

Abstract
R2‐like ligand‐binding oxidase (R2lox) is a ferritin‐like protein that harbours a heterodinuclear manganese–iron active site. Although R2lox function is yet to be established, the enzyme binds a fatty acid ligand coordinating the metal centre and catalyses the formation of a tyrosine–valine ether cross‐link in the protein scaffold upon O2 activation. Here, we characterized the ligands copurified with R2lox by mass spectrometry‐based metabolomics. Moreover, we present the crystal structures of two new homologs of R2lox, from Saccharopolyspora erythraea and Sulfolobus acidocaldarius, at 1.38 Å and 2.26 Å resolution, respectively, providing the highest resolution structure for R2lox, as well as new insights into putative mechanisms regulating the function of the enzyme.
Keywords: aldehyde deformylating oxygenase, ferritin‐like protein, hydroxy fatty acids, long‐chain fatty acids, R2‐like ligand‐binding oxidase, R2lox
Two new structures of R2‐like ligand‐binding oxidase (R2lox), together with the characterization of the ligands copurified with R2lox, give new insights into the substrate and the regulating mechanisms of the enzyme.
Abbreviations
ADO, aldehyde deformylating oxygenase
APCI‐LCMS, atmospheric pressure chemical ionization liquid chromatography‐mass spectrometry
ESI‐LCMS, electrospray ionization liquid chromatography‐mass spectrometry
GkR2loxI, Geobacillus kaustophilus R2lox first homolog
HFA, hydroxy fatty acid
MtR2lox, Mycobacterium tuberculosis R2lox
PEG, polyethylene glycol
R2, ribonucleotide reductase R2 subunit
R2lox, R2‐like ligand‐binding oxidase
RNR, ribonucleotide reductase
SaR2loxI, Sulfolobus acidocaldarius R2lox first homolog
SaR2loxII, Sulfolobus acidocaldarius R2lox second homolog
SeR2lox, Saccharopolyspora erythraea R2lox
The ferritin‐like superfamily is composed by a large diversity of proteins sharing a common fold, constituted of an α‐helix bundle core housing a dinuclear metal centre [1, 2]. Despite their similar architecture, members of this superfamily differ largely in sequence and perform a variety of functions. Well‐studied members of this family include the soluble methane monooxygenase which catalyses the conversion of methane to methanol [3], the ribonucleotide reductase (RNR) that reduces ribonucleotides to deoxyribonucleotides [4], the aldehyde deformylating oxygenase (ADO) which produces alkanes from fatty aldehydes [5], and fatty acid desaturases involved in the synthesis of many unsaturated lipids essential to cells [6].
The metalloenzyme R2‐like ligand‐binding oxidase (R2lox) was discovered due to its sequence resemblance with the R2 subunit of the class Ic RNR (R2c) [7, 8]. Both R2lox and R2c belong to the ferritin‐like superfamily and harbour a heterodinuclear manganese–iron centre [9, 10, 11]. R2lox has been identified in various organisms, and a number of organisms encode several isoforms, for example, Sulfolobus acidocaldarius encodes four R2lox homologues [9]. In R2c, the high‐valent Mn(IV)/Fe(III) centre catalyses a single‐electron transfer to produce a free radical essential to the synthesis of deoxyribonucleotides [12]. In R2lox, the mature cofactor was characterized by spectroscopy as a Mn(III)/Fe(III) centre bridged by a μ‐hydroxo/bis‐μ‐carboxylato network [13]. The assembly of the R2lox active site takes place upon dioxygen (O2) activation and involves the formation of high‐valent intermediates which, unlike R2c, catalyse a two‐electron reaction to generate a tyrosine–valine ether cross‐link in the proximity of the metal centre [14, 15]. Different types of covalent cross‐links have been observed in proteins to serve different purposes [16, 17, 18], including in another di‐metal ferritin‐like protein where a valine–phenylalanine cross‐link has been proposed to stabilize the metal centre [19]. In addition, it was shown that the redox state of the cofactor impacts R2lox structure, leading to the formation of likely routes for O2 and substrate access to the active site [20]. Despite these findings, the physiological substrate and function of R2lox remain unknown.
To date, crystal structures of R2lox proteins from three organisms have been solved, that is, from Mycobacterium tuberculosis (MtR2lox), Geobacillus kaustophilus (GkR2loxI) and Sulfolobus acidocaldarius (SaR2loxI) (PDB ID: 3EE4, 4HR0 and 6QRZ, respectively) [8, 14, 21]. The structures show that, although the R2‐protein fold is conserved, R2loxes are extensively remodelled to accommodate an unexpected ligand in interaction with the dinuclear metal centre. An additional study showed by mass spectrometry that GkR2loxI copurifies with a mixture of mainly C16 and C18 hydroxy fatty acids (HFAs) when the recombinant protein is produced in Escherichia coli [20].
Here, using mass spectrometry‐based metabolomics, we further characterized some of the ligands that copurify with GkR2loxI and SaR2loxI. In addition, we solved two novel R2lox structures by X‐ray crystallography, the highest resolution structure of an R2lox to date from Saccharopolyspora erythraea (SeR2lox) at 1.38 Å resolution and the structure of a second R2lox homolog from Sulfolobus acidocaldarius (SaR2loxII, 54% sequence identity with SaR2loxI) at 2.26 Å resolution, which exhibits unusual features. Using a comparative structural approach combined with bioinformatics, we obtained new insights into the potential regulatory mechanisms of R2lox and discuss the function of this enigmatic enzyme.
