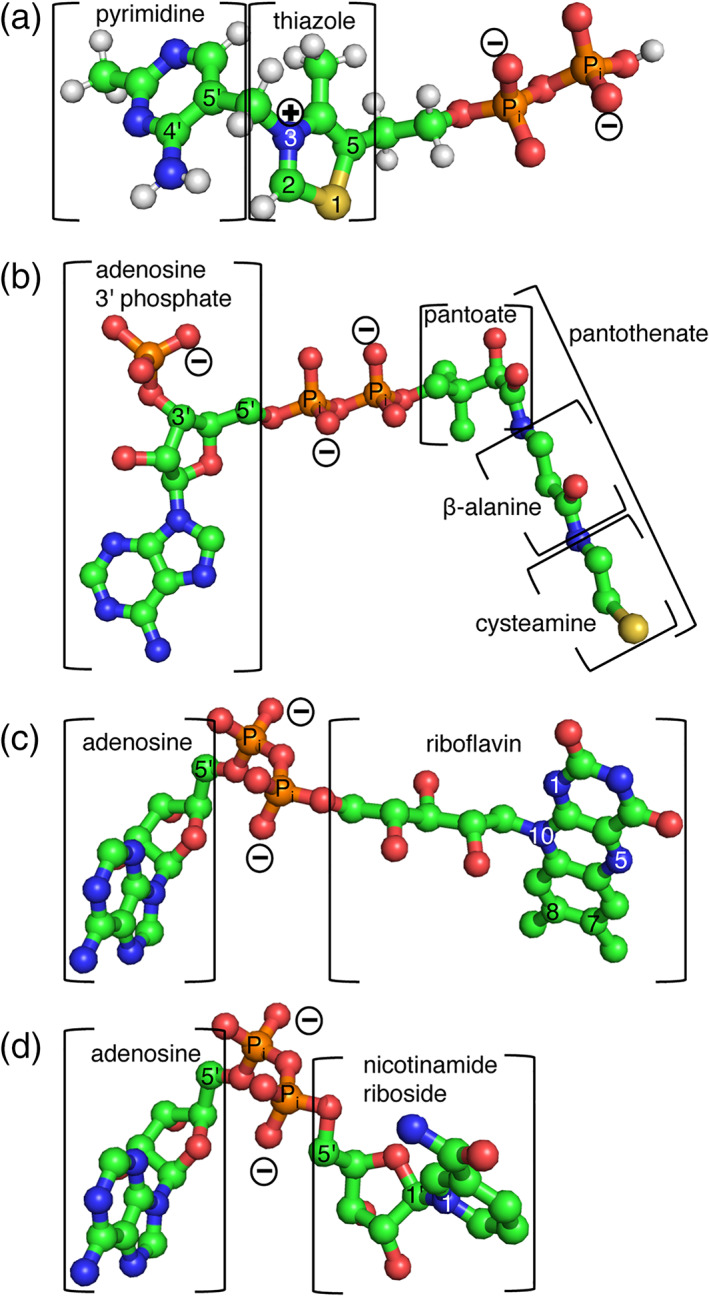
Coenzymes represented in ball‐and‐stick. (a) Thiamine pyrophosphate, (b) coenzyme A, (c) FAD and (d) NAD+. These cofactors have nucleotide and nucleotide‐like moieties: coenzyme A, FAD and NAD+ contain an adenosine, whereas ThPP lacks the ribose but contains a pyrimidine. All four of them contain a pyrophosphate
The substrates of many human, S. cerevisiae and A. thaliana MCs have been identified through an approach comprising their recombinant expression, purification and reconstitution into liposomes that are used in transport assays (EPRA method). 36 , 50 , 51 Besides determining the substrate specificity, the transport assays have been used to identify mitochondrial carrier inhibitors, characterize their kinetic parameters and their modes of transport, that is, uniport, symport and antiport, of which the latter is most common, 52 quantify the importance of each residue along their structure as well as to hypothesize their physiological roles. 16 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 To understand the physiological role of MCs, their characterized transport function is integrated with the knowledge of the identified substrates in metabolism, position in metabolic pathways, subcellular location of the substrates and enzymes using them. Many important clues also come from human diseases caused by mutations in MCs. 61 , 62 , 63 , 64 By using this kind of analysis, the importance for mitochondrial and general metabolism of MC‐mediated transport of the B‐vitamin derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD+ is demonstrated in this review.
2. THIAMINE PYROPHOSPHATE
Thiamine pyrophosphate (ThPP), also known as thiamine diphosphate, is an essential cofactor that is required in the general metabolism of all organisms. It is a derivative of thiamine (vitamin B1 or aneurin) that consists of a six‐membered pyrimidine ring linked through a methylene to a five‐membered thiazole ring, in which the carbon‐5 substitution may be covalently linked to one, two or three phosphates, forming thiamine monophosphate (ThMP), diphosphate (i.e., ThPP, Figure 1a) or triphosphate (ThTP), respectively. ThPP is the active cofactor form and corresponds to about 80% of thiamine found in the human body as free thiamine, ThMP, ThPP, and ThTP. 65 When ThPP is bound to enzymes its phosphates are coordinated by Mg2+ and function as a handle to keep the cofactor in the right position in the active site. The reactivity of ThPP depends on the activation of carbon‐2 (C2) between the nitrogen and the sulfur in the thiazole ring that, upon donation of a hydrogen ion, forms a carbanion. This acts as a nucleophile attacking a carbonyl‐carbon of the first substrate. 66 In most reactions, the C2‐proton is donated to the N′4 amino group of the pyrimidine moiety, being in proximity in the v‐conformation of ThPP (Figure 1a). The metastable reaction intermediate formed by a covalent bond, between the thiazole‐C2 of ThPP and the carbonyl‐carbon of the first substrate, is then resolved by reacting with the second substrate and giving rise to the subsequent formation of the products. In this second “half”‐reaction ThPP thiazole C2 gets re‐protonated and returns to its initial state (Figure 1a). Although ThPP‐dependent enzymes have different 2‐oxocarboxylate or 2‐hydroxycarboxylate substrates and catalyse diverse reactions that break or make C—C, C—N, C—O, C—H, and C—S bonds, from a mechanistic point of view their reactions may all be categorized into decarboxylations and non‐decarboxylations (i.e., transferase or transketolase reactions). 67 Active site residues and additional cofactors or prosthetic groups further contribute to ThPP reactivity, catalysis and substrate binding.
2.1. Biosynthesis and distribution of ThPP
Thiamine, which is necessary for ThPP formation, is synthesized de novo in most prokaryotes and some eukaryotes, such as fungi and plants, but not in animals. The biosynthesis of thiamine requires different enzymes in different species; however, the thiazole and pyrimidine moieties are always synthesized by separate pathways before they are conjugated, as described in detail in several reviews. 68 , 69 , 70 In prokaryotes, the thiazole and pyrimidine rings are linked by thiamine phosphate synthase to form ThMP. ThMP is subsequently phosphorylated by thiamine phosphate kinase to form ThPP. In bacteria, cytoplasmic ThPP is used directly by, for example, transketolase, and other enzymes, that in eukaryotes are located in mitochondria, such as the E1 components of pyruvate dehydrogenase, 2‐oxoglutarate dehydrogenase and acetolactate synthase. In yeast too, the thiazole ring and the pyrimidine ring are unified by thiamine phosphate synthase to ThMP. 68 ThMP is then dephosphorylated to thiamine, which is diphosphorylated to produce ThPP. Whereas the yeast enzymes involved in ThMP biosynthesis are all located in the cytoplasm, in plants ThMP production takes place in chloroplasts and only the last two reactions needed to convert ThMP into ThPP take place in the cytoplasm, as in yeast. 70 In all thiamine‐producing organisms additional enzymes taking part in thiamine salvage pathways have been identified, that is, various bacteria have several enzymes for thiazole, pyrimidine and thiamine recuperation 69 as well as active and facilitated transporters for thiamine uptake 71 , 72 ; yeast may import thiamine from the environment by Thi7p (Thi10p) 73 and convert it into ThPP 68 ; Arabidopsis too have various thiamine salvage pathways that bypass the energetic costs of de novo biosynthesis of ThPP. 70 In thiamine‐synthesizing organisms, the expression levels of several enzymes involved in ThPP biosynthesis are regulated by feed‐back inhibition mechanisms involving mRNA riboswitches, which sense increased concentrations of ThPP leading to reduced transcription and translation. 74
Animals, which are all unable of ThPP biosynthesis, rely on the absorption of the essential vitamin thiamine and its derivatives from the diet (RDA about 1.2 mg for adults), mainly depending on plant‐produced thiamine and animal/gut microflora‐synthesized ThPP. 75 In humans, it has been estimated that about 95% of the thiamine intake (normally <2 mg) is absorbed by the body. Most of thiamine absorption is thought to take place in the proximal part of the small intestine by the major high‐affinity thiamine transporters SLC19A2 (ThTR‐1) 76 , 77 and SLC19A3 (ThTR‐2), 78 , 79 which have about 48% sequence identity and belong to the 12 transmembrane α‐helix major facilitator family of membrane transporters. Furthermore, a homolog of these two proteins, SLC19A1, which is mainly a folate transporter, may contribute to the transport of ThMP in the gastrointestinal tract to some extent. 80 In colon, thiamine may also be taken up by the unspecific organic cation transporters (OCTs), and ThPP specifically by SLC44A44. 81 , 82 The thiamine and thiamine derivatives absorbed across the apical membrane of intestinal epithelial cells are exported through the basolateral membrane into the blood, probably via some of the above‐mentioned transporters. Subsequently, thiamine is distributed throughout the body by erythrocytes, which assimilate >80% of the total blood thiamine (total body content of thiamine and its derivatives about 25–30 mg) and is found especially in skeletal muscles, heart, brain, liver and kidneys. 83 To import thiamine or its derivatives, cells are thought to use several different transporters, many of which are expressed in numerous cell types at different levels; these transporters are SLC19A2 (highly expressed in skeletal muscle), 76 , 77 SLC19A3 (adipose tissue, breast and placenta), SLC19A1 (blood and brain), SLC35F3 (brain) 84 and SLC22A1 (OCT1, liver) 85 (Figure 2). Excess thiamine from intestinal absorption and cell secretion (9–18 days turnover time) is eliminated from circulation mainly through urine by glomerular filtration and in minor quantities in faeces and sweat in the form of free thiamine, ThMP, ThPP or more than 20 different degradation intermediates. 75 , 86 , 87
FIGURE 2.
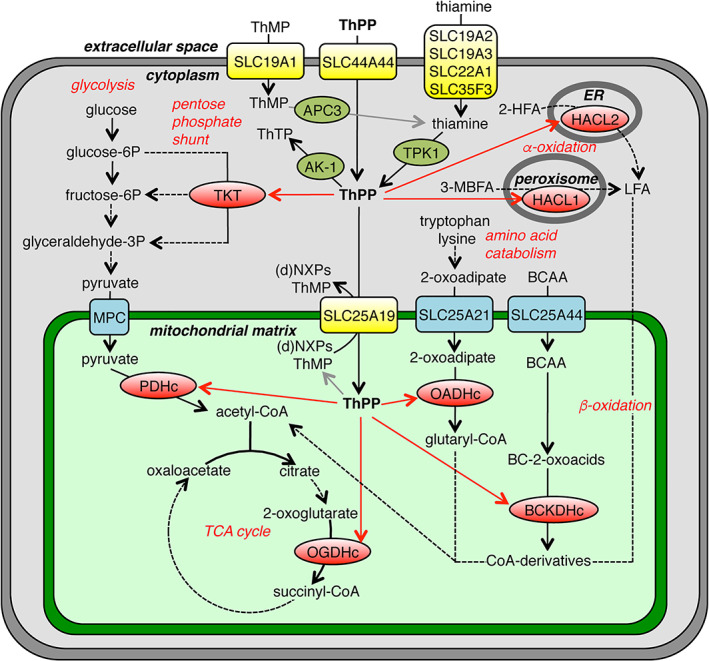
Thiamine metabolism and transport in a typical human cell. The schematic overview shows thiamine/thiamine‐derivative transporters (yellow), enzymes using ThPP as a cofactor (red), enzymes metabolizing ThPP (olive green), enzymatic reactions (black arrows), reactions with unidentified enzymes (grey arrows) and distribution (red arrows). 2‐HFA, 2‐hydroxy‐fatty acids; 3‐MBFA, 3‐methyl‐branched‐chain fatty acids; AK‐1, adenylate kinase 1; APC3, prostatic acid phosphatase; BC‐2‐oxoacids, branched‐chain 2‐oxoacids; BCAA, branched‐chain amino acids; BCKDHc, branched‐chain α‐ketoacid dehydrogenase; (d)NXPs, (deoxy)nucleotides; HACL1 and HACL2, 2‐hydroxyacyl‐CoA lyase 1 and 2; LFA, long fatty acids; MPC, mitochondrial pyruvate carrier; OADHc, 2‐oxoadipate dehydrogenase complex; OGDHc, 2‐oxoglutarate dehydrogenase complex; P, phosphate; PDHc, pyruvate dehydrogenase complex; ThMP, thiamine monophosphate; ThPP, thiamine pyrophosphate; ThTP, thiamine triphosphate; TKT, transketolase; TPK1, thiamine pyrophosphokinase
Once imported into the cytoplasm of animal cells, thiamine is transformed into ThPP by thiamine pyrophosphokinase (TPK1, thiamine pyrophosphotransferase or thiamine diphosphokinase) by transfer of a pyrophosphate from ATP onto thiamine (Figure 2). 88 Most probably cell‐imported ThMP is converted into free thiamine and then to ThPP as in yeast and plants. ThPP is rapidly bound to proteins or imported into organelles leaving only a small cytoplasmic fraction free, which has a fast turnover although not being very reactive by itself. 89 In most eukaryotic cells, cytosolic ThPP is a cofactor of transketolase (TKT), which is an enzyme of the pentose phosphate shunt. 90 Cytosolic ThPP may also be used by adenylate kinase 1 (AK‐1) to form ThTP, which corresponds to approximately 5–10% of total thiamine in the human body and has a regulatory role activating the chloride channels and acting as a phosphate donor in some phosphorylation reactions. 89 , 91 , 92 Moreover, in plant and yeast, cytosolic ThPP is used as a cofactor by pyruvate decarboxylases (PDC) for the non‐oxidative conversion of pyruvate (and other 2‐oxo acids) to acetaldehydes (and other aldehydes) and CO2. 93 , 94 The existence of other ThPP‐dependent enzymes and riboswitches in many non‐mammalian organisms have made them interesting targets for drugs/pesticides. 95
In eukaryotic cells, as mentioned above, cytoplasmic ThPP is distributed into the subcellular compartments; in most human cells, about 50% of total thiamine is found in cytoplasm, 35% in mitochondria, 10% in nucleus and 5% in ER 75 although in rat brain a much higher thiamine level has been measured in mitochondria versus cytoplasm. 96 In fact, ThPP is known to be a cofactor not only of enzymatic reactions in cytoplasm but also in ER, peroxisomes and mitochondria (Figure 2) as well as in chloroplasts. In ER, ThPP is required for 2‐hydroxyacyl‐CoA lyase 2 (HACL2 or ILVBL) in the phytosphingosine (and other 2‐hydroxy‐fatty acids) α‐oxidation degradation pathway. 97 In animal and probably also in plant peroxisomes, ThPP is a coenzyme of 2‐hydroxyacyl‐CoA lyase 1 (HACL1), which catalyses the cleavage of phytanic acid (and other 3‐methyl branched fatty acids) in α‐oxidation. 98 Mitochondria require import of ThPP for the following enzymes: pyruvate dehydrogenase (PDH), which converts pyruvate to acetyl‐CoA which enters the tricarboxylic acid (TCA) cycle 99 ; 2‐oxoglutarate dehydrogenase (OGDH), an enzyme of the TCA cycle 100 ; 2‐oxoadipate dehydrogenase (OADH) 101 ; branched‐chain α‐ketoacid dehydrogenase (BCKDH in animals and plants), which is involved in the degradation of BCAA 102 (Figure 2); and acetolactate synthase (ALS in yeast), which is involved in BCAA biosynthesis. In plants, ThPP serves as a cofactor for chloroplastic pyruvate dehydrogenase to produce acetyl‐CoA and NADH for fatty acid synthesis, transketolase, an enzyme in the Calvin cycle, 2‐deoxyxylulose 5‐phosphate synthase (DXPS) for the biosynthesis of isoprenoids via the mevalonate‐independent pathway, and aceto‐hydroxyacid synthase (AHAS, similar to ALS) for BCAA production. 70 Whereas no transporters for ThPP or other forms of thiamine have been identified for their import into ER, peroxisomes or chloroplasts, mitochondrial ThPP transporters have been identified in yeast, insect, human and plants.
