

Abstract
Prostate cancer (PCa) is a tumor with a great heterogeneity, both at a molecular and clinical level. Despite its global good prognosis, cases can vary from indolent to lethal metastatic and scientific efforts are aimed to discern those with worse outcomes. Current prognostic markers, as Gleason score, fall short when it comes to distinguishing these cases. Identification of new early biomarkers to enable a better PCa distinction and classification remains a challenge. In order to identify new genes implicated in PCa progression we conducted several differential gene expression analyses over paired samples comparing primary PCa tissue against healthy prostatic tissue of PCa patients. The results obtained show that this approach is a serious alternative to overcome patient heterogeneity. We were able to identify 250 genes whose expression varies along with tissue differentiation—healthy to tumor tissue, 161 of these genes are described here for the first time to be related to PCa. The further manual curation of these genes allowed to annotate 39 genes with antitumoral activity, 22 of them described for the first time to be related to PCa proliferation and metastasis. These findings could be replicated in different cohorts for most genes. Results obtained considering paired differential expression, functional annotation and replication results point to: CGREF1, UNC5A, C16orf74, LGR6, IGSF1, QPRT and CA14 as possible new early markers in PCa. These genes may prevent the progression of the disease and their expression should be studied in patients with different outcomes.
Keywords: antitumoral genes, biomarkers, prostate cancer
What's new?
Prostate cancer presents with great molecular and clinical heterogeneity, and the identification of new early biomarkers to discern outcomes remains a challenge. By comparing the differential gene expression of prostate tumour and healthy tissues using paired samples, here the authors found that patients with low Gleason scores already show molecular changes related to metastasis and proliferation. Altogether, 161 genes were described for the first time to be prostate cancer‐related. Moreover, 22 of them had anti‐tumour activity, which could explain the generally low progression of prostate cancer. Seven genes are proposed as potential new early biomarkers.
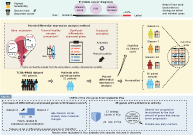
Abbreviations
- DEA
differential expression analysis
- PCA
principal component analysis
- PCa
prostate cancer
- PSA
prostate antigen levels
- TCGA
The Cancer Genome Atlas
- TNM
trimmed mean of m method
1. INTRODUCTION
After lung cancer, prostate cancer (PCa) is the second most commonly diagnosed tumor in males worldwide, with 1 276 106 new cases in 2018. 1 In recent years, an increase in cancer cases has been observed in Europe, with PCa the most frequent cancer in males and the third most common cause of cancer mortality. 2
PCa is the neoplasia with the highest hereditary component. 3 Up to 15% of all PCa is attributable to high‐risk hereditary factors. 4 Moreover, numerous genetic susceptibility markers for PCa have been identified using different approaches such as family‐based studies, candidate gene association studies, genome‐wide association studies and emerging RNA‐Seq technology. 5 Some gene examples of the most widely identified in the expression profile of PCa include BRCA1, BRCA2, PTEN and HOXB13. 6 , 7 , 8 However, only a few have been associated with cancer outcomes and often with inconsistent results. 9 Specifically, antitumor genes could reveal new insights into molecular mechanisms of PCa pathogenesis and be a promising approach for gene therapy of PCa. 10 , 11 However, compared to other cancers such as breast or colorectal cancers, 12 , 13 little has been described regarding antitumor genes in PCa.
These knowledge gaps could be due to the high intra‐ and intertumor heterogeneity of PCa, both at a molecular and clinical level. 14 , 15 In this sense, the clinical behavior of PCa ranges from indolent to lethal metastatic disease, with the Gleason score as the strongest prognostic marker identified to date. 16 , 17 However, Gleason grading presents moderate reproducibility when used for the pathological classification of PCa in routine clinical practice. 18 Thus, the development of new biomarkers to enable better PCa distinction and classification is a sizable challenge. 19 , 20 Identifying gene expression profiles could be useful in this context to overcome individual tumor variability by comparing healthy prostate tissue as control. 21 , 22
In this article, we analyze the genomic expression of PCa to identify genes implicated in the proliferation and metastasis progression of PCa by comparing gene expression of paired samples between prostate tumoral and healthy tissues. The final objective was to find new candidates for early PCa markers.
2. MATERIAL AND METHODS
2.1. Dataset description
The TCGA‐PRAD dataset 23 was downloaded from The Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov/) 24 with TCGAbiolinks package 25 version 2.14.1. This dataset included RNA‐Seq expression data from 56 499 genes for 551 samples of primary PCa adenocarcinoma, from 495 patients, in addition to social patient data, Gleason related measures, prostate antigen levels (PSA), mutational landscape information and sample collection data. Procedures of samples are specified in the original paper. 23 Methodological, biological gene expression and vial (aliquots) variability were assessed by principal component analysis (PCA) using the NOISeq package, 26 version 2.30.0. Sample variability assessment by PCA dismissed batch effect and biological variability but showed an aliquot variability, thus using only vial A samples for further analysis (Figure S1).
2.2. Differential expression analysis
Gene expression analysis was performed using R language version 3.6.2 (https://cran.r-project.org/bin/windows/) and R Studio 1.2.5019; ggplot2 package version 3.3.3 (https://rstudio.com/) 27 and pheatmap version 1.0.12 was used for graphical representation. For the discovery process, only paired samples (51) were selected from the TCGA‐PRAD dataset. Additionally, this set was divided into three further subsets according to the Gleason Score (“Gleason 6,” “Gleason 7” and “Gleason ≥8”) (Figure 1).
FIGURE 1.