Materials and methods
Cloning
Constructs encoding the full‐length Sulfolobus acidocaldarius R2lox homolog II (accession number WP_011277966) and full‐length Saccharopolyspora erythraea R2lox (accession number WP_009945174) were amplified using polymerase chain reaction from genomic DNA obtained from DSMZ (DSM number 639 and 40517, respectively). Then, constructs were inserted into pET‐46 Ek/LIC (Novagen, Merck KGaA, Darmstadt, Germany) in frame with the N‐terminal 6xHis tag using the following primers: 5’‐GACGACGACAAGATGGTATTAAATTTCGAAGAATACAAACATACG‐3’ (forward) and 5’‐GAGGAGAAGCCCGGTTATAAACTCTCTATCACATCCAAGTCCTTTG‐3’ (reverse) for SaR2loxII, and 5’ GACGACGACAAGATGACGAGCACCGCGACC‐3’ and 5’‐GAGGAGAAGCCCGGTTAGGCCTTTTCCAGCGCG‐3’ for SeR2lox. The constructs encoding the N‐terminally His‐tagged full‐length R2loxI from Geobacillus kaustophilus (accession number yp_148624) and R2loxI from S. acidocaldarius (accession number WP_011278976) were cloned as described in [14] and [21], respectively.
Protein production and purification
Expression of SaR2loxII and SeR2lox was carried out in Escherichia coli BL21(DE3) (Novagen) in a benchtop bioreactor system (Harbinger). Cells expressing SaR2loxII were cultured in terrific broth medium (Formedium) supplemented with ampicillin (50 μg·mL−1). Cells expressing SeR2lox were cultured in minimal media (Formedium) supplemented with ampicillin (50 μg·mL−1), 0.25 mm MnCl2 and 0.25 mm (NH4)2Fe(SO4)2, initially at 37 °C. Expression was induced with 0.5 mm isopropyl β‐D‐1‐thiogalactopyranoside at an OD600 of 0.8 for 15 h at ambient temperature. Cells were then harvested by centrifugation and stored at −80 °C. Cells expressing SaR2loxII and SeR2lox were disrupted by high‐pressure homogenization (Avestin Emulsiflex C3), after resuspension in lysis buffer (25 mm Hepes‐Na pH 7.0, 300 mm NaCl and 10 mm imidazole). The lysate was cleared by centrifugation and the supernatant applied to a nickel–nitrilotriacetic acid agarose (Protino) gravity flow column. The sample was washed and eluted using washing buffer (lysis buffer containing 20 mm imidazole) and elution buffer (lysis buffer containing 250 mm imidazole). The protein was concentrated using Vivaspin 20 centrifugal concentrators with a 30,000 molecular weight cut‐off polyethersulfone membrane (Sartorius) and applied to a HiLoad 16/60 Superdex 200 prep grade size exclusion column (GE Healthcare Sverige AB, Danderyd, Stockholm, Sweden) equilibrated in a final buffer of 25 mm Hepes‐Na pH 7.0 and 150 mm NaCl. Fractions corresponding to the pure protein were pooled, concentrated (12.5 mg·mL−1 and 25.6 mg·mL−1 for SaR2loxII and SeR2lox, respectively), aliquoted, flash‐frozen in liquid nitrogen and stored at −80 °C. The protein concentrations were calculated using theoretical molecular weight for SaR2loxII (37010.62) and extinction coefficient at 280 nm of 44350 m −1 cm−1, and SeR2lox (37249.35) and extinction coefficient at 280 nm of 53190 m −1 cm−1, as calculated by ProtParam [22]. SaR2loxI and the metal‐free GkR2loxI were produced and purified as described in [21] and [14], respectively.
Crystallization, X‐ray data collection and structure refinement
SeR2lox and SaR2loxII were crystallized by sitting‐drop vapour diffusion method using a Mosquito nanolitre pipetting robot (SPT Labtech, Melbourn, Hertfordshire, UK). For SeR2lox, a volume of 200 nL protein solution at 25.6 mg·mL−1 in 25 mm Hepes‐Na pH 7.0 and 150 mm NaCl was mixed with 200 nL of reservoir condition composed of 200 mm lithium sulfate, 100 mm sodium acetate pH 4.6 and 50% (v/v) polyethylene glycol 400, resulting in crystals appearing overnight at 21 °C. For SaR2loxII, a volume of 75 nL of a protein solution at 12.5 mg·mL−1 in 25 mm Hepes‐Na pH 7.0 and 150 mm NaCl was mixed with 150 nL of reservoir solution consisting of 24% (w/v) polyethylene glycol 1500, 5% (v/v) formamide, 40 mm sodium propionate, 20 mm sodium cacodylate trihydrate and 40 mm bis‐tris propane (pH 9.0). A single crystal grew in a week at 21 °C. Of note, crystallization conditions were optimized from initial hits found using commercial screens, that is, from condition A1 in JCSG plus (Molecular Dimensions) for SeR2lox and from condition C3 from PACT premier (Molecular Dimensions) supplemented by condition H5 from Additive Screen (Hampton Research) for SaR2loxII. SeR2lox and SaR2loxII crystals were flash‐cooled in liquid nitrogen without additional cryoprotecting agent, and X‐ray diffraction data were collected at cryogenic temperature on the beamlines PX14.1 at BESSY (Berlin, Germany) and X06SA at the Swiss Light Source (Villigen, Switzerland), respectively. Data reduction and scaling were performed using XDS [23]. The high‐resolution cut‐off was determined based on a combination of I/σ(I), R meas and CC1/2.
Phasing was performed by molecular replacement using Phaser [24], using the atomic coordinates of GkR2loxI (PDB ID: 4HR0 [14]) and MtR2lox (PDB ID: 3EE4 [8]) edited with Sculptor [25], as a starting search model for SaR2loxII and SeR2lox, respectively. A well‐contrasted solution was obtained with one molecule per asymmetric unit in space group P64 for SaR2loxII, and P43212 for SeR2lox. Refinement was performed with phenix.refine [26]. Model examination and manual modifications were iteratively conducted with Coot [27]. When all residues visible in the electron density map were built, metal ions and waters were added, and the model was further refined. Restraints for metal coordination were generated using elbow [28] and manually modified based on previous studies on R2lox structures [8, 14]. Metal ions were modelled as Mn(III) and Fe(III) based on previous studies on R2lox [13]. For SaR2loxII, the metal ions were refined at an occupancy of 0.35; no electron density could be observed for a putative tyrosine–valine ether cross‐link between Tyr170 and Val78. Similarly, no clear electron density could support the presence of a ligand in the protein binding pocket. The protein was modelled from residue 20 to the last protein residue, that is, 308. Residues 126 to 130 could not be modelled in the electron density. The SeR2lox structure was modelled from residue Ala6 to Asp296, the metal ions were refined to an occupancy of 1, and electron density connecting the phenolic oxygen of Tyr167 and the Cβ of Val76 was observed. This ether cross‐link was restrained to an ideal distance of 1.5 ± 0.05 Å. Quality of models was evaluated using MolProbity [29]. Data reduction and model refinement statistics were compiled using phenix.table_one. Anomalous difference maps were computed with phenix.maps using Friedel pair reflections unmerged to preserve the contribution of anomalous scattering atoms. All phenix programmes belong to the PHENIX suite version 1.19_4092 [26]. Figures were prepared using Inkscape 1.0 and the pymol Molecular Graphics System, version 2.4, Schrödinger, LLC. Electrostatic protein contact potentials plotted on molecular surfaces shown in Fig. 3 were generated with the vacuum electrostatics tool in pymol.