2.2. Mitochondrial transporters of ThPP
In S. cerevisiae, a mitochondrial transporter for ThPP has been identified and named the ThPP carrier (Tpc1p), which is encoded by YGR096w and belongs to the MC family. 7 Tpc1p is responsible for the import of cytosol‐synthesized ThPP into the mitochondrial matrix, where it is required as a cofactor for a few enzymes.
Several pieces of evidence from in vivo and in vitro experiments led to the annotation of this function to this carrier. 7 The localization of Tpc1p to the mitochondrial inner membrane was demonstrated by immunoblot of mitochondria and co‐fractionation with an inner membrane protein marker. 7 The function of Tpc1p was especially associated with anaerobic metabolism in yeast, because of its higher expression levels on fermentable than on non‐fermentable carbon sources. In fact, only on synthetic minimal media (SM) supplemented with fermentable carbon sources, the yeast TPC1 knockout strain (tpc1Δ) displayed a requirement for thiamine or BCAA for growth. This finding was interpreted as an indication that Tpc1p was involved in mitochondrial ThPP import to supply the cofactor for ALS. Therefore, ALS and OGDH activities were measured in mitochondrial extracts from the tpc1Δ strain grown under the above‐mentioned conditions (SM with a fermentable carbon source without thiamine) and found to be 20% and 25%, respectively, of those of the wild‐type extracts. Interestingly, both the activities could be restored by adding ThPP to the reaction. In contrast, the activity of PDC, which is localized in the cytoplasm, was equal in tpc1Δ and wild‐type cells. The levels of ThPP were eight times higher in mitochondria of wild‐type cells compared with tpc1Δ cells, whereas the cytosolic levels were similar in both of them. It is noteworthy that no growth defect of the tpc1Δ strain was observed with non‐fermentable carbon sources, although substantial mitochondrial requirement for ThPP would also be expected. This finding could indicate that in aerobic conditions another mitochondrial ThPP transporter exists or that an uncharacterized mitochondrial ThPP salvage pathway is activated. In conclusion, these in vivo experiments suggest that Tpc1p is involved in ThPP import into yeast mitochondria.
The EPRA method was applied to confirm the identification of Tpc1p and characterize its transport properties by several sets of experiments 7 : (a) substrate homo‐exchange (with the proteoliposomes loaded with a substrate and the same radioactively‐labelled substrate added externally) to identify transportable radioactive substrates; (b) hetero‐exchange (with different substrates loaded in the proteoliposomes and an identified transportable radioactive substrate added externally) to identify exchangeable substrates (without the need of having all substrates radioactively labelled); (c) and efflux (with the identified transportable radioactive substrate loaded into the proteoliposomes and various externally added unlabeled substrates). Furthermore, the conditions of these experiments were also varied, for example, times of reaction (for time‐courses of transport), concentrations of substrate (for kinetic parameter determinations), pH values, omission of the non‐radioactive substrate (to study uniport), addition of competitive substrates or various inhibitors (specific inhibitors or general protein modifiers). At the time of Tpc1p characterization by the EPRA method, no radioactive ThPP was available. However, radioactive dATP was shown to be transported by Tpc1p in the experimental set (a) and exchanged for ThPP in the (b) and (c) procedures. 7 Furthermore, non‐radioactive ThPP efflux from proteoliposomes was assayed enzymatically. Taken together, the in vitro results obtained by the EPRA method demonstrated that Tpc1p is able to transport ThPP, ThMP, and to a lower extent PAP, various ribonucleotides and deoxyribonucleotides, but not many compounds such as thiamine, adenosine, CoA or S‐adenosylmethionine (SAM). The Tpc1p‐catalysed ThPP transport was observed to be faster in antiport than in uniport mode. The kinetic parameters Km and Vmax for dATP uniport uptake were determined to be 0.5 mM and about 40 μmol/g protein per minute at 25°C, respectively. Competitive dATP transport inhibition by addition of ThPP, ThMP, dADP, ADP and AMP was also studied to determine their constants (Ki), which are about 0.2, 0.3, 0.6, 0.7, and 0.6 mM, respectively. Based on these results, it was suggested that the main function of Tpc1p is to import ThPP into the mitochondrial matrix by uniport transport or in exchange for ThMP or one of the other substrates. 7
In H. sapiens the closest homolog of yeast Tpc1p is called SLC25A19 (TPC). To this carrier was originally, even before the characterization of Tpc1p, attributed deoxynucleotide transport ability (and therefore named deoxynucleotide carrier, DNC). 103 Deoxynucleotides were investigated by the EPRA method because phylogenetic analyses showed that TPC clusters together with the adenine nucleotide carrier. The conclusion of the initial experiments was that TPC transports deoxynucleoside diphosphates and ADP very efficiently, GDP, CDP, UDP and dideoxynucleoside triphosphates less effectively, and NTPs, dNTPs, dNMPs and phosphate at low levels, whereas no transport was observed with NMPs. 103 Due to the findings with S. cerevisiae Tpc1p, which has 28% sequence identity with human TPC, the transport capability of TPC was investigated further. 8 SLC25A19 knockout mice displayed elevated 2‐oxoglutarate in the amniotic fluid and did only survive the early embryonic stage, but their isolated mitochondria exhibited normal NTP and dNTP levels. On the contrary, mitochondrial ThPP and ThMP levels were reduced, whereas their cytosolic levels were increased in knockout compared to wild‐type cells. In the knockout cells, PDH and OGDH activities were diminished and could be rescued by addition of ThPP. Furthermore, EPRA approach results with human TPC showed that SLC25A19 (TPC) is a strict antiporter with transport capacity for dATP (radioactive form available), ThPP and ThMP. Thus, it was clear that TPC can import ThPP into mitochondria; however, given its strict antiport mode of transport, what mitochondrial matrix substrate is ThPP exchanged with, among all the substrates transported by TPC? It should be noted that it had been observed previously that isolated rat‐liver mitochondria may convert ThPP to ThMP 104 ; however, a mitochondrial phosphatase responsible for this reaction has not been identified yet. In addition, in certain physiological conditions mitochondria require a net import of ThPP necessitating another matrix counter‐substrate than ThMP. Therefore, also human TPC has a similar function as yeast Tpc1p: mitochondrial import of ThPP in exchange for ThMP or another of its substrate (Figure 2). 8
A mitochondrial ThPP carrier in Drosophila melanogaster, DmTpc1p, has also been characterized by the EPRA method. 105 DmTpc1p, which has 53 and 24% sequence identity with human TPC and yeast Tpc1p, respectively, transports ThPP and, to a lesser extent, (d)NTPs, (d)NDPs and pyrophosphate, whereas ThMP, thiamine, (d)NMPs or phosphate are not transported. Furthermore, DmTpc1p expression complements the growth defects of the S. cerevisiae tpc1Δ strain grown on fermentable carbon sources. These results suggest that also DmTpc1p has the function of importing ThPP into mitochondria in exchange for another of its substrates.
In A. thaliana, two isoforms of mitochondrial ThPP carrier were found, AtTpc1 (At3g21390) and AtTpc2 (At5g48970) that share 74% sequence identity. They were characterized together with their maize homologs, ZmTpc1 (GRMZM2G118515) and ZmTpc2 (GRMZM2G124911) 106 ; and all of these four plant Tpcs display 23–24%, 34–36% and 29–32% sequence identity with the mitochondrial ThPP carrier from S. cerevisiae, H. sapiens and D. melanogaster, respectively. Consistently, in phylogenetic analysis, the four plant proteins cluster together with the human, Drosophila and yeast ThPP carriers. 32 Their subcellular localization was determined by two approaches; the dual import assay, 107 in which radioactively‐labelled protein is incubated with isolated plant chloroplast or mitochondria and then treated with proteases, and confocal microscopy of suspension‐cultured tobacco BY‐2 cells in which GFP‐fused carrier constructs were expressed. 108 Both approaches indicated that all four proteins are mitochondrial and not localized to chloroplasts, peroxisomes or any other organelle. 106 Unfortunately, although AtTpc1, AtTpc2 and ZmTpc1 were expressed in E. coli with the purpose of being used for characterization by the EPRA method, no activity was detected under any of the reconstitution and transport conditions tested. However, the expression of each of the four proteins in the yeast tpc1Δ strain rescued the growth rate on fermentable carbon sources to wild‐type levels. Furthermore, ThPP transport in mitochondria isolated from the yeast tpc1Δ strain was restored by the expression of each of the four plant proteins. In addition, ALS activity was rescued in the AtTpc‐ and ZmTpc‐complemented tpc1Δ strain. Taken together, although the biochemical characterization of the isolated proteins remains to be determined, the above‐mentioned results strongly support the notion that AtTpc1, AtTpc2, ZmTpc1 and ZmTpc2 have a similar physiological function to those of the mitochondrial ThPP carriers identified in other organisms.
2.3. Role of ThPP in mitochondria
As illustrated in Figure 2, in animal cells, ThPP is involved in important enzyme reactions in carbohydrate, lipid and amino acid catabolism. ThPP imported into mitochondria is used by four different 2‐oxoacid dehydrogenase complexes (PDHc, OGDHc, OADHc and BCKDHc) that, although function in distinct metabolic pathways, have similar macromolecular assembly structures and catalyse quite similar decarboxylation reactions. 109 The E1 components (also called PDH, OGDH, OADH and BCKDH) of the four complexes bind ThPP and the initial 2‐oxoacid substrates, whereas the other components E2 and E3 bind the other cofactors: lipoate, CoA, FAD and NAD+. In all four enzymes, ThPP is directly involved in the decarboxylation of the incoming substrate and the transfer of the remaining decarboxylated acyl intermediate to lipoate bound to E2. The subsequent reaction steps lead to the final formation of acyl‐CoA while the electrons are transferred onto NAD+. The role of ThPP in PDH will be described in the next paragraph and, subsequently, compared with that in the other enzyme complexes. In yeast, mitochondria contain another ThPP‐dependent enzyme, ALS.
PDHc exists in all known organisms and links glycolysis to the TCA cycle by catalysing the conversion of pyruvate into acetyl‐CoA and CO2 (Figure 2). 110 In humans, this large complex consists of multiple copies of the E1, E2 (dihydrolipoyl transacetylase) and E3 (dihydrolipoyl dehydrogenase) components, with a total of 114 polypeptides and a mass of 9 MDa. Each human E1 component is a heterotetramer of two α‐ and two β‐subunits (PDHA1/2 and PDHB). ThPP is bound in the active site of PDH (E1 component) with the pyrimidine ring hydrogen‐bonded at the interface between the α‐ and β‐subunits and the pyrophosphate moiety penetrating into the α‐subunit where a Mg2+ is bound leaving the reactive thiazole at the centre of the active pocket where the substrate pyruvate enters. 111 In all known ThPP‐requiring enzymes, a special motif takes part in binding the cofactor: in 166GDGX26NN196 of the α‐subunit of human PDH Asp167 and Asn196 coordinate the Mg2+, and Gly168 and Ala169 make hydrogen bonds with the pyrophosphate moiety. 111 , 112 The dissociation constant of ThPP in pig heart PDH has been estimated to be about 7 μM, which supposedly is above the mitochondrial concentration of ThPP. 113 ThPP is directly involved in two consecutive steps of the PDH‐catalysed reaction: (a) the deprotonated thiazole‐C2 attacks the 2‐oxo‐carbon of pyruvate leading to the decarboxylation of pyruvate and the formation of a covalently linked intermediate, (b) the transfer of this intermediate in the reductive acetylation onto the lipoyl group bound to E2 and the return of ThPP to its normal protonated state. 110 Subsequently, E2 catalyses the transfer of the acetyl‐moiety to CoA to form acetyl‐CoA, and E3 oxidizes the E2 lipoyl group by electron transfer to FAD and then to NAD+. The binding of ThPP in PDH orders α‐subunit loops localized at the entrance of the substrate‐binding pocket containing serines, which are phosphorylated/dephosphorylated by kinases/phosphatases and play an important role in the metabolic regulation of PDH activity. 110
OGDH is the E1 component of OGCHc, which catalyses the conversion of 2‐oxoglutarate into succinyl‐CoA, a reaction that is most commonly known for its role in the TCA cycle, where it is a rate‐limiting step (Figure 2). 114 Besides from carbohydrate metabolism, 2‐oxoglutarate may also derive from amino acid catabolism. ThPP is bound to OGDH in a very similar way as to PDH and the reaction mechanism is also very similar: deprotonated C2 of the ThPP thiazole ring attacks the 2‐oxo‐carbon of 2‐oxoglutarate, CO2 leaves and the metastable intermediate is resolved by the transfer of succinyl via lipoate onto CoA by E2 (dihydrolipoyl succinyltransferase), while the electrons are handled by E3 (dihydrolipoyl dehydrogenase).
In animals and maybe in plants but not in yeast, OADHc is involved in the catabolism of lysine and tryptophan (Figure 2), 114 from which 2‐oxoadipate is derived directly in mitochondria or imported into mitochondria via ODC (SLC25A21). 115 OADHc has its own specific E1 component (encoded by DHTKD1‐dehydrogenase E1 and transketolase domain‐containing protein 1), whereas the E2 and E3 components are identical to those of OGDHc. 109 The substrate 2‐oxoadipate and product glutaryl‐CoA are only one CH2 longer than those of OGDHc, 2‐oxoglutarate and succinyl‐CoA, which is reflected by the slightly larger substrate‐binding pocket in OADHc. The reaction mechanism of OADHc is thought to be very similar to that of OGDHc and PDHc.
