

Summary
Background
Hepatitis delta virus (HDV), which causes the most severe form of viral hepatitis, is an obligated hepatitis B (HBV) satellite virus that can either infect naïve subjects simultaneously with HBV (co‐infection), or chronically infect HBV carriers (super‐infection). An estimated 12 million people are infected by HDV worldwide.
Aims
To summarise the most relevant aspects of the molecular biology of HDV, and to discuss the latest understanding of the induced pathology, interactions with the immune system, as well as both approved and investigational treatment options.
Methods
References for this review were identified through searches of PubMed with the terms “HDV” “viral hepatitis” “co‐infection” and “super‐infection,” published between 1980 and October 2021
Results
The limited access to the HDV‐infected liver has hampered the investigation of the intrahepatic compartment and our understanding of the mechanisms of HDV pathogenesis. In the absence of standardised and sensitive diagnostic tools, HDV is often underdiagnosed and owing to its strong dependence on host cellular factors, the development of direct antiviral agents has been challenging. New therapeutic agents targeting different steps of the viral cycle have recently been investigated, among which bulevirtide (which was conditionally approved by EMA in July 2020) and lonafarnib; both drugs having received orphan drug designation from both the EMA and FDA.
Conclusions
The HBV cure programme potentially offers a unique opportunity to enhance HDV treatment strategies. In addition, a more comprehensive analysis of the intrahepatic compartment is mandated to better understand any liver‐confined interaction of HDV with the host immune system.
Keywords: fine‐needle aspiration, HDV, immune system, intrahepatic compartment, viral hepatitis
The diagrams show the time‐course of the most common outcomes of (A) HBV‐HDV co‐infection and (B) HDV super‐infection of an HBV carrier. Biochemical and serological parameters are indicated.

1. INTRODUCTION
1.1. Hepatitis delta virus biology
Hepatitis delta virus (HDV) causes the most severe form of viral hepatitis, currently infecting an estimated 12 million people worldwide. 1 HDV is the smallest virus known to infect humans (only 36 nm in diameter). 2 First described in 1977 by Mario Rizzetto as a new antigen–antibody system associated with hepatitis B virus (HBV) infection, 3 , 4 HDV is a negative‐sense single‐stranded RNA virus (Deltaviridae family, genus Deltavirus).
The first step in HDV replication is the synthesis of multimeric copies of a complementary RNA, the antigenome, by the DNA‐dependent RNA‐polymerase II in the nucleus. 5 An autocatalytic cleavage mediated by ribozymes that are included in both the genomic and anti‐genomic strands produces monomeric molecules of either polarity of approximatively 1700 nucleotides. 6 These monomers are then ligated to form circular RNA with a high degree of self‐complementarity that mimics a dsDNA molecule. 7 The HDV genome encodes a structural protein (the hepatitis delta antigen—HDAg) produced in two different isoforms (small—S‐HDAg and large—L‐HDAg) following the editing of the anti‐genomic molecule in position 1012 by the host deaminase ADAR1, which converts the UAG stop codon into a UGG tryptophan codon, allowing a C‐terminal extension of the translated protein. 8 , 9 HDV RNA and both isoforms of HDAg interact to form the ribonucleoprotein (RNP). S‐HDAg is produced in the first stages of the viral cycle and is necessary for the initiation of viral replication and HDV RNA accumulation. L‐HDAg is synthesised later during infection and its 19 extra amino acids confer unique functional properties, such as inhibition of viral replication and viral assembly. 10 , 11 , 12 Both antigens undergo post‐translational modifications catalysed by host enzymes; particularly relevant is the prenylation on Cys211 in the C‐terminal portion of L‐HDAg, necessary for the interaction with HBV envelope proteins (HBsAg) expressed in the same cell, allowing the assembly of the viral envelope and the formation of infectious particles. 13 , 14 Therefore, even though HDV genome replication and RNP formation are HBV independent, HDV can productively complete its infective cycle only in hepatocytes expressing HBsAg. 3 , 15 HDV is, in effect, an obligated HBV satellite virus that can either infect naïve patients simultaneously with HBV (co‐infection), or chronically infected HBV carriers (super‐infection). Human species‐specificity and liver tropism of HDV have been ascribed to the HBV envelope proteins (Figure 1), where HDV and HBV have been shown to share the same entry mechanism to infect hepatocytes through the Na+/taurocholate co‐transporting polypeptide (hNTCP). 16 , 17 However, glycoproteins from HBV‐unrelated viruses have recently been shown to have the capability to package HDV RNPs in vitro, and HCV's ability to propagate HDV infection in humanised mice has been reported, although this remains a subject of debate. 18 Moreover, the clinical relevance of these findings remains uncertain. A recent study that involved 323 HCV RNA‐positive and HBsAg‐negative patients could only detect HDV markers in eight HBV core antibody (anti‐HBcore)‐positive patients, representing prior HBV infections, but not among the remaining anti‐HBcore antibody‐negative patients, suggesting the occurrence of replicative HDV infections in HCV mono‐infected patients is rare or unlikely. 19 Another study investigating a cohort of 160 patients from Venezuela infected with HCV (in the absence of molecular markers for HBV) detected two patients with anti‐HDAg antibodies, and one patient with low‐level circulating HDV RNA, 20 also indicating the occurrence of replicative HDV infections in HCV mono‐infected patients an unlikely event.
FIGURE 1.
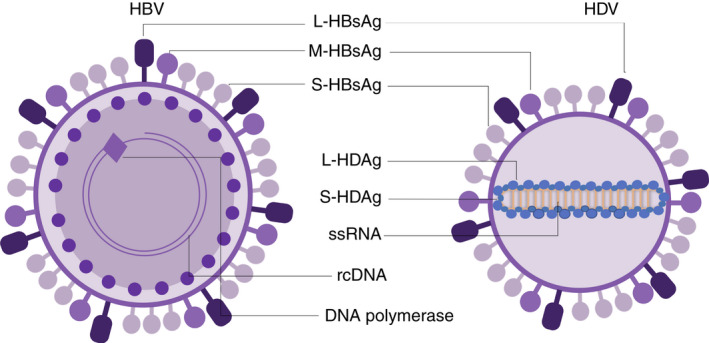
Schematic representation of HBV and HDV virions and their components. Figures not drawn to scale. HBV, hepatitis B virus; HDV, hepatitis delta virus; rcDNA, relaxed circular DNA of HBV; ssRNA, single‐stranded RNA
Despite depending on HBV for new virion formation, HDV can survive in non‐dividing human hepatocytes in the absence of HBV as observed in patients post liver transplantation. Importantly, these observations were confirmed both in in vitro and in vivo experimental settings in which HDV infection and replication was maintained in dividing human hepatoma cell lines and proliferating primary human hepatocytes in the presence of entry inhibitors, propagating among daughter cells for several weeks. 21 Thus, HDV demonstrates unique persistence capacity which underlines why intrahepatic clearance is so rarely achieved.
1.2. Genotypes and epidemiology
Eight different genotypes of HDV have been characterised to date, each showing a specific geographic distribution with the exception of the ubiquitous clade HDV‐1. 22 The severity of the clinical manifestation is heterogeneous (with patients from South America reporting the most severe liver disease) and is potentially related to the infecting HDV genotype (HDV‐1 and HDV‐3 might be associated with greater severity, although studies are limited), or to genetic and environmental factors. 23 Notably, few studies are available in patients of African origin with the less common genotypes (HDV‐5 to HDV‐8). 24
Limited data are available about the specificity of the combination between HBV and HDV genotypes, in all cases restricted to defined geographical areas. The hypothesis of an association driven more by the relative abundance than by a higher affinity between HBV and HDV genotypes is currently accepted. 25 , 26
It is estimated that 257–300 million people are chronically infected with HBV, and approximately 4.5% have been exposed to HDV (anti‐HDV antibody positive), 1 however, this may be an underestimate of the true prevalence of HDV, due to limited testing worldwide and thus the actual prevalence of HDV may be much higher. The risk factors for HDV infection are the same as those for HBV and include intravenous drug use and high‐risk sexual behaviour. However, in areas where HDV remains endemic, or where migratory patterns from such countries is significant, perinatal or early childhood transmission accounts for a significant proportion of new infections. 27 Even though HBV vaccination programmes have reduced the global prevalence of HDV infection, there are regional differences in vaccine coverage, with the African, Eastern Mediterranean and European regions below the global average. 28
2. HOST–VIRUS INTERACTIONS
2.1. Animal models
Since the HDV envelope is composed of the small, medium and large HBV surface proteins (S‐, M‐ and L‐HBsAg's), both viruses share the same species‐specificity and hepatotropism, exploiting the same entry mechanism. The three isoforms of HBsAg share the C‐terminal portion (corresponding to the S protein) and differ in their N‐termini. Following an early attachment step mediated by heparansulphate proteoglycans (HSPG), 29 the pre‐S1 domain of the L‐HBsAg specifically interacts with hNTCP, a bile salt transporter expressed in the basolateral membrane of the hepatocytes, and the residues 157–165 were identified to be critical for binding and entry for both HBV and HDV infection. Differences in residues within this motif between homologous NTCP in mammals determine the species‐specificity of these pathogens. 16
Humanised mice represent a useful tool for the study of HDV: livers of immune‐deficient mice are engrafted with human hepatocytes, susceptible to HBV and HDV infection. These models have helped to elucidate some aspects of hepatitis delta virology, however, they do not permit the study of the interaction between the virus and the host adaptive immune system, although they do provide an understanding of innate immune responses. 30 , 31
More recently, mouse models expressing the human transporter hNTCP or its humanised version were developed. hNTCP transgenic mice are able to support a transient single‐round HDV infection of about 3% of hepatocytes in an age‐dependent manner. 32 Both neonate and adult mice carrying the humanised receptor are susceptible to HDV infection, but surprisingly they are not able to support HBV infection, so that co‐infection or super‐infection cannot be studied. 33 Moreover, the short‐term transient infection achieved in these two models does not lead to liver damage, representing an important limitation for the study of HDV‐related liver pathology.