Fig. 3.
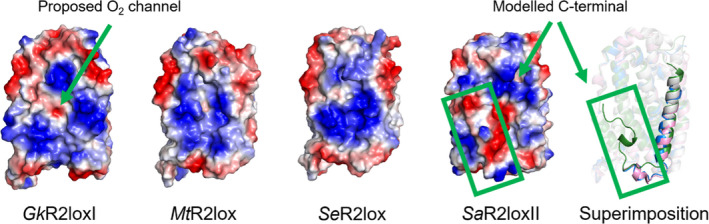
The positive patch of SaR2loxII is disrupted by the ordered C‐terminus. A comparison of the electrostatic surface (positive in blue and negative in red) of GkR2loxI, MtR2lox, SeR2lox and SaR2loxII and a superimposition of the four structures highlighting their modelled C‐terminus (PDB ID: 4HR0, 3EE4, 7QBP and 7QBK, respectively). GkR2loxI is coloured in white, MtR2lox in pink, SeR2lox in blue and SaR2loxII in green. The C‐terminus in the SaR2loxII structure shields the positive electrostatic patch hypothesized to be an interaction surface with protein partners or the cell membrane.
Ligand identification by ESI‐MS and APCI‐MS
GkR2loxI samples were mixed with 60 μL GC‐grade chloroform (Sigma‐Aldrich, St. Louis, MO, USA) and vortexed for 30 s. A volume of 30 μL of LC‐grade methanol (Sigma‐Aldrich, St. Louis, MO, USA) was subsequently added, and the samples were vortexed for other 30 s and centrifugated at 1000× g for 10 min. The chloroform phase was finally extracted and dried under N2 gas in an HPLC vial, before being stored at −80 °C. The frozen samples were re‐suspended before analysis in 20 μL methanol, HPLC‐grade from Fischer Scientific (Waltham, MA, USA), and Milli‐Q water before LCMS analysis. Each batch of samples was first analysed in positive mode and finally in negative.
The chromatographic separation was performed on an Agilent 1290 Infinity UHPLC‐system (Agilent Technologies, Waldbronn, Germany). A total of 2 μL of each sample were injected onto an Acquity UPLC HSS T3, 2.1 × 50 mm, 1.8 μm C18 column in combination with a 2.1 mm × 5 mm, 1.8 μm VanGuard precolumn (Waters Corporation, Milford, MA, USA) held at 40 °C. The gradient elution buffers were A (H2O, 0.1% formic acid) and B (75/25 MeOH:2‐propanol, 0.1% formic acid), and the flow rate was 0.5 mL min‐1. The compounds were eluted with a linear gradient consisting of 0.1–10% B over 2 min; B was increased to 99% over 5 min and held at 99% for 2 min; B was decreased to 0.1% for 0.3 min, and the flow rate was increased to 0.8 mL min‐1 for 0.5 min; these conditions were held for 0.9 min, after which the flow rate was reduced to 0.5 mL min‐1 for 0.1 min before the next injection.
The compounds were detected with an Agilent 6546 Q‐TOF mass spectrometer equipped with a jet stream electrospray ion source operating in positive or negative ion mode. The settings were kept identical between the modes, with exception of the capillary voltage. A reference interface was connected for accurate mass measurements; the reference ions purine (4 μm) and HP‐0921 (Hexakis(1H, 1H, 3H‐tetrafluoropropoxy)phosphazine) (1 μm) were infused directly into the MS at a flow rate of 0.05 mL min‐1 for internal calibration, and the monitored ions were purine m/z 121.05 and m/z 119.03632; HP‐0921 m/z 922.0098 and m/z 966.000725 for positive and negative modes, respectively. The gas temperature was set to 150 °C, the drying gas flow to 8 L min‐1 and the nebulizer pressure 35 psig. The sheath gas temp was set to 350 °C, and the sheath gas flow 11 L min‐1. The capillary voltage was set to 4000 V in both positive and negative ion modes. The nozzle voltage was 300 V. The fragmentor voltage was 120 V, the skimmer 65 V and the OCT 1 RF Vpp 750 V. The collision energy was set to 0 V. The m/z range was 70–1700, and data were collected in centroid mode with an acquisition rate of 4 scans s‐1 (1977 transients/spectrum).
For APCI ionization, all chromatographic settings were kept the same. The APCI source settings were as follows: the gas temperature was set to 170 °C, the vaporizer to 250 °C, the gas flow 6 L min‐1, and the nebulizer pressure 40 psig. The capillary voltage was set to 3000 V in both positive and negative ion modes. The Corona current was 4 μA in both modes. The fragmentor voltage was 120 V, the skimmer 65 V and the OCT 1 RF Vpp 750 V. The collision energy was set to 0 V. The m/z range was 70–1700, and data were collected in centroid mode with an acquisition rate of 4 scans s‐1 (1977 transients/spectrum).
All data processing was performed using the Agilent Masshunter Qual version B.07.00 (Agilent Technologies Inc., Santa Clara, CA, USA). Reference and tuning standards: purine, 4 μm, Agilent Technologies (Santa Clara, CA, USA) HP‐0921 (Hexakis(1H, 1H, 3H‐tetrafluoropropoxy)phosphazine), 1 μm, Agilent Technologies (Santa Clara, CA, USA) Calibrant, ESI‐TOF, ESI‐L Low Concentration Tuning Mix, Agilent Technologies (Santa Clara, CA, USA) HP‐0321 (Hexamethoxyphosphazine) and 0.1 mm, Agilent Technologies (Santa Clara, CA, USA).