BCKDHc, which is found in human and plants but probably not in yeast, is involved in the catabolism of BCAA (leucine, isoleucine and valine), which are imported into mitochondria by SLC25A44 43 and deaminated into their corresponding 2‐oxoacids (2‐oxoisocaproate, 2‐oxo‐3‐methylvalerate and 2‐oxovalerate) (Figure 2). These 2‐oxoacids react with ThPP bound to component E1 (2‐oxoisovalerate dehydrogenase) of BCKDHc, and the reactions catalysed by E2 (dihydrolipoyl transacylase) and E3 (dihydrolipoyl dehydrogenase) lead to the formation of isovaleryl‐CoA, 2‐methyl‐buteryl‐CoA and isobutyryl‐CoA, which are further transformed to metabolic intermediates and acetyl‐CoA that may enter the TCA cycle. 116
ALS (encoded by ILV2 and ILV6 in S. cerevisiae) is a ThPP‐requiring enzyme, which catalyses the first step out of four in the biosynthetic pathway of BCAA in yeast. 117 Whereas yeast ALS is found in the mitochondrial matrix and the corresponding enzyme AHAS of plants is localized to chloroplasts and that of bacteria in the cytoplasm, no counterpart of these enzymes exists in animals. ALS condenses two molecules of pyruvate producing CO2 and 2‐acetolactate (which is used for the synthesis of valine and leucine) or one molecule of pyruvate and one of 2‐oxobutyrate forming 2‐aceto‐2‐hydroxybutyrate for isoleucine synthesis. Thus, unlike all the other mitochondrial above‐mentioned 2‐oxoacid dehydrogenase complexes that have similar supramolecular structures, ALS is different and belongs, together with PDC, to the pyruvate oxidase protein family. In fact, ALS consists of a catalytic subunit (CSU, ILV2), containing the cofactors ThPP, Mg2+ and FAD, and a regulatory subunit (RSU, ILV6). 118
2.4. Diseases related to thiamine and ThPP
Several diseases, both acquired and genetic, caused by alterations in genes encoding thiamine transporters, thiamine‐converting and ThPP‐dependent enzymes, have been described. Insufficient intake of thiamine causes beriberi and Wernicke's encephalopathy. 119 Disease‐causing mutations have been found in SLC19A2, SLC19A3, SLC44A44 and TPK1 as well as in the E1 subunits of the mitochondrial ThPP‐dependent enzymes PDHc, OGDHc, OADHc and BCKDHc. 120 SLC25A19 (TPC) deficiency is caused by mutations in the gene encoding the mitochondrial ThPP importer. Until now, two autosomal recessive disorders have been disclosed: Amish microcephaly (OMIM 607196) and thiamine metabolism dysfunction syndrome 4 (progressive polyneuropathy type, OMIM 613710). Amish microcephaly is the more severe form and is caused by the mutation p.Gly177Ala, which affects a highly conserved residue of the second signature motif sequence. The recombinant protein harbouring this mutation was found to display a negligible transport activity in proteoliposomes by the EPRA method. 8 , 121 This mutation has been traced back several generations in Amish family in Pennsylvania. 121 Amish microcephaly symptoms are severe congenital microcephaly, 2‐oxoglutaric aciduria and premature death in many patients. In an attempt to create an animal model for SLC25A19 deficiency, mice with the single knockout of this gene appeared normal, whereas the double knockout displayed mitochondrial ThPP and ThMP depletion, embryonic lethality, brain malformations and anaemia. 8 The diminished mitochondrial ThPP content caused reduced OGDHc and PDHc activities resulting in 2‐oxoglutaric aciduria and accumulation of lactate. Thiamine metabolism dysfunction syndrome 4 is caused by mutations of non‐conserved residues (p.Gly125Ser and p.Gln192His, inside the substrate translocation pore, and p.Ser194Pro and p.Leu290Gln outside the pore) as well as by the mutation p.Glu304Lys, which affects a conserved residue of the cytoplasmic gate (for a review see Reference 64). This disease is characterized by increased lactate levels in cerebrospinal fluid or serum, chronic progressive polyneuropathy and fever‐triggered acute episodes of flaccid paralysis and encephalopathy with bilateral striatal necrosis. 122 , 123 , 124 , 125 In contrast to Amish microcephaly, patients with this disorder have normal head circumference, development and cognitive skills, and oral thiamine administration seems to improve the clinical situation. 123 , 125 The severe neuronal syndromes including encephalopathy associated with thiamine metabolism dysfunction syndrome 4 are also symptoms linked to mutations in SLC19A3, TPK1 and PDHc. Notably, all these genetic variants affect four enzymatic steps on the linear pathway from thiamine absorption to ThPP‐dependent PDHc activity (Figure 2). 120
3. COENZYME A
Coenzyme A (CoA, SHCoA, CoASH) is a cofactor present in all organisms of the three domains of life: Archaea, Bacteria and Eukarya. It is involved in approximately 4% of all enzymatic reactions, most of which play key roles in the metabolism of fatty acids, carbohydrates, amino acids and ketone bodies. 126 CoA is composed of 3′‐phosphoadenosine‐5′diphosphate linked to pantothenate (vitamin B5), which consists of pantoate amide bonded to β‐alanine, which in turn is connected to cysteamine by a second amide bond (Figure 1b). The terminal thiol group of cysteamine has the property of forming high‐energy thioester bonds with carboxylated intermediates leading, for example, to the formation of acetyl‐CoA and various size acyl‐CoA, which can be transferred to various C, O, N and S acceptors in enzymatic reactions, where the CoA moiety is used as a handle. Due to its size and hydrophilic nature, CoA does not penetrate membranes.
3.1. Biosynthesis and distribution of CoA
Pantothenate (vitamin B5), which is essential for the formation of the CoA molecule, is synthesized de novo in plants, some bacteria and fungi. In these organisms, pantoate‐β‐alanine ligase (PanC) catalyses the condensation of β‐alanine with pantoate to form pantothenate. 127 In non‐pantothenate‐producing bacteria and fungi, pantothenate is imported by transporters of the major facilitator family with 12 transmembrane helices, such as PanF and Fen2p. In animals, which are unable to synthesize pantothenate, the uptake of this vitamin is essential. It is found in food such as meat, vegetables, cereal grains, legumes, eggs, and milk as well as is produced by the intestinal flora. Intestinal CoA is hydrolysed into the intermediates dephospho‐CoA, 4′‐phosphopantetheine and pantetheine. 128 In the small intestine, pantothenate is taken up by the sodium‐dependent multivitamin transporter SLC5A6 predicted to contain 13 transmembrane segments. 129 This transporter is also expressed in the cellular plasma membrane of heart, brain, placenta, lung, liver, skeletal muscle, kidney and pancreas, and is therefore probably responsible for the import of pantothenate in most cells (Figure 3). 129 In all living organisms pantothenate, cysteine and four molecules of ATP are used to synthesize CoA in a five‐step process (Figure 3): (a) pantothenate is phosphorylated to 4′‐phosphopantothenate by pantothenate kinase (PANK1), which is the committed step in CoA biosynthesis, requires ATP and is regulated by product inhibition; (b) cysteine is added to 4′‐phosphopantothenate by phosphopantothenoylcysteine synthetase (PPCS) to form 4′‐phospho‐N‐pantothenoylcysteine in an ATP‐dependent reaction; (c) the latter metabolite is decarboxylated by phosphopantothenoylcysteine decarboxylase (PPC‐DC) to 4′‐phosphopantetheine; (d) to which AMP is added by phosphopantetheine adenylyl transferase (PPAT or COASY) to form dephospho‐CoA; (e) which is phosphorylated by dephosphocoenzyme A kinase (DPCK or COASY) to form CoA. 130 In humans, a single polypeptide chain constitutes CoA synthase COASY, a bifunctional enzyme catalysing both PPAT and DPCK (step [d] and [e]) activities. In humans, all of these enzymes are localized outside mitochondria with the exception of PANK2, one of several PANK isoforms, which has been suggested to be present in the intermembrane space of mitochondria. 131 Furthermore, the subcellular localization of COASY is under debate, because it has been found in association with mitochondrial outer membrane, 132 , 133 in mitochondrial matrix 134 , 135 , 136 and partly in the cytosol. 134 , 137 Later, it was found that COASY is expressed as two splice variants: α, which is expressed ubiquitously, and β, which is predominantly expressed in brain. 136 Unfortunately, in these later studies, the cellular localization of these two variants was not investigated. However, looking at the pre‐sequences, it should be noted that α‐COASY has a predicted N‐terminal mitochondrial targeting peptide, whereas β‐COASY has a 29 residue N‐terminal extension preceding the above‐mentioned peptide rendering the latter variant of COASY to be predicted non‐mitochondrial. It has been suggested that CoA may also be synthesized through alternate routes when its intracellular levels are reduced and the de novo pathway is impaired. 138 Furthermore, Daugherty et al. 139 identified a human gene on chromosome 17, named DCAKD, encoding a putative monofunctional human DPCK enzyme (Accession Numbers: gi13623688 and Q8WVC6 [UniProt]), which shared 35% sequence identity with the E. coli DPCK over its entire length. Orthologs of monofunctional DCAKD are also present in D. melanogaster (Q9VI57) and C. elegans (P34558). 140 Interestingly, monofunctional DCAKDs display a greater sequence identity to the E. coli and other bacterial DPCK sequences than to the paralogous DPCK portions of the bifunctional PPAT/DPCK sequences in each organism. Submitochondrial fractionation studies revealed that most of the DCAKD enzymatic activity is associated with the outer membrane of mitochondria. 134
FIGURE 3.
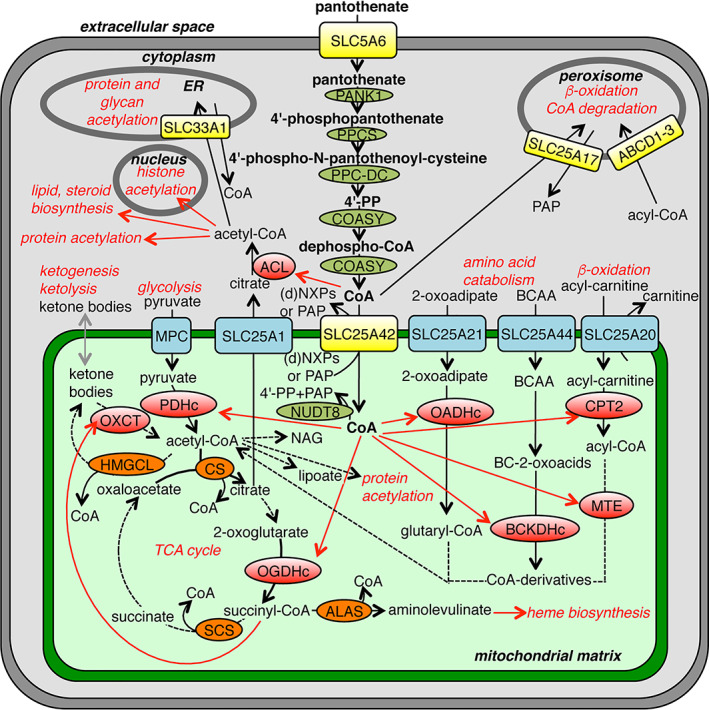
CoA metabolism and transport in a typical human cell. The schematic overview shows CoA/CoA‐derivative transporters (yellow), enzymes adding CoA on a metabolite (red), enzymes taking away CoA from a metabolite (orange), enzymes metabolizing CoA (olive green), enzymatic reactions (black arrows), reactions with unidentified enzymes (grey arrows) and distribution (red arrows). (d)NXPs, (deoxy)nucleotides; ACL, ATP‐citrate lyase; ALAS, aminolevulinate synthase; BC‐2‐oxoacids, branched‐chain 2‐oxoacids; BCAA, branched‐chain amino acids; BCKDHc, branched‐chain α‐ketoacid dehydrogenase; COASY, coenzyme A synthase; CPT2, carnitine O‐palmitoyltransferase 2; CS, citrate synthase; HMGCL, Hydroxymethylglutaryl‐CoA lyase; MPC, mitochondrial pyruvate carrier; MTE, mitochondrial trifunctional enzyme; NAG, N‐acetyl‐glutamate; OADHc, 2‐oxoadipate dehydrogenase complex; OGDHc, 2‐oxoglutarate dehydrogenase complex; OXCT, 3‐oxoacid‐CoA transferase; PANK1, pantothenate kinase 1; PAP, adenosine 3′,5′‐diphosphate; PDHc, pyruvate dehydrogenase complex; PPC‐DC, phosphopantothenoylcysteine decarboxylase; PPCS, phosphopantothenoylcysteine synthetase; SCS, succinyl‐CoA synthetase
Excess CoA may be degraded intracellularly, mainly by mitochondrial and peroxisomal NUDIX (nucleoside diphosphate‐linked moiety X) hydrolases, or extracellularly, which leads to its conversion into pantothenate that may be recycled by cellular uptake and de novo CoA synthesis. 141
CoA is distributed within the mammalian cells between the different compartments: cytosol, nucleus, ER, peroxisomes, lysosomes and mitochondria (Figure 3). Cytosolic/nucleic concentrations of CoA are estimated between 0.11 and 0.14 mM in liver and heart, in peroxisomes 0.3–0.7 mM CoA, whereas estimated mitochondrial CoA concentrations are significantly higher, about 5 mM. 141 Although these estimates are dependent on the purity and integrity of the subcellular fractionations, they suggest that mitochondria contain 80–95% of total cellular CoA. In the cytosol and nucleus, CoA is used by ATP‐citrate lyase to form acetyl‐CoA for fatty acid and steroid synthesis, and for post‐translational acetylation of thousands of sites on proteins, for example, histones. 142 The ratio of acetyl‐CoA to CoA controls protein acetylation influencing key cellular processes, such as cell growth, regulated cell death and autophagy. 143 In the ER membrane, SLC33A1 imports acetyl‐CoA in exchange for CoA for acetylation of proteins and glycans. 144 In peroxisomes, ABC transporters ABCD1‐3 import a variety of acyl‐CoAs, 145 and SLC25A17 (as well as its homologue PXN in plants) transport CoA, which is used, for example, in β‐oxidation. 146 , 147 In mitochondria, CoA is imported into the matrix by SLC25A42, 10 where it plays key roles in metabolic processes, such as the oxidation of pyruvate, oxoglutarate, BCAA and fatty acids (Figure 3).
3.2. Mitochondrial transport of CoA
The human mitochondrial carrier for CoA is SLC25A42, which clusters together with SLC25A16 as well as yeast Leu5p and YPR011cp, and Arabidopsis AT1G14560 and AT4G26180. 32 The EPRA approach was employed to characterize SLC25A42. Among the radioactive substrates tested, ATP, ADP and AMP were transported in homo‐exchange (type i) experiments; subsequently, radioactive ADP was used in hetero‐exchange (type ii) and efflux (type iii) transport experiments. The results showed that SLC25A42 transports CoA, adenosine 3′,5′‐diphosphate (PAP), dephospho‐CoA and (deoxy‐)adenine nucleotides by an obligatory counter‐exchange mechanism. 10 Furthermore, radioactive ADP uptake by reconstituted SLC25A42 was inhibited competitively by PAP (Ki of 27 μM), ADP (Ki of 54 μM), CoA and dephospho‐CoA (Ki about 110 μM for both), the last Ki corresponding approximately to the cytosolic concentration of CoA. In addition, the expression of human SLC25A42 in a yeast strain with deleted Leu5, which had been suggested previously to be a mitochondrial CoA carrier, 148 complemented the growth defects on non‐fermentable carbon sources that require mitochondrial CoA for their oxidation. 10 Finally, a fusion construct of SLC25A42‐GFP expressed in CHO cells showed that the protein is localized to mitochondria. 10 SLC25A42 is expressed in almost all tissues. 149 Based on these results and assuming a cytosolic localization of the COASY, it was suggested that the main function of SLC25A42 is to import cytosolic CoA or dephospho‐CoA into the mitochondrial matrix in exchange for PAP, which is generated from CoA in the mitochondrial matrix, or another substrate. 10 An alternative scenario should be taken into consideration if COASY were localized in the matrix. In this case, SLC25A42 could be involved in the export of CoA, dephospho‐CoA or PAP from the mitochondria, possibly by an exchange reaction with ADP. 10 Outside the mitochondria, dephospho‐CoA would be converted to CoA by the monofunctional DCAKD localized in the mitochondrial outer membrane. 134 This hypothetical route might also be operating in D. melanogaster, where the SLC25A42 ortholog transports dephospho‐CoA (and not CoA, see below) and DCAKD is present too. Although SLC25A42 can bi‐directionally exchange CoA or dephospho‐CoA with [14C]ADP in vitro, 10 no data are available about the direction of this exchange reaction in vivo until now. Answering this question may be of utmost importance in the light of the conflicting results on the sub‐mitochondrial localization of COASY.
The functions of some of the SLC25A42 homologues have also been investigated. It is likely that yeast Leu5p is a mitochondrial transporter for CoA or one of its precursors, because deletion of its gene reduces mitochondrial CoA content and diminishes the activity of mitochondrial CoA‐dependent enzymes. 148 The same conclusion was drawn for the human SLC25A16, which partly complements the alterations observed in the Leu5‐knockout strain. 148 However, the substrate specificities of these two proteins have not yet been investigated. By using the EPRA method, yeast YPR011cp has been shown to transport not CoA or PAP but phosphate, sulfate, adenosine 5′‐phosphosulfate (APS) and 3′‐phospho‐adenosine 5′‐phosphosulfate (PAPS), the last two being structurally similar to PAP. 150 The homologue of SLC25A42 in D. melanogaster has been characterized using the EPRA method and shown to transport dephospho‐CoA (and (deoxy‐)adenine nucleotides) but not CoA; it also complements the growth defects of the yeast Leu5‐deletion strain. 151 Arabidopsis AT1G14560 and AT4G26180 (and their two maize homologues), which are localized to mitochondria as shown by the dual import assay and confocal microscopy of tobacco BY‐2 cells with GFP‐fused proteins, also rescue the growth defects and the reduced mitochondrial CoA content of the yeast Leu5‐deletion strain. 152 Taken together, these findings suggest that the SLC25A42 homologues, with the exception of YPR011cp, have a similar physiological function in importing CoA and/or dephospho‐CoA into mitochondria.