Recombinant adeno‐associated viral vectors have been used to successfully mimic co‐infection in adult immunocompetent mice, including the editing of the HDV anti‐genome, the expression of both S‐ and L‐HDAg, in addition to long‐lasting detectable viremia. Importantly, liver damage, inflammation and induction of the innate immune response were observed together with the upregulation of genes involved in hepatocellular carcinoma (HCC), cirrhosis, fibrosis, cell death and proliferation; processes which are exacerbated in HDV‐infected patients. In this model, the HDAgs, but not the host immune response, are considered to be cytotoxic and induce liver injury. 34 , 35 , 36 , 37
A similar induction of cell‐intrinsic and innate immune response was also observed in both immunodeficient and immunocompetent HBV1.3× hNTCP dual transgenic mice, able to produce both isoforms of HDAg and release viral particles after HDV infection. However, in this immunocompetent mouse model, HDV viremia lasted only 14 days and neither liver damage nor histological alterations were observed, suggesting a role for lymphocytes in counteracting HDV infection and in the clearance of HDV‐infected hepatocytes. 38
Conflicting results from the most recent aforementioned mouse models highlight the need for more in‐depth studies in HBV‐HDV co‐infected patients and specifically the study of the antiviral immune response.
2.2. Current understanding from clinical samples
Current knowledge about the immune response against HDV is largely restricted to the analysis of peripheral blood mononuclear cells (PBMCs) in patients with chronic HDV infection.
In comparison to patients with HBV and HCV infections, a high frequency of cytotoxic perforin‐positive CD4+ T cells have been found in the blood of patients with HDV chronic infection, 39 with some MHC‐I and MHC‐II epitopes identified to date. 40 , 41 , 42 , 43 The HDV‐specific T cell response generated in patients with chronic HDV infection is, however, weak and insufficient to contain the infection, similar to that seen in HBV and HCV chronic infections. 44 One study showed IL‐2 and interferon‐gamma (IFN‐γ) production from HDV‐specific T cells, as well as an IP‐10 response exerted by activated monocytes, contributing to the inflammatory environment. 45
Broadly directed low‐level HDV‐specific CD4+ and CD8+ T‐cell responses were detected after in vitro expansion in a number of patients with chronic HDV infection. No correlation was observed between the magnitude of HDV‐specific CD4+ or CD8+ T‐cell responses and the level of viremia or clinical status of patients; similarly, no difference was observed in patients with spontaneous or treatment‐induced HDV PCR negativity. In the same study, one HBV patient super‐infected with HDV was monitored during the acute phase of HDV infection, showing a strong CD8+ T cell response during the first month followed by a decrease in HDV viral load, suggesting a role for T cell responses in the control of HDV viremia, analogous to HBV mono‐infection. 42
Another study identified six CD8+ T cell HDAg epitopes stimulating PBMCs from lonafarnib/ritonavir‐treated HDV patients; in this case, the ex vivo activation state correlated with transaminase activity and the production of IFN‐γ after peptide stimulation correlated inversely with HDV titre. The majority of the HDV‐specific CD8+ T cells presented with a memory‐like phenotype (PD‐1+ CD127+). These cells also expressed the activation marker CD38, and the transcription factor TCF1 (known to be important for the formation of memory CD8+ T cells), being capable of a low‐level IFN‐γ response (Figure 2A). 46
FIGURE 2.
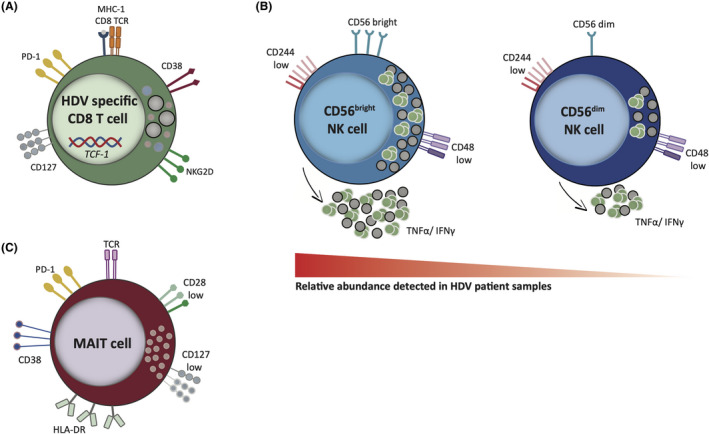
Characteristics of PBMCs subtypes in HDV patients. A, HDV specific CD8+ T cells presenting with a memory‐like phenotype along with increased expression of the innate‐like receptor NKG2D. B, The NK cell population is mainly composed of CD56bright cells with low cytotoxic potential and high cytokine production; both the CD56bright and the CD56dim subsets are characterised by a low expression of the CD244 and CD48 surface markers. C, Residual MAIT cells in HDV patients show an exhausted phenotype and a downregulation of costimulatory molecules. Shaded icons represent low expression. HDV, hepatitis delta virus; MAIT, mucosal‐associated invariant T; NK, natural killer; PBMC, peripheral blood mononuclear cell
Recently, the ex vivo characterisation of paired blood samples and liver biopsies (LB) from patients with chronic HDV in comparison with non‐viral controls has revealed that both HDV‐specific and total liver CD8+ T cells express the innate‐like receptor NKG2D, associated with TCR‐independent activation. In the patients analysed in this study, the percentage of total CD8+ T cells expressing NKG2D correlate with liver inflammation, suggesting a role for cytokine‐mediated TCR‐independent activation of non‐antigen specific bystander CD8+ T cells in liver inflammation and disease. 47
Since the observed specific adaptive immune response in HDV patients appears weak, more attention has recently been paid to innate immune responses. As in patients with HBV and HCV mono‐infection, HBV‐HDV co‐infected patients show a higher frequency of natural killer (NK) cells, but with a less activated phenotype (low expression of the activating receptors CD244 and CD48, resulting in impaired cytolytic activity and reduced cytokine production). In addition, the proportion of the CD56bright subset (less mature and immunoregulatory in nature) is increased in relation to the more mature and cytolytic CD56dim subset (Figure 2B). Inhibition of NK cell activity thus seems to be a common escape mechanism of hepatitis viruses, relying more on the inflammatory environment than on virus‐specific factors. 48 Moreover, the same study showed that NK cells from patients with HDV produced the highest amounts of IFN‐γ and TNF‐α among the different hepatitis virus infections, and this finding could in part explain the more aggressive liver damage observed in these patients. However, no correlation between NK cell phenotype and viral load was found, arguing against a direct effect specific to any single hepatitis virus on NK cells. 48 Pegylated IFN‐α (PEG‐IFN‐α‐2a) treatment caused a significant change in NK cell differentiation status, with an enrichment in immature circulating NK cells. Conversely, peripheral blood NK cells of untreated HDV‐infected patients were phenotypically similar to healthy controls, suggesting that the HDV effect on NK cells may be local and limited to the liver. 49 While in previous studies increased NK cell activity in HBV patients was linked to liver injury, Lunemann et al. reported no correlation between NK phenotype and disease severity in the HDV untreated cohort. Instead, high frequency of CD56dim NK cells before treatment was associated with responsiveness to PEG‐IFN‐α‐2a. 49
Similarly, a loss of mucosal‐associated invariant T (MAIT) cells was observed in the blood and LB of patients with chronic HDV but not of patients with HBV mono‐infection in comparison with healthy controls; the reduction occurred preferentially in the CD8+ compartment and did not affect non‐MAIT cells. The reduction in this cell type correlates with the concentration of IL‐12 and IL‐18 in patient serum (higher than in patients with HBV and healthy controls), suggesting that dysregulating cytokine levels may contribute to MAIT cell activation and loss in patients with chronic HDV. Moreover, residual MAIT cells in patients with chronic HDV presented with an abnormal phenotype, characterised by upregulation of activation and exhaustion markers (CD38, HLA‐DR and PD‐1), downregulation of co‐stimulatory molecules (CD28, CD127), and altered expression of the transcription factor Helios (Figure 2C), resulting in reduced functionality. 50
2.3. HDV and the intrahepatic compartment
The common histopathology features of LB from HDV patients with chronic hepatitis are similar to other types of viral hepatitis, with piecemeal necrosis, portal inflammation, cytoplasmic dissociation, sanded nuclei (loaded with viral antigens), and the presence of apoptotic bodies. The degree of inflammation and necrosis is enhanced in co‐infection or super‐infection, and the presence of delta antigen (mainly with a nuclear localisation) can be determined immunohistochemically. 51
The liver is enriched in innate immune cells like NK, natural killer T cells (NKT), ILC and γδT cells (with NK and NKT cells constituting about 50% of total hepatic lymphocytes), and presents with a reversal of the CD4:CD8 ratio when compared with that of the peripheral compartment (CD8+ T cells being more abundant in the liver). 52 , 53 Liver‐resident lymphocytes share many phenotypic and transcriptional characteristics with other tissue‐resident lymphocytes. Despite this, the specific tissue microenvironment can have an effect on their phenotype and function, providing unique properties, which may be key determinants in liver‐related diseases and infections. 53 This implies that what is known about peripheral blood lymphocytes may not be representative of the intrahepatic compartment; however, the analysis of the cells in the blood of HBV‐HDV‐infected patients is still the most accessible surrogate for the study of the intrahepatic immune response, since the use of LB is limited in many centres. Nevertheless, direct liver sampling is still irreplaceable for the study of the immune response, virological markers and gene expression profiling. 54 , 55 Some data now exist delineating tissue‐specific immune responses in HDV, but these still require further evaluation to fully understand innate and adaptive immune interactions in the liver, requiring liver sampling.
In this context, fine‐needle aspiration (FNA) has been shown to be a safe and well‐tolerated technique that allows a broad analysis of the intrahepatic lymphocyte compartment, with significant correlation with the results obtained by LB and confirming the differences previously reported with PBMC composition. Furthermore, its reduced invasiveness could potentially allow repeated sampling and monitoring of the kinetics of the immune response during different disease stages or during treatment. 56 , 57 This technique has a proven track record in longitudinal monitoring of HCV infected patients, 58 , 59 and its suitability to quantify HBV antigen‐expressing hepatocytes by flow cytometry has also been reported. 60 More recently, FNAs were demonstrated to be able to sample tissue‐resident subsets of T 61 and NK cells 62 in the context of HBV infection, as well as to provide viable hepatocytes and myeloid cells. 63 It would also be of interest to explore the role of other non‐parenchymal cell types (hepatic stellate cells and liver sinusoid epithelial cells) and their response to the cytokines present in the HDV‐infected liver. At present, FNAs are not able to be utilised for diagnostic purposes for liver disease, thus remain a research tool, but can still provide detailed relevant scientific information which may aid future translational research advances.