Ligand pocket comparison
The PDB entry 4HR0 of GkR2loxI and the refined structures of SaR2loxII and SeR2lox published in this work were run on the ConSurf server (https://consurf.tau.ac.il) [30, 31] using the homolog search algorithm HMMER, 1 iteration, UniRef90, number of putative homologous sequences to 150 and PSI‐BLAST E‐value cut‐off to 0.0001. The maximal %ID between sequences was set to 95% and the minimal %ID for homologs at 35%. The alignment method was set to MAFFT‐L‐INS‐i.
Results
GkR2loxI and SaR2loxI copurify with a mixture of long‐chain HFAs
The ligand copurifying with GkR2loxI was previously identified by mass spectrometry as a mixture of long‐chain HFAs. However, the position of the hydroxy group could not be determined [20]. In this study, we analysed the pool of ligands in GkR2loxI and SaR2loxI with electrospray ionization liquid chromatography‐mass spectrometry (ESI‐LCMS) and compared them to a pool of HFA standards to identify the position of the hydroxy group. Our first observation was that both proteins copurify with a similar mixture of ligands (Fig. S1). We performed ESI‐LCMS analysis on the copurified ligands of GkR2loxI and can confirm that they mainly are HFAs with a hydroxy group further away from the carboxyl group and in some cases includes a double bond. Chen et al. (2016) and Yang et al. (2013) show that the position of the hydroxy group and any double bond have a large impact on the fragmentation patterns of HFAs [32, 33]. We identified two major liquid chromatography peaks at 271.23 m·z−1 and 297.24 m·z−1. The peak at 271.23 m·z−1 corresponds to a C16 HFA, but the MSMS spectra acquired give poor structural information. The fragmentation spectra only confirm the presence of a signal at 225.22 m·z−1 corresponding to the loss of a carboxylic group (Fig. 1A). The peak at 297.24 m·z−1 corresponds to a C18 HFA with one double bond. The fragment 251.24 m·z−1 corresponds to the loss of the carboxylic acid (Fig. 1B). The presence of a peak at 183.01 m·z−1 indicates that the location of the hydroxy group is on position C12 by comparison with the fragmentation of the standard 12‐hydroxy‐9(E)‐octadecenoic acid at 20 eV (Fig. 1C). However, the ligand shows a slightly different fragmentation pattern than 12‐hydroxy‐9(E)‐octadecenoic acid at 40 eV (Fig. 1D). This indicates that the double bond is in a different position between the hydroxy and the carboxyl groups. In addition, we performed atmospheric pressure chemical ionization liquid chromatography‐mass spectrometry (APCI‐LCMS) to identify possible aldehydes, but were unable to find other compounds than the ones already visible in the ESI‐LC chromatogram. Finally, in order to search for eventual enzyme products formed upon redox chemistry, we chemically reduced and oxidized GkR2loxI and analysed the ligands with ESI‐LCMS and APCI‐LCMS. Regrettably, no new compounds were identified following this procedure (Fig. S2).
Fig. 1.
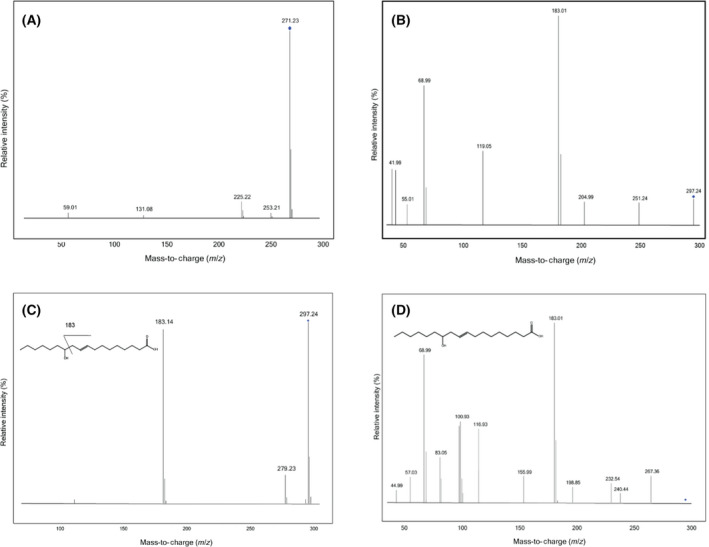
ESI‐LCMS fragmentation of copurified ligands from GkR2loxI and the standard 12‐hydroxy‐9(E)‐octadecenoic acid. (A) Fragmentation of ion 271 m·z−1 at 20 eV. The signal is weak and hard to compare to any standard. The only identifiable ion at 225 m·z−1 indicates the loss of the carboxyl group. (B) Fragmentation of ion 297 m·z−1 at 40 eV. The fragment 183.01 m·z−1 indicates the fragmentation around the hydroxy group, which was identified by comparing with the (C) fragmentation spectra of the standard 12‐hydroxy‐9(E)‐octadecenoic acid at 20 eV, which shows that fragmentation around the hydroxy group in position C12 gives a peak at 183.14 m·z−1. (D) Fragmentation spectra of the standard 12‐hydroxy‐9(E)‐octadecenoic acid at 40 eV which, however, shows a different fragmentation to the R2lox ligand in Fig. 1B. The ligand is, therefore, most likely a 12‐hydroxy‐octadecenoic acid, but the position of the double bond was not identifiable.