3.3. Role of CoA in mitochondria
Mitochondrial CoA is used by a long list of matrix enzymes involved in carbohydrate, lipid, amino acid and ketone body catabolism to form various length acyl‐chain‐CoA intermediates, most of which eventually end up as acetyl‐CoA (Figure 3). Glycolysis‐derived and mitochondrially‐imported pyruvate is transformed into acetyl‐CoA by PDHc (as described in Section 2.3); the E2 component transfers the acetyl‐moiety (originally donated from pyruvate in E1) from the lipoyl group onto CoA, followed by re‐oxidation of lipoate by the E3 component. 110 In the TCA cycle, CoA is used by the E2 component of OGDHc to form succinyl‐CoA. In the mitochondrial β‐oxidation, CoA substitutes the carnitine moiety of mitochondrially‐imported carnitine‐linked fatty acid chains by the action of carnitine O‐palmitoyltransferase 2, and functions as a handle in the subsequent steps catalysed by acyl‐CoA dehydrogenase, enoyl‐CoA hydratase, 3‐hydroxyacyl‐CoA dehydrogenase and β‐ketothiolase, of which the latter adds a new CoA molecule to the remaining acyl chain after the exit of the terminal acetyl‐CoA. In amino acid catabolism, the E2 component (lipoamide acyltransferase) of BCKDHc adds CoA to the 2‐oxoacids derived from BCAAs; branched‐chain specific acyl‐CoA dehydrogenases convert the carbon skeletons into acetyl‐CoA; and the E2 component of OADHc adds CoA onto 2‐oxoadipate, which is derived from lysine and tryptophan, and is further converted to acetyl‐CoA. Finally, ketone bodies, such as acetoacetate and β‐hydroxybutyrate, are utilized for ATP production by conversion into acetyl‐CoA in the mitochondrial matrix of non‐hepatic tissues when glucose and glycogen stores are diminished. 153
Once mitochondrial acetyl‐CoA has been formed it may be used in various ways. When the cellular demand for ATP is high, most acetyl‐CoA enters the TCA cycle by citrate synthase, which condenses acetyl and oxaloacetate into citrate, leaving CoA free. In the same metabolic cycle, succinyl‐CoA is converted into succinate and free CoA by the action of succinyl‐CoA synthetase. Matrix succinyl‐CoA may also be condensed with glycine by 5‐aminolevulinate synthase to form free CoA and aminolevulinate, which is the first step in heme synthesis. 154 Especially in liver during fasting, mitochondrial acetyl‐CoA derived from β‐oxidation may be used for the biosynthesis of the ketone bodies acetoacetate and β‐hydroxybutyrate, which are activated and oxidized in other tissues with the production of ATP. 153 Furthermore, acetyl‐CoA (via malonyl‐CoA) is utilized in mitochondrial type II fatty acid synthesis for the production of lipoate, which acts as a cofactor in the E2 component of the matrix dehydrogenase complexes. 155 In addition, mitochondrial acetyl‐CoA may be used for regulatory purposes through acetylation of proteins and the production of the urea cycle regulator N‐acetylglutamate. 156 , 157
The above‐mentioned mitochondrial enzymes associate or disassociate CoA from acyl chains without changing the CoA moiety itself. However, mitochondria also contain the NUDIX protein NUDT8, which hydrolyses CoA (or acyl‐CoAs) to phosphopantetheine (or acyl‐phosphopantetheine) and PAP, exhibiting the same function in CoA degradation as its peroxisomal homologues NUDT7 and NUDT19. 158 It seems most likely that NUDT8 is localized in the matrix, but it cannot be excluded that it is in the intermembrane space. 141 The fact that also COASY may be located in the matrix implies that the resulted phosphopantetheine may be recycled into CoA; in this case, however, NUDT8 and COASY activities must be regulated to avoid a futile cycle of CoA synthesis and breakdown. 141 , 158 Moreover, mitochondrial type II fatty acid synthesis of lipoate requires acyl carrier protein (ACP), which is modified covalently with a phosphopantetheinylate group derived from CoA, by phosphopantetheinyl transferase producing PAP as a leaving group. 155 In yeast, phosphopantetheinyl transferase is localized in mitochondria, whereas the corresponding enzyme in human appears to be cytoplasmic. Besides being an intermediate in lipoate synthesis, acetylated ACP (via the phosphopantetheine group) seems to have a role in mitochondrial translation, iron–sulfur cluster biogenesis and assembly of the respiratory chain complexes. 155 Thus, if human NUDT8 and phosphopantetheinyl transferase are located in the mitochondrial matrix, PAP, the breakdown product of CoA, may be exported from the mitochondrial matrix by SLC25A42. 10
3.4. Diseases related to CoA
In humans, pantothenate deficiency due to low intake of the vitamin (RDA 5 to 10 mg) is a very rare disorder that causes asthenia, headaches, nauseas and vomiting, as well as some forms of neuropathies similar to those of other vitamin B deficiencies. 159 Mutations in PANK2 and COASY give rise to neurodegeneration with brain iron accumulation. 160 Homozygous mutations in SLC25A42 cause variable symptoms with wide spectrum of severity, even among patients with the same mutation in the same family, including lactic acidosis, mitochondrial myopathy with muscle weakness, encephalopathy, developmental regression and epilepsy. 161 , 162 , 163 The identified disease‐causing mutations are a splice site and a point mutation (p.Asn291Asp) close to the substrate‐binding site (for a review, see Reference 64).
4. FLAVIN ADENINE DINUCLEOTIDE (FAD)
FAD is an important cofactor in redox reactions catalysed by flavoproteins, which contain FAD, or its mononucleotide form FMN, and play key roles in cell metabolism. FAD consists of a riboflavin moiety (vitamin B2 or previously vitamin G), a flavin isoalloxazine ring bound to the reduced pentose ribitol, that with one phosphate attached forms FMN or two phosphates and adenosine forms FAD (Figure 1c). Two nitrogen atoms (in positions 1 and 5) in the conjugated 3 six‐membered ring system of riboflavin have the ability of accepting a proton and an electron, each giving the fully reduced form of FAD and FMN. FAD works as a prosthetic group in flavoproteins, to which it is most often bound non‐covalently.
4.1. FAD biosynthesis and distribution
The FAD precursor riboflavin is not synthesized by animals but by bacteria, fungi and plants where its synthesis starts from GTP and ribulose‐5‐phosphate. 164 In humans riboflavin is mainly taken up in small intestine from food such as meat, cereals, fish, dark‐green vegetables, egg, nuts, mushrooms, legumes, milk and dairy products. 165 Food processing and exposure to UV light may reduce riboflavin content. In the small intestine, FAD and FMN are hydrolysed to riboflavin, which is mainly absorbed by SLC52A3 (RFVT3) in the proximal small intestine and, to some extent, in the large intestine. 166 Also, the homologues of this transporter, SLC52A1 and SLC52A2, with 86% sequence identity between them and about 42% with SLC52A3, may contribute to intestinal absorption, enterocyte export, blood cell import/export and cellular uptake of riboflavin (Figure 4). 167 , 168 In blood riboflavin is transported bound to albumin or in erythrocytes and leukocytes.
FIGURE 4.
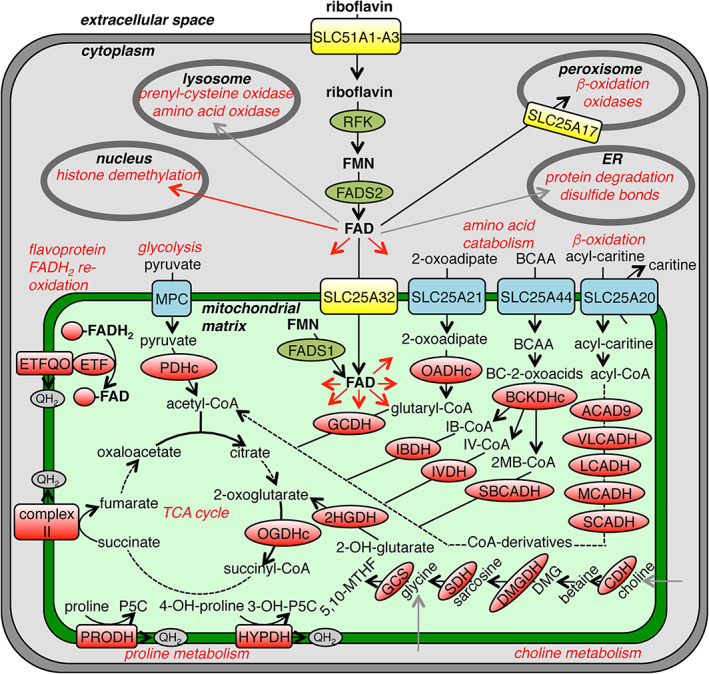
FAD metabolism and transport in a typical human cell. The schematic overview shows FAD/FMN/riboflavin transporters (yellow), flavoenzymes (red), enzymes metabolizing FAD (olive green), enzymatic reactions (black arrows), reactions with unidentified enzymes (grey arrows) and distribution (red arrows). 2HGDH, 2‐hydroxyglutarate dehydrogenase; 2‐MB‐CoA, 2‐methylbutyryl‐CoA; 2‐OH‐glutarate, 2‐hydroxyglutarate; 3‐OH‐P5C, Δ1‐pyrroline‐3‐hydroxy‐5‐carboxylate; 4‐OH‐proline, 4‐hydroxyproline; 4'‐PP, 4′‐phosphopantetheine; 5,10‐MTHF, 5,10‐methylenetetrahydrofolate; ACAD9, acyl‐CoA dehydrogenase 9; BC‐2‐oxoacids, branched‐chain 2‐oxoacids; BCAA, branched‐chain amino acids; BCKDHc, branched‐chain α‐ketoacid dehydrogenase; CDH, choline dehydrogenase; DMG, dimethylglycine; DMGDH, dimethylglycine dehydrogenase; ETF, electron transfer flavoprotein; ETFQO, electron transfer flavoprotein ubiquinone oxidoreductases; FADS, FAD synthase; GCDH glutaryl‐CoA dehydrogenase; GCS, glycine cleavage system; HYPDH, hydroxyproline dehydrogenase; IB‐CoA, isobutyryl‐CoA; IBDH, isobutyryl‐CoA dehydrogenase; IV‐CoA, isovaleryl‐CoA; IVDH, isovaleryl‐CoA dehydrogenase; LCADH, long‐chain acyl‐CoA dehydrogenase; MCADH, medium‐chain acyl‐CoA dehydrogenase; MPC, mitochondrial pyruvate carrier; OADHc, 2‐oxoadipate dehydrogenase complex; OGDHc, 2‐oxoglutarate dehydrogenase complex; PDHc, pyruvate dehydrogenase complex; P5C, Δ1‐pyrroline‐5‐carboxylate; PRODH, proline dehydrogenase; QH2, reduced ubiquinone; RFK, riboflavin kinase; SBCADH, short branched‐chain acyl‐CoA dehydrogenase; SCADH, short‐chain acyl‐CoA dehydrogenase; SDH, sarcosine dehydrogenase; VLCADH, very‐long‐chain acyl‐CoA dehydrogenase
Imported cellular riboflavin is phosphorylated by riboflavin kinase (RFK, ATP‐riboflavin 5′phosphotransferase) to form FMN, which is subsequently adenylated by FAD synthase (FADS, ATP‐FMN adenylyltransferase) to FAD (Figure 4). 169 Whereas RFK is thought to be cytoplasmic, FADS has been suggested to have two isoforms in human, one cytoplasmic and one mitochondrial. 170 In yeast, there is only one isoform of FADS, which may be localized both to the cytoplasm and mitochondria due to a cis‐acting mitochondrial localization motif present in the 3′‐untranslated region of its mRNA. 171 , 172 FAD is distributed in different cellular compartments: cytoplasm, peroxisomes, ER, nucleus, lysosomes and mitochondria. Very limited examples of processes in which FAD is involved are: cytochrome P450 reductase and nitric oxide synthase 2 in the cytosol 173 , 174 ; β‐oxidation of fatty acids, D‐amino acid and sarcosine oxidases in peroxisomes 145 ; degradation of proteins and formation of disulfide bonds in the ER 175 , 176 ; histone lysine demethylases in the nucleus 177 ; amino acid and prenyl‐cysteine oxidases in lysosomes 178 , 179 ; and many enzymes involved in carbohydrate, fatty acid and amino acid catabolism, such as succinate dehydrogenase (complex II), in mitochondria. 168 Moreover, in chloroplasts, FAD is used by acetolactate synthase and for fatty acid biosynthesis. 70 These compartmentalized FAD‐requiring processes suggest that FAD, or one of its precursors, has to be imported into these organelles. SLC25A17 and SLC25A32 have been identified as transporters for FAD (and/or FMN) in peroxisomes, 146 and in mitochondria, 180 , 181 respectively. SLC25A17 is better characterized than SLC25A32 biochemically; it transports FAD and FMN along with CoA and other substrates as shown by the EPRA method. In plants, the mitochondrial carriers NDT1 and NDT2, characterized by the EPRA method, display the ability of transporting FAD and FMN although at a lower extent than their other substrates NAD+, NAAD, NMN, ADP and AMP (described in more detail in Section 5.2). 182
4.2. Mitochondrial FAD transport
FLX1 (flavin exchange) has been suggested to import FAD into mitochondria and hence be the yeast mitochondrial carrier for FAD 5 for the following reasons: (a) a respiratory defective mutant strain of S. cerevisiae was shown to have low mitochondrial FAD/FMN ratio; (b) the gene responsible for this phenotype was identified as FLX1; (c) mitochondrial vesicles from wild‐type cells displayed more efficient transport of FAD than those from the flx1 mutant strain; and (d) yeast FADS was thought to be located in the cytoplasm.
The closest human homologue of FLX1, SLC25A32 (29% sequence identity), was originally suggested to transport folate (vitamin B9) and named mitochondrial folate carrier (MFT), because of its ability to complement glycine auxotrophy of a Chinese hamster ovary cell line (CHO GlyB cells) with diminished mitochondrial folate levels. 6 Later, human SLC25A32 was shown to rescue growth defects and succinate dehydrogenase activity of the flx1 mutant strain. 180 The same authors suggested that mitochondrial FAD is necessary for dimethylglycine dehydrogenase and sarcosine dehydrogenase, which are involved in one‐carbon metabolism in connection to the folate cycle, catalysing the conversion of choline to glycine. Furthermore, FAD is also the cofactor of other enzymes involved in folate metabolism such as methylene THF reductase or the glycine clevage system. These grounds might explain the previous observations and the suggestion that SLC25A32 is a folate transporter. Also an Arabidopsis homologue of SLC25A32, At5g66380, which is located in chloroplasts, has been suggested to be a folate transporter based on complementation of CHO GlyB cells. 183 Unfortunately, no direct evidence for transport of FAD, FMN, folate or other substrates has yet been reported with isolated yeast FLX1, human SLC25A32 or Arabidopsis At5g66380. Therefore, it is not known whether these transporters are uniporters or antiporters and, given the possibility that FADS is located both in cytoplasm and mitochondria, 170 , 171 it is also not clear whether they have a role in mitochondrial FAD (or FMN) export.
4.3. Role of FAD in mitochondria
FAD is an important cofactor for several mitochondrial matrix enzymes, mostly catabolic dehydrogenases. In these reactions, FAD is reduced to FADH2 by accepting two electrons and two protons (Figure 4). The electrons of FADH2 in integral membrane flavoproteins are directly transferred onto ubiquinone, sometimes via other electron transfer groups. One example of membrane flavoprotein is complex II, succinate dehydrogenase, which catalyses the conversion of succinate to fumarate in the TCA cycle and to which FAD is covalently linked to a histidine residue, 184 whereas complex I NADH dehydrogenase contains FMN. Two other inner membrane bound mitochondrial flavoproteins, involved in proline/hydroxy‐proline catabolism, are proline dehydrogenase (PRODH), which converts proline to Δ1‐pyrroline‐5‐carboxylate, and hydroxyproline dehydrogenase (HYPDH), which transforms hydroxyproline, derived from collagen, to Δ1‐pyrroline‐3‐hydroxy‐5‐carboxylate. 185 , 186 FADH2 of soluble flavoproteins is re‐oxidized to FAD by electron transfer flavoprotein (ETF) onto ETF ubiquinone oxidoreductases (ETFQO), which transfers the electrons onto ubiquinone for the inner membrane‐embedded electron transport chain. One example of a soluble matrix flavoprotein is the E3 component (dihydrolipoyl dehydrogenase, DLD), which is identical for all four different 2‐oxoacid dehydrogenase complexes (PDHc, OGDHc, OADHc and BCKDHc) as discussed in Section 2.3. Dihydrolipoyl dehydrogenase is also part of the glycine cleavage system (GCS), involved in glycine metabolism. 187 Nine other soluble mitochondrial FAD‐dependent enzymes are involved in (a) fatty acid β‐oxidation, (b) BCAA catabolism and (c) amino acid catabolism. These include: (a) acyl‐CoA dehydrogenases for various acyl‐chain lengths (short‐, medium‐, long‐, and very‐long‐chain; SCADH, MCADH, LCADH and VLCADH) as well as ACAD9 (acyl‐CoA dehydrogenase 9); (b) isovaleryl‐CoA, isobutyryl‐CoA, and short branched‐chain acyl‐CoA dehydrogenases (IVDH, IBDH and SBCADH); and (c) glutaryl‐CoA dehydrogenase (GCDH). 168 Other mitochondrial soluble flavoenzymes are l‐ and D‐2‐hydroxyglutarate dehydrogenases (2HGDH), 188 , 189 which are keeping the levels of toxic 2‐hydroxyglutarate low, as well as choline dehydrogenase (CDH), dimethylglycine dehydrogenase (DMGDH) 190 and sarcosine dehydrogenase (SDH), 191 which are involved in choline metabolism. In addition, mitochondrial FAD is involved in heme biosynthesis (protoporphyrinogen oxidase PPOX), redox control (thioredoxin reductase TRXR2), sulfur metabolism (SQOR), complex I assembly/subcomplex (FXRD1, NDUFA10, NDUA9 and ACAD9), ubiquinone biosynthesis (COQ6), adenosylcobalamin biosynthesis (MMAB) and mitochondrial tRNA modifications (MTO1).