Due to the complex cellular composition of the liver, only a comprehensive analysis of the site of infection can lead to a better understanding of all the players involved in HBV‐HDV co‐infection and their specific roles in the observed pathology.
3. CLINICAL ASPECTS
3.1. Diagnosis
European Association for the Study of the Liver (EASL) recommends HDV screening for all patients diagnosed with HBV infection, whereas AASLD recommends risk‐based screening. However, for various reasons, these recommendations are not always followed in the real‐world clinical setting, and in some areas, there is limited access to robust diagnostics. This impedes a more accurate estimation of the prevalence of HDV infection, in addition to a more precise definition of the natural history of the disease.
Clinical differentiation between co‐infection and super‐infection can be challenging. Co‐infection is based on the simultaneous presence of HBsAg and HDV RNA in serum. High‐titre anti‐HBcore IgM antibodies are a distinguishing feature of acute hepatitis delta co‐infection since they are absent in chronic hepatitis. HBV‐HDV co‐infection is also characterised by an increase in serum aminotransferases, high serum HDV RNA levels and hepatitis B viremia, dependent on the degree of inhibition exerted by HDV on HBV. Anti‐HDAg IgG antibodies appear late and at low titres. Conversely, since anti‐HBcore IgM is not present during chronic HBV infection, the concomitant presence of high‐level HDV viremia, high‐titre and persistent anti‐HDAg antibodies in the absence of anti‐HBcore IgM indicates super‐infection. In this clinical profile, the antibody for HBcAg is usually IgG, and HBV DNA titre is more commonly low or undetectable. 64 , 65 Anti‐HDAg IgM antibodies can increase and correlate with disease activity during chronic infection and frequently do not allow distinction between acute and chronic infection. Early studies showed that anti‐HDAg IgM persists in patients with progressive disease, while it declines and subsequently disappears in carriers with disease resolution; the kinetics of total antibodies (combined IgM and IgG) is similar but slower and appears less accurate. 66 Levels of anti‐HDAg IgM were found to positively correlate with biochemical and histological activity, but not with serum HDV RNA levels, and to associate with the clinical long‐term outcome. 67 Anti‐HDAg IgM is, therefore, a good indicator of disease activity and has been suggested as a prognostic marker, to help stratify which patients may benefit most from treatment. Due to the anti‐HDAg IgM association with disease activity, it can assist the differential diagnosis of acute co‐infection (where its detection is more limited in time) and super‐infection (where its detection is more persistent).
The majority of patients with anti‐HDAg antibodies are HBeAg negative, but no difference has been found in the long‐term outcome of the disease between HBeAg‐positive and ‐negative patients. 23 , 68
Hepatitis delta virus RNA quantification is used as a diagnostic marker as well as to monitor response to treatment. In 2012, the first World Health Organisation International Standard for HDV RNA was made available; however, quantification is often performed with in‐house nucleic acid amplification protocols, impairing comparability between results from different centres. 69 HDV RNA is the best indicator for the presence of replicating virus, but as long as standardised quantification is not widely implemented, anti‐HDAg IgM or total anti‐HDAg antibodies are still important tools for the diagnosis of HDV infection.
Figure 3 shows a simple algorithm for the diagnosis and management of patients with HDV.
FIGURE 3.
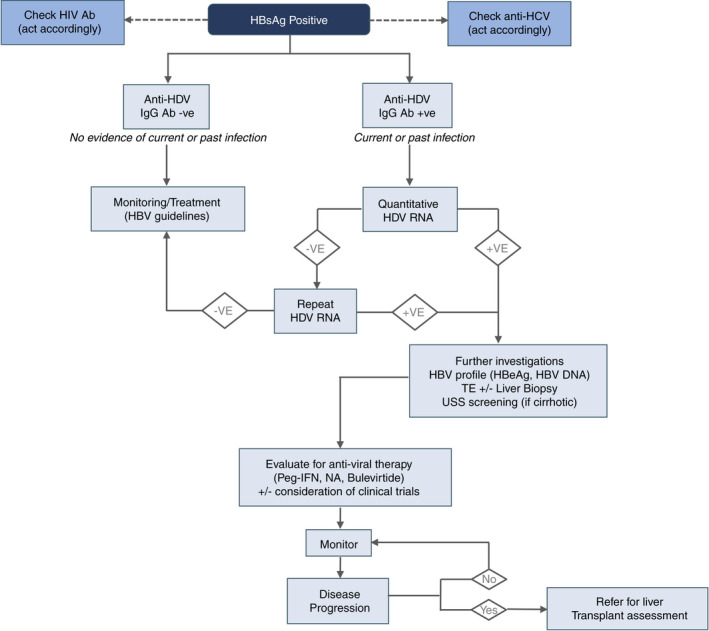
Management algorithm for the diagnosis and management of HDV infection. Flow chart indicating management of patient with HBV infection. Outlined are the required investigations and how to proceed according to the investigation results. +VE, positive; −VE, negative; Ab, antibody; HBsAg, hepatitis B surface antigen; HBeAg, hepatitis B all envelope antigen; HBV, hepatitis B virus; HCV, hepatitis C virus; HDV, hepatitis delta virus; NA, nucleos(t)ide analogue; peg‐IFN, pegylated interferon; TE, transient elastography; USS, ultrasound screening
3.2. Natural history
The clinical course of HDV infection largely depends on the underlying status of the HBV infection and on the infecting HDV genotype. The simultaneous infection with HBV and HDV in a susceptible individual usually leads to an acute self‐limiting hepatitis similar to acute hepatitis B mono‐infection, with a similar rate of progression to chronicity (between 2% and 5%). In 95% of cases, spontaneous resolution is observed, but it remains an important differential diagnosis for severe or fulminant hepatitis, nonetheless. Super‐infection, on the other hand, can cause fulminant hepatitis, but chronicity rates exceed 80%, with a higher risk of developing cirrhosis and HCC. 64 , 65
In the case of co‐infection, the incubation period of HDV is dependent on the titre of the co‐infecting HBV. HDV infection depends on the virulence of the concomitant HBV infection, since a limited expression of HBsAg may result in abortive HDV infection, while it has been proposed that an abundant expression will ensure successful HDV propagation and pathogenicity. The clinical outcome of co‐infection varies from mild to severe or even fulminant hepatitis. More recently, HDV infection has been associated with a milder course than in the past, when fulminant hepatitis and rapid progression to cirrhosis was more commonly reported; this was most likely due to more pathogenic emerging strains of HDV rapidly circulating among HBeAg‐positive subjects. 70 Acute hepatitis can be either monophasic or biphasic depending on the relative titres of the two viruses, the first peak usually being caused by HBV and the second by HDV. 64
In the setting of a super‐infection, HDV takes advantage of the pre‐existing HBsAg and immediately establishes infection. It can cause an exacerbation of the pre‐existing chronic hepatitis B (CHB) or lead to an emergent hepatitis in a previously asymptomatic HBsAg carrier. Even though most of the chronic HBsAg carriers superinfected by HDV develop progressive chronic HDV, a minority of them will experience self‐limited hepatitis and clear HBV. 64
Spontaneous fluctuation of both HBV and HDV viral markers has also been observed, and a recent retrospective study of untreated HDV patients showed that one‐quarter of them achieved a spontaneous HDV RNA decline associated with HBsAg and HBV DNA decreases. 71 Clinical details of HDV infection are summarised in Table 1 and Figure 4.
TABLE 1.
| Infection type | Incubation (wk) | Serology | Clinical features |
|---|---|---|---|
| Acute HDV co‐infection | 3–7 |
|
|
| Acute HDV super‐infection | <3 |
|
|
| Chronic HDV infection |
|
|
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDAg, hepatitis delta antigen; HDV, hepatitis delta virus: KC, Kupffer cells: wk, weeks
FIGURE 4.
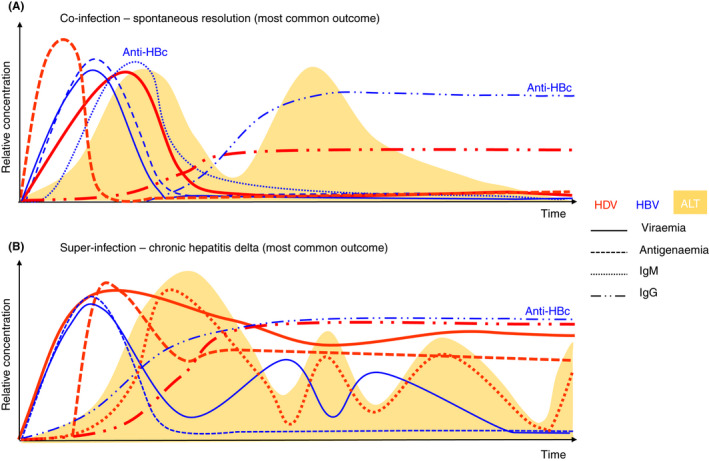
Natural history of hepatitis delta infection. Schematic representation indicating the time‐course of (A) co‐infection and (B) super‐infection. Biochemical and serological parameters are indicated
3.3. Disease pathogenesis
The exact mechanisms of the pathogenesis of HDV infection are poorly understood; HDV is commonly considered a non‐cytopathic virus since delta viremia has no correlation with the extent of liver damage. 72 Thus HDV‐associated hepatic damage is thought to be immune‐mediated, akin to HBV and HCV infection. 73 , 74 However, early studies, both in vitro and on autopsy specimens, suggest the possibility of a direct cytopathic effect of the viral components. 75 , 76 In particular, in vitro studies suggested that S‐HDAg expression can be responsible for direct cytotoxicity, significantly contributing to the hepatocyte injury observed during HDV infection. 75 On the contrary, prenylated L‐HDAg was demonstrated to activate TGF‐β and AP‐1 transduction signal in vitro, both alone and synergistically with the Hepatitis virus X protein, suggesting a mechanism by which HDV may contribute to fibrosis and cirrhosis, and that may explain why HDV super‐infection in patients with HBV accelerates the progression of liver disease. 77
The inhibitory activity of HDV over HBV replication is well recognised, with most patients with chronic HDV showing low or undetectable levels of circulating HBV DNA. HBV replication is inhibited during the acute phase of HDV infection, and the two viruses can have fluctuating patterns of predominance over time. The exact mechanisms of the inhibitory effect of HDV on HBV are thought to be independent of the adaptive immune system since the phenomenon is reproducible in vitro. Inhibition of HBV replication can be due to a direct interaction between the two viruses inside the cells: HDV needs HBsAg for packaging and release, and it has been speculated that HDV is able to repress HBV replication maintaining surface protein production, for instance competing for RNA pol‐II recruitment. The dominant role of HDV has been explained by in vitro data showing that both S‐ and L‐HDAg can inhibit the activity of HBV enhancers; moreover, innate immune responses triggered by HDV infection represent an indirect mechanism of HBV suppression. Some patients, however, have both HBV and HDV replication, and fluctuations in their viral profile suggest a more complex interaction between the two viruses at the intrahepatic level. 78 , 79 , 80
In chronic HDV infection, following the acute phase, HDV RNA levels may subside (and become undetectable) in the presence of HDAg positivity. These patients, in the absence of ongoing HDV replication, can have normal serum transaminases and HBV replication may remain low, akin to HBeAg‐negative chronic infection. There are limited studies in determining if these patients are at risk of reactivating HDV and thus their management remains uncertain. As there is no active HDV replication, anti‐HDV therapy is not indicated, however, they may already have significant liver fibrosis depending on previous HDV activity and the host immune response. Data are also limited on whether other non‐invasive viral markers in these patients are similar to those with chronic HBV infection (e.g. HBV RNA, HBsAg). 81 Further studies elucidating the natural history of these subjects compared to those with active HDV replication and HBV mono‐infection are needed.