SeR2lox high‐resolution structure
The gene coding for SeR2lox was recombinantly expressed in E. coli using minimal medium supplemented with equimolar amounts of Mn(II) and Fe(II). The crystal structure of the protein was solved to 1.38 Å resolution (Table S1) and currently represents the highest resolution for an R2lox structure. SeR2lox exhibits a canonical ferritin‐like fold, along with a ligand‐binding pocket accommodating a putative fatty acid molecule (Fig. 2A). The metal site is identical to previously solved GkR2loxI and MtR2lox structures. Mn(III) and Fe(III) ions were modelled at full occupancy in sites 1 and 2, respectively, based on X‐ray anomalous diffraction data (Fig. S3A) consistently with previous structural studies [8, 14]. The metal ions are bridged by a μ‐hydroxo ligand and coordinated by the carboxyl group of the fatty acid ligand. Electron density corresponding to the ether cross‐link between the phenolic oxygen of Tyr167 and the Cβ of Val76 is also clearly observed (Fig. 2B). Palmitic acid (C16) was modelled into the binding pocket, although weak electron density extending further away from the pocket possibly suggests the presence of a fatty acid with a longer chain, such as C18, at low occupancy (Fig 2C and S3B). However, similar to other R2lox structures, no clear electron density allowed us to model a hydroxy group in position C12.
Fig. 2.
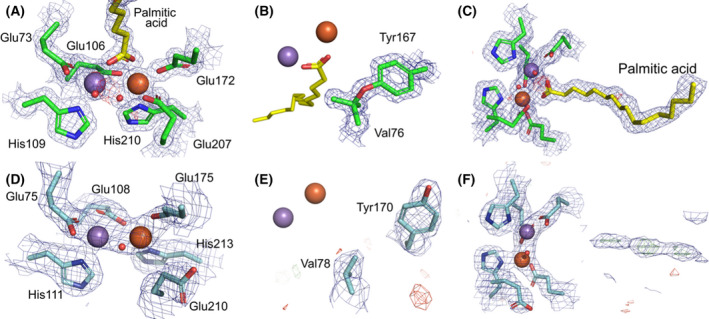
SeR2lox and SaR2loxII crystal structures. SeR2lox displays a well‐defined metal centre (A), tyrosine–valine ether cross‐link (B), and ligand modelled as palmitic acid in the binding pocket (C). SaR2loxII exhibits a metal centre at low occupancy (D), no cross‐link between Tyr170 and Val78 (E), and its binding pocket is mostly ligand‐free (F). Refined electron density maps are shown in mesh: 2Fo–Fc maps are coloured in blue and contoured at 2 σ in (A) and (B) and at 1 σ in (C) to (F); Fo–Fc maps are in green (positive) and red (negative) and contoured at 4 σ. Manganese and iron ions are depicted as purple and orange spheres, respectively.
SaR2loxII is mainly ligand‐free and lacks the tyrosine–valine ether cross‐link
Recombinant SaR2loxII was produced and purified from E. coli cultured in rich medium. The crystal structure of SaR2loxII was solved to 2.26 Å resolution (Table S1). The protein displays the ferritin‐like fold and ligand‐binding pocket conserved among R2lox. Neither the first 19 residues of the protein nor residues 126 to 130 were visible in the electron density. In contrast, the C‐terminus was modelled until the last residue. Besides, although the first coordination sphere residues of the metal centre are conserved, Glu210 exhibits an unusual outward conformation and the protein appears to be mainly metal‐free, as the Mn(III) and Fe(III) ions were both refined to an occupancy of 0.35 (Fig. 2D). Furthermore, the protein does not show a tyrosine–valine ether cross‐link, as Tyr170 displays an uncommon conformation, facing away from Val78 (Fig. 2E). Another atypical feature of SaR2loxII is that no clear electron density could support the presence of a fatty acid ligand at high occupancy in the protein binding pocket (Fig. 2F). In similar expression conditions in E. coli cultured in rich media, MtR2lox, GkR2loxI and SaR2loxI are copurified with putative fatty acid ligands [8, 14, 34].
Ordered C‐terminus in SaR2loxII modifies the surface electrostatics of the protein
Two apparently recurrent features of R2lox crystal structures are the disordered C‐terminal tail and a positively charged patch on the enzyme’s surface proposed to be an interface for interaction with either a protein partner or with the cell membrane [20, 35]. In the structure of SaR2loxII, the C‐terminus forms a well‐defined coil which covers the positive patch (Fig. 3). In addition, the outward conformation of the conserved metal‐binding residue Glu210 (Fig. 2D) extends towards the surface of the protein and contributes to further disrupting this positive patch.
The closing loop of the ligand‐binding tunnel conserves its flexibility
Griese et al. (2015) proposed that a flexible loop gates the entrance of the ligand pocket in GkR2loxI [20]. By superimposing the structures, we can see that the configuration of this loop, located between helices G and H, varies between the different R2lox proteins, compared with the rest of the scaffold (Fig. 4A). However, a common aspect is that this loop seems to display a high flexibility in the previously solved structures of GkR2loxI, MtR2lox and SaR2loxI as well as in our new structures of SeR2lox and SaR2loxII. Indeed, this loop always appears among the most flexible segments of the protein, as is evident when representing the atomic B factors of the structures (Fig. 4B). Interestingly, a motif of three sequential proline residues is found on this gating loop in the structures of SaR2loxI (Pro257, Pro262 and Pro267), SaR2loxII (Pro257, Pro263 and Pro268) and SeR2lox (Pro251, Pro255 and Pro259). Similarly, MtR2lox also exhibits a pair of proline residues (Pro253 and Pro254). However, no proline residues are observed on this loop in GkR2loxI, probably leading to a difference in flexibility as prolines are known to introduce rigidity in flexible regions [36].
Fig. 4.
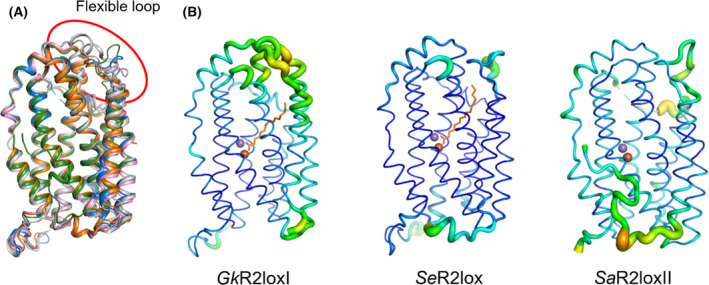
The closing loop of the ligand‐binding tunnel is highly flexible. (A) Superimposition of GkR2loxI in white, MtR2lox in pink, SaR2loxI in orange, SeR2lox in blue and SaR2loxII in green (PDB ID: 4HR0, 3EE4, 6QRZ, 7QBP and 7QBK, respectively). The highlighted portion of the structure shows the highly flexible loop covering the entrance to the fatty acid ligand channel. (B) B factor representation of GkR2loxI, SeR2lox and SaR2loxII. B factor values are depicted by the thickness of the ribbon and by the colour range from blue to orange. Structures in (A) and (B) are shown with identical orientation.