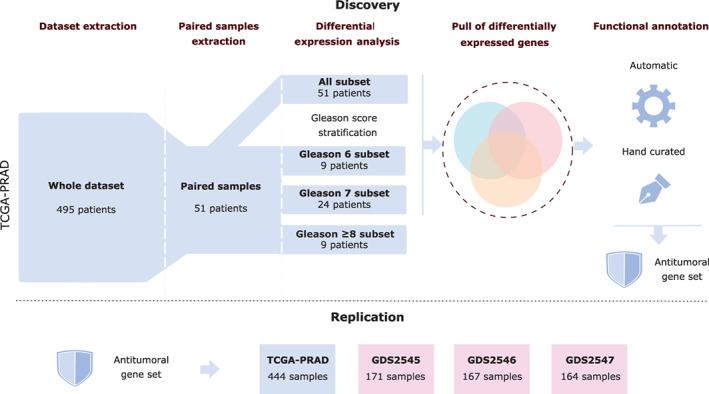
Methodology flowchart [Color figure can be viewed at wileyonlinelibrary.com]
The four subsets, the whole of the 51 paired samples and the three subsets after stratifying by Gleason (Figure 1) were independently analyzed using gene filtering with filterByExpr from the edgeR package with default parameters, 28 version 3.28.1. Normalization was done using the trimmed mean of m method (TMM) with the NOISeq package, and values were later transformed in the logarithmic scale. Differentially expressed genes between tumor and healthy adjacent samples for each subset were obtained using a two groups single‐channel experimental design from the limma package, 29 version 3.42.2. Inclusion criteria included a false discovery rate‐adjusted P‐value <.01 and at least a 2‐fold change.
2.3. Functional annotation
Annotation was carried out over the total pool of differential expressed genes obtained from the four subsets described in Section 2.2 (Figure 1).
Automatic annotation and enrichment analysis was done using ConsensusPathDB 30 , 31 across all available databases and selecting Gene Ontology categories at level 4.
Manually curated annotation was done using own‐made scripts for mining the Pubmed database (https://www.ncbi.nlm.nih.gov/pubmed/). The search terms for each gene differentially expressed were: “[‘prostate cancer’ AND ‘gen symbol’],” “[‘prostate adenocarcinoma’ AND ‘gen symbol’].” If no significant results were found, a secondary search was performed using the terms: [“cancer” AND “gen symbol.”] The articles retrieved were manually inspected, and gene functions were categorized according to known Prostate Cancer Hallmarks 32 and stored in a database.
2.4. Replication datasets
No available datasets with paired data were found for PCa primary tumor. Consequently, to replicate our findings, three independent cohorts from the GEO database (http://www.ncbi.nlm.nih.gov/geo): GDS2545, GDS2546, GDS2547; were used. They included data from healthy tissue, healthy adjacent tissue, primary PCa tissue and metastatic tissue with 171, 167 and 164 samples, respectively (Table 1). Only normalized gene expression data was available, but data could undergo logarithmic transformation. Moreover, TCGA‐PRAD remaining samples were normalized and scaled, using the same parameters as described in Section 2.2 (Figure 1). Replication analysis was done with all samples, but the 51 paired samples were used for discovering the antitumoral differentially expressed gene set.
TABLE 1.
Study and validation datasets number of samples
| Source | Healthy prostate tissue | Healthy prostate adjacent tissue | Primary prostate tissue | Metastatic prostate tissue | |
|---|---|---|---|---|---|
| Discovery dataset | TCGA‐PRAD | – | 51 | 51 | – |
|
Replication datasets |
TCGA‐PRAD | – | – | 444 | – |
| GDS2545 | 18 | 63 | 65 | 25 | |
| GDS2546 | 17 | 59 | 66 | 25 | |
| GDS2547 | 17 | 58 | 64 | 25 |
Replication samples' antitumoral expression tendencies were analyzed for different tumor stages. Statistical tests were performed using the statistical rstatix package version 0.6.0, 33 Welch's ANOVA with Bonferroni correction was used for groups' comparison. Differences were considered statistically significant at the P < 0.01 level. Joint plots from TCGA‐PRAD and GEO datasets showing expression changes and statistical analysis are in Supporting Information 1.
3. RESULTS
As previously described in Section 2.2, in order to identify clear expression patterns related to PCa progression only paired samples were used for the differential expression analysis (DEA) (Figure 1). Paired samples analysis involves comparing healthy adjacent and tumor samples from the same patient, avoiding interpersonal variability interferences in detecting differential gene expressions due to disease progress. DEA analysis between healthy and tumoral samples were performed overall for 51 patients, subset named “All,” and for three additional subsets resulting from the stratification of the 51 patients according to their Gleason score and named: “Gleason 6” subset, “Gleason 7” and “Gleason ≥8” (Figure 1).
DEA over the subset “All” showed 195 differentially expressed genes, from which 107 were overexpressed and 88 underexpressed in tumoral tissue with regard to healthy adjacent tissue (Table S1). The subset “Gleason 6” showed 54 differentially expressed genes, 45 of them overexpressed. The number of differentially expressed genes was much higher in the subset “Gleason 7” reaching 192 genes, 115 overexpressed. However, the subset “Gleason ≥8” did not present any significant differentially expressed genes between tumoral tissue and adjacent tissue. The differentially expressed genes found in subsets “All,” “Gleason 6” and “Gleason 7” were represented in heatmaps and clustered samples by healthy and tumor tissue (Figures S2‐S4). In all cases, differential expression between healthy tissue and the corresponding subgroup tumoral tissue is visible. The interpersonal variability in expression independent of the tissue, is also noteworthy.