3.4. Prevention and treatment
The vaccination campaign against HBV has contributed to a significant decrease in HDV prevalence in endemic areas in Southern Europe, Italy being a prime example where HDV is expected to disappear from the domestic population in the near future. 82 However, in high‐income countries that have benefited from the vaccination programme, the HDV‐positive population consists of a cohort of older native patients, some with advanced fibrosis or cirrhosis, with new infections often being introduced by younger immigrants from areas where HDV remains endemic. 83
Owing to HDV's strong dependence on host cell factors, the development of direct antiviral treatments is challenging. Since HDV does not encode any viral RNA polymerase, the only viral targets available are the HDAgs and the ribozyme. Specific inhibition of the ribozyme activity has been achieved in vitro with small molecules as well as with small interfering RNA (siRNA) strategies, avoiding multimeric RNA cleavage, functional genomic and antigenomic RNA production. 84 , 85
One of the strategies used to date is the suppression of HBV replication and the induction of a strong immune response against the helper virus, with the aim of developing a protective titre of anti‐HBsAg antibodies. Current treatments for chronic HBV infection comprise PEG‐IFN‐α‐2a and nucleo(t)ide analogues, but only PEG‐IFN‐α‐2a was shown to be an effective therapeutic strategy against HDV infection.
Alternative therapeutic approaches are currently being developed, exploiting molecules involved in three different mechanisms: stimulation of the innate immune response, inhibition of viral entry via competitive binding to hNTCP, and assembly and release of viral particles. Thus, IFN, entry inhibitors and prenylation inhibitors are under evaluation in large scale clinical trials (Table 2). 86
TABLE 2.
Currently employed and under development options for the treatment of HDV infection
| Name | Mechanism of action | Endpoint | Side effects | Duration of treatment | Status |
|---|---|---|---|---|---|
| PEG‐IFN‐α‐2a | Immune modulator; enhancement of innate immunity (induction of interferon‐stimulated genes) | Change from baseline in HDV viral load; normalisation of ALT | Flu‐like syndrome, myalgia, headache, fatigue, weight loss, depression, hair loss and local reactions at the site of injection. | Minimum 24–48 wk | Recommended by international guidelines; not approved by regulatory authorities |
| Nucleotide analogues a (entecavir, tenofovir disoproxil fumarate and tenofovir alafenamide) | Inhibitors of HBV replication | HBV DNA undetectable | Gastrointestinal, nephropathy, Fanconi syndrome, osteomalacia, lactic acidosis | Long‐term | Approved by FDA and EMA for the treatment of HBV, no efficacy for HDV |
| PEG‐IFN‐λ | Immune modulator; enhancement of innate immunity (induction of interferon‐stimulated genes) | Change from baseline in HDV viral load; normalisation of ALT | Flu‐like symptoms, gastrointestinal, loss of appetite, back pain, dizziness, dry mouth, taste changes (milder than with PEG‐IFN‐α‐2a) | 48 wk | Phase 2 clinical trials; monotherapy or in combination with Lonafarnib or Ritonavir |
| Bulevirtide | Entry inhibitor | Change from baseline in HDV viral load | Raised levels of bile salts in the blood | 24 wk in studies, long‐term therapy (maintenance) | Conditional marketing authorisation by EMA; Phase 2 and 3 clinical trials with either Bulevertide alone or in combination with PEG‐IFN‐α‐2a |
| Lonafarnib | Prenylation inhibitor (assembly inhibitor) | Change from baseline in HDV viral load | Gastrointestinal | Not yet determined | Phase 2 and 3 clinical trials with Ritonavir/ PEG‐IFN‐α‐2a vs. with Ritonavir |
| Nucleic acid polymers | Multiple: attachment inhibitor, HBsAg release inhibitor, assembly inhibitor | Change from baseline in HDV viral load | None reported so far for REP 2139‐Ca and REP 21‐39‐Mg | 24 or 48 wk | Phase 2 clinical trials in combination with tenofovir disoproxil fumarate and PEG‐IFN‐α‐2a |
| siRNA a , b | Inhibitors of viral replication | HBsAg loss | Injection site reactions | Not yet determined | Phase 2 clinical trials for HBV monoinfection. Different siRNA’s are combined with nucleotide analogues +/− PEG‐IFN‐α‐2a or +/− capsid assembly modulators |
Abbreviations: ALT, alanine aminotransferase; EMA, European Medicine Agency; HBV, hepatitis B virus; HDV, hepatitis delta virus; HBsAg, hepatitis B virus surface antigen; wk, weeks.
Do not have a direct antiviral effect on HDV, but they reduce the formation rate of new HDV virions by inhibiting HBV replication and therefore limiting HBsAg availability.
siRNA have been studied so far only in clinical trials for HBV monoinfection, where they inhibit transcription from the cccDNA, therefore, reducing HBsAg production
The Food and Drug Administration (FDA) proposed that “drugs that are intended to be used as chronic suppressive therapy, a greater than or equal to 2‐log10 decline in HDV‐viral load and ALT normalisation on‐treatment could be considered an acceptable surrogate endpoint reasonably likely to predict clinical benefit”. 87 An important issue to be considered, however, is the performances of the available assays for HDV RNA‐viral load quantification. A recent international quality‐control study concluded that most of the assays dramatically underestimated or failed to detect/quantify positive HDV RNA samples, especially from patients infected with strains of African origin (HDV‐1 and HDV‐5 to −8), highlighting the lack of robust tools to routinely monitor HDV RNA for the therapeutic management of infected patients. 88 A suitable quantification assay must have a good sensitivity to be able to detect early rebounds, regardless of the high genetic variability of the HDV‐infected strains reported earlier. Therefore, it remains a subject of debate in the field whether a 2‐log decrease in the HDV‐RNA‐VL, would be a relevant goal in the monitoring of on‐treatment HDV‐infected patients.
3.4.1. Immune modulators
Pegylated‐interferon‐α‐2a
At present, PEG‐IFN‐α‐2a is the only available drug that has been proven to have long‐term antiviral efficacy and fibrosis regression against chronic HDV infection, showing on‐treatment virologic response rates between 17% and 47%, 89 but late relapses of HDV replication beyond week 24 following treatment cessation occurred in excess of 50% of responders. Long‐term PEG‐IFN‐α‐2a was proven effective in a group of patients for whom treatment extension was individually tailored based on HBsAg status. No liver‐related deaths were recorded among patients achieving virological response (58% of participants), indicating that successful PEG‐IFN‐α‐2a treatment can change the natural history of the disease and potentially reduce all‐cause mortality by influencing chronic inflammatory status. 90 Furthermore, PEG‐IFN‐α‐2a led to the regression of fibrosis in patients with advanced fibrosis. 91 Treatment of longer duration (up to 96 weeks) of PEG‐IFN‐α‐2a alone or in combination with tenofovir, showed relapse in 36%–39% of responding patients. 27 , 89 Although PEG‐IFN‐α‐2a has not been approved by the FDA or the European Medicine Agency (EMA) for the treatment of chronic HDV infection, PEG‐IFN‐α‐2a 180 μg/wk for 48 weeks has been recommended by several international guidelines. 27 , 89 , 92 , 93 , 94 , 95 , 96
Pegylated‐interferon‐lambda
Efficacy and tolerability of pegylated interferon lambda (PEG‐IFN‐λ) monotherapy or its combination with lonafarnib (prenylation inhibitor) and ritonavir (protease inhibitor and CYP3A4 inhibitor) have been evaluated in two phase 2 clinical trials (NCT02765802 and NCT03600714); IFN‐λ partially shares biochemical pathways with IFN‐α, inducing a common set of downstream genes, but with different kinetics. IFN‐λ showed better tolerability than IFN‐α, which is likely due to the narrower distribution of its receptor. 97
3.4.2. Direct‐acting antivirals
Bulevirtide
Bulevirtide (Hepcludex®; previously Myrcludex®) is a myristoylated synthetic peptide of 47 amino acids derived from the S1 domain of HBsAg that inhibits viral entry by interfering with viral binding to hNTCP, thus avoiding novel infection of hepatocytes. Hepcludex® was given conditional marketing authorisation by EMA in July 2020. Two clinical trials showed that a drug regimen of 2 mg/d is well tolerated and effective in reducing HDV RNA serum levels, alone or in combination with either PEG‐IFN‐α‐2a or tenofovir, but the durability of response was observed in only a small fraction of responders (NCT03546621). 98 , 99 Furthermore, bulevirtide has demonstrated early virological efficacy and safety in a real world‐setting confirming the potential of this new treatment. Further results to demonstrate long‐term clinical benefits will be key in the wider use of bulevirtide. 100 The EMA approved Hepcludex® at a dose of 2 mg subcutaneous per day for the treatment of chronic HDV infection in adult patients with compensated liver disease and positive HDV viremia. The optimal treatment duration has not been determined and treatment should be continued if a clinical benefit is observed with bulevirtide administration. Should the treatment be associated with HBsAg seroconversion for at least 6 months or where sustained virological and biochemical responses are observed, treatment discontinuation could be considered.