R2lox ligand‐binding pockets are highly diverse within the orthologues
A comparison of the R2lox ligand‐binding pockets reveals a large diversity in length and shape across the orthologues (Fig. 5). Furthermore, we compared the evolutionary conservation of each residue of the ligand‐binding pocket with the ConSurf server (https://consurf.tau.ac.il) [30, 31]. Our results show that residues are highly conserved in proximity to the metal cofactor (dark purple in Fig. 5), but the remaining length of the pocket, especially in the vicinity of the entrance, is highly variable (dark green in Fig. 5).
Fig. 5.
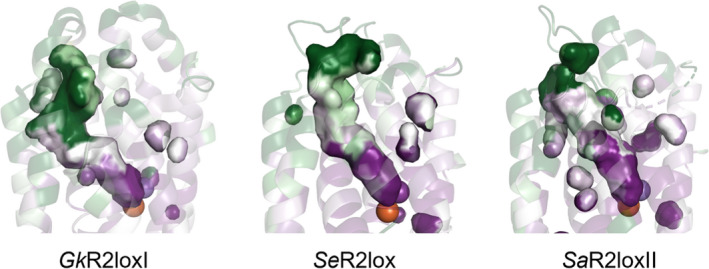
The ligand pocket of R2lox with conservation scores for each residue. The highly conserved residues of the binding pocket of GkR2loxI, SeR2lox and SaR2loxII are shown in dark purple, and the not conserved residues are shown in dark green (PDB ID: 4HR0, 7QBP and 7QBK, respectively).
Discussion
In this study, we have shown by mass spectrometry that GkR2loxI and SaR2loxI copurify with a similar mixture of HFAs, mainly C16 and C18. The fact that we did not observe the formation of a product after subjecting GkR2loxI to redox cycles might indicate that the fatty acids bound are not substrates of the enzyme. However, it seems that these ligands mimic the binding of R2lox substrates and thus are trapped in the binding pocket of recombinant proteins from the heterologous expression host E. coli where C16 and C18 fatty acids are abundant [37, 38]. A similar feature can be observed in the ferritin‐like enzyme ADO which also copurifies with a mixture of C16 and C18 fatty acids that are not substrates of the enzyme [39]. Instead, ADO catalyses the conversion of Cn fatty aldehydes into the corresponding Cn–1 alkanes or alkenes [5] for a large range of chain length, including C4 to C18 [39]. Unlike ADO, fatty aldehydes do not seem to be the substrates of R2lox because MtR2lox and SeR2lox do not show ADO activity as reported by a recent study (proteins #65 and #75 in this study, respectively) [40]. In addition, fatty acids copurifying with ADO are not hydroxylated, and thereby the hydroxy group found in R2lox ligands might be physiologically relevant. Nonetheless, the variety of copurified HFAs evidenced by mass spectrometry in our study suggests a putative promiscuous substrate specificity of R2lox, possibly comparable to ADO. Besides, the diversity of binding pocket shapes, previously discussed by Xu et al. for a smaller subset of structures [21], together with the low residue conservation in its distal part might indicate distinct substrate specificity between R2lox homologs.
The structure of SaR2loxII solved in this study exhibits several unusual features for R2lox that could shed some light on the enzymatic mechanisms of R2lox. First, we observe outward conformations for Tyr170, canonically forming an ether cross‐link with Val78 in R2loxes, as well as for Glu210 which usually coordinates the metal centre. These characteristics cannot be fully explained by the low occupancy of the metal ions, as metal‐free GkR2loxI does not display such outward conformations [14]. Instead, it could represent a mechanism regulating the access of O2 and/or metal ions allowing the correct maturation of the cofactor under specific conditions. Moreover, the fact that SaR2loxII did not trap a fatty acid ligand unlike SaR2loxI although both recombinant proteins were produced under similar conditions, that is, in E. coli cultured in terrific broth media [34], suggests that paralogous R2loxes might have different substrate specificity and thus could be active under different environmental conditions.
SaR2loxII displays an ordered C‐terminus shielding a conserved positively charged patch on the protein surface that could potentially be involved in protein–protein or protein–membrane interactions. A comparable feature was previously observed in only one particular structure of MtR2lox (PDB ID: 4AC8), although here the C‐terminal segment forms a long α helix [35]. Even if we cannot exclude a crystallographic artefact, the apparent dynamic behaviour between an ordered and disordered state of the R2lox C‐terminal tail likely plays a role in the physiological function of the enzyme, as previously suggested [35]. A parallel can be drawn with the phylogenetically related RNR R2 subunit, in which the C‐terminus is disordered in crystal structures of R2 alone, but it is crucial for interaction with its partner R1 [41]. Similarly, we can hypothesize that the C‐terminal tail of R2lox might modulate the interaction with a putative partner or the cell membrane, for example, for the delivery of long‐chain aliphatic compounds that are poorly water‐soluble. For comparison, ADO interacts with an acyl–acyl carrier protein reductase producing fatty aldehydes which are transferred to ADO via a putative channel between the two proteins in order to efficiently synthesize alkanes [42]. In R2lox, such hypothetical transfer could occur through reorganization of the conserved flexible loop closing the substrate‐binding tunnel. Although a pH contribution on the flexibility and conformation of the loop from the different crystallization conditions cannot be ruled out, a comparison of the R2lox structures crystallized at similar pH suggests that pH is not the determining factor for the differences in loop conformation. The fact that this loop structure shows the largest variability between structures hints that it is involved in substrate recognition and/or gating.