Functional annotation was done in a two‐step process. First using automatic annotation and then a manually curated process. The annotation was performed over the differentially expressed genes found in the subsets “All”, “Gleason 6” and “Gleason 7” (Figure 2), which made a total of 250 genes, 140 overexpressed and 110 underexpressed in tumor tissue vs paired healthy samples. Most of the 250 genes were protein‐coding genes (82.8%), followed by long noncoding RNA, lncRNA, (14%) and pseudogenes (3.2%).
FIGURE 2.
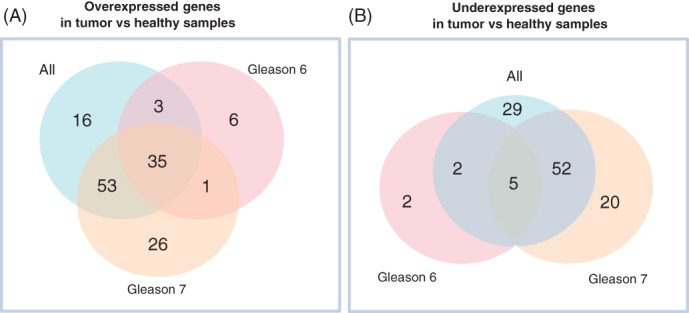
Venn diagram of the 250 significant (P‐value <.01) differentially expressed genes between paired samples—healthy and tumor. The figure shows results found in the subsets “All,” “Gleason 6” and “Gleason 7.” (A) Results across the three datasets and their overlap for overexpressed genes; (B) Results across the three datasets and their overlap for underexpressed genes [Color figure can be viewed at wileyonlinelibrary.com]
Automatic functional analysis evidenced that, in general, PCa is related to the endocrine process and that over and underexpressed genes are associated with different biological processes. Moreover, most of the differentially expressed genes detected are specific to a particular Gleason score and are related to different biological processes. For example, concerning the six overexpressed genes from the subset “Gleason 6,” they were linked to cell cycle (G2/M transition, checkpoints and Rho GTPases); the 26 specific genes to subset “Gleason 7” were related to lipase activity and rhodopsin‐like receptors.
In the case of underexpressed genes, the differences were significant in the number of specific genes related to the Gleason score. While “Gleason 6” presented two exclusive underexpressed genes, “Gleason 7” presented 20. The two genes specific for “Gleason 6” could not be related to any biological process so far (C16orf74 and PHYHIPL), while the 20 genes of the “Gleason 7” were involved in androgen receptor, skeletal system development and neuron fate commitment pathways. Additional information to Figure 2 is available in Table S2.
Results obtained with the automatic approach highlight that PCa, regardless of the Gleason score, is associated with changes in the endocrine system. Moreover, bring the insight that genes differentially expressed in patients with Gleason score 6 are related to proliferation while in the case of Gleason score 7 they are mainly related to the progression of the disease.
Taking into account the delay in information flow from published resources to the specialized databases, we performed a manually curated functional annotation (Supporting Information 2). This approach allowed us to have a detailed understanding of the DEA genes' functions, and classify them according to cancer hallmarks for PCa and their antitumoral activity. It is noteworthy that using manually curated functional annotation there is still little information or a complete lack of information for 45 of these genes in relation to their function or specific role in cancer development (Figure 3). Moreover, we found that 43 genes had been previously described to be related to cancer (eg, omics experiments), but there was not enough information related to specific cancer hallmarks. However, there were 162 genes for which the manually curated annotation encountered new information and allowed their classification according to those hallmarks. From these 162 genes, 89 were directly linked to Prostate Cancer Hallmarks, whereas 73 were found to be related to hallmarks described in other cancers (eg, breast cancer, colorectal cancer or cancer in general). Most of 162 genes were related to metastasis (96) and proliferation (75), followed by resisting cell death 24 (Figure 3). There were 110 genes strictly related to one hallmark and 52 linked to more than one.
FIGURE 3.
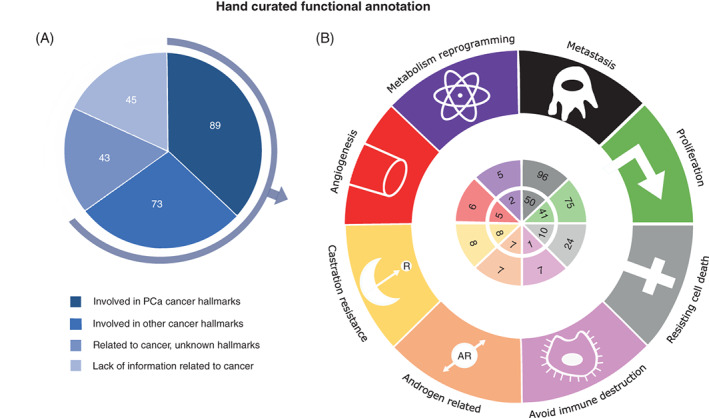
Results of the manually curated functional annotation over the 250 significant (P‐value <.01) differentially expressed genes between paired samples—healthy and tumor. (A) Percentage of genes merged by the available information linked to their activity related to cancer and prostate cancer. (B) Number of genes whose activity could be pooled into cancer hallmarks in prostate cancer (inner circle) and cancer in general (outer circle), which includes prostate cancer. Genes could be related to more than one hallmark [Color figure can be viewed at wileyonlinelibrary.com]
Manually curated functional annotation revealed that, among the 162 genes with information, there were 39 antitumoral genes (24%). From these genes, 17 were identified to have known antitumoral activity in PCa by manual functional annotation (Table S5). Remarkably, the remaining 22 antitumoral genes found in our study have not been previously related to PCa, though all of them have been previously described to have antitumoral activity in other cancers (Table 2).