At present, bulevirtide efficacy and safety are being assessed in three ongoing phase 2 and phase 3 clinical trials exploring different doses employed alone or in combination with PEG‐IFN‐α‐2a (NCT02888106, NCT03852719 and NCT03852433).
Lonafarnib
Prenylation of the last four C‐terminal aa's of L‐HDAg are known to be necessary for the interaction with HBsAg and virion assembly. 13 , 14 Safety, tolerability, and efficacy of the farnesyltransferase inhibitor, lonafarnib, previously used as an anti‐cancer drug, were assessed during 4 weeks of treatment. Interestingly, in November 2020, the U.S. FDA approved Zokinvy® (lonafarnib) capsules to reduce the risk of death due to Hutchinson‐Gilford progeria syndrome and for the treatment of certain processing‐deficient progeroid laminopathies in patients 1 year of age and older. 101
Preliminary results showed a significant reduction of hepatitis delta viremia, even though virological rebound was observed following treatment cessation. No mutation of L‐HDAg was detected in the non‐responders, and gastrointestinal (GI) side effects, previously reported with lonafarnib treatment for other purposes, were reported. 102 More recent clinical studies have investigated optimal lonafarnib regimens, exploring different doses, combination with PEG‐IFN‐α‐2a, PEG‐IFN‐λ or ritonavir and different treatment durations (NCT02511431, NCT03600714, NCT02430194 and NCT02527707). Low doses of lonafarnib (100 mg twice‐daily) in combination with Ritonavir showed a greater antiviral activity with less GI side effects than lonafarnib monotherapy (up to 300 mg twice‐daily), and similar results were obtained in combination with PEG‐IFN‐α‐2a. 103
Nucleic acid polymers
Nucleic acid polymers (NAPs) have been demonstrated to inhibit HIV‐1 and HCV entry in a sequence‐independent and size‐dependent manner. 104 , 105 Antiviral activity of this class of compound was shown in vitro, having entry‐ and post‐entry‐inhibitor properties. The mechanisms underlying the antiviral activity of NAPs include inhibition of the interaction between HBsAg and HSPG on the cell surface, inhibition of HBsAg release and inhibition of subviral particle assembly; a direct interaction between NAPs and the HDAgs has been hypothesised. 106 , 107 , 108
Targeting HBsAg is a promising strategy to achieve HBV and HDV cure. 109 Safety and efficacy results of 48‐week treatments with two different HBsAg‐targeting NAPs, REP‐2139‐Mg or REP‐2165‐Mg, combined with tenofovir and PEG‐IFN‐α‐2a, were reported in CHB patients 110 with around half of the patients achieving HBsAg loss/HBsAg seroconversion. These promising results need to be confirmed in larger studies. 110 , 111
A first clinical trial using the lead compound REP 2139‐Ca has been completed with a limited number of patients with chronic HDV. 112 , 113 , 114 A follow‐up period of 3.5 years, confirmed the durability of HDV functional cure in seven out of nine responders, four of whom also achieved HBV functional cure after a REP 2139‐Ca and PEG‐IFN‐λ combination treatment. 115 HBsAg loss was often accompanied by ALT normalisation, suggesting an alteration of immune function; a direct immune‐modulatory effect of NAPs has not been confirmed to date, but specific analysis of B and T cell functionality will be introduced in future trials. 116
Even if direct‐acting antivirals likely do not interact with the host immune system, it is believed that they can restore the HBV‐specific antiviral immune responses by lowering the viral antigen load, leading to long‐term recovery of the immune dysfunction in patients with HBV‐HDV. In addition to the described therapeutic targets against HDV, a number of molecules in the quest for HBV functional cure are also in the clinical trial pipeline, including siRNA's, anti‐sense oligonucleotides (ASO's), checkpoint inhibitors, immune stimulators used alone or more likely in combination approaches. 117 , 118 It remains to be determined if these agents potentially offering HBsAg loss and HBV functional cure, can also be employed effectively to treat HDV.
Figure 5 indicates the HDV life cycle and the therapeutic interactions within it.
FIGURE 5.
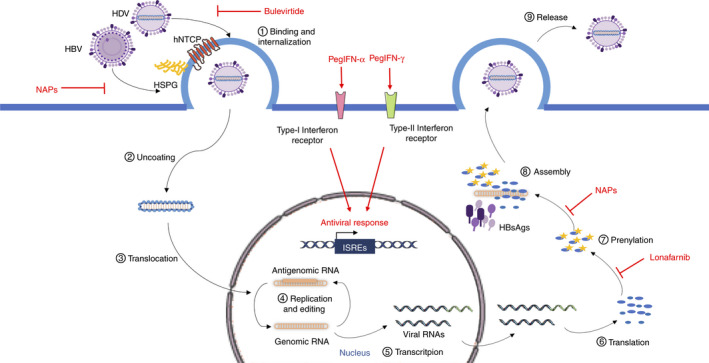
HDV life cycle and drug targets. HDV and HBV entry into the hepatocytes is inhibited by Bulevirtide through competitive binding to hNTCP; NAPs interfere with the first attachment of the virus to the cell surface mediated by HSPG. Lonafarnib inhibits post‐translational modification on L‐HDAg, while NAPs interfere with viral particle assembly, both preventing the production of new virions. Pegylated forms of IFN‐α and IFN‐λ, upon binding with their specific receptor on the cell surface, initiate intracellular signalling cascades leading to the expression of antiviral genes. Parts of the figure were drawn by using pictures from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Commons Attribution 3.0 Unported Licence (https://creativecommons.org/licenses/by/3.0/). HBV, hepatitis B virus; HCV, hepatitis C virus; HDAg, hepatitis delta antigen; HDV, hepatitis delta virus; hNTCP, human Na+/taurocholate co‐transporting polypeptide; HSPG, heparansulphate proteoglycans; IFN, interferon; ISRE, Interferon‐sensitive response element; NAP, nucleic acid polymer
For all new treatment approaches, we need a long‐term clinical response which would include an improvement in survival with a reduction in the development of cirrhosis, decompensation events and the development of HCC. Finally, we can expect a reasonable and achievable short to medium‐term endpoint: virological and biochemical response. While an ideal and more robust clinical endpoint would be HBsAg loss, it is recognised that this will be more challenging to achieve and should be considered a medium to long‐term endpoint. However, it will be equally important to ascertain if novel therapies which can reduce HBsAg levels may also result in restoration of the immune response with better treatment outcomes in HDV. 109
4. CONCLUSIONS
Due to its relatively low frequency with higher prevalence in less economically developed countries, HDV infection has been a neglected disease. The implementation of the prophylactic HBV vaccine remains the best preventative strategy to limit HDV spread, which should be combined with a wider accessibility to reliable diagnostics. The need for widely available standardised and more sensitive diagnostic tools in addition to the fact that many HBV‐positive patients are not screened for HDV, 28 raise the possibility that the actual global HDV prevalence could be significantly underestimated.
The lack of a specific antiviral treatment to date has provided a path for bulevirtide and lonafarnib to receive orphan drug designation from both the EMA and FDA. However, since the best preliminary results to date were obtained when a direct‐acting antiviral (bulevirtide or lonafarnib) were combined with an immune‐modulator, PEG IFN is likely to have a continued role in HDV management until more effective and well‐tolerated immune modulators become available. Moreover, network meta‐analysis will be required to assess the safety and efficacy of bulevirtide and other antivirals used alone or in combination with PEG‐IFN‐α‐2a in comparison with PEG‐IFN‐α‐2a monotherapy.
This review highlights that our understanding of HDV pathogenesis is limited by access to HDV‐infected liver tissue. In particular, important gaps in our knowledge concern the proportion of HBV‐positive hepatocytes also infected by HDV, the relevance of integrated HBV DNA in HDV co‐infection and super‐infection, and the extent of HDV direct toxicity in the liver. We believe a more comprehensive analysis of the intrahepatic compartment is necessary to determine the presence of liver‐specific immune cells or any liver‐confined effect of HDV on the host immune response, given the partially contradictory observations reported between immune cell phenotype, viral load and the degree of liver injury observed in HDV patients. This remains a major unmet need and requires further investigation in tandem with the exploration of new therapies to manage HDV infection, while simultaneously exploring the efficacy of novel HBV therapies in the management of HDV.
AUTHORSHIP
Guarantor if the article: Patrick T. Kennedy.
Author contributions: CU, USG and ACR: literature research; CU and USG: figures; CU: draft of the manuscript; USG, TA and PK: critical review of the manuscript; PK: guarantor of the article. All authors contributed to and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
Declaration of personal interests: Tarik Asselah has acted as a speaker and/or advisor board and/or investigator for Abbvie, Eiger Biopharmaceutical, Janssen, Gilead, Myr Pharmaceutical, Roche, and Merck. Patrick Kennedy has served as a speaker, a consultant/advisory board member for Aligos, Antios Therapeutics, Assembly Biosciences, Gilead Sciences, Janssen, GlaxoSmithKline, Immunocore and Drug Farm, and has received research funding from Gilead Sciences.
Declaration of funding interests: None.