4.4. Diseases related to FAD
The RDA of riboflavin is about 0.9–1.3 mg/day for adults: a daily intake less than 0.6 mg may lead to vitamin B2 deficiency (ariboflavinosis), which is manifested by generalized weakness, angular stomatitis, glossitis, cheilosis, corneal vascularization, photophobia and, in severe cases, generalized seborrhoeic dermatitis. 192 , 193 , 194 Disease‐causing mutations have been identified in the genes encoding the riboflavin transporters SLC52A1–3, mitochondrial SLC25A32, FADS1, ETFα, ETFβ and ETFQO. 168 Mutations in the latter three proteins, which are involved in the mitochondrial re‐oxidation of FAD (Figure 4), give rise to a group of three severe disorders called multiple acyl‐CoA dehydrogenase deficiencies (MADD I‐III, respectively). These disorders involve multiple organs such as muscle, heart and liver, and are characterized by the accumulation of organic acids in urine, acylcarnitines and amino acids in plasma due to their compromised catabolism. 168
SLC25A32 deficiency (OMIM 616839) has been found in two patients, of which one displayed milder symptoms and the other more severe. The milder case exhibited recurrent late‐onset exercise intolerance and biochemical features similar to those of MADD patients. 181 The patient had compound heterozygous SLC25A32 mutations: a premature stop codon and a conservative missense mutation (p.Arg147His) in the second positively charged residue of the second signature motif sequence. The expression of the missense mutant protein could complement the defects in the yeast flx1 mutant strain only partly. 181 The more severe case had a neuromuscular phenotype that manifested early onset ataxia, myoclonia and dysarthria besides muscle weakness, exercise intolerance and MADD metabolites. 195 This patient had a homozygous indel mutation causing loss of the translational start codon and consequent loss of SLC25A32 expression or the use of an alternative start codon and production of a dysfunctional SLC25A32. If the former possibility is true, it might be that matrix FAD is not only provided through this carrier. For MAADIII and the disorders caused by mutations in riboflavin/FAD transporters SLC25A32 and SLC52A1–3, diet supplemented with riboflavin is a quite effective treatment improving the prognosis of patients. 168
5. NICOTINAMIDE ADENINE DINUCLEOTIDE (NAD +)
Nicotinamide adenine dinucleotide (NAD+) is a redox cofactor used by hundreds of enzymes because of its capacity to act as an electron donor and acceptor in its reduced (NADH) and oxidized (NAD+) form, respectively. In addition, NAD+ is also used by non‐redox enzymes, such as sirtuins and poly(ADP‐ribose) polymerases for protein deacylation and ADP‐ribosylation reactions, respectively. It is formed by AMP and nicotinamide mononucleotide (NMN) linked through their phosphates (Figure 1). NMN consists of a riboside, which is linked to nicotinamide through its 1′‐carbon (together forming nicotinamide riboside), and a 5′‐phosphate. Nicotinamide and nicotinamide riboside together with nicotinate (nicotinic acid or niacin) are different forms of vitamin B3. The oxidized form NAD+ has a positive charge on N1 of nicotinamide (along with the two negative charges on the phosphates), whereas the reduced form NADH has accepted two electrons and one proton (together a hydride ion), giving rise to neutralized N1 and an additional hydrogen added onto C4 of the nicotinamide ring. Unlike FAD, which is most often tightly bound to their flavoproteins, NAD+ is loosely bound to their proteins, and in many cases the NAD+ binding domain has a Rossman fold.
5.1. NAD + biosynthesis and distribution
There are several routes of NAD+ biosynthesis in animals that require the absorption of at least one of the essential precursors: the different forms of vitamin B3 or the essential amino acid tryptophan. Intestinal absorption of vitamin B3 derivatives is mediated by SLC5A8, SLC22A13, SLC12A8 and SLC29 family members, whereas tryptophan is taken up by SLC7A5, SLC36A4 and other transporters (Figure 5). 196 , 197 The majority of NAD+ is produced by the Preiss–Handler pathway initiating from nicotinate. 198 NAD+ may also be synthesized de novo from tryptophan by the kynurenine pathway, which is thought to operate mainly in liver producing nicotinamide that can be exported to many different tissues and converted into NAD+ by the NAD+ salvage pathway. Nicotinamide is also formed by the action of extracellular NAD+ glycohydrolases (NADases: CD38 and CD73/CD157) and by the intracellular non‐redox NAD+‐dependent enzymes. 199 Furthermore, NAD+ may be repristinated by other salvage pathways from NMN and nicotinamide riboside. 198 It is noteworthy that NAD+ homeostasis has been shown to be crucial in health, and several diseases are associated with its unbalance. 200 , 201
FIGURE 5.
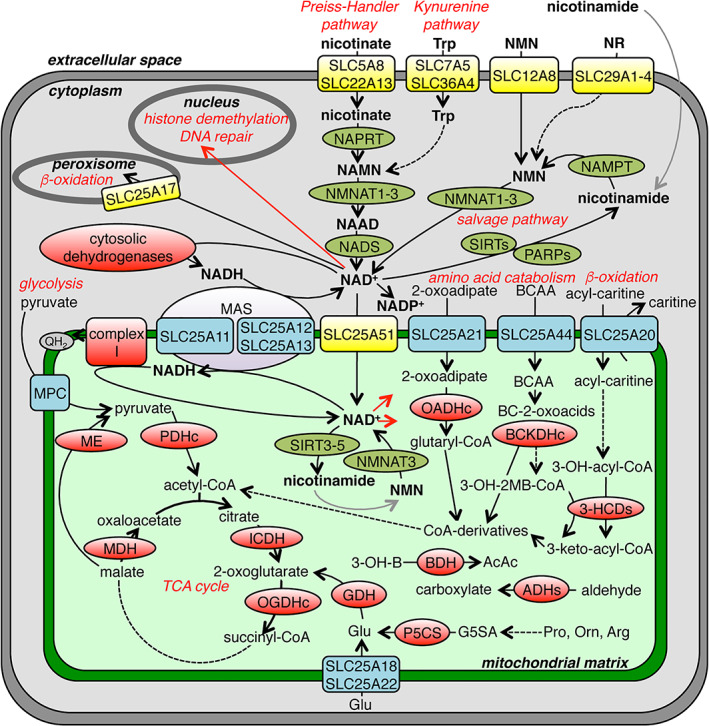
NAD+ metabolism and transport in a typical human cell. The schematic overview shows NAD+, vitamin B3 and tryptophan transporters (yellow), NAD+‐using enzymes (red), enzymes metabolizing NAD+ (olive green), enzymatic reactions (black arrows), reactions with unidentified enzymes (grey arrows) and distribution (red arrows). 3‐HCDHs, 3‐hydroxyacyl‐CoA dehydrogenases; 3‐OH‐2 MB‐CoA, 3‐hydroxy‐2‐methylbutyryl‐CoA; 3‐OH‐acyl‐CoA, 3‐hydroxy‐acyl‐CoA; 3‐OH‐B, 3‐hydroxybutyrate; AcAc, acetoacetate; ADHs, aldehyde dehydrogenases; BC‐2‐oxoacids, branched‐chain 2‐oxoacids; BCAA, branched‐chain amino acids; BCKDHc, branched‐chain α‐ketoacid dehydrogenase; GDH, glutamate dehydrogenase; ICDH, isocitrate dehydrogenase; MAS, malate–aspartate shuttle; MDH, malate dehydrogenase; ME, malic enzyme; MPC, mitochondrial pyruvate carrier; NAAD, nicotinic acid adenine dinucleotide; NADS, NAD+ synthase; NAMN, nicotinate mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NAPRT, nicotinate phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NMNAT, NMN adenylyl transferase; NR, nicotinamide ribonucleotide; OADHc, 2‐oxoadipate dehydrogenase complex; OGDHc, 2‐oxoglutarate dehydrogenase complex; PARPs, poly(ADP‐ribose) polymerases; PDHc, pyruvate dehydrogenase complex; P5CS, Δ1‐pyrroline‐5‐carboxylate synthase; QH2, reduced ubiquinone; SIRTs, sirtuins
Intracellular NAD+ is especially involved in reactions catalysed by dehydrogenases in multiple catabolic pathways, including glycolysis, glutaminolysis and fatty acid β‐oxidation (Figure 5). The reducing equivalents of cytosolic NADH, produced, for example, from glycolysis, are shuttled across the mitochondrial membrane by the classic malate–aspartate and glycerol‐3‐phosphate shuttles, 202 , 203 , 204 to intramitochondrial NAD+, which is re‐oxidized to NAD+ by complex I of the respiratory chain. NAD+ is distributed in all cellular compartments; measures indicate a cytosolic free NAD+ concentration of about 0.1 mM and twice as much in mitochondria. 205 However, other measures indicate 0.3–0.8 mM total cellular NAD+. 206 In the nucleus, NAD+ is required for the de‐acetylation of histones mediated by sirtuins and poly(ADP‐ribose) polymerases, which are involved in DNA damage repair. 198 MC family members have been identified to be responsible for mitochondrial and peroxisomal import of NAD+ from the cytoplasm. In peroxisomes, human SLC25A17 and plant PXN have been shown to transport NAD+ along with other nucleotides and cofactors. 146 , 147 , 207 In animal mitochondria, net import of NAD+ is catalysed by NAD+ transporters that will be described briefly here, because they have been treated thoroughly in a recent review. 49
5.2. Mitochondrial NAD + transport
The EPRA method has been used to identify and characterize two NAD+ transporter isoforms of the MC family in S. cerevisiae (Ndt1p and Ndt2p) 9 and in A. thaliana (AtNDT1 and AtNDT2), 182 whereas genetic approaches and indirect biochemical and complementation assays were used to identify the human SLC25A51 as NAD+ transporter. 11 , 12 , 13
Yeast Ndt1p and Ndt2p localize to mitochondria, as shown by GFP‐fusion protein constructs. Ndt1p transports NAD+ (Km of 0.38 mM) as well as (d)AMP, (d)GMP, NAAD and several other nucleotides to a lesser extent. 9 Both Ndt1p and Ndt2p deletion strains display growth defects on non‐fermentable carbon sources and reduced mitochondrial NAD+ levels; in double knockout cells these levels are even further decreased. 9 , 208 Based on these results, it was suggested that the main physiological role of Ndt1p and Ndt2p is mitochondrial NAD+ import in exchange for (d)AMP or (d)GMP. 9
The plant AtNDT1 and AtNDT2, which have 26–34% sequence identity with their yeast homologues, transport NAD+ (Km values of 0.25 and 0.15 mM, respectively), NAAD, NMN, ADP and AMP, and less efficiently NAMN, FAD, FMN, and other nucleotides. 182 AtNDT1 and AtNDT2 are localized to mitochondria, and their expression in yeast Ndt1p‐ and Ndt2p‐deletion strains complements the growth defect and mitochondrial NAD+ transport. 182 , 209 The plant mitochondrial NAD+ carriers are particularly expressed in energetically active and developing tissues, and their depletion leads to impaired reproduction. 182 , 209 , 210 Therefore, AtNDT1 and AtNDT2 most likely mediate the import of NAD+ into mitochondria, possibly in exchange for ADP or AMP. 182
Given that the closest human homologue of the yeast and plant NAD+ carriers, SLC25A32, does not transport NAD+ 211 and the subcellular localization of the NAD+ biosynthesis enzymes in animals is rather uncertain, the existence of an animal mitochondrial NAD+ transporter has been enigmatic for quite a long time. Several pieces of evidence have recently pointed towards SLC25A51 as a mitochondrial NAD+ importer (Figure 5), although this carrier has not yet been characterized by direct transport measurements. That SLC25A51, which is ubiquitously expressed in humans and has an isoform with 96% sequence identity (SLC25A52), has a central role in metabolism was demonstrated by SLC25A51 knockout and knockdown in several cell lines causing defects in mitochondrial respiration, TCA cycle and fatty acid oxidation. 11 , 12 , 13 Furthermore, (a) its functional role was linked to other mitochondrial cofactor transporters, the plasma membrane glucose transporter SLC2A1 as well as to one‐carbon and glutathione metabolism, (b) SLC25A51 knockdown led to a reduction of mitochondrial NAD+ levels and transport, whereas overexpression of the protein had opposite effects, and (c) reduced mitochondrial respiration and NAD+ levels of the human SLC25A51 knockout cells were rescued by expression of yeast Ndt1p and, vice versa, the growth defect, diminished mitochondrial NAD+ levels and mitochondrial transport of NAD+ was complemented by SLC25A51 expression in the yeast Ndt1/Ndt2 double knockout. 11 , 12 , 13 SLC25A51 lacks several of the conserved residues found in almost all MCs, and in particular, at variance with nucleotide‐transporting MCs, it does not have G[IVLM] in contact point II nor does it cluster together with them in phylogenetic trees. 49 These sequence features make SLC25A51 different from the NAD+ carriers of yeast and Arabidopsis as well as from the other MCs for B‐vitamin‐derived cofactors. Until now, no disease has been associated with SLC25A51. Based on the above‐described results, SLC25A51 most likely mediates mitochondrial NAD+ import.
5.3. Role of NAD + in mitochondria
In the mitochondrial matrix, NAD+ is used as a cofactor in redox reactions catalysed by numerous enzymes (Figure 5), for example, the E3 component (di‐hydrolipoamide dehydrogenase, DLDH), which is common to all the 2‐oxoacid dehydrogenase complexes (PDHc, OGDHc, OADHc and BCKDHc) discussed in Section 2.3; other enzymes of, or connected to, the TCA cycle: isocitrate dehydrogenase, malate dehydrogenase (also involved in the malate–aspartate shuttle) and malic enzyme; of amino acid catabolism: glutamate dehydrogenase 2, δ‐1‐pyrroline‐5‐carboxylate dehydrogenase (ALDH4A1) and 3‐hydroxyacyl‐CoA dehydrogenases (HCD2, DHB8 and HCDH); and of ketone body and alcohol metabolism: aldehyde dehydrogenases (ALDH2, AL1B1) and 3‐hydroxybutyrate dehydrogenase (BDH). NADH, obtained from most of these reactions, is then re‐oxidized to NAD+ by complex I, which transfers two electrons to the electron transfer chain.