TABLE 2.
Antitumoral genes described for the first time to be involved in PCa
| Gene | Function | Cancer hallmark | PCa exp. |
|---|---|---|---|
| CGREF1 | Inhibits AP‐1, c‐Jun, c‐Fos, p42/44 and p38 suppressing proliferation | Prol. | OE |
| FFAR2 | Its loss promotes colon cancer by an epigenetic dysregulation of inflammation and its OE induced apoptosis in leukemia | A.I.D. | OE |
| SRARP | Its expression is inversely correlated with genes that promote cell proliferation | Prol. | OE |
| UNC5A | UE in most cancers. Reduces apoptosis when unbound to its ligand | R.C.D. | OE |
| TGM3 | Its OE acts repressing EMT in colorectal cancer but promoting it in hepatocellular carcinoma | Met. | OE |
| TOX3 | Inhibits cancer migration and invasion via transcriptional regulation of SNAI1 and SNAI2 but seems to play a critical role in progression of breast cancer | Met. | OE |
| PGM5‐AS1 | Its downregulation inhibits the proliferation and metastasis via increasing miRNAs expression in colorectal cancer and esophageal squamous cell carcinoma | Met. Prol. | UE |
| CRABP2 | Its loss reduces viability and proliferation and induces apoptosis, cytotoxicity and interferon‐signaling in malignant peripheral nerve sheath tumors | Prol., R.C.D. | UE |
| C16orf74 | OE in pancreatic cancer, plays a crucial role in proliferation and invasion | Met., Prol. | UE |
| P2RX6 | Promotes renal cancer cells migration and invasion modulating ERK1/2 phosphorylation and MMP9 signaling pathway | Met. | UE |
| LGR6 | Downregulation inhibits proliferation and invasion and increases apoptosis by increased expression of Bcl‐2 and caspase‐3 and inhibition of PI3K/AKT | Met., Prol. | UE |
| MSLN | OE in multiple cancers, activates the NFB, MAPK and PI3K pathways and subsequently induce resistance to apoptosis or promote cell proliferation, migration and metastasis | Met., Prol., R.C.D. | UE |
| PDE1C | Silencing PDE1C significantly mitigates proliferation and EMT in glioblastoma | Met., Prol. | UE |
| ACTC1 | ACTC1‐negative glioma had better prognosis than ACTC1 positive, and it was related to epithelial mesenchymal transition (EMT) | Met. | UE |
| EMX2OS | Induces proliferation, invasion and sphere formation in ovarian cancer via regulating the mir‐654‐3P/AKT3/PD‐L1 axis | Met., Prol. | UE |
| LINC00958 | Its OE facilitates cell proliferation, migration and invasion | Met., Prol. | UE |
| IGSF1 | Its knockdown could inhibit cell proliferation and significantly impair the migration and invasion in vitro in thyroid cancer | Met., Prol. | UE |
| SYT8 | Inhibition was correlated with decreased invasion, migration and fluorouracil resistance in gastric cancer | Met. | UE |
| CHP2 | Its OE promotes proliferation by activating AKT and suppression of FOXO3 transcription factor in breast cancer | Prol. | UE |
| CRABP2 | Its loss reduces viability and proliferation and induces apoptosis, cytotoxicity and interferon‐signaling in malignant peripheral nerve sheath tumors | Prol., R.C.D. | UE |
| QPRT | It suppresses spontaneous cell death by inhibiting overproduction of active‐caspase‐3 | R.C.D. | UE |
| PON3 | Enhances cell death resistance | R.C.D. | UE |
| CA14 | It is usually upregulated in cancer and linked with deadification | M.R. | UE |
The antitumoral activity was scored according to genetic manipulation experiments and pathways activity annotations (Tables S3 and S4). From the 39 antitumoral genes, 34 are protein‐coding genes and 5 are regulatory lncRNAs, 29 presented differential underexpression.
The expression pattern found in the paired sample analysis for most of the 39 antitumoral genes agrees with the expression pattern (over‐ or underexpressed) previously described in the literature for other cancers. However, there were eight genes (TRPM8, CCN5, TGM3, TOX3, ACTC1, EMX2OS, PGM5‐AS1 and CRABP2) whose activity can be antitumoral or carcinogenic depending on the cancer type (Tables S3 and S4). For example, the overexpression of TGM3 (Table 2) in colorectal cancer presents antitumoral activity, while, in the case of hepatocellular carcinoma, it has a carcinogenic effect. We also found an increase in the overall number of antitumoral genes with cancer progression, Gleason 6 vs Gleason 7, as shown in Figure 4, and most were related to proliferation and metastasis inhibition.
FIGURE 4.