Usai C, Gill US, Riddell AC, Asselah T, Kennedy PT. Review article: emerging insights into the immunopathology, clinical and therapeutic aspects of hepatitis delta virus. Aliment Pharmacol Ther. 2022;55:978–993. doi: 10.1111/apt.16807
The Handling Editor for this article was Professor Geoffrey Dusheiko, and this uncommissioned review was accepted for publication after full peer‐review.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Stockdale AJ, Kreuels B, Henrion MYR, et al. The global prevalence of hepatitis D virus infection: systematic review and meta‐analysis. J Hepatol. 2020;73:523‐532. doi: 10.1016/j.jhep.2020.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rizzetto M, Hoyer B, Canese MG, Shih JW, Purcell RH, Gerin JL. Delta agent: association of delta antigen with hepatitis B surface antigen and RNA in serum of delta‐infected chimpanzees. Proc Natl Acad Sci USA. 1980;77:6124‐6128. doi: 10.1073/pnas.77.10.6124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen PJ, Kalpana G, Goldberg J, et al. Structure and replication of the genome of the hepatitis delta virus. Proc Natl Acad Sci USA. 1986;83:8774‐8778. doi: 10.1073/pnas.83.22.8774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rizzetto M, Canese MG, Aricò S, et al. Immunofluorescence detection of new antigen‐antibody system (delta/anti‐delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18:997‐1003. doi: 10.1136/gut.18.12.997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greco‐Stewart VS, Miron P, Abrahem A, Pelchat M. The human RNA polymerase II interacts with the terminal stem–loop regions of the hepatitis delta virus RNA genome. Virology. 2007;357:68‐78. doi: 10.1016/j.virol.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 6. Kuo MY‐P, Sharmeen L, Dinter‐Gottlieb G, Taylor J. Characterization of self‐cleaving RNA sequences on the genome and antigenome of human Hepatitis Delta virus. J Virol. 1988;62:4439‐4444. http://jvi.asm.org/content/62/12/4439.full.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang KS, Choo QL, Weiner AJ, et al. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature. 1986;323:508‐514. doi: 10.1038/323508a0 [DOI] [PubMed] [Google Scholar]
- 8. Casey JL, Gerin JL. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J Virol. 1995;69:7593‐7600. doi: 10.1128/jvi.69.12.7593-7600.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong SK, Lazinski DW. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc Natl Acad Sci USA. 2002;99:15118‐15123. doi: 10.1073/pnas.232416799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chao M, Hsieh SY, Taylor J. Role of two forms of hepatitis delta virus antigen: evidence for a mechanism of self‐limiting genome replication. J Virol. 1990;64:5066‐5069. doi: 10.1128/JVI.64.10.5066-5069.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee CH, Chang SC, Wu CHH, Chang MF. A novel chromosome region maintenance 1‐independent nuclear export signal of the large form of Hepatitis Delta antigen that is required for the viral assembly. J Biol Chem. 2001;276:8142‐8148. doi: 10.1074/jbc.M004477200 [DOI] [PubMed] [Google Scholar]
- 12. Sato S, Cornillez‐Ty C, Lazinski DW. By inhibiting replication, the large Hepatitis Delta antigen can indirectly regulate Amber/W editing and its own expression. J Virol. 2004;78:8120‐8134. doi: 10.1128/jvi.78.15.8120-8134.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Malley B, Lazinski DW. Roles of carboxyl‐terminal and farnesylated residues in the functions of the large hepatitis delta antigen. J Virol. 2005;79:1142‐1153. doi: 10.1128/JVI.79.2.1142-1153.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glenn JS, Watson JA, Havel CM, White JM. Identification of a prenylation site in delta virus large antigen. Science. 1992;256:1331‐1333. doi: 10.1126/science.1598578 [DOI] [PubMed] [Google Scholar]
- 15. Bonino F, Heermann KH, Rizzetto M, Gerlich WH. Hepatitis Delta virus: protein composition of Delta antigen and its hepatitis B virus‐derived envelope. J Virol. 1986;58:945‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. elife. 2012;1:e00049. doi: 10.7554/eLife.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ni Y, Lempp FA, Mehrle S, et al. Hepatitis B and D viruses exploit sodium taurocholate co‐transporting polypeptide for species‐specific entry into hepatocytes. Gastroenterology. 2014;146:1070‐1083.e6. doi: 10.1053/j.gastro.2013.12.024 [DOI] [PubMed] [Google Scholar]
- 18. Perez‐Vargas J, Amirache F, Boson B, et al. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat Commun. 2019;10:2098. doi: 10.1038/s41467-019-10117-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pflüger LS, Schulze zur Wiesch J, Polywka S, Lütgehetmann M. Hepatitis delta virus propagation enabled by hepatitis C virus—scientifically intriguing, but is it relevant to clinical practice? J Viral Hepat. 2021;28:213‐216. doi: 10.1111/jvh.13385 [DOI] [PubMed] [Google Scholar]
- 20. Chemin I, Pujol FH, Scholtès C, et al. Preliminary evidence for Hepatitis Delta virus exposure in patients who are apparently not infected with hepatitis B virus. Hepatology. 2021;73:861‐864. doi: 10.1002/hep.31453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giersch K, Bhadra OD, Volz T, et al. Hepatitis delta virus persists during liver regeneration and is amplified through cell division both in vitro and in vivo. Gut. 2019;68:150‐157. doi: 10.1136/gutjnl-2017-314713 [DOI] [PubMed] [Google Scholar]
- 22. Le Gal F, Gault E, Ripault MP, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis. 2006;12:1447‐1450. doi: 10.3201/eid1209.060112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wranke A, Pinheiro Borzacov LM, Parana R, et al. Clinical and virological heterogeneity of Hepatitis Delta in different regions world‐wide: the Hepatitis Delta international network (HDIN). Liver Int. 2018;38:842‐850. doi: 10.1111/liv.13604 [DOI] [PubMed] [Google Scholar]
- 24. Roulot D, Brichler S, Layese R, et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic Hepatitis Delta. J Hepatol. 2020;73:1046‐1062. doi: 10.1016/j.jhep.2020.06.038 [DOI] [PubMed] [Google Scholar]
- 25. Nguyen HM, Sy BT, Trung NT, et al. Prevalence and genotype distribution of hepatitis delta virus among chronic hepatitis B carriers in Central Vietnam. PLoS One. 2017;12:1‐15. doi: 10.1371/journal.pone.0175304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Melo Da Silva E, Kay A, Lobato C, et al. Non‐F HBV/HDV‐3 coinfection is associated with severe liver disease in Western Brazilian Amazon. J Med Virol. 2019;91:1081‐1086. doi: 10.1002/jmv.25411 [DOI] [PubMed] [Google Scholar]
- 27. Terrault NA, Lok ASF, McMahon BJ, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67:1560‐1599. doi: 10.1002/hep.29800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. World Health Organization . Global Hepatitis Report, 2017. World Health Organization; 2017. https://www.who.int/hepatitis/publications/global‐hepatitis‐report2017/en/. . [Google Scholar]
- 29. Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein‐dependent binding to heparan sulfate proteoglycans. Hepatology. 2007;46:1759‐1768. doi: 10.1002/hep.21896 [DOI] [PubMed] [Google Scholar]
- 30. Dandri M, Burda MR, Török E, et al. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology. 2001;33:981‐988. doi: 10.1053/jhep.2001.23314 [DOI] [PubMed] [Google Scholar]
- 31. Kremsdorf D, Strick‐Marchand H. Modeling hepatitis virus infections and treatment strategies in humanized mice. Curr Opin Virol. 2017;25:119‐125. doi: 10.1016/j.coviro.2017.07.029 [DOI] [PubMed] [Google Scholar]
- 32. He W, Ren B, Mao F, et al. Hepatitis D virus infection of mice expressing human sodium taurocholate co‐transporting polypeptide. PLOS Pathog. 2015;11:e1004840. doi: 10.1371/journal.ppat.1004840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. He W, Cao Z, Mao F, et al. Modification of three amino acids in sodium taurocholate Cotransporting polypeptide renders mice susceptible to infection with hepatitis D virus in vivo. J Virol. 2016;90:8866‐8874. doi: 10.1128/JVI.00901-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang YH, Fang CC, Tsuneyama K, et al. A murine model of hepatitis B‐associated hepatocellular carcinoma generated by adeno‐associated virus‐mediated gene delivery. Int J Oncol. 2011;39:1511‐1519. doi: 10.3892/ijo.2011.1145 [DOI] [PubMed] [Google Scholar]
- 35. Lucifora J, Salvetti A, Marniquet X, et al. Detection of the hepatitis B virus ( HBV ) covalently‐closed‐circular DNA ( cccDNA ) in mice transduced with a recombinant AAV‐HBV vector. Antivir Res. 2017;145:14‐19. doi: 10.1016/j.antiviral.2017.07.006 [DOI] [PubMed] [Google Scholar]
- 36. Suárez‐Amarán L, Usai C, Di Scala M, et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN‐β induction. J Hepatol. 2017;67:669‐679. doi: 10.1016/j.jhep.2017.05.010 [DOI] [PubMed] [Google Scholar]
- 37. Usai C, Maestro S, Camps G, et al. TNF‐alpha inhibition ameliorates HDV‐induced liver damage in a mouse model of acute severe infection. JHEP Rep. 2020;3:100098. doi: 10.1016/j.jhepr.2020.100098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Winer BY, Shirvani‐Dastgerdi E, Bram Y, et al. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for hepatitis delta virus infection. Sci Transl Med. 2018;10:1‐11. doi: 10.1126/scitranslmed.aap9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aslan N, Yurdaydin C, Wiegand J, et al. Cytotoxic CD4+ T cells in viral hepatitis. J Viral Hepat. 2006;13:505‐514. doi: 10.1111/j.1365-2893.2006.00723.x [DOI] [PubMed] [Google Scholar]