Sirtuins 3–5 are mitochondrial non‐redox enzymes that require NAD+ to remove various modifications (e.g., acetylations, lipoylations, biotinylations, succinylations and malonylations) on lysine residues of proteins producing nicotinamide as a byproduct. 212 Given that the above‐mentioned modifications generally cause inhibition of the protein activities, these regulatory sirtuins usually act as activators of enzymes, such as PDHc and LCAD, but also others involved in fatty acid oxidation, BCAA catabolism, TCA cycle, urea cycle, ATP production and ketone body synthesis. In humans, the nicotinamide produced by the sirtuins of the mitochondrial matrix might be recuperated into NAD+ by a salvage pathway; an NMN adenylyl transferase (NMNAT3) has been localized to the matrix, but no nicotinamide phosphoribosyltransferase that would have been required for NAD+ recycling. 213 , 214 These observations evoke the questions of the other fates of mitochondrial nicotinamide and NMN (are they also transported?) and an alternative role of NMNAT3. 49
6. CONCLUSIONS/PERSPECTIVES
In the last two decades, a substantial progress has been made concerning the distribution of the vitamin B‐derived cofactors and their metabolism/recycling enzymes in the various cellular compartments, and especially about their transporters across the cellular plasma membrane and the membranes of the intracellular organelles. In particular, a considerable advancement has been reached in the knowledge about how thiamine pyrophosphate, coenzyme A, FAD and NAD+ are transported into mitochondria, used by enzymes in alternative ways and recycled. However, as noted in this review, many pieces in the puzzle are missing to comprehend mitochondrial transport and metabolism of vitamin B‐derived coenzymes. Some of the key points listed below should be explored to improve our understanding of this matter.
Some mitochondrial enzymes involved in cofactor metabolism/recycling have not yet been identified or their subcellular localization is unclear. These uncertainties have important consequences on the comprehension of the transporter functions in vivo. For example, in the case that ThPP phosphatase, which transforms ThPP in ThMP, does not exist in mitochondria, ThMP would be an unlikely counter‐substrate for TPC‐mediated ThPP mitochondrial import. Similarly, if nicotinamide phosphoribosyltransferase, which converts nicotinamide in NMN, is not localized within the mitochondria, a mitochondrial nicotinamide and/or NMN transporter would be required. Another example is COASY: if COASY is only localized to the cytoplasm, SLC25A42 most likely would mediate mitochondrial CoA import; if COASY is dually localized in mitochondrial matrix and cytoplasm, the matrix variant could take part in a CoA salvage pathway; and, finally, if COASY is localized only to the matrix, 4′‐phosphopantetheine would have to be imported into mitochondria and SLC25A42 would probably export CoA to the cytoplasm.
The substrate specificities and modes of transport (uniport/antiport) have not been characterized for the mitochondrial FAD carrier SLC25A32 and NAD+ carrier SLC25A51 by direct transport measurements (e.g., the EPRA method) using purified proteins. Given the presence of FADS1 in mitochondria, one could hypothesize that SLC25A32 catalyses FMN import and FAD export. Under the current state of knowledge, several hypotheses can be made about which NAD+‐derivatives are transported by SLC25A51 into the mitochondria and about which, if any, are the counter‐substrates.
Other transporters for the same coenzymes in the inner mitochondrial membrane may exist, especially because other members of the MC family have been shown to have overlapping substrate specificity. There are already some clues suggesting that this is the case also for some of the vitamin B‐derived coenzyme transporters. The close phylogenetic relationship between SLC25A42 and SLC25A16 as well as between SLC25A51, SLC25A52 and SLC25A53 might suggest similar substrates within each of these two groups of carriers. Furthermore, the fact that no growth defect of the S. cerevisiae tpc1Δ strain was observed when grown on non‐fermentable carbon sources suggests the existence of another mitochondrial ThPP transporter. However, there is no close homologue of Tpc1p in yeast, which might indicate that a more distant member of the MC family or a transporter belonging to another protein family is involved in the import of ThPP into mitochondria of yeast cells grown on non‐fermentable carbon sources.
The precise physiological roles of the vitamin B‐derived coenzyme transporters in vivo remain to be investigated. These studies, which have already started by analyses of single gene overexpression or knockout in cells and model organisms, and are being much helped by the biochemical characterization of the purified transporters and their metabolic context, would benefit from the use of combined pairs/sets of gene knockouts and multiple‐parameter analyses of their phenotypes. Furthermore, an improved and enlarged analysis of the phenotypes of the genetic variants causing vitamin B‐related pathologies could contribute to a better understanding of the physiological roles of the transporters as well as the pathogenetic mechanisms underlying the major symptoms of these diseases. Such knowledge may further be used for future improvement of disease diagnosis and development of new therapeutic protocols.
For the vitamin B‐derived coenzymes pyridoxal phosphate, biotin and cobalamin, no mitochondrial transporter has been identified yet. This is another interesting area of future research that might involve members of transporter families different from the MC family.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
Research in the authors' laboratories was supported by (a) PRIN 2017 (2017PAB8EM) and PRIN 2020 (2020RRJP5L) grants from the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR), and (b) Centre of Excellence on Comparative Genomics (CEGBA). Open Access Funding provided by Universita degli Studi di Bari Aldo Moro within the CRUI‐CARE Agreement.
Palmieri F, Monné M, Fiermonte G, Palmieri L. Mitochondrial transport and metabolism of the vitamin B‐derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD +, and related diseases: A review. IUBMB Life. 2022;74:592–617. 10.1002/iub.2612
Ferdinando Palmieri and Magnus Monné contributed equally to this work.
Funding information Centre of Excellence on Comparative Genomics (CEGBA); Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR), Grant/Award Numbers: PRIN 2020 (2020RRJP5L), PRIN 2017 (2017PAB8EM)
REFERENCES
- 1. Solmonson A, DeBerardinis RJ. Lipoic acid metabolism and mitochondrial redox regulation. J Biol Chem. 2018;293:7522–7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piel RB, Dailey HA, Medlock AE. The mitochondrial heme metabolon: Insights into the complex(ity) of heme synthesis and distribution. Mol Genet Metab. 2019;128:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hargreaves I, Heaton RA, Mantle D. Disorders of human coenzyme Q10 metabolism: An overview. Int J Mol Sci. 2020;21:E6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braymer JJ, Freibert SA, Rakwalska‐Bange M, Lill R. Mechanistic concepts of iron‐sulfur protein biogenesis in biology. Biochim Biophys Acta Mol Cell Res. 2021;1868:118863. [DOI] [PubMed] [Google Scholar]
- 5. Tzagoloff A, Jang J, Glerum DM, Wu M. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J Biol Chem. 1996;271:7392–7397. [DOI] [PubMed] [Google Scholar]
- 6. Titus SA, Moran RG. Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J Biol Chem. 2000;275:36811–36817. [DOI] [PubMed] [Google Scholar]
- 7. Marobbio CMT, Vozza A, Harding M, Bisaccia F, Palmieri F, Walker JE. Identification and reconstitution of the yeast mitochondrial transporter for thiamine pyrophosphate. EMBO J. 2002;21:5653–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindhurst MJ, Fiermonte G, Song S, et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci USA. 2006;103:15927–15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Todisco S, Agrimi G, Castegna A, Palmieri F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J Biol Chem. 2006;281:1524–1531. [DOI] [PubMed] [Google Scholar]
- 10. Fiermonte G, Paradies E, Todisco S, Marobbio CMT, Palmieri F. A novel member of solute carrier family 25 (SLC25A42) is a transporter of coenzyme a and adenosine 3′,5′‐diphosphate in human mitochondria. J Biol Chem. 2009;284:18152–18159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Girardi E, Agrimi G, Goldmann U, et al. Epistasis‐driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat Commun. 2020;11:6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kory N, Uit de Bos J, van der Rijt S, et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci Adv. 2020;6:eabe5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luongo TS, Eller JM, Lu M‐J, et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature. 2020;588:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palmieri F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. 2004;447:689–709. [DOI] [PubMed] [Google Scholar]
- 15. Palmieri F, Pierri CL. Structure and function of mitochondrial carriers ‐ role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010;584:1931–1939. [DOI] [PubMed] [Google Scholar]
- 16. Palmieri F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol Aspects Med. 2013;34:465–484. [DOI] [PubMed] [Google Scholar]
- 17. Picault N, Hodges M, Palmieri L, Palmieri F. The growing family of mitochondrial carriers in Arabidopsis. Trends Plant Sci. 2004;9:138–146. [DOI] [PubMed] [Google Scholar]
- 18. Saraste M, Walker JE. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982;144:250–254. [DOI] [PubMed] [Google Scholar]
- 19. Palmieri F, Bisaccia F, Capobianco L, et al. Transmembrane topology, genes, and biogenesis of the mitochondrial phosphate and oxoglutarate carriers. J Bioenerg Biomembr. 1993;25:493–501. [DOI] [PubMed] [Google Scholar]
- 20. Palmieri F. Mitochondrial carrier proteins. FEBS Lett. 1994;346:48–54. [DOI] [PubMed] [Google Scholar]
- 21. Bisaccia F, Capobianco L, Brandolin G, Palmieri F. Transmembrane topography of the mitochondrial oxoglutarate carrier assessed by peptide‐specific antibodies and enzymatic cleavage. Biochemistry. 1994;33:3705–3713. [DOI] [PubMed] [Google Scholar]
- 22. Capobianco L, Bisaccia F, Michel A, Sluse FE, Palmieri F. The N‐ and C‐termini of the tricarboxylate carrier are exposed to the cytoplasmic side of the inner mitochondrial membrane. FEBS Lett. 1995;357:297–300. [DOI] [PubMed] [Google Scholar]
- 23. Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem. 2010;47:37–52. [DOI] [PubMed] [Google Scholar]
- 24. Pebay‐Peyroula E, Dahout‐Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ‐M, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. [DOI] [PubMed] [Google Scholar]
- 25. Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ, Kunji ERS. Structures of yeast mitochondrial ADP/ATP carriers support a domain‐based alternating‐access transport mechanism. Proc Natl Acad Sci USA. 2014;111:E426–E434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruprecht JJ, King MS, Zogg T, et al. The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell. 2019;176:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klingenberg M. The ADP,ATP shuttle of the mitochondrion. Trends Biochem Sci. 1979;4:249–252. [Google Scholar]
- 28. Krämer R, Palmieri F. Metabolite carriers in mitochondria. In: Ernster L, editor. Molecular mechanisms in bioenergetics. New Comprehensive Biochemistry: Elsevier, Amsterdam, 1992; p. 359–384. [Google Scholar]
- 29. Palmieri F, Indiveri C, Bisaccia F, Krämer R. Functional properties of purified and reconstituted mitochondrial metabolite carriers. J Bioenerg Biomembr. 1993;25:525–535. [DOI] [PubMed] [Google Scholar]
- 30. Indiveri C, Tonazzi A, Palmieri F. The reconstituted carnitine carrier from rat liver mitochondria: Evidence for a transport mechanism different from that of the other mitochondrial translocators. Biochim Biophys Acta. 1994;1189:65–73. [DOI] [PubMed] [Google Scholar]
- 31. Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta. 2008;1778:1978–2021. [DOI] [PubMed] [Google Scholar]
- 32. Palmieri F, Pierri CL, De Grassi A, Nunes‐Nesi A, Fernie AR. Evolution, structure and function of mitochondrial carriers: A review with new insights. Plant J. 2011;66:161–181. [DOI] [PubMed] [Google Scholar]
- 33. Pietropaolo A, Pierri CL, Palmieri F, Klingenberg M. The switching mechanism of the mitochondrial ADP/ATP carrier explored by free‐energy landscapes. Biochim Biophys Acta. 2016;1857:772–781. [DOI] [PubMed] [Google Scholar]
- 34. Ruprecht JJ, Kunji ERS. The SLC25 mitochondrial carrier family: Structure and mechanism. Trends Biochem Sci. 2020;45:244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tonazzi A, Giangregorio N, Console L, Palmieri F, Indiveri C. The mitochondrial carnitine acyl‐carnitine carrier (SLC25A20): Molecular mechanisms of transport, role in redox sensing and interaction with drugs. Biomolecules. 2021;11:521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palmieri F, Monné M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim Biophys Acta. 2016;1863:2362–2378. [DOI] [PubMed] [Google Scholar]
- 37. Marobbio CMT, Di Noia MA, Palmieri F. Identification of a mitochondrial transporter for pyrimidine nucleotides in Saccharomyces cerevisiae: Bacterial expression, reconstitution and functional characterization. Biochem J. 2006;393:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vozza A, Parisi G, De Leonardis F, et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA. 2014;111:960–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Porcelli V, Fiermonte G, Longo A, Palmieri F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J Biol Chem. 2014;289:13374–13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Noia MA, Todisco S, Cirigliano A, et al. The human SLC25A33 and SLC25A36 genes of solute carrier family 25 encode two mitochondrial pyrimidine nucleotide transporters. J Biol Chem. 2014;289:33137–33148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monné M, Daddabbo L, Gagneul D, et al. Uncoupling proteins 1 and 2 (UCP1 and UCP2) from Arabidopsis thaliana are mitochondrial transporters of aspartate, glutamate, and dicarboxylates. J Biol Chem. 2018;293:4213–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gorgoglione R, Porcelli V, Santoro A, et al. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim Biophys Acta Bioenerg. 2019;1860:724–733. [DOI] [PubMed] [Google Scholar]
- 43. Yoneshiro T, Wang Q, Tajima K, et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature. 2019;572:614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palmieri F. Energy metabolism, mitochondrial transporters of the solute carrier 25 (SLC25) superfamily. In: Jez J, editor. Encyclopedia of biological chemistry III. 3rd ed. Oxford, UK: Elsevier, 2021; p. 213–243. [Google Scholar]
- 45. Robinson AJ, Kunji ERS. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc Natl Acad Sci USA. 2006;103:2617–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marobbio CMT, Giannuzzi G, Paradies E, Pierri CL, Palmieri F. α‐Isopropylmalate, a leucine biosynthesis intermediate in yeast, is transported by the mitochondrial oxalacetate carrier. J Biol Chem. 2008;283:28445–28553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Monné M, Miniero DV, Daddabbo L, Robinson AJ, Kunji ERS, Palmieri F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J Biol Chem. 2012;287:7925–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tonazzi A, Console L, Giangregorio N, Indiveri C, Palmieri F. Identification by site‐directed mutagenesis of a hydrophobic binding site of the mitochondrial carnitine/acylcarnitine carrier involved in the interaction with acyl groups. Biochim Biophys Acta. 2012;1817:697–704. [DOI] [PubMed] [Google Scholar]
- 49. Ziegler M, Monné M, Nikiforov A, Agrimi G, Heiland I, Palmieri F. Welcome to the family: Identification of the NAD+ transporter of animal mitochondria as member of the solute carrier family SLC25. Biomolecules. 2021;11:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fiermonte G, Walker JE, Palmieri F. Abundant bacterial expression and reconstitution of an intrinsic membrane‐transport protein from bovine mitochondria. Biochem J. 1993;294:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Palmieri F, Agrimi G, Blanco E, et al. Identification of mitochondrial carriers in Saccharomyces cerevisiae by transport assay of reconstituted recombinant proteins. Biochim Biophys Acta. 2006;1757:1249–1262. [DOI] [PubMed] [Google Scholar]