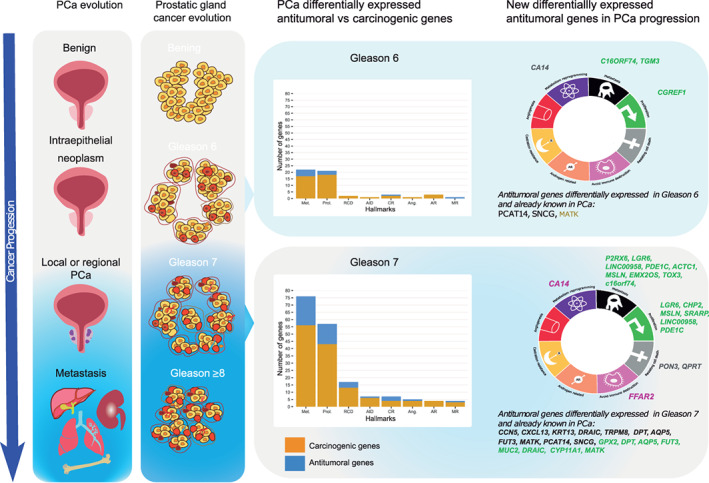
Results summary of antitumoral differentially expressed genes found in “Gleason 6” and “Gleason 7” subsets merged by cancer hallmarks manually curated annotation [Color figure can be viewed at wileyonlinelibrary.com]
To corroborate our findings, we studied the expression levels of these antitumoral genes in the remaining tumoral samples from TCGA‐PRAD dataset not used for discovery and in the GEO datasets GDS2545, GDS2546 and GDS2547 (Figure 1). We could replicate the results in the remaining TCGA‐PRAD nonpaired samples in all cases but one, SYT7.
Regarding replicate analysis in the GEO samples, 31 of the 39 antitumoral genes were present in at least one of the three GEO replicate datasets (Table S6). Unfortunately, none of these experiments were paired samples. The replication percentage was lower in the case of the GEO expression datasets but still reached more than 74% replication in all samples where the gene was present. As an example of variable replication, we have PDE1C and ACTC1, where the expression profile of one replication sample agreed with TCGA_PRAD paired analysis results, but the other did not. In other cases, differences were so small between healthy adjacent tissue and primary PCa tissue that no conclusions could be made. However, in most cases, the tendency was to agree with the TCGA_PRAD paired sample analysis results. Results of all antitumoral genes are provided in Supporting Information 3.
Considering paired differential expression, manually curated functional annotation and replication results, we propose seven new potential PCa early‐stage antitumoral genes: CGREF1, UNC5A, C16orf74, LGR6, IGSF1, QPRT and CA14. Taking into account that some of the remaining genes could not be assessed in replica datasets, thus not being discarded for future analysis. Additionally, we found three genes—TGM3, TOX3 and CRABP—that present significant differential expressions between healthy and tumor samples and have been proved to play a role in other cancer processes (eg, colorectal, breast cancer). However, this evidence shows contradictory results (antitumoral or carcinogenic activity) depending on the type of cancer and thus the role of these three genes in PCa should be further investigated.
4. DISCUSSION
The use of differential gene expression between tumoral and healthy tissue over paired samples points to this approach as a serious alternative to overcome patient heterogeneity. It allowed the identification of 250 genes whose expression varies along with tissue differentiation—healthy to tumor tissue—, 161 of these genes are described here for the first time to be related to PCa. All these genes were found to be implied in diverse molecular mechanisms related to cancer. It is noteworthy that 15% of these 250 genes could be related to antitumoral activity, allowing the identification of new potential early PCa markers.
Paired DEA strategy highlighted clear differences between healthy and tumor tissue when considering all available paired samples. These differences persisted when samples were stratified by Gleason score. There were no genes differentially expressed between the “healthy tissue” and “Gleason ≥8” tumor when using this analysis. This result is not surprising due to the “field effect” that occurs within the normal epithelium adjacent to cancer. The absence of differentially expressed genes in this comparison could be explained by the histological changes and altered gene expression in the healthy tissue, an effect that has been already described to be more pronounced in aggressive PCa disease. 34 , 35 , 36 Thus, we focused on the paired comparisons of healthy tissue with earlier differentiation stage tumors (“Gleason 6” and “Gleason 7”) in order to identify potential early PCa markers. This stratification revealed that the number of differentially expressed genes depended on the tumor differentiation stage, with a higher number in “Gleason 7” compared to “Gleason 6.” This result is in line with previous findings for PCa and breast cancer, relating gene expression changes to different tumor profiles and behavior. 37 , 38
Automatic functional analysis over the identified genes did not bring other insights than those previously known to be related to PCa in the literature. For example, the well‐known role of the endocrine process in the development and progression of PCa. 39 The “Gleason 6” subset was related to proliferation mechanisms, and the “Gleason 7” subset with progression indicating a loss of local cell control in the latter. These functions agree with described changes in behavior between Gleason 6 stage and Gleason 7, specifically in Gleason 7 (4 + 3) in PCa. 40
The use of automatic functional annotation implies some drawbacks due to the delay of information flow between published results and databases. 41 Thus, manually curated functional analysis allowed us, on the one hand, to have a deeper and actualized insight about the newly identified genes, and on the other hand, to link their activity, when data was available, to cancer hallmarks. This strategy identified the antitumoral activity of many newly discovered differentially expressed genes in PCa, such as the underexpressed C16orf74. There was no available information for this gene when using automatic functional annotation, but manual annotation showed that it plays a crucial role in proliferation and invasion when overexpressed in pancreatic cancer. This strategy also revealed that almost 20% of the genes were not protein‐coding but regulatory genes, lncRNA and pseudogenes. This finding is in line with the increasingly recognized role of noncoding genes in the course of PCa, 42 and it supports the integration of these genes into precision medicine. However, to date, their role has been reserved only for research purposes and not for translational medicine. 43
More interesting is that this exhaustive approach revealed a high number of genes with potential antitumoral activity (n = 39) based on findings in the literature using manually curated annotation. Among them, we found 22 genes that have been identified previously as antitumoral in other cancers (eg, breast and colorectal cancer). However, they are described here for the first time to be differentially expressed between healthy and tumor tissue and in different grades of PCa differentiation. For example, CGREF1, whose overexpression in cancer suppresses proliferation. This does not necessarily mean that the 22 new antitumoral genes will act in PCa in the same way as described in the literature in other cancers. In fact, among these 22 genes, there are six (TGM3, TOX3, ACTC1, EMX2OS, PGM5‐AS1 and CRABP2) whose activity varies in the literature between carcinogenic or antitumoral, depending on the cancer type. However, this opens a new line of study for potential antitumoral genes in PCa in addition to the 17 already reported for this cancer.