Interestingly, genomic studies of haloarchaea revealed that R2lox is often coded in a cluster of genes putatively encoding ferredoxin, sterol carrier protein and sterol‐binding protein in these organisms [43]. Notably, ADO activity is supported by a ferredoxin–ferredoxin reductase system to provide electrons required for catalysis [44]. Besides, it was also shown that gene coding for R2lox is up‐regulated in the hydrocarbon‐degrading haloarchaeon Halorientalis hydrocarbonoclasticus when cultured in the presence of hexadecane, thereby suggesting a role for R2lox in the biodegradation of this C16 alkane [43]. According to our present work, a substrate such as hexadecane, or a related compound, is conceivable as the length is compatible with our mass spectrometry and crystallography data.
Still, R2lox’s function and substrate remain to be determined. Despite copurifying with C16 and C18 HFAs, we believe that these ligands occupy the binding pocket by being trapped from the heterologous expression host organism and could be misleading in the search for R2lox’s actual substrate. One of the biggest challenges in the discovery of R2lox’s function is the identification of its physiological substrate. The best way to achieve this goal might be expressing R2lox in their native organisms. The subsequent in vitro activation of the enzyme and identification of the product should finally become a more achievable task. The substrate identification might also be tackled by finding a putative partner of R2lox. Finally, the physiological proximity of R2lox to the lipid membrane should be tested since the positive patch on the enzyme surface might indicate the interaction with the negative membrane surface.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
RD contributed to protein production, mass spectrometry, bioinformatics and structural analysis and manuscript writing; VS contributed to protein production and crystallization, data processing and analysis; AJ contributed to mass spectrometry and data analysis; AN contributed to mass spectrometry and data analysis; JJG contributed to cloning; HL contributed to protein production and crystallization, data processing and analysis, and manuscript writing; MH contributed to project design, data analysis and manuscript writing.
Supporting information
F ig. S1. Extracted ion chromatograms (EICs) for GkR2loxI and SaR2loxI showing the presence of the same ligands in both proteins.
Fig. S2. Extracted ion chromatograms (EICs) of the ligands identified in GkR2loxI after reduction‐oxidation cycle.
Fig. S3. Metal center in SeR2lox and ligands modeled in R2lox proteins.
Table S1. Data collection and refinement statistics.
Acknowledgements
We thank the staff from the beamline X06SA at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland, and beamline 14.1 of BESSY, Berlin, Germany, as well as members of Martin Högbom’s and Pål Stenmark’s groups for their help with data collection. We acknowledge financial support from the Knut and Alice Wallenberg Foundation (2017.0275 and 2019.0436), the Swedish Research Council (2017‐04018) and the European Research Council (HIGH‐GEAR 724394). The funding sources had no involvement in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Edited by Dietmar Manstein
Contributor Information
Hugo Lebrette, Email: hugo.lebrette@dbb.su.se.
Martin Högbom, Email: hugo.lebrette@dbb.su.se, Email: hogbom@dbb.su.se.
Data accessibility
The atomic coordinates of the SeR2lox and SaR2loxII structures are deposited in the Protein Data Bank under accession codes 7QBP and 7QBK, respectively. Other raw data are available upon request.
References
- 1. Lundin D, Poole AM, Sjöberg B‐M, Högbom M. Use of structural phylogenetic networks for classification of the ferritin‐like superfamily. J Biol Chem. 2012;287:20565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fox BG. Diiron enzyme structure and catalysis. In: Comprehensive Coordination Chemistry III. Elsevier. 2021;455–99. [Google Scholar]
- 3. Banerjee R, Jones JC, Lipscomb JD. Soluble methane monooxygenase. Ann Rev. Biochem. 2019;88:409–31. [DOI] [PubMed] [Google Scholar]
- 4. Högbom M, Sjöberg B‐M, Berggren G. Radical Enzymes. In: eLS. Wiley. 2020;375–93. [Google Scholar]
- 5. Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB. Microbial biosynthesis of alkanes. Science. 2010;329:559–62. [DOI] [PubMed] [Google Scholar]
- 6. Lee JM, Lee H, Kang S, Park WJ. fatty acid desaturases, polyunsaturated fatty acid regulation, and biotechnological advances. Nutrients. 2016;8:E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Högbom M, Stenmark P, Voevodskaya N, McClarty G, Gräslund A, Nordlund P. The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science. 2004;305:245–8. [DOI] [PubMed] [Google Scholar]
- 8. Andersson CS, Högbom M. A Mycobacterium tuberculosis ligand‐binding Mn/Fe protein reveals a new cofactor in a remodeled R2‐protein scaffold. Proc Natl Acad Sci USA. 2009;106:5633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Högbom M. The manganese/iron‐carboxylate proteins: what is what, where are they, and what can the sequences tell us? J Biol Inorg Chem. 2010;15:339–49. [DOI] [PubMed] [Google Scholar]
- 10. Kutin Y, Srinivas V, Fritz M, Kositzki R, Shafaat HS, Birrell J, et al. Divergent assembly mechanisms of the manganese/iron cofactors in R2lox and R2c proteins. J Inorg Biochem. 2016;162:164–77. [DOI] [PubMed] [Google Scholar]
- 11. Kutin Y, Kositzki R, Branca RMM, Srinivas V, Lundin D, Haumann M, et al. Chemical flexibility of heterobimetallic Mn/Fe cofactors: R2lox and R2c proteins. J Biol Chem. 2019;294:18372–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, et al. A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science. 2007;316:1188–91. [DOI] [PubMed] [Google Scholar]
- 13. Shafaat HS, Griese JJ, Pantazis DA, Roos K, Andersson CS, Popović‐Bijelić A, et al. Electronic structural flexibility of heterobimetallic Mn/Fe cofactors: R2lox and R2c proteins. J Am Chem Soc. 2014;136:13399–409. [DOI] [PubMed] [Google Scholar]