- 52. Monné M, Palmieri F. Antiporters of the mitochondrial carrier family. Curr Top Membr. 2014;73:289–320. [DOI] [PubMed] [Google Scholar]
- 53. Indiveri C, Tonazzi A, Stipani I, Palmieri F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria: Electrical nature and coupling of the exchange reaction with H+ translocation. Biochem J. 1997;327:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palmieri L, Vozza A, Hönlinger A, et al. The mitochondrial dicarboxylate carrier is essential for the growth of Saccharomyces cerevisiae on ethanol or acetate as the sole carbon source. Mol Microbiol. 1999;31:569–577. [DOI] [PubMed] [Google Scholar]
- 55. Palmieri L, Runswick MJ, Fiermonte G, Walker JE, Palmieri F. Yeast mitochondrial carriers: Bacterial expression, biochemical identification and metabolic significance. J Bioenerg Biomembr. 2000;32:67–77. [DOI] [PubMed] [Google Scholar]
- 56. Palmieri L, Lasorsa FM, Vozza A, et al. Identification and functions of new transporters in yeast mitochondria. Biochim Biophys Acta. 2000;1459:363–369. [DOI] [PubMed] [Google Scholar]
- 57. Indiveri C, Iacobazzi V, Tonazzi A, et al. The mitochondrial carnitine/acylcarnitine carrier: Function, structure and physiopathology. Mol Aspects Med. 2011;32:223–233. [DOI] [PubMed] [Google Scholar]
- 58. Cappello AR, Curcio R, Miniero DV, et al. Functional and structural role of amino acid residues in the even‐numbered transmembrane alpha‐helices of the bovine mitochondrial oxoglutarate carrier. J Mol Biol. 2006;363:51–62. [DOI] [PubMed] [Google Scholar]
- 59. Cappello AR, Miniero DV, Curcio R, et al. Functional and structural role of amino acid residues in the odd‐numbered transmembrane alpha‐helices of the bovine mitochondrial oxoglutarate carrier. J Mol Biol. 2007;369:400–412. [DOI] [PubMed] [Google Scholar]
- 60. Miniero DV, Cappello AR, Curcio R, Ludovico A, Daddabbo L, et al. Functional and structural role of amino acid residues in the matrix α‐helices, termini and cytosolic loops of the bovine mitochondrial oxoglutarate carrier. Biochim Biophys Acta. 2011;1807:302–310. [DOI] [PubMed] [Google Scholar]
- 61. Palmieri F. Diseases caused by defects of mitochondrial carriers: A review. Biochim Biophys Acta. 2008;1777:564–578. [DOI] [PubMed] [Google Scholar]
- 62. Stanley CA, Palmieri F, Bennett MJ. Disorders of the mitochondrial carnitine shuttle. In: Valle D, Vogelstein B, Kinzler K, et al., editors. Online molecular and metabolic basis of inherited disease. New York, NY: McGraw Hill, 2013. [Google Scholar]
- 63. Palmieri F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J Inherit Metab Dis. 2014;37:565–575. [DOI] [PubMed] [Google Scholar]
- 64. Palmieri F, Scarcia P, Monné M. Diseases caused by mutations in mitochondrial carrier genes SLC25: A review. Biomolecules. 2020;10:655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Losa R, Sierra MI, Fernández A, Blanco D, Buesa JM. Determination of thiamine and its phosphorylated forms in human plasma, erythrocytes and urine by HPLC and fluorescence detection: A preliminary study on cancer patients. J Pharm Biomed Anal. 2005;37:1025–1029. [DOI] [PubMed] [Google Scholar]
- 66. Andrews FH, McLeish MJ. Substrate specificity in thiamin diphosphate‐dependent decarboxylases. Bioorg Chem. 2012;43:26–36. [DOI] [PubMed] [Google Scholar]
- 67. Frank RAW, Leeper FJ, Luisi BF. Structure, mechanism and catalytic duality of thiamine‐dependent enzymes. Cell Mol Life Sci. 2007;64:892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kowalska E, Kozik A. The genes and enzymes involved in the biosynthesis of thiamin and thiamin diphosphate in yeasts. Cell Mol Biol Lett. 2008;13:271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jurgenson CT, Begley TP, Ealick SE. The structural and biochemical foundations of thiamin biosynthesis. Annu Rev Biochem. 2009;78:569–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fitzpatrick TB, Chapman LM. The importance of thiamine (vitamin B1) in plant health: From crop yield to biofortification. J Biol Chem. 2020;295:12002–12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rodionov DA, Hebbeln P, Eudes A, et al. A novel class of modular transporters for vitamins in prokaryotes. J Bacteriol. 2009;191:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jaehme M, Singh R, Garaeva AA, Duurkens RH, Slotboom D‐J. PnuT uses a facilitated diffusion mechanism for thiamine uptake. J Gen Physiol. 2018;150:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Enjo F, Nosaka K, Ogata M, Iwashima A, Nishimura H. Isolation and characterization of a thiamin transport gene, THI10, from Saccharomyces cerevisiae. J Biol Chem. 1997;272:19165–19170. [DOI] [PubMed] [Google Scholar]
- 74. Bocobza SE, Aharoni A. Switching the light on plant riboswitches. Trends Plant Sci. 2008;13:526–533. [DOI] [PubMed] [Google Scholar]
- 75. Turck D, Bresson J‐L, Burlingame B, et al. Dietary reference values for thiamin. EFSA J. 2016;14:e04653. [Google Scholar]
- 76. Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. The gene mutated in thiamine‐responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22:305–308. [DOI] [PubMed] [Google Scholar]
- 77. Dutta B, Huang W, Molero M, et al. Cloning of the human thiamine transporter, a member of the folate transporter family. J Biol Chem. 1999;274:31925–31929. [DOI] [PubMed] [Google Scholar]
- 78. Rajgopal A, Edmondnson A, Goldman ID, Zhao R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta. 2001;1537:175–178. [DOI] [PubMed] [Google Scholar]
- 79. Said HM, Balamurugan K, Subramanian VS, Marchant JS. Expression and functional contribution of hTHTR‐2 in thiamin absorption in human intestine. Am J Physiol Gastrointest Liver Physiol. 2004;286:G491–G498. [DOI] [PubMed] [Google Scholar]
- 80. Zhao R, Gao F, Goldman ID. Reduced folate carrier transports thiamine monophosphate: An alternative route for thiamine delivery into mammalian cells. Am J Physiol Cell Physiol. 2002;282:C1512–C1517. [DOI] [PubMed] [Google Scholar]
- 81. Lemos C, Faria A, Meireles M, Martel F, Monteiro R, Calhau C. Thiamine is a substrate of organic cation transporters in Caco‐2 cells. Eur J Pharmacol. 2012;682:37–42. [DOI] [PubMed] [Google Scholar]
- 82. Nabokina SM, Inoue K, Subramanian VS, Valle JE, Yuasa H, Said HM. Molecular identification and functional characterization of the human colonic thiamine pyrophosphate transporter. J Biol Chem. 2014;289:4405–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gangolf M, Czerniecki J, Radermecker M, et al. Thiamine status in humans and content of phosphorylated thiamine derivatives in biopsies and cultured cells. PLoS One. 2010;5:e13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang K, Huentelman MJ, Rao F, et al. Genetic implication of a novel thiamine transporter in human hypertension. J Am Coll Cardiol. 2014;63:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen L, Shu Y, Liang X, et al. OCT1 is a high‐capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc Natl Acad Sci USA. 2014;111:9983–9988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Alexander B, Landwehr G, Mitchell F. Studies of thiamine metabolism in man. II. Thiamine and pyrimidine excretion wtih special reference to the relationship between injected and excreted thiamine in normal and abnormal subjects. J Clin Invest. 1946;25:294–303. [PubMed] [Google Scholar]
- 87. Ariaey‐Nejad MR, Balaghi M, Baker EM, Sauberlich HE. Thiamin metabolism in man. Am J Clin Nutr. 1970;23:764–778. [DOI] [PubMed] [Google Scholar]
- 88. Nosaka K, Onozuka M, Kakazu N, et al. Isolation and characterization of a human thiamine pyrophosphokinase cDNA. Biochim Biophys Acta. 2001;1517:293–297. [DOI] [PubMed] [Google Scholar]
- 89. Bettendorff L, Wins P. Thiamin diphosphate in biological chemistry: New aspects of thiamin metabolism, especially triphosphate derivatives acting other than as cofactors. FEBS J. 2009;276:2917–2925. [DOI] [PubMed] [Google Scholar]
- 90. Boyle L, Wamelink MMC, Salomons GS, et al. Mutations in TKT are the cause of a syndrome including short stature, developmental delay, and congenital heart defects. Am J Hum Genet. 2016;98:1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lakaye B, Makarchikov AF, Antunes AF, et al. Molecular characterization of a specific thiamine triphosphatase widely expressed in mammalian tissues. J Biol Chem. 2002;277:13771–13777. [DOI] [PubMed] [Google Scholar]
- 92. Manzetti S, Zhang J, van der Spoel D. Thiamin function, metabolism, uptake, and transport. Biochemistry. 2014;53:821–835. [DOI] [PubMed] [Google Scholar]
- 93. Kürsteiner O, Dupuis I, Kuhlemeier C. The pyruvate decarboxylase1 gene of Arabidopsis is required during anoxia but not other environmental stresses. Plant Physiol. 2003;132:968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Romagnoli G, Luttik MAH, Kötter P, Pronk JT, Daran J‐M. Substrate specificity of thiamine pyrophosphate‐dependent 2‐oxo‐acid decarboxylases in Saccharomyces cerevisiae. Appl Environ Microbiol. 2012;78:7538–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bunik VI, Tylicki A, Lukashev NV. Thiamin diphosphate‐dependent enzymes: From enzymology to metabolic regulation, drug design and disease models. FEBS J. 2013;280:6412–6442. [DOI] [PubMed] [Google Scholar]
- 96. Bettendorff L, Wins P, Lesourd M. Subcellular localization and compartmentation of thiamine derivatives in rat brain. Biochim Biophys Acta. 1994;1222:1–6. [DOI] [PubMed] [Google Scholar]
- 97. Kitamura T, Seki N, Kihara A. Phytosphingosine degradation pathway includes fatty acid α‐oxidation reactions in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2017;114:E2616–E2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Foulon V, Antonenkov VD, Croes K, et al. Purification, molecular cloning, and expression of 2‐hydroxyphytanoyl‐CoA lyase, a peroxisomal thiamine pyrophosphate‐dependent enzyme that catalyzes the carbon‐carbon bond cleavage during alpha‐oxidation of 3‐methyl‐branched fatty acids. Proc Natl Acad Sci USA. 1999;96:10039–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kato M, Wynn RM, Chuang JL, et al. Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: Role of disordered phosphorylation loops. Structure. 2008;16:1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Armstrong CT, Anderson JLR, Denton RM. Studies on the regulation of the human E1 subunit of the 2‐oxoglutarate dehydrogenase complex, including the identification of a novel calcium‐binding site. Biochem J. 2014;459:369–381. [DOI] [PubMed] [Google Scholar]
- 101. Danhauser K, Sauer SW, Haack TB, et al. DHTKD1 mutations cause 2‐aminoadipic and 2‐oxoadipic aciduria. Am J Hum Genet. 2012;91:1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Evarsson AA, Chuang JL, Wynn RM, Turley S, Chuang DT, Hol WG. Crystal structure of human branched‐chain alpha‐ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure. 2000;8:277–291. [DOI] [PubMed] [Google Scholar]
- 103. Dolce V, Fiermonte G, Runswick MJ, Palmieri F, Walker JE. The human mitochondrial deoxynucleotide carrier and its role in the toxicity of nucleoside antivirals. Proc Natl Acad Sci USA. 2001;98:2284–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Barile M, Valenti D, Brizio C, Quagliariello E, Passarella S. Rat liver mitochondria can hydrolyse thiamine pyrophosphate to thiamine monophosphate which can cross the mitochondrial membrane in a carrier‐mediated process. FEBS Lett. 1998;435:6–10. [DOI] [PubMed] [Google Scholar]
- 105. Iacopetta D, Carrisi C, De Filippis G, et al. The biochemical properties of the mitochondrial thiamine pyrophosphate carrier from Drosophila melanogaster. FEBS J. 2010;277:1172–1181. [DOI] [PubMed] [Google Scholar]
- 106. Frelin O, Agrimi G, Laera VL, et al. Identification of mitochondrial thiamin diphosphate carriers from Arabidopsis and maize. Funct Integr Genomics. 2012;12:317–326. [DOI] [PubMed] [Google Scholar]
- 107. Rudhe C, Chew O, Whelan J, Glaser E. A novel in vitro system for simultaneous import of precursor proteins into mitochondria and chloroplasts. Plant J. 2002;30:213–220. [DOI] [PubMed] [Google Scholar]
- 108. Brandizzi F, Irons S, Kearns A, Hawes C. BY‐2 cells: Culture and transformation for live cell imaging. Curr Protoc Cell Biol. 2003;19:1.7.1–1.7.16. 10.1002/0471143030.cb0107s19 [DOI] [PubMed] [Google Scholar]
- 109. Bezerra GA, Foster WR, Bailey HJ, et al. Crystal structure and interaction studies of human DHTKD1 provide insight into a mitochondrial megacomplex in lysine catabolism. IUCrJ. 2020;7:693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: Structure‐based function and regulation. J Biol Chem. 2014;289:16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ciszak EM, Korotchkina LG, Dominiak PM, Sidhu S, Patel MS. Structural basis for flip‐flop action of thiamin pyrophosphate‐dependent enzymes revealed by human pyruvate dehydrogenase. J Biol Chem. 2003;278:21240–21246. [DOI] [PubMed] [Google Scholar]
- 112. Hawkins CF, Borges A, Perham RN. A common structural motif in thiamin pyrophosphate‐binding enzymes. FEBS Lett. 1989;255:77–82. [DOI] [PubMed] [Google Scholar]
- 113. Walsh DA, Cooper RH, Denton RM, Bridges BJ, Randle PJ. The elementary reactions of the pig heart pyruvate dehydrogenase complex. A study of the inhibition by phosphorylation. Biochem J. 1976;157:41–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nemeria NS, Zhang X, Leandro J, et al. Toward an understanding of the structural and mechanistic aspects of protein‐protein interactions in 2‐oxoacid dehydrogenase complexes. Life (Basel). 2021;11:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fiermonte G, Dolce V, Palmieri L, et al. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J Biol Chem. 2001;276:8225–8230. [DOI] [PubMed] [Google Scholar]
- 116. Adeva‐Andany MM, López‐Maside L, Donapetry‐García C, Fernández‐Fernández C, Sixto‐Leal C. Enzymes involved in branched‐chain amino acid metabolism in humans. Amino Acids. 2017;49:1005–1028. [DOI] [PubMed] [Google Scholar]
- 117. Liu Y, Li Y, Wang X. Acetohydroxyacid synthases: Evolution, structure, and function. Appl Microbiol Biotechnol. 2016;100:8633–8649. [DOI] [PubMed] [Google Scholar]
- 118. Lonhienne T, Low YS, Garcia MD, et al. Structures of fungal and plant acetohydroxyacid synthases. Nature. 2020;586:317–321. [DOI] [PubMed] [Google Scholar]
- 119. Crook MA, Sriram K. Thiamine deficiency: The importance of recognition and prompt management. Nutrition. 2014;30:953–954. [DOI] [PubMed] [Google Scholar]
- 120. Marcé‐Grau A, Martí‐Sánchez L, Baide‐Mairena H, Ortigoza‐Escobar JD, Pérez‐Dueñas B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J Inherit Metab Dis. 2019;42:581–597. [DOI] [PubMed] [Google Scholar]
- 121. Rosenberg MJ, Agarwala R, Bouffard G, et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175–179. [DOI] [PubMed] [Google Scholar]
- 122. Spiegel R, Shaag A, Edvardson S, et al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–424. [DOI] [PubMed] [Google Scholar]
- 123. Ortigoza‐Escobar JD, Alfadhel M, Molero‐Luis M, et al. Thiamine deficiency in childhood with attention to genetic causes: Survival and outcome predictors. Ann Neurol. 2017;82:317–330. [DOI] [PubMed] [Google Scholar]
- 124. Bottega R, Perrone MD, Vecchiato K, et al. Functional analysis of the third identified SLC25A19 mutation causative for the thiamine metabolism dysfunction syndrome 4. J Hum Genet. 2019;64:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gowda VK, Srinivasan VM, Jehta K, Bhat MD. Bilateral striatal necrosis with polyneuropathy with a novel SLC25A19 (mitochondrial thiamine pyrophosphate carrier OMIMI*606521) mutation: Treatable thiamine metabolic disorder‐a report of two Indian cases. Neuropediatrics. 2019;50:313–317. [DOI] [PubMed] [Google Scholar]
- 126. Begley TP, Kinsland C, Strauss E. The biosynthesis of coenzyme a in bacteria. Vitam Horm. 2001;61:157–171. [DOI] [PubMed] [Google Scholar]
- 127. Leonardi R, Jackowski S. Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus. 2007;2. 10.1128/ecosalplus.3.6.3.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Shibata K, Gross CJ, Henderson LM. Hydrolysis and absorption of pantothenate and its coenzymes in the rat small intestine. J Nutr. 1983;113:2107–2115. [DOI] [PubMed] [Google Scholar]