In global terms, PCa has a good prognosis, with 5‐year survival data reaching more than 90%, but some cases have the potential to progress to a lethal outcome. 44 , 45 In fact, among the 495 patients in the TCGA‐PRAD subset with primary PCa, only 2% (10 cases) died: Gleason 6 (1 case) or Gleason 7 (4 cases 3 + 4, 1 case 4 + 3), Gleason ≥8 (2 cases), and unclassified (2 cases). Until now, it remains unclear how to identify between “low” and “high” risk subgroups through traditional clinical and pathologic criteria. 46 , 47 The search for new biomarkers has been suggested as a possible new alternative for classifying PCa risk. 48 , 49 We hypothesize that the antitumoral differentially expressed genes identified in our study could be playing a relevant role in the body defense against the tumoral process. We found an increase of antitumoral gene's expression with higher Gleason scores, mainly related to cancer hallmarks such as proliferation, metastasis, resisting cell death and castration resistance (Figure 4). Thus, these genes may be potential early biomarkers of changes in the clinical course of the disease.
A limitation of the present study is the low number of paired samples we used to identify new genes involved in PCa. However, this approach has been shown to overcome interpersonal PCa heterogeneity, and results could help find other ways of classifying the risk of PCa patients. 50 A major challenge was to find replication datasets containing information about the identified genes; however, the results could be replicated in GEO databases up to 74%, and in the remaining TCGA‐PRAD samples could be achieved for all but one gene (SYT8). Another strength of our study is the use of manually curated functional annotation that allowed the discovery of antitumoral activity of genes differentially expressed in PCa with no annotation in current databases (eg, C16orf74).
In conclusion, taking into account paired differential expression, functional annotation and replication results, we propose the following genes as possible early markers in PCa: CGREF1, UNC5A, C16orf74, LGR6, IGSF1, QPRT and CA14. These genes may prevent the progression of the disease, and their expression should be studied in patients with different outcomes. Moreover, currently Gleason 6 and Gleason 7 treatment consists mainly in active surveillance, but these results already show molecular changes related to cancer proliferation and progression in these stages. Further studies with suitable datasets from large consortia are needed to confirm the role of these genes as early biomarkers.
CONFLICT OF INTEREST
The authors declare no potential conflict of interests.
AUTHOR CONTRIBUTIONS
Elisa Díaz de la Guardia‐Bolívar contributed to the conceptualization, collection of data, programming, preparation of the figures and tables, draft writing and editing of the manuscript. Rocío Barrios‐Rodríguez contributed writing, review and editing of the manuscript. Igor Zwir contributed reviewing and editing the manuscript and resources. José Juan Jiménez‐Moleón contributed drafting and reviewing the manuscript. Coral del Val acted as main supervisor, conceptualization, visualization and preparation of figures, draft, writing, review and edition of the manuscript. All authors read and approved the final manuscript. The work reported in the article has been performed by the authors, unless clearly specified in the text.
Supporting information
Appendix S1. Supporting Information.
ACKNOWLEDGEMENTS
This work was supported by the Spanish Ministry of Science and Technology with projects RTI2018‐098983‐B‐100 and PI15/00914. Elisa Díaz de la Guardia‐Bolívar was funded by a doctoral fellowship, PRE2019‐089807, from the Spanish Ministry of Science and Innovation. Universidad de Granada/CBUA funded open access charges.
Díaz de la Guardia‐Bolívar E, Barrios‐Rodríguez R, Zwir I, Jiménez‐Moleón JJ, del Val C. Identification of novel prostate cancer genes in patients stratified by Gleason classification: Role of antitumoral genes. Int. J. Cancer. 2022;151(2):255‐264. doi: 10.1002/ijc.33988
Elisa Díaz de la Guardia‐Bolívar and Rocío Barrios‐Rodríguez contributed equally to this work.