- 14. Griese JJ, Roos K, Cox N, Shafaat HS, Branca RMM, Lehtio J, et al. Direct observation of structurally encoded metal discrimination and ether bond formation in a heterodinuclear metalloprotein. Proc Natl Acad Sci USA. 2013;110:17189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miller EK, Trivelas NE, Maugeri PT, Blaesi EJ, Shafaat HS. Time‐Resolved Investigations of Heterobimetallic Cofactor Assembly in R2lox Reveal Distinct Mn/Fe Intermediates. Biochemistry. 2017;56:3369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie L, van der Donk WA. Homemade cofactors: self‐processing in galactose oxidase. Proc Natl Acad Sci USA. 2001;98:12863–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jakopitsch C, Kolarich D, Petutschnig G, Furtmüller PG, Obinger C. Distal side tryptophan, tyrosine and methionine in catalase‐peroxidases are covalently linked in solution. FEBS Lett. 2003;552(2–3):135–40. [DOI] [PubMed] [Google Scholar]
- 18. Nicolussi A, Auer M, Sevcnikar B, Paumann‐Page M, Pfanzagl V, Zámocký M, et al. Posttranslational modification of heme in peroxidases ‐ Impact on structure and catalysis. Arch Biochem Biophys. 2018;2:14–23. [DOI] [PubMed] [Google Scholar]
- 19. Cooley RB, Rhoads TW, Arp DJ, Karplus PA. A diiron protein autogenerates a valine‐phenylalanine cross‐link. Science. 2011;332:929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griese JJ, Kositzki R, Schrapers P, Branca RMM, Nordström A, Lehtiö J, et al. Structural basis for oxygen activation at a heterodinuclear manganese/iron cofactor. J Biol Chem. 2015;290:25254–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu H, Lebrette H, Clabbers MTB, Zhao J, Griese JJ, Zou X, et al. Solving a new R2lox protein structure by microcrystal electron diffraction. Sci Adv. 2019;5:4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. p. 571–607. [Google Scholar]
- 23. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bunkóczi G, Read RJ. Improvement of molecular‐replacement models with Sculptor. Acta Crystallogr D Biol Crystallogr. 2011;67:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, et al. Macromolecular structure determination using X‐rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019;75(Pt 10):861–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moriarty NW, Grosse‐Kunstleve RW, Adams PD. Electronic Ligand Builder and Optimization Workbench ( eLBOW ): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009;65:1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, et al. MolProbity: More and better reference data for improved all‐atom structure validation. Protein Sci. 2018;27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, et al. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucl Acids Res. 2005;33(Web Server):W299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen YY, Liang NY, Curtis JM, Gänzle MG. Characterization of Linoleate 10‐Hydratase of Lactobacillus plantarum and Novel Antifungal Metabolites. Front Microbiol. 2016;7:1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang N‐Y, Yang Y‐F, Li K. Analysis of Hydroxy Fatty Acids from the Pollen of Brassica campestris L. var. oleifera DC. by UPLC‐MS/MS. J Pharm (Cairo). 2013;2013:874875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu H, Zou X, Högbom M, Lebrette H. Redetermination of the first unknown protein MicroED structure by high resolution X‐ray diffraction. bioRxiv; 2021. Available from: http://biorxiv.org/lookup/doi/ 10.1101/2021.04.07.438860 [DOI]
- 35. Andersson CS, Berthold CL, Högbom M. A dynamic C‐terminal segment in the Mycobacterium tuberculosis Mn/Fe R2lox protein can adopt a helical structure with possible functional consequences. Chem Biodivers. 2012;9:1981–8. [DOI] [PubMed] [Google Scholar]
- 36. Yu H, Zhao Y, Guo C, Gan Y, Huang H. The role of proline substitutions within flexible regions on thermostability of luciferase. Biochim Biophys Acta. 2015;1854:65–72. [DOI] [PubMed] [Google Scholar]
- 37. Lu X, Vora H, Khosla C. Overproduction of free fatty acids in E. coli: implications for biodiesel production. Metab Eng. 2008;10:333–9. [DOI] [PubMed] [Google Scholar]
- 38. Kassab E, Mehlmer N, Brueck T. GFP Scaffold‐Based Engineering for the Production of Unbranched Very Long Chain Fatty Acids in Escherichia coli With Oleic Acid and Cerulenin Supplementation. Front Bioeng Biotechnol. 2019;7:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khara B, Menon N, Levy C, Mansell D, Das D, Marsh ENG, et al. Production of propane and other short‐chain alkanes by structure‐based engineering of ligand specificity in aldehyde‐deformylating oxygenase. ChemBioChem. 2013;14:1204–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mak WS, Wang X, Arenas R, Cui Y, Bertolani S, Deng WQ, et al. Discovery, design, and structural characterization of alkane‐producing enzymes across the ferritin‐like superfamily. Biochemistry. 2020;59:3834–43. [DOI] [PubMed] [Google Scholar]
- 41. Kang G, Taguchi AT, Stubbe J, Drennan CL. Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science. 2020;368:424–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao Y, Zhang H, Fan M, Jia C, Shi L, Pan X, et al. Structural insights into catalytic mechanism and product delivery of cyanobacterial acyl‐acyl carrier protein reductase. Nat Commun. 2020;11:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar S, Zhou J, Li M, Xiang H, Zhao D. Insights into the metabolism pathway and functional genes of long‐chain aliphatic alkane degradation in haloarchaea. Extremophiles. 2020;24:475–83. [DOI] [PubMed] [Google Scholar]
- 44. Crépin L, Barthe M, Leray F, Guillouet SE. Alka(e)ne synthesis in Cupriavidus necator boosted by the expression of endogenous and heterologous ferredoxin–ferredoxin reductase systems. Biotechnol Bioeng. 2018;115:2576–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
F ig. S1. Extracted ion chromatograms (EICs) for GkR2loxI and SaR2loxI showing the presence of the same ligands in both proteins.
Fig. S2. Extracted ion chromatograms (EICs) of the ligands identified in GkR2loxI after reduction‐oxidation cycle.
Fig. S3. Metal center in SeR2lox and ligands modeled in R2lox proteins.
Table S1. Data collection and refinement statistics.
Data Availability Statement
The atomic coordinates of the SeR2lox and SaR2loxII structures are deposited in the Protein Data Bank under accession codes 7QBP and 7QBK, respectively. Other raw data are available upon request.