- 40. Nisini R, Paroli M, Accapezzato D, et al. Human CD4+ T‐cell response to hepatitis delta virus: identification of multiple epitopes and characterization of T‐helper cytokine profiles. J Virol. 1997;71:2241‐2251. doi: 10.1128/JVI.71.3.2241-2251.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang Y‐H. Identification of novel HLA‐A*0201‐restricted CD8+ T‐cell epitopes on hepatitis delta virus. J Gen Virol. 2004;85:3089‐3098. doi: 10.1099/vir.0.80183-0 [DOI] [PubMed] [Google Scholar]
- 42. Landahl J, Bockmann JH, Scheurich C, et al. Detection of a broad range of Low‐level major histocompatibility complex class II–restricted, Hepatitis Delta virus (HDV)–specific T‐cell responses regardless of clinical status. J Infect Dis. 2019;219:568‐577. doi: 10.1093/infdis/jiy549 [DOI] [PubMed] [Google Scholar]
- 43. Karimzadeh H, Kiraithe MM, Oberhardt V, et al. Mutations in hepatitis D virus allow it to escape detection by CD8+ T cells and evolve at the population level. Gastroenterology. 2019;156:1820‐1833. doi: 10.1053/j.gastro.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schirdewahn T, Grabowski J, Owusu Sekyere S, et al. The third signal cytokine interleukin 12 rather than immune checkpoint inhibitors contributes to the functional restoration of hepatitis D virus–specific T cells. J Infect Dis 2017;215:139–149. doi: 10.1093/infdis/jiw514 [DOI] [PubMed] [Google Scholar]
- 45. Grabowski J, Yurdaydìn C, Zachou K, et al. Hepatitis D virus‐specific cytokine responses in patients with chronic hepatitis delta before and during interferon alfa‐treatment. Liver Int. 2011;31:1395‐1405. doi: 10.1111/j.1478-3231.2011.02593.x [DOI] [PubMed] [Google Scholar]
- 46. Kefalakes H, Koh C, Sidney J, et al. Hepatitis D virus‐specific CD8+ T cells have a memory‐like phenotype associated with viral immune escape in patients with chronic hepatitis D virus infection. Gastroenterology. 2019;156:1805‐1819.e9. doi: 10.1053/j.gastro.2019.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kefalakes H, Horgan X, Jung M, et al. Liver‐resident bystander CD8+ T cells contribute to liver disease pathogenesis in chronic hepatitis D virus infection. Gastroenterology July 2021. doi: 10.1053/j.gastro.2021.07.027, 161, 1567, 1583.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lunemann S, Malone DFG, Hengst J, et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J Infect Dis. 2014;209:1362‐1373. doi: 10.1093/infdis/jit561 [DOI] [PubMed] [Google Scholar]
- 49. Lunemann S, Malone DFGG, Grabowski J, et al. Effects of HDV infection and pegylated interferon α treatment on the natural killer cell compartment in chronically infected individuals. Gut. 2015;64:469‐482. doi: 10.1136/gutjnl-2014-306767 [DOI] [PubMed] [Google Scholar]
- 50. Dias J, Hengst J, Parrot T, et al. Chronic hepatitis delta virus infection leads to functional impairment and severe loss of MAIT cells. J Hepatol. 2019;71:301‐312. doi: 10.1016/j.jhep.2019.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ishak KG. Pathologic features of chronic hepatitis. A review and update. Am J Clin Pathol. 2000;113:40‐55. doi: 10.1309/42D6-W7PL-FX0A-LBXF [DOI] [PubMed] [Google Scholar]
- 52. Norris S, Collins C, Doherty DG, et al. Resident human hepatic lymphocytes are phenotypically different from circulating lymphocytes. J Hepatol. 1998;28:84‐90. doi: 10.1016/S0168-8278(98)80206-7 [DOI] [PubMed] [Google Scholar]
- 53. Wang Y, Zhang C. The roles of liver‐resident lymphocytes in liver diseases. Front Immunol. 2019;10:1‐13. doi: 10.3389/fimmu.2019.01582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boeijen LL, van Oord GW, Hou J, et al. Gene expression profiling of human tissue‐resident immune cells: comparing blood and liver. J Leukoc Biol. 2019;105:603‐608. doi: 10.1002/JLB.6AB0718-278R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gill US, Pallett LJ, Kennedy PTF, Maini MK. Liver sampling: a vital window into HBV pathogenesis on the path to functional cure. Gut. 2018;67:767‐775. doi: 10.1136/gutjnl-2017-314873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sprengers D, Van Der Molen RG, Kusters JG, et al. Flow cytometry of fine‐needle‐aspiration biopsies: a new method to monitor the intrahepatic immunological environment in chronic viral hepatitis. J Viral Hepat. 2005;12:507‐512. doi: 10.1111/j.1365-2893.2005.00626.x [DOI] [PubMed] [Google Scholar]
- 57. Pembroke T, Gallimore A, Godkin A. Tracking the kinetics of intrahepatic immune responses by repeated fine needle aspiration of the liver. J Immunol Methods. 2015;424:131‐135. doi: 10.1016/j.jim.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pembroke T, Christian A, Jones E, et al. The paradox of NKp46+ natural killer cells: drivers of severe hepatitis C virus‐induced pathology but in‐vivo resistance to interferon α treatment. Gut. 2014;63:515‐524. doi: 10.1136/gutjnl-2013-304472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spaan M, Van Oord GW, Janssen HLA, De Knegt RJ, Boonstra A. Longitudinal analysis of peripheral and intrahepatic NK cells in chronic HCV patients during antiviral therapy. Antivir Res. 2015;123:86‐92. doi: 10.1016/j.antiviral.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 60. van der Laan LJW, Taimr P, Kok A, et al. Flowcytometric quantitation of hepatitis B viral antigens in hepatocytes from regular and fine‐needle biopsies. J Virol Methods. 2007;142:189‐197. doi: 10.1016/j.jviromet.2007.01.027 [DOI] [PubMed] [Google Scholar]
- 61. Pallett LJ, Davies J, Colbeck EJ, et al. IL‐2 high tissue‐resident T cells in the human liver: sentinels for hepatotropic infection. J Exp Med. 2017;214:1567‐1580. doi: 10.1084/jem.20162115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stegmann KA, Robertson F, Hansi N, et al. CXCR6 marks a novel subset of T‐bet lo Eomes hi natural killer cells residing in human liver. Sci Rep. 2016;6:1‐10. doi: 10.1038/srep26157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gill U S, Pallett LJ, Thomas N, et al. Fine needle aspirates comprehensively sample intrahepatic immunity. Gut. 2019;68:1493‐1503. 10.1136/gutjnl-2018-317071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Farci P, Niro G. Clinical features of hepatitis D. Semin Liver Dis. 2012;32:228‐236. doi: 10.1055/s-0032-1323628 [DOI] [PubMed] [Google Scholar]
- 65. Negro F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb Perspect Med. 2014;4:a021550. doi: 10.1101/cshperspect.a021550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Farci P, Gerin JL, Aragona M, et al. Diagnostic and prognostic significance of the IgM antibody to the Hepatitis Delta virus. J Am Med Assoc. 1986;255:1443‐1446. doi: 10.1001/jama.1986.03370110065022 [DOI] [PubMed] [Google Scholar]
- 67. Wranke A, Heidrich B, Ernst S, et al. Anti‐HDV IgM as a marker of disease activity in hepatitis delta. PLoS One. 2014;9:1‐13. doi: 10.1371/journal.pone.0101002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Heidrich B, Serrano BC, Idilman R, et al. HBeAg‐positive hepatitis delta: virological patterns and clinical long‐term outcome. Liver Int. 2012;32:1415‐1425. doi: 10.1111/j.1478-3231.2012.02831.x [DOI] [PubMed] [Google Scholar]
- 69. Stelzl E, Ciesek S, Cornberg M, et al. Reliable quantification of plasma HDV RNA is of paramount importance for treatment monitoring: a European multicenter study. J Clin Virol. 2021;142:104932. doi: 10.1016/J.JCV.2021.104932 [DOI] [PubMed] [Google Scholar]
- 70. Gençdal G, Zeybel M, Yurdaydin C. Editorial: natural history of hepatitis delta virus‐induced liver disease—less severe today but still needs attention. Aliment Pharmacol Ther. 2021;54:519‐520. doi: 10.1111/apt.16527 [DOI] [PubMed] [Google Scholar]
- 71. Palom A, Sopena S, Riveiro‐Barciela M, et al. One‐quarter of chronic hepatitis D patients reach HDV‐RNA decline or undetectability during the natural course of the disease. Aliment Pharmacol Ther. 2021;54:462‐469. doi: 10.1111/apt.16485 [DOI] [PubMed] [Google Scholar]
- 72. Zachou K, Yurdaydin C, Drebber U, et al. Quantitative HBsAg and HDV‐RNA levels in chronic delta hepatitis. Liver Int. 2010;30:430‐437. doi: 10.1111/j.1478-3231.2009.02140.x [DOI] [PubMed] [Google Scholar]
- 73. Bang BR, Elmasry S, Saito T. Organ system view of the hepatic innate immunity in HCV infection. J Med Virol. 2016;88:2025‐2037. doi: 10.1002/jmv.24569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bocher WO, Herzog‐HauffF S, Herr W, et al. Regulation of the neutralizing anti‐hepatitis B surface (HBs) antibody response in vitro in HBs vaccine recipients and patients with acute or chronic hepatitis B virus (HBV) infection. Clin Exp Immunol. 1996;105:52‐58. doi: 10.1046/j.1365-2249.1996.d01-732.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cole SM, Gowans EJ, Macnaughton TB, De La M, Hall P, Burrell CJ. Direct evidence for cytotoxicity associated with expression of hepatitis delta virus antigen. Hepatology. 1991;13:845‐851. doi: 10.1002/hep.1840130508 [DOI] [PubMed] [Google Scholar]
- 76. Popper H, Thung SN, Gerber MA, et al. Histologic studies of Severe Delta agent infection in Venezuelan Indians. Hepatology. 1983;3:906‐912. doi: 10.1002/hep.1840030603 [DOI] [PubMed] [Google Scholar]
- 77. Choi SH, Jeong SH, Hwang SB. Large Hepatitis Delta antigen modulates transforming growth factor‐β signaling cascades: implication of Hepatitis Delta virus‐induced liver fibrosis. Gastroenterology. 2007;132:343‐357. doi: 10.1053/j.gastro.2006.10.038 [DOI] [PubMed] [Google Scholar]
- 78. Morante AL, De La Cruz F, De Lope CR, Echevarria S, Rodriguez GM, Pons‐Romero F. Hepatitis B virus replication in hepatitis B and D coinfection. Liver. 1989;9:65‐70. doi: 10.1111/j.1600-0676.1989.tb00381.x [DOI] [PubMed] [Google Scholar]