Funding information Spanish Ministry of Science and Innovation, Grant/Award Number: PRE2019‐089807; Spanish Ministry of Science and Technology, Grant/Award Numbers: PI15/00914, RTI2018‐098983‐B‐100; Universidad de Granada/CBUA
DATA AVAILABILITY STATEMENT
Only publicly available data were used in our study, and data sources and handling of these data are described in the Materials and Methods. Further information is available from the corresponding author upon request.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Colombet M, Soerjomataram I, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries and 25 major cancers in 2018 [Internet]. Eur J Cancer. 2018;103:356‐387. https://pubmed.ncbi.nlm.nih.gov/30100160/. Accessed May 17, 2021 [DOI] [PubMed] [Google Scholar]
- 3. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland [Internet]. N Engl J Med. 2000;343:78‐85. https://pubmed.ncbi.nlm.nih.gov/10891514/. Accessed May 17, 2021 [DOI] [PubMed] [Google Scholar]
- 4. Heidegger I, Tsaur I, Borgmann H, et al. Hereditary prostate cancer—primetime for genetic testing? [Internet]. Cancer Treat Rev. 2019;81:101927 https://pubmed.ncbi.nlm.nih.gov/31783313/. Accessed May 17, 2021 [DOI] [PubMed] [Google Scholar]
- 5. Rebbeck TR. Prostate cancer genetics: variation by race, ethnicity, and geography [Internet]. Semin Radiat Oncol. 2017;27:3‐10. https://pubmed.ncbi.nlm.nih.gov/27986209/. Accessed May 17, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng S, Yang S, Shi Y, Shi R, Yeh Y, Yu X. Neuroendocrine prostate cancer has distinctive, non‐prostatic HOX code that is represented by the loss of HOXB13 expression. Sci. Rep. 2021;11(1):2778. doi: 10.1038/s41598-021-82472-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel VL, Busch EL, Friebel TM, et al. Association of genomic domains in BRCA1 and BRCA2 with prostate cancer risk and aggressiveness. Cancer Res. 2020;80:624‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao D, Cai L, Lu X, et al. Chromatin regulator chd1 remodels the immunosuppressive tumor microenvironment in pten‐deficient prostate cancer. Cancer Discov. 2020;10:1374‐1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z, Gerke T, Bird V, Prosperi M. Trends in gene expression profiling for prostate cancer risk assessment: a systematic review [Internet]. Biomed Hub. 2017;2:1‐15. https://pubmed.ncbi.nlm.nih.gov/31988908/. Accessed May 17, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kato M, Kurozumi A, Goto Y, et al. Regulation of metastasis‐promoting LOXL2 gene expression by antitumor microRNAs in prostate cancer. J Hum Genet. 2017;62:123‐132. [DOI] [PubMed] [Google Scholar]
- 11. Yang YF, Xue SY, Lu ZZ, et al. Antitumor effects of oncolytic adenovirus armed with PSA‐IZ‐CD40L fusion gene against prostate cancer. Gene Ther. 2014;21:723‐731. [DOI] [PubMed] [Google Scholar]
- 12. Shan Z, An N, Qin J, Yang J, Sun H, Yang W. Long non‐coding RNA Linc00675 suppresses cell proliferation and metastasis in colorectal cancer via acting on miR‐942 and Wnt/β‐catenin signaling [Internet]. Biomed Pharmacother. 2018;101:769‐776. https://pubmed.ncbi.nlm.nih.gov/29524886/. Accessed May 3, 2021 [DOI] [PubMed] [Google Scholar]
- 13. Jeong G, Bae H, Jeong D, et al. A Kelch domain‐containing KLHDC7B and a long non‐coding RNA ST8SIA6‐AS1 act oppositely on breast cancer cell proliferation via the interferon signaling pathway. Sci Rep. 2018;8:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fraser M, Berlin A, Bristow RG, Van der Kwast T. Genomic, pathological, and clinical heterogeneity as drivers of personalized medicine in prostate cancer. Urol Oncol Semin Orig Investig. 2015;33:85‐94. [DOI] [PubMed] [Google Scholar]
- 15. Tolkach Y, Kristiansen G. The heterogeneity of prostate cancer: a practical approach. Pathobiology. 2018;85:108‐116. [DOI] [PubMed] [Google Scholar]
- 16. Lysenko I, Mori K, Mostafaei H, et al. Prognostic value of Gleason score at positive surgical margin in prostate cancer: a systematic review and meta‐analysis. Clin Genitourin Cancer. 2020;18:e517‐e522. [DOI] [PubMed] [Google Scholar]
- 17. Van den Broeck T, van den Bergh RCN, Arfi N, et al. Prognostic value of biochemical recurrence following treatment with curative intent for prostate cancer: a systematic review. Eur Urol. 2019;75:967‐987. [DOI] [PubMed] [Google Scholar]
- 18. Ozkan TA, Eruyar AT, Cebeci OO, Memik O, Ozcan L, Kuskonmaz I. Interobserver variability in Gleason histological grading of prostate cancer. Scand J Urol. 2016;50:420‐424. [DOI] [PubMed] [Google Scholar]
- 19. Hadavand MA, Mayer D, Chen W, Wnorowski A, Siddiqui MM. Role of metabolic imaging in diagnosis of primary, metastatic, and recurrent prostate cancer. Curr Opin Oncol. 2020;32:223‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spahn M, Boxler S, Joniau S, Moschini M, Tombal B, Karnes RJ. What is the need for prostatic biomarkers in prostate cancer management? Curr Urol Rep. 2015;16:624‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tang KL, Yao WJ, Li TH, Li YX, Cao ZW. Cancer classification from the gene expression profiles by discriminant kernel‐pls. J Bioinform Comput Biol. 2010;8:147‐160. [DOI] [PubMed] [Google Scholar]
- 22. Thibodeau SN, French AJ, McDonnell SK, et al. Identification of candidate genes for prostate cancer‐risk SNPs utilizing a normal prostate tissue eQTL data set. Nat Commun. 2015;6:8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Wspolczesna Onkol. 2015;1A:A68‐A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data [Internet]. Nucleic Acids Res. 2016;44:e71 https://pubmed.ncbi.nlm.nih.gov/26704973/. Accessed May 14, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tarazona S, Furió‐Tarí P, Turrà D, et al. Data quality aware analysis of differential expression in RNA‐seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43(21):e140. doi: 10.1093/nar/gkv711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wickham H. ggplot2: Elegant Graphics for Data Analysis, New York: Springer‐Verlag; 2016. Available at: https://ggplot2.tidyverse.org. [Google Scholar]
- 28. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data [Internet]. Bioinformatics. 2009;26:139‐140. http://bioconductor.org. Accessed May 17, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies [Internet]. Nucleic Acids Res. 2015;43:e47 https://pubmed.ncbi.nlm.nih.gov/25605792/. Accessed May 17, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB—a database for integrating human functional interaction networks. Nucleic Acids Research. 2009;37(suppl_1):D623‐D628. doi: 10.1093/nar/gkn698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology [Internet]. Nucleic Acids Res. 2011;39:D712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Datta D, Aftabuddin M, Gupta DK, Raha S, Sen P. Human Prostate Cancer Hallmarks Map [Internet]. Sci Rep. 2016;6:1‐14. http://www.nature.com/scientificreports/. Accessed May 17, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kassambara A. rstatix: Pipe‐Friendly Framework for Basic Statistical Tests [Internet]; 2020. https://cran.r-project.org/package=rstatix. Accessed May 14, 2021
- 34. Chandran UR, Dhir R, Ma C, Michalopoulos G, Becich M, Gilbertson J. Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer. 2005;5(1):45. doi: 10.1186/1471-2407-5-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Magi‐Galluzzi C, Maddala T, Falzarano SM, et al. Gene expression in normal‐appearing tissue adjacent to prostate cancers are predictive of clinical outcome: evidence for a biologically meaningful field effect. Oncotarget. 2016;7:33855‐33865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nonn L, Ananthanarayanan V, Gann PH. Evidence for field cancerization of the prostate. Prostate. 2009;69:1470‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yeong J, Thike AA, Tan PH, Iqbal J. Identifying progression predictors of breast ductal carcinoma in situ [Internet]. J Clin Pathol. 2017;70:102‐108. https://pubmed.ncbi.nlm.nih.gov/27864452/. Accessed April 27, 2021 [DOI] [PubMed] [Google Scholar]
- 38. Alberti C. Prostate cancer progression and surrounding microenvironment [Internet]. Int J Biol Markers. 2006;21:88‐95. https://pubmed.ncbi.nlm.nih.gov/16847811/. Accessed April 27, 2021 [DOI] [PubMed] [Google Scholar]
- 39. Vazquez JP, Pulido EG, Aparicio LMA. Cytokine and endocrine signaling in prostate cancer [Internet]. Med Oncol. 2012;29:1956‐1963. https://pubmed.ncbi.nlm.nih.gov/21858553/. Accessed April 27, 2021 [DOI] [PubMed] [Google Scholar]
- 40. Sato S, Kimura T, Onuma H, Fukuda Y, Egawa S, Takahashi H. Combination of total length of Gleason pattern 4 and number of Gleason score 3 + 4 = 7 cores detects similar outcome group to Gleason score 6 cancers among cases with ≥5% of Gleason pattern 4 [Internet]. Pathol Int. 2020;70:992‐998. https://pubmed.ncbi.nlm.nih.gov/32997878/. Accessed April 30, 2021 [DOI] [PubMed] [Google Scholar]
- 41. Griesemer M, Kimbrel JA, Zhou CE, Navid A, D'haeseleer P. Combining multiple functional annotation tools increases coverage of metabolic annotation. BMC Genomics. 2018;19(1). doi: 10.1186/s12864-018-5221-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu Y‐H, Deng J‐L, Wang G, Zhu Y‐S. Long non‐coding RNAs in prostate cancer: Functional roles and clinical implications. Cancer Lett. 2019;464:37‐55. doi: 10.1016/j.canlet.2019.08.010 [DOI] [PubMed] [Google Scholar]
- 43. Khurana E, Fu Y, Chakravarty D, Demichelis F, Rubin MA, Gerstein M. Role of non‐coding sequence variants in cancer [Internet]. Nat Rev Genet. 2016;17:93‐108. https://pubmed.ncbi.nlm.nih.gov/26781813/. Accessed April 30, 2021 [DOI] [PubMed] [Google Scholar]
- 44. Alonso‐Molero J, Molina AJ, Jiménez‐Moleón JJ, et al. Cohort profile: the MCC‐Spain follow‐up on colorectal, breast and prostate cancers: study design and initial results. BMJ Open. 2019;9(11):e031904. doi: 10.1136/bmjopen-2019-031904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang AJ, Autio KA, Roach M, Scher HI. High‐risk prostate cancer‐classification and therapy [Internet]. Nat Rev Clin Oncol. 2014;11:308‐323. https://pubmed.ncbi.nlm.nih.gov/24840073/. Accessed May 7, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patel HD, Tosoian JJ, Carter HB, Epstein JI. Adverse pathologic findings for men electing immediate radical prostatectomy. JAMA Oncol. 2018;4(1):89. doi: 10.1001/jamaoncol.2017.1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koo KM, Mainwaring PN, Tomlins SA, Trau M. Merging new‐age biomarkers and nanodiagnostics for precision prostate cancer management. Nat Rev Urol. 2019;16:302‐317. [DOI] [PubMed] [Google Scholar]
- 48. Johnston WL, Catton CN, Swallow CJ. Unbiased data mining identifies cell cycle transcripts that predict non‐indolent Gleason score 7 prostate cancer. BMC Urol. 2019;19(1). doi: 10.1186/s12894-018-0433-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klotz L. Active surveillance in intermediate‐risk prostate cancer. BJU Int. 2020;125(3):346‐354. doi: 10.1111/bju.14935 [DOI] [PubMed] [Google Scholar]
- 50. Platz EA, De Marzo AM, Giovannucci E. Prostate cancer association studies: pitfalls and solutions to cancer misclassification in the PSA era. J Cell Biochem. 2004;91:553‐571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Data Availability Statement
Only publicly available data were used in our study, and data sources and handling of these data are described in the Materials and Methods. Further information is available from the corresponding author upon request.