- 129. Wang H, Huang W, Fei YJ, et al. Human placental Na+−dependent multivitamin transporter. Cloning, functional expression, gene structure, and chromosomal localization. J Biol Chem. 1999;274:14875–14883. [DOI] [PubMed] [Google Scholar]
- 130. Leonardi R, Zhang Y‐M, Rock CO, Jackowski S. Coenzyme a: Back in action. Prog Lipid Res. 2005;44:125–153. [DOI] [PubMed] [Google Scholar]
- 131. Alfonso‐Pecchio A, Garcia M, Leonardi R, Jackowski S. Compartmentalization of mammalian pantothenate kinases. PLoS One. 2012;7:e49509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tahiliani AG, Neely JR. Mitochondrial synthesis of coenzyme a is on the external surface. J Mol Cell Cardiol. 1987;19:1161–1167. [DOI] [PubMed] [Google Scholar]
- 133. Zhyvoloup A, Nemazanyy I, Panasyuk G, et al. Subcellular localization and regulation of coenzyme a synthase. J Biol Chem. 2003;278:50316–50321. [DOI] [PubMed] [Google Scholar]
- 134. Skrede S, Halvorsen O. Mitochondrial pantetheinephosphate adenylyltransferase and dephospho‐CoA kinase. Eur J Biochem. 1983;131:57–63. [DOI] [PubMed] [Google Scholar]
- 135. Rhee H‐W, Zou P, Udeshi ND, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dusi S, Valletta L, Haack TB, et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2014;94:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Robishaw JD, Berkich D, Neely JR. Rate‐limiting step and control of coenzyme a synthesis in cardiac muscle. J Biol Chem. 1982;257:10967–10972. [PubMed] [Google Scholar]
- 138. Sibon OCM, Strauss E. Coenzyme a: To make it or uptake it? Nat Rev Mol Cell Biol. 2016;17:605–606. [DOI] [PubMed] [Google Scholar]
- 139. Daugherty M, Polanuyer B, Farrell M, et al. Complete reconstitution of the human coenzyme a biosynthetic pathway via comparative genomics. J Biol Chem. 2002;277:21431–21439. [DOI] [PubMed] [Google Scholar]
- 140. O'Toole N, Barbosa JARG, Li Y, Hung L‐W, Matte A, Cygler M. Crystal structure of a trimeric form of dephosphocoenzyme a kinase from Escherichia coli. Protein Sci. 2003;12:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Naquet P, Kerr EW, Vickers SD, Leonardi R. Regulation of coenzyme a levels by degradation: The “ins and outs.”. Prog Lipid Res. 2020;78:101028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. [DOI] [PubMed] [Google Scholar]
- 143. Pietrocola F, Galluzzi L, Bravo‐San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme a: A central metabolite and second messenger. Cell Metab. 2015;21:805–821. [DOI] [PubMed] [Google Scholar]
- 144. Kanamori A, Nakayama J, Fukuda MN, et al. Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O‐acetylated ganglioside: A putative acetyl‐CoA transporter. Proc Natl Acad Sci USA. 1997;94:2897–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chornyi S, IJlst L, van Roermund CWT, Wanders RJA, Waterham HR. Peroxisomal metabolite and cofactor transport in humans. Front Cell Dev Biol. 2021;8:613892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Agrimi G, Russo A, Scarcia P, Palmieri F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme a, FAD and NAD+. Biochem J. 2012;443:241–247. [DOI] [PubMed] [Google Scholar]
- 147. Agrimi G, Russo A, Pierri CL, Palmieri F. The peroxisomal NAD+ carrier of Arabidopsis thaliana transports coenzyme a and its derivatives. J Bioenerg Biomembr. 2012;44:333–340. [DOI] [PubMed] [Google Scholar]
- 148. Prohl C, Pelzer W, Diekert K, et al. The yeast mitochondrial carrier Leu5p and its human homologue Graves' disease protein are required for accumulation of coenzyme a in the matrix. Mol Cell Biol. 2001;21:1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Haitina T, Lindblom J, Renström T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics. 2006;88:779–790. [DOI] [PubMed] [Google Scholar]
- 150. Todisco S, Di Noia MA, Castegna A, Lasorsa FM, Paradies E, Palmieri F. The Saccharomyces cerevisiae gene YPR011c encodes a mitochondrial transporter of adenosine 5′‐phosphosulfate and 3′‐phospho‐adenosine 5′‐phosphosulfate. Biochim Biophys Acta. 2014;1837:326–334. [DOI] [PubMed] [Google Scholar]
- 151. Vozza A, De Leonardis F, Paradies E, et al. Biochemical characterization of a new mitochondrial transporter of dephosphocoenzyme a in Drosophila melanogaster. Biochim Biophys Acta. 2017;1858:137–146. [DOI] [PubMed] [Google Scholar]
- 152. Zallot R, Agrimi G, Lerma‐Ortiz C, et al. Identification of mitochondrial coenzyme a transporters from maize and Arabidopsis. Plant Physiol. 2013;162:581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Cox TC, Sadlon TJ, Schwarz QP, et al. The major splice variant of human 5‐aminolevulinate synthase‐2 contributes significantly to erythroid heme biosynthesis. Int J Biochem Cell Biol. 2004;36:281–295. [DOI] [PubMed] [Google Scholar]
- 155. Masud AJ, Kastaniotis AJ, Rahman MT, Autio KJ, Hiltunen JK. Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim Biophys Acta Mol Cell Res. 2019;1866:118540. [DOI] [PubMed] [Google Scholar]
- 156. Zhao G, Jin Z, Allewell NM, Tuchman M, Shi D. Crystal structure of the N‐acetyltransferase domain of human N‐acetyl‐L‐glutamate synthase in complex with N‐acetyl‐L‐glutamate provides insights into its catalytic and regulatory mechanisms. PLoS One. 2013;8:e70369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pougovkina O, te Brinke H, Ofman R, et al. Mitochondrial protein acetylation is driven by acetyl‐CoA from fatty acid oxidation. Hum Mol Genet. 2014;23:3513–3522. [DOI] [PubMed] [Google Scholar]
- 158. Kerr EW, Shumar SA, Leonardi R. Nudt8 is a novel CoA diphosphohydrolase that resides in the mitochondria. FEBS Lett. 2019;593:1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Otten JJ, Hellwig JP, Meyers LD. Dietary reference intakes: The essential guide to nutrient requirements. 1st ed. Washington, D.C: Natl Academy Pr, 2006. [Google Scholar]
- 160. Di Meo I, Carecchio M, Tiranti V. Inborn errors of coenzyme a metabolism and neurodegeneration. J Inherit Metab Dis. 2019;42:49–56. [DOI] [PubMed] [Google Scholar]
- 161. Shamseldin HE, Smith LL, Kentab A, et al. Mutation of the mitochondrial carrier SLC25A42 causes a novel form of mitochondrial myopathy in humans. Hum Genet. 2016;135:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Almannai M, Alasmari A, Alqasmi A, et al. Expanding the phenotype of SLC25A42‐associated mitochondrial encephalomyopathy. Clin Genet. 2018;93:1097–1102. [DOI] [PubMed] [Google Scholar]
- 163. Iuso A, Alhaddad B, Weigel C, et al. A homozygous splice site mutation in SLC25A42, encoding the mitochondrial transporter of coenzyme a, causes metabolic crises and epileptic encephalopathy. JIMD Rep. 2019;44:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Bacher A, Eberhardt S, Fischer M, Kis K, Richter G. Biosynthesis of vitamin b2 (riboflavin). Annu Rev Nutr. 2000;20:153–167. [DOI] [PubMed] [Google Scholar]
- 165. Suwannasom N, Kao I, Pruß A, Georgieva R, Bäumler H. Riboflavin: The health benefits of a forgotten natural vitamin. Int J Mol Sci. 2020;21:E950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yoshimatsu H, Yonezawa A, Yao Y, et al. Functional involvement of RFVT3/SLC52A3 in intestinal riboflavin absorption. Am J Physiol Gastrointest Liver Physiol. 2014;306:G102–G110. [DOI] [PubMed] [Google Scholar]
- 167. Barile M, Giancaspero TA, Leone P, Galluccio M, Indiveri C. Riboflavin transport and metabolism in humans. J Inherit Metab Dis. 2016;39:545–557. [DOI] [PubMed] [Google Scholar]
- 168. Mereis M, Wanders RJA, Schoonen M, Dercksen M, Smuts I, van der Westhuizen FH. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int J Biochem Cell Biol. 2021;132:105899. [DOI] [PubMed] [Google Scholar]
- 169. Brizio C, Galluccio M, Wait R, et al. Over‐expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem Biophys Res Commun. 2006;344:1008–1016. [DOI] [PubMed] [Google Scholar]
- 170. Torchetti EM, Brizio C, Colella M, et al. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion. 2010;10:263–273. [DOI] [PubMed] [Google Scholar]
- 171. Bafunno V, Giancaspero TA, Brizio C, et al. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: Involvement of the Flx1p carrier in FAD export. J Biol Chem. 2004;279:95–102. [DOI] [PubMed] [Google Scholar]
- 172. Bruni F, Giancaspero TA, Oreb M, et al. Subcellular localization of Fad1p in Saccharomyces cerevisiae: A choice at post‐transcriptional level? Life. 2021;11:967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three‐dimensional structure of NADPH‐cytochrome P450 reductase: Prototype for FMN‐ and FAD‐containing enzymes. Proc Natl Acad Sci USA. 1997;94:8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Nishiya T, Matsumoto K, Maekawa S, et al. Regulation of inducible nitric‐oxide synthase by the SPRY domain‐ and SOCS box‐containing proteins. J Biol Chem. 2011;286:9009–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Cabibbo A, Pagani M, Fabbri M, et al. ERO1‐L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J Biol Chem. 2000;275:4827–4833. [DOI] [PubMed] [Google Scholar]
- 176. Riemer J, Appenzeller‐Herzog C, Johansson L, Bodenmiller B, Hartmann‐Petersen R, Ellgaard L. A luminal flavoprotein in endoplasmic reticulum‐associated degradation. Proc Natl Acad Sci USA. 2009;106:14831–14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Lin Y, Wu Y, Li J, et al. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine‐specific demethylase 1. EMBO J. 2010;29:1803–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tschantz WR, Zhang L, Casey PJ. Cloning, expression, and cellular localization of a human prenylcysteine lyase. J Biol Chem. 1999;274:35802–35808. [DOI] [PubMed] [Google Scholar]
- 179. Boulland M‐L, Marquet J, Molinier‐Frenkel V, et al. Human IL4I1 is a secreted L‐phenylalanine oxidase expressed by mature dendritic cells that inhibits T‐lymphocyte proliferation. Blood. 2007;110:220–227. [DOI] [PubMed] [Google Scholar]
- 180. Spaan AN, Ijlst L, van Roermund CWT, Wijburg FA, Wanders RJA, Waterham HR. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl‐CoA dehydrogenase deficiency. Mol Genet Metab. 2005;86:441–447. [DOI] [PubMed] [Google Scholar]
- 181. Schiff M, Veauville‐Merllié A, Su CH, et al. SLC25A32 mutations and riboflavin‐responsive exercise intolerance. N Engl J Med. 2016;374:795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Palmieri F, Rieder B, Ventrella A, et al. Molecular identification and functional characterization of Arabidopsis thaliana mitochondrial and chloroplastic NAD+ carrier proteins. J Biol Chem. 2009;284:31249–31259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Bedhomme M, Hoffmann M, McCarthy EA, et al. Folate metabolism in plants: An Arabidopsis homolog of the mammalian mitochondrial folate transporter mediates folate import into chloroplasts. J Biol Chem. 2005;280:34823–34831. [DOI] [PubMed] [Google Scholar]
- 184. Kim HJ, Winge DR. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim Biophys Acta. 2013;1827:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Summitt CB, Johnson LC, Jönsson TJ, Parsonage D, Holmes RP, Lowther WT. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem J. 2015;466:273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Zareba I, Palka J. Prolidase‐proline dehydrogenase/proline oxidase‐collagen biosynthesis axis as a potential interface of apoptosis/autophagy. Biofactors. 2016;42:341–348. [DOI] [PubMed] [Google Scholar]
- 187. Carothers DJ, Pons G, Patel MS. Dihydrolipoamide dehydrogenase: Functional similarities and divergent evolution of the pyridine nucleotide‐disulfide oxidoreductases. Arch Biochem Biophys. 1989;268:409–425. [DOI] [PubMed] [Google Scholar]
- 188. Achouri Y, Noël G, Vertommen D, Rider MH, Veiga‐Da‐Cunha M, Van Schaftingen E. Identification of a dehydrogenase acting on D‐2‐hydroxyglutarate. Biochem J. 2004;381:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Rzem R, Van Schaftingen E, Veiga‐da‐Cunha M. The gene mutated in l‐2‐hydroxyglutaric aciduria encodes l‐2‐hydroxyglutarate dehydrogenase. Biochimie. 2006;88:113–116. [DOI] [PubMed] [Google Scholar]
- 190. Augustin P, Hromic A, Pavkov‐Keller T, Gruber K, Macheroux P. Structure and biochemical properties of recombinant human dimethylglycine dehydrogenase and comparison to the disease‐related H109R variant. FEBS J. 2016;283:3587–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Bar‐joseph I, Pras E, Reznik‐Wolf H, et al. Mutations in the sarcosine dehydrogenase gene in patients with sarcosinemia. Hum Genet. 2012;131:1805–1810. [DOI] [PubMed] [Google Scholar]
- 192. Powers HJ. Riboflavin (vitamin B‐2) and health. Am J Clin Nutr. 2003;77:1352–1360. [DOI] [PubMed] [Google Scholar]
- 193. Pinto JT, Zempleni J. Riboflavin. Advances in Nutrition. 2016;7:973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kennedy DO. B vitamins and the brain: Mechanisms, dose and efficacy—A review. Nutrients. 2016;8:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hellebrekers DMEI, Sallevelt SCEH, Theunissen TEJ, et al. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur J Hum Genet. 2017;25:886–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50:284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kropotov A, Kulikova V, Nerinovski K, et al. Equilibrative nucleoside transporters mediate the import of nicotinamide riboside and nicotinic acid riboside into human cells. Int J Mol Sci. 2021;22:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22:119–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Gasparrini M, Sorci L, Raffaelli N. Enzymology of extracellular NAD metabolism. Cell Mol Life Sci. 2021;78:3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Katsyuba E, Romani M, Hofer D, Auwerx J. NAD+ homeostasis in health and disease. Nat Metab. 2020;2:9–31. [DOI] [PubMed] [Google Scholar]
- 201. Zapata‐Pérez R, Wanders RJA, van Karnebeek CDM, Houtkooper RH. NAD+ homeostasis in human health and disease. EMBO Mol Med. 2021;13:e13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Bücher T, Klingenberg M. Wege des Wasserstoffs in der lebendigen Organisation. Angewandte Chemie. 1958;70:552–570. [Google Scholar]
- 203. Indiveri C, Krämer R, Palmieri F. Reconstitution of the malate/aspartate shuttle from mitochondria. J Biol Chem. 1987;262:15979–15983. [PubMed] [Google Scholar]
- 204. Palmieri L, Pardo B, Lasorsa FM, et al. Citrin and aralar1 are ca(2+)‐stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Cambronne XA, Stewart ML, Kim D, et al. Biosensor reveals multiple sources for mitochondrial NAD+ . Science. 2016;352:1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Bernhardt K, Wilkinson S, Weber APM, Linka N. A peroxisomal carrier delivers NAD+ and contributes to optimal fatty acid degradation during storage oil mobilization. Plant J. 2012;69:1–13. [DOI] [PubMed] [Google Scholar]
- 208. Agrimi G, Brambilla L, Frascotti G, et al. Deletion or overexpression of mitochondrial NAD+ carriers in Saccharomyces cerevisiae alters cellular NAD and ATP contents and affects mitochondrial metabolism and the rate of glycolysis. Appl Environ Microbiol. 2011;77:2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. de Souza Chaves I, Feitosa‐Araújo E, Florian A, et al. The mitochondrial NAD+ transporter (NDT1) plays important roles in cellular NAD+ homeostasis in Arabidopsis thaliana. Plant J. 2019;100:487–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Feitosa‐Araujo E, de Souza Chaves I, Florian A, et al. Downregulation of a mitochondrial NAD+ transporter (NDT2) alters seed production and germination in Arabidopsis. Plant Cell Physiol. 2020;61:897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. VanLinden MR, Dölle C, Pettersen IKN, et al. Subcellular distribution of NAD+ between cytosol and mitochondria determines the metabolic profile of human cells. J Biol Chem. 2015;290:27644–27659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Carrico C, Meyer JG, He W, Gibson BW, Verdin E. The mitochondrial Acylome emerges: Proteomics, regulation by Sirtuins, and metabolic and disease implications. Cell Metab. 2018;27:497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Barile M, Passarella S, Danese G, Quagliariello E. Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochem Mol Biol Int. 1996;38:297–306. [PubMed] [Google Scholar]
- 214. Nikiforov A, Dölle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem. 2011;286:21767–21778. [DOI] [PMC free article] [PubMed] [Google Scholar]