- 79. Pollicino T, Raffa G, Santantonio T, et al. Replicative and transcriptional activities of hepatitis B virus in patients coinfected with hepatitis B and Hepatitis Delta viruses. J Virol. 2011;85:432‐439. doi: 10.1128/JVI.01609-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Giersch K, Dandri M. Hepatitis B and Delta virus: advances on studies about interactions between the two viruses and the infected hepatocyte. J Clin Transl Hepatol. 2015;3:220‐229. doi: 10.14218/JCTH.2015.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ricco G, Popa DC, Cavallone D, et al. Quantification of serum markers of hepatitis B (HBV) and Delta virus (HDV) infections in patients with chronic HDV infection. J Viral Hepat. 2018;25:911‐919. doi: 10.1111/jvh.12895 [DOI] [PubMed] [Google Scholar]
- 82. Stroffolini T, Sagnelli E, Sagnelli C, et al. Hepatitis delta infection in Italian patients: towards the end of the story? Infection. 2017;45:277‐281. doi: 10.1007/s15010-016-0956-1 [DOI] [PubMed] [Google Scholar]
- 83. Rizzetto M, Hamid S, Negro F. The changing context of hepatitis D. J Hepatol. 2021;74:1200‐1211. doi: 10.1016/j.jhep.2021.01.014 [DOI] [PubMed] [Google Scholar]
- 84. Yen L. Identification of inhibitors of ribozyme self‐cleavage in mammalian cells via high‐throughput screening of chemical libraries. RNA. 2006;12:797‐806. doi: 10.1261/rna.2300406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chang J, Taylor JM. Susceptibility of human Hepatitis Delta virus RNAs to small interfering RNA action. J Virol. 2003;77:9728‐9731. doi: 10.1128/JVI.77.17.9728-9731.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Loureiro D, Castelnau C, Tout I, et al. New therapies for hepatitis delta virus infection. Liver Int. 2021;41:30‐37. doi: 10.1111/liv.14838 [DOI] [PubMed] [Google Scholar]
- 87. Chronic Hepatitis D Virus Infection: Developing Drugs for Treatment Guidance for Industry. FDA. 2019; https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/chronic‐hepatitis‐d‐virus‐infection‐developing‐drugs‐treatment‐guidance‐industry . [Google Scholar]
- 88. Le Gal F, Brichler S, Sahli R, et al. First international external quality assessment for hepatitis delta virus RNA quantification in plasma. Hepatology. 2016;64:1483‐1494. doi: 10.1002/hep.28772 [DOI] [PubMed] [Google Scholar]
- 89. European Association for the Study of the Liver . EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67:370‐398. [DOI] [PubMed] [Google Scholar]
- 90. Hercun J, Kim GE, Da BL, et al. Durable virological response and functional cure of chronic hepatitis D after long‐term peginterferon therapy. Aliment Pharmacol Ther. 2021;54:176‐182. doi: 10.1111/apt.16408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Farci P, Roskams T, Chessa L, et al. Long‐term benefit of interferon α therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology. 2004;126:1740‐1749. doi: 10.1053/J.GASTRO.2004.03.017 [DOI] [PubMed] [Google Scholar]
- 92. Sarin SK, Kumar M, Lau GK, et al. Asian‐Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int. 2016;10:1‐98. doi: 10.1007/s12072-015-9675-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wedemeyer H, Yurdaydìn C, Dalekos GN, et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med. 2011;364:322‐331. doi: 10.1056/NEJMoa0912696 [DOI] [PubMed] [Google Scholar]
- 94. Heidrich B, Yurdaydın C, Kabaçam G, et al. Late HDV RNA relapse after peginterferon alpha‐based therapy of chronic hepatitis delta. Hepatology. 2014;60:87‐97. doi: 10.1002/hep.27102 [DOI] [PubMed] [Google Scholar]
- 95. Heller T, Rotman Y, Koh C, et al. Long‐term therapy of chronic delta hepatitis with peginterferon alfa. Aliment Pharmacol Ther. 2014;40:93‐104. doi: 10.1111/apt.12788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real‐world experience. Antivir Ther. 2014;19:463‐468. doi: 10.3851/IMP2728 [DOI] [PubMed] [Google Scholar]
- 97. Chan HLY, Ahn SH, Chang TT, et al. Peginterferon lambda for the treatment of HBeAg‐positive chronic hepatitis B: a randomized phase 2b study (LIRA‐B). J Hepatol. 2016;64:1011‐1019. doi: 10.1016/j.jhep.2015.12.018 [DOI] [PubMed] [Google Scholar]
- 98. Wedemeyer H, Bogomolov P, Blank A, et al. Final results of a multicenter, open‐label phase 2b clinical trial to assess safety and efficacy of Myrcludex B in combination with tenofovir in patients with chronic HBV/HDV co‐infection. J Hepatol. 2018;68:S3. doi: 10.1016/s0168-8278(18)30224-1 [DOI] [Google Scholar]
- 99. Bogomolov P, Alexandrov A, Voronkova N, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: first results of a phase Ib/IIa study. J Hepatol. 2016;65:490‐498. doi: 10.1016/j.jhep.2016.04.016 [DOI] [PubMed] [Google Scholar]
- 100. Asselah T, Loureiro D, Le Gal F, et al. Early virological response in six patients with hepatitis D virus infection and compensated cirrhosis treated with Bulevirtide in real‐life. Liver Int. 2021;41:1509‐1517. doi: 10.1111/liv.14950 [DOI] [PubMed] [Google Scholar]
- 101. FDA Approves First Treatment for Hutchinson‐Gilford Progeria Syndrome and some Progeroid Laminopathies. FDA. 2020; https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐first‐treatment‐hutchinson‐gilford‐progeria‐syndrome‐and‐some‐progeroid‐laminopathies . [Google Scholar]
- 102. Koh C, Canini L, Dahari H, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof‐of‐concept randomised, double‐blind, placebo‐controlled phase 2A trial. Lancet Infect Dis. 2015;15:1167‐1174. doi: 10.1016/S1473-3099(15)00074-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yurdaydin C, Keskin O, Kalkan Ç, et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: the LOWR HDV‐1 study. Hepatology. 2018;67:1224‐1236. doi: 10.1002/hep.29658 [DOI] [PubMed] [Google Scholar]
- 104. Matsumura T, Hu Z, Kato T, et al. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology. 2009;137:673‐681. doi: 10.1053/j.gastro.2009.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vaillant A, Juteau JM, Lu H, et al. Phosphorothioate oligonucleotides inhibit human immunodeficiency virus type 1 fusion by blocking gp41 core formation. Antimicrob Agents Chemother. 2006;50:1393‐1401. doi: 10.1128/AAC.50.4.1393-1401.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Beilstein F, Blanchet M, Vaillant A, Sureau C. Nucleic acid polymers are active against Hepatitis Delta virus infection in vitro. J Virol. 2018;92:e01416‐e01417. doi: 10.1128/JVI.01416-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Blanchet M, Sinnathamby V, Vaillant A, Labonté P. Inhibition of HBsAg secretion by nucleic acid polymers in HepG2.2.15 cells. Antivir Res. 2019;164:97‐105. doi: 10.1016/j.antiviral.2019.02.009 [DOI] [PubMed] [Google Scholar]
- 108. Shamur MM, Peri‐naor R, Mayer R, Vaillant A. Interaction of nucleic acid polymers with the large and small forms of the hepatitis delta antigen protein. Hepatol AASLD Abstr. 2017;66:504‐505. [Google Scholar]
- 109. Tout I, Loureiro D, Mansouri A, Soumelis V, Boyer N, Asselah T. Hepatitis B surface antigen seroclearance: immune mechanisms, clinical impact, importance for drug development. J Hepatol. 2020;73:409‐422. doi: 10.1016/J.JHEP.2020.04.013 [DOI] [PubMed] [Google Scholar]
- 110. Bazinet M, Pântea V, Placinta G, et al. Safety and efficacy of 48 weeks REP 2139 or REP 2165, tenofovir disoproxil, and pegylated interferon alfa‐2a in patients with chronic HBV infection Naïve to Nucleos(t)ide therapy. Gastroenterology. 2020;158:2180‐2194. doi: 10.1053/J.GASTRO.2020.02.058 [DOI] [PubMed] [Google Scholar]
- 111. Durantel D, Asselah T. Nucleic acid polymers are effective in targeting hepatitis B surface antigen, but more trials are needed. Gastroenterology. 2020;158:2051‐2054. doi: 10.1053/J.GASTRO.2020.04.020 [DOI] [PubMed] [Google Scholar]
- 112. Al‐Mahtab M, Bazinet M, Vaillant A. Safety and efficacy of nucleic acid polymers in monotherapy and combined with immunotherapy in treatment‐naive Bangladeshi patients with HBeAg+ chronic hepatitis B infection. PLoS One. 2016;11:e0156667. doi: 10.1371/journal.pone.0156667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Guillot C, Hantz O, Vaillant A, Chemin I. Antiviral effects of nucleic acid polymers on hepatitis B virus infection. J Hepatol. 2015;62:S523. doi: 10.1016/S0168-8278(15)30763-7 [DOI] [Google Scholar]
- 114. Bazinet M, Pântea V, Cebotarescu V, et al. Safety and efficacy of REP 2139 and pegylated interferon alfa‐2a for treatment‐naive patients with chronic hepatitis B virus and hepatitis D virus co‐infection (REP 301 and REP 301‐LTF): a non‐randomised, open‐label, phase 2 trial. Lancet Gastroenterol Hepatol. 2017;2:877‐889. doi: 10.1016/S2468-1253(17)30288-1 [DOI] [PubMed] [Google Scholar]
- 115. Bazinet M, Pântea V, Cebotarescu V, et al. Persistent control of hepatitis B virus and Hepatitis Delta virus infection following REP 2139‐ca and pegylated interferon therapy in chronic hepatitis B virus/Hepatitis Delta virus coinfection. Hepatol Commun. 2021;5:189‐202. doi: 10.1002/hep4.1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vaillant A. REP 2139: antiviral mechanisms and applications in achieving functional control of HBV and HDV infection. ACS Infect Dis. 2019;5:675‐687. doi: 10.1021/acsinfecdis.8b00156 [DOI] [PubMed] [Google Scholar]
- 117. Gill US, Kennedy PTF. The impact of currently licensed therapies on viral and immune responses in chronic hepatitis B: considerations for future novel therapeutics. J Viral Hepat. 2019;26:4‐15. doi: 10.1111/jvh.13040 [DOI] [PubMed] [Google Scholar]
- 118. Sandmann L, Cornberg M. Experimental drugs for the treatment of hepatitis D. J Exp Pharmacol. 2021;13:461‐468. doi: 10.2147/JEP.S235550